



95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol. , 20 January 2012
Sec. Molecular Bacterial Pathogenesis
Volume 1 - 2011 | https://doi.org/10.3389/fcimb.2011.00027
This article is part of the Research Topic Burkholderia cepacia complex: an embarrassment of niches View all 11 articles
In recent times, increased attention has been given to evaluating the efficacy of phage therapy, especially in scenarios where the bacterial infectious agent of interest is highly antibiotic resistant. In this regard, phage therapy is especially applicable to infections caused by the Burkholderia cepacia complex (BCC) since members of the BCC are antibiotic pan-resistant. Current studies in BCC phage therapy are unique from many other avenues of phage therapy research in that the investigation is not only comprised of phage isolation, in vitro phage characterization and assessment of in vivo infection model efficacy, but also adapting aerosol drug delivery techniques to aerosol phage formulation delivery and storage.
The Burkholderia cepacia complex (BCC) is a group of 17 genetically diverse, but phenotypically similar Gram-negative rod-shaped bacteria (Papaleo et al., 2010). These opportunistic pathogens are medically significant, as they cause respiratory infections in cystic fibrosis (CF) patients. BCC infections can significantly reduce the life expectancy of CF patients and in severe cases can lead to “cepacia syndrome,” which is characterized by rapid deterioration of pulmonary function (Isles et al., 1984). BCC infections are difficult to treat because they are highly antibiotic resistant, making alternative treatments such as phage therapy attractive. Phage therapy utilizes bacteriophages (phages), viruses that specifically target particular species of bacteria, to combat the infection. This review outlines the origins of phage therapy as well as highlights current research applicable to phage therapy for combating BCC infections.
Bacteriophages were discovered independently by Twort (1915) at Browns Veterinary Hospital in London and by d’Herelle (1917) at the Institut Pasteur in Paris. Phages are viruses that exclusively attack and lyse a bacteria (often of a particular species or strain), which prompted D’Herelle to develop the term “bacteriophage,” meaning “bacteria-eater” (d’Herelle, 1917; Bradbury, 2004). Shortly after the discovery of phages, d’Herelle proposed using them as a possible method of combating infection and used phages to treat patients with dysentery, bubonic plague, and cholera with some success. Although there were reports of successful phage treatment, not all phage therapy testing was effective. These variable results were most likely due to a lack of knowledge in the area of phage biology, a lack of understanding regarding phage specificity, the use of phage to treat conditions not caused by a bacterial infection, improper phage preparation leading to phage inactivation, and an inability to produce high volumes of purified phage (reviewed in Sulakvelidze and Morris, 2001; Bradbury, 2004). The discovery of chemical antibiotics, combined with a perceived inconsistency in phage therapy performance, led to the discontinuation of phage therapy in the West by the 1940s (Merril et al., 2003).
In Eastern Europe, particularly Georgia, phage therapy research has continued at the Eliava Institute. During the Soviet Era, the Eliava Institute was responsible for producing phage preparations for both prophylactic uses as well as for the treatment of infections in patients across the Soviet Union. The Eliava Institute has reported many successes, treating patients with a wide range of infections including skin wounds, and eye, respiratory, and gastrointestinal infections (Sulakvelidze and Morris, 2001). Unfortunately, these reports are anecdotal in nature and have rarely been published in scientific literature, especially in the English language. With the advent of antibiotic resistant infections and an improved knowledge of phage biology and bacterial identification, phage therapy is now being revisited by researchers in the West.
Current evaluation of phage therapy is often performed using animal models, as phage therapy must be proven with controlled studies before its efficacy in a clinical setting can be evaluated. One of the most commonly used animal models is the mouse. Mice are an attractive animal model for many reasons. From a practical standpoint, their small size and low maintenance costs allow for comprehensive studies with many subjects (Nadithe et al., 2003). Mice have been well-characterized and their immune systems have similarities to the human immune system (Nadithe et al., 2003).
The applications of phage therapy are numerous and many of these have been and are currently being evaluated, including the treatment of thermal injury infection (McVay et al., 2007; Kumari et al., 2009) and systemic infections (Cerveny et al., 2002; Capparelli et al., 2007), including those that are antibiotic resistant (Biswas et al., 2002). The applications of phage therapy in veterinary medicine are also being assessed. There is an increased interest in finding alternatives to antibiotics for use in animal husbandry as more restrictive regulations against antibiotic use in livestock and fowl are put in place (Johnson et al., 2008). Such research has included the evaluation of treating enterotoxigenic Escherichia coli infections in pigs, calves, and lambs (Smith and Huggins, 1983) and airsacculitis in chickens (Huff et al., 2002).
Recently, three studies (Merabishvili et al., 2009; Rhoads et al., 2009; Wright et al., 2009) have either outlined the steps required to produce a phage preparation useful in clinical studies or evaluated phage therapy in controlled human studies. Merabishvili et al. (2009) outline the production and quality control methods required to produce a cocktail of Pseudomonas aeruginosa and Staphylococcus aureus phages (BFC-1) for use in burn wound infections. At the time of press, BFC-1 had been tested on eight patients with no reported adverse effects (Merabishvili et al., 2009). Rhoads et al. (2009) completed a phase I trial evaluating the safety of WPP-201, a phage cocktail active against P. aeruginosa, S. aureus, and E. coli, when applied to venous leg ulcers. This study did not evaluate the efficacy of the phages, but demonstrated the safety of WPP-201 with no significant difference in the adverse side effects reported in comparison to the control group. Wright et al. (2009) evaluated the efficacy of treating chronic otitis caused by antibiotic resistant P. aeruginosa in a phase I/II clinical trial. Patients were given a single treatment of either a placebo or Biophage-PA, a cocktail of six phages, and monitored for 42 days. The Biophage-PA treated group saw significant clinical improvement, and phage activity was retained on average for 23 days after application. The safety of the product was also demonstrated in that no adverse side effects were reported that could be attributed to the treatment. This phage cocktail should be further evaluated in phase III clinical trials (AmpliPhi, 2011).
The main principle upon which phage therapy rests is that after replication within a bacterium, phages lyse and kill their host cell. The best candidates for phage therapy are lytic phages (Skurnik and Strauch, 2006). The basic steps of lytic phage infection for phages of the order Caudovirales (composed of three families of tailed phages: Siphoviridae, Myoviridae, and Podoviridae) are as follows (Figure 1): First, a phage binds to a receptor on the surface of a bacterium. On a Gram-negative bacterium, phage receptors can include lipopolysaccharide (both smooth and rough types) and outer membrane components such as porins, transport proteins, enzymes, and structural proteins (Rakhuba et al., 2010). Phage interaction with a surface receptor may be enhanced through the production of a phage-associated enzyme capable of degrading exopolysaccharide or biofilm components. Once bound to the receptor, the phage injects its nucleic acid into the bacterial host. The phage early genes are expressed, disrupting bacterial systems and inducing replication of the phage genome. Late phage genes are then expressed, allowing production of proteins required for phage assembly and host lysis. Finally, mature phage particles are assembled and the host is lysed, causing release of the newly assembled phages (Skurnik and Strauch, 2006).
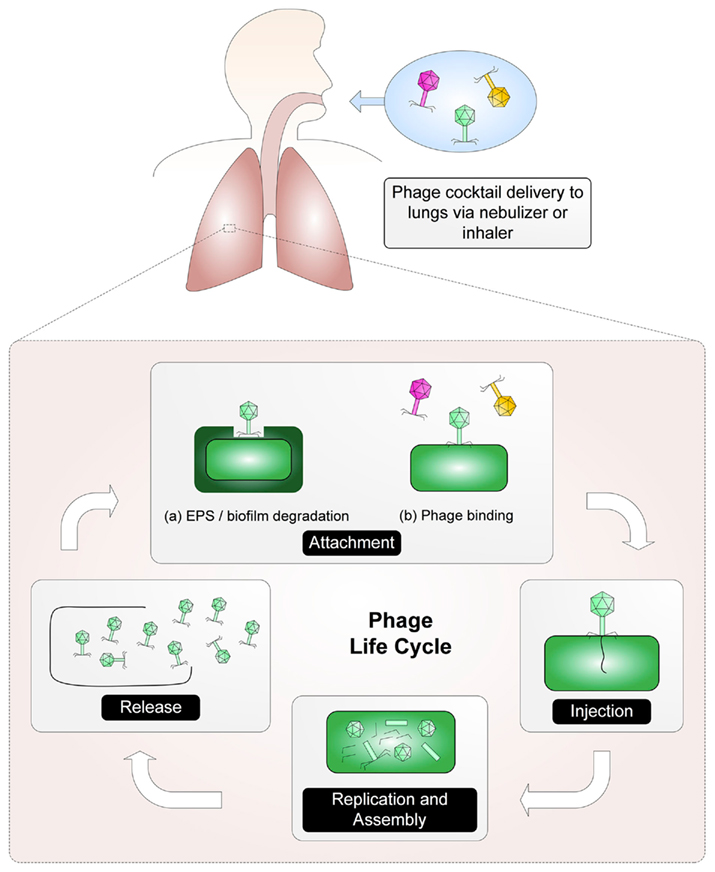
Figure 1. A phage cocktail is delivered via an aerosol to the site of the infection where the phage life cycle can be established. First, phage attach to receptors on the host. (Phage may need to degrade exopolysaccharide [EPS] in order to reach the phage receptor). After binding, the phage are able to transfer their nucleic acid into the host. Host systems are disrupted and the replication of the phage genome begins. Once mature phage particles have been assembled, the host is lysed and the phages are released.
Some phages are capable of a temperate or lysogenic lifestyle in which they integrate into the host genome or exist as a plasmid and remain dormant, simply being replicated along with normal bacterial cell division. Such phages will remain in this state until conditions trigger the phage to enter a lytic lifecycle. This dormancy can be problematic when using phages in a phage therapy strategy because additional new phages are not being released (Kropinski, 2006). Temperate phages are also not optimal candidates for phage therapy because they do not cause the rapid host death required for effective phage therapy treatment (Skurnik and Strauch, 2006). Additional concerns include lysogenic conversion, superinfection immunity, and transduction. Lysogenic conversion is the utilization of prophage genes by the host which may render the host more virulent than prior to infection. Superinfection immunity may develop in situations where the host cell becomes resistant to subsequent infection by a similar phage. Finally, transduction occurs when bacterial DNA (possibly containing virulence factor genes) is packaged into the phage capsid and transferred to a new host via the phage (reviewed by Lynch et al., 2010a). Although lytic phages remain the optimal choice for phage therapy, in situations where the use of lytic phages is not possible, the efficacy of phage therapy using temperate (or putatively temperate) virions has also been demonstrated (Capparelli et al., 2007; Seed and Dennis, 2009; Carmody et al., 2010; Lynch et al., 2010a; Paul et al., 2011).
There are three basic steps in treating a clinical infection with phage therapy. First, the cause of the infection must be isolated and characterized in order to assess strain sensitivity. Second, a phage preparation must be chosen that is active against the appropriate host. Preferably, the preparation will be a high titer cocktail of phages with different host receptors, thereby increasing the activity of the preparation. Finally, the treatment must be appropriately administered to the patient according to the location of the infection (Bradbury, 2004). Many different methods of phage administration have been described in the literature including oral, rectal, topical, intravenous, and via an aerosol (Sulakvelidze and Morris, 2001).
The pharmacokinetics of phage therapy are quite different from any drug currently employed. With a chemical drug, the concentration decreases after administration. The opposite is true of phage. After the originally administered phage replicate, they provide an increasing phage dose that will continue until the infection is cleared. This phenomenon is known as active phage therapy (Payne and Jansen, 2003). Passive phage therapy is also possible if the initial phage dose is high enough to clear the infection without phage replication (Payne and Jansen, 2003). Although the concept of active phage therapy suggests that a low phage titer is all that is required for successful treatment, this is not necessarily the case. Using a mathematical model simulating the pharmacokinetics of phage therapy, Payne and Jansen (2003) demonstrated that selecting the correct phage concentration required for successful treatment should be based on the bacterial density of the infection. A sufficiently dense bacterial community will allow for active phage therapy. However, a less dense population will not be able to support active phage therapy because the phage will not be able to reproduce quickly enough to yield and maintain the high in vivo concentrations required to sustain a high bacterial kill rate. In situations like this, multiple high titer phage treatments, administered with a similar schedule to antibiotic therapy, would be required in order to completely eliminate the infection (Levin and Bull, 2004).
In addition to titer, timing in phage administration is critical. Using mathematical modeling, Payne and Jansen (2003) demonstrated that a delay in treatment would prove detrimental to combating an infection. These modeled data have also been demonstrated in a mouse model (Cerveny et al., 2002). Interestingly, Payne and Jansen (2003) also demonstrated that a treatment delivered too early may be detrimental to clearing of the infection. This detrimental effect is related to low bacterial density. Sparse bacterial densities, in which the probability of a phage encountering a bacterial host is low, do not lend themselves to sustainable active phage therapy. If treatment is delayed until active phage therapy is possible, it will likely be more effective.
Although there are some concerns regarding phage therapy (see below), there are a large number of potentially positive aspects to phage therapy as well. Phages are active against bacterial cells using completely different mechanisms than classical antibiotics, allowing them to be active against antibiotic resistant bacteria (Bradbury, 2004). Phage activity against antibiotic resistant bacterial strains is likely the most promising feature of phage therapy, as phages can be used in instances where antibiotic treatment is no longer possible. Phages have many positive qualities as antimicrobials, as shown in Table 1. In direct contrast to antibiotics, which are often broad spectrum, phages show more host specificity, and in most cases show specificity to a single species or strain of bacteria. Using phages would allow for the treatment of an infection without harming the natural microflora of a patient (Mattey and Spencer, 2008). Phages do not cause negative side effects to patients, as demonstrated in a study in which human volunteers drank water containing the E. coli phage T4 (Bruttin and Brüssow, 2005). In this study there were no reported adverse affects caused by the phage, as well as no T4-specific immune response in the volunteers. These three key features demonstrate phage therapy’s potential as an effective tool in combating bacterial infections.
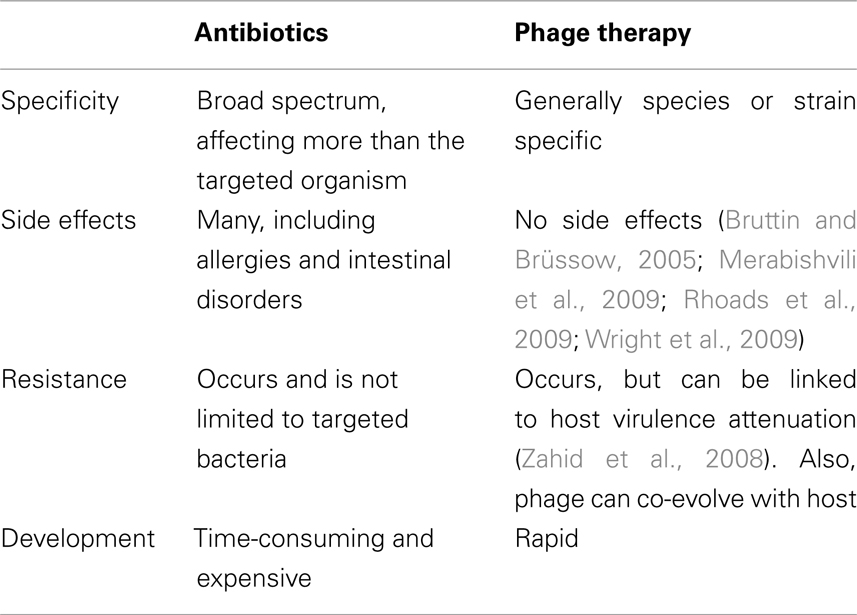
Table 1. A comparison of some of the main advantages and disadvantages of antibiotics and phage therapy.
The specificity of phages, which is certainly an advantage, can also provide one of the challenges that must be overcome when using phage therapy as a treatment method. Before an infection can be treated, the causative agent must be characterized or typed in order to ensure that the correct phage is being used for its treatment. Strain characterization can be time-consuming and is especially troublesome in cases that must be treated rapidly. In order to overcome such problems, phage cocktails can be developed that are active against a broad range of strains or even different bacterial species (Levin and Bull, 2004). A potentially serious complication to phage therapy that must be taken into consideration is that bacterial strains can develop phage resistance or semi-resistance. Phage resistance may pose significant problems, especially if a phage must undergo rigorous and time-consuming testing before it is approved for clinical use (Mattey and Spencer, 2008). In some situations, partial phage resistance can occur. Although phage can still infect semi-resistant bacteria, their infection rate is far lower than in sensitive bacteria, rendering phage therapy less effective (Levin and Bull, 2004). Semi-resistance is most often seen in mucoid colonies, as the mucoid barrier provides an additional obstacle to phage contacting the appropriate receptor. Phage resistance generally occurs by a mutation in the receptor to which the phage binds. Unlike semi-resistant bacteria, receptor mutations prevent phages from binding to the cell, thereby preventing the initial step in phage infection. Phage resistance and semi-resistance can be minimized by treating infections with a phage cocktail containing a mixture of phages that will target different binding receptors on the bacterial cell. This allows the cocktail to be effective, even if a receptor is altered. Interestingly, receptor mutation leading to phage resistance may not always be a bad thing. In some instances, bacteria that develop phage resistance are also less competitive and less virulent, or even avirulent. This phenomenon is most noticeable in phage resistant bacteria where the phage receptor lost is a capsule or other virulence factor (Levin and Bull, 2004), as was demonstrated by Zahid et al. (2008) who found that Vibrio cholerae cultured with phage in a nutrient medium quickly became phage resistant, but also lost their O1 antigen. Another form of resistance that must be considered is restriction endonuclease resistance. In this case, the bacterium encodes restriction endonucleases active against the phage genome, causing destruction of the phage’s genetic material and inhibiting phage replication. Although restriction endonucleases must be taken into consideration, it may not be of significant importance as many phages are able to evade restriction (Levin and Bull, 2004).
Phage therapy critics have suggested that the presence of clustered regularly interspaced short palindromic repeats (CRISPR) in 40% of sequenced bacterial genomes is a detriment to phage therapy. Briefly, CRISPRs are genomic sequences of alternating spacers and palindromic repeats. The spacers are sequences of phage or plasmid DNA that the host had previously obtained. Identical matches between the spacers and foreign DNA entering the cell alert the bacterium to the presence of a potential predator, which triggers the degradation of the foreign DNA. CRISPRs have been reviewed in further detail by Horvath and Barrangou (2010). It should be noted, however, that if the phage has even a single point mutation in the sequence to be matched to the CRISPR sequence, the phage will be able to evade the CRISPR system and proceed with host infection. Additionally, CRISPRs are not a pressing concern in regards to BCC phage therapy as only one of the analyzed BCC species (B. ambifaria AMMD) contains a confirmed CRISPR locus (Grissa et al., 2007).
Research into phages infecting and lysogenizing BCC species is a relatively recent endeavor, particularly when compared to the almost century of work related to Enterobacteriaceae phages. The goal of early BCC phage studies was not to identify phage therapy candidates, but instead to isolate generalized transducing phages that could be used for the genetic manipulation of BCC strains (Cihlar et al., 1978; Matsumoto et al., 1986; Nzula et al., 2000). Cihlar et al. (1978) were the first to publish a description of a BCC phage in 1978 (Bcep781 was also isolated in 1978, but its sequence was not published until 2006; Summer et al., 2006). This group identified a temperate phage of P. cepacia 249 (now B. multivorans ATCC 17616) named CP1. This myovirus could infect P. cepacia 104, 382, and 383 (now B. cepacia ATCC 17478, B. cepacia ATCC 17759, and B. lata ATCC 17760, respectively), but not 249. Host range mutants of CP1 were able to infect P. pickettii 472 (now Ralstonia pickettii), but not P. aeruginosa, P. fluorescens, or P. phaseolicola (now P. syringae pathovar phaseolicola). Matsumoto et al. (1986) were the second group to characterize a BCC phage and the first to identify one capable of generalized transduction. CP75 is a temperate phage of P. cepacia PCT1, an uncharacterized clinical isolate. This myovirus has a ∼52 kbp genome and a broad host range, infecting 46 out of 105 P. cepacia strains screened (although the identity and relatedness of these strains were not discussed). More recently, Nzula et al. (2000) identified two generalized transducing phages, NS1 and NS2. These are ∼48 kbp temperate phages of B. vietnamiensis ATCC 29424 and B. multivorans ATCC 17616, respectively. Interestingly, both CP1 and NS2 (in addition to KS5, discussed below) are temperate myoviruses of ATCC 17616, but they were found to have differing generalized transduction efficiencies and sensitivity to chloroform (Cihlar et al., 1978; Nzula et al., 2000). Similar to KS5, both NS1 and NS2 are predicted to use LPS as a receptor (Nzula et al., 2000; Lynch et al., 2010a). NS1 and NS2 can infect strains with different serotypes, which may be a result of minimal phage interaction with the O-antigen or changes in the strains that were screened (Kenna et al., 2003). These phages also have a broad host range, particularly NS2: each could infect strains of B. cepacia, B. multivorans, B. cenocepacia, B. vietnamiensis, and P. aeruginosa (Nzula et al., 2000). Certain BCC strains were also found to be susceptible to infection by P. aeruginosa phages, specifically E79, B3, F116L, and G101. While phages that infect both BCC and Pseudomonas strains may be extremely useful with respect to phage therapy development, it remains a concern that generalized transducing phages with such a broad host range could facilitate the exchange of virulence genes between Burkholderia and Pseudomonas (Nzula et al., 2000).
One BCC phage that has been putatively shown to transfer resistance genes between distinct species is BcP15. BcP15, a temperate siphovirus of the environmental isolate B. cepacia DR11, was found to infect only Shigella flexneri Pl-35 (a plasmid-cured isolate of S. flexneri NK1925; Hens et al., 2005, 2006). Putative Pl-35 lysogens were resistant to co-trimoxazole, trimethoprim, and erythromycin, and it was suggested that the integrated BcP15 DNA was responsible for this phenotype (Hens et al., 2006). Further characterization of BcP15, its hosts, and the mechanisms of gene transfer and resistance are needed in order to assess the possible clinical significance of these results.
In order for phage therapy to be a viable alternative treatment for BCC infections, BCC-specific phages appropriate for clinical use must first be isolated and/or developed. Selection criteria for such phages include a broad host range (in particular clinically prevalent species and/or strains), an obligately lytic lifestyle, and the absence of genes encoding potential virulence factors. Full genome sequencing is required for the most complete characterization of these traits. To date, one of the major challenges in BCC phage therapy development has been finding phages that satisfy all of the above criteria. Analysis of BCC phages possessing only some of these characteristics remains an important exercise because it provides (a) essential data for studying BCC comparative genomics, (b) potential clues as to the development of BCC virulence, and (c) candidate phages for phage therapy that fit these criteria more closely through the use of genetic modification.
Two major studies describing the isolation of novel BCC phages were published by Langley et al. (2003) and Seed and Dennis (2005). Langley et al. (2003) isolated phages from both known BCC strains (belonging to B. cepacia, B. multivorans, B. cenocepacia, and B. stabilis) and the environment (soil, rhizosphere, and sediment samples). Many of these phages have broad host ranges, infecting multiple BCC species and even P. aeruginosa or B. gladioli in some cases. These phages belong to either the Siphoviridae or the Myoviridae. Similarly, Seed and Dennis (2005) isolated putatively lytic phages from onion soil and temperate phages from B. multivorans, B. cenocepacia, B. ambifaria, and B. pyrrocinia. These phages had narrow to broad host ranges within the BCC and in some cases could infect B. gladioli (but not P. aeruginosa). These phages also belong to either the Siphoviridae or the Myoviridae.
An issue common to many BCC phage studies is the identification and characterization of identical or nearly identical temperate phages by multiple groups. This issue is best illustrated by phages BcepMu, DK4, and KS4/KS4-M. BcepMu was isolated and fully sequenced by Summer et al. (2004). This phage is a 36,748 bp transposable myovirus found as a prophage in B. cenocepacia J2315, BC7, C5424, and other B. cenocepacia ET12 strains (excluding K56-2). BcepMu shows relatedness to transposable phages of Salmonella typhi (SalMu), Photorhabdus luminescens (PhotoMu), and Chromobacterium violaceum (ChromoMu). DK4, isolated and described by Langley et al. (2003) as a temperate phage of J2315, was later identified as BcepMu based on sequencing of PCR amplicons and RFLP analysis (Langley et al., 2005). Like BcepMu, KS4 was isolated from a culture of J2315 and propagated on K56-2 (Seed and Dennis, 2005). Repeated passage in broth culture yielded a liquid-clearing variant of this phage named KS4-M (Seed and Dennis, 2009). Despite its temperate nature, this phage may be a candidate for clinical use as it has been shown to be (a) active against K56-2 in vivo and (b) stable during nebulization, freeze-drying/lyophilization, and spray-drying (as discussed below; Golshahi et al., 2008, 2011; Seed and Dennis, 2009; Matinkhoo et al., 2011). As phage characteristics such as host range and apparent virion dimensions may be recorded differently among various research groups, the determination of partial or complete genome sequences remains a critical first step upon isolation of novel BCC phages.
Although most of the following studies are only peripherally related to phage genomics, they provide a wealth of information regarding the distribution of prophages in BCC genomes and their patterns of expression under different environmental conditions.
Using in vivo expression technology (IVET), Nishiyama et al. (2010) examined the expression of B. multivorans ATCC 17616 genes in soil. They found that several phage genes were induced under these conditions, including BMULJ_03661–03667 (KS5 genes 21–27, encoding a translational regulator and tail morphogenesis proteins). In that same year, Ronning et al. (2010) identified putative prophage regions in several Burkholderia species, including B. multivorans. These included two regions in B. multivorans CGD1, three regions in B. multivorans CGD2, and three regions in ATCC 17616 [including those containing Bcep176 (PI 17616-3) and KS5 (PI 17616-4)].
To identify genomic differences between the highly pathogenic B. cenocepacia and other less pathogenic BCC species, Bernier and Sokol (2005) used suppression-subtractive hybridization with B. multivorans C5393, B. cenocepacia K56-2, and B. stabilis LMG 14294. Several putative phage or phage-related genes were identified that were unique to B. cenocepacia. When the complete genome sequence of J2315 was published in 2009, at least five prophages were identified: BcenGI1 (25.0 kbp) and BcenGI7 (37.7 kbp; KS10) on chromosome 1, BcenGI12 (46.8 kbp) and BcenGI13 (46.2 kbp) on chromosome 2, and BcenGI14 (36.7 kbp; BcepMu) on chromosome 3 (Holden et al., 2009). BcenGI7 (discussed below) and BcenGI14 (discussed above) have also been sequenced independently of J2315 (Summer et al., 2004; Goudie et al., 2008). Two studies profiling the transcription patterns of B. cenocepacia strains under different conditions have identified prophage genes as part of their analysis. O’Grady et al. (2009) collected microarray data for stationary phase B. cenocepacia K56-2 quorum sensing mutants (cepR, cciR, and cepRcciIR) and found that 24, 5, and 20 phage genes, respectively, had different expression in the mutants compared to wild type K56-2. Many of these genes corresponded to BcenGI12. When Peeters et al. (2010) exposed J2315 biofilms to hydrogen peroxide for extended periods (30–60 min) and then performed microarray analysis, they observed increased gene expression from BcenGI14 (specifically the region containing BcepMu genes 1–16), with some genes showing an increase in expression greater than 10-fold as compared to unstressed biofilm cells not treated with hydrogen peroxide.
Although there are many BCC phages with sequences that have been deposited into GenBank, this review will focus on those that are both in the database and published. All BCC phages sequenced to date belong to the order Caudovirales, containing the three families of tailed phages: Siphoviridae (with long non-contractile tails), Myoviridae (with contractile tails), and Podoviridae (with short non-contractile tails; Ackermann, 2001).
The only BCC siphovirus sequence published to date is that of KS9, a 39,896 bp temperate phage of B. pyrrocinia LMG 21824 (Seed and Dennis, 2005; Lynch et al., 2010a). This phage is most closely related to the temperate phages ΦE125 of B. thailandensis and Φ1026b of B. pseudomallei and to a prophage element in B. cenocepacia PC184. KS9-lysogenized K56-2 is no more virulent in the Galleria mellonella model than wild type K56-2, suggesting that this phage does not increase host virulence. This phage was used in a proof-of-principle experiment (Lynch et al., 2010a) to show that stable lysogeny could be prevented by knocking out the phage repressor gene, thus making the phage a better candidate for clinical use.
Summer et al. (2006) published the first and thus far only genomic characterization of obligately lytic BCC phages isolated from the environment: myoviruses BcepB1A (47,399 bp), Bcep43 (48,024 bp), Bcep1 (48,177 bp), and Bcep781 (48,247 bp). These phages have narrow host ranges, only infecting one or two BCC strains each. The latter three phages are very closely related, with percent identities between 87.4 and 97.6% for pairwise comparisons.
Similar to BcepMu, Goudie et al. (2008) characterized a second transposable Mu-like temperate myovirus of B. cenocepacia J2315. KS10 is 37,635 bp in length and infects B. cenocepacia PC184, B. stabilis 18870, and B. ambifaria LMG 19467. Like BcepMu, it has a relatively wide distribution in B. cenocepacia, lysogenizing K56-2, J2315, C5424, and seven clinical isolates. Although this distribution suggests that KS10 may be selected for in B. cenocepacia, it was not found to encode any proteins that could be considered virulence factors.
Lynch et al. (2010b) characterized three BCC myoviruses that belong to the P2-like phage genus. Although all of these phages are temperate, they were isolated from both BCC cultures and/or environmental samples. Their genome size range is broad: 32,317 bp for KS14, 37,236 bp for KS5, and 40,555 bp for KL3. These phages encode a number of interesting features, including an insertion sequence and reverse transcriptase in KS5 and a DNA methylation, restriction, and repair module in KL3. Phages KL3 and KS5 were found to integrate as prophages into a threonine tRNA gene and an AMP nucleosidase gene, respectively. KS14 has been shown to be active in vivo and is stable upon spray drying (Seed and Dennis, 2009; Matinkhoo et al., 2011).
The only BCC podovirus sequences published to date, Bcep22 (63,882 bp) and BcepIL02 (62,714 bp), are closely related and belong to a single recently identified phage type: the Bcep22-like phages (Gill et al., 2011). Both phages encode multiple copies of their tail fiber proteins (four for BcepIL02 and three for Bcep22) and a large protein of ∼4600 amino acids with soluble lytic transglycosylase, helicase, and methylase domains. Although BcepIL02 – which has been shown to be active in vivo – was initially thought to be lytic, it was found that both of these phages encode proteins for integration but cannot form stable lysogens (Carmody et al., 2010; Gill et al., 2011). BcepIL02 carries a putative lipid A modification gene which may modulate bacterial host virulence. The presence of such a gene in BCC phages is rare (an obvious advantage for the development of phage therapy), as the only other sequenced BCC phage with a putative virulence gene is BcepMu (Summer et al., 2004). It has been suggested that bacterial virulence genes are not frequently found in the BCC phage population because BCC bacteria are not classical bacterial pathogens, but inhabitants of both the environment and immunocompromised individuals (Summer et al., 2007). A manuscript describing DC1, a third Bcep22-like phage with a relatively broad host range, is currently in press (Lynch et al., in press).
Although aerosol phage therapy has not been reported in the literature a great deal, it remains a popular option for treating respiratory infections and many of the techniques used for aerosol drug delivery are being adapted to phage therapy. Aerosol drug delivery has been successfully employed for many pharmaceuticals including bronchodilators, antibiotics, mucolytics, and anesthetics (Heslop and Harkawat, 2000), as it enables a drug to reach the diseased site rapidly and requires lower doses of medication than other delivery methods (Thorsson and Geller, 2005). As phages require direct contact with their bacterial hosts in order to be active, aerosol phage delivery appears to be a logical method of delivering phages rapidly to the site of a pulmonary infection. There are many aspects of aerosol phage therapy to be evaluated in order to ensure that it can be used as a reliable treatment method. These research areas include phage preparation, storage, and delivery methods, as well as demonstration of activity in vitro and effective treatment of an infection in vivo.
There are three case studies that mention the use of inhalation phage therapy as a clinical treatment method. Unfortunately, one of these papers is not written in English and therefore is difficult to review (Garsevanishvili, 1974). The other two papers (Hoeflmayr, 1963; Kutateladze and Adamia, 2008) are case studies describing the treatment and recovery rates of patients treated by inhalation phage therapy. Although these studies do not directly address the treatment of BCC infections, they are still of interest because they outline situations in which aerosol phage therapy has been employed in the past. The first clinical study (Hoeflmayr, 1963) evaluated the recovery of 29 respiratory infection patients treated by an aerosolized phage cocktail. The patients were chosen because they could not be treated by conventional methods such as expectorant, secretolytic, or antibiotic therapies. Approximately two-thirds of the patients had Streptococcus infections, while the remaining patients had Staphylococcus infections. The phage cocktail used was Diriphagen, a commercially available cocktail containing 180–200 phages specific for different bacterial species. In addition to phage, Diriphagen also contained an “aimed antimicrobic,” which targeted bacterial strains that became phage resistant during the course of the treatment. No mention was made as to what this aimed antimicrobic was or how it functioned. This particular phage cocktail was chosen because of the large number of different phages it contained. During the course of the study, the patients received daily aerosolized phage treatments. The treatments lasted 10–15 min. The average number of treatments per patient was 11, while some patients received as few as three treatments and one patient received as many as 40 treatments. After treatment, the symptoms of the 29 patients were classed into three groups: 55% were reported to have made a complete recovery, 35% showed considerable improvements and 10% showed no change. When the bacterial load in the sputum cultures was determined, it yielded contradictory results. All patients had detectable levels of either Staphylococcus or Streptococcus, with 30% of the patients showing considerable improvement, 55% showing some improvement and 10% showing no improvement. Interestingly, at least one patient showed no change in symptoms. However, her bacterial counts decreased substantially. Unfortunately, there were many aspects of this study that were not properly described or quantified. There was no information given as to what exact infection each of the patients had (as it was not uniform throughout the group), making it difficult to determine if the phage cocktail was more effective against certain species or strains. No mention was made of what criteria were given to class the patients’ symptoms as “considerably improved” and neither were there values associated with a “considerable decrease” or “some decrease” in bacterial load in the sputum. This study provides some anecdotal evidence that phage therapy may be an effective method of treating respiratory infections, but cannot provide any conclusive results.
A more recent case study (Kutateladze and Adamia, 2008) described the treatment of a 5-year-old CF patient at the Eliava Institute in Tbilisi, Georgia. This patient had both S. aureus and P. aeruginosa respiratory infections that were resistant to antibiotic treatment. These strains were sensitive to pyophage, a commercially available phage cocktail active against S. aureus, P. aeruginosa, Streptococcus, Proteus, and E. coli. The patient completed a treatment regimen of three multi-day treatments administered at 1 month intervals. During the first treatment period, pyophage was administered by nebulizer daily over a 6-day period. In addition to the pyophage treatment, the patient received vitamins and an antimucosal treatment. After this treatment, the patient’s condition was described as improved; this characterization was based on weight gain (the patient had gained no weight in the previous year and gained 1 kg after the treatment), facilitation of expectoration and lack of sputum. No mention was made to indicate if the bacterial load in the lungs had changed after this treatment. A second 10-day treatment was performed a month after the initial treatment. The concentration of S. aureus increased while the P. aeruginosa concentration did not change during this treatment period. The second treatment was not effective and based on the increase in S. aureus concentration, it was assumed that the S. aureus had become pyophage resistant. The third and final treatment administered in this case study was a combination of pyophage and tetracycline. After this treatment S. aureus and P. aeruginosa could not be detected in the patient. At the time of publication the patient was still receiving phage therapy on occasion as respiratory infections were identified. It is unfortunate that this particular case study was presented as an example of successful inhalation phage therapy as the evidence is anecdotal and not particularly convincing. A quantitative change in respiratory bacterial load before and after the treatments was not reported. Instead, the efficacy of the treatment was generally evaluated based on the change in patient symptoms. The S. aureus and P. aeruginosa strains appeared to become phage resistant during the second treatment and did not decrease to undetectable levels until the phage treatment was combined with antibiotics, suggesting that the antibiotic treatment or the combination therapy at best, but not the phage therapy alone, cleared the infection. In order to demonstrate that phage therapy is an effective medical treatment, full clinical studies with control groups will be required and this case study emphasizes this point.
Methods of aerosol drug delivery are continually evolving and so it is important to assess these methods with respect to the efficacy of aerosol phage delivery, ensuring that the phages are delivered to the location of the infection and also that they are delivered intact and able to infect and lyse their host. In treating a respiratory infection, one must ensure that the phage aerosol has the correct characteristics required to impact into the area of the lung where the infection resides. Phage liquids can be delivered using a nebulizer. Two common types of nebulizers in use in a clinical setting are the jet nebulizer, such as the Collison or the LC Star, and the ultrasonic nebulizer, such as the eFLOW (Golshahi et al., 2008). The key feature of a nebulizer is that it must generate particles of the appropriate size to impact in the correct location within the pulmonary system. The typical particle size generated by a nebulizer is generally 1–5 μm, which allows the aerosol to reach the smaller bronchioles (Golshahi et al., 2008). One study has been performed to assess the feasibility of aerosolizing phage using a nebulizer, which is commonly used for delivering drugs via an inhalation route (Golshahi et al., 2008). This in vitro method employed a computer controlled breath simulator to mimic the natural breathing patterns of an adult. A BCC phage, KS4-M, able to infect B. cenocepacia strain K56-2 was aerosolized using one of two commonly used nebulizers: the LC star (a jet nebulizer) or the eFLOW (an ultrasonic nebulizer). The aerosol size distribution of the particles was measured as the aerosol exited the nebulizer using phase Doppler anemometry. A filter placed in line with the breath simulator collected the aerosolized phage, allowing for the quantification of phage exiting the nebulizer, which was classified in the study as the inhaled phage. The collected phage were quantified by plating using a soft agar overlay method. As this method only quantified viable phage, it only took into account the number of phage that were able to survive the aerosolization process (and would therefore theoretically be active within a host). In addition to enumerating the number of phage able to survive aerosolization, these data were also employed in mathematical models to determine the regional lung deposition of the phage aerosol based on a numerical lung deposition model. Using this model, calculations were performed to compare the phage deposition in the extrathoracic, tracheobronchial, and alveolar regions of the lung with both of the nebulizers. The results for both nebulizers were quite similar suggesting that either class of nebulizer would be effective in generating phage aerosols. The LC star and eFLOW produced aerosols with similar properties with mass median diameters of 4.98 and 5.83 μm, respectively and geometric standard deviation (the measure of particle monodispersity) of 1.48 and 1.44 μm, respectively. The inhaled phage concentrations produced by both of the nebulizers was also quite comparable with the LC star aerosolizing 1.06 × 108 plaque forming units (PFU) and the eFLOW aerosolizing 1.15 × 108 PFU. There was a one order of magnitude decrease from the inhaled phage concentration (approximately 108 PFU) to the predicted concentrations deposited in each of the three lung areas (approximately 107 PFU) for both of the nebulizers tested. However, this is still a significant phage concentration being delivered to the lungs and the particle deposition in each region of the lung was similar in both cases. The results of this study add credibility to aerosol phage delivery as a potential treatment method for respiratory infection. While mathematical modeling provides an indication as to the outcome of an experiment, it is not a replacement for biological experimental data. This study serves as an excellent starting point for experimentally determining phage deposition within the lungs during aerosol phage therapy.
A number of recent studies have been performed to investigate phage preparation methods for use in phage aerosols, including lyophilization and spray-drying (Puapermpoonsiri et al., 2009; Golshahi et al., 2011; Matinkhoo et al., 2011). Freeze-drying and spray-drying allow the phage stock to be converted to a dry powder, which is delivered to a patient via an inhaler. Delivering phage as a powder would be a rather simple method of treating patients. A dry powder would also be simpler to handle and easier to transport than the liquid phage suspension required for nebulization. There are a number of advantages to dry powder phage preparations including easier transport and longer-term storage capabilities. Treatments can be delivered using a dry powder inhaler in a single breath, making it a faster delivery method than nebulization. A recent study (Puapermpoonsiri et al., 2009) outlines a method for lyophilizing (or freeze-drying) and encapsulating phage in biodegradable poly(DL-lactic-co-glycolic acid) microspheres. The microspheres can be delivered directly to the lungs via a dry powder inhaler and have been previously approved for use in humans (Lü et al., 2009). This proof-of-concept was performed to determine if phages specific to S. aureus and P. aeruginosa could be freeze-dried and disseminated as well as to determine the shelf-life of the phages after freeze-drying and encapsulation. The microcapsules were developed for use in a dry powder inhaler. The encapsulation efficiency for S. aureus phage was 18%, while the efficiency for the P. aeruginosa phage was 27%. In dispersion tests, 55% of the total released contents of the S. aureus phage microcapsules was released within 30 min and release of the phage continued for 6 h. Similarly, within 30 min 63% of the total released P. aeruginosa phage was dispersed.
Both of the phages tested could survive lyophilization and remain at high titers for at least 3 days, at 4°C as well as at 22°C, which would also allow for easier handling and transport. However, after 7 days the phages were no longer viable. Unfortunately, the phage titers dropped quickly enough that counts of “too numerous to count” could not be replated for accurate counts and had to be reported in a semi-quantitative fashion, so only trends could be reported. This study demonstrated that freeze-drying followed by encapsulation into microspheres shows promise, especially as a dispersal technique. However the phages would need to be further stabilized for increased survivability.
An additional study (Golshahi et al., 2011) took a different approach to developing phage aerosol powders, formulating powders for both KS4-M, a BCC phage, and ΦKZ, a P. aeruginosa phage. Instead of encapsulating the phage powder in a microsphere, the endotoxin-removed phage stocks were freeze-dried in a mixture of carriers that help to stabilize the phage as well as enhance the dispersibility of the powder. The latter is an important consideration for the future use of phage powders as a therapeutic because dispersibility affects the efficacy with which the phages will reach the site of infection. Golshahi et al. (2011) demonstrated that a combination of lactose/lactoferrin 60: 40 w/w was an effective carrier for both phage stability and dispersibility. Lactose is an excipient that is currently used in dry powder inhalers in the United States (Telko and Hickey, 2005), while lactoferrin was chosen for its effect on particle size as well as for its antimicrobial properties. The combination of lactoferrin and phages has also been previously demonstrated to be more effective in treating infection in mice than either treatment separately (Zimecki et al., 2008). The lyophilization process caused a reduction in phage titer, with a reduction of two orders of magnitude for KS4-M and one order of magnitude for ΦKZ. This finding should not pose a problem for lyophilizing these phages because both can be grown to a high initial titer. Additionally, in contrast to freeze-dried, encapsulated phages (Puapermpoonsiri et al., 2009), both the freeze-dried KS4-M and ΦKZ phages were able to maintain viability for long periods of time. Both phage titers remained essentially constant at the same order of magnitude (approximately 108 PFU/100 mg) for 3 months, regardless of storage temperature (both 4 and 22°C were tested). After a year of storage at 4°C, ΦKZ remained at 108 PFU/100 mg while KS4-M dropped to 107 PFU/100 mg. Even at 22°C ΦKZ fared well with the titer remaining at 107 PFU/100 mg, while KS4-M dropped to 102 PFU/100 mg (unpublished results). This lyophilization method shows great promise for long-term phage storage. In addition to testing phage stability, the phage powders were tested in a commercially available Aerolizer® dry powder inhaler. When attached to a mouth–throat replica known as the “Alberta Idealized Geometry,” the efficacy with which the freeze-dried phages could be delivered to the site of a pulmonary infection was evaluated. This study also provided promising results with less than an order of magnitude drop from the phage load in the inhaler to deposition within the lungs. This study demonstrated that phage freeze-drying is a viable method of preserving phages for long-term storage. The powdered phages also show a great deal of promise as a therapeutic delivery method in aerosol phage therapy and should be evaluated further.
Using a different approach, Matinkhoo et al. (2011) evaluated spray-drying rather than lyophilization as a method of developing phage powders. Spray-drying is another method of producing pharmaceutical powders which has the added advantage of allowing for a greater selection in particle characteristics (Vehring, 2008). As with Golshahi et al. (2011), KS4-M and ΦKZ were used in the study, but an additional BCC phage, KS14 (discussed above) was also tested. The spray-drying process was less damaging to the phages than lyophilization with one-half to one order of magnitude drop in titer for each of the phages, depending on the excipients used. These findings show an improvement over the two orders of magnitude drop in titer after freeze-drying. Although the long-term viability of the spray-dried phages was not determined in this study and therefore cannot be compared to freeze-dried phages, the phage powders were tested in the same mouth–throat apparatus as the freeze-dried phage in the previously described study (Golshahi et al., 2011). The spray-dried phages outperformed the freeze-dried phages with respect to lung deposition with two times more spray-dried phage delivered to the lungs in comparison to the freeze-dried phage. Also, more than 50% of the total inhaler dose was delivered to the lungs. In comparison, a survey of 12 commercially available inhalers showed a range of delivery efficiencies from 6 to 41% (Newman and Busse, 2002). These data demonstrate the plausibility of using spray-drying as a method of developing phage therapeutics, provided that the spray-dried phages can remain stable for long periods of time.
To date, there are two major in vivo models that have been used for evaluating phage therapy against BCC infections: the G. mellonella (Greater wax moth) larvae model (Seed and Dennis, 2009; Lynch et al., 2010a) and the mouse lung infection model (Carmody et al., 2010). The G. mellonella larvae infection model is used to determine the efficacy of phages to rescue infected larvae from death. This model is relatively simple to work with: the larvae are injected first with bacteria, then phage (either immediately after infection or after a set duration) and the percentage of deaths is then calculated 48 h post-infection. Prior to the use of G. mellonella as a phage therapy model, the pathogenicity of 23 BCC strains in G. mellonella was compared to pathogenicity in alfalfa seedlings, in rats, and in Caenorhabditis elegans. It was demonstrated that G. mellonella results are comparable to those in other in vivo models (Seed and Dennis, 2008). Seed and Dennis (2009) initially evaluated this model for BCC infections using two strains of B. cenocepacia, K56-2 and C6433, given at lethal concentrations. The survival rate of the larvae increased with increased phage multiplicity of infection (MOI) for KS12. A decrease in duration between infection and treatment also rescued more larvae. Using the same model, Lynch et al. (2010a) were able to compare the efficacy of a genetically modified lytic phage and the wild type phage in rescuing infected G. mellonella larvae. Gene 41 of KS9, a temperate phage isolated from B. pyrrocinia LMG 21824, was identified to putatively encode the phage repressor. By disrupting the gene, KS9 became a lytic variant (named KS9c). KS9c was tested in the G. mellonella model to determine if the conversion from a temperate phage to a lytic phage would allow it to be a more effective phage therapeutic. Unexpectedly, although the KS9c variant did not stably lysogenize the bacterial host and was active in vivo, it performed similarly to the wild type phage KS9. These results demonstrate the value in having a simple animal model for testing hypotheses such as the one described. Both of these papers demonstrate the usefulness of G. mellonella as an initial test for evaluating the efficacy of a phage against the BCC in vivo.
The mouse lung infection model involves delivering first bacteria and then phage to the lungs of mice. The bacterial or phage suspensions are often delivered intranasally by placing small volumes of liquid on the nares of anesthetized mice, allowing the mice to inhale the liquid. Intranasal sample delivery is used to mimic aerosol delivery to a human via a nebulizer. This method was used for evaluating the use of aerosol phage therapy to treat a BCC infection (Carmody et al., 2010). In this study, mice were infected with B. cenocepacia via tracheotomy and 24 h post-infection were treated with phage BcepIL02 delivered by either intranasal inhalation or intraperitoneal injection. Forty-eight hours after treatment the mice were euthanized and the lungs assayed for bacterial and phage titer. Interestingly, the mice that received the intranasal phage treatment had approximately a one order of magnitude drop in BCC bacterial load while the mice that received the intraperitoneal phage treatment had a two log drop in BCC bacterial load. Although neither of the described phage delivery methods produced substantial therapeutic results, the authors suggest that phages delivered indirectly are more effective than phages applied directly to the infection site. It should be noted that although intranasal delivery of liquids to the lung is meant to mimic aerosol delivery via a nebulizer, it is not an optimal replacement for actual aerosol delivery in mice. Intranasal delivery in mice has been demonstrated to be far more variable and less reproducible than aerosol delivery (Halperin et al., 1988). Additionally, aerosol particle deposition within the lung was shown to have improved uniformity in comparison to intranasal delivery. These data suggest that a study with similar methodologies to that of Carmody et al. (2010) should be performed using an aerosol delivery method and the results compared to determine if there is a difference between intranasal and aerosol phage delivery in mice.
Although these studies are not directly comparable due to different host–phage interactions, it is interesting to contrast the findings of Carmody et al. (2010) with the results found by Debarbieux et al. (2010) and Morello et al. (2011) when treating P. aeruginosa respiratory infections in mice. Both studies infected and treated the mice via intranasal inhalation. Debarbieux et al. (2010) treated the mice 2, 4, or 6 h post-infection and tracked mortality for 72 h. At the endpoint, 100% of mice treated 2 h post-infection, 75% of mice treated 4 h post-infection, and 25% of mice treated 6 h post-infection survived. The mice treated 2 h post-infection had a bacterial load six orders of magnitude lower than untreated mice 24 h after treatment. Similarly, Morello et al. (2011) demonstrated that mice infected with a strain of P. aeruginosa isolated from a CF patient and treated with a sufficiently high MOI 2 h post-infection and euthanized 20 h post-infection had a reduction in bacterial load of two orders of magnitude. Interestingly, they were also able to demonstrate, using immunohistochemistry, that the remaining bacteria in the treated mice were mainly in the macrophages in the lungs while the untreated mice had bacteria throughout the macrophages, alveolae, and extracellular spaces within the lungs. These findings indicate that phage therapy was able to work with the immune system in order to effectively clear the P. aeruginosa infection. These conflicting results between Carmody et al. (2010) and the results obtained by Debarbieux et al. (2010) and Morello et al. (2011) demonstrate the necessity for further evaluation of aerosol phage therapy.
In both P. aeruginosa studies, the effect of phage as a prophylactic was also evaluated. Debarbieux et al. (2010) demonstrated that phage administered 24 h prior to infection allowed for 100% survival, while all mice in the control group died within 2 days. Morello et al. (2011) reported a similar trend with a pretreatment occurring 4 days prior to infection. Although generally phage therapy is thought of as an infection treatment method, these results suggest that aerosol phage therapy may also be a useful method for preventing BCC infection.
Aerosol phage therapy and phage therapy against the BCC are two fields of study that have not been investigated in great detail until recently. Through the use of two different in vivo models, the G. mellonella model and the mouse lung infection model, phages have recently been demonstrated to be effective against the BCC. Using a mechanical lung model it was demonstrated that BCC phages could successfully be delivered to the human lung, remaining viable after deposition within the lung. Additionally, dry powder aerosol phage delivery appears to be a promising alternative to liquid phage nebulization. BCC phages have been successfully freeze-dried and spray-dried and can be successfully stored and aerosolized after processing. With advancing research, BCC phage therapy continues to show promise as an alternative antimicrobial therapy.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Jonathan J. Dennis gratefully acknowledges funding from Cystic Fibrosis Canada, and Canadian Institutes of Health Research (CIHR Team on Aerosol Phage Therapy). Karlene H. Lynch thanks Cystic Fibrosis Canada, Alberta Innovates – Health Solutions, the Killam Trusts, and the Natural Sciences and Engineering Research Council of Canada for studentship funding.
Ackermann, H.-W. (2001). Frequency of morphological phage descriptions in the year 2000. Arch. Virol. 146, 843–857.
AmpliPhi. (2011). Product Pipeline. Available at: http://www.ampliphibio.com/index.php/pipeline/product_pipeline [accessed December 4, 2011].
Bernier, S. P., and Sokol, P. A. (2005). Use of suppression-subtractive hybridization to identify genes in the Burkholderia cepacia complex that are unique to Burkholderia cenocepacia. J. Bacteriol. 187, 5278–5291.
Biswas, B., Adhya, S., Washart, P., Paul, B., Trostel, A. N., Powell, B., Carlton, R., and Merril, C. R. (2002). Bacteriophage therapy rescues mice bacteremic from a clinical isolate of vancomycin-resistant Enterococcus faecium. Infect. Immun. 70, 204–210.
Bradbury, J. (2004). “My enemy’s enemy is my friend” – using phages to fight bacteria. Lancet 363, 624–625.
Bruttin, A., and Brüssow, H. (2005). Human volunteers receiving Escherichia coli phage T4 orally: a safety test of phage therapy. Antimicrob. Agents Chemother. 49, 2874–2878.
Capparelli, R., Parlato, M., Borriello, G., Salvatore, P., and Iannelli, D. (2007). Experimental phage therapy against Staphylococcus aureus in mice. Antimicrob. Agents Chemother. 51, 2765–2773.
Carmody, L. A., Gill, J. J., Summer, E. J., Sajjan, U. S., Gonzalez, C. F., Young, R. F., and LiPuma, J. J. (2010). Efficacy of bacteriophage therapy in a model of Burkholderia cenocepacia pulmonary infection. J. Infect. Dis. 201, 264–271.
Cerveny, K. E., DePaola, A., . Duckworth, D. H., and Gulig, P. A. (2002). Phage therapy of local and systemic disease caused by Vibrio vulnificus in iron-dextran-treated mice. Infect. Immun. 70, 6251–6262.
Cihlar, R. L., Lessie, T. G., and Holt, S. C. (1978). Characterization of bacteriophage CP1, an organic solvent sensitive phage associated with Pseudomonas cepacia. Can. J. Microbiol. 24, 1404–1412.
Debarbieux, L., Leduc, D., Maura, D., Morello, E., Criscuolo, A., Grossi, O., Balloy, V., and Touqui, L. (2010). Bacteriophages can treat and prevent Pseudomonas aeruginosa lung infections. J. Infect. Dis. 201, 1096–1104.
d’Herelle, F. (1917). An invisible antagonist microbe of dysentery bacilli. C. R. Acad. Sci. Paris 165, 373–375.
Garsevanishvili, T. I. (1974). Certain methodological aspects of the use of inhalation of a polyvalent bacteriophage in the treatment of pneumonia of young children. Pediatriia 53, 65–66.
Gill, J., Summer, E., Russell, W., Cologna, S., Carlile, T., Fuller, A., Kitsopoulos, K., Mebane, L. M., Parkinson, B. N., Sullivan, D., Carmody, L. A., Gonzalez, C. F., LiPuma, J. J., and Young, R. (2011). Genome and characterization of phages Bcep22 and BcepIL02, founders of a novel phage type in Burkholderia cenocepacia. J. Bacteriol.193, 5300–5313.
Golshahi, L., Lynch, K., Dennis, J., and Finlay, W. (2011). In vitro lung delivery of bacteriophages KS4-M and ΦKZ using dry powder inhalers for treatment of Burkholderia cepacia complex and Pseudomonas aeruginosa infections in cystic fibrosis. J. Appl. Microbiol. 110, 106–117.
Golshahi, L., Seed, K. D., Dennis, J. J., and Finlay, W. H. (2008). Toward modern inhalational bacteriophage therapy: nebulization of bacteriophages of Burkholderia cepacia complex. J. Aerosol Med. Pulm. Drug Deliv. 21, 351–360.
Goudie, A. D., Lynch, K. H., Seed, K. D., Stothard, P., Shrivastava, S., Wishart, D. S., and Dennis, J. J. (2008). Genomic sequence and activity of KS10, a transposable phage of the Burkholderia cepacia complex. BMC Genomics 9, 615.
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8, 172.
Halperin, S., Heifetz, S., and Kasina, A. (1988). Experimental respiratory infection with Bordetella pertussis in mice: comparison of two methods. Clin. Invest. Med. 11, 297–303.
Hens, D. K., Chatterjee, N. C., and Kumar, R. (2006). New temperate DNA phage BcP15 acts as a drug resistance vector. Arch. Virol. 151, 1345–1353.
Hens, D. K., Ghosh, A. N., and Kumar, R. (2005). A new small temperate DNA phage BcP15 isolated from Burkholderia cepacia DR11. Arch. Virol. 150, 2421–2428.
Heslop, K., and Harkawat, R. (2000). Nebulizer therapy from a practical perspective. Eur. Respir. Rev. 10, 213–215.
Hoeflmayr, J. (1963). Inhalation Therapy Using Bacteriophages in Therapy-Resistant Infections. Frederick, MD: Army Biological Laboratories.
Holden, M. T. G., Seth-Smith, H. M. B., Crossman, L. C., Sebaihia, M., Bentley, S. D., Cerdeño-Tárraga, A. M., Thomson, N. R., Bason, N., Quail, M. A., Sharp, S., Cherevach, I., Churcher, C., Goodhead, I., Hauser, H., Holroyd, N., Mungall, K., Scott, P., Walker, D., White, B., Rose, H., Iversen, P., Mil-Homens, D., Rocha, E. P., Fialho, A. M., Baldwin, A., Dowson, C., Barrell, B. G., Govan, J. R., Vandamme, P., Hart, C. A., Mahenthiralingam, E., and Parkhill, J. (2009). The genome of Burkholderia cenocepacia J2315, an epidemic pathogen of cystic fibrosis patients. J. Bacteriol. 91, 261–277.
Horvath, P., and Barrangou, R. (2010). CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170.
Huff, W. E., Huff, G. R., Rath, N. C., Balog, J. M., and Donoghue, A. M. (2002). Prevention of Escherichia coli infection in broiler chickens with a bacteriophage aerosol spray. Poult. Sci. 81, 1486–1491.
Isles, A., Maclusky, I., and Corey, M. (1984). Pseudomonas cepacia infection in cystic fibrosis: an emerging problem. J. Pediatr. 104, 206–210.
Johnson, R. P., Gyles, C. L., Huff, W. E., Ojha, S., Huff, G. R., Rath, N. C., and Donoghue, A. M. (2008). Bacteriophages for prophylaxis and therapy in cattle, poultry and pigs. Anim. Health Res. Rev. 9, 201–215.
Kenna, D. T., Barcus, V. A., Langley, R. J., Vandamme, P., and Govan, J. R. W. (2003). Lack of correlation between O-serotype, bacteriophage susceptibility and genomovar status in the Burkholderia cepacia complex. FEMS Immunol. Med. Microbiol. 35, 87–92.
Kropinski, A. M. (2006). Phage therapy – everything old is new again. Can. J. Infect. Dis. Med. Microbiol. 17, 297–306.
Kumari, S., Harjai, K., and Chhibber, S. (2009). Efficacy of bacteriophage treatment in murine burn wound infection induced by Klebsiella pneumoniae. J. Microbiol. Biotechnol. 19, 622–628.
Kutateladze, M., and Adamia, R. (2008). Phage therapy experience at the Eliava institute. Med. Mal. Infect. 38, 426–430.
Langley, R., Kenna, D. T., Vandamme, P., Ure, R., and Govan, J. R. W. (2003). Lysogeny and bacteriophage host range within the Burkholderia cepacia complex. J. Med. Microbiol. 52, 483–490.
Langley, R. J., Kenna, D., Bartholdson, J., Campopiano, D. J., and Govan, J. R. W. (2005). Temperate bacteriophages DK4 and BcepMu from Burkholderia cenocepacia J2315 are identical. FEMS Immunol. Med. Microbiol. 45, 349–350.
Levin, B. R., and Bull, J. J. (2004). Population and evolutionary dynamics of phage therapy. Nat. Rev. Microbiol. 2, 166–173.
Lü, J., Wang, X., Marin-Muller, C., Wang, H., Lin, P. H., Yao, Q., and Chen, C. (2009). Current advances in research and clinical applications of PLGA-based nanotechnology. Expert Rev. Mol. Diagn. 9, 325–341.
Lynch, K. H., Seed, K. D., Stothard, P., and Dennis, J. J. (2010a). Inactivation of Burkholderia cepacia complex phage KS9 gp41 identifies the phage repressor and generates lytic virions. J. Virol. 84, 1276–1288.
Lynch, K. H., Stothard, P., and Dennis, J. J. (2010b). Genomic analysis and relatedness of P2-like phages of the Burkholderia cepacia complex. BMC Genomics 11, 599.
Lynch, K. H., Stothard, P., and Dennis, J. J. (in press). Characterization of DC1, a broad host range Bcep22-like podovirus. Appl. Environ. Microbiol.
Matinkhoo, S., Lynch, K. H., Dennis, J. J., Finlay, W. H., and Vehring, R. (2011). Spray-dried respirable powders containing bacteriophages for the treatment of pulmonary infections. J. Pharm. Sci. 100, 5197–5205.
Matsumoto, H., Itoh, Y., Ohta, S., and Terawaki, Y. (1986). A generalized transducing phage of Pseudomonas cepacia. J. Gen. Microbiol. 132, 2583–2586.
Mattey, M., and Spencer, J. (2008). Bacteriophage therapy – cooked goose or phoenix rising? Curr. Opin. Biotechnol. 19, 608–612.
McVay, C. S., Velasquez, M., and Fralick, J. A. (2007). Phage therapy of Pseudomonas aeruginosa infection in a mouse burn wound model. Antimicrob. Agents Chemother. 51, 1934–1938.
Merabishvili, M., Pirnay, J., Verbeken, G., Chanishvili, N., Tediashvili, M., Lashkhi, N., Glonti, T., Krylov, V., Mast, J., Van Parys, L., Lavigne, R., Volckaert, G., Mattheus, W., Verween, G., De Corte, P., Rose, T., Jennes, S., Zizi, M., and De Vos, D. (2009). Quality-controlled small-scale production of a well-defined bacteriophage cocktail for use in human clinical trials. PLoS ONE 4, e4944.
Merril, C., Scholl, D., and Adhya, S. (2003). The prospect for bacteriophage therapy in Western medicine. Nat. Rev. Drug Discov. 2, 489–497.
Morello, E., Saussereau, E., Maura, D., Huerre, M., Touqui, L., and Debarbieux, L. (2011). Pulmonary bacteriophage therapy on Pseudomonas aeruginosa cystic fibrosis strains: first steps towards treatment and prevention. PLoS ONE 6, e16963.
Nadithe, V., Rahamatalla, M., Finlay, W. H., Mercer, J. R., and Samuel, J. (2003). Evaluation of nose-only aerosol inhalation chamber and comparison of experimental results with mathematical simulation of aerosol deposition in mouse lungs. J. Pharm. Sci. 92, 1066–1076.
Newman, S. P., and Busse, W. W. (2002). Evolution of dry powder inhaler design, formulation, and performance. Respir. Med. 96, 293–304.
Nishiyama, E., Ohtsubo, Y., Nagata, Y., and Tsuda, M. (2010). Identification of Burkholderia multivorans ATCC 17616 genes induced in soil environment by in vivo expression technology. Environ. Microbiol. 12, 2539–2558.
Nzula, S., Vandamme, P., and Govan, J. R. W. (2000). Sensitivity of the Burkholderia cepacia complex and Pseudomonas aeruginosa to transducing bacteriophages. FEMS Immunol. Med. Microbiol. 28, 307–312.
O’Grady, E. P., Viteri, D. F., Malott, R. J., and Sokol, P. A. (2009). Reciprocal regulation by the CepIR and CciIR quorum sensing systems in Burkholderia cenocepacia. BMC Genomics 10, 441.
Papaleo, M. C., Perrin, E., Maida, I., Fondi, M., Fani, R., and Vandamme, P. (2010). Identification of species of the Burkholderia cepacia complex by sequence analysis of the hisA gene. J. Med. Microbiol. 59, 1163–1170.
Paul, V. D., Sundarrajan, S., Rajagopalan, S. S., Hariharan, S., Kempashanaiah, N., Padmanabhan, S., Sriram, B., and Ramachandran, J. (2011). Lysis-deficient phages as novel therapeutic agents for controlling bacterial infection. BMC Microbiol. 11, 195.
Payne, R. J. H., and Jansen, V. A. A. (2003). Pharmacokinetic principles of bacteriophage therapy. Clin. Pharmacokinet. 42, 315–325.
Peeters, E., Sass, A., Mahenthiralingam, E., Nelis, H., and Coenye, T. (2010). Transcriptional response of Burkholderia cenocepacia J2315 sessile cells to treatments with high doses of hydrogen peroxide and sodium hypochlorite. BMC Genomics 11, 90.
Puapermpoonsiri, U., Spencer, J., and van der Walle, C. F. (2009). A freeze-dried formulation of bacteriophage encapsulated in biodegradable microspheres. Eur. J. Pharm. Biopharm. 72, 26–33.
Rakhuba, D., Kolomiets, E., Szwajcer Dey, E., and Novik, G. (2010). Bacteriophage receptors, mechanisms of phage adsorption and penetration into the host cell. Pol. J. Microbiol. 59, 145–155.
Rhoads, D. D., Wolcott, R. D., Kuskowski, M. A., Wolcott, B. M., Ward, L. S., and Sulakvelidze, A. (2009). Bacteriophage therapy of venous leg ulcers in humans: results of a phase I safety trial. J. Wound Care 18, 237–238; 240–243.
Ronning, C. M., Losada, L., Brinkac, L., Inman, J., Ulrich, R. L., Schell, M., Nierman, W. C., and DeShazer, D. (2010). Genetic and phenotypic diversity in Burkholderia: contributions by prophage and phage-like elements. BMC Microbiol. 10, 202.
Seed, K. D., and Dennis, J. J. (2005). Isolation and characterization of bacteriophages of the Burkholderia cepacia complex. FEMS Microbiol. Lett. 251, 273–280.
Seed, K. D., and Dennis, J. J. (2008). Development of Galleria mellonella as an alternative infection model for the Burkholderia cepacia complex. Infect. Immun. 76, 1267–1275.
Seed, K. D., and Dennis, J. J. (2009). Experimental bacteriophage therapy increases survival of Galleria mellonella larvae infected with clinically relevant strains of the Burkholderia cepacia complex. Antimicrob. Agents Chemother. 53, 2205–2208.
Skurnik, M., and Strauch, E. (2006). Phage therapy: facts and fiction. Int. J. Med. Microbiol. 296, 5–14.
Smith, H., and Huggins, M. (1983). Effectiveness of phages in treating experimental Escherichia coli diarrhea in calves, piglets and lambs. J. Gen. Microbiol. 129, 2659–2675.
Sulakvelidze, A., and Morris, J. G. (2001). Bacteriophages as therapeutic agents. Ann. Med. 33, 507–509.
Summer, E. J., Gill, J. J., Upton, C., Gonzalez, C. F., and Young, R. (2007). Role of phages in the pathogenesis of Burkholderia, or “where are the toxin genes in Burkholderia phages?” Curr. Opin. Microbiol. 10, 410–417.
Summer, E. J., Gonzalez, C. F., Bomer, M., Carlile, T., Embry, A., Kucherka, A. M., Lee, J., Mebane, L., Morrison, W. C., Mark, L., King, M. D., LiPuma, J. J., Vidaver, A. K., and Young, R. (2006). Divergence and mosaicism among virulent soil phages of the Burkholderia cepacia complex. J. Bacteriol. 188, 255–268.
Summer, E. J., Gonzalez, C. F., Carlisle, T., Mebane, L. M., Cass, A. M., Savva, C. G., LiPuma, J. J., and Young, R. (2004). Burkholderia cenocepacia phage BcepMu and a family of Mu-like phages encoding potential pathogenesis factors. J. Mol. Biol. 340, 49–65.
Thorsson, L., and Geller, D. (2005). Factors guiding the choice of delivery device for inhaled corticosteroids in the long-term management of stable asthma and COPD: focus on budesonide. Respir. Med. 99, 836–849.
Wright, A., Hawkins, C. H., Änggård, E. E., and Harper, D. R. (2009). A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa: a preliminary report of efficacy. Clin. Otolaryngol. 34, 349–357.
Zahid, M. S. H., Udden, S. M. N., Faruque, A. S. G., Calderwood, S. B., Mekalanos, J. J., and Faruque, S. M. (2008). Effect of phage on the infectivity of Vibrio cholerae and emergence of genetic variants. Infect. Immun. 76, 5266–5273.
Keywords: Burkholderia, Burkholderia cepacia complex, bacteriophage, phage therapy, animal infection models, lung infections, aerosols, dry powder inhalers
Citation: Semler DD, Lynch KH and Dennis JJ (2012) The promise of bacteriophage therapy for Burkholderia cepacia complex respiratory infections. Front. Cell. Inf. Microbio. 1:27. doi: 10.3389/fcimb.2011.00027
Received: 20 October 2011; Accepted: 31 December 2011;
Published online: 20 January 2012.
Edited by:
Joanna Goldberg, University of Virginia Health System, USAReviewed by:
Baolin Sun, University of Science and Technology of China, ChinaCopyright: © 2012 Semler, Lynch and Dennis. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jonathan J. Dennis, Department of Biological Sciences, Centennial Centre for Interdisciplinary Science, University of Alberta, Edmonton, AB, Canada T6G 2E9. e-mail:am9uLmRlbm5pc0B1YWxiZXJ0YS5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.