 Can Jin
Can Jin Jin-Xin Ma
Jin-Xin Ma Hao Wang
Hao Wang Jing Si
Jing Si- Institute of Microbiology, School of Ecology and Nature Conservation, Beijing Forestry University, Beijing, China
Sanghuangporus, also known as “Sanghuang” in China, is a well-known genus of traditional Chinese medicinal macrofungi. To make more effective use of Sanghuangporus resources, we completed the first genome assembly and annotation of a monokaryon strain of S. weigelae in the present study. A 33.96-Mb genome sequence was assembled as 13 contigs, leading to prediction of 9377 protein-coding genes. Phylogenetic and average nucleotide identity analyses indicated that the S. weigelae genome is closely related to those of other Sanghuangporus species in evolutionary tree, which clustered in one clade. Collinearity analysis revealed a high level of collinearity of S. weigelae with S. baumii, S. vaninii, and S. sanghuang. Biosynthesis pathways potentially involved in medicinal properties, including terpenoid and polysaccharide synthesis, were identified in S. weigelae, while polysaccharides were identified as the main medicinal metabolites in S. weigelae, with flavonoids more important in Sanghuangporus than other medicinal mushroom groups. Genes encoding 332 carbohydrate-active enzymes were identified in the S. weigelae genome, including major glycoside hydrolases and glycosyltransferases predicted, revealing the robust lignocellulose degradation capacity of S. weigelae. Further, 130 genes, clustered in seven classes were annotated to encode cytochromes P450 in the S. weigelae genome. Overall, our results reveal the remarkably medicinal capacity of S. weigelae and provide new insights that will inform the study of evolution and medicinal application of S. weigelae. The data are a reference resource for the formulation of scientific and rational ecological protection policies for Sanghuangporus species.
1 Introduction
Sanghuangporus is a traditional Chinese medicine that has been used in China to prevent and treat diseases for thousands of years (Dai and Cui, 2014). The long history of consumption and medicinal use of Sanghuangporus has brought health benefits for Chinese people and led to development of a unique traditional Chinese medicine culture surroundings Sanghuangporus, in the ancient ways of the Yellow River (Zhou et al., 2016). As a valuable class of medicinal mushroom, Sanghuangporus species are comparable with the famous Chinese medicine “DongchongXiacao” (Ophiocordyceps sinensis) (Wu et al., 2016). In the Compendium of Materia Medica compiled by Li Shizhen, it is recorded in Chinese that “Sanghuang” can “Li wu zang, Xuan chang qi”, meaning that it can benefit the five internal organs of the body (heart, spleen, liver, lungs, and kidneys) and promote gastrointestinal function (Huang et al., 2023). Separate from Chinese traditional medicine, Sanghuangporus species have been shown to be effective for treating neurodegenerative diseases, such as Parkinson’s and Alzheimer’s (Fang et al., 2020). Further, there is evidence that Sanghuangporus mycelia secrete high levels of bioactive substances, including polysaccharides, flavonoids, polyphenols, pyrones, terpenes, proteins, lipids, alkaloids, minerals, vitamins, and other substances (Wang X. T., et al., 2023). These bioactive compounds have prophylactic and therapeutic benefits, including tumor suppression, antioxidant, bacteriostatic, anti-inflammatory, blood glucose, liver protection, and immune regulation properties (Cai et al., 2019; Wei et al., 2023).
In recent years, the utility of traditional Chinese medicine, and Sanghuangporus in particular, has attracted increasing attention in spheres from folklore to scientific and industrial fields (Zheng et al., 2023). There are 18 known species of Sanghuangporus, ten of which are widely distributed in China, including S. weigelae, which mainly grows in subtropical warm temperate regions, at the base of or inside wood; the fruiting body is usually flat and inverted, with a light brown surface, and small amounts are available for sale on the market (Wu et al., 2016). S. weigelae ranks the second among eight Sanghuangporus strains with the highest polysaccharide yields (Wang H. et al., 2023), along with species such as S. sanghuang, S. vaninii, and S. baumii. Further, S. weigelae has superior 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical scavenging ability, triterpenoid content, and ascorbic acid content. These results are consistent with those of an important study (Meng et al., 2016), demonstrating that, in liquid culture, S. weigelae has significantly higher capacity to generate polyphenols, malonaldehyde content, superoxide dismutase activity, total antioxidant capacity, and DPPH radical scavenging ability than that of Perenniporia robiniophila, as well as slightly higher capacity than S. sanghuang. Hence, S. weigelae is undoubtedly an important traditional Chinese medicinal resource.
With advances in DNA sequencing technologies, medicinal studies of wood-inhabiting macrofungi have gradually generated genome sequences, in addition to data on the metabolites produced (Kiss et al., 2019). Comparison of fungal genomes can reveal important information on the production of various bioactive compounds and metabolic pathways (Min et al., 2018). Genome assemblies of several Sanghuangporus mushrooms have recently been reported, including S. sanghuang (Jiang et al., 2021), S. vaninii (Song et al., 2021), and S. baumii, which are closely related to S. weigelae, and have generated several scaffold-scale genome sequences. Other medicinal mushrooms, such as Ganoderma sinense, Pleurotus giganteus, and Oudemansiella raphanipes, have been also subjected to whole genome sequencing. This sequencing and analysis of various fungi genomes has improved understanding of fungal growth and development. Nevertheless, data on Sanghuangporus on genome and transcriptome sequences are largely lacking, and the lack of a complete S. weigelae genome sequence has impeded deeper understanding of the mechanisms related to biosynthesis of bioactive compounds, such as terpenoids and polysaccharides, hindering further studies based on genome editing. Genome editing techniques have the potential to overcome oxidation issues in mushrooms, creating opportunities for developing improved strains. Only three whole genome sequences of Sanghuangporus species are available, which could hinder application of the unique medicinal properties of each Sanghuangporus species, delaying further applications for permissions to develop commercial products (Brandenburger et al., 2018).
Here, to promote the medicinal use and industrial development of S. weigelae from a genomic perspective, we assembled and annotated the high-level genome of S. weigelae and compared it with other related genomes. Besides information for medicinal application, our genomic analyses, particularly comparisons with genomes of other related fungal species, also reveal evolutionary information regarding the Sanghuangporus genus. Our genomic analyses will help to elucidate the medicinal properties, and biosynthetic pathways of these medicinal mushrooms, which will further facilitate their medicinal use and commercial development.
2 Materials and methods
2.1 Strain culture and DNA isolation
The monokaryotic strain of S. weigelae used in this study was isolated from a wild fruiting body collected from the fallen trunk of a Weigela florida specimen in Jinfoshan Forest Park, Chongqing, and has been deposited at Beijing Forestry University. Mycelia were harvested after growing on sterile cellophane covering potato dextrose agar (PDA) culture medium plates at 28°C for 5−7 d. Mycelia were collected from the liquid medium, packaged in aluminum paper, frozen in liquid nitrogen, and stored at -80°C for DNA extraction (Figure 1). S. weigelae was inoculated on PDA plates covered with cellophane. Mycelia were scraped from the cellophane after they covered the entire plate. High-quality DNA was extracted using the QIAGEN® Genomic kit.

Figure 1 Monokaryotic mycelia of Sanghuangporus weigelae.
2.2 Genome sequencing and assembly
Libraries were constructed and raw sequencing data were obtained by single-molecule real-time fluorescence DNA sequencing using a Pacbio Sequel series sequencer (Wick et al., 2019). MGISEQ2000 platform genome sequencing data were processed using fastp v.0.20.0. Sequel and Sequel II downstream data were obtained using Pacbio proprietary Smrtlink software at the Nextomics Biosciences Co., Ltd (Wuhan, China), which uses high quality region finder to identify the longest region of a singly loaded enzyme that maintains its activity, and signal noise ratio to filter low quality regions and obtain subreads. Downlinked data had no single-base quality values and all base quality values were recorded, to maintain format integrity, but were not of practical significance. The read quality of reads that passed the filter was set at 0.8, and those that did not pass were labeled 0. For quality control, of HiFi reads, subreads were converted to HiFi reads using CCS software, and reads > 1000 bp filtered as pass reads, which were directly used for assembly. Since HiFi read sequences are highly accurate (≥ 99%), they are suitable for genome assembly using Hifiasm (parameter: -n 5). Subsequently, second-generation data were filtered using fastp (-n 0), and the genome polished using the filtered second-generation data. Four iterations of correction with Nextpolish were conducted to obtain the final genome sequence.
2.3 Gene prediction and annotation
Gene structure prediction was performed using a combination of three methods: ab initio prediction using AUGUSTUS (Stanke et al., 2008), homology-based prediction with GeMoMa (Keilwagen et al., 2016), and transcriptome prediction using PASA (Haas et al., 2003). The results were integrated with EvidenceModeler software (Haas et al., 2008), and final structural annotation were obtained by removing genes containing transposable elements using TransposonPSI (Urasaki et al., 2017). Interspersed repetitive sequences were predicted using RepeatMasker (Bedell et al., 2000). Tandem repeats were analyzed using GMATA (Wang and Wang, 2016) and Tandem Repeats Finder (Benson, 1999). tRNA genes were predicted using tRNAscan-SE (Lowe and Eddy, 1997). Gene functions were predicted with reference to these databases which were downloaded from the official web site and localized to compare and annotate the whole genome: Gene Ontology (GO) database (http://geneontology.org/), Kyoto Encyclopedia of Genes and Genomes (KEGG) database (https://www.kegg.jp/), Eukaryotic Orthologous Group (KOG) database (https://www.creative-proteomics.com/services/kog-annotation-analysis-service.htm), Non-Redundant Protein (NR) database (https://www.ncbi.nlm.nih.gov/prote-in), SwissProt database (https://www.uniprot.org/), Pfam database (http://pfam.xfam.org/), Fungal Cytochrome P450 (CYP) database (http://p450.riceblast.snu.ac.kr/cyp.php), and Carbohydrate-Active Enzyme (CAZyme) database (http://www.cazy.org). The whole genome predicted coding genes were aligned with these databases with cut-off values of E-value ≤ 1 × 10-5, identity ≥ 40%, and coverage ≥ 40% by DIAMOND (Buchfink et al., 2015). When a single gene retrieved from the database with more than one result meeting the cut-off values, the gene was annotated by the best score. The antiSMASH program (Medema et al., 2011) with default parameters was employed to predict gene clusters encoding secondary metabolites.
2.4 Comparative genomics analysis
Pairwise average nucleotide identity (ANI) values between genomes were determined using FastANI software (Jain et al., 2018). To explore the dynamics of speciation in Sanghuangporus, genome sequences of S. weigelae, S. sanghuang, S. vaninii, and S. baumii were aligned pairwise using MCScanX software, based on location information from Sanghuangporus GFF3 files (Wang et al., 2012). Based on the resulting blocks, a genomic synteny map among the four species was drawn using TBtools (Chen et al., 2020) supported in the jcvi package in Python 3. To identify the differences and similarities in medicinal applications of Sanghuangporus with a uniform standard, the genome sequences of S. sanghuang, S. baumii, and S. vaninii were reannotated using the same pipeline. Then, genome structure and protein-coding genes related to medicinal use were compared among these four species. In addition, the numbers of genes encoding various families of CAZymes in the four species were clustered in heatmaps using TBtools v.2.0, using the log scale option. To explore the evolutionary dynamics of S. weigelae, the genome sequences of 22 additional fungal species were downloaded from the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/genbank/) for phylogenomics (Supplementary Table S1). Single-copy orthologous genes from the 23 fungal species were inferred using OrthoFinder software (Emms and Kelly, 2019) with the mafft option for subsequent multiple sequence alignment. The species tree was visualized using iTOL software (Zhou et al., 2023). Two Ascomycetous species (Neurospora crassa and Tuber melanosporum) were selected as outgroup taxa from a further 21 Basidiomycetous species (Supplementary Table S1).
3 Results
3.1 Genome sequence assembly and annotation
The first whole genome sequence of the medicinal fungus S. weigelae was generated using the PacBio Sequel II platform. Clean reads were obtained and used for K-mer analysis and genome polishing. GenomeScope v.1.0 was used to generate a histogram of sequencing depth distribution (k = 17) (Figure 2A). A single K-mer coverage peak was observed and the heterozygosity rate was 0.58%. The integrity of the genome was evaluated using BUSCO software (Simão et al., 2015) and determined to be 94.99%. A 33.96-Mb genome sequence was assembled from 10267 Mb raw data and mapped to 13 contigs, with a GC content of 47.93%. Of the 13 contigs, the longest was 4.49 Mb, while N50 length was 2.77 Mb; specific sequence length distribution data are presented in Table 1; Supplementary Figure S1.
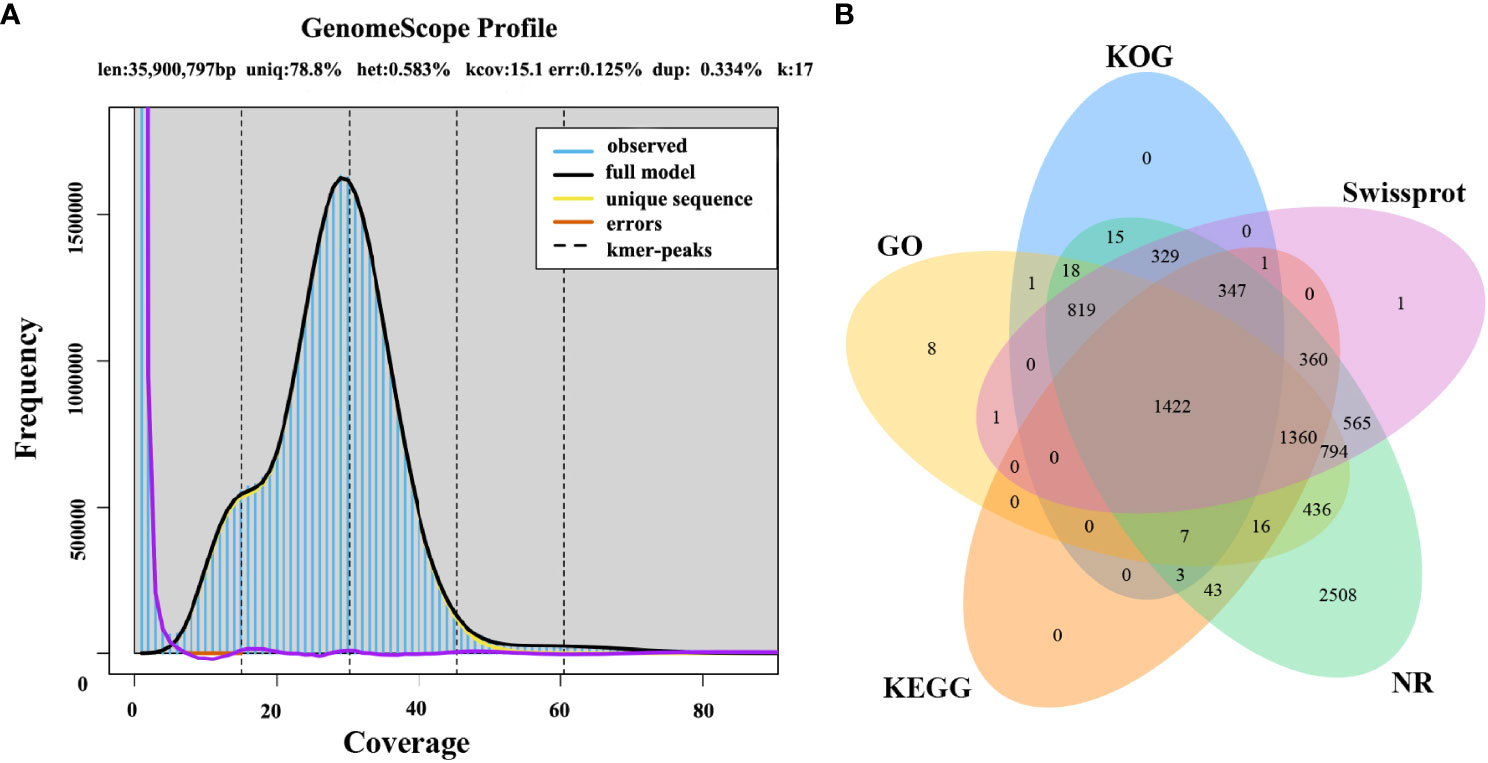
Figure 2 (A) Histogram of the depth distribution of Sanghuangporus weigelae sequencing. (B) Genes were annotated using information from five databases.
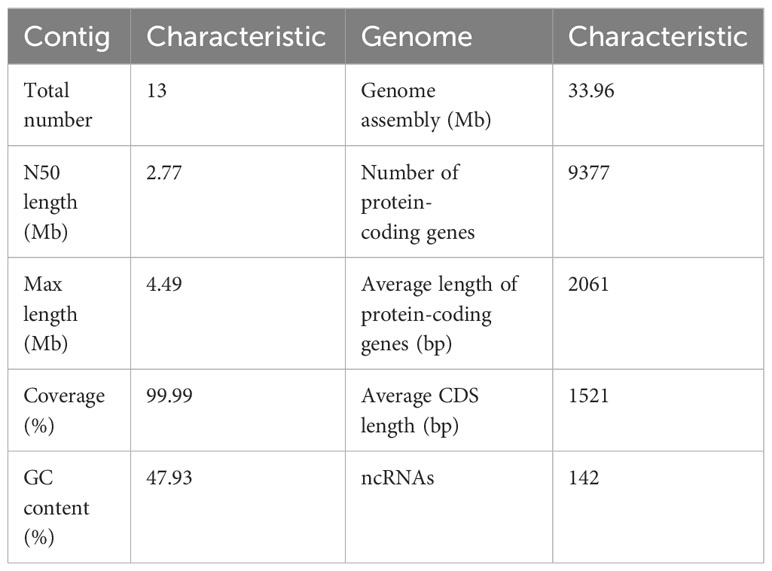
Table 1 Genome assembly and features of Sanghuangporus weigelae.
The size and GC content of the S. weigelae genome were similar to those of other Sanghuangporus species; it was slightly larger than the S. baumii and S. sanghuang genomes, but smaller than that of S. vaninii. Analysis of GC skew did not reveal an obvious distribution pattern across the whole genome. Repeat sequences were identified using RepeatMasker, based on homology alignment and ab initio prediction, and accounted for 16.91% of the S. weigelae genome (Table 2; Supplementary Figure S2). The majority of repetitive sequences were long terminal repeats (LTR) (12.22%), where 1.62% and 1.16% of repeat elements were DNA transposons and miniature inverted-repeat transposable elements (MITEs). Long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs) made up 0.06% and 0.01% of the S. weigelae genome, respectively.
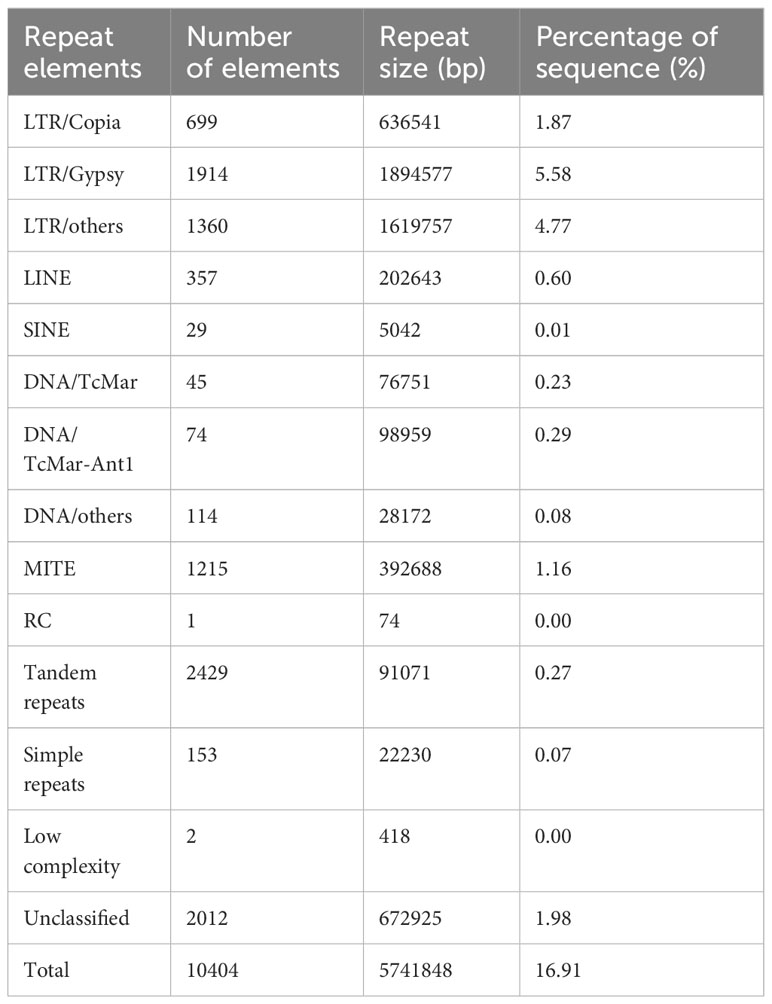
Table 2 Repeat element analysis in the Sanghuangporus weigelae genome.
A total of 9377 protein-coding genes were predicted (mean length, 2061 bp; mean coding sequences (CDS) length, 1521 bp) (Table 1), while 142 non-coding RNAs (ncRNAs) were predicted, accounting for 0.02% of the whole genome sequence. Of the identified genes, 9042 (96.43%) and 5999 (63.98%) (Figure 2B) were annotated in the NR and SwissProt databases, respectively. The completeness of S. weigelae gene prediction was also evaluated using BUSCO software with fungi_odb10. Together, these data indicate that the S. weigelae genome sequence assembly is high quality.
3.2 Terpenoid biosynthesis
In total, 16 genes encoding key enzymes involved in terpenoid backbone biosynthesis via the mevalonate (MVA) pathway were identified in S. weigelae genome sequence (Supplementary Figure S3). Among them, protein-S-isoprenylcysteine O-methyltransferase was encoded by a triple-copy gene, and both farnesyl diphosphate synthase and protein farnesyltransferase subunit β were encoded by double-copy genes, while the other 12 enzymes were encoded by single-copy genes (Supplementary Table S2). In addition to the 16 key enzymes in the MVA pathway, genes indirectly related to terpenoid biosynthesis were also identified: one gene (ID: C02.848) encoding farnesyl-diphosphate farnesyltransferase, one gene (ID: C02.604) involved in the biosynthesis of sesquiterpenoids and triterpenoids (Supplementary Figure S4), and the β-apo-4′-carotenal oxygenase, encoded by a single copy gene (ID: C04.738) involved in carotenoid biosynthesis. For example, the mevalonate kinase existed in terpenoid backbone biosynthesis in S. sanghuang but not in S. weigelae. The diphosphomevalonate decarboxylase, hydroxymethylglutaryl-CoA reductase, and protein-S-isoprenylcysteine O-methyltransferase were only found in S. weigelae. The four species of Sanghuangporus had a comparable number of genes involved in terpenoid backbone synthesis pathways (15 genes in S. baumii; 14 genes in S. sanghuang; 15 genes in S. vaninii; 18 genes in S. weigelae) (Table 3).
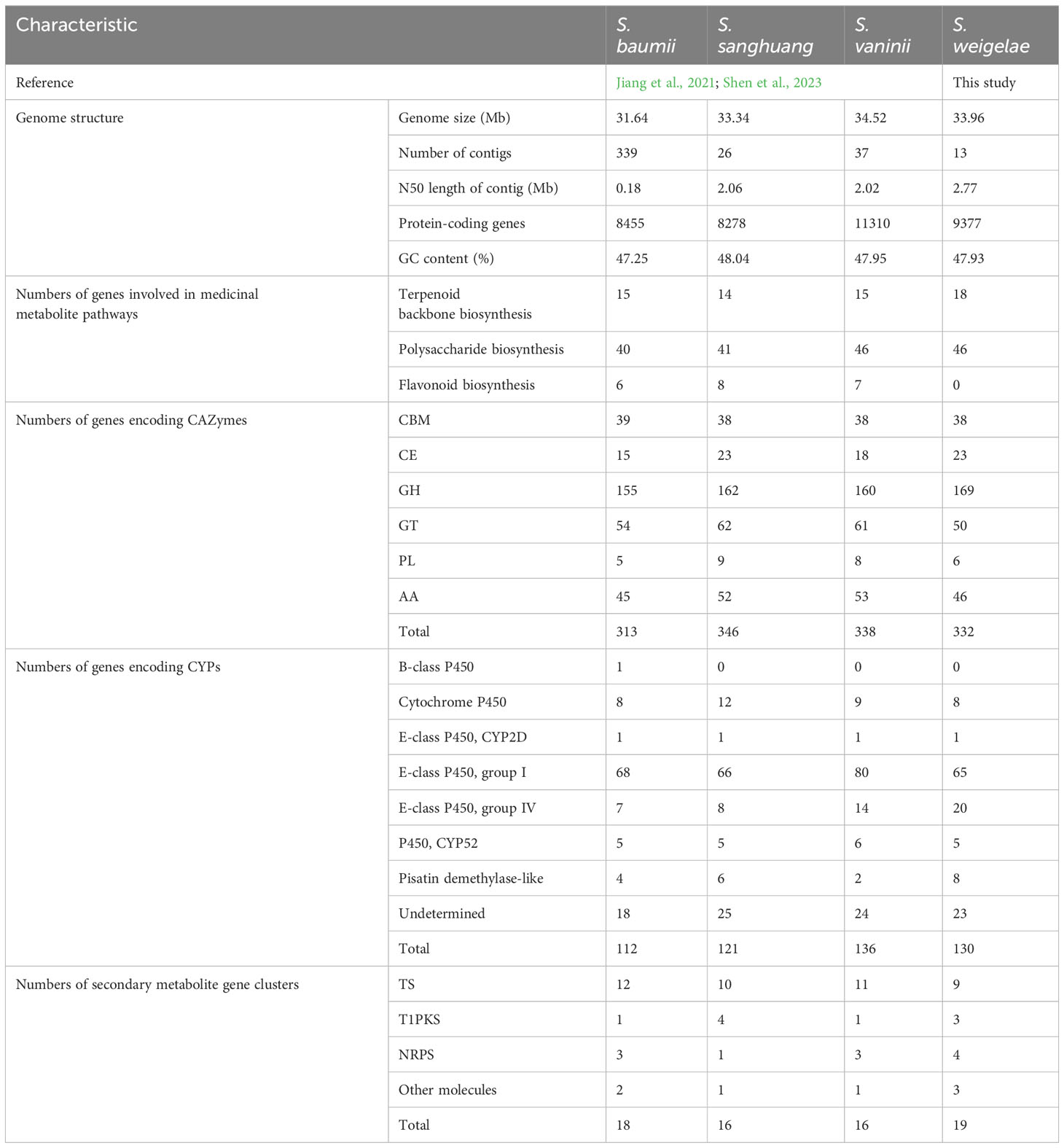
Table 3 Comparative genomics analyses of Sanghuangporus baumii, S. sanghuang, S. vaninii, and S. weigelae.
3.3 Polysaccharide biosynthesis
A total of 19 enzymes, encoded by 48 genes, involved in the biosynthesis of polysaccharides (starch and sucrose metabolism) were identified in S. weigelae genome (Supplementary Figure S5; Supplementary Table S3). Most of these enzymes were encoded by single- or double-copy genes, while the endoglucanase, glucan 1,3-β-glucosidase and cellulose 1,4-β-cellobiosidase, and β-glucosidase were encoded by four, five, six, and nine-genes, respectively. The four species of Sanghuangporus had similar numbers of genes involved in polysaccharide biosynthesis pathways, namely 40 genes in S. baumii, 41 genes in S. sanghuang, 46 genes in S. vaninii, and 46 genes in S. weigelae (Table 3). The differences were focused on two genes encoding 1,3-β-glucosidase and seven genes encoding β-glucosidase. Among them, genes encoding polysaccharide biosynthesis were identified and remarkably higher in the genome of S. weigelae than those of S. vaninii and S. sanghuang (Supplementary Table S3).
3.4 Flavonoid biosynthesis
No key enzymes directly related to flavone and flavonoid biosynthesis pathways were identified in the S. weigelae genome; however, six putative genes involved in flavonoid compound biosynthesis were annotated, including P-type H+-ATPase, anthocyanin 3-O-6″-O-coumaroylglucoside: glucosyltransferase, anthocyanin 3-O-6″-O-coumaroylglucoside: glucosyltransferase, acetyl-CoA carboxylase, glutathione S-transferase, and flavonoid 3-O-glucosyltransferase (Supplementary Table S4).
3.5 Carbohydrate-active enzymes
CAZymes play important roles in drug metabolism, xenobiotic detoxification, and steroid biosynthesis. In S. weigelae, a total of 332 genes were predicated to encode six classes of CAZymes, including 38 genes encoding carbohydrate-binding modules (CBMs), 23 carbohydrate esterases (CEs), 169 glycoside hydrolases (GHs), 50 glycosyltransferases (GTs), 6 polysaccharide lyases (PLs), and 46 auxiliary activities (AAs) (Table 3). Among these six classes, GHs were encoded by the most genes and are mainly involved in degradation of celluloses (GH5 and GH7), hemicelluloses (GH43), pectins (GH28), chitins (GH18), and starches (GH31). Families encoded by > 10 genes included GH5 (23), CBM1 (18), GT2 (15), AA3 (14), AA2 (14), GH16 (12), GT4 (11), and AA1 (11). The total numbers of genes encoding CAZymes were similar among the four species of Sanghuangporus (S. baumii 313; S. sanghuang 346; S. vaninii 338), as was the number of genes encoding each of the six classes of CAZymes (Table 3). Only a few differences were detected at the level of gene families encoding CAZymes (Figure 3). Based on the numbers of genes belonging to different CAZyme families, S. weigelae appears to have a more similar strategy for use of woody substrates to that of S. sanghuang than that of S. baumii.
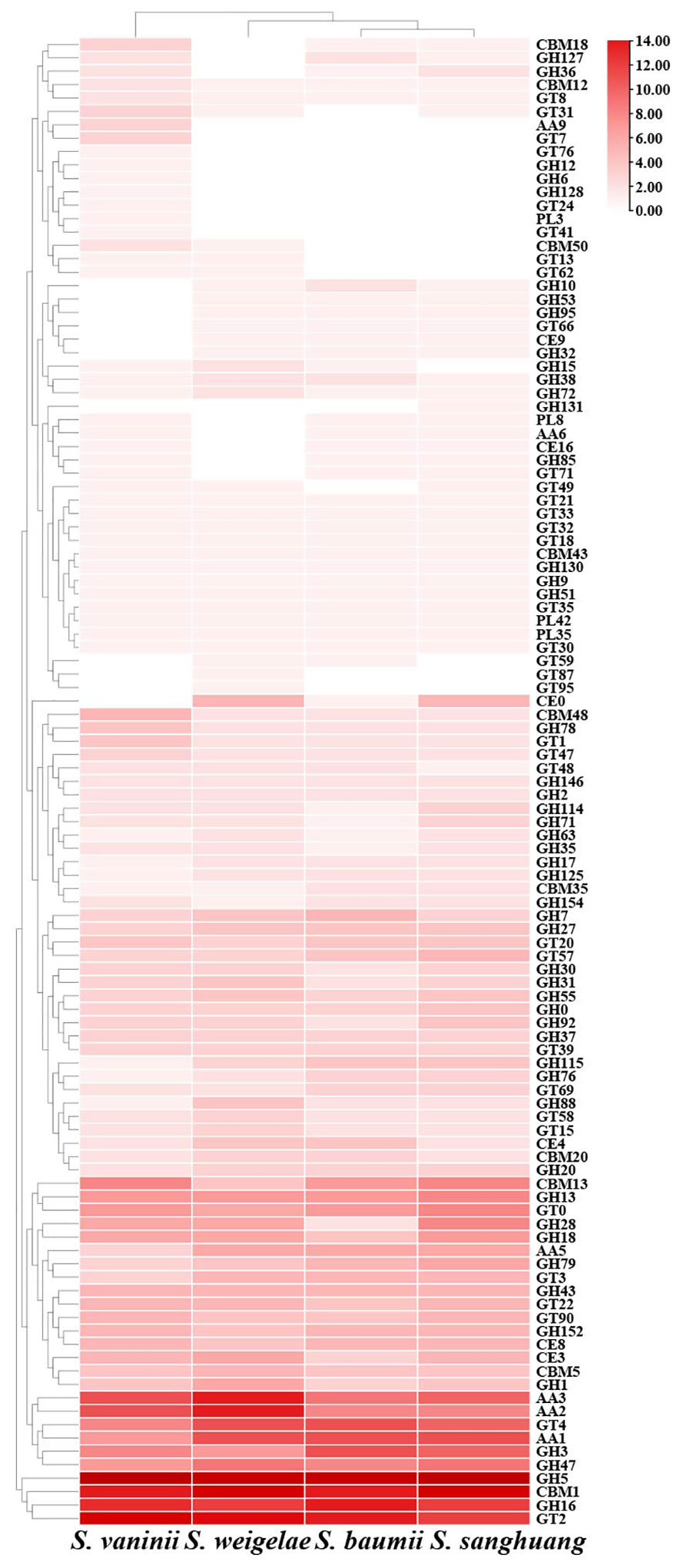
Figure 3 Heatmap of CAZyme families found in Sanghuangporus vaninii, S. weigelae, S. baumii, and S. sanghuang. The x and y axes represent species and CAZyme families, respectively. Boxes are colored according to log values of gene numbers encoding CAZyme families, where box color from white to red indicates an increase in gene numbers encoding CAZyme families.
3.6 Cytochromes P450
CYPs are among the most important gene families in the fungal genome. A total of 130 genes were annotated to encode CYPs in S. weigelae (Table 3). Of these genes, eight were classified to encode CYPs belonging to the ‘Cytochrome P450’ class, one to the ‘E-class P450, CYP2D’ class, 65 to the ‘E-class P450, group I’ class, 20 to the ‘E-class P450, group IV’ class, five to the ‘P450, the CYP52’ class, and eight to the ‘pisatin demethylase-like’ class, while 23 genes were determined to encode CYPs not belonging to any known class. The ‘E-class P450, group I’ (Figure 4) and ‘E-class P450, group IV’ classes encoded by the most genes in S. weigelae, are involved in oxidation-reduction reactions, while the CYP classes encoded by the next largest number of genes are generally responsible for signal transduction of metabolic processes. The numbers of genes encoding CYPs in S. weigelae (130) did not differ significantly at the class level from those in the other three species S. baumii (112), S. sanghuang (121), and S. vaninii (136). The differences are focused on the numbers of genes encoding ‘E-class P450, group I’ class, with the highest in S. sanghuang (80), followed by other three species fluctuating around 65. The numbers of genes encoding other classes including ‘B-class P450’, ‘Cytochrome P450’, ‘E-class P450, CYP2D’, ‘E-class P450, group IV’, ‘P450, CYP52’, ‘Pisatin demethylase-like’, and ‘Undetermined’ were almost consistent (Table 3).

Figure 4 A combined plot of evolutionary tree, motif, structural domain, and gene structure of 55 genes with the conserved motifs of E-class P450, group I class of CYP family proteins.
3.7 Gene clusters
Nineteen gene clusters were predicted in the S. weigelae genome, of which nine were identified to encode terpene synthases (TSs), three to encode iterative type I polyketide synthases (T1PKSs), four to encode nonribosomal peptide synthetases (NRPSs), and three to encode other molecules (Table 3). Among the four species of Sanghuangporus, the numbers of gene clusters involved in TS synthesis were similar, while S. weigelae had more gene clusters involved in T1PKS and NRPS synthesis (Table 3).
3.8 Comparative genomics
Sanghuangporus are rare medicinal mushrooms with long history of controversial species classification and are not easily distinguished from several species of similar brown rot fungi, which has seriously affected the use of Sanghuangporus as a medicinal resource. To assess the evolutionary relationships of S. weigelae, we conducted comparative analysis of the S. weigelae genome and those of 22 fully sequenced fungi (20 Basidiomycetes and 2 Ascomycetes). A total of 1647 orthologous groups, including 451 single-copy genes, were identified in all 22 studied fungal species. A phylogenetic tree constructed based on conserved single copy orthologous gene alignment showed that S. weigelae had a close evolutionary relationship with other Sanghuangporus species (Figure 5), particularly S. vaninii. Indeed, the fruiting bodies of the four species share similar characteristics, including sessile ascospores; field-grown ascospores, mostly superposed; fresh ascospores, corky; and sclerotized after drying.
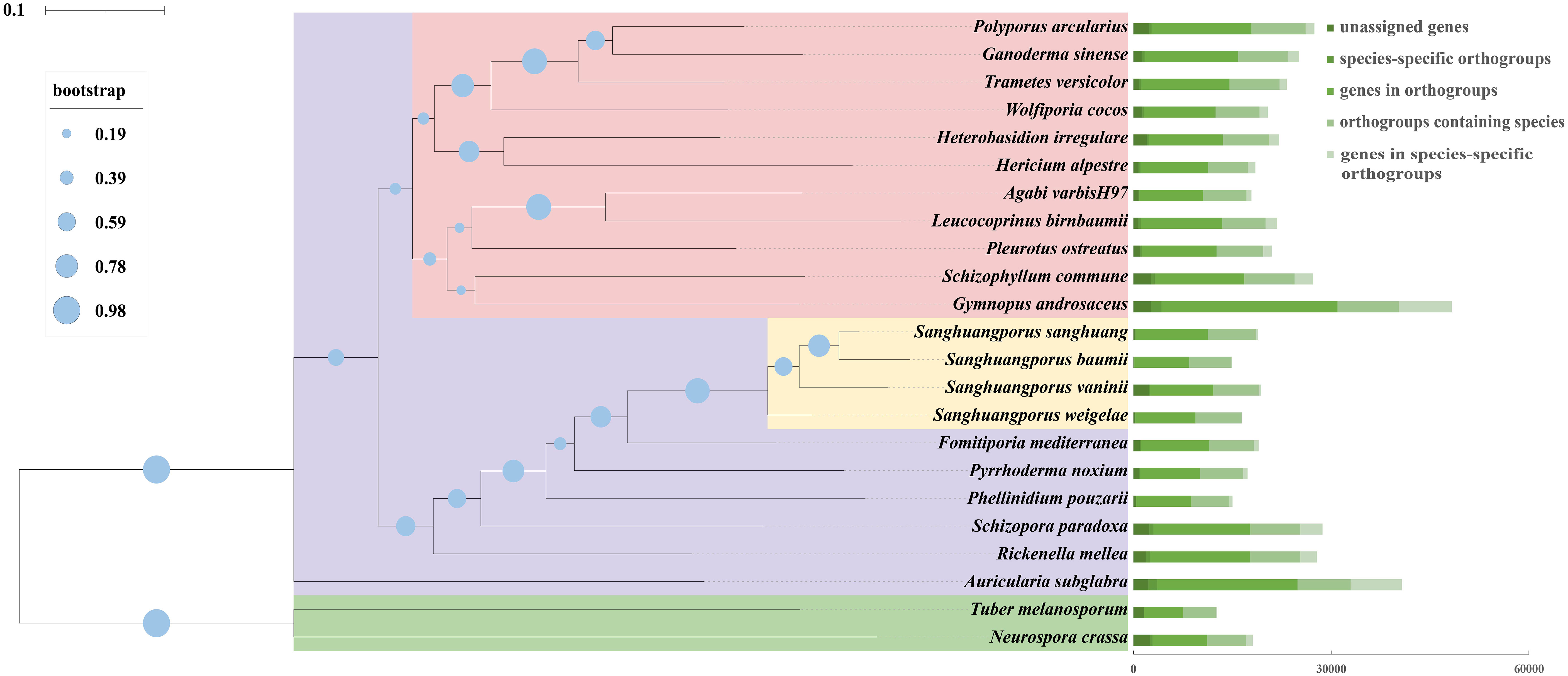
Figure 5 Comparison of genomes among Sanghuangporus weigelae and 22 other fungal species (20 Basidiomycetes and 2 Ascomycetes). Evolutionary relationship analysis and phylogenetic tree construction were conducted based on 451 single-copy orthologous genes using OrthoFinder. Single-copy orthologs were defined as orthologs present as a single-copy gene in all 23 species. Numbers of different orthologous gene types were calculated in each fungal species and are indicated by different colors. Tree scale = 0.1.
Next, we analyzed orthogroups among the four Sanghuangporus species. The S. weigelae genome sequence had a higher similarity to that of S. sanghuang (90.32%), S. baumii (90.28%), and S. vaninii (89.73%); however, while 5412 orthologous groups were identified in the four Sanghuangporus species, S. weigelae shared more orthologous groups with S. sanghuang (125) and S. vaninii (102) than with S. baumii (32) (Supplementary Figure S6). In addition, S. vaninii had the largest number of unique orthologous groups (219), followed by S. weigelae (164) (Supplementary Figure S6). As shown in Supplementary Figure S6, S. weigelae and S. sanghuang shared 125 orthogroups, which were higher than those shared with S. baumii (32 orthogroups). S. weigelae and S. sanghuang have similar fruiting bodies. Therefore, we predict that these 125 orthogroups may be related to fruiting body shape and other properties. The underlying detailed molecular mechanisms require investigation by differential analysis of gene or protein expression in these species.
The ANI analysis method facilitates high-resolution classification and is widely used for research. To further understand the evolutionary relationships among these mushrooms, ANI analysis was performed to estimate genomic differences and relatedness between Sanghuangporus species, G. sinense, and Wolfifiporia cocos. Sanghuangporus species in the species tree showed lower genomic similarities (73% to 75%) with G. sinense and W. cocos, but higher genomic similarities with one another (88% to 100%) (Figure 6). In summary, these results confirm that S. weigelae belong to the Sanghuangporus genus, consistent with its current classification. Collinearity analysis of S. weigelae and other three medicinal mushrooms, which have high levels of genome similarity, was performed using MCScanX software (Figure 7). The results revealed high levels of collinearity between S. weigelae and S. sanghuang. The connection of these contigs could be further confirmed using PCR or HiC technology.
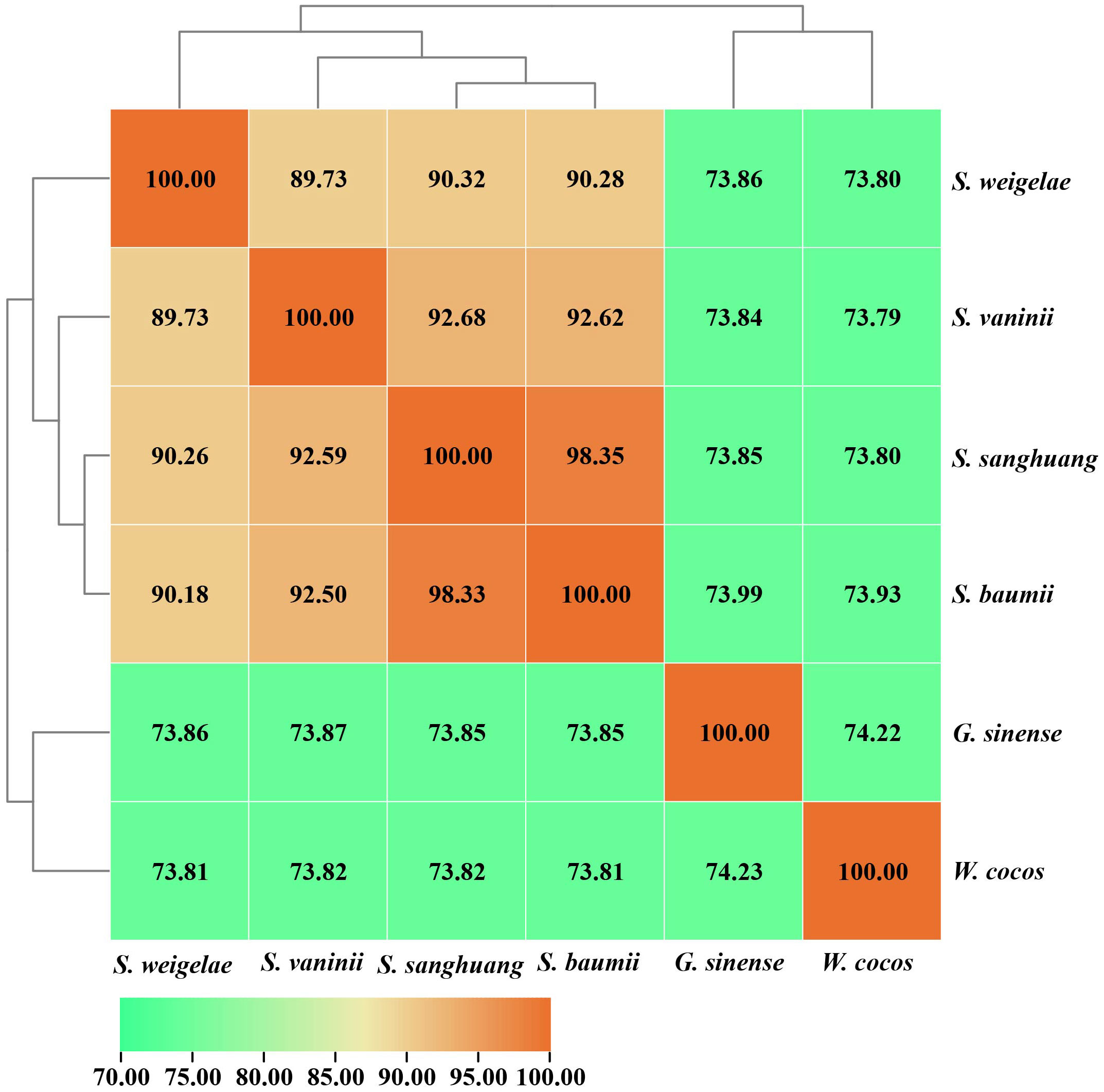
Figure 6 ANI values based on the fastANI algorithm generated-matrix for Sanghuangporus, Ganoderma sinense, and Wolfifiporia cocos genomes. Clustering was conducted using Euclidean distance matrix.

Figure 7 Genome collinearity among Sanghuangporus sanghuang, S. vaninii, S. weigelae, and S. baumii. Each line connects a pair of collinearity blocks between two genomes (S. sanghuang and S. vaninii, 16141 of collinear genes, 73.50%; S. vaninii and S. weigelae, 15797 of collinear genes, 78.28%; S. weigelae and S. baumii, 12794 of collinear genes, 71.53%).
4 Discussion
4.1 Cultural origins of Sanghuangporus weigelae genome sequencing
Chinese medicine, the treasure of ancient Chinese science (He et al., 2023), is also known as “the key to unlocking the treasury of Chinese civilization” (Bai et al., 2022), and has become an important part of cultural exchanges between China and other countries, spreading and gaining influence around the world, and as well as the promotion of exchanges and mutual understanding between the civilizations of the East and the West (Guo et al., 2023). “Sanghuang” (Sanghuangporus), a valuable traditional Chinese medicine, known as “forest gold” (Cao et al., 2020), can be compared with the Chinese medicines, “Lingzhi” (Ganoderma lucidum), “Zizhi” (G. sinensis), and “Fuling” (W. cocos), which have been the focus of intense research. Among Sanghuangporus species, S. weigelae is a medicinal mushroom used in traditional Chinese medicines (Wang et al., 2022; Zhang et al., 2022), and has been recognized as possessing important medicinal properties, such as polysaccharide yields, DPPH radical scavenging ability, and triterpenoid content. To date, the taxonomic status of 18 Sanghuangporus species have been described in the NCBI database, and 15 Sanghuangporus species are considered to be “Sanghuang”, with S. weigelae among those with superior pharmacological activity (Wang H. et al., 2023). As a potentially important medicinal fungal resource, the genome sequence of S. weigelae has not previously been published. In the current study, we generated the first whole genome sequence of S. weigelae (Figure 8) to provide theoretical support and basic research content to advance understanding of traditional Chinese medicine and the development of the macrofungal industry.
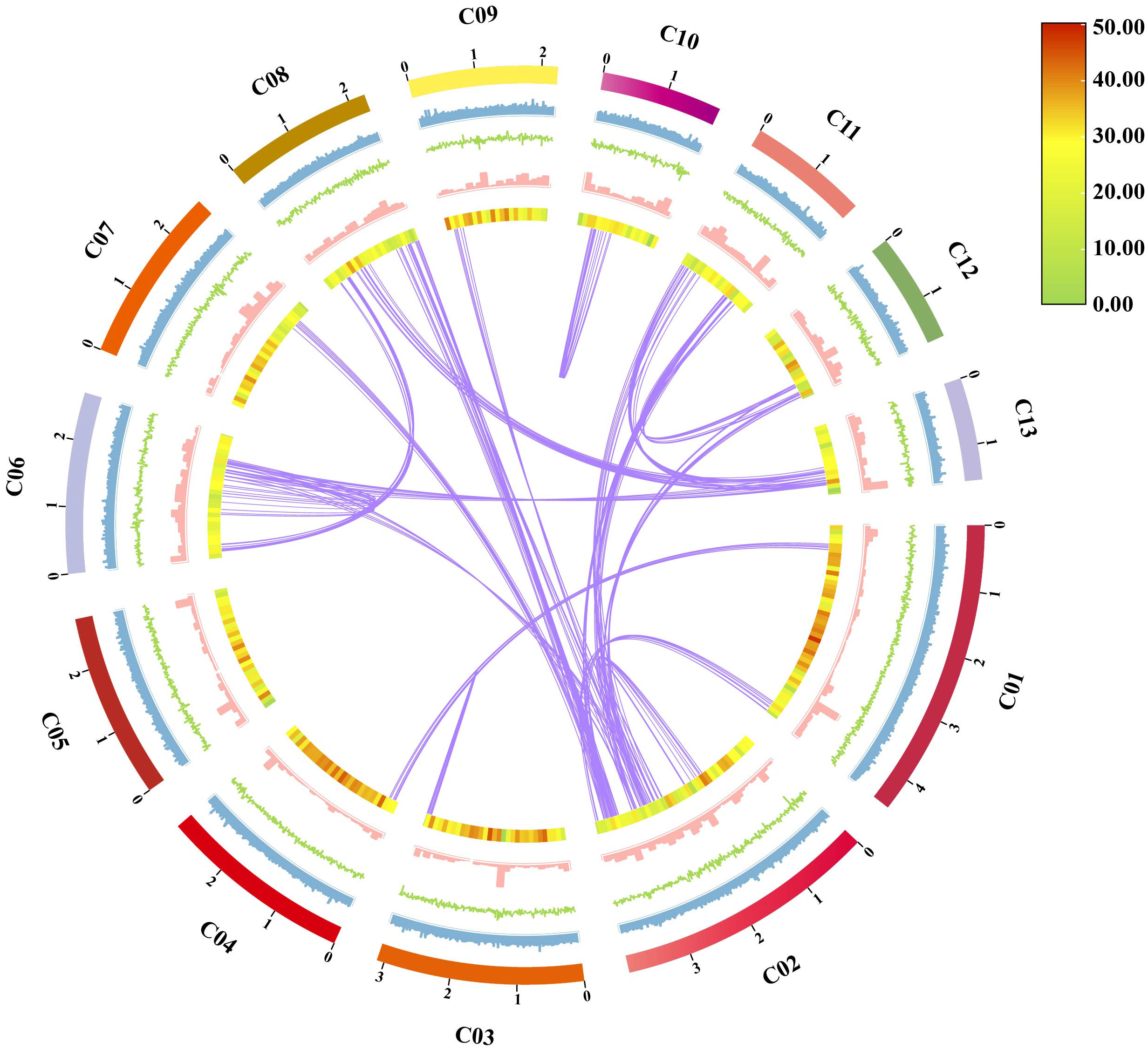
Figure 8 Characteristics of Sanghuangporus weigelae genomic assembly features. From outside to inside: (1) Contigs (> 1 Mb in length); (2) GC ratio, calculated as the percentage of G + C in 1 kb non-overlapping windows; (3) GC skew, calculated as the percentage of (G − C)/(G + C) in 1 kb non-overlapping windows; (4) Repeat sequence density per window; (5) Gene density per window; (6) Collinearity in the whole genome, calculated as 1e-5E-value, 5 number of hits, and 5 number of aligns connected by purple lines.
4.2 Characteristics of Sanghuangporus weigelae genome
Whole-genome similarity analysis is among the best ways to decipher strain taxonomy and generate genetic information on the molecular mechanisms underlying fungal growth and breeding (Yu et al., 2022). In this study, we conducted the whole genome sequencing to explore the evolutionary status and information about functional genes in S. weigelae. We assembled the S. weigelae genome into 13 contigs, with much higher quality sequencing data than those reported for the closely related species strains, S. baumii, S. vaninii, and S. sanghuang. Genome sizes were relatively similar among the four Sanghuangporus species, while the numbers of predicted protein-coding genes were much higher in S. vaninii and S. baumii (Table 3). ANI values indicated that S. weigelae and its closely related strains are interspecies (90 < ANI < 95) (Jain et al., 2018), demonstrating that high-quality genome sequencing of different species can help to clarify their phylogenetic and evolution status. To comprehensively analyze the relationships among S. weigelae and related species from other genera, 20 Basidiomycota and 2 Ascomycota fungal species were included in the phylogenetic analysis, which demonstrated that S. weigelae is closely related to S. vaninii, S. sanghuang, and S. baumii. All of these Agaricomycetes commonly have fruiting bodies, which are good resources for studying genomic changes underlying incomplexity levels in mushroom fruiting bodies (Coelho et al., 2017). Understanding the genetic basis of fruiting body evolution may directly contribute to the improvement of medicinal fungal culture and production. Furthermore, synteny analysis revealed high collinearity between the S. weigelae and S. sanghuang genomes, suggesting that S. sanghuang can likely serve as a reference mode for cultivation and breeding of S. weigelae during largescale industrialization.
4.3 Pharmacological properties of Sanghuangporus weigelae from a genomic perspective
Aqueous extracts of “Sanghuang” can induce apoptosis of cancer cells, as demonstrated by researchers from Japan and Korea, and its anticancer efficacy has been reported (Ayeka, 2018). The substances with anticancer effects are due to the polysaccharide components of the fruiting bodies, which enhanced by our new perspective on Sanghuangporus secondary metabolites (Seçme et al., 2018). Notably, terpenoids and polysaccharides are two major classes of chemical compounds among the medicinal metabolites in Sanghuangporus that exhibit multiple pharmacological properties (Cai et al., 2019). Among the secondary metabolite gene clusters, those encoding T1PKS and NRPS are frequently detected in filamentous fungi, whereas TS-encoding gene clusters are more common in Basidiomycetes (Shao et al., 2020). Consistently, the four Sanghuangporus species analyzed here contained more TS gene clusters that participate in terpenoid biosynthesis (Table 3). Triterpenoids extracted from Sanghuangporus species have high antioxidant activity in the presence of added fungal polysaccharide inducers or ultrasound stimulation (Cai et al., 2019). Genes of the conserved MVA pathway are involved in terpenoid skeleton biosynthesis. In the current study, we show that S. baumii, S. sanghuang, S. vaninii, and S. weigelae share similar genotypes and gene numbers annotated in the MVA pathway (Table 3), indicating that triterpenoids from these species have similar medicinal potential. More studies of the biological properties of triterpenoids in S. baumii, S. sanghuang, S. vaninii, and S. weigelae are warranted.
We also identified numerous essential genes related to secondary metabolites, which endow S. weigelae with biological activities that promote its survival in a specific environment and defense responses to pathogens. Genes involved in regulation of terpenes and NRPSs were found in the S. weigelae genome. Terpenes are among the largest groups of bioactive natural products identified and play crucial roles in the biological functions identified in wild edible mushrooms (Quin et al., 2014; Min et al., 2018; Liang et al., 2020). For example, triterpenes from G. lucidum have significant anti-tumor effects and their potential for anti-tumor application has been assessed in the context of cancer treatment (Li et al., 2020). S. weigelae has a large number of terpene synthesis genes relative to reports regarding those in other medicinal mushrooms (Table 3). NRPSs are considered key factors in host-pathogen interactions of medicinal mushrooms, yet their specific functions have not been identified (Oide and Turgeon, 2020; Ilyukhin et al., 2022). Further, no function of NRPSs in Basidiomycetes has been reported to date (Duan et al., 2022). The numbers of terpenes and NRPSs in S. weigelae may indicate potential for medicinal development of the pathways generating biologically active chemicals in Sanghuangporus, and further research is urgently needed to investigate this area.
Besides genes involved in triterpenoid biosynthesis, several genes involved in sesquiterpenoid synthesis were also annotated in the S. weigelae genome. The results were similar to those of previous studies, indicating that pathways potentially involved in biosynthesis of terpenoids other than triterpenoids occur in Sanghuangporus (Jiang et al., 2021). Notably, sesquiterpenoids were extracted from Sanghuangporus and showed antibacterial, antifungal, and cytotoxic activities (Rajachan et al., 2020). These results suggest that sesquiterpenoids may be among common bioactive secondary metabolites in Sanghuangporus.
Polysaccharides are the most widely studied class of compounds among fungal secondary metabolites (Meng et al., 2016; Zheng et al., 2017; Wang et al., 2021; Wang et al., 2022). Differences in molecular weight, branching configuration, conformation, and chemical modification of polysaccharides provide the basis for their diverse biological activities, such as antitumor, antioxidant, and anti-inflammatory effects (Maity et al., 2021). These medicinal functions have also been detected in Sanghuangporus (Jiang et al., 2021; Jiang et al., 2022). Here, for the first time, we identified a polysaccharide biosynthesis pathway in S. weigelae, namely the starch and sucrose metabolism pathway, which involved 48 genes (Supplementary Figure S3; Supplementary Table S2). Similar genes were revealed to be involved in polysaccharide biosynthesis pathways in S. baumii, S. sanghuang, and S. vaninii (Table 3). The higher numbers of genes involved in polysaccharide biosynthesis, relative to other medicinal pathways, in Sanghuangporus suggest that polysaccharides may be the major medicinal metabolites in Sanghuangporus. The dominant numbers of genes encoding the water-soluble 1,3-β-and 1,6-β-glucans, the most active immunomodulatory and antioxidant compounds (Shao et al., 2020), accounted for the pharmaceutical potential of S. weigelae. However, further experiments are expected and required to test this hypothesis.
Unlike terpenoids and polysaccharides, flavonoids have received little attention in mushrooms, partly because they comprise a low proportion of total metabolites. Only six related flavoprotein genes were identified in S. weigelae, consistent with previous findings that the absence of chalcone isomerase 1 in the flavonoid biosynthesis pathway suggests that this mushroom synthesizes flavonoids by a different mechanism from that in plants (Shao et al., 2020). Flavonoids in mushrooms also deserve to become the focus of increased attention in medicinal studies, as these gene clusters may confer favorable medicinal properties in S. weigelae. In addition, differential expression of genes associated with medicinal properties may be responsible for the variation in medicinal properties among Sanghuangporus species. Transcriptomic and metabolic data could help to address this question. Further explorative work including elucidation of the effects of number and diversity of genes encoding related enzymes on the medicinal values will assist in cultivation of medicinal mushrooms and development of health care products.
4.4 Specification and uniqueness in Sanghuangporus
A major reason for the lack of widespread and large-scale utilization of mushrooms is the shortage of mature fruiting bodies, and the corresponding practical solution to this restriction is artificial cultivation (Saini et al., 2022). Genomics data has potential to provide clues to facilitate the cultivation of S. weigelae. Recent study has shown that CAZymes are crucial for the growth and development of wood-inhabiting fungi and for them to thrive in environments rich in carbohydrates, particularly lignocellulose and cellulose (Sharma et al., 2022; He et al., 2023). In the S. weigelae genome, the main CAZymes were GHs, GTs, and AAs, while PLs, CEs, and CMBs were in the minority. The number of GH genes, the most abundant family in the S. weigelae genome, was four times than that of GT genes, possibly due to the fact that lignocellulose degradation capacity is necessary for S. weigelae survival, as most GH-related genes encode proteins involved in starch degradation. These data reveal that GH gene enrichment has contributed to the diversification of nutrient substrate utilization in S. weigelae. Moreover, compared with other mushrooms, S. weigelae has the most AA genes, which are important for lignin degradation. The CAZymes of S. weigelae were analyzed in comparison with those of S. vaninii, S. baumii, and S. sanghuang. In all four species, genes encoding GH5, GH16, AAs, and GH18 were relatively abundant (Figure 3). GH5, one of the largest GH families, historically known as “cellulase family A”, has a wide range of specificities, is extremely abundant in various ecological niches, and is often found encoded as part of microbial communities (Aspeborg et al., 2012). GH16 members are widely distributed in all areas of life, in which they play various biological roles, including in the degradation of xyloglucan (Viborg et al., 2019). AA9, a class of copper-dependent oxidases that act on crystalline cellulose, enhances the hydrolytic activity of cellulase hydrolases (Vaaje-Kolstad et al., 2010). GH18 catalyzes the biodegradation of the β-1,4 glycosidic bond in aminoglycans through a substrate-assisted retention mechanism, and is involved in various physiological processes, including tissue degradation and remodeling, nutrient uptake, invasion, and pathogenesis, as well as immune response regulation (Sørbotten et al., 2005). The enrichment of genes encoding all of the above-mentioned CAZymes in Sanghuangporus highlights their importance in lignocellulose degradation, and it can be assumed that these genes are core genes required for efficient nutrient utilization. Meanwhile, our data provide a theoretical basis for screening and utilization of this genetic resource, to breed new varieties of Sanghuangporus with excellent qualities. Certain CAZyme families, including GTs, are encoded by more genes in S. weigelae than in S. baumii and S. sanghuang (Figure 4), indicating that S. weigelae has unique nutritional strategy that warrants further study.
CYPs are an important class of monooxygenases with vital roles in various biological activities (Črešnar and Petrič, 2011; Zeng et al., 2016); however, fungal CYPs have been little studied relative to those of mammals and plants (Črešnar and Petrič, 2011; Qhanya et al., 2015). In the current study, 23 of 130 CYPs identified in S. weigelae, compared with 18 of 112 CYPs in S. baumii, 25 of 121 CYPs in S. sanghuang, and 24 of 136 CYPs in S. vaninii could not be categorized into any known class (Table 3). It was speculated that the 23 genes might participate in several types of secondary metabolic processes, including biodegradation of xenobiotics, carbohydrate metabolism, and biosynthesis of antibiotics. Furthermore, there are specific types of CYPs in basidiomycetous biotrophic plant pathogens that allow the mushrooms to adapt to a wide range of ecological niches. Therefore, these 23 S. weigelae specific CYPs may be involved in important adaptive mechanisms specific to the host plant. In addition, similar uncharacterized CYPs were detected in S. baumii, S. vaninii, and S. sanghuang, and hypothesized to be associated with host adaptation. Functional experiments are required to clarify the roles of these genes in Sanghuangporus evolution and their relationship with host adaptation. In future, comparative transcriptomics between Sanghuangporus species at the primordium and fruiting body formation stages will reveal the key genes involved in these processes and thus facilitate cultivation of S. weigelae.
5 Conclusions
Here, we generated the first whole genome sequence of medicinal mushroom S. weigelae, represented by a monokaryotic strain. Integrity, completeness, and collinearity analyses revealed the high quality of our genome assembly. Comparative genomic and phylogenetic analyses indicated clearly that S. weigelae should be classified in the Sanghuangporus genus. In addition, identification of CAZyme-encoding genes revealed that S. weigelae has robust lignocellulose degradation capacity. Overall, the S. weigelae genome provides insights useful for basic research into the nutrition and medicinal utility of this mushroom. Comprehensive understanding of the S. weigelae genome has potential to provide a basis for its future application in pharmacological and industrial fields. Future generation of transcriptomic and metabolic data will further facilitate the appropriate application of S. weigelae.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: BioProject PRJNA1030531.
Author contributions
CJ: Data curation, Formal Analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. JM: Formal Analysis, Investigation, Software, Validation, Writing – original draft, Writing – review & editing. HW: Investigation, Validation, Writing – original draft. LT: Investigation, Validation, Writing – original draft. YY: Investigation, Validation, Writing – original draft. XL: Investigation, Validation, Writing – original draft. JS: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Beijing Nova Program (20230484322) and the National Natural Science Foundation of China (32270016 and 32070016).
Acknowledgments
We express our gratitude to Drs. Jie Song (Guangdong Eco-Engineering Polytechnic, China) and Hai-Jiao Li (National Institute of Occupational Health and Poison Control, Chinese Center for Disease Control and Prevention, China) for sample collection and data analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1325418/full#supplementary-material
References
Aspeborg, H., Coutinho, P. M., Wang, Y., Brumer, H., Henrissat, B. (2012). Evolution, substrate specificity and subfamily classification of glycoside hydrolase family 5 (GH5). BMC Evol. Biol. 12, 186. doi: 10.1186/1471-2148-12-186
Ayeka, P. A. (2018). Potential of mushroom compounds as immunomodulators in cancer immunotherapy: A review. Evid.-Based Complement. Alternat. Med. 2018, 7271509. doi: 10.1155/2018/7271509
Bai, X., Liu, Z. J., Tang, T., Yu, S. J., Liu, D., Liu, G. M., et al. (2022). An integrative approach to uncover the components, mechanisms, and functions of traditional Chinese medicine prescriptions on male infertility. Front. Pharmacol. 13. doi: 10.3389/fphar.2022.794448
Bedell, J. A., Korf, I., Gish, W. (2000). MaskerAid: a performance enhancement to RepeatMasker. Bioinformatics 16 (11), 1040–1041. doi: 10.1093/bioinformatics/16.11.1040
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27 (2), 573–580. doi: 10.1093/nar/27.2.573
Brandenburger, E., Braga, D., Kombrink, A., Lackner, G., Gressler, J., Künzler, M., et al. (2018). Multi-genome analysis identifies functional and phylogenetic diversity of basidiomycete adenylate-forming reductases. Fungal Genet. Biol. 112, 55–63. doi: 10.1016/j.fgb.2016.07.008
Buchfink, B., Xie, C., Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12 (1), 59–60. doi: 10.1038/nmeth.3176
Cai, C. S., Ma, J. X., Han, C. R., Jin, Y., Zhao, G. Z., He, X. W. (2019). Extraction and antioxidant activity of total triterpenoids in the mycelium of a medicinal fungus, Sanghuangporus sanghuang. Sci. Rep. 9, 7418. doi: 10.1038/s41598-019-43886-0
Cao, Z. T., Gan, L. Q., Jiang, W., Wang, J. L., Zhang, H. B., Zhang, Y., et al. (2020). Protein binding affinity of polymeric nanoparticles as a direct indicator of their pharmacokinetics. ACS Nano 14 (3), 3563–3575. doi: 10.1021/acsnano.9b10015
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13 (8), 1194–1202. doi: 10.1101/289660
Coelho, M. A., Bakkeren, G., Sun, S., Hood, M. E., Giraud, T. (2017). Fungal sex: the basidiomycota. Microbiol. Spectr. 5 (3), FUNK–0046-2016. doi: 10.1128/microbiolspec.FUNK-0046-2016
Črešnar, B., Petrič, Š. (2011). Cytochrome P450 enzymes in the fungal kingdom. BBA-Proteins Proteom. 1814 (1), 29–35. doi: 10.1016/j.bbapap.2010.06.020
Dai, Y. C., Cui, B. K. (2014). Progress on the species of medicinal fungus Inonotus sanghuang. J. Beijing For. U. 36 (5), 1–7. doi: 10.13332/j.cnki.jbfu.2014.05.017
Duan, Y. C., Han, H. Y., Qi, J. Z., Gao, J. M., Xu, Z. C., Wang, P. C., et al. (2022). Genome sequencing of Inonotus obliquus reveals insights into candidate genes involved in secondary metabolite biosynthesis. BMC Genomics 23 (1), 314. doi: 10.1186/s12864-022-08511-x
Emms, D. M., Kelly, S. (2019). OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20 (1), 238. doi: 10.1186/s13059-019-1832-y
Fang, M., Wang, X. E., Chen, Y., Wang, P., Lu, L. X., Lu, J., et al. (2020). Genome sequence analysis of Auricularia heimuer combined with genetic linkage map. J. Fungi 6 (1), 37. doi: 10.3390/jof6010037
Guo, J. F., Zhang, J., Liu, Q. Y., Yang, N. N., Huang, Y., Hu, T. T., et al. (2023). Research progress on components and mechanisms of neurotoxicity induced by traditional Chinese medicine. J. Appl. Toxicol. 43 (3), 338–349. doi: 10.1002/jat.4396
Haas, B. J., Delcher, A. L., Mount, S. M., Wortman, J. R., Smith, R. K., Jr., Hannick, L. I., et al. (2003). Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31 (19), 5654–5666. doi: 10.1093/nar/gkg770
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9 (1), R7. doi: 10.1186/gb-2008-9-1-r7
He, H. J., Sun, W. J., Chang, J., Hu, S. Y., Yang, J., Yi, X., et al. (2023). Multi-component immune knockout: A strategy for studying the effective components of traditional Chinese medicine. J. Chromatogr. A 1692, 463853. doi: 10.1016/j.chroma.2023.463853
Huang, L. Q., Hong, J. R., Wang, Y. (2023). Traditional Chinese medicine for non-motor symptoms in Parkinson disease: A systematic review and meta-analysis of RCTs. Medicine 102 (30), e34425. doi: 10.1097/MD.0000000000034425
Ilyukhin, E., Markovskaja, S., Elgorban, A. M., Al-Rejaie, S. S., Maharachchikumbura, S. S. N. (2022). Genomic characteristics and comparative genomics analysis of Parafenestella ontariensis sp. nov. J. Fungi 8 (7), 732. doi: 10.3390/jof8070732
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114. doi: 10.1038/s41467-018-07641-9
Jiang, J. H., Wu, S. H., Zhou, L. W. (2021). The first whole genome sequencing of Sanghuangporus sanghuang provides insights into its medicinal application and evolution. J. Fungi 7 (10), 787. doi: 10.3390/jof7100787
Jiang, X., Li, S. Q., Feng, X. R., Li, L. Z., Hao, J., Wang, D., et al. (2022). Mushroom polysaccharides as potential candidates for alleviating neurodegenerative diseases. Nutrients 14 (22), 4833. doi: 10.3390/nu14224833
Keilwagen, J., Wenk, M., Erickson, J. L., Schattat, M. H., Grau, J., Hartung, F. (2016). Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44 (9), e89. doi: 10.1093/nar/gkw092
Kiss, E., Hegedüs, B., Virágh, M., Varga, T., Merényi, Z., Kószó, T., et al. (2019). Comparative genomics reveals the origin of fungal hyphae and multicellularity. Nat. Commun. 10, 4080. doi: 10.1038/s41467-019-12085-w
Li, P., Liu, L. X., Huang, S., Zhang, Y. H., Xu, J. H., Zhang, Z. Q. (2020). Anti-cancer effects of a neutral triterpene fraction from Ganoderma lucidum and its active constituents on SW620 human colorectal cancer cells. Anti-Cancer Agent. Med. Chem. 20 (2), 237–244. doi: 10.2174/1871520619666191015102442
Liang, Y., Lu, D. X., Wang, S., Zhao, Y. H., Gao, S. H., Han, R. B., et al. (2020). Genome assembly and pathway analysis of edible mushroom Agrocybe cylindracea. Genom. Proteom. Bioinf. 18 (3), 341–351. doi: 10.1016/j.gpb.2018.10.009
Lowe, T. M., Eddy, S. R. (1997). tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25 (5), 955–964. doi: 10.1093/nar/25.5.955
Maity, P., Sen, I. K., Chakraborty, I., Mondal, S., Bar, H., Bhanja, S. K., et al. (2021). Biologically active polysaccharide from edible mushrooms: A review. Int. J. Biol. Macromol. 172, 408–417. doi: 10.1016/j.ijbiomac.2021.01.081
Medema, M. H., Blin, K., Cimermancic, P., de Jager, V., Zakrzewski, P., Fischbach, M. A., et al. (2011). antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 39, W339–W346. doi: 10.1093/nar/gkr466
Meng, G., Zheng, F., Liu, H. X., Si, J., Cui, B. K. (2016). Antioxidant activity of Inonotus weigelae during the liquid cultivation. Biotechnol. Bull. 32 (9), 140–148. doi: 10.13560/j.cnki.biotech.bull.1985.2016.09.019
Min, B., Kim, S., Oh, Y. L., Kong, W. S., Park, H., Cho, H., et al. (2018). Genomic discovery of the hypsin gene and biosynthetic pathways for terpenoids in Hypsizygus marmoreus. BMC Genomics 19, 789. doi: 10.1186/s12864-018-5159-y
Oide, S., Turgeon, B. G. (2020). Natural roles of nonribosomal peptide metabolites in fungi. Mycoscience 61 (3), 101–110. doi: 10.1016/j.myc.2020.03.001
Qhanya, L. B., Matowane, G., Chen, W. P., Sun, Y. X., Letsimo, E. M., Parvez, M., et al. (2015). Genome-wide annotation and comparative analysis of cytochrome P450 monooxygenases in Basidiomycete biotrophic plant pathogens. PloS One 10 (11), e0142100. doi: 10.1371/journal.pone.0142100
Quin, M. B., Flynn, C. M., Schmidt-Dannert, C. (2014). Traversing the fungal terpenome. Nat. Prod. Rep. 31 (10), 1449–1473. doi: 10.1039/C4NP00075G
Rajachan, O. A., Sangdee, A., Kanokmedhakul, K., Tontapha, S., Amornkitbamrung, V., Kanokmedhakul, S. (2020). Cyclofarnesane sesquiterpenoids from the fungus Sanghuangporus sp. Phytochem. Lett. 37, 17–20. doi: 10.1016/j.phytol.2020.03.007
Saini, S., Kumar, A., Singhal, B., Kuhad, R. C., Sharma, K. K. (2022). Fungal oxidoreductases and CAZymes effectively degrade lignocellulosic component of switchgrass for bioethanol production. Fuel 328, 125341. doi: 10.1016/j.fuel.2022.125341
Seçme, M., Kaygusuz, O., Eroglu, C., Dodurga, Y., Colak, O. F., Atmaca, P. (2018). Potential anticancer activity of the parasol mushroom, Macrolepiota procera (Agaricomycetes), against the A549 human lung cancer cell line. Int. J. Med. Mushrooms 20 (11), 1075–1086. doi: 10.1615/IntJMedMushrooms.2018028589
Shao, Y., Guo, H. W., Zhang, J. P., Liu, H., Wang, K., Zuo, S., et al. (2020). The genome of the medicinal macrofungus Sanghuang provides insights into the synthesis of diverse secondary metabolites. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.03035
Sharma, G., Kaur, B., Raheja, Y., Agrawal, D., Basotra, N., Di Falco, M., et al. (2022). Lignocellulolytic enzymes from Aspergillus allahabadii for efficient bioconversion of rice straw into fermentable sugars and biogas. Bioresour. Technol. 360, 127507. doi: 10.1016/j.biortech.2022.127507
Shen, Z. Q., Jiang, J. H., Li, C. T., Li, Y., Zhou, L. W. (2023). Genome re-annotation and transcriptome analyses of Sanghuangporus sanghuang. J. Fungi 9 (5), 505. doi: 10.3390/jof9050505
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31 (19), 3210–3212. doi: 10.1093/bioinformatics/btv351
Song, T. T., Xu, F., Shen, Y. Y., Fan, L. J., Cai, W. M. (2021). The complete mitochondrial genome of Sanghuangporus vaninii Zhehuang-1 (Hymenochaetales, Basidiomycota). Mitochondrial DNA B 6 (3), 1096–1097. doi: 10.1080/23802359.2020.1832592
Sørbotten, A., Horn, S. J., Eijsink, V. G. H., Vårum, K. M. (2005). Degradation of chitosans with chitinase B from Serratia marcescens - Production of chito-oligosaccharides and insight into enzyme processivity. FEBS J. 272 (2), 538–549. doi: 10.1111/j.1742-4658.2004.04495.x
Stanke, M., Diekhans, M., Baertsch, R., Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24 (5), 637–644. doi: 10.1093/bioinformatics/btn013
Urasaki, N., Takagi, H., Natsume, S., Uemura, A., Taniai, N., Miyagi, N., et al. (2017). Draft genome sequence of bitter gourd (Momordica charantia), a vegetable and medicinal plant in tropical and subtropical regions. DNA Res. 24 (1), 51–58. doi: 10.1093/dnares/dsw047
Vaaje-Kolstad, G., Westereng, B., Horn, S. J., Liu, Z. L., Zhai, H., Sørlie, M., et al. (2010). An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330 (6001), 219–222. doi: 10.1126/science.1192231
Viborg, A. H., Terrapon, N., Lombard, V., Michel, G., Czjzek, M., Henrissat, B., et al. (2019). A subfamily roadmap of the evolutionarily diverse glycoside hydrolase family 16 (GH16). J. Biol. Chem. 294 (44), 15973–15986. doi: 10.1074/jbc.RA119.010619
Wang, H., Ma, J. X., Wu, D. M., Gao, N., Si, J., Cui, B. K. (2023). Identifying bioactive ingredients and antioxidant activities of wild Sanghuangporus species of medicinal fungi. J. Fungi 9 (2), 242. doi: 10.3390/jof9020242
Wang, H., Ma, J. X., Zhou, M., Si, J., Cui, B. K. (2022). Current advances and potential trends of the polysaccharides derived from medicinal mushrooms sanghuang. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.965934
Wang, H., Qian, K., Si, J., Cui, B. K. (2021). Research advances on polysaccharides from sanghuang. Mycosystema 40 (4), 895–911. doi: 10.13346/j.mycosystema.200364
Wang, X. T., Sun, J., Wang, S. X., Sun, T. T., Zou, L. (2023). Salicylic acid promotes terpenoid synthesis in the fungi Sanghuangporus baumii. Microb. Biotechnol. 16 (6), 1360–1372. doi: 10.1111/1751-7915.14262
Wang, X. W., Wang, L. (2016). GMATA: an integrated software package for genome-scale SSR mining, marker development and viewing. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01350
Wang, Y. P., Tang, H. B., DeBarry, J. D., Tan, X., Li, J. P., Wang, X. Y., et al. (2012). MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40 (7), e49. doi: 10.1093/nar/gkr1293
Wei, J. S., Liu, L. Y., Yuan, X. L., Wang, D., Wang, X. Y., Bi, W., et al. (2023). Transcriptome analysis reveals the putative polyketide synthase gene involved in hispidin biosynthesis in Sanghuangporus sanghuang. Mycobiology 51 (5), 360–371. doi: 10.1080/12298093.2023.2257999
Wick, R. R., Judd, L. M., Holt, K. E. (2019). Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 20, 129. doi: 10.1186/s13059-019-1727-y
Wu, S. H., Huang, G. Z., Chen, Y. P., Dai, Y. C., Zhou, L. W. (2016). Taxonomy and development prospects of Sanghuang (Sanghuangporus sanghuang). J. Fungal Res. 14 (4), 187–200. doi: 10.13341/j.jfr.2014.6401
Yu, H. L., Zhang, M. Y., Sun, Y. T., Li, Q. Z., Liu, J. Y., Song, C. Y., et al. (2022). Whole-genome sequence of a high-temperature edible mushroom Pleurotus giganteus (zhudugu). Front. Microbiol. 13. doi: 10.3389/fmicb.2022.941889
Zeng, X. Y., Zhu, Y. J., Li, Y., Sun, C. (2016). Research of cytochrome P450 in Ganoderma sinense based on analysis of genomic data. Mod. Chin. Med. 12 (18), 1555–1559. doi: 10.13313/j.issn.1673-4890.2016.12.005
Zhang, H. N., Jiang, F. C., Li, L. Q., Liu, X. Z., Yan, J. K. (2022). Recent advances in the bioactive polysaccharides and other key components from Phellinus spp. and their pharmacological effects: A review. Int. J. Biol. Macromol. 222, 3108–3128. doi: 10.1016/j.ijbiomac.2022.10.085
Zheng, F., Meng, G., An, Q., Tian, X. M., Si, J. (2017). Antioxidant activities of medicinal fungus Sanghuangporus sanghuang during liquid cultivation. Mycosystema 36 (1), 98–111. doi: 10.13346/j.mycosystema.160011
Zheng, T., Jiang, T. T., Huang, Z. L., Ma, H. X., Wang, M. X. (2023). Role of traditional Chinese medicine monomers in cerebral ischemia/reperfusion injury: a review of the mechanism. Front. Pharmacol. 14. doi: 10.3389/fphar.2023.1220862
Zhou, L. W., Vlasák, J., Decock, C., Assefa, A., Stenlid, J., Abate, D., et al. (2016). Global diversity and taxonomy of the Inonotus linteus complex (Hymenochaetales, Basidiomycota): Sanghuangporus gen. nov., Tropicoporus excentrodendri and T. guanacastensis gen. et spp. nov., and 17 new combinations. Fungal Divers. 77 (1), 335–347. doi: 10.1007/s13225-015-0335-8
Keywords: Sanghuang, medicinal properties, secondary metabolites, cytochromes P450, carbohydrate-active enzymes, evolution
Citation: Jin C, Ma J-X, Wang H, Tang L-X, Ye Y-F, Li X and Si J (2024) First genome assembly and annotation of Sanghuangporus weigelae uncovers its medicinal functions, metabolic pathways, and evolution. Front. Cell. Infect. Microbiol. 13:1325418. doi: 10.3389/fcimb.2023.1325418
Received: 21 October 2023; Accepted: 18 December 2023;
Published: 09 January 2024.
Edited by:
Gang Ding, Peking Union Medical College, ChinaReviewed by:
Kunlong Yang, Jiangsu Normal University, ChinaLili Shu, Shenyang Agricultural University, China
JinPing Yan, Kunming University of Science and Technology, China
Qiang Li, Chengdu University, China
Copyright © 2024 Jin, Ma, Wang, Tang, Ye, Li and Si. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Si, amluZ3NpMTc4OEAxMjYuY29t