 Fu-Ying Qin1†
Fu-Ying Qin1† Yan-Yi Chen
Yan-Yi Chen Jiao-Jiao Zhang
Jiao-Jiao Zhang Yong-Xian Cheng
Yong-Xian Cheng- 1Institute for Inheritance-Based Innovation of Chinese Medicine, School of Pharmaceutical Sciences, Health Science Center, Shenzhen University, Shenzhen, China
- 2Guangdong Key Laboratory for Functional Substances in Medicinal Edible Resources and Healthcare Products, School of Life Sciences and Food Engineering, Hanshan Normal University, Chaozhou, China
(±)-Dimercochlearlactones A−J (1–10), ten pairs of novel meroterpenoid dimers and one known spirocochlealactone A (11), were isolated from Ganoderma mushrooms. The structural elucidation of new compounds, including their absolute configurations, depends on spectroscopic analysis and electronic circular dichroism (ECD) calculations. Biological studies showed that (+)- and (–)-2, (–)-3, and (+)- and (–)-11 are cytotoxic toward human triple negative breast cancer (TNBC) cells (MDA-MB-231) with IC50 values of 28.18, 25.65, 11.16, 8.18, and 13.02 μM, respectively. Wound healing assay revealed that five pairs of meroterpenoids (±)-5−(±)-8 and (±)-10 could significantly inhibit cell mobility at 20 μM in MDA-MB-231 cells. The results provide a new insight into the biological role of Ganoderma meroterpenoids in TNBC.
Introduction
Triple negative breast cancer (TNBC), a subgroup of breast cancer, is often found as high grade of invasive ductal carcinoma with aggressive behavior (O'Reilly et al., 2021). The incidence rate of TNBC accounts for approximately 15%–20% of breast cancers (Chen et al., 2021; Chowdhury et al., 2021). “Triple negative” is regarded as the absence of the expression of three receptors, estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor 2 receptor (HER2). It causes an about threefold shorter median overall survival (OS) comparing with other breast cancers (O'Reilly et al., 2021). In addition, the relapse and metastasis rate of TNBC is high, the relapse commonly found within 3 years, and the metastasis often occurs in visceral and brain (O'Reilly et al., 2021; Chen et al., 2021; Chowdhury et al., 2021). Due to the difficulties in treating TNBC, more potential molecules are needed.
Ganoderma is a traditional Chinese medicine and has been discovered numerous bioactivities as hypoglycemic effect, cardiovascular protection, anti-tumor, antioxidant, and brain injury prevention (Lin and Deng, 2019; Lin and Sun, 2019; Liu and Tie, 2019; Meng and Yang, 2019; Quan et al., 2019). All along, Ganoderma triterpenoids have been considered as the main active components with anti-tumor effects. In recent years, Ganoderma meroterpenoids, which process phenol moiety and terpene moiety, have been continuously excavated significant anti-tumor activities, such as toward human cancer cell lines (A549, KYSE30, BT549, and MDA-MB-231) (Qin et al., 2018; Cai et al., 2021; Zhang et al., 2021). For the purpose of discovering active agents toward TNBC from natural sources, eleven meroterpenoid dimers including ten novel ones were isolated from Ganoderma (Figure 1). This paper deals with their isolation, structural elucidation, and biological evaluation for cytotoxicity and cell migration inhibition in TNBC cells.
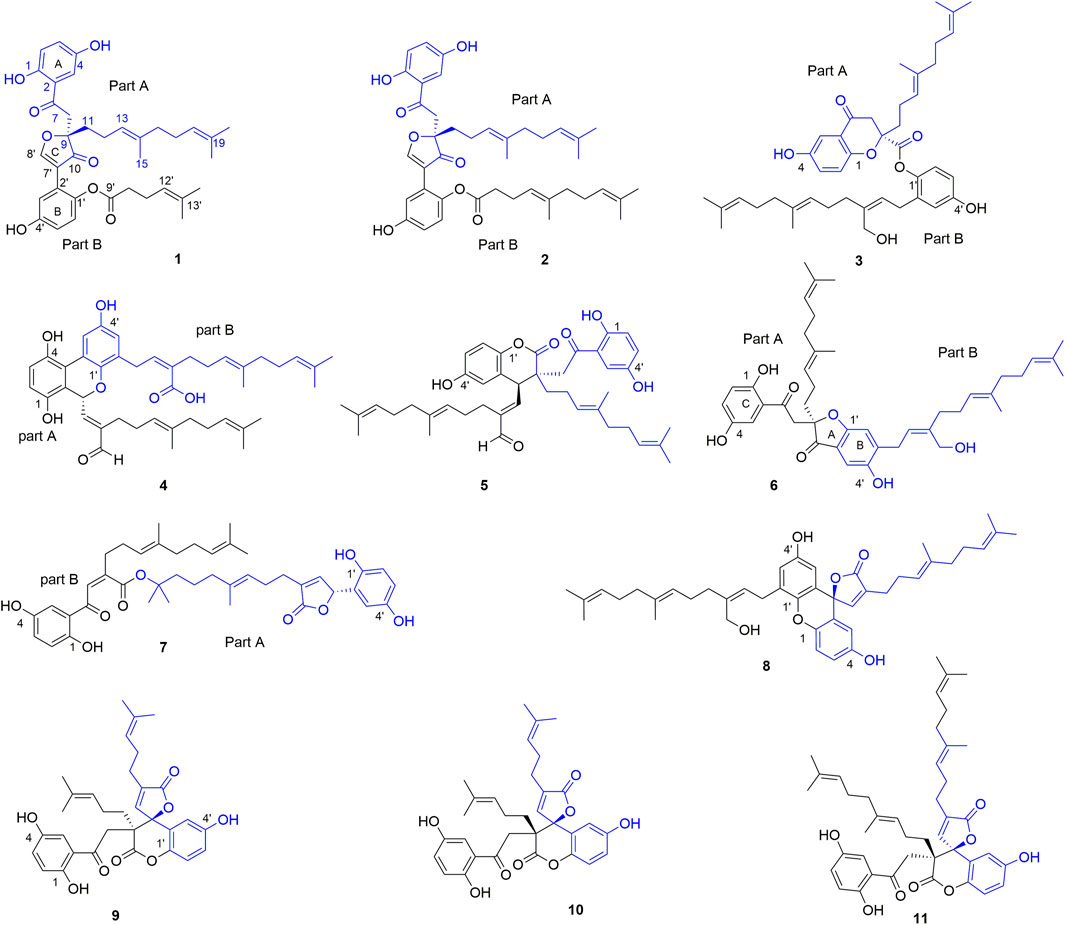
FIGURE 1. The structures of 1–11.
Materials and Methods
General
An Anton Paar MCP-100 digital polarimeter was used to collect optical rotations data. UV and CD spectra were measured on a Chirascan instrument. NMR spectra were collected by a Bruker Avance Ⅲ 600 MHz or a 500-MHz spectrometer, and internal standard is TMS. HRESIMS were recorded on a Waters Xevo G2-XS QTOF or a Shimazu LC-20AD AB Sciex X500R MS spectrometer (Shimadzu Corporation, Tokyo, Japan). C-18 silica gel (40–60 μm; Daiso Co., Japan), MCI gel CHP 20P (75–150 μm, Mitsubishi Chemical Industries, Tokyo, Japan), Sephadex LH-20 (Amersham Pharmacia, Uppsala, Sweden), and Silica gel (Qingdao Marine Chemical Inc., Qingdao, China) were used for column chromatography. Preparative HPLC was carried out using a Chuangxin-Tongheng chromatograph equipped with a Thermo Hypersil GOLD-C18 column (250 mm × 21.2 mm, i.d., 5 μm). Semi-preparative HPLC was taken on a SEP-LC52 chromatograph with a YMC-Pack ODS-A column (250 mm × 10 mm, i.d., 5 μm). Chiral HPLC analysis was taken on an Agilent 1260 or SEP-LC52 chromatograph with a Daicel Chiralpak column (IC, 250 mm × 10 mm, i.d., 5 μm).
Fungal Material
Ganoderma cochlear were purchased from Guangzhou Tongkang Pharmaceutical Co. Ltd. (Guangdong Province, China) in July 2014. Ganoderma lucidum were collected from Dayao County, Yunnan Province, China, in April 2018. Prof. Zhu-Liang Yang from Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, China, authenticated these fungi. The voucher specimens (CHYX-0589 for G. cochlear and CHYX-0615 for G. lucidum) are deposited at the School of Pharmaceutical Sciences, Shenzhen University Health Science Center, China.
Extraction and Isolation
Powdered fruiting bodies of G. cochlear (200 kg) were extracted using refluxing 80% EtOH (3 × 120 L, 4, 3, 3 h) to yield a crude extract. An aliquot (8 kg of the residue corresponding to 95 kg fungal material) was suspended in H2O and extracted three times with EtOAc. The EtOAc soluble residue (4 kg) was then cut into four parts (Fr.1–Fr.4) by a silica gel column with increasing acetone in petroleum ether (10:1–0:1). Fr.2 (860 g) was separated by an MCI gel CHP 20P column (aqueous MeOH, 50%–100%) to get six subfractions (Fr.2.1–Fr.2.6). Of which, the subfraction of Fr.2.2 (120.0 g) was submitted to an RP-18 column (aqueous MeOH, 40%–100%) to obtain five parts (Fr.2.2.1−Fr.2.2.5). Fr.2.2.2 (12.4 g) was fractionated into three parts (Fr.2.2.2.1–Fr.2.2.2.3) by an MCI gel CHP 20P column (aqueous MeOH, 40%–100%). Among them, the last part (7.4 g) was purified by using Sephadex LH-20 (MeOH) and then separated by semi-preparative HPLC (MeOH/H2O, containing 0.05% TFA in H2O, 78%, flow rate: 3 ml/min) to get compounds 9 (tR = 17.0 min, 30.2 mg) and 10 (tR = 23.5 min, 36.5 mg).
Fr.2.5 (70 g) was fractionated into four parts (Fr.2.5.1–Fr.2.5.6) by a silica gel column eluted by increasing acetone in petroleum ether (10:1–0:1). Of which, Fr.2.5.4 (12.0 g) was divided into three parts (Fr.2.5.4.1–Fr.2.5.4.3) by an MCI gel CHP 20P column (aqueous MeOH, 70%–100%). The second part (6.0 g) was first gel filtrated over Sephadex LH-20 (MeOH), then cut by preparative HPLC (aqueous MeOH, 65%–100%) to get five parts (Fr.2.5.4.2.1–Fr.2.5.4.2.5). Among them, Fr.2.5.4.2.4 (500.0 mg) was further purified by semi-preparative HPLC (aqueous MeOH containing 0.05% TFA, 93%, flow rate: 3 ml/min) to afford 3 (tR = 15.6 min, 10.0 mg) and 4 (tR = 24.8 min, 25.6 mg).
Fr.2.5.5 (15.0 g) was fractionated into five parts (Fr.2.5.5.1–Fr.2.5.5.5) by a C-18 column (aqueous MeOH, 70%–100%). Of which, Fr.2.5.5.3 (3.0 g) was first filtrated by using Sephadex LH-20 (MeOH), then divided into six parts (Fr.2.5.5.3.1–Fr.2.5.5.3.6) by preparative HPLC (aqueous MeOH, 65%–100%). Fr.2.5.5.3.5 (150.0 mg) was purified via semi-preparative HPLC (aqueous MeOH containing 0.05% TFA, 92%, flow rate: 3 ml/min) to afford 1 (tR = 12.5 min, 8.0 mg), 8 (tR = 13.3 min, 8.6 mg), 5 (tR = 15.8 min, 0.8 mg), and the impure part was further purified by semi-preparative HPLC to afford 2 (tR = 25.8 min, 1.8 mg) (acetonitrile/H2O, 85% containing 0.05% TFA, flow rate: 3 ml/min). The subfraction of Fr.2.5.5.3.4 (200.0 mg) was purified by semi-preparative HPLC (aqueous MeOH containing 0.05% TFA, 88%, flow rate: 3 ml/min) to afford 6 (tR = 14.8 min, 10.0 mg).
Fr.3 (780.0 g) was divided into eight subfractions (Fr.3.1–Fr.3.8) by an MCI gel CHP 20P column (aqueous MeOH, 40%–100%). Of which, Fr.3.6 (282.0 g) was first purified by Sephadex LH-20 (MeOH) to provide three parts (Fr.3.6.1–Fr.3.6.3). The last part (146.8 g) was frationated into four parts (Fr.3.6.3.1–Fr.3.6.3.4) using a C-18 column (aqueous MeOH, 50%–100%). Of them, Fr.3.6.3.2 (81.0 g) was also first filtrated by Sephadex LH-20 (MeOH), and then cut by an MCI gel CHP 20P column (aqueous MeOH, 40%–100%) to provide five parts (Fr.3.6.3.2.1–Fr.3.6.3.2.5). Among them, Fr.3.6.3.2.4 (23.0 g) filtrated by Sephadex LH-20 (MeOH) followed by a C-18 column (elution solvent: aqueous MeOH, 50%–100%) to get seven parts (Fr.3.6.3.2.4.1-Fr.3.6.3.2.4.7). Fr.3.6.3.2.4.5 (3.9 g) was filtrated by using Sephadex LH-20 (MeOH), then purified by semi-preparative HPLC (aqueous acetonitrile, 78% containing 0.05% TFA, flow rate: 3 ml/min) to obtain 7 (7.5 mg, tR = 20.6 min).
The dried fruiting bodies of G. lucidum (30.0 kg) were powdered and extracted with 95% EtOH under percolation (240 L) at room temperature to afford a crude extract (2.1 kg), which was partitioned between water and EtOAc for three times to obtain an EtOAc extract (1.1 kg). The extract was submitted to an MCI gel CHP 20P column (aqueous MeOH, 40%–100%) to obtain 13 fractions (Fr.1–Fr.13). Fr.13 (228.0 g) was cut by a silica gel column eluted by increasing acetone in petroleum ether (10:1−3:1) to give two parts (Fr.13.1 and Fr.13.2). The second part (50.9 g) was submitted to Sephadex LH-20 (MeOH) to obtain three parts (Fr.13.2.1–Fr.13.2.3). Fr.13.2.2 (2.5 g) was separated by vacuum liquid chromatography (VLC) with increasing acetone in petroleum ether (10:1–3:1) to give five parts (Fr.13.2.2.1–Fr.13.2.2.5). Among them, Fr.13.2.2.3 (456.0 mg) was purified by preparative HPLC (aqueous AcCN, 57%–95%) to afford 11 (39.6 mg).
Compounds 1–11 are racemics, further purification by chiral column (Daicel Chiralpak IC, 250 mm × 10 mm, i.d., 5 μm) (flow rate: 3.0 ml/min) afforded their enantiomers (+)-1 (3.50 mg, tR = 14.8 min) and (–)-1 (3.60 mg, tR = 19.9 min) (n-hexane/ethanol, 90:10); (+)-2 (0.75 mg, tR = 8.6 min) and (–)-2 (0.79 mg, tR = 10.7 min) (n-hexane/ethanol, 85:15); (+)-3 (4.70 mg, tR = 20.6 min) and (–)-3 (4.50 mg, tR = 18.1 min) (n-hexane/ethanol, 92:8); (+)-4 (10.50 mg, tR = 18.1 min) and (–)-4 (10.80 mg, tR = 20.8 min) (n-hexane/ethanol, 95:5); (+)-5 (0.35 mg, tR = 12.3 min) and (–)-5 (0.38 mg, tR = 13.2 min) (n-hexane/ethanol, 90:10); (+)-6 (4.70 mg, tR = 24.4 min) and (–)-6 (4.50 mg, tR = 21.8 min) (n-hexane/ethanol, 90:10); (+)-7 (3.60 mg, tR = 17.6 min) and (–)-7 (3.40 mg, tR = 20.9 min) (n-hexane/ethanol, 90:10); (+)-8 (3.70 mg, tR = 11.9 min) and (–)-8 (3.50 mg, tR = 13.8 min) (n-hexane/ethanol, 90:10); (+)-9 (1.32 mg, tR = 19.7 min) and (–)-9 (1.33 mg, tR = 24.1 min) (n-hexane/ethanol, 95:5), (+)-10 (0.98 mg, tR = 22.6 min) and (–)-10 (1.01 mg, tR = 20.3 min) (n-hexane/ethanol, 95:5); (+)-11 (18.8 mg, tR = 17.2 min) and (–)-11 (18.5 mg, tR = 15.0 min) (n-hexane/ethanol, 96:4).
Compound Characterization
Dimercochlearlactone A (1): yellowish gum; [α]D20 +18.9 (c 0.09, MeOH); CD (MeOH) Δε370 + 0.9, Δε310 –2.5, Δε260 + 2.8, Δε216 –3.1; (+)-1; [α]D20 –11.0 (c 0.10, MeOH); CD (MeOH) Δε371 –0.4, Δε310 + 2.0, Δε260 –2.2, Δε215 + 1.5; (–)-1; UV (MeOH) λmax (logε) 369 (3.42), 284 (58), 254 (3.95), 233 (4.15), 209 (4.31) nm; HRESIMS m/z 625.2774 [M + Na]+ (calcd for C36H42NaO8, 625.2777). 1H and 13C NMR data, see Tables 1 and 2.

TABLE 1. 1H NMR data of 1–5 (δ in ppm, J in Hz).
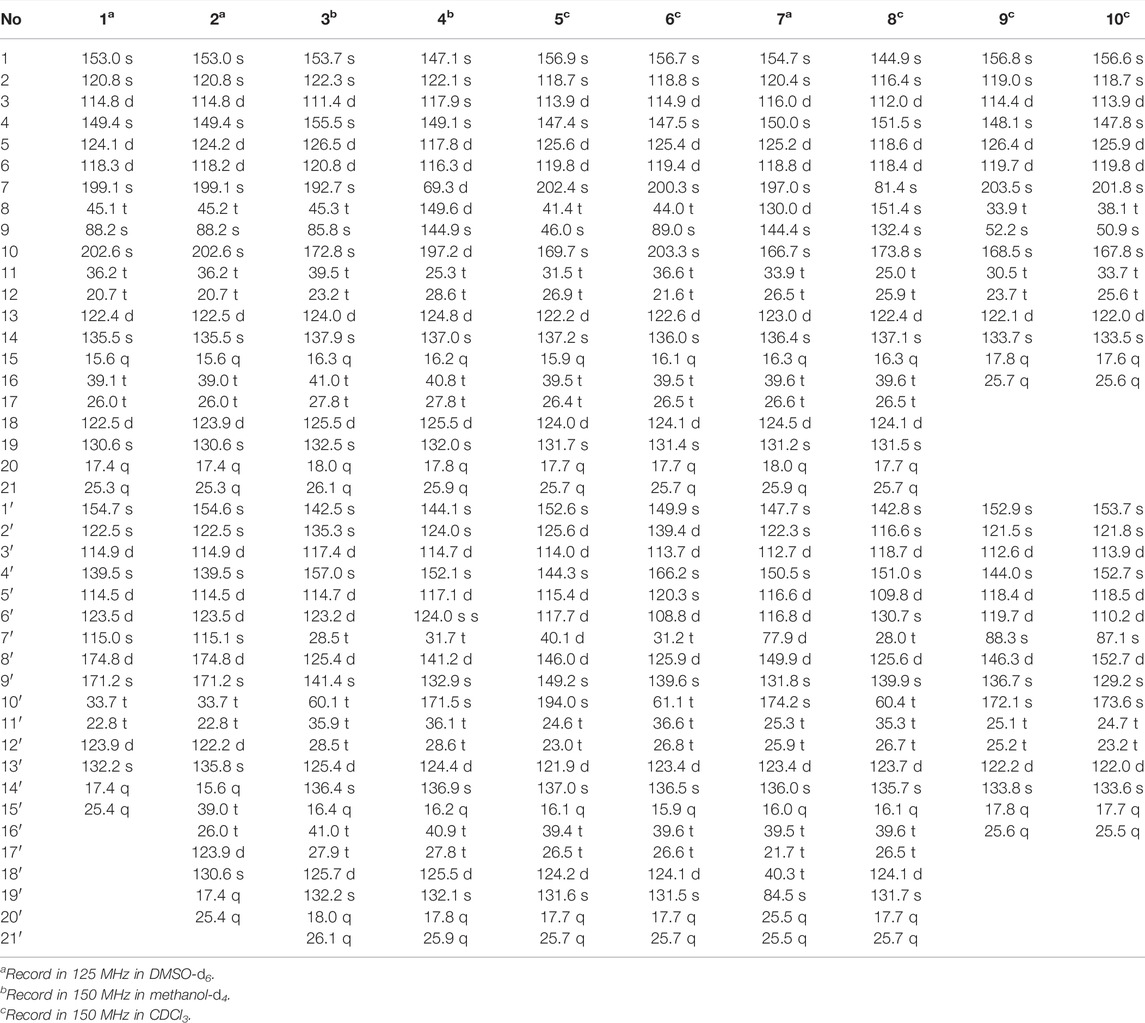
TABLE 2. 13C NMR data of 1–10 (δ in ppm, J in Hz).
Dimercochlearlactone B (2): yellowish gum; [α]D20 +17.0 (c 0.11, MeOH); CD (MeOH) Δε372 + 1.0, Δε311 –3.3, Δε260 + 3.5, Δε209 –6.3; (+)-2; [α]D20 –14.7 (c 0.11, MeOH); CD (MeOH) Δε369 –0.4, Δε309 + 1.7, Δε260 –2.0, Δε214 + 2.8; (–)-2; UV (MeOH) λmax (logε) 369 (3.31), 285 (3.49), 259 (3.87), 233 (4.10), 201 (4.39) nm; HRESIMS m/z 693.3406 [M + Na]+ (calcd for C41H50NaO8, 693.3403). 1H and 13C NMR data, see Tables 1 and 2.
Dimercochlearlactone C (3): yellowish gum; [α]D20 +30.6 (c 0.12, MeOH); CD (MeOH) Δε329 + 3.5 Δε229 –11.6, ; (+)-3; [α]D20 –57.6 (c 0.12, MeOH); CD (MeOH) Δε330 – 4.2, Δε231 + 7.8; (–)-3; UV (MeOH) λmax (logε) 356 (3.55), 283 (3.50), 254 (3.96), 221 (4.48), 203 (4.81) nm; HRESIMS m/z 669.3777 [M – H]– (calcd for C42H53O7, 669.3797). 1H and 13C NMR data, see Tables 1 and 2.
Dimercochlearlactone D (4): yellowish gum; [α]D20 +4.5 (c 0.17, MeOH); CD (MeOH) Δε350 + 4.6, Δε279 –9.2, Δε220 + 13.6, Δε205 –8.5; (+)-4; [α]D20 –4.7 (c 0.22, MeOH); CD (MeOH) Δε352 –5.5, Δε280 + 9.1, Δε220 –14.4, Δε204 + 8.8; (–)-4; UV (MeOH) λmax (logε) 350 (3.62), 225 (4.44) nm; HRESIMS m/z 667.3620 [M – H]– (calcd for C42H51O7, 667.3640). 1H and 13C NMR data, see Tables 1 and 2.
Dimercochlearlactone E (5): yellowish gum; [α]D20 +26.9 (c 0.30, MeOH); CD (MeOH) ε368 + 0.9, Δε279 + 1.5, Δε265 –1.0, Δε250 + 1.7, Δε232 –7.6, Δε216 + 2.9; Δε205 –3.0; (+)-5; [α]D20 –19.0 (c 0.32, MeOH); CD (MeOH) ε368 –0.9, Δε280 –1.5, Δε264 + 1.4, Δε249 –1.4, Δε233 + 7.9, Δε216 –4.0, Δε204 + 3.7 (–)-5; UV (MeOH) λmax (logε) 369 (3.36), 285 (3.34), 255 (3.87), 222 (4.28), 203 (4.50) nm; HRESIMS m/z 781.3580 [M + CF3COO]– (calcd for C44H52F3O9, 781.3569). 1H and 13C NMR data, see Tables 1 and 2.
Dimercochlearlactone F (6): yellowish gum; [α]D20 +38.2 (c 0.10, MeOH); CD (MeOH) ε381 –5.7, Δε319 + 5.5, Δε255 + 3.6, Δε215 –3.5; (+)-6; [α]D20 –16.4 (c 0.11, MeOH); CD (MeOH) ε380 + 4.7, Δε320 –4.0, Δε256 –3.8 Δε214 + 2.0; (–)-6; UV (MeOH) λmax (logε) 368 (3.94), 262 (4.24), 229 (4.40), 203 (4.64) nm; HRESIMS m/z 783.3727 [M + CF3COO]– (calcd for C44H54F3O9, 783.3725). 1H and 13C NMR data, see Tables 2 and 3.

TABLE 3. 1H NMR data of 6–10 (δ in ppm, J in Hz).
Dimercochlearlactone G (7): yellowish gum; [α]D20 +18.6 (c 0.09, MeOH); CD (MeOH) Δε209 + 12.1; (+)-7; [α]D20 –20.2 (c 0.10, MeOH); CD (MeOH) Δε207 –14.2; (–)-7; UV (MeOH) λmax (logε) 380 (3.42) 259 (3.95) nm; HRESIMS m/z 723.3501 [M + Na]+ (calcd for C42H52NaO9, 723.3509). 1H and 13C NMR data, see Tables 2 and 3.
Dimercochlearlactone H (8): yellowish gum; [α]D20 +17.6 (c 0.09, MeOH); CD (MeOH) Δε230 + 1.7; Δε210 –8.4; (+)-8; [α]D20 –18.9 (c 0.10, MeOH); CD (MeOH) Δε228 –2.0; Δε207 + 6.4; (–)-8; UV (MeOH) λmax (logε) 316 (3.85), 242 (4.33), 203 (4.79) nm; HRESIMS m/z 765.3633 [M + CF3COO]– (calcd for C44H52F3O8, 765.3620). 1H and 13C NMR data, see Tables 2 and 3.
Dimercochlearlactone I (9): yellowish gum; [α]D20 +10.1 (c 0.08, MeOH); CD (MeOH) Δε244 –17.7, Δε213 –9.4; (+)-9; [α]D20 −5.1 (c 0.07, MeOH); CD (MeOH) Δε239 + 13.9, Δε209 + 13.76; (–)-9; UV (MeOH) λmax (logε) 373 (3.39), 296 (3.35), 254 (3.82), 209 (4.42) nm; HRESIMS m/z 569.2154 [M + Na]+ (calcd for C32H34NaO8, 569.2152). 1H and 13C NMR data, see Tables 2 and 3.
Dimercochlearlactone J (10): yellowish gum; [α]D20 +58.5 (c 0.09, MeOH); CD (MeOH) Δε260 −4.4, Δε226 + 31.5; (+)-10; [α]D20 –47.1 (c 0.10, MeOH); CD (MeOH) Δε258 + 7.5, Δε226 −31.8; (–)-10; UV (MeOH) λmax (logε) 372 (3.66), 297 (3.57), 254 (4.08), 203 (4.45) nm; HRESIMS m/z 569.2149 [M + Na]+ (calcd for C32H34NaO8, 569.2152). 1H and 13C NMR data, see Tables 2 and 3.
Biological Activity Assay on TNBC Cell Lines (MDA-MB-231)
TNBC cell line MDA-MB-231 was purchased from Procell (Procell Life Science & Technology Co. Ltd., Wuhan, China). Cell culture, cell viability, and wound healing assays were conducted following the reported protocols (Cai et al., 2021).
Results and Discussion
Dimercochlearlactone A (1) was determined to have a molecular formula as C36H42O8 from its positive HRESIMS (m/z 625.2774 [M + Na]+, calcd for C36H42NaO8, 625.2777). Two typical ABX spin systems (δH 7.14, d, J = 3.0 Hz, H-3; δH 6.98, dd, J = 8.9, 3.0 Hz, H-5; δH 6.80, d, J = 8.9 Hz, H-6; δH 7.10, d, J = 2.9 Hz, H-3′; δH 6.71, dd, J = 8.8, 2.9 Hz, H-5′; δH 6.93, d, J = 8.8 Hz, H-6′) were observed by its 1H NMR data (Table 1). The 13C NMR (Table 2) and DEPT spectra show 36 carbon signals, which were assigned as 5 methyl, 7 methylene, 10 methine, and 14 nonprotonated carbons (10 aromatic including 4 oxygenated, 1 oxygenated aliphatic, 2 ketones, and 1 carbonyl). When consideration of the NMR data of the previous reported meroterpenoids (Qin et al., 2018), the above signals suggest that dimercochlearlactone A (1) might be a dimeric meroterpenoid. The structure of dimercochlearlactone A (1) contains two parts (Parts A and B in Figure 1), which were mainly determined by 2D NMR spectra. For the structure of part A, the 1H-1H COSY correlations from H2-12 (δH 1.97 and 1.83) to H2-11 (δH 1.86 and 1.74) and H-13 (δH 5.03), and from H2-17 (δH 2.00) to H2-16 (δH 1.91) and H-18 (δH 5.07), along with the HMBC correlations (Figure 2) from H3-20 (δH 1.54) and H3-21 (δH 1.63) to C-18 (δC 122.5) and C-19 (δC 130.6), from H3-20 to C-21 (δC 25.3), from H3-15 (δH 1.52) and H2-16 to C-13 (δC 122.4) and C-14 (δC 135.5), from H3-15 to C-16 (δC 39.1), and from H2-12 to C-14 suggest the presence of two isoprenyl moieties in dimercochlearlactone A (1). In addition, the HMBC correlations from H-8 (δH 3.92 and 3.57) to C-7 (δC 199.1), C-9 (δC 88.2) and C-10 (δC 202.6) and from H2-11 to C-9 and C-10 imply another isoprenyl residue. Furthermore, HMBC correlation between H-3 (δH 7.14) and C-7 indicates that the sesquiterpeniod moiety is attached to the ring A via C-7 to C-2. Thus, the structure of part A was determined as shown.
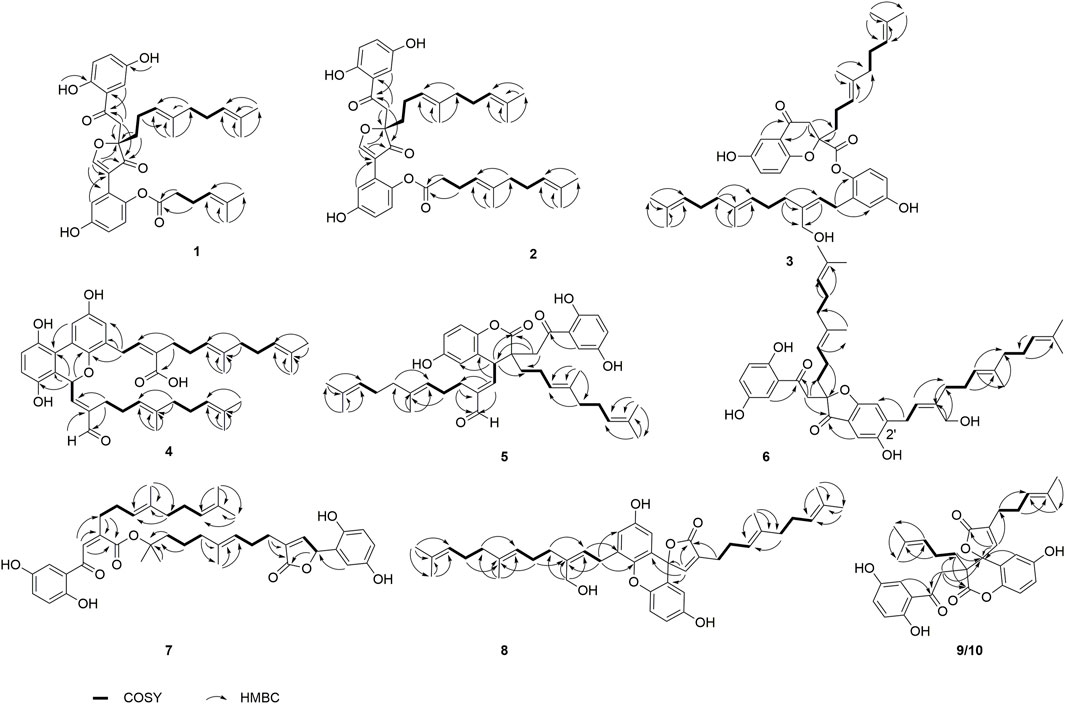
FIGURE 2. Key COSY and HMBC correlations of 1–10.
Part B was also elucidated by 2D NMR experiments (1H-1H COSY, HSQC and HMBC). The HMBC correlations from H-3′ (δH 7.10) to C-7′, from H-8′ (δH 8.77) to C-2′ (δC 122.5), C-7′ (δC 115.0), C-9 and C-10 and the above-mentioned HMBC correlations from H-8 to C-9 and C-10 not only imply the presence of ring B but also indicate that C-2′ is attached to the ring C. The structure of side chain in part B was confirmed by 1H-1H COSY correlations observed from H2-11′(δH 2.26) to H2-10′ (δH 2.51) and H-12′(δH 5.09) and HMBC correlations from H3-14′ (δH 1.55) and H3-15′ (δH 1.63) to C-12′ (δC 123.9) and C-13′ (δC 132.2), and from H2-11′ and H2-10′ to C-9′ (δC 171.2). Since the carboxyl group (C-9′) in the side chain of part B needs form an ester with the phenolic hydroxyl group to meet the molecular formula requirement, the position of the side chain in part B linkage can be determined by the following evidence. In the 2D NMR experiments, the HMBC correlations of 1-OH (δH 10.75)/C-1 (δC 153.0), 4-OH (δH 9.17)/C-4 (δC 149.4), and 4′-OH (δH 9.50)/C-4′ (δC 139.5) and ROESY correlations between 1-OH with H-6, 4-OH with H-3, and 4′-OH with H-6 led to the determination of the side chain linkage at the C-1′ position. This conclusion was further secured by the ROESY correlation (Figure 3) between H2-10′ and H-6′. Thus, the 2D structure of 1 was assigned.
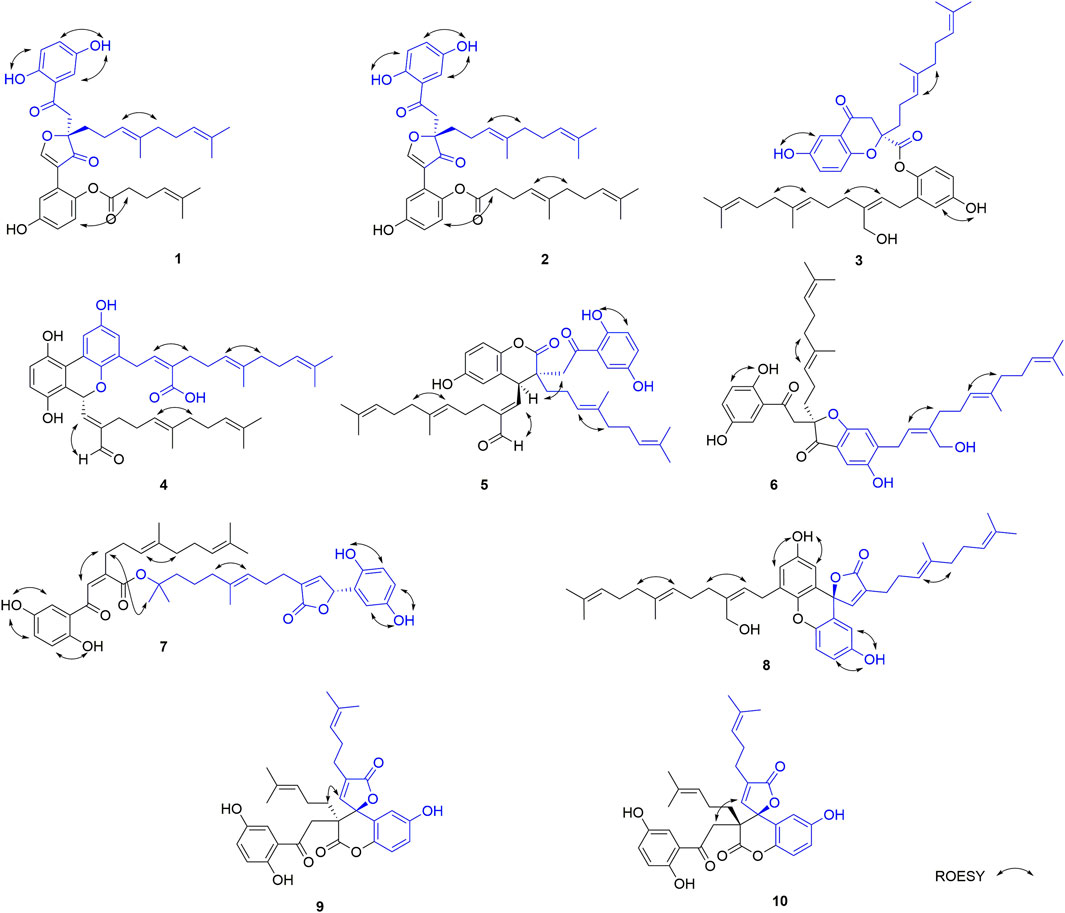
FIGURE 3. Key ROESY correlations of 1–10.
In the ROESY spectrum, the correlation between H-13 and H2-16 demonstrates that the Δ13(14) double bond is E configuration. Dimercochlearlactone A (1) was found to be a racemate, the separation by chiral-phase HPLC afforded enantiomers (+)-dimercochlearlactone A (1) and (–)-dimercochlearlactone A (1). Computational ECD spectral methods at time-dependent density functional theory (TDDFT) were employed to define the absolute configurations of (+)-dimercochlearlactone A (1) and (–)-dimercochlearlactone A (1). Due to the structure flexibility of dimercochlearlactone A (1), the model compound (1a) was constructed to ECD calculations. The result showed that the experimental CD spectrum of (+)-dimercochlearlactone A (1) exhibited similar Cotton effects with calculated ECD spectrum (Figure 4) of (9R)-1a. Accordingly, the absolute configurations as 9R for (+)-dimercochlearlactone A (1) and 9S for (–)-dimercochlearlactone A (1) were determined.
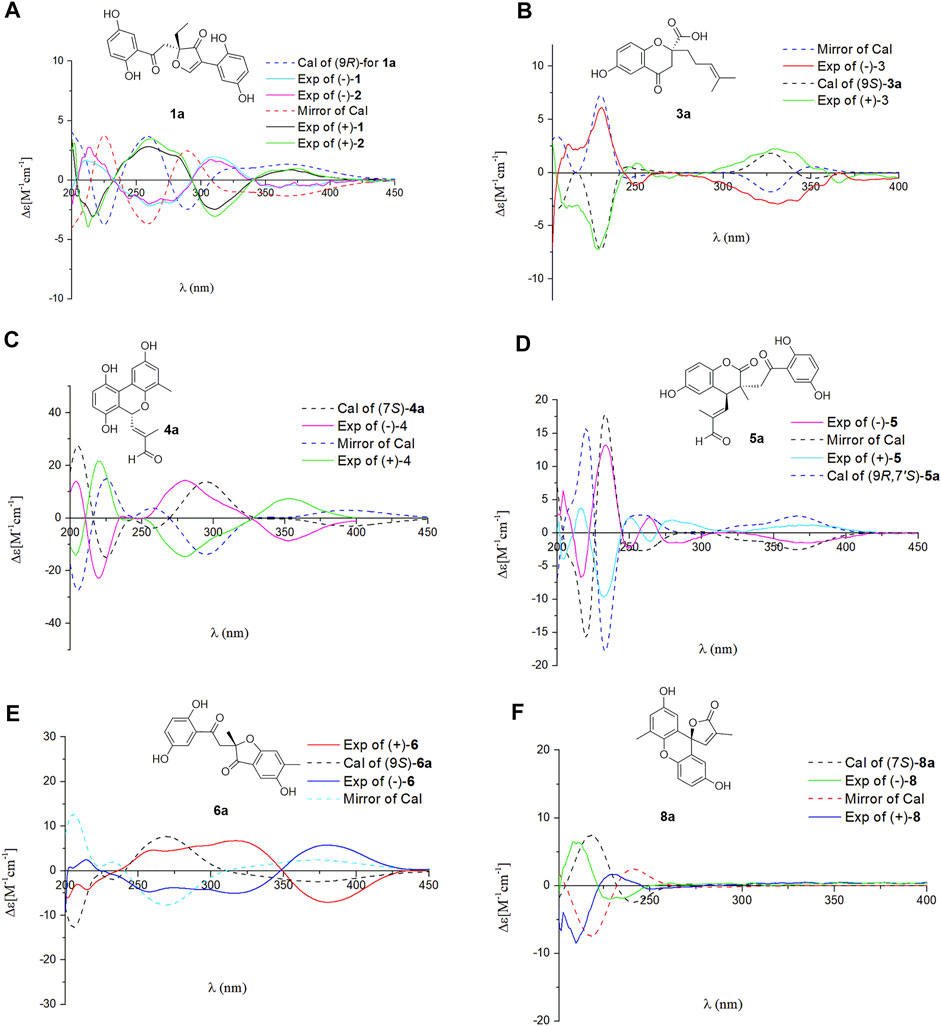
FIGURE 4. Comparison of the calculated ECD spectra with the experimental CD spectra in MeOH. (A) The calculated ECD spectrum of (10S)-1a at APFD/6-311+G (2d,p) level, σ = 0.30 eV; shift = –12 nm. (B) The calculated ECD spectrum of (9S)-3a at B3LYP/6-31G (d,p) level, σ = 0.20 eV; shift = +20 nm. (C) The calculated ECD spectrum of (7S)-4a at B3LYP/6-31G (d,p) level, σ = 0.30 eV; shift = +0 nm. (D) The calculated ECD spectrum of (9R,7′S)-5a at B3LYP/6-31G (d,p) level, σ = 0.20 eV; shift = +0 nm. (E) The calculated ECD spectrum of (9S)-6a at APFD/6-311+G (2d,p) level, σ = 0.40 eV; shift = –10 nm. (F) The calculated ECD spectrum of (7S)-8a at B3LYP/6-31G (d,p) level, σ = 0.30 eV; shift = –30 nm.
The NMR data of dimercochlearlactone B (2) resemble those of dimercochlearlactone A (1) revealing that the structure of dimercochlearlactone B (2) similar to that of dimercochlearlactone A (1). Only difference appears at the their side chains, which is a 7-carbon side chain in dimercochlearlactone A (1) was attached an isopentenyl to form a 12-carbon side chain in dimercochlearlactone B (2), supporting by the 1H-1H COSY correlations from H2-16′ (δH 2.00) to H2-15′ (δH 1.92) and H-17′ (δH 5.04) and the HMBC correlations (Figure 2) from H3-19′ (δH 1.54) and H3-20′ (δH 1.62) to C-17′ (δC 123.9) and C-18′ (δC 130.0), from H2-14′ (δH 1.56) to C-12′ (δC 122.2), C-13′ (δC 135.8) and C-15′ (δC 39.0). In the ROESY experiment, correlations between H-13 and H2-16 and between H-12′ and H2-15′ suggest that both double bonds Δ13(14) and Δ12′(13′) are E-from configurations (Figure 3). Racemic dimercochlearlactone B (2) was separated by chiral HPLC to yield (+)-dimercochlearlactone B (2) and (–)-dimercochlearlactone B (2). Their absolute configurations were deduced as 9R for (+)-dimercochlearlactone B (2) and 9S for (–)-dimercochlearlactone B (2) by using the above-mentioned ECD calculations (Figure 4).
Compounds 1 and 2 bear a same skeleton, which are different from the previously isolated Ganoderma meroterpenoids, a plausible pathway for the biogenesis of 2 was proposed (Figure 5). At first, fornicin C (Niu et al., 2006) undergoes a series of oxidation, ring formation, and reduction reactions to form intermediates A and B, respectively, which further form intermediate C via aldol condensation reaction. Intermediate C undergoes a reduction and substitution reaction to form D, which then give hemiacetal E via a substitution addition reaction. After a decarboxylation reaction, hemiacetal E can produce intermediate F. Finally, F undergoes intermediates G and H through dehydration and C-C bond cracking to form 2.
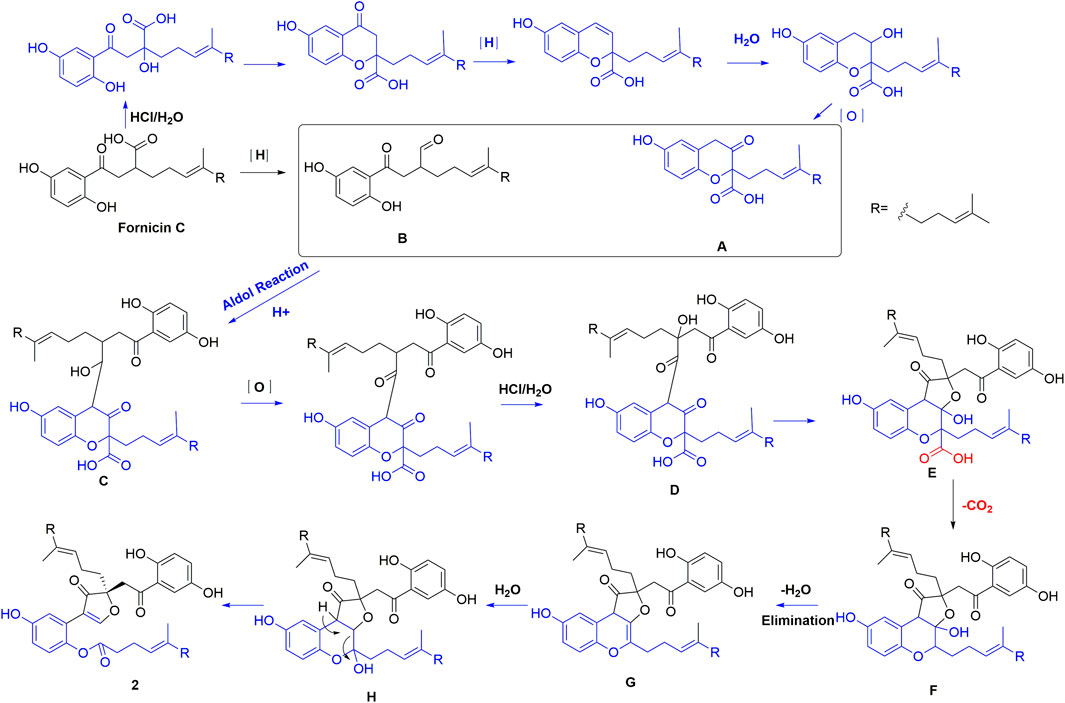
FIGURE 5. Plausible pathway for the biogenesis of 2.
Dimercochlearlactone C (3) has the molecular formula C42H54O7 (16 degrees of unsaturation) deduced by the HRESIMS analysis at m/z 669.3777 [M-H]-(calcd for C42H53O7, 669.3797). The 1H NMR spectrum of dimercochlearlactone C (3) exhibits signals for two typical ABX systems (δH 7.16, d, J = 3.0 Hz, H-3; δH 7.06, dd, J = 8.9, 3.0 Hz, H-5; δH 7.01, d, J = 8.9 Hz, H-6; δH 6.57, d, J = 2.7 Hz, H-3′; δH 6.54, dd, J = 8.6, 2.7 Hz, H-5′; δH 6.51, d, J = 8.6 Hz, H-6′). It was found 42 carbon signals including 6 methyl, 11 methylene, 11 methine, and 14 nonprotonated carbons (11 aromatic including 4 oxygenated, 1 oxygenated aliphatic, 1 ketone, and 1 carbonyl) by analyzing its 13C NMR and DEPT spectra. Like compound dimercochlearlactone A (1), the NMR data of dimercochlearlactone C (3) suggest a meroterpenoid dimer. The data of part A are very similar to those of ganotheaecolumol A (Luo et al., 2018), differing in that C-20 is a hydroxymethylene in ganotheaecolumol A, while the same position in part A of dimercochlearlactone C (3) is a methyl group. This deduction is supported by the HMBC correlations between H3-20 (δH 1.58) with C-18 (δC 125.5), C-19 (δC 132.5), and C-21 (δC 26.1).
The analysis of 2D NMR spectra (See Supplementary Figures S30–S33) of dimercochlearlactone C (3) reveals that the structure of part B is similar to that of ganomycin F (Cheng et al., 2018). Thus, there are four possibilities for the connection between part A and part B of dimercochlearlactone C (3), C-4-O-C-1′, C-4-O-C-4′, C-9-O-C-1′, and C-9-O-C-4′. In the 1H NMR spectrum, signals of two phenolic hydroxyl groups (δH 9.51, s and δH 9.41, s) are observed, it could be concluded that the connections between part A and part B in dimercochlearlactone C (3) are C-9-O-C-1′ or C-9-O-C-4′. Furthermore, the ROESY correlations (observed in DMSO-d6) between 4-OH with H-3 and 4′-O with/H-3′ are indicative of phenolic hydroxyl attaching to C-4 and C-4′, which indicate that C-9 and C-1′ are connected via oxygen atom to form phenolic ester. The ROESY correlation between H-8′ (δH 5.13) and H2-11′ (δH 2.14) suggests that the double bond Δ8′(9′) is Z-form configuration. Furthermore, ROESY correlations between H-13 with H2-16 and H-13′ with H2-16′ demonstrate that both Δ13(14) and Δ13′(14′) double bonds are E configuration. Racemic dimercochlearlactone C (3) was segregated into (+)-dimercochlearlactone C (3) and (–)-dimercochlearlactone C (3) by using chiral HPLC. Since the calculated ECD curve of (9S)-3a (Model structure) agrees with the experimental CD spectrum of (+)-dimercochlearlactone C (3) (Figure 4), the absolute configurations at the stereogenic center were established as 9S for (+)-dimercochlearlactone C (3) and 9R for (–)-dimercochlearlactone C (3).
The molecular formula of dimercochlearlactone D (4) was deduced as C42H52O7 by its negative HRESIMS. In 1H NMR spectrum of dimercochlearlactone D (4), the signals at (δH 6.70, d, J = 8.5 Hz, H-6; 6.61, d, J = 8.5 Hz, H-5; δH 8.00, d, J = 3.0 Hz, H-3′; δH 6.57, d, J = 3.0 Hz, H-5′) suggest that a 1,2,3,4-tetrasubstituted benzene ring and a 1,3,4,5-tetrasubstituted benzene ring in the structure of dimercochlearlactone D (4). The 13C NMR and DEPT spectra of dimercochlearlactone D (4) show 42 carbons including 6 methyl, 9 methylene, 12 methine, and 15 nonprotonated carbons (14 aromatic including 4 oxygenated and 1 carbonyl). The structure of dimercochlearlactone D (4) was mainly determined by 2D NMR spectra. The observation correlations from H2-12 to H2-11 and H-13, and from H2-17 to H2-16 and H-18 in 1H-1H COSY spectrum, along with the HMBC correlations from H3-20 and H3-21 to C-18 and C-19, from H3-20 to C-21, from H3-15 and H-16 to C-13 and C-14, and from H3-15 to C-16 indicate the presence of two isoprenyl moieties. Another isoprenyl residue is supported by the observation of 1H-1H COSY correlation between H-7 with H-8 and the HMBC correlations from H-10 and H-11 to C-8 and C-9, from H-10 to C-11, and from H-7 to C-9. Further observation of HMBC correlations from H-7 and H-8 to C-2 suggests that C-7 is connected to C-2. Similarly, the 1H-1H COSY correlations from H2-7′ to H-8′, from H2-12′ to H2-11′ and H-13′, and from H2-17′ to H2-16′ and H-18′ along with the HMBC correlations from H3-20′, and H3-21′ to C-18′ and C-19′, from H-20′ to C-21′, from H3-15′ and H-16′ to C-13′ and C-14′, from H3-15′ to C-16′, from H-10′ and H-11′ to C-8′ and C-9′, and from H-10′ to C-11′ indicates substructure consisting of three isoprenyl groups in part B of 4. Moreover, the correlation from H-6′ to C-7′ in HMBC spectrum suggests that the side chain and benzene ring of part B are linked via C-7′-C-6′. Finally, the two meroterpenoids are linked via C-2′-C-3 and C-8-O-C-1′ supported by the key HMBC correlations from H-3′ to C-2 and from H-8 to C-1′. The ROESY correlations from H-8 to H-10, from H-13 to H-16, and from H-13′ to H-16′ indicate that three double bonds (Δ8(9), Δ13(14), and Δ13′(14′)) are E-form, and that between H-8′ and H2-11′ suggests Δ8′(9′) double bond is Z form. Racemic 4 was submitted to chiral HPLC to afford their enantiomers. The absolute configurations were determined to be 7S for (–)-4 and 7R for (+)-4 by using computational ECD methods (Figure 4).
The NMR data of dimercochlearlactone E (5) are similar to those of the known spirocochlealactone A (Qin et al., 2018). Careful analysis of their structures showed that compound 5 is formed by the reduction of nonprotonated carbon (δC 88.3) and a carbonyl group (δC 173.0) in spirocochlealactone A to a methine (δC 40.1) and an aldehyde group (δC 194.0), respectively. The 1H-1H COSY correlation between H-7′ and H-8′, and the HMBC correlations from H-7′ to C-8′ and C-9′, and from H-8′ to C-9′, C-10′ and C-11′ supports the above conclusions. In the ROESY spectrum, correlations from H-8′ to H-10′, from H-13′ to H-16′, and from H-13 to H-16 indicate that three double bonds (Δ8′(9′), Δ13′(14′), and Δ13(14)) are E-form. Furthermore, the relative configuration of dimercochlearlactone E (5) was assigned as 9R*,7S*, gaining support from the ROESY correlation of Hb-8/H-7′. Dimercochlearlactone E (5) was also separated by chiral HPLC to afforded (+)-dimercochlearlactone E (5) and (–)-dimercochlearlactone E (5). Their absolute configurations were assigned as 9R,7S for (+)-dimercochlearlactone E (5) and 9S,7R for (–)-dimercochlearlactone E (5) by comparing their CD curves with the calculated ones. Thus, the structure of 5 was determined.
Dimercochlearlactone F (6) has the molecular formula C42H54O7 based on HRESIMS analysis (m/z 783.3727 [M + CF3COO]-; calcd for 783.3725). The 1H NMR spectrum of 6 contains signals for one typical ABX system (δH 7.11, d, J = 2.7 Hz, H-3; δH 6.99, dd, J = 8.9, 2.7 Hz, H-5; δH 6.80, d, J = 8.9 Hz, H-6) and a 1,2,4,5-tetrasubstituted benzene ring (δH 6.58, s, H-3′ and δH 7.08 s, H-5′). The 13C NMR and DEPT spectra contain the resonances for 42 carbons including 6 methyl, 11 methylene, 10 methine, and 15 nonprotonated carbons (12 aromatic including 4 oxygenated, 1 oxygenated aliphatic, 2 ketones). The above signals suggest that compound 6 is also a meroterpenoid dimer, and its structure consists of parts A and B. The substructure of part A in 6 is similar to that of part A in 1 as they have very similar NMR data. The substructure of part B is very similar to ganomycin F (Cheng et al., 2018). The difference is that the 3-position of the benzene ring in part A is connected to the other additional substructures, which is a hydrogen atom in ganomycin F. The HMBC correlation from H-6′ to C-10 suggests that C-10 is connected to C-5′. Although no HMBC correlations are observed to support C-9-O-C-3 fragment, the presence of ring A is confirmed due to the observation of characteristic chemical shift of C-4 (δC 166.2) in the benzene ring (Ring B). Same phenomenon was observed in other such kind of benzofuran structures, such as cochlearol I and spiroapplanatumines A–Q (Luo et al., 2017; Wang et al., 2019). The ROESY correlations from H-13′ to H-16′ and from H-13 to H-16 indicate that both double bonds (Δ13′(14′) and Δ13(14)) are E-form. Further correlation from H-8′ to H-11′ observed in ROESY spectrum suggests a E-form double bond (Δ8′(9′)). Chiral separation by HPLC afforded (+)-6 and (–)-6. Their absolute configurations were determined as 9S and 9R, respectively, when comparing their experimental CD curves with the calculated ones. As a result, the structure of 6 was assigned.
Dimercochlearlactone G (7) has a molecular formula C42H52O9 based on its HRESIMS, 13C NMR, and DEPT data. Two ABX aromatic coupling systems at δH 7.07 (d, J = 3.0 Hz, H-3), 7.01 (dd, J = 8.9, 3.0 Hz, H-5), 6.82 (d, J = 8.9 Hz, H-6), 6.35 (d, J = 2.9 Hz, H-3′), 6.57 (dd, J = 8.6, 2.9 Hz, H-5′), and 6.69 (d, J = 8.6 Hz, H-6′) were observed in its 1H NMR spectrum. Its 13C NMR and DEPT spectra reveal the presence of 42 carbons ascribed to 6 methyl, 9 methylene, 12 methine, and 15 nonprotonated carbons. Analysis of the NMR data of 7 found that part A is similar to dayaolingzhiol K (Zhang et al., 2021). The main difference is that the chemical shift of C-19 is 71.5 ppm in dayaolingzhiol K, while in compound 7 the chemical shift of C-19′ is downfield to 84.5 ppm. In addition, the NMR data of part B resembles that of ganodercin A implying that they have similar structure (Qin et al., 2021). Since no tailing behavior is observed in TLC, the carboxyl group in the structure of part B must be esterified. The structure of 7 is further confirmed by 1H NMR, HMBC, and ROESY spectra in DMSO-d6. In the 1H NMR spectrum, there are four free phenolic hydroxyl signals at δH 11.20 (s, 1-OH), 9.18 (s, 4-OH), 9.21 (s, 1′-OH), and 8.77 (s, 4′-OH). The ROESY correlations from 1-OH to H-6, from 4-OH to H-3, from 1′-OH to H-6′, and from 4′-OH to H-3′ fix position of phenolic hydroxyl. Thus, it can be inferred that the carboxyl group can satisfy the requirement of molecular weight only when it is esterified with C-19′. This conjecture was further secured by the ROESY correlation between H3-21′ and H2-11 and the above-mentioned downfield chemical shift of C-19′. Therefore, the planar structure of 7 was deduced. The ROESY correlation between H-8 and H-11 suggests that the double bond Δ8(9) is Z-form configuration. Furthermore, ROESY correlations from H-13 to H-16 and from H-13′ to H-16′ imply Δ13(14) and Δ13′(14′) double bonds are both E-form. The absolute configurations of 7 were assigned as 7′R for (+)-7 and 7′S for (–)-7 by comparing experimental CD spectra with those of (+)- and (–)-zizhine A (Cao et al., 2016).
The molecular formula of dimercochlearlactone H (8) was assigned as C42H52O6 by the analysis of its HRESIMS. Its 1H NMR spectrum contains one ABX aromatic coupling system with the signals at δH 6.57 (d, J = 3.0 Hz, H-3), 6.88 (dd, J = 8.9, 3.0 Hz, H-5), and 7.12 (d, J = 8.9 Hz, H-6). The signals at δH 6.44 (d, J = 2.9 Hz, H-3′) and δH 6.77 (d, J = 2.9 Hz, H-5′) demonstrate the existence of a 1,3,4,5-tetrasubstituted benzene ring in 8. Analysis of its 13C NMR and DEPT spectra resulted in 42 carbons, including 6 methyl, 10 methylene, 11 methine, and 15 nonprotonated carbons (13 sp2 including 4 oxygenated, 1 oxygenated aliphatic, and 1 carbonyl). Compound 8 is a meroterpenoids dimer consisting of two parts. The data of part A resemble those of ganomycin I (El Dine et al., 2009); the difference is that C-7 (δC 81.4) is a nonprotonated carbon in 8. The HMBC correlations from H-3 and H-8 to C-7 and from H-8 to C-9 and C-10 support the structure of part A. The structure of part B is very similar to ganomycin F, with the difference appearing at C-2′ being a nonprotonated carbon. The HMBC correlations from H-3′ to C-7 and from H-8 to C-2′ suggest that C-7 is connected to C-2′. The molecular weight and the unsaturation of the molecule need to form another ring to be satisfied. There are three possible ring formations, which are C-1-O-C-1′ C-1′-O-C-10 or C-1-O-C-10. In the 1H NMR spectrum (DMSO-d6), there are two free phenolic hydroxyl signals at δH 9.42 (s, 4-OH), 9.29 (s, 4′-OH). The ROESY correlations from 4-OH to H-3 and H-5 and from 4′-OH to H-3′ and H-5′ fix phenolic hydroxyl at C-4′ and C-4′, confirming the formation of ring C. Furthermore, the ROESY correlations from H-13′ to H-16′ and from H-13 to H-16 suggest that double bonds Δ13′(14′) and Δ13(14) are E-form. Additional ROESY correlation between H-8′ and H-11′ suggests a E-form double bond Δ8′(9′). The absolute configurations of 8 were determined to be 7R for (+)-8 and 7S for (–)-8 by using ECD calculation methods.
The molecular formula of 9 was specified as C32H34O8 based on the analysis of its positive HRESIMS ([M + Na]+, m/z 569.2154, calcd 569.2151). The 1H NMR spectrum exhibits two typical ABX spin systems δH 7.14, d, J = 2.9 Hz, H-3; 7.08, dd, J = 8.9, 2.9 Hz, H-5; 6.84 d, J = 8.9 Hz, H-6; δH 6.98, d, J = 8.9 Hz, H-6′; 6.85, dd, J = 8.9, 2.9 Hz, H-5′; 6.45, d, J = 2.9 Hz, H-3′. The 13C NMR and DEPT spectra of 9 display 4 methyl, 5 methylene, 9 olefifinic methine, 14 nonprotonated carbons (11 aromatic including 4 oxygenated, 2 carbonyl, and a ketone group). These data are very similar to those of spirocochlealactone B (Qin et al., 2018). The difference between 9 and spirocochlealactone B is that a sesquiterpenoid residue in spirocochlealactone B is disappeared instead of a monoterpenoid residue in 9. This inference is supported by the 1H-1H COSY correlations from H2-12′ to H2-11′ and H-13′ and the HMBC correlations from H3-15′ and H3-16′ to C-13′ and C-14′, from H-8′ to C-9′, C-10′ and C-11′, and from H-11′ to C-12′ and C-10′. The relative configurations at the chiral centers were determined to be 9S*,7′S* based on the observation of the ROESY correlation H-8′(δH 7.06)/Ha-11 (δH 2.21). The absolute configurations of 9 were determined as 9R,7′R for (+)-9 and 9S,7′S for (−)-9 by comparing the experimental CD spectra with those of (+)-spirocochlealactone B and (−)-spirocochlealactone B (Qin et al., 2018). Thus, the structure of 9 was identified and named as dimercochlearlactone I.
The molecular formula of compound dimercochlearlactone J (10) is similar to that of 9. Careful examination of 2D NMR (see Supplementary Figures S81–S84, S91−S94) data between 10 and 9 indicates that they have same planar structure. The observed ROESY correlation (see Supplementary Figure S94) of H-8′ (δH 7.16)/H-8 (δH 3.68, 3.07) in 10 suggests 9R*,7S′* relative configurations at chiral centers. The absolute configurations of 10 were determined as 9S,7′R for (+)-dimercochlearlactone J (10) and 9R,7′S for (−)-dimercochlearlactone J (10) by comparing their experimental CD spectra to (+)-spirocochlealactone A and (−)-spirocochlealactone A (Qin et al., 2018). Thus, the structure of 10 was assigned.
Compound 11 was identified as spirocochlealactone A by comparing its NMR and MS data with the literature data (Qin et al., 2018). This compound has been isolated by us from 10 kg of G. cochlear, and in this experiment, it was isolated from G. lucidum.
To investigate the anti-TNBC effects of the isolated compounds, we used the MDA-MB-231 cells for our analyses. All 22 dimer meroterpenoid enantiomers were evaluated for their suppressive effect toward MDA-MB-231 cells. It was found that (+)-2, (–)-2, (–)-3, (+)-11, and (–)-11 significantly decreased the cell viability in MDA-MB-231 cells (Figure 6A). Moreover, the morphological and density changes were observed in MDA-MB-231 cells upon exposure of the compounds (Supplementary Figure S98). To further analyze the effects of the compounds on MDA-MB-231 cells, the dose-response studies were performed. The cell viability of MDA-MB-231 cells was substantially inhibited by treatment with compounds for 48 h in a dose-dependent manner. As shown in Figure 6B, similar to the positive control (cisplatin), compound treatment dose-dependently inhibits MDA-MB-231 cells growth. The IC50 of compounds for MDA-MB-231 cells are 28.18, 25.65, 11.16, 8.18, and 13.02 μM, respectively. The remaining compounds showed negligible inhibitory effects on cell viability at 20 μM (Figure 6A). Interestingly, although all the remaining compounds have rather low cytotoxicities toward MDA-MB-231 cells, ten of the isolates (+)-5, (–)-5, (+)-6, (–)-6, (+)-7, (–)-7, (+)-8, (–)-8, (+)-10, and (–)-10 significantly inhibit the migration ability of MDA-MB-231 cells (Figure 7), suggesting that they might be promising lead compounds for the development of anti-cancer drugs against metastasis of TNBC.
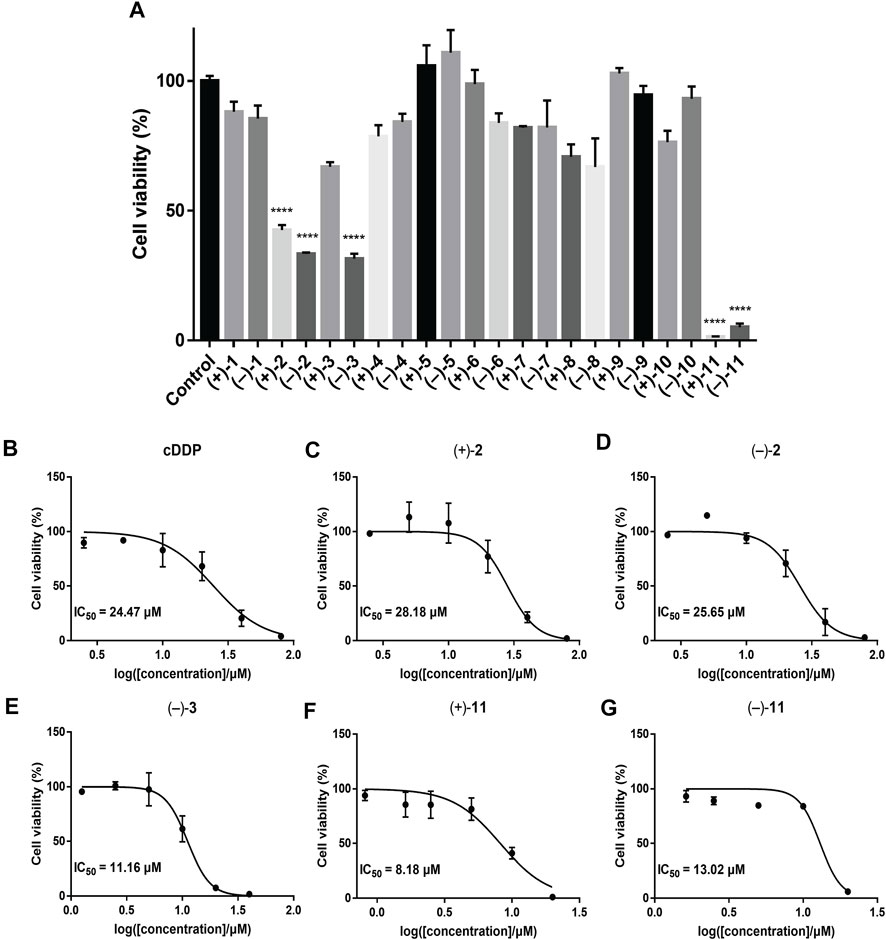
FIGURE 6. Cell viability of the compounds in MDA-MB-231 cells by CCK-8 assay. The results are represented as mean ± SD of triplicates. ****p < 0.0001 vs. control group. (A) Primary screening at 20 μM. (B–G) Dose-response curves of cisplatin (positive control) and the tested compounds showing IC50 values.
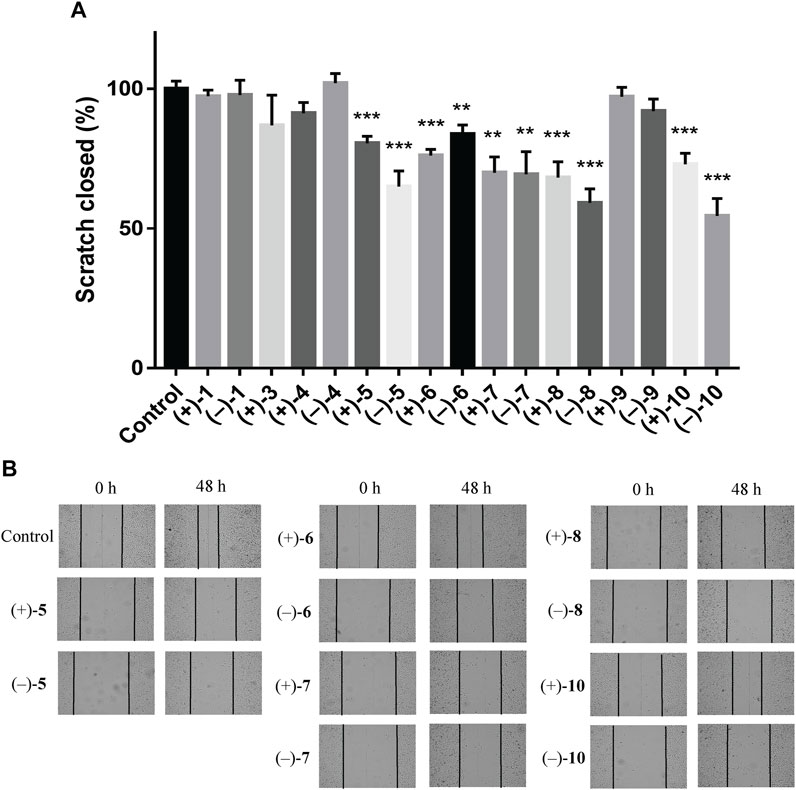
FIGURE 7. Compounds suppress the migratory ability of MDA-MB-231 cells by wound healing assay. (A) Quantification of the migratory ability of compounds against MDA-MB-231 cells at 20 μM. The results were represented as mean ± SD of triplicates. **p < 0.01, ***p < 0.001 (vs. control group). (B) Representative images of the wound healing assay in MDA-MB-231 cells.
Conclusion
To conclude, this study resulted in the isolation of ten pairs of novel meroterpenid dimers and one pair of known compounds from Ganoderma species. Biological results revealed the importance of (+)-2, (–)-2, (–)-3, (+)-11, and (–)-11 in the development of the anti-TNBC drugs. Furthermore, (+)-5, (–)-5, (+)-6, (–)-6, (+)-7, (–)-7, (+)-8, (–)-8, (+)-10, and (–)-10 significantly inhibit the migration ability of MDA-MB-231 cells, thereby providing promising compounds for the development of anti-TNBC drugs.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
Y‐XC designed the research. FQ conducted chemical experiments (isolated compounds 1–10). Y‐YC conducted biological experiments in vitro. JZ isolated compound 11. FQ, Y‐YC, JZ and Y‐XC analyzed data. FQ and Y‐XC wrote and revised the manuscript. All authors discussed the results and commented on the manuscript at all stages.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank National Natural Science Foundation of China (82030115), Shenzhen Fundamental Research Program (JCYJ20200109113803838), NSFC-Joint Foundation of Yunnan Province (U1702287), National Science Fund for Distinguished Young Scholars (81525026), National Natural Science Foundation of China (82104036), Guangdong Key Laboratory for Functional Substances in Medicinal Edible Resources and Healthcare Products (2021B1212040015), and SZU Top Ranking Project (86000000210) for financial supports.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.888371/full#supplementary-material
References
Cai, D., Zhang, J. J., Wu, Z. H., Qin, F. Y., Yan, Y. M., Zhang, M., et al. (2021). Lucidumones B-H, Racemic Meroterpenoids That Inhibit Tumor Cell Migration from Ganoderma lucidum. Bioorg. Chem. 110, 104774. doi:10.1016/j.bioorg.2021.104774
Cao, W. W., Luo, Q., Cheng, Y. X., and Wang, S. M. (2016). Meroterpenoid Enantiomers from Ganoderma sinensis. Fitoterapia 110, 110–115. doi:10.1016/j.fitote.2016.03.003
Chen, H., Yang, J. P., Yang, Y. L., Zhang, J. P., Xu, Y., and Lu, X. L. (2021). The Natural Products and Extracts: Anti-triple-negative Breast Cancer In Vitro. Chem. Biodivers 18, e2001047. doi:10.1002/cbdv.202001047
Cheng, L. Z., Qin, F. Y., Ma, X. C., Wang, S. M., Yan, Y. M., and Cheng, Y. X. (2018). Cytotoxic and N-Acetyltransferase Inhibitory Meroterpenoids from Ganoderma cochlear. Molecules 23, 1797. doi:10.3390/molecules23071797
Chowdhury, P., Ghosh, U., Samanta, K., Jaggi, M., Chauhan, S. C., and Yallapu, M. M. (2021). Bioactive Nanotherapeutic Trends to Combat Triple Negative Breast Cancer. Bioact. Mater. 6, 3269–3287. doi:10.1016/j.bioactmat.2021.02.037
El Dine, R. S., El Halawany, A. M., Ma, C. M., and Hattori, M. (2009). Inhibition of the Dimerization and Active Site of HIV-1 Protease by Secondary Metabolites from the Vietnamese Mushroom Ganoderma colossum. J. Nat. Prod. 72, 2019–2023. doi:10.1021/np900279u
Lin, Z., and Deng, A. (2019). Antioxidative and Free Radical Scavenging Activity of Ganoderma (Lingzhi). Adv. Exp. Med. Biol. 1182, 271–297. doi:10.1007/978-981-32-9421-9_12
Lin, Z., and Sun, L. (2019). Antitumor Effect of Ganoderma (Lingzhi) Mediated by Immunological Mechanism and its Clinical Application. Adv. Exp. Med. Biol. 1182, 39–77. doi:10.1007/978-981-32-9421-9_2
Liu, Q., and Tie, L. (2019). Preventive and Therapeutic Effect of Ganoderma (Lingzhi) on Diabetes. Adv. Exp. Med. Biol. 1182, 201–215. doi:10.1007/978-981-32-9421-9_8
Luo, Q., Wei, X. Y., Yang, J., Luo, J. F., Liang, R., Tu, Z. C., et al. (2017). Spiro Meroterpenoids from Ganoderma applanatum. J. Nat. Prod. 80, 61–70. doi:10.1021/acs.jnatprod.6b00431
Luo, Q., Li, M. K., Luo, J. F., Tu, Z. C., and Cheng, Y. X. (2018). COX-2 and JAK3 Inhibitory Meroterpenoids from the Mushroom Ganoderma theaecolum. Tetrahedron 74, 4259–4265. doi:10.1016/j.tet.2018.06.053
Meng, J., and Yang, B. (2019). Protective Effect of Ganoderma (Lingzhi) on Cardiovascular System. Adv. Exp. Med. Biol. 1182, 181–199. doi:10.1007/978-981-32-9421-9_7
Niu, X. M., Li, S. H., Sun, H. D., and Che, C. T. (2006). Prenylated Phenolics from Ganoderma fornicatum. J. Nat. Prod. 69, 1364–1365. doi:10.1021/np060218k
O'Reilly, D., Sendi, M. A., and Kelly, C. M. (2021). Overview of Recent Advances in Metastatic Triple Negative Breast Cancer. Wjco 12, 164–182. doi:10.5306/wjco.v12.i3.164
Qin, F. Y., Yan, Y. M., Tu, Z. C., and Cheng, Y. X. (2018). Meroterpenoid Dimers from Ganoderma cochlear and Their Cytotoxic and COX-2 Inhibitory Activities. Fitoterapia 129, 167–172. doi:10.1016/j.fitote.2018.06.019
Qin, F. Y., Zhang, J. J., Wang, D. W., Xu, T., Cai, D., and Cheng, Y. X. (2021). Direct Determination of E and Z Configurations for Double Bond in Bioactive Meroterpenoids from Ganoderma Mushrooms by Diagnostic 1H NMR Chemical Shifts and Structure Revisions of Previous Analogues. J. Funct. Foods 87, 104758. doi:10.1016/j.jff.2021.104758
Quan, Y., Ma, A., and Yang, B. (2019). Preventive and Therapeutic Effect of Ganoderma (Lingzhi) on Brain Injury. Adv. Exp. Med. Biol. 1182, 159–180. doi:10.1007/978-981-32-9421-9_6
Wang, X. L., Wu, Z. H., Di, L., Zhou, F. J., Yan, Y. M., and Cheng, Y. X. (2019). Renoprotective Meroterpenoids from the Fungus Ganoderma cochlear. Fitoterapia 132, 88–93. doi:10.1016/j.fitote.2018.12.002
Keywords: Ganoderma cochlear, Ganoderma lucidum, meroterpenoid dimers, dimercochlearlactone A−J, triple negative breast cancer
Citation: Qin F-Y, Chen Y-Y, Zhang J-J and Cheng Y-X (2022) Meroterpenoid Dimers from Ganoderma Mushrooms and Their Biological Activities Against Triple Negative Breast Cancer Cells. Front. Chem. 10:888371. doi: 10.3389/fchem.2022.888371
Received: 02 March 2022; Accepted: 28 March 2022;
Published: 03 May 2022.
Edited by:
Guigen Li, Texas Tech University, United StatesReviewed by:
Jungui Dai, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaSrinath Pashikanti, Idaho State University, United States
Copyright © 2022 Qin, Chen, Zhang and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong-Xian Cheng, eXhjaGVuZ0BzenUuZWR1LmNu
†These authors have contributed equally to this work