 Jianzhan Yang
Jianzhan Yang Wanna Cai
Wanna Cai Fan Wu
Fan Wu Shengmei Xu
Shengmei Xu Xiaoqi Zhang
Xiaoqi Zhang Bo Liu
Bo Liu Fangfang Xu
Fangfang Xu- 1Guangdong Provincial Key Laboratory of Research on Emergency in TCM, the Second Clinical College of Guangzhou University of Chinese Medicine, Guangzhou, China
- 2Department of Pharmacy, The First Affiliated Hospital of Shantou University Medical College, Shantou, China
- 3Guangdong Provincial Engineering Research Center for Modernization of TCM, College of Pharmacy, Jinan University, Guangzhou, China
Sixteen ceanothane-type triterpenoids, including four new compounds—hovendulcisic acids A–D (1–4) —were purified from the stems of Hovenia dulcis Thunb. The structures of 1–4 were confirmed by comprehensive means including ECD and quantum chemical calculations. Putative biosynthetic pathways of 1–16 were proposed, and 3, 5, and 15 exhibited antitumor activity on A549 and MDA-MB-231 cells.
Introduction
Hovenia dulcis Thunb (Rhamnaceae family) is a traditional Chinese medicine, and many parts of this herb, including seeds, fruit, leaves, and stem, have been used to relieve alcohol toxicity and protect the liver in ancient medical books (Wang, 1958; Su, 1985; Flora of China Editorial Committee, 1982; Chinese materia medica editorial board, 1999; Wu, 2000). Research on the Hovenia genus has revealed its anti-tumor efficacy (Ji, 2003; Ji, et al., 2003; Zhang, 2017), with triterpenoids and flavonoids as their main bioactive constituents (Yoshikawa et al., 1992; Yoshikawa et al., 1998a; Kang et al., 2017; Xu, et al., 2020a; Xu, et al., 2020b; Cai, et al., 2021).
Previous studies revealed that the triterpenoid saponins from H. dulcis inhibited Nrf2 expression (Cai, et al., 2021), indicated potential anti-tumor sensitization activity (Lin et al., 2020). This discovery piqued our interest about the triterpenoid constituents of the Hovenia genus. We found the stems of Hovenia genus to be rich in triterpenoids by LC-MS experimentation. The chemical constituents from the stem of H. dulcis were thus isolated and identified in this study. As a result, four new triterpenoids of hovendulcisic acid A-D (1–4) were isolated from 70% alcohol extract by multi-column chromatography and HPLC methods. The activity of 1–4 on Nrf2 were measured by luciferase reporter gene assay, and the cell viability of 1–4 on MDA-MB-231 and A549 cells were tested by CCK8 method. This research serves as a reference for further study on the pharmacological effects and drug active substance basis of H. dulcis.
Materials and methods
General experimental procedures
NMR data were obtained with a Bruker AMX-600 (Germany). HRESIMS was performed on a Thermo LTQ Orbitrap-Discovery (United States). Acid hydrolysis results were analyzed by Agilent GC 7890A/5975C (United States). UV was recorded on a Hitachi U-2910 (Japan). IR were acquired using a Perkin Elmer Spectrum 400 (United States). Preparative HPLC used an Agilent 1260 instrument equipped with a Cosmosil ODS C18 column (5 μm, 10 mm × 250 mm). Optical rotations were measured on a Rodolph Autopol Ⅰ Polarimeter (United States); column chromatographies were performed with 80–100 and 200–300 mesh silica gel (Qingdao Haiyang) and Sephadex LH-20 (Pharmacia, United States). TLC was carried out on GF254 plates (Qingdao Haiyang). Human MDA-MB-231 cells were purchased from ATCC (VA, United States). D-(+)-glucose and tert-butylhydroquinone (tBHQ) were bought from Sigma Chemical (MO, United States).
Plant materials
The stems of H. dulcis Thunb. were collected in December 2020 in Zhen-an City, Shanxi Province and identified by pharmacist Ganshu She in the Guangdong Provincial Hospital of Chinese Medicine. The samples were authenticated as voucher number (202012001) and stored at the TCM storehouse in the Second Clinical College of the Guangzhou University of Chinese Medicine.
Extraction and Isolation
A total of 30.0 kg of air-dried and mechanically powdered stems of H. dulcis were extracted by reflux using ethanol-H2O solvent (70:30, v/v) thrice and then concentrated to obtain residue. The crude extract was then suspended with water and successively extracted by EtOAc and n-BuOH. The EtOAc fraction (382.9 g) was isolated on a silica gel column and eluted with CHCl2/MeOH solvent system to obtain seven fractions (E1 – E7). Fraction E1 (0.77 g) was isolated via ODS column [MeOH/H2O (40 : 60–100 : 0)], obtaining four subfractions (E1−1 − E1-4). Subfraction E1-2 was separated on a semi-prepared HPLC [aqueous acetonitrile (3.0 mL/min, 99 : 1, v/v)] and obtained 15 (2 mg, tR = 18.9 min). Fraction E2 (62.37 g) was isolated on a silica gel (300–400 mesh) column [PE/EtOAc (1:0, 10:1, 9:1, 6:1, 5:1, 4:1, 3:1, 2:1, 1:1, and 0:1)] with 15 subfractions (E2−1 − E2-15). E2-2 was repeatedly dissolved and recrystallized in CHCl2 and MeOH to obtain compound 16 (35.1 mg). E2-4 (201.0 mg) was separated on a Sephadex LH-20 column (MeOH) and then separated on a ODS column [MeOH/H2O solvent (2:3–1:0)] to divide into four subfractions (E2-4–1 − E2-4–4). Compound 3 (19.1 mg, tR = 12.1 min) was isolated from subfraction E2-4-2 by semi-prepared HPLC using aqueous acetonitrile (3.0 mL/min, 99 : 1, v/v). E2-6 (781.0 mg) was separated on a Sephadex LH-20 column (MeOH), and compound 5 (66 mg) was obtained by MeOH redissolution. E2-9 (1.11 g) fractions were separated by a silica gel column with CHCl2/MeOH (100:0, 250:1, 200:1, 150:1, 100:1, 50:1, 30:1, 10:1, 0:100). Compound 11 (38 mg, tR = 20.5 min) was obtained from E2-9-6 by semi-prepared HPLC (acetonitrile: water, 65:35, v/v). E2-10 (2.60 g) was separated on a Sephadex LH-20 column (MeOH), obtaining five subfractions E2-10–1 − E2-10–5. E2-10–2 (268 mg) was isolated on an ODS column eluted with gradient MeOH/H2O solvent (20:80–100:0, v/v) to divide into six subfractions (E2-10-2–1 − E2-10-2–6). Compound 12 (2.2 mg, tR = 9.0 min) was obtained from E2-10–2-2 by semi-prepared HPLC (acetonitrile: water, 67 : 33, v/v), and compound 8 (9.1 mg, tR = 18.4 min) was obtained from E2-10–2-5 by semi-prepared HPLC (acetonitrile: water, 70 : 30, v/v). E2-12 (2.65 g) was separated on an ODS column [MeOH/H2O solvent (30:70–100:0, v/v)] to divide into six subfractions (E2-12–1 − E2-12–6). Compound 2 (5.1 mg, tR = 10.6 min) was obtained from E2-12-4 by semi-prepared HPLC (acetonitrile: water, 75 : 25, v/v). Compound 7 (54 mg) was obtained from E2-12-5 by repeatedly dissolving and recrystallizing in MeOH. E2-13 (5.86 g) was separated on an ODS column [MeOH/H2O solvent (20 : 80–100 : 0, v/v)] to divide into eight subfractions (E2-13–1 − E2-13–8). Compound 9 (228.1 mg, tR = 8.0 min) was obtained from E2-13-4 by semi-prepared HPLC (acetonitrile: water, 60 : 40, v/v). E2-13–6 was separated by silica gel column chromatography (CHCl2/MeOH, 100:0, 50:1, 40:1, 25:1, 15:1, 10:1, 5:1), obtaining five subfractions (E2-13-6–1 – E2-13-6–5). We obtained 10 (33.2 mg, tR = 7.2 min) by semi-prepared HPLC (acetonitrile: water, 65:35, v/v) from E2-13-6–3. Fraction E3 (41.53 g) was separated on a silica gel (300–400 mesh) column eluted by CHCl2/MeOH (100:0, 25:1, 15:1, 10:1, 6:1, 4:1, 0:100) to obtain ten subfractions (E3−1 − E3-15). Subfraction E3-6 (2.73 g) was further separated on a Sephadex LH-20 column (MeOH) into eight subfractions (E3-6–1 − E3-6–8). Subfraction E3-6–2 (519.1 mg) was purified on an ODS column eluted with gradient MeOH/H2O (1:4–1:0), followed by semi-prepared HPLC eluted with acetonitrile/0.1% formic acid water (3.0 mL/min, 57 : 43, v/v) to obtain 4 (9.0 mg, tR = 12.8 min). Compound 1 (5.1 mg) was isolated from subfraction E3-6–3 (591.1 mg) by an ODS column eluted with MeOH/H2O (1:4–1:0). Compound 13 (2.2 mg) was obtained from E3-6-5 by repeated dissolving and recrystallizing in MeOH. E3-8 (2.47 g) was separated on a Sephadex LH-20 column (MeOH) and semi-prepared HPLC (acetonitrile: water, 70 : 30, v/v), obtaining compound 14 (13.2 mg, tR = 16 min).
Spectroscopic data
Hovendulcisic acid A (1): white powder; m.p. 340–342°C; [α]25D +11.20 (c 0.8, MeOH); UV (MeOH) λmax: 201.0 nm; IR (νmax cm−1): 3330.0, 2955.3, 2868.3, 1457.3, 1392.8, 1241.0, 1715.6, 1682.6, 1644.6, 1174.0, and 883.5. HR-ESI-MS: m/z 533.3124 [M-H]- (C30H45O8, calcd. for 533.3109). 1D NMR see Table 1 and Table 2.
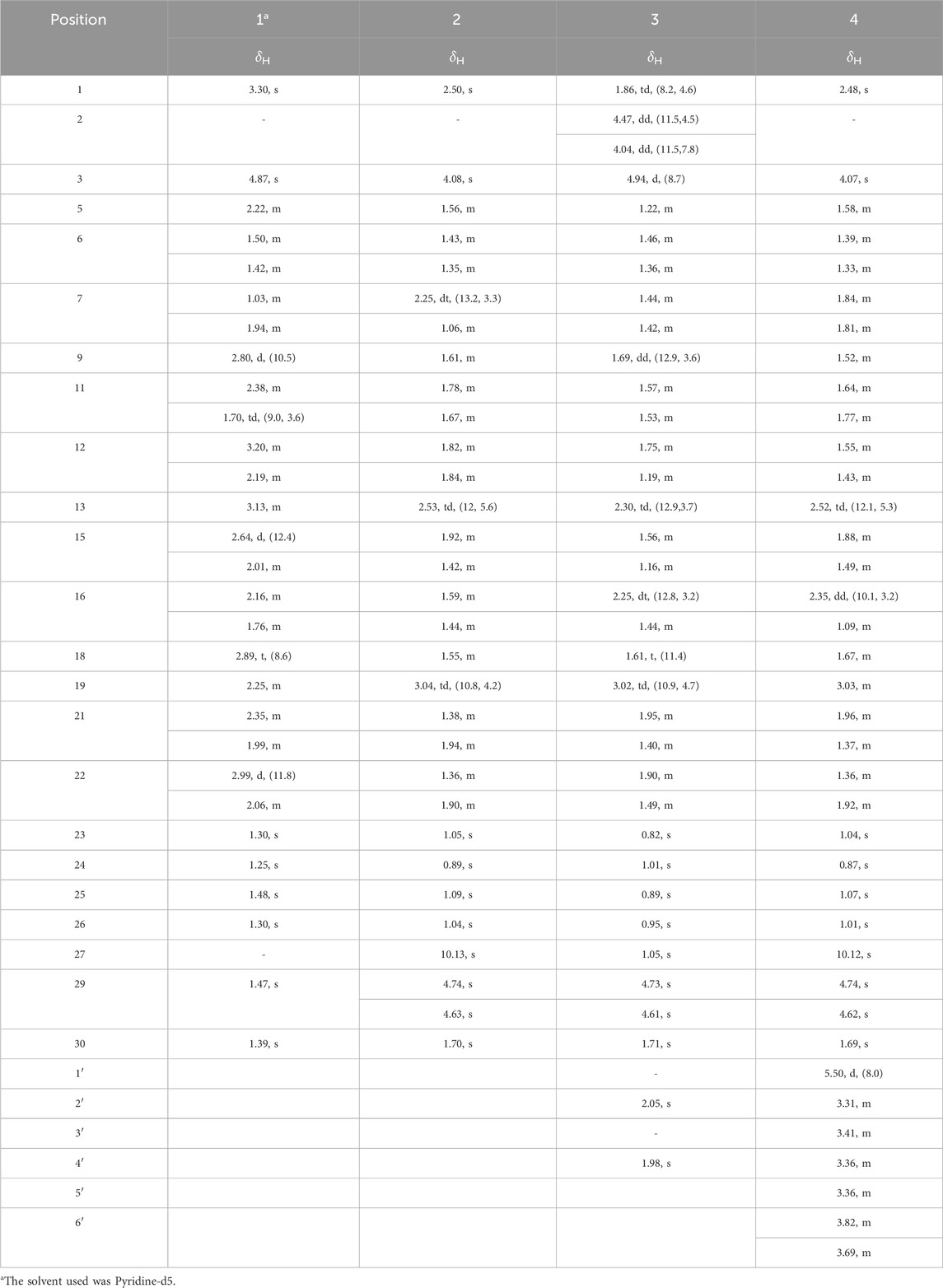
Table 1. 1H NMR data of 1–4 (CD3OD, 600 MHz, δ in ppm, J in Hz).
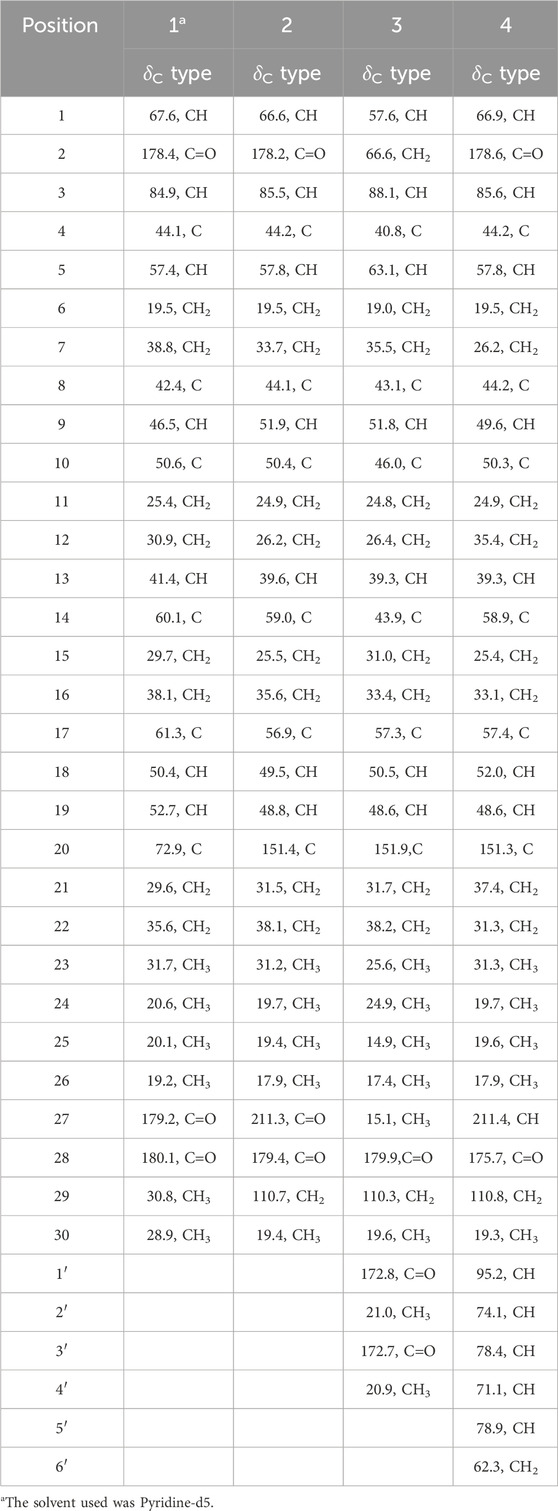
Table 2. 13C NMR data of 1–4 (CD3OD, 150 MHz, δ in ppm).
Hovendulcisic acid B (2): white powder; m.p. 324–326°C; [α]25D +16.0 (c 0.1, MeOH); UV (MeOH) λmax: 203.5 nm; IR (νmax cm−1): 3475.3, 2968.4, 2937.8, 2868.3, 1454.1, 1402.4, 1379.8, 1722.0, 1718.9, 1687.9, 1644.6, 1235.4, and 883.5. HR-ESI-MS: m/z 499.3060 [M-H]- (C30H43O6, calcd. for 499.3054). 1D NMR see Table 1 and Table 2.
Hovendulcisic acid C (3): white powder; m.p. 326–328°C; [α]25D −11.8 (c 0.1, MeOH); UV (MeOH) λmax: 204.0 nm; IR (νmax cm−1): 3397.0, 2944.8, 2869.0, 1739.6, 1701.3, 1458.2, 1375.0, 1235.1, 1186.2, 1029.0, and 774.7. HR-ESI-MS: m/z 555.3699 [M-H]- (C34H51O6, calcd. for 555.3680). 1D NMR see Table 1 and Table 2.
Hovendulcisic acid D (4): white powder; m.p. 243.6–245.1°C; [α]25D +15.12 (c 0.5, MeOH); UV (MeOH) λmax: 202.2 nm; IR (νmax cm−1): 3391.4, 2947.8, 2868.3, 1748.0, 1722.0, 1698.9, 1641.4, 1065.5, 1028.0, and 892.3. HR-ESI-MS: m/z 661.3597 [M-H]- (C36H53O11, calcd. for 661.3582). 1D NMR see Table 1 and Table 2.
Acid hydrolysis and GC analysis
Acid hydrolysis and GC analysis of the glycoside were carried out according to Cai, et al. (2021). First, Compound 4 (2.1 mg) was hydrolyzed in HCl solution (2 M, 10 mL) in an oven (90°C, 4 h), and then evaporated to dryness. H2O was added to the residue and CHCl3 was used for extraction twice, then sugar residue was obtained from the H2O layer. The sugar fraction was reacted with L-cysteine methyl ester hydrochloride (in pyridine, 1 mL) in an oven (60°C, 2 h).
After concentration and drying, the residue was reacted with 1-(trimethylsilyl) imidazole (0.2 mL) in an oven (60°C, 1 h) and then extracted with n-hexane. The n-hexane fraction was acquired for GC analysis. The sugar was identified as D-glucose (tR/min) 25.170, reference D-glucose (tR/min) 25.172.
Computational section
All calculations and processing were conducted using ORCA 5.0.4 and Python 3.10.6. The optimization of the structures used for CD and NMR calculations was performed in pyridine solvents and B3LYP-D3BJ/6–31 g (d, p) levels in the CPCM model, using tight criteria and checking for the absence of virtual frequencies. NMR calculations were performed using the SMD model and at the revTPSS/PCSSEG −1 level (10.1021/ACS.JCTC. 1C00604 indicates that this is a good level, and ECD calculations were performed using TDDFT under the SMD model. We calculated 90 excited states at the ωb97x-d4/def2-TZVP level to cover the excited levels as much as possible, and the rotor intensity and excitation wavelength were expanded using the Gauss function. The FWHM was set at 10 nm to draw the spectrum.
Bioactivity assays
Luciferase reporter gene assay
The cells used in this experiment were MDA-MB-231 stable reporter cell lines transfected with ARE-luciferase plasmid in the previous study (Chen, et al., 2016). The cells were cultured in medium containing puromycin (1.5 mg mL−1) in 48 well plates for 24 h. The isolated constituents 1–4 (1–10 μM) were added to the cells for 24 h. The luciferase activities were tested using the manufacturer’s protocol after the digestion of the cells. TBHQ was used as positive control.
Cytotoxic activity assay
The CCK8 method was applied in a cell viability assay in human MDA-MB-231 cells and human A549 cells. Compounds 1–4 (10–80 μM) were added to the cells for 24 h. The medium was discarded, and CCK8 solution was added and then cultured for 90 min. Absorbance at 450 nm was recorded and used to calculate cell viability.
Results and discussion
Structure elucidation
The stems of H. dulcis were extracted with 70% EtOH, and EtOAc extraction fraction was further isolated by multi-separation methods, acquiring four novel ceanothane-type triterpenoid hovendulcisic acids A-D (1–4) and twelve known: methylceanothate (5) (Leal, et al., 2010), ceanothic acid (6) (Ganapaty, et al., 2006), epiceanothic acid (7) (Li, et al., 2007), 3-O-vanillylceanothic acid (8) (Ganapaty, et al., 2006), 27-hydroxyceanothic acid (9) (Li, et al., 1997), ceanothetric acid (10) (Li, et al., 1997), ceanothanolic acid (11) (Lee, et al., 1997), ceanothetric acid 2-methyl ester (12) (Lee, et al., 1997), ceanothic acid 28β-glucosylester (13) (Lee, et al., 1991), hovetrichoside H (14) (Yoshikawa, et al., 1998), zizyberenalic acid (15) (Zhang, et al., 2012), and ceanothenic acid (16) (Jou, et al., 2004) (Figure 1).
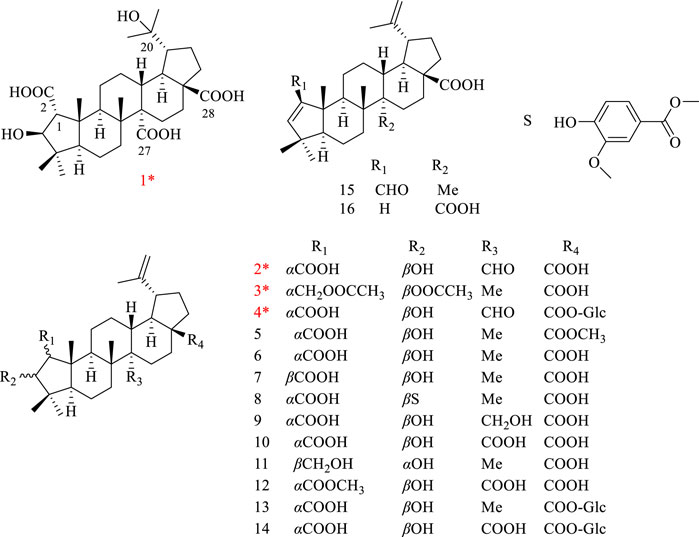
Figure 1. Structures of compounds 1–4.
Compound 1 was a white amorphous powder, and its formula of C30H45O8 was deduced by HR-ESI-MS peaks [M-H]- at m/z 533.3124 (calcd. for C30H45O8, 533.3109). IR data showed absorption bands for OH (3330.0 cm−1), C=O (1715.6, 1682.6 cm−1), and C=C (1644.6 cm−1). 1H NMR spectrum (Table 1) exhibited six methyl groups [δH 1.48(3H, s, H3-25), 1.30 (3H, s, H3-23), 1.30 (3H, s, H3-26), 1.25 (3H, s, H3-24), 1.47 (3H, s, H3-29), and 1.39 (3H, s, H3-30)]. 13C NMR and DEPT-135 data (Table 2) exhibited 30 carbon signals, including six methyls, eight methylenes, seven methines, and nine quaternary carbons (including three carboxylic acid carbons at δC 180.1, 179.2, 178.4). The hydrogen signals and relevant carbon atoms were assigned by HSQC spectrum, and the above information suggested that 1 possessed a triterpenoid skeleton. The NMR data of 1 was similar to those of the known compound ceanothetric acid but without the isoallyl signals (Yoshikawa et al., 1998b). Compared with ceanothetric acid, the molecular weight of 1 added 18 Da and the unsaturation degree decreased by 1, indicating that the double bond between C-20 and C-30 of ceanothetric acid was oxidized and a hydroxyl group was added. The correlations in the HMBC spectrum between C-20 (72.9) and H-29 (δH 1.47), H-30 (δH 1.39) revealed that 1 lost a double bond between C-20 and C-30, which was distinct from the typical ceanothane-type triterpenoid. The HMBC correlations between C-20 (δC 72.9) and H-18 (δH 2.89), H-19 (δH 2.25) also confirmed the position of quaternary carbon (Figure 2). Three carboxylic acid signals (δC 180.1, 179.2, 178.4) were attributed to C-28, C-27, and C-2 according to the HMBC spectra and literature data (Kang et al., 2017).
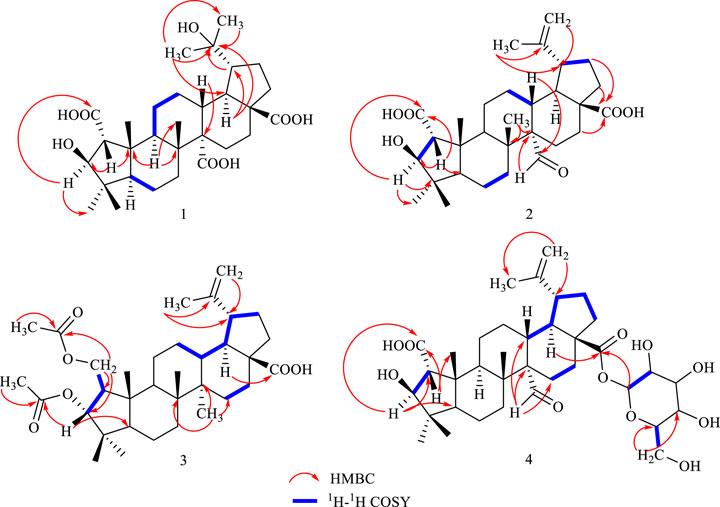
Figure 2. Key 1H−1H COSY and HMBC correlations in compounds 1–4.
NOESY correlations between H−1 (δH 3.30) and δH H-25 (1.48), H-3 (δH 4.87) and H-5 (δH 2.22) suggested the α-configuration of C-2 and β-configuration of C-3 (Li et al., 1997). NOESY correlations between δH 2.25 (H-19) and δH 3.13 (H-13) indicated α-configuration at C-20 (Figure 3). The 13C NMR chemical shift calculations for the 1S, 5R, 7R, and 23R of 1 agreed well with the experimental data, and the correlation coefficient R2 was 99.64% (Figure 4A). Finally, the absolute configuration of 1 (1S, 5R, 7R, and 23R) was determined according to the comparison experimental data with calculated ECD spectra (Figure 5A).
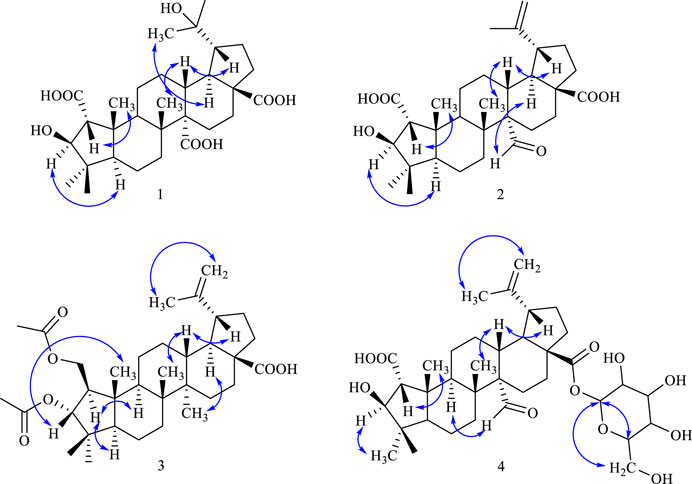
Figure 3. Key NOESY correlations in compounds 1–4.
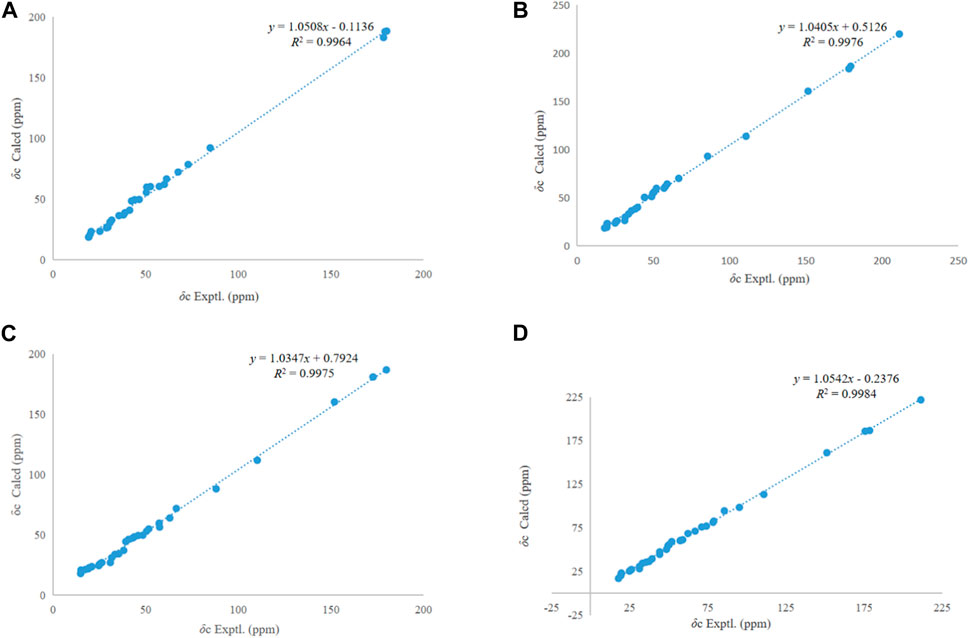
Figure 4. Regression analysis of experimental and calculated 13C NMR chemical shifts of 1–4 (A–D).
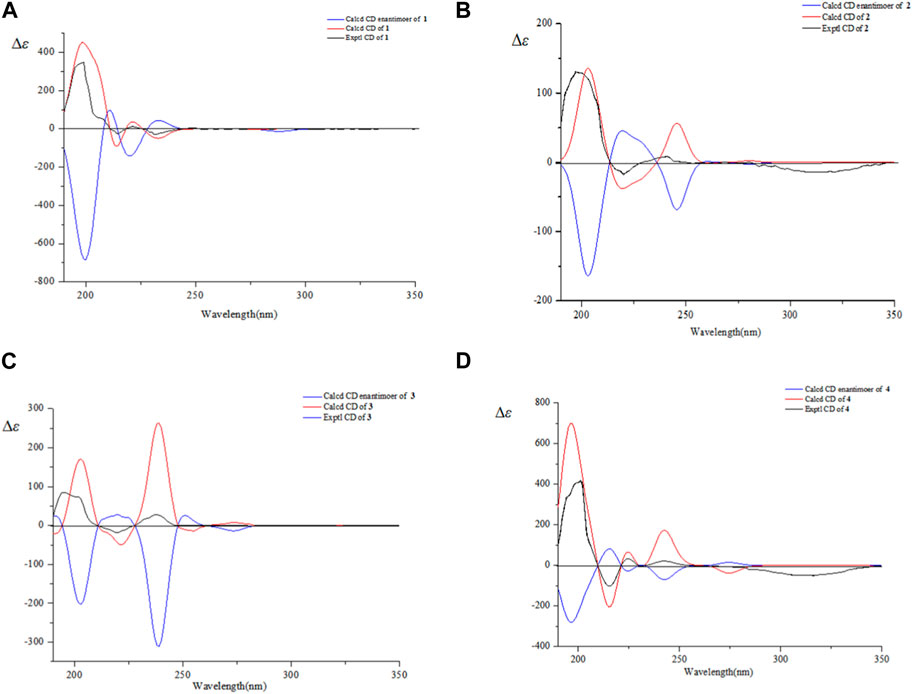
Figure 5. Experimental and calculated ECD spectra of 1–4 (A–D).
Taken together, compound 1 was novel and was identified as 2α-carboxy-3β, 20-dihydroxy-A (1)-norlup-27, 28-dioic acid and named “hovendulcisic acid A”.
Compound 2 was obtained as a white amorphous powder, and the formula was observed to be C30H44O6 according to HR-ESI-MS data [M-H]- at m/z 499.3060 (calcd for C30H43O6, 499.3054). The IR absorption bands exhibited OH (3475.3 cm−1), C=O (1722.0, 1718.9, 1688.0 cm−1), C=C (1644.6 cm−1), and C-O (1235.4 cm−1). The 1H and 13C NMR spectral data of 2 are similar to those of ceanothetric acid, but added a aldehyde group instead of carboxylic acid carbons (Yoshikawa et al., 1998). The correlations in the HMBC spectrum between C-2 (δC 178.2) and H−1 (δH 2.50), H-3 (δH 4.08), C-28 (δC 179.4), and H-16 (δH 1.59), H-21 (δH 1.38), H-22 (δH 1.90) confirmed the position of the carboxylic acid groups. The HMBC correlations between C-2 (δC 178.2), C-4 (δC 44.2), and H-3 (δH 4.08) also indicated that the hydroxy was located at C-3. The HMBC correlations between the C-14 (δC 59.0) and aldehyde proton signal H-27 (δH 10.13), C-27 (δC 211.3) and H-13 (δH 2.53), H-15 (1.92) indicated that the aldehyde group was located at C-27 (Figure 2). NOESY correlations between δH 1.55 (H-18) and δH 10.13 (H-27) suggested that the α-configuration was at C-27 (Figure 3). The 13C NMR chemical shift calculations for the 1S, 5R, 7R, and 23R of 2 correlated well with the experimental result, and the correlation coefficient R2 was 99.76% (Figure 4B). The absolute configuration of 2 was determined by ECD method (Figure 5B). Taken together, compound 2 was identified as 2α-carboxy-3β-hydroxy-A (1)-norlup-20 (29)-en-27-aldehydo-28-oic acid and named “hovendulcisic acid B”.
Compound 3 was obtained as a white amorphous powder, and its formula was C34H52O6 though HR-ESI-MS data [M-H]- at m/z 555.3699 (calcd for C34H51O6, 555.3680). The IR data exhibited the existence of OH (3397.0 cm−1), C=O (1739.6, 1701.3 cm−1), C=C (1644.6 cm−1), and C-O (1186.2, 1029.0 cm−1). The 13C NMR and DEPT spectra exhibited 34 carbon resonances, including eight methyls, ten methylenes (including an oxygenated methylene group at δC 66.56), seven methines (including an oxygenated methine group at δC 88.07), and nine quaternary carbons (including a carboxylic acid carbons at δC 179.91 and two ester groups at δC 172.82, 172.70). The 1D NMR data of 3 were similar to those of 2 but different from the existence of two oxygenated methylene protons [δH 4.47 (1H, dd, J = 11.5, 4.5 Hz, H-2α), 4.04 (1H, dd, J = 11.5, 7.8 Hz, H-2β)] and one oxygenated methine proton [δH 4.94(1H, d, J = 8.7 Hz, H-3)] (Tables 1 and 2). The above data indicated that 3 was a ceanothane-type triterpenoid with two acetyl groups. HMBC correlation of oxygenated methylene proton [δH 4.47, 4.04 (H-2)] with δC 172.8 (C-1′) indicated that one acetyl group was linked at the C-2. HMBC correlation between oxygenated methine proton δH 4.94 (H-3) and δC 172.7 (C-3′), suggesting that another acetyl group was located at C-3 (Figure 2). According to the correlations in the HMBC spectrum between C-1’ (δC 172.8) and H-2 ′ (δH 2.05), C-3’ (δC 172.7) and H-4’ (δH 1.98), the position of the methyl proton can be assigned. The position of the isopropenyl group can be determined according to the HMBC correlations between C-19 (δC 48.6), C-20 (δC 151.9), and [δH 4.73, 4.61 (H-29)], H-30 (δH 1.71). NOESY correlations between δH 1.86 (H−1) and δH 1.22 (H-5), δH 1.69 (H-9), between δH 4.94 (H-3) and δH 0.89 (H-25) indicated the β-configuration of C-2 and α-configuration of C-3’ (Figure 3).
The 13C NMR chemical shift calculations confirmed the relative configuration of 3 (Figure 4C) while the experimental and calculated ECD data confirmed its absolute configuration (Figure 5C). Therefore, compound 3 was identified as 2β, 3α-diacetyl-A (1)-norlup-20 (29)-en-28- oic acid and named “hovendulcisic acid C”.
Compound 4 was isolated as a white amorphous powder with formula C36H54O11 according to HR-ESI-MS data [M-H]- at m/z 661.3597 (calcd for C36H53O11, 661.3582). IR absorption bands exhibited the existence of OH (3391.4 cm−1), C=O (1748.0, 1722.0, 1699.0 cm−1), C=C (1641.4 cm−1) ,and C-O (1065.5, 1028.0 cm−1). The NMR data of 4 were similar to those of 2 but added a sugar residue [δH 5.50 (1H, d, J = 8.0 Hz, H-1′ in Glu), proton signals at δH 3.32–3.82] (Tables 1 and 2). The sugar residue was finally identified as D-glucose according to acid hydrolysis and GC analysis. HMBC correlations between C-28 (δC 175.7) and H-1′ in Glu (δH 5.50) indicated that the glycosidic bonds were located at C-28. The HMBC correlations between the aldehyde proton signal H-27 (δH 10.12) and C-14 (δC 58.9), C-13 (δC 39.3), C-15 (δC 25.4) indicated that the aldehyde group was located at C-27. The position of the isopropenyl group can be determined according to the HMBC correlations between [δH 4.74, 4.62 (H-29)] and C-19 (δC 48.6), C-30 (δC 19.3).
The relative configuration of 4 was deduced by the experimental and calculated 13C NMR method, while the absolute configuration of 4 was approved by experimental and calculated ECD data (Figures 4D, 5D). Therefore, 4 was elucidated as 2α-carboxy-3β-hydroxy-A (1)-norlup-20 (29)-en-27-aldehydo-28-oic acid (hovendulcisic acid B) 28-β-D-glucopyranoside and named “hovendulcisic acid D”.
Putative biosynthetic pathways analysis
The plausible biosynthetic pathway of compounds 1–16 is shown in Scheme 1. The precursor ceanothanolic acid (11), which was first obtained from Paliurus hemsleyanus (Lee, et al., 1997), was oxidated at C-2 to give ceanothic acid (6) and epiceanothic acid (7), diacetylated at C-2 and C-3 to afford hovendulcisic acid C (3). Then ceanothic acid (6) was vanillylated with OH at C-3 to give 3-O-vanillylceanothic acid (8), while ceanothic acid (6) was methyl esterificated and glycosylated of C-28 to afford methylceanothate (5) and ceanothic acid 28β-glucosylester (13), respectively. Epiceanothic acid (7) was oxidated to 27-hydroxyceanothic acid (9), hovendulcisic acid B (2), ceanothetric acid (10), and hovendulcisic acids A (1) with different levels. Hovendulcisic acid B (2) and ceanothetric acid (10) were further glycosylated to hovendulcisic acids D (4) and hovetrichoside H (14), respectively. Epiceanothic acid (7) was dehydrated and reduced to zizyberenalic acid (15), which was further decarboxylated and oxygenated to ceanothenic acid (16).
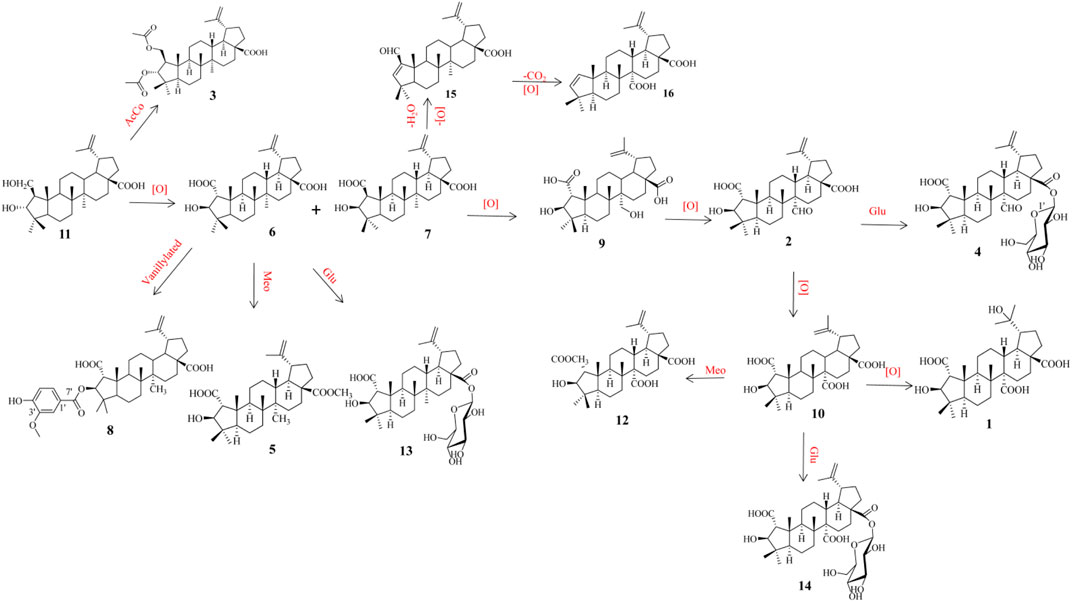
Scheme 1. Putative biosynthetic pathways to 1–16.
Bioactivity results
The antitumor effects of isolated compounds 1–16 were investigated. The results of the ARE luciferase reporter gene showed that 1–4 showed strong inhibitory effect at low concentrations of 1 to 5 μM (Figure 6). Generally, MDA-MB-231 cells appeared to be more sensitive than A549 cells. Compounds 3, 5, and 15 exhibited significant inhibitory activity against A549 cells (Figure 7A), and 3, 5, 12, and 15 exhibited significant inhibitory activity against MDA-MB-231 cells (Figure 7B). The structure–activity relationships analysis revealed that the acetyl and aldehyde groups played an important role in anti-tumor activity. The above results indicated that 3 suppressed tumor activity by inhibiting Nrf2 expression.
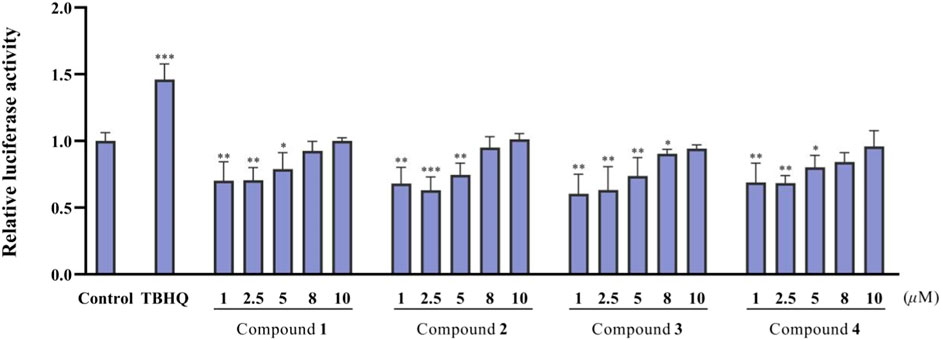
Figure 6. ARE-dependent luciferase activity of the isolated compounds 1–4 (n = 3).
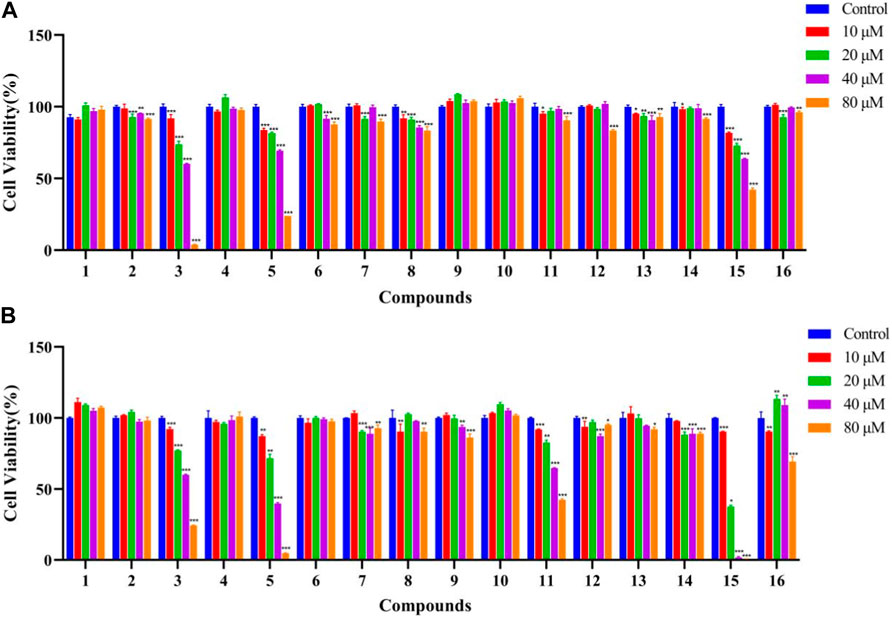
Figure 7. Effects of compounds 1–16 against A549 cells (A) and MDA-MB-231 cells (B) (n = 3). The data represent the mean ± SD of three experiments. *p < 0.05 and **p < 0.01 vs. the control group.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Author contributions
JY: methodology and writing–original draft. WC: writing–original draft. FW: investigation, methodology, and writing–original draft. SX: investigation, methodology, supervision, and writing–review and editing. XZ: conceptualization and writing–review and editing. BL: writing–review and editing. FX: writing–original draft and writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (Nos 82173700 and U1801287), the Science and Technology Planning Project of Guangdong Province (Nos 2022A1515010103 and 2023B1212060063; 2023B1212060062), The Special Foundation of Guangzhou Key Laboratory (Nos 2024A04J9997, 202201020488), the Research Fund for Bajian Talents of Guangdong Provincial Hospital of Chinese Medicine (No. BJ2022KY08), and the Special Funds for State Key Laboratory of Dampness Syndrome of Chinese Medicine (No. SZ2023ZZ13).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2024.1383886/full#supplementary-material
References
Cai, W. N., Xu, S. M., Ma, T., Zhang, X. Q., Liu, Bo., and Xu, F. F. (2021). Five novel triterpenoid saponins from Hovenia dulcis and their Nrf2 inhibitory activities. Arab J Chem 14 (8), 103292. doi:10.1016/j.arabjc.2021.103292
Chen, W., Li, S., Li, J., Zhou, W., Wu, S. H., Xu, S. M., et al. (2016). Artemisitene activates the Nrf2-dependent antioxidant response and protects against bleomycin-induced lung injury. FASEB J. 30 (7), 2500–2510. doi:10.1096/fj.201500109R
Chinese materia medica editorial board (1999). Chinese herbs. Shanghai: Shanghai Scientific and Technical Publishers, 238.
Ganapaty, S., Thomas, P., Ramana, K., and KaragianisWaterman, G. P. G. (2006). Dammarane and ceanothane triterpenes from Zizyphus glabrata. Z Naturforsch B 61 (1), 87–92. doi:10.1515/znb-2006-0118
Ji, Y. (2003). The study of cytotoxic effect and antitumor effect by Hovenia dulcis Thunb. Chin. Arch. Tradit. Chin. Med. (04), 538–543. doi:10.13193/j.archtcm.2003.04.58.jiy.030
Ji, Y., Di, Y. M., Tao, S. Y., and Zhang, K. R. (2003). Experimental study on toxicity and antitumor effect of Hovenia acerba. Chin. J. Tradit. Med. Sci. Technol. (02), 98–99.
Jou, S., Chen, C., Guh, J., Lee, C. N., and Lee, S. S. (2004). Flavonol glycosides and cytotoxic triterpenoids from Alphitonia Philippinensis. J. Chin. Chem. Soc. 51, 827–834. doi:10.1002/jccs.200400124
Kang, K. B., Jun, J. B., Kim, J. W., Kim, H. W., and Sung, S. H. (2017). Ceanothane- and lupane-type triterpene esters from the roots of Hovenia dulcis and their antiproliferative activity on HSC-T6 cells. Phytochemistry 142, 60–67. doi:10.1016/j.phytochem.2017.06.014
Leal, I. C., dos Santos, K. R., Júnior, I. I., Porzel, O. A. C., and Wessjohann, A. (2010). Ceanothane and lupane type triterpenes from Zizyphus joazeiro, an anti-staphylococcal evaluation. Planta Med. 76, 47–52. doi:10.1055/s-0029-1185947
Lee, S., Shy, S., and Liu, K. C. (1997). Triterpenes from Paliurus hemsleyanus. Phytochemistry 46, 549–554. doi:10.1016/s0031-9422(97)00313-0
Lee, S., Su, W., Liu, K., and Liu, K. C. (1991). Two new triterpene glucosides from Paliurus ramosissimus. J. Nat. Prod. 54 (2), 615–618. doi:10.1021/np50074a047
Li, W. H., Zhang, X. M., Tian, R. R., Zheng, Y. T., Zhao, W. M., and Qiu, M. H. (2007). A new anti-HIV lupane acid from Gleditsia sinensis Lam. J. Asian Nat. Prod. Res. 9 (6-8), 551–555. doi:10.1080/10286020600883419
Li, X. C., Cai, L., and Wu, C. D. (1997). Antimicrobial compounds from Ceanothus americanus against oral pathogens. Phytochemistry 46 (1), 97–102. doi:10.1016/S0031-9422(97)00222-7
Lin, H., Qiao, Y., Yang, H., Nan, Q., Qu, W., Feng, F., et al. (2020). Small molecular Nrf2 inhibitors as chemosensitizers for cancer therapy. Future Med. Chem. 12, 243–267. doi:10.4155/fmc-2019-0285
Wang, H. Y. (1958). The benediction of the peace [M]. First edition. Beijing: People’s Medical Publishing House, 1196.
Wu, Y. L. (2000). Herbal medicine renews [M]. First edition. Shanghai: Shanghai Scientific and Technical Publishers, 308.
Xu, F. F., Gao, M. H., Li, H., Han, X. D., Zhang, X. Q., Li, Y. L., et al. (2020). Three new bisflavonols from the seeds of Hovenia dulcis Thunb. and their anti-RSV activities. Fitoterapia 143, 104587. doi:10.1016/j.fitote.2020.104587
Xu, F. F., Liu, B., and Zhang, X. Q. (2020). Research progress on chemical constituents and pharmacological activities of Hovenia. China J. Chin. Mater Med. 45 (20), 4827–4835. doi:10.19540/j.cnki.cjcmm.20200706.602
Yoshikawa, K., Eiko, K., Mimura, N., Kondo, Y., and Arihara, S. (1998). Hovetrichosides C-G, Five new glycosides of two auronols, two neolignans, and a pheylpropanoid from the bark of Hovenia trichocrea. J. Nat. Prod. 61, 78–79.
Yoshikawa, K., Kondo, Y., Kimura, S., and Arihara, S. (1998). A lupane-triterpene and a 3(2→1) abeolupane glucoside from Hoveina trichocarea. Phtochemistry 49 (7), 2057–2060. doi:10.1016/s0031-9422(98)00409-9
Yoshikawa, K., Shinichi, T., Kazuko, Y., and Arihara, S. (1992). Antisweet natural products VII. Hodulosides I, II, III, IV, and V from the leaves of Hovenia dulcis Thunb. Chem. Pharm. Bull. 40 (9), 2287–2291. doi:10.1248/cpb.40.2287
Zhang, Q. (2017). Isolation and purification of anti-tumor active compound SIPI-ZQ2 from Hovenia dulcis Thunb and its in vitro and in vivo antitumor activity evaluation and acute toxicity [D]. Shanghai: Shanghai institute of pharmaceutical industry.
Keywords: Hovenia dulcis Thunb, ceanothane-type triterpenoids, hovendulcisic acid, antitumor activity, quantum chemical calculations
Citation: Yang J, Cai W, Wu F, Xu S, Zhang X, Liu B and Xu F (2024) Hovendulcisic acid A-D: four novel ceanothane-type triterpenoids from Hovenia dulcis stems with anticancer properties. Front. Chem. 12:1383886. doi: 10.3389/fchem.2024.1383886
Received: 08 February 2024; Accepted: 12 April 2024;
Published: 14 May 2024.
Edited by:
Xuetao Xu, Wuyi University, ChinaReviewed by:
Li-She Gan, Zhejiang Chinese Medical University, ChinaXiaoxiao Huang, Shenyang Pharmaceutical University, China
Copyright © 2024 Yang, Cai, Wu, Xu, Zhang, Liu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fangfang Xu, eHVkdWJmYW5nQDE2My5jb20=; Bo Liu, ZG9jdGxpdUAyNjMubmV0; Xiaoqi Zhang, dHpoeHEwMUBqbnUuZWR1LmNu
†These authors have contributed equally to this work