 Xingcen Chen
Xingcen Chen Ruyi Peng1,2,3
Ruyi Peng1,2,3 Dongzi Peng
Dongzi Peng Deliang Liu
Deliang Liu Rong Li
Rong Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol. , 08 December 2023
Sec. Bacteria and Host
Volume 13 - 2023 | https://doi.org/10.3389/fcimb.2023.1282956
This article is part of the Research Topic Reviews in Microbiome in Health & Disease View all 13 articles
Helicobacter pylori (H. pylori) infection is thought to impact various extragastric diseases, including nonalcoholic fatty liver disease (NAFLD), the most common chronic liver disease. Meanwhile, the pathogenesis of NAFLD needs further research, and effective treatment for this disease remains elusive. In this mini-review, we enumerate and ponder on the evidence demonstrating an association between H. pylori infection and NAFLD. Primarily, we delve into high-quality meta-analyses and clinical randomized controlled trials focusing on the association studies between the two. We also discuss clinical studies that present opposite conclusions. In addition, we propose a mechanism through which H. pylori infection aggravates NAFLD: inflammatory cytokines and adipocytokines, insulin resistance, lipid metabolism, intestinal barrier and microbiota, H. pylori outer membrane vesicles and H. pylori-infected cell-extracellular vesicles. This mini-review aims to further explore NAFLD pathogenesis and extragastric disease mechanisms caused by H. pylori infection.
Helicobacter pylori (H. pylori) is a gram-negative bacillus that colonizes the human stomach. It is microaerophilic and has the ability to metabolize urea into ammonia and carbon dioxide. H. pylori is transmitted through the oral-oral and fecal-oral routes, and it infects approximately 4.4 billion people worldwide, with a prevalence rate of 44.3% (95% CI: 40.9-47.7) (Hooi et al., 2017; Sjomina et al., 2018; Zamani et al., 2018). An epidemiological survey based on family units revealed that the prevalence of H. pylori in China was approximately 40.66%, 43.45% in adults, and 20.55% in children and adolescents (Zhou et al., 2023). Multiple studies have substantiated that H. pylori infection serves as the initiating factor in the progression of chronic gastritis to gastric cancer (Correa, 1992; Uemura et al., 2001; Kusters et al., 2006). In addition to peptic ulcers, gastritis, gastric cancer, and other gastric diseases, a variety of extragastric diseases, such as stroke, Alzheimer’s disease, and nonalcoholic fatty liver disease (NAFLD), are closely related to H. pylori infection (Santos et al., 2020).
NAFLD refers to a spectrum of diseases, including simple hepatocellular steatosis, nonalcoholic steatohepatitis (NASH), NASH-associated cirrhosis, and hepatocellular carcinoma. With changes in diet and lifestyle, NAFLD is the the most common chronic liver disease (Chalasani et al., 2018; Younossi et al., 2020). Moreover, in the United States, NAFLD is the second leading cause of liver transplantation after alcoholic liver disease (Kim et al., 2018). The overall global prevalence of NAFLD was estimated to be approximately 29.1% (95% CI: 26.8-31.5), with the highest prevalence in Latin America (44.4%) and the lowest in Western Europe (24.6%). Moreover, the global prevalence of NAFLD is progressively increasing, rising from 24.4% in 1991-2006 to 36.0% in 2016-2020 (Henry et al., 2022). However, the pathogenesis of NAFLD remains unknown. The multiple-hit pathogenesis reviewed by Buzzetti et al. is widely accepted in academia and includes insulin resistance (IR), hormones secreted fromadipose tissue, nutritional factors, and gut microbiota (Buzzetti et al., 2016). No agents have been approved for the clinical treatment of NAFLD, and their treatment options rely mainly on weight loss through dietary modification and physical exercise (Wang and Malhi, 2018). Therefore, it is necessary to correctly discern the pathogenesis of NAFLD and propose targeted treatment options for NAFLD.
Since the initial report of H. pylori DNA being detected in the liver of NAFLD patients (Cindoruk et al., 2008), numerous studies have investigated the relationship between H. pylori infection and NAFLD (Kim et al., 2017; He et al., 2018; Yu et al., 2022). Based on the pandemic and the rate of H. pylori infection and NAFLD worldwide, a large proportion of patients have comorbid diseases. Despite the increasing severity of antibiotic-resistant forms of H. pylori, the eradication rate with multiple first-line treatment regimens remains more than 80% (Rokkas et al., 2021). Compared with NAFLD, for which no effective medical therapy is currently available, H. pylori can be eradicated in a large proportion of patients with comorbidities. In that case, it can delay or improve the progression of NAFLD and maximize the benefit of patients with comorbid conditions while greatly relieving the disease burden of NAFLD. However, there remains controversy about whether H. pylori infection is clinically associated with NAFLD, with some extensive multicenter clinical studies suggesting an association (Sumida et al., 2015; Kim et al., 2017; Doulberis et al., 2020) and others suggesting no association (Jamali et al., 2013; Okushin et al., 2015; Baeg et al., 2016). Even if they are relevant, mechanistic studies are needed to conduct research on how H. pylori infection impacts NAFLD. This mini-review intends to review the association between H. pylori and NAFLD and propose a hypothesis according to the summarized literature: H. pylori infection may exacerbate the development of NAFLD. Moreover, the authors explore the mechanism of the occurrence and development of NAFLD and the mechanism of extragastric diseases caused by H. pylori.
Basic research on H. pylori infection and NAFLD is rare, and no precise mechanism has been found. He et al. (He et al., 2018) established a mouse model of H. pylori infection, fed a high-fat diet (HFD) and a chow diet for six months, which showed that HFD plus H. pylori-infected mice had significantly increased abdominal circumference, fasting blood glucose (FBG), low-density lipoprotein cholesterol (LDL-C), and alanine aminotransferase (ALT) compared with HFD controls, and showed more severe hepatic steatosis, which was consistent with our hypothesis. Liver fibrosis is a progressive manifestation of NAFLD (Friedman et al., 2018). H. pylori infection has demonstrated to promote CCl4-induced liver fibrosis in animal models (Goo et al., 2009), and that the proinflammatory signaling pathways may occur through transforming growth factor-beta1 (TGF-β1) (Ki et al., 2010).
The progress of experimental research on the association between H. pylori infection and NAFLD has been slow, which could be attributed to the following factors: (1) challenges in mimicking the complex hepatic physiological environment in cellular and molecular experiments; (2) H. pylori infection does not promote NAFLD through direct pathways; and (3) H. pylori infection is not associated with NAFLD. More experimental studies in line with human physiology, such as hepatic organoids, are needed to demonstrate whether H. pylori infection is associated with NAFLD.
Table 1 summarizes nine meta-analyses of clinical research on H. pylori infection with NAFLD (Wijarnpreecha et al., 2018; Liu et al., 2019; Mantovani et al., 2019; Ning et al., 2019; Zhou et al., 2019; Wei and Ding, 2021; Heydari et al., 2022; Ma et al., 2022; Xu et al., 2023). These studies included a minimum of 38,622 and a maximum of 218,573 participants. Most clinical studies included in these meta-analyses were conducted in the Asian region (China, Japan, and South Korea), and a few involved Europe, the United States, and Egypt. The results of the subgroup analysis suggested that H. pylori infection was stable and associated with NAFLD in Asian regions (P < 0.01), and there were no uniform results in other regions due to different inclusion studies in each meta-analysis (Mantovani et al., 2019; Ning et al., 2019; Zhou et al., 2019; Wei and Ding, 2021; Xu et al., 2023). This may be due to differences in the cytotoxin-associated gene A (Cag A) status of H. pylori and strain virulence between Asia and other regions (Yamaoka, 2009; Park et al., 2018). The heterogeneity of these meta-analyses was generally high, which may be related to the differences in the region, population, and methods of diagnosis included in the study. Only one meta-analysis had publication bias (Ning et al., 2019), which could be due to the small sample size of the studies. After removing two studies with small sample sizes, publication bias was eliminated, and H. pylori infection remained significantly associated with NAFLD. All meta-analyses demonstrated a positive association between H. pylori infection and NAFLD. The odds ratio (OR) ranged from 1.14 to 1.53, which meant that the proportion of H. pylori infection in NAFLD patients was 1.14 - 1.53 times higher than that in their non-NAFLD counterparts. We hypothesize that H. pylori infection alone may have difficulty causing NAFLD, but H. pylori infection combined with a fast food diet and lifestyle disorders may exacerbate NAFLD levels.
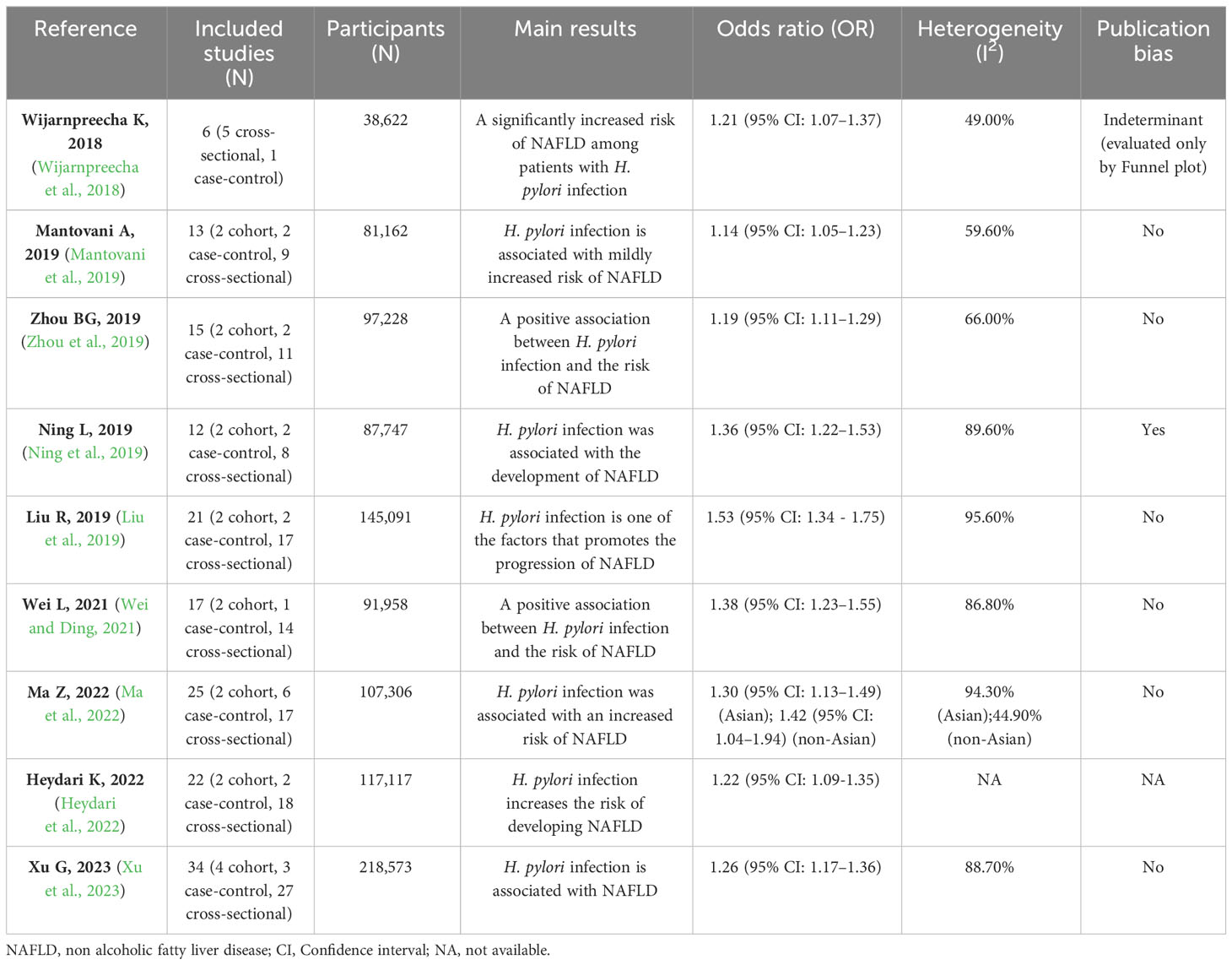
Table 1 Meta-analyses of the association between H. pylori infection and NAFLD.
The meta-analyses performed subgroup analyses according to race, region, diagnostic methods, and different study types to make the results more robust and credible and to reduce overall heterogeneity. However, most included studies were cross-sectional and could not illustrate the causal relationship between H. pylori and NAFLD. Second, correcting the effects of confounding factors such as hygiene level, dietary habits, physiological activity, and genetics from the clinical data collected by meta-analyses was difficult. With the specification of the NAFLD definition, the nomenclature of new fatty liver disease- metabolic dysfunction-associated steatotic liver disease (MASLD) (Rinella et al., 2023) will provide more accurate and high-quality clinical studies on the relationship between H. pylori infection and NAFLD/MASLD.
The above meta-analyses results suggest that H. pylori infection is positively associated with NAFLD. However, some clinical studies are controversial to this conclusion. We collected 13 clinical studies on H. pylori infection that had no bearing on NAFLD, including one bidirectional Mendelian randomization (MR) study (Liu et al., 2022), two clinical trials (Jamali et al., 2013; Polyzos et al., 2014), and ten cross-sectional studies (Okushin et al., 2015; Baeg et al., 2016; Cai et al., 2018; Fan et al., 2018; Kang et al., 2018; Lu et al., 2018; Han et al., 2021; Rahman et al., 2021; Wang et al., 2022a; Wernly et al., 2022), as shown in Table 2. Then, we provide a detailed dialectical discussion of clinical trials on studies needing revised or improved designs. A detailed discussion of the clinical randomized controlled trials (RCTs) by Jamali et al. (Jamali et al., 2013) and Polyzos et al. (Polyzos et al., 2014) is provided in Section 2.3.
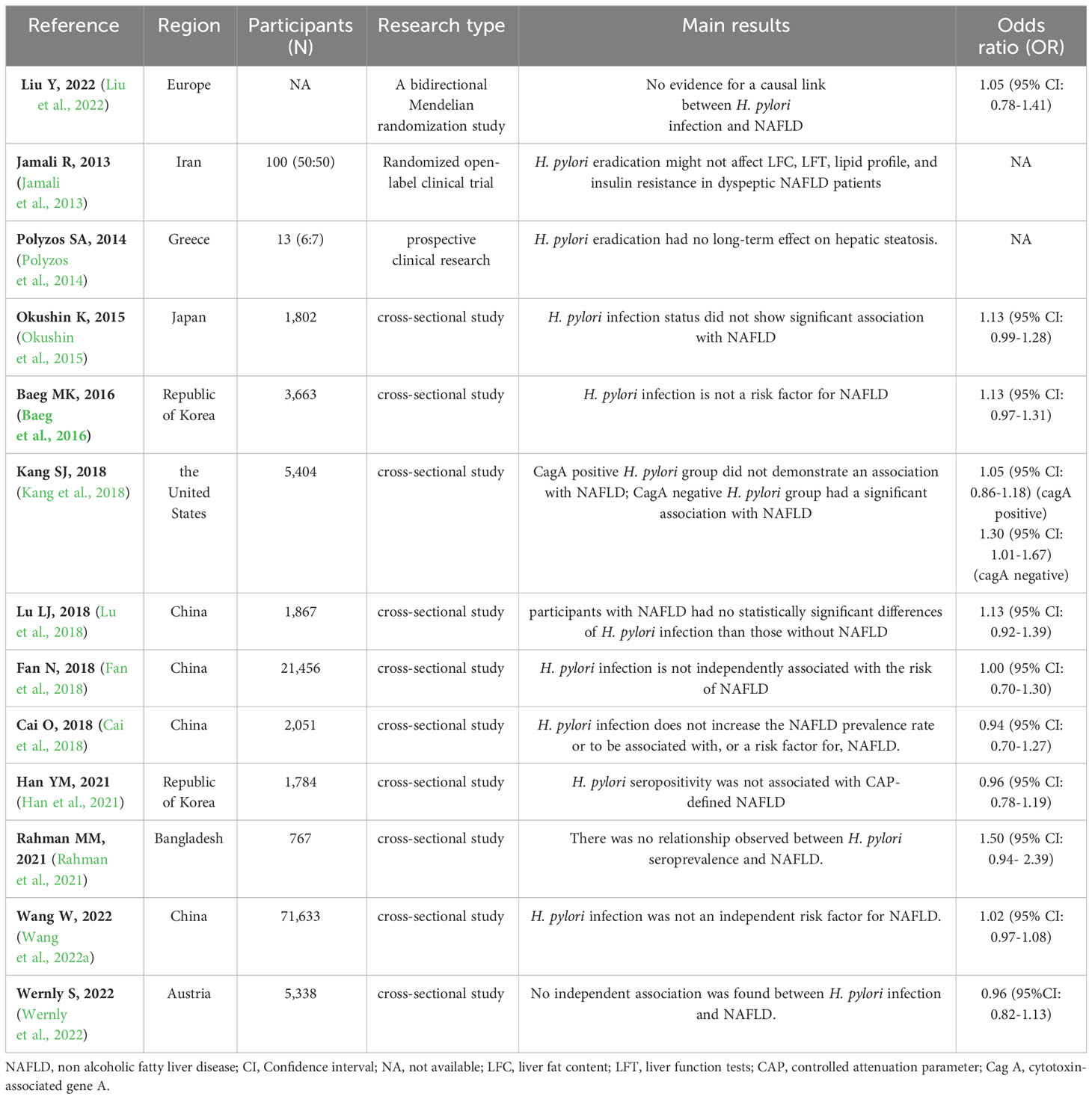
Table 2 Clinical studies on H. pylori infection is not associated with NAFLD.
MR, which uses single nucleotide polymorphisms as instrumental variables to investigate the causal relationship between exposure factors and disease, have been widely used in epidemiological causal inferences in recent years (Davey Smith and Hemani, 2014). Liu et al. (Liu et al., 2022) showed no causal link between H. pylori infection and NAFLD. Moreover, H. pylori infection was not significantly associated with triglycerides (TG), LDL-C, high-density lipoprotein cholesterol (HDL-C), or FBG. However, in this study, H. pylori infection diagnosis was based on serological testing and only involved European participants, which could have made the results potentially over-evaluated.
The cross-sectional study results were quite different. The lower 95% CI of the OR for some studies (Okushin et al., 2015; Baeg et al., 2016; Rahman et al., 2021; Wang et al., 2022a) was very close to 1, suggesting that the statistical results have questionable reliability in practice. This may be due to (1) different H. pylori strains circulating in various regions; (2) H. pylori infection not being diagnosed uniformly, such as 13C- urea breath test and serum H. pylori antibody detection; (3) NAFLD being diagnosed differently, such as ultrasonography and hepatic steatosis index (HSI) and NAFLD liver fat score (NAFLD-LFS); and (4) differences in race and living routine between regions. Fan et al.(Fan et al., 2018) showed a significant association between H. pylori infection and NAFLD after adjusting for age and sex (OR = 1.1, 95% CI: 1.0 - 1.1, P = 0.004). Nevertheless, after adjusting for body mass index (BMI) and systolic and diastolic blood pressure, there was no significant relationship between them (OR = 0.9, 95% CI: 0.9 - 1.0, P = 0.097). Finally, fasting plasma glucose, hemoglobin A1C (HbA1c), triglycerides, total cholesterol, HDL-C, LDL-C, and serum creatinine were adjusted (OR = 1.0, 95% CI: 0.7 - 1.3, P = 0.753). After adjusting for the metabolic index, H. pylori infection was not associated with NAFLD, which suggested that H. pylori infection may lead to metabolic disturbances rather than directly causing NAFLD. Our cross-sectional study of 16,942 participants also presents a significant link between H. pylori infection and metabolic index in humans (unpublished data), consistent with existing findings (Buzás, 2014).
Clinical RCTs are the gold standard for assessing the theoretical efficacy of clinical interventions. We collected recent RCTs on H. pylori eradication therapy in H. pylori-positive NAFLD patients (Jamali et al., 2013; Polyzos et al., 2014; Abdel-Razik et al., 2018; Maharshi et al., 2020; Yu et al., 2022). According to clinical RCTs, we speculated that eradicating H. pylori in H. pylori -positive NAFLD patients contributes to improving metabolic parameters.
The first randomized open-label clinical trial (Jamali et al., 2013) finally enrolled 100 patients with NAFLD randomly divided into a lifestyle modification group and a lifestyle modification plus H. pylori eradication group. After six months, it was found that liver fat content, liver function tests, lipid profile, and IR were improved in both groups compared with baseline levels. Nonetheless, there was no statistically significant difference between the two groups. Notably, the results of a recent randomized controlled trial of H. pylori eradication in NAFLD patients were the opposite. Yu et al. (Yu et al., 2022) enrolled 191 NAFLD patients with H. pylori infection and randomly divided them into untreated (health education and lifestyle guidance) and treated (health education and lifestyle guidance plus 14 days of H. pylori quadruple therapy) groups. One year later, the patient’s metabolic index and FibroScan controlled attenuation parameter (CAP) values improved compared to those before treatment. The metabolic index [FBG, HbA1c (%), homeostatic model assessment of insulin resistance (HOMA-IR), TG, and BMI], CAP value, and inflammatory parameters [white blood cells, high-sensitivity C-reactive protein, interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α)] of the treated group were significantly improved compared with those of the untreated group. These two prospective studies came to a paradoxical conclusion, which may be mainly due to differences in inclusion criteria and follow-up times. Jamali et al. included NAFLD patients with ALT and AST greater than the upper limit of normal (ULN) who developed dyspeptic symptoms. Yu et al. included NAFLD patients with ALT and AST < 2 times the ULN and no gastrointestinal disease symptoms. In other words, Jamali et al. included patients with moderate and severe NAFLD; Yu et al. included patients with mild NAFLD. There was no difference in H. pylori eradication between the two groups in the former and a significant improvement between the two groups in the latter, illustrating the necessity of early eradication of H. pylori in NAFLD patients with H. pylori infection. Polyzos et al. (Polyzos et al., 2014) recruited 13 patients with biopsy-proven NAFLD who were divided into H. pylori (+) and H. pylori (−) groups according to whether they had H. pylori infection, both of whom received standard instructions for diet and exercise, and the H. pylori (+) group received H. pylori eradication therapy. Hepatic steatosis, HSENSI [Homocysteine, serum glutamic oxaloacetic transaminase, Erythrocyte sedimentation rate, and Nonalcoholic Steatohepatitis Index] were assessed twelve months later. The results indicated that H. pylori eradication had no long-term effect on magnetic resonance imaging -assessed hepatic steatosis. However, they suggested a trend toward improved NAFLD fibrosis score and HSENSI with H. pylori eradication. Interestingly, a retrospective study by the Polyzos team (Doulberis et al., 2020) suggested that active H. pylori infection was significantly associated with liver function, HOMA-IR, and liver fibrosis stage. The inclusion criteria for this study were patients with NAFLD demonstrated by liver biopsy and divided into H. pylori (+) and H. pylori (−) groups according to the presence or absence of H. pylori infection demonstrated by gastric biopsy. The findings of the same team seem paradoxical, probably because prospective studies included insufficient participants.
Abdel-Razik et al. (Abdel-Razik et al., 2018) followed 369 NAFLD patients (171 H. pylori-positive and 198 H. pylori-negative) for 24 months. They found that H. pylori eradication significantly reduced IR, lipid profile, HSI, and NAFLD-LFS and increased HDL. Maharshi et al. (Maharshi et al., 2020) followed 64 NAFLD patients (36 H. pylori-positive and 28 H. pylori-negative) for six months and found that H. pylori eradication improved hepatic steatosis and metabolic parameters. These findings also coincide with the results of another prospective RCT (Gen et al., 2010): beneficial impacts of H. pylori eradication therapy on IR, atherogenic lipid abnormalities, and low-grade inflammation.
Currently, there is no direct experimental mechanistic evidence that H. pylori infection impacts NAFLD, and the primary explanation focuses on IR, inflammatory cytokines or adipocytokines, lipid metabolism, and the intestinal barrier (Li et al., 2013; Cheng et al., 2017; Doulberis et al., 2021). We summarize the latest experimental evidence on the effects of H. pylori on the above factors and propose a new pathway: extracellular vesicles (EVs) or H. pylori outer membrane vesicles (H. pylori-OMVs), as shown in Figure 1. It should be noted that these possible mechanisms do not act independently on the development of NAFLD, such as multiple inflammatory cytokines that can also be involved in IR, lipid metabolism disorders, and intestinal barrier dysfunction. These factors are jointly engaged in multiple hits in NAFLD.
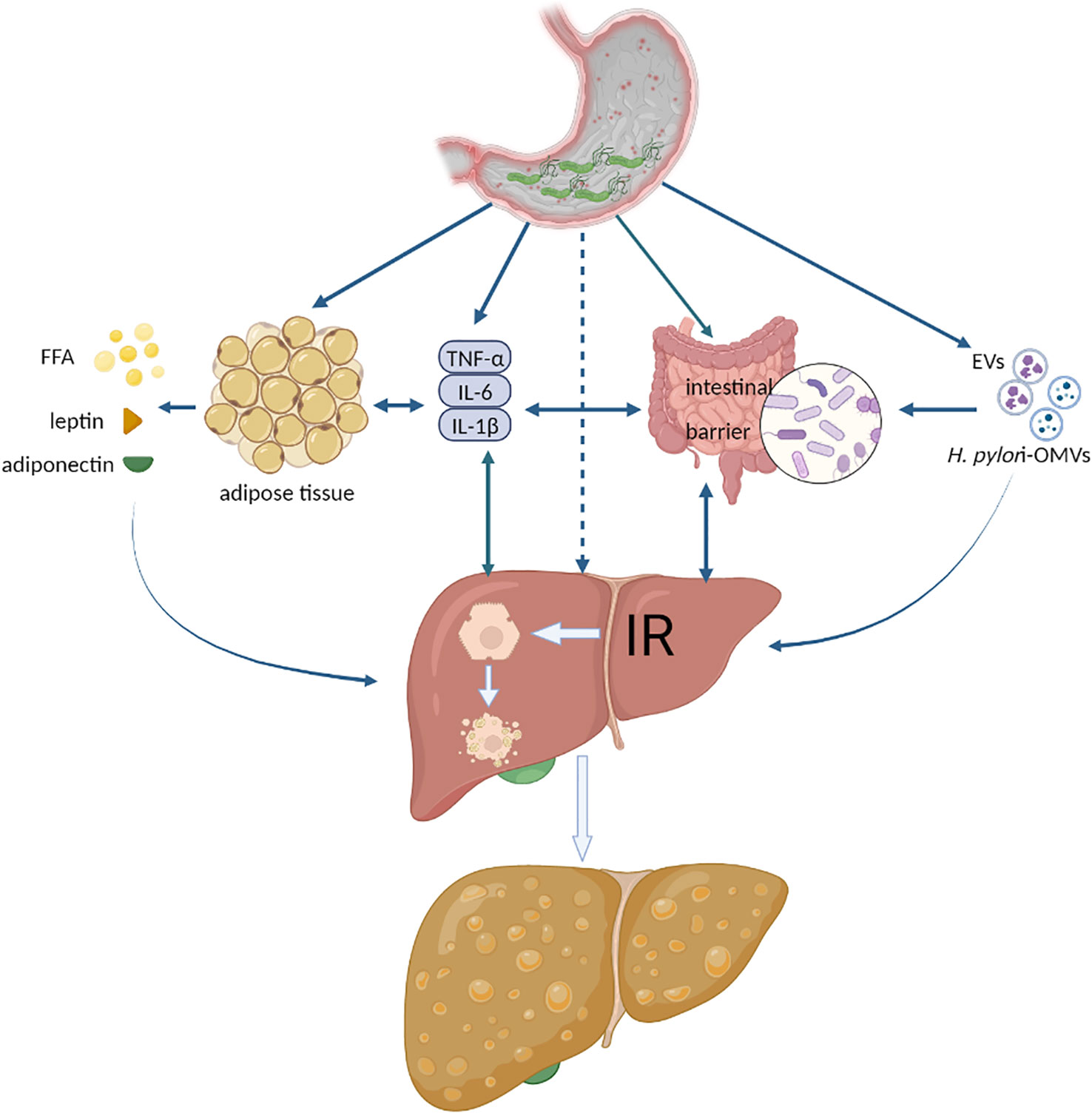
Figure 1 The possible mechanisms of H. pylori infection exacerbating NAFLD. FFA, free fatty acid; IR, insulin resistance; EVs, extracellular vesicles; H. pylori-OMVs, H. pylori- outer membrane vesicles. Authors hypothesize that systemic low-grade systemic inflammation caused by H. pylori infection aggravates NAFLD. Various inflammatory cytokines act directly or indirectly on adipose tissue, liver, and intestine to trigger or aggravate IR, disrupt the intestinal barrier, lead to hepatocyte steatosis, or activate liver fibrosis. Another possible mechanism is extracellular vesicles released by H. pylori-infected host cells or outer membrane vesicles secreted by H. pylori, which act directly with the liver and promote the development of NAFLD.
Inflammatory cytokines play a critical role in the pathology of H. pylori infection and NAFLD. In patients with persistent H. pylori infection, the body may lead to a chronic low-grade inflammation state and increased levels of the NOD-like receptor protein 3 (NLRP3) inflammasome (Pérez-Figueroa et al., 2016) and inflammatory cytokines, such as interleukin-1beta (IL-1β), IL-6, and TNF-α (Buzás, 2014). Inflammasomes and inflammatory cytokines are secreted by H. pylori-infected gastric epithelial cells and mucosal and circulating monocytes (Algood and Cover, 2006), reaching the liver via the circulatory system. NLRP3 and IL-1β participat in the whole process of liver inflammation, including IR and liver fibrosis (Tilg et al., 2016). IL-1β is directly involved in IL-1β/TNF-induced hepatocyte necrosis (Shen et al., 2020), promoting hepatic steatosis (Stienstra et al., 2010; Negrin et al., 2014), with positive feedback amplification of inflammation-induced IL-1β and TNF-α (Petrasek et al., 2012). In addition, researchers have found that IL-1β inhibits the fibroblast growth factor 21 (FGF 21) coreceptor beta-Klotho (KLB) to suppress FGF21 physiological effects (Zhao et al., 2016), which is crucial in regulating hepatic lipid metabolism and anti-inflammation (Xu et al., 2009). Mitsuyoshi et al. found that NLRP3, procaspase-1, IL-1β, and IL-18 mRNA levels were significantly increased in the livers of NAFLD patients compared to healthy controls (Mitsuyoshi et al., 2017). Accordingly, choline-deficient amino acid-defined diet-induced hepatomegaly, liver inflammation, and fibrosis were significantly ameliorated in Nlrp3 knockout mice compared with wild-type mice (Wree et al., 2014). Similar to IL-1β and NLRP3, TNF-α, and IL-6 play a direct or indirect role in the progression of NAFLD. TNF-α directly increases the expression of mast cell proteinase 1, Tgfb1, and tissue inhibitor of metalloproteinase 1 in hepatocytes, participating in hepatic lipid metabolism, inflammation, and liver fibrosis processes (Kakino et al., 2018). It is well-known that TNF-α also phosphorylates the serine of insulin receptor substrate-1 (IRS-1) to induce IR (Hotamisligil et al., 1996). Studies have demonstrated that IL-6 is involved in NASH progression through the IL-6/signal transducer and activator of transcription 3 (STAT3) signaling pathway (Cai et al., 2016; Li et al., 2022).
Visceral white adipose tissue (WAT), when infiltrated by inflammatory cells, releases adipocytokines (including adiponectin, leptin, and resistin) and inflammatory factors (such as TNF-α and IL-6), which are involved in regulating IR and inflammation via endocrine or paracrine mechanisms (Kershaw and Flier, 2004; Stojsavljević et al., 2014). Adiponectin plays a role in alleviating IR and reducing intrahepatic triglyceride accumulation and is well known as a protective factor for NAFLD. On the one hand, adiponectin activates AMP-activated kinase (AMPK) by binding to adiponectin receptor 2 on the surface of hepatocytes, inhibits acetyl-CoA carboxylase, and decreases malonyl-CoA production, thereby increasing β-oxidation of fatty acids (FAs). On the other hand, adiponectin inhibits glycogenolysis and gluconeogenesis by inhibiting glucose-6-phosphatase and phosphoenolpyruvate carboxyl kinase mRNA expression (Combs and Marliss, 2014). Normally, hepatic leptin activates the phosphatidylinositol-3 kinase/Akt (protein kinase B)/mammalian target of rapamycin (mTOR) pathway mainly by binding to hepatocyte surface leptin receptor b, suppressing hepatic glucose production and improving insulin sensitivity. In addition, leptin can also activate the Janus kinase 2/STAT3 signaling pathway in the liver and regulate the function of suppressors of cytokine signaling 3 (SOSC3) function, a negative feedback regulatory molecule of the leptin receptor signaling pathway (Polyzos et al., 2015). However, the association of H. pylori infection with serum adipocytokines remains debated, as clinical observational studies have reached contradictory conclusions. Chen et al. found no significant difference in circulating leptin and adiponectin levels between H. pylori-positive and H. pylori-negative patients (Chen et al., 2015). Abdel-Razik et al. found a significant decrease in serum leptin and no significant change in adiponectin levels after H. pylori eradication therapy (Abdel-Razik et al., 2018), while Ando et al. found a significant increase in serum adiponectin levels after H. pylori eradication therapy (Ando et al., 2013). Future large-scale, prospective studies are needed to find specific links between H. pylori infection and adipocytokines.
IR, a central cause of NAFLD development, plays a significant role in hepatic triglyceride deposition, the inflammatory response, and hepatic fibrosis progression (Watt et al., 2019). Numerous studies have shown that H. pylori infection is an independent risk factor for IR. A cross-sectional study involving 1107 participants found that IR patients had a significantly higher prevalence rate of H. pylori infection than non-IR patients, even after adjusting for sex, age, BMI, waist circumference, visceral and subcutaneous adipose tissue, smoking status, alcohol consumption, dietary habits, and physical activity (Gunji et al., 2009). In H. pylori-infected patients, fasting glucose, fasting insulin, HbA1c, and HOMA-IR values decreased significantly after H. pylori eradication compared with those before treatment (Dogan et al., 2015). In addition, multiple meta-analyses have also indicated a link between H. pylori infection and IR (Polyzos et al., 2011; Azami et al., 2021). Animal studies have shown that HFD-fed mice with H. pylori infection developed more severe IR than HFD-fed mice alone, and mice fed a HFD for 12 weeks plus H. pylori infection were obese similar to mice fed a HFD for 24 weeks (He et al., 2016). Further studies revealed that H. pylori infection increases the expression of the inflammation-related transcription factor c-Jun. C-Jun can bind to the promoter region of the miR-203 gene to inhibit miR-203 expression, an inhibitor of the insulin negative feedback regulator SOCS3, and ultimately promote hepatic IR through the c-Jun/miR-203/SOCS3 pathway (Zhou et al., 2015).
Hepatocyte steatosis is the primary pathological manifestation of NAFLD; its essence is lipid metabolism disorder in hepatocytes (Powell et al., 2021). Clinical studies have suggested that H. pylori infection affects lipid metabolism (Buzás, 2014; Watanabe et al., 2021). A large-cohort propensity score-matched analysis revealed that eradicating H. pylori could alleviate the deterioration of lipid metabolism but not return to uninfected levels. Specifically, HDL-C continued to decrease, LDL-C continued to rise in H. pylori-infected patients after H. pylori eradication therapy, and their lipid changes were significantly greater than those in participants with persistent H. pylori-negative status and significantly smaller than those in patients with persistent H. pylori-positive status (Wang et al., 2022b).
Increased triglyceride synthesis [fatty acid uptake, de novo lipogenesis (DNL)] and decreased consumption [fatty acid β-oxidation, very low-density lipoprotein (VLDL) transport] in the liver are the leading causes of triglyceride deposition (Kawano and Cohen, 2013). Donnelly et al. (Donnelly et al., 2005) demonstrated hepatic triglyceride deposition in NAFLD patients, 59% from plasma nonesterified fatty acids [also called free fatty acids (FFAs)], 26% from de novo lipogenesis, and 15% from the diet. FFAs in plasma are mainly derived from adipose tissue lipolysis. There is no direct linkage to the evidence that H. pylori infection increases adipose tissue lipolysis, whereas chronic systemic inflammation triggered by H. pylori infection induces WAT lipolysis (Xu et al., 2003). It has been found that fatty acid synthase and ATP-citrate lyase, critical enzymes of DNL in the gastric mucosa of H. pylori-infected patients, are upregulated, which means that DNL in the gastric mucosa of H. pylori-infected patients is increased (Chen et al., 2020), but whether H. pylori infection directly affects DNL in the liver needs further experimental research. Moreover, H. pylori infection affects the intestinal barrier and gut microbiota, affecting diet-derived lipid metabolism (see Section 4.4). VLDL is a carrier for transporting TGs synthesized by the liver to peripheral organs or circulation. When hepatic lipid production is excessive and VLDL transport capacity is not matched, hepatic steatosis and lipotoxicity produced by hepatic triglyceride accumulation. It causes dyslipidemia, on the other hand (Heeren and Scheja, 2021). Circulating VLDL is significantly higher in H. pylori-infected patients than in non-infected patients (Işıktaş Sayılar et al., 2015), which may result indirectly from IR caused by H. pylori infection or endoplasmic reticulum stress in hepatocytes.
Substantial evidence has demonstrated that H. pylori infection impacts intestinal barrier function. Under pathological conditions such as hypoxia, inflammatory response, and intestinal microbiota disorders, intestinal bacteria, and their metabolites pass through the damaged intestinal barrier, enter the circulation and participate in the development of NAFLD (Sanduzzi Zamparelli et al., 2016).
A clinical study matching participants’ sex, age, BMI, alcohol consumption, smoking, proton pump inhibitor usage, history of peptic ulcer disease, and dietary habits found that H. pylori infection was associated with alterations in the fecal microbiota and increased overall diversity of fecal microbes (Frost et al., 2019). Experimental studies have found that mice fed a HFD plus H. pylori infection have increased intestinal abundance of Helicobacter and decreased abundance of Lactobacillus, with a loss of diversity. Meanwhile, H. pylori infection also aggravated HFD-induced hyperglycemia, which could not be restored even with H. pylori eradication (Peng et al., 2021). Another study in mice fed a HFD plus H. pylori infection found that the expression of tight junction components, such as occludin, zonula occludens-1, and claudin-1, which are important components of the intestinal barrier, was significantly decreased, indicating that H. pylori infection directly affects intestinal barrier function (He et al., 2016). Further studies revealed that CagA -containing exosomes increased intestinal permeability by upregulating Claudin-2 expression through activation of CDX2 (Caudal-related homeodomain transcription 2) (Guo et al., 2022). Intestinal barrier dysfunction gives rise to (1) increased intestinal permeability, intestinal bacteria and their metabolites (such as dimethylamine, trimethylamine) and lipopolysaccharide (LPS) in the liver, triggering a liver inflammatory response, hepatocyte damage, and liver fibrosis (Cui et al., 2019); (2) intestinal epithelial cells release inflammatory cytokines to promote NAFLD (Wigg et al., 2001); and (3) intestinal nutrient absorption dysfunction and metabolic substances such as choline deficiency (Spencer et al., 2011).
H. pylori-OMVs are bilayer membrane spherical vesicles released from H. pylori and contain various bacterial elements, such as LPS, outer membrane proteins (OMPs), and virulence proteins [such as CagA and Vacuolating cytotoxin A (Vac A)]. H. pylori-OMVs can influence bacterial survival, transmit toxins and virulence factors, and regulate the host immune response and genetic material transfer (Parker and Keenan, 2012). EVs are membranous vesicles secreted by H. pylori-infected host cells that maintain intercellular communication, promote inflammatory responses, and manipulate the lesion microenvironment (González et al., 2021). Extracellular vesicles are divided into two categories according to diameter: exosomes and microvesicles, the former of which have been most studied for their biological characteristics.
H. pylori-OMVs and H. pylori-infected host cell-derived EVs have been found to accelerate the development of various diseases (Chmiela et al., 2018; González et al., 2021; Qiang et al., 2022). CagA-containing exosomes have been demonstrated in the circulation of both CagA-positive H. pylori-infected patients and mice (Shimoda et al., 2016; Xia et al., 2020). Exosomes in the blood circulation reach the liver directly, impair endothelial function, activate hepatic Kupffer cells, and promote the hepatic inflammatory response and hepatocyte damage (Kazankov et al., 2019; Xia et al., 2020). Additionally, Zahmatkesh et al. (Zahmatkesh et al., 2022) found that exosomes derived from H. pylori-OMVs-infected hepatocytes activated hepatic stellate cells and upregulated the overexpression of liver fibrosis markers (vimentin, cadherin 1, and catenin beta 1). However, the role of H. pylori-OMVs and EVs in NAFLD needs further exploration.
H. pylori infection and NAFLD are chronic diseases, and their prolonged progression may lead to irreversible damage to the body. If H. pylori eradication therapy can delay or improve the physiological status of H. pylori positive NAFLD patients, it will significantly alleviate the disease burden of NAFLD. Numerous clinical and animal studies have suggested a link between H. pylori infection and NAFLD. We summarize here meta-analyses investigating the association between H. pylori infection and NAFLD, analyze some studies that oppose the association between them, and enumerate a number of high-quality evidence.
Understanding the relationship between H. pylori infection and NAFLD contributes to comprehending the mechanisms by which H. pylori leads to extragastric disease. This comprehension helps clinicians better understand and manage NAFLD and facilitates NAFLD patients with H. pylori infection to benefit from H. pylori eradication. Future multicenter prospective studies are needed to illustrate how H. pylori eradication will improve the physiological status of NAFLD patients or whether H. pylori eradication has additional benefits for NAFLD patients. In experimental studies, it is critical to determine whether H. pylori infection affects the progression of NAFLD in a direct (e.g., EVs or H. pylori-OMVs) or indirect (e.g., systemic chronic low-grade inflammation, IR) manner.
XC: Writing – original draft, Conceptualization, Data curation, Visualization. RP: Writing – original draft, Data curation, Visualization. DP: Writing – original draft, Data curation, Visualization. JX: Writing – original draft, Data curation, Visualization. DL: Writing – review & editing, Methodology, Supervision. RL: Writing – review & editing, Methodology, Supervision.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Natural Science Foundation of Hunan Province under Grant 211087206069.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdel-Razik, A., Mousa, N., Shabana, W., Refaey, M., Elhelaly, R., Elzehery, R., et al. (2018). Helicobacter pylori and non-alcoholic fatty liver disease: A new enigma. Helicobacter 23, e12537. doi: 10.1111/hel.12537
Algood, H. M., Cover, T. L. (2006). Helicobacter pylori persistence: an overview of interactions between H. pylori and host immune defenses. Clin. Microbiol. Rev. 19, 597–613. doi: 10.1128/CMR.00006-06
Ando, T., Ishikawa, T., Takagi, T., Imamoto, E., Kishimoto, E., Okajima, A., et al. (2013). Impact of Helicobacter pylori eradication on circulating adiponectin in humans. Helicobacter 18, 158–164. doi: 10.1111/hel.12028
Azami, M., Baradaran, H. R., Dehghanbanadaki, H., Kohnepoushi, P., Saed, L., Moradkhani, A., et al. (2021). Association of Helicobacter pylori infection with the risk of metabolic syndrome and insulin resistance: an updated systematic review and meta-analysis. Diabetol. Metab. Syndr. 13, 145. doi: 10.3748/wjg.v22.i8.2592
Baeg, M. K., Yoon, S. K., Ko, S. H., Noh, Y. S., Lee, I. S., Choi, M. G. (2016). Helicobacter pylori infection is not associated with nonalcoholic fatty liver disease. World J. Gastroenterol. 22, 2592–2600. doi: 10.3748/wjg.v22.i8.2592
Buzás, G. M. (2014). Metabolic consequences of Helicobacter pylori infection and eradication. World J. Gastroenterol. 20, 5226–5234. doi: 10.3748/wjg.v20.i18.5226
Buzzetti, E., Pinzani, M., Tsochatzis, E. A. (2016). The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metab. Clin. Exp. 65, 1038–1048. doi: 10.1016/j.metabol.2015.12.012
Cai, X., Fang, C., Hayashi, S., Hao, S., Zhao, M., Tsutsui, H., et al. (2016). Pu-erh tea extract ameliorates high-fat diet-induced nonalcoholic steatohepatitis and insulin resistance by modulating hepatic IL-6/STAT3 signaling in mice. J. Gastroenterol. 51, 819–829. doi: 10.1007/s00535-015-1154-0
Cai, O., Huang, Z., Li, M., Zhang, C., Xi, F., Tan, S. (2018). Association between helicobacter pylori infection and nonalcoholic fatty liver disease: A single-center clinical study. Gastroenterol. Res. Pract. 2018, 8040262. doi: 10.1155/2018/8040262
Chalasani, N., Younossi, Z., Lavine, J. E., Charlton, M., Cusi, K., Rinella, M., et al. (2018). The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67, 328–357. doi: 10.1002/hep.29367
Chen, L. W., Chien, C. Y., Yang, K. J., Kuo, S. F., Chen, C. H., Chien, R. N. (2015). Helicobacter pylori Infection Increases Insulin Resistance and Metabolic Syndrome in Residents Younger than 50 Years Old: A Community-Based Study. PloS One 10, e0128671. doi: 10.1371/journal.pone.0128671
Chen, P., Li, L., Wang, H., Zhao, J., Cheng, Y., Xie, J., et al. (2020). Omeprazole, an inhibitor of proton pump, suppresses De novo lipogenesis in gastric epithelial cells. BioMed. Pharmacother. 130, 110472. doi: 10.1016/j.biopha.2020.110472
Cheng, D. D., He, C., Ai, H. H., Huang, Y., Lu, N. H. (2017). The possible role of helicobacter pylori infection in non-alcoholic fatty liver disease. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00743
Chmiela, M., Walczak, N., Rudnicka, K. (2018). Helicobacter pylori outer membrane vesicles involvement in the infection development and Helicobacter pylori-related diseases. J. Biomed. Sci. 25, 78. doi: 10.1186/s12929-018-0480-y
Cindoruk, M., Cirak, M. Y., Unal, S., Karakan, T., Erkan, G., Engin, D., et al. (2008). Identification of Helicobacter species by 16S rDNA PCR and sequence analysis in human liver samples from patients with various etiologies of benign liver diseases. Eur. J. Gastroenterol. Hepatol. 20, 33–36. doi: 10.1097/MEG.0b013e3282efa4f2
Combs, T. P., Marliss, E. B. (2014). Adiponectin signaling in the liver. Rev. Endocr. Metab. Disord. 15, 137–147. doi: 10.1007/s11154-013-9280-6
Correa, P. (1992). Human gastric carcinogenesis: a multistep and multifactorial process–First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 52, 6735–6740.
Cui, Y., Wang, Q., Chang, R., Zhou, X., Xu, C. (2019). Intestinal barrier function-non-alcoholic fatty liver disease interactions and possible role of gut microbiota. J. Agric. Food Chem. 67, 2754–2762. doi: 10.1021/acs.jafc.9b00080
Davey Smith, G., Hemani, G. (2014). Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 23, R89–R98. doi: 10.1093/hmg/ddu328
Dogan, Z., Sarikaya, M., Ergul, B., Filik, L. (2015). The effect of Helicobacter pylori eradication on insulin resistance and HbA1c level in people with normal glucose levels: a prospective study. BioMed. Pap Med. Fac Univ Palacky Olomouc Czech Repub 159, 242–245. doi: 10.5507/bp.2014.036
Donnelly, K. L., Smith, C. I., Schwarzenberg, S. J., Jessurun, J., Boldt, M. D., Parks, E. J. (2005). Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 115, 1343–1351. doi: 10.1172/JCI23621
Doulberis, M., Papaefthymiou, A., Srivastava, D. S., Exadaktylos, A. K., Katsinelos, P., Kountouras, J., et al. (2021). Update on the association between non-alcoholic fatty liver disease and Helicobacter pylori infection. Int. J. Clin. Pract. 75, e13737. doi: 10.1111/ijcp.13737
Doulberis, M., Srivastava, S., Polyzos, S. A., Kountouras, J., Papaefthymiou, A., Klukowska-Rötzler, J., et al. (2020). Active helicobacter pylori infection is independently associated with nonalcoholic steatohepatitis in morbidly obese patients. J. Clin. Med. 9, 933. doi: 10.3390/jcm9040933
Fan, N., Peng, L., Xia, Z., Zhang, L., Wang, Y., Peng, Y. (2018). Helicobacter pylori infection is not associated with non-alcoholic fatty liver disease: A cross-sectional study in China. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.00073
Friedman, S. L., Neuschwander-Tetri, B. A., Rinella, M., Sanyal, A. J. (2018). Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 24, 908–922. doi: 10.1038/s41591-018-0104-9
Frost, F., Kacprowski, T., Rühlemann, M., Bang, C., Franke, A., Zimmermann, K., et al. (2019). Helicobacter pylori infection associates with fecal microbiota composition and diversity. Sci. Rep. 9, 20100. doi: 10.1038/s41598-019-56631-4
Gen, R., Demir, M., Ataseven, H. (2010). Effect of Helicobacter pylori eradication on insulin resistance, serum lipids and low-grade inflammation. South. Med. J. 103, 190–196. doi: 10.1097/SMJ.0b013e3181cf373f
González, M. F., Díaz, P., Sandoval-Bórquez, A., Herrera, D., Quest, A. (2021). Helicobacter pylori Outer Membrane Vesicles and Extracellular Vesicles from Helicobacter pylori-Infected Cells in Gastric Disease Development. Int. J. Mol. Sci. 22, 4823. doi: 10.3390/ijms22094823
Goo, M. J., Ki, M. R., Lee, H. R., Yang, H. J., Yuan, D. W., Hong, I. H., et al. (2009). Helicobacter pylori promotes hepatic fibrosis in the animal model. Lab. Invest. 89, 1291–1303. doi: 10.1038/labinvest.2009.90
Gunji, T., Matsuhashi, N., Sato, H., Fujibayashi, K., Okumura, M., Sasabe, N., et al. (2009). Helicobacter pylori infection significantly increases insulin resistance in the asymptomatic Japanese population. Helicobacter 14, 144–150. doi: 10.1111/j.1523-5378.2009.00705.x
Guo, Y., Xu, C., Gong, R., Hu, T., Zhang, X., Xie, X., et al. (2022). Exosomal CagA from Helicobacter pylori aggravates intestinal epithelium barrier dysfunction in chronic colitis by facilitating Claudin-2 expression. Gut Pathog. 14, 13. doi: 10.1186/s13099-022-00486-0
Han, Y. M., Lee, J., Choi, J. M., Kwak, M. S., Yang, J. I., Chung, S. J., et al. (2021). The association between Helicobacter pylori with nonalcoholic fatty liver disease assessed by controlled attenuation parameter and other metabolic factors. PloS One 16, e0260994. doi: 10.1371/journal.pone.0260994
He, C., Cheng, D., Wang, H., Wu, K., Zhu, Y., Lu, N. (2018). Helicobacter pylori infection aggravates diet-induced nonalcoholic fatty liver in mice. Clin. Res. Hepatol. Gastroenterol. 42, 360–367. doi: 10.1016/j.clinre.2017.12.008
He, C., Yang, Z., Cheng, D., Xie, C., Zhu, Y., Ge, Z., et al. (2016). Helicobacter pylori infection aggravates diet-induced insulin resistance in association with gut microbiota of mice. EBioMedicine 12, 247–254. doi: 10.1016/j.ebiom.2016.09.010
Heeren, J., Scheja, L. (2021). Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol. Metab. 50, 101238. doi: 10.1016/j.molmet.2021.101238
Henry, L., Paik, J., Younossi, Z. M. (2022). Review article: the epidemiologic burden of non-alcoholic fatty liver disease across the world. Aliment. Pharmacol. Ther. 56, 942–956. doi: 10.1111/apt.17158
Heydari, K., Yousefi, M., Alizadeh-Navaei, R., Lotfi, P., Sheydaee, F., Raei, M., et al. (2022). Helicobacter pylori infection and non-alcoholic fatty liver disease: A systematic review and meta-analysis. Turk J. Gastroenterol. 33, 171–181. doi: 10.5152/tjg.2022.21467
Hooi, J., Lai, W. Y., Ng, W. K., Suen, M., Underwood, F. E., Tanyingoh, D., et al. (2017). Global prevalence of helicobacter pylori infection: systematic review and meta-analysis. Gastroenterology 153, 420–429. doi: 10.1053/j.gastro.2017.04.022
Hotamisligil, G. S., Peraldi, P., Budavari, A., Ellis, R., White, M. F., Spiegelman, B. M. (1996). IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 271, 665–668. doi: 10.1126/science.271.5249.665
Işıktaş Sayılar, E., Çelik, B., Dumlu, Ş. (2015). Relationship between Helicobacter pylori infection and metabolic syndrome. Turk J. Gastroenterol. 26, 468–473. doi: 10.5152/tjg.2015.0197
Jamali, R., Mofid, A., Vahedi, H., Farzaneh, R., Dowlatshahi, S. (2013). The effect of helicobacter pylori eradication on liver fat content in subjects with non-alcoholic Fatty liver disease: a randomized open-label clinical trial. Hepat Mon 13, e14679. doi: 10.5812/hepatmon.14679
Kakino, S., Ohki, T., Nakayama, H., Yuan, X., Otabe, S., Hashinaga, T., et al. (2018). Pivotal role of TNF-α in the development and progression of nonalcoholic fatty liver disease in a murine model. Horm. Metab. Res. 50, 80–87. doi: 10.1055/s-0043-118666
Kang, S. J., Kim, H. J., Kim, D., Ahmed, A. (2018). Association between cagA negative Helicobacter pylori status and nonalcoholic fatty liver disease among adults in the United States. PloS One 13, e0202325. doi: 10.1371/journal.pone.0202325
Kawano, Y., Cohen, D. E. (2013). Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 48, 434–441. doi: 10.1007/s00535-013-0758-5
Kazankov, K., Jørgensen, S., Thomsen, K. L., Møller, H. J., Vilstrup, H., George, J., et al. (2019). The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 16, 145–159. doi: 10.1038/s41575-018-0082-x
Kershaw, E. E., Flier, J. S. (2004). Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 89, 2548–2556. doi: 10.1210/jc.2004-0395
Ki, M. R., Goo, M. J., Park, J. K., Hong, I. H., Ji, A. R., Han, S. Y., et al. (2010). Helicobacter pylori accelerates hepatic fibrosis by sensitizing transforming growth factor-β1-induced inflammatory signaling. Lab. Invest. 90, 1507–1516. doi: 10.1038/labinvest.2010.109
Kim, W. R., Lake, J. R., Smith, J. M., Schladt, D. P., Skeans, M. A., Harper, A. M., et al. (2018). OPTN/SRTR 2016 annual data report: liver. Am. J. Transplant. 18 Suppl 1, 172–253. doi: 10.1111/ajt.14559
Kim, T. J., Sinn, D. H., Min, Y. W., Son, H. J., Kim, J. J., Chang, Y., et al. (2017). A cohort study on Helicobacter pylori infection associated with nonalcoholic fatty liver disease. J. Gastroenterol. 52, 1201–1210. doi: 10.1007/s00535-017-1337-y
Kusters, J. G., van Vliet, A. H., Kuipers, E. J. (2006). Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 19, 449–490. doi: 10.1128/CMR.00054-05
Li, H., Liu, N. N., Li, J. R., Wang, M. X., Tan, J. L., Dong, B., et al. (2022). Bicyclol ameliorates advanced liver diseases in murine models via inhibiting the IL-6/STAT3 signaling pathway. BioMed. Pharmacother. 150, 113083. doi: 10.1016/j.biopha.2022.113083
Li, M., Shen, Z., Li, Y. M. (2013). Potential role of Helicobacter pylori infection in nonalcoholic fatty liver disease. World J. Gastroenterol. 19, 7024–7031. doi: 10.3748/wjg.v19.i41.7024
Liu, R., Liu, Q., He, Y., Shi, W., Xu, Q., Yuan, Q., et al. (2019). Association between Helicobacter pylori infection and nonalcoholic fatty liver: A meta-analysis. Med. (Abingdon) 98, e17781. doi: 10.1097/MD.0000000000017781
Liu, Y., Xu, H., Zhao, Z., Dong, Y., Wang, X., Niu, J. (2022). No evidence for a causal link between Helicobacter pylori infection and nonalcoholic fatty liver disease: A bidirectional Mendelian randomization study. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.1018322
Lu, L. J., Hao, N. B., Liu, J. J., Li, X., Wang, R. L. (2018). Correlation between helicobacter pylori infection and metabolic abnormality in general population: A cross-sectional study. Gastroenterol. Res. Pract. 2018, 7410801. doi: 10.1155/2018/7410801
Ma, Z., Chu, X., Yan, X., Wang, W. (2022). Association between Helicobacter pylori infection and non-alcoholic fatty liver disease for Asian and non-Asian population: A systematic review and meta-analysis. Front. Public Health 10. doi: 10.3389/fpubh.2022.1062942
Maharshi, V., Gupta, P., Kumar, V. L., Upadhyay, A. D., Das, P., Yadav, R., et al. (2020). Effect of Helicobacter pylori-eradication therapy on hepatic steatosis in patients with non-alcoholic fatty liver disease: a randomized-controlled pilot study. Gastroenterol. Rep. (Oxf) 8, 104–110. doi: 10.1093/gastro/goz058
Mantovani, A., Turino, T., Altomari, A., Lonardo, A., Zoppini, G., Valenti, L., et al. (2019). Association between Helicobacter pylori infection and risk of nonalcoholic fatty liver disease: An updated meta-analysis. Metab. Clin. Exp. 96, 56–65. doi: 10.1016/j.metabol.2019.04.012
Mitsuyoshi, H., Yasui, K., Hara, T., Taketani, H., Ishiba, H., Okajima, A., et al. (2017). Hepatic nucleotide binding oligomerization domain-like receptors pyrin domain-containing 3 inflammasomes are associated with the histologic severity of non-alcoholic fatty liver disease. Hepatol. Res. 47, 1459–1468. doi: 10.1111/hepr.12883
Negrin, K. A., Roth Flach, R. J., DiStefano, M. T., Matevossian, A., Friedline, R. H., Jung, D., et al. (2014). IL-1 signaling in obesity-induced hepatic lipogenesis and steatosis. PloS One 9, e107265. doi: 10.1371/journal.pone.0107265
Ning, L., Liu, R., Lou, X., Du, H., Chen, W., Zhang, F., et al. (2019). Association between Helicobacter pylori infection and nonalcoholic fatty liver disease: a systemic review and meta-analysis. Eur. J. Gastroenterol. Hepatol. 31, 735–742. doi: 10.1097/MEG.0000000000001398
Okushin, K., Takahashi, Y., Yamamichi, N., Shimamoto, T., Enooku, K., Fujinaga, H., et al. (2015). Helicobacter pylori infection is not associated with fatty liver disease including non-alcoholic fatty liver disease: a large-scale cross-sectional study in Japan. BMC Gastroenterol. 15, 25. doi: 10.1186/s12876-015-0247-9
Park, J. Y., Forman, D., Waskito, L. A., Yamaoka, Y., Crabtree, J. E. (2018). Epidemiology of helicobacter pylori and cagA-positive infections and global variations in gastric cancer. Toxins (Basel) 10, 163. doi: 10.3390/toxins10040163
Parker, H., Keenan, J. I. (2012). Composition and function of Helicobacter pylori outer membrane vesicles. Microbes Infect. 14, 9–16. doi: 10.1016/j.micinf.2011.08.007
Peng, C., Xu, X., He, Z., Li, N., Ouyang, Y., Zhu, Y., et al. (2021). Helicobacter pylori infection worsens impaired glucose regulation in high-fat diet mice in association with an altered gut microbiome and metabolome. Appl. Microbiol. Biotechnol. 105, 2081–2095. doi: 10.1007/s00253-021-11165-6
Pérez-Figueroa, E., Torres, J., Sánchez-Zauco, N., Contreras-Ramos, A., Alvarez-Arellano, L., Maldonado-Bernal, C. (2016). Activation of NLRP3 inflammasome in human neutrophils by Helicobacter pylori infection. Innate Immun. 22, 103–112. doi: 10.1177/1753425915619475
Petrasek, J., Bala, S., Csak, T., Lippai, D., Kodys, K., Menashy, V., et al. (2012). IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Invest. 122, 3476–3489. doi: 10.1172/JCI60777
Polyzos, S. A., Kountouras, J., Mantzoros, C. S. (2015). Leptin in nonalcoholic fatty liver disease: a narrative review. Metab. Clin. Exp. 64, 60–78. doi: 10.1016/j.metabol.2014.10.012
Polyzos, S. A., Kountouras, J., Zavos, C., Deretzi, G. (2011). The association between Helicobacter pylori infection and insulin resistance: a systematic review. Helicobacter 16, 79–88. doi: 10.1111/j.1523-5378.2011.00822.x
Polyzos, S. A., Nikolopoulos, P., Stogianni, A., Romiopoulos, I., Katsinelos, P., Kountouras, J. (2014). Effect of Helicobacter pylori eradication on hepatic steatosis, NAFLD fibrosis score and HSENSI in patients with nonalcoholic steatohepatitis: a MR imaging-based pilot open-label study. Arquivos gastroenterol. 51, 261–268. doi: 10.1590/s0004-28032014000300017
Powell, E. E., Wong, V. W., Rinella, M. (2021). Non-alcoholic fatty liver disease. Lancet 397, 2212–2224. doi: 10.1016/S0140-6736(20)32511-3
Qiang, L., Hu, J., Tian, M., Li, Y., Ren, C., Deng, Y., et al. (2022). Extracellular vesicles from helicobacter pylori-infected cells and helicobacter pylori outer membrane vesicles in atherosclerosis. Helicobacter 27, e12877. doi: 10.1111/hel.12877
Rahman, M. M., Kibria, M. G., Sultana, N., Akhter, M., Begum, H., Haque, M. A., et al. (2021). Seroprevalence of Helicobacter pylori and its association with metabolic syndrome in a rural community of Bangladesh. JGH Open 5, 64–72. doi: 10.1002/jgh3.12448
Rinella, M. E., Lazarus, J. V., Ratziu, V., Francque, S. M., Sanyal, A. J., Kanwal, F., et al. (2023). A multi-society Delphi consensus statement on new fatty liver disease nomenclature. J. Hepatol. S0168-8278(23)00418-X. doi: 10.1016/j.jhep.2023.06.003
Rokkas, T., Gisbert, J. P., Malfertheiner, P., Niv, Y., Gasbarrini, A., Leja, M., et al. (2021). Comparative effectiveness of multiple different first-line treatment regimens for helicobacter pylori infection: A network meta-analysis. Gastroenterology 161, 495–507.e4. doi: 10.1053/j.gastro.2021.04.012
Sanduzzi Zamparelli, M., Compare, D., Coccoli, P., Rocco, A., Nardone, O. M., Marrone, G., et al. (2016). The metabolic role of gut microbiota in the development of nonalcoholic fatty liver disease and cardiovascular disease. Int. J. Mol. Sci. 17, 1225. doi: 10.3390/ijms17081225
Santos, M., de Brito, B. B., da Silva, F., Sampaio, M. M., Marques, H. S., Oliveira E Silva, N., et al. (2020). Helicobacter pylori infection: Beyond gastric manifestations. World J. Gastroenterol. 26, 4076–4093. doi: 10.3748/wjg.v26.i28.4076
Shen, Y., Malik, S. A., Amir, M., Kumar, P., Cingolani, F., Wen, J., et al. (2020). Decreased hepatocyte autophagy leads to synergistic IL-1β and TNF mouse liver injury and inflammation. Hepatology 72, 595–608. doi: 10.1002/hep.31209
Shimoda, A., Ueda, K., Nishiumi, S., Murata-Kamiya, N., Mukai, S. A., Sawada, S., et al. (2016). Exosomes as nanocarriers for systemic delivery of the Helicobacter pylori virulence factor CagA. Sci. Rep. 6, 18346. doi: 10.1038/srep18346
Sjomina, O., Pavlova, J., Niv, Y., Leja, M. (2018). Epidemiology of Helicobacter pylori infection. Helicobacter 23 Suppl 1, e12514. doi: 10.1111/hel.12514
Spencer, M. D., Hamp, T. J., Reid, R. W., Fischer, L. M., Zeisel, S. H., Fodor, A. A. (2011). Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 140, 976–986. doi: 10.1053/j.gastro.2010.11.049
Stienstra, R., Saudale, F., Duval, C., Keshtkar, S., Groener, J. E., van Rooijen, N., et al. (2010). Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology 51, 511–522. doi: 10.1002/hep.23337
Stojsavljević, S., Gomerčić Palčić, M., Virović Jukić, L., Smirčić Duvnjak, L., Duvnjak, M. (2014). Adipokines and proinflammatory cytokines, the key mediators in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 20, 18070–18091. doi: 10.3748/wjg.v20.i48.18070
Sumida, Y., Kanemasa, K., Imai, S., Mori, K., Tanaka, S., Shimokobe, H., et al. (2015). Helicobacter pylori infection might have a potential role in hepatocyte ballooning in nonalcoholic fatty liver disease. J. Gastroenterol. 50, 996–1004. doi: 10.1007/s00535-015-1039-2
Tilg, H., Moschen, A. R., Szabo, G. (2016). Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 64, 955–965. doi: 10.1002/hep.28456
Uemura, N., Okamoto, S., Yamamoto, S., Matsumura, N., Yamaguchi, S., Yamakido, M., et al. (2001). Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 345, 784–789. doi: 10.1056/NEJMoa001999
Wang, W., Fan, M., Gong, R., Zhang, Y., Zeng, J., Xu, S., et al. (2022a). Helicobacter pylori infection is not an independent risk factor of non-alcoholic fatty liver disease in China. BMC Gastroenterol. 22, 81. doi: 10.1186/s12876-022-02148-6
Wang, X. J., Malhi, H. (2018). Nonalcoholic fatty liver disease. Ann. Intern. Med. 169, ITC65–ITC80. doi: 10.7326/AITC201811060
Wang, Z., Wang, W., Gong, R., Yao, H., Fan, M., Zeng, J., et al. (2022b). Eradication of Helicobacter pylori alleviates lipid metabolism deterioration: a large-cohort propensity score-matched analysis. Lipids Health Dis. 21, 34. doi: 10.1186/s12944-022-01639-5
Watanabe, J., Hamasaki, M., Kotani, K. (2021). The effect of helicobacter pylori eradication on lipid levels: A meta-analysis. J. Clin. Med. 10, 904. doi: 10.3390/jcm10050904
Watt, M. J., Miotto, P. M., De Nardo, W., Montgomery, M. K. (2019). The liver as an endocrine organ-linking NAFLD and insulin resistance. Endocr. Rev. 40, 1367–1393. doi: 10.1210/er.2019-00034
Wei, L., Ding, H. G. (2021). Relationship between Helicobacter pylori infection and nonalcoholic fatty liver disease: What should we expect from a meta-analysis. Med. (Abingdon) 100, e26706. doi: 10.1097/MD.0000000000026706
Wernly, S., Wernly, B., Semmler, G., Völkerer, A., Rezar, R., Semmler, L., et al. (2022). Non-alcoholic fatty liver disease is not independently associated with Helicobacter pylori in a central European screening cohort. Minerva Med. 113, 936–949. doi: 10.23736/S0026-4806.22.07928-9
Wigg, A. J., Roberts-Thomson, I. C., Dymock, R. B., McCarthy, P. J., Grose, R. H., Cummins, A. G. (2001). The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 48, 206–211. doi: 10.1136/gut.48.2.206
Wijarnpreecha, K., Thongprayoon, C., Panjawatanan, P., Manatsathit, W., Jaruvongvanich, V., Ungprasert, P. (2018). Helicobacter pylori and risk of nonalcoholic fatty liver disease: A systematic review and meta-analysis. J. Clin. Gastroenterol. 52, 386–391. doi: 10.1097/MCG.0000000000000784
Wree, A., McGeough, M. D., Peña, C. A., Schlattjan, M., Li, H., Inzaugarat, M. E., et al. (2014). NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. 92, 1069–1082. doi: 10.1007/s00109-014-1170-1
Xia, X., Zhang, L., Chi, J., Li, H., Liu, X., Hu, T., et al. (2020). Helicobacter pylori infection impairs endothelial function through an exosome-mediated mechanism. J. Am. Heart Assoc. 9, e014120. doi: 10.1161/JAHA.119.014120
Xu, H., Barnes, G. T., Yang, Q., Tan, G., Yang, D., Chou, C. J., et al. (2003). Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 112, 1821–1830. doi: 10.1172/JCI19451
Xu, J., Lloyd, D. J., Hale, C., Stanislaus, S., Chen, M., Sivits, G., et al. (2009). Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 58, 250–259. doi: 10.2337/db08-0392
Xu, G., Ma, S., Dong, L., Mendez-Sanchez, N., Li, H., Qi, X. (2023). Relationship of helicobacter pylori infection with nonalcoholic fatty liver disease: A meta-analysis. Can. J. Gastroenterol. Hepatol. 2023, 5521239. doi: 10.1155/2023/5521239
Yamaoka, Y. (2009). Helicobacter pylori typing as a tool for tracking human migration. Clin. Microbiol. infect. 15, 829–834. doi: 10.1111/j.1469-0691.2009.02967.x
Younossi, Z. M., Stepanova, M., Younossi, Y., Golabi, P., Mishra, A., Rafiq, N., et al. (2020). Epidemiology of chronic liver diseases in the USA in the past three decades. Gut 69, 564–568. doi: 10.1136/gutjnl-2019-318813
Yu, Y. Y., Tong, Y. L., Wu, L. Y., Yu, X. Y. (2022). Helicobacter pylori infection eradication for nonalcoholic fatty liver disease: a randomized controlled trial. Sci. Rep. 12, 19530. doi: 10.1038/s41598-022-23746-0
Zahmatkesh, M. E., Jahanbakhsh, M., Hoseini, N., Shegefti, S., Peymani, A., Dabin, H., et al. (2022). Effects of exosomes derived from helicobacter pylori outer membrane vesicle-infected hepatocytes on hepatic stellate cell activation and liver fibrosis induction. Front. Cell Infect. Microbiol. 12. doi: 10.3389/fcimb.2022.857570
Zamani, M., Ebrahimtabar, F., Zamani, V., Miller, W. H., Alizadeh-Navaei, R., Shokri-Shirvani, J., et al. (2018). Systematic review with meta-analysis: the worldwide prevalence of Helicobacter pylori infection. Aliment. Pharmacol. Ther. 47, 868–876. doi: 10.1111/apt.14561
Zhao, Y., Meng, C., Wang, Y., Huang, H., Liu, W., Zhang, J. F., et al. (2016). IL-1β inhibits β-Klotho expression and FGF19 signaling in hepatocytes. Am. J. Physiol. Endocrinol. Metab. 310, E289–E300. doi: 10.1152/ajpendo.00356.2015
Zhou, X., Liu, W., Gu, M., Zhou, H., Zhang, G. (2015). Helicobacter pylori infection causes hepatic insulin resistance by the c-Jun/miR-203/SOCS3 signaling pathway. J. Gastroenterol. 50, 1027–1040. doi: 10.1007/s00535-015-1051-6
Zhou, X. Z., Lyu, N. H., Zhu, H. Y., Cai, Q. C., Kong, X. Y., Xie, P., et al. (2023). Large-scale, national, family-based epidemiological study on Helicobacter pylori infection in China: the time to change practice for related disease prevention. Gut. 72, 855–869. doi: 10.1136/gutjnl-2022-328965
Keywords: Helicobacter pylori, extragastric disease, nonalcoholic fatty liver disease, meta-analysis, pathogenesis
Citation: Chen X, Peng R, Peng D, Xiao J, Liu D and Li R (2023) An update: is there a relationship between H. pylori infection and nonalcoholic fatty liver disease? why is this subject of interest? Front. Cell. Infect. Microbiol. 13:1282956. doi: 10.3389/fcimb.2023.1282956
Received: 25 August 2023; Accepted: 21 November 2023;
Published: 08 December 2023.
Edited by:
Leena Malayil, University of Maryland, College Park, United StatesReviewed by:
Giuseppe Losurdo, University of Bari Medical School, ItalyCopyright © 2023 Chen, Peng, Peng, Xiao, Liu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rong Li, eHlsdWxyQGNzdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.