 Wenli Wang1†
Wenli Wang1† Yuxin Li1†Ye He2†Xing Jiang1Ying Yi1Xihan Zhang1Shiyu Zhang1Guangtong Chen1Min Yang1Jia-Lie Luo1*
Yuxin Li1†Ye He2†Xing Jiang1Ying Yi1Xihan Zhang1Shiyu Zhang1Guangtong Chen1Min Yang1Jia-Lie Luo1* Boyi Fan1*
Boyi Fan1*- 1School of Pharmacy, Nantong University, Nantong, China
- 2Jinghua Pharmaceutical Group Co., Ltd., Nantong, China
Resin glycosides, mainly distributed in plants of the family Convolvulaceae, are a class of novel and complex glycolipids. Their structural complexity and significant biological activities have received much attention from synthetic chemists, and a number of interesting resin glycosides have been synthesized. The synthesized resin glycosides and their analogues not only helped in structural verification, structural modification, and further biological activity exploration but also provided enlightenment for the synthesis of glycoside compounds. Herein, the present review summarizes the application of various efforts toward the synthesis of resin glycosides in the last decade.
Introduction
Natural products have significantly contributed to the development of new drugs for human disorders (Newman and Cragg, 2020). Resin glycosides are a class of novel and complex natural glycolipids mainly discovered in plants of the family Convolvulaceae. Because of the diversified structures and various bioactivities, resin glycosides have attracted plenty of interests from organic chemists and pharmacologists (Pereda-Miranda et al., 2010; Fan et al., 2022). Up to now, hundreds of resin glycosides and their derivatives have been identified and reported, some of which exhibited significant biological activities, such as cytotoxic (Fan et al., 2014; Ono et al., 2019), anti-viral (Ono et al., 2014), anticonvulsant (Castro-García et al., 2018), anti-tumor migration (Chen et al., 2017), sedative (León-Rivera et al., 2014), vasorelaxant (León-Rivera et al., 2014), and α-glucosidase inhibitory effects (Pan et al., 2015), as well as multidrug-resistant reversal effects on both microbial pathogens and mammalian cancer cells (Figueroa-González et al., 2012; Wang et al., 2018; Lira-Ricárdez and Pereda-Miranda. 2020), indicating their potential as lead compounds for drug discovery. However, not enough in-depth biological exploitation has been taken place. Possible reasons could be the difficulties on the isolation and purification.
The structures of resin glycosides consist of three ingredients: oligosaccharide chain, long-chain fatty acid aglycone, and modified acyl groups (Pereda-Miranda et al., 2010; Fan et al., 2022). The sugar units of oligosaccharides mostly range from three to seven, with D-glucose (Glc), L-rhamnose (Rha), D-fucose (Fuc), D-quinovose (Qui), D-xylose (Xyl), and D-galactose (Gal) as the monosaccharide unit. The aglycones are usually monohydroxy to trihydroxy C14–C17 long-chain fatty acids, which often link through an intramolecular esterification with the oligosaccharide chain to form a macrolactone ring. The oligosaccharide residues are also modified with some fatty acids (Fan et al., 2022). Due to the variation in the oligosaccharide units and sequence, aglycone, macrolactone site, acylated residues, and the formation of the dimers, the structural complexity of resin glycosides has received much attention from synthetic chemists.
The first total synthesis of resin glycosides was published in the 1990s (Lu et al., 1997), and then many resin glycosides have been synthesized successfully. The construction of the macrolide structures and oligosaccharide chains presented a major challenge to develop methodologies for their synthesis. In 2010, Pereda-Miranda and coworkers reviewed the state of resin glycoside synthesis to early 2009 (Pereda-Miranda et al., 2010). Since then, more total synthesis research studies on resin glycosides have been achieved and published. Therefore, to obtain a comprehensive perspective, the present review provides a systematic summary of synthetic strategies for the total synthesis of resin glycosides over the period from 2009 to the end of 2021. Several electronic databases, including the Web of Science, PubMed, Scopus, and Google Scholar, were searched using the keyword “resin glycoside” paired with “synthesis” or “total synthesis.” A total of 13 resin glycosides, as shown in Figure 1, have been synthesized during this period, including ipomoeassins A–F (1–6), batatoside L (7), murucoidins IV (8) and V (9), merremoside D (10), tricolorins A (11) and F (12), and batatin VI (13).
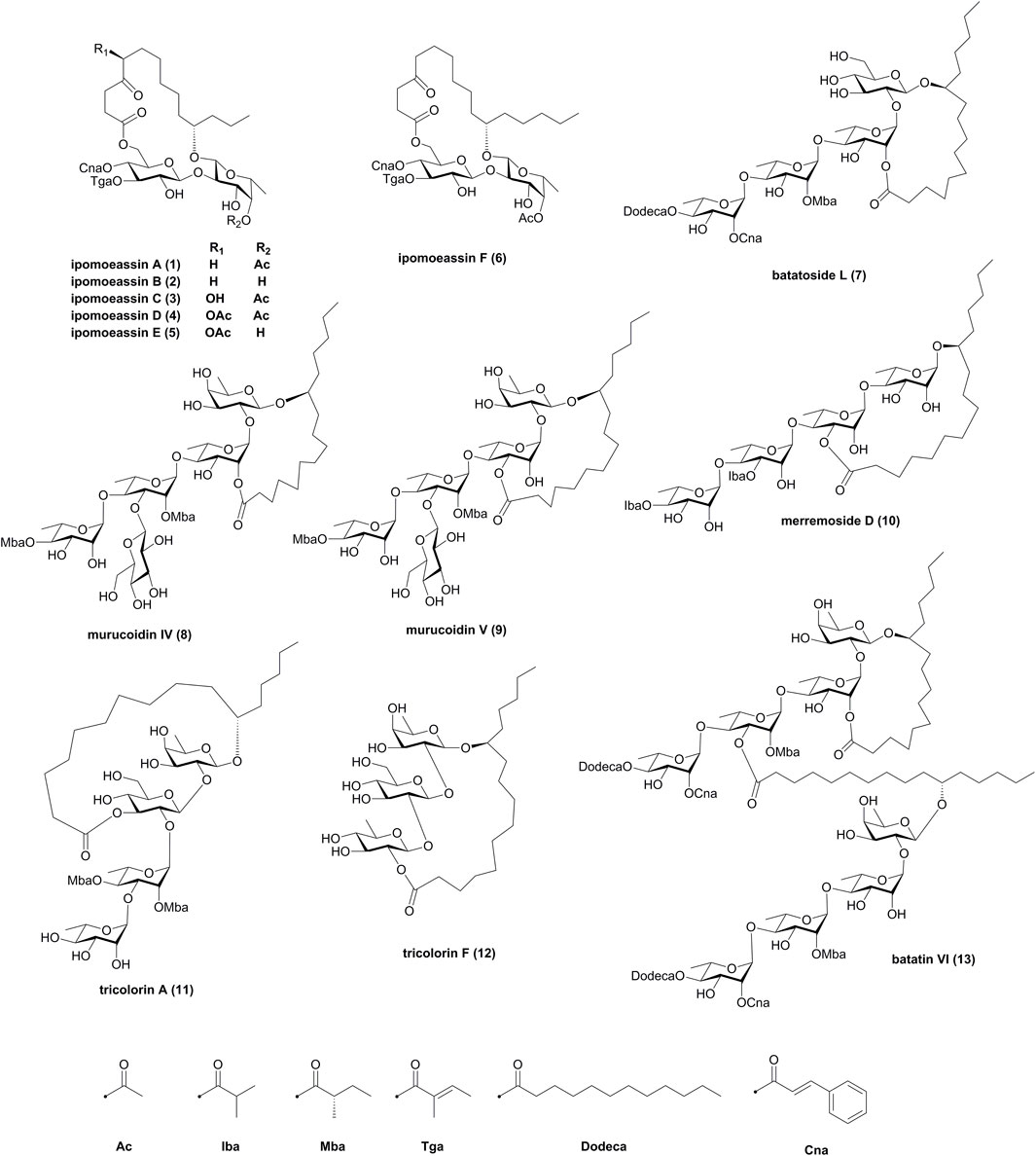
FIGURE 1. Structures of resin glycosides 1–13. Iba, isobutyryl; Mba, 2S-methylbutyryl; Tga, tiglyl; Dodeca, n-dodecanoyl; Cna, trans-cinnamyl.
Total synthesis of ipomoeassins A–F (1–6)
Ipomoeassins A–F (1–6) are a kind of disaccharide resin glycosides isolated from the leaves of Ipomoea squamosa in the Suriname rainforest (Cao et al., 2005; Cao et al., 2007). Among them, ipomoeassins D (4) and F (6) showed exceptionally potent low cytotoxicity to single-digit nanomolar IC50 values against several cancer cell lines. Therefore, these natural glycoconjugates quickly inspired synthetic chemists to tackle their total synthesis.
Approach by Fürstner’s group
Fürstner’s group has reported the first total synthesis of ipomoeassins B (2) and E (5) based on a ring-closing olefin metathesis (RCM) reaction for the formation of the macrocycle at the C8–C9 bond of the aglycone (Fürstner and Nagano. 2007). They also provided access to all naturally occurring ipomoeassins and a few analogues, together with the SAR of these compounds (Nagano et al., 2009). Although the total synthesis of ipomoeassins E (5) by Fürstner’s group has been reviewed by Pereda-Miranda et al. (2010), here we give a more comprehensive review on the Fürstner’s work.
As shown in Figure 2, the route by Fürstner’s group relied on the use of compound 19 as a new cinnamic acid surrogate, which was hydrogenation-resistant over Wilkinson’s catalyst and allowed the unsaturated acids to be attached at an early phase of the synthesis. The key disaccharide 16 was assembled by a BF3·Et2O-mediated glycosidation method and was esterified with tiglic acid at the more nucleophilic O-3 site of β-glucoside to form 17. Then, the reductive opening of the substituted benzylidene acetal using NaBH3CN in combination with TMSCl furnished a mixture of 6-OPMB ether (18) and 4-OPMB ether in up to 84% combined yield with a 4:1 isomeric ratio. Alcohol 18 was esterified with 19 under Yamaguchi conditions, which was then subjected to oxidative PMB cleavage followed by the attachment of the 4-oxo-8-nonenoic acid ester segment. The resulting diene 21 was treated with catalytic amounts of ruthenium alkylidene complex 22 for RCM macrolactonization and then was hydrogenated with the aid of [RhCl(PPh3)3] to afford macrolactone 23. After cleavage of the C-silyl and O-silyl groups and deprotection of isopropylidene acetal, ipomoeassin B (2) was obtained. Ipomoeassin A (1) was converted from ipomoeassin B (2) by treating with MeC(OEt)3 in the presence of camphersulfonic acid, followed by an HOAc-induced rearrangement. The synthesis of ipomoeassins D (4) and E (5) was very similar with ipomoeassins A (1) and B (2), respectively, with the only difference on the use of a synthetic chiral acid 26 (Figure 3) for the esterification of the primary alcohol in 20. The PMB-protected acid 27 (Figure 3) was used to form diene in the synthesis of ipomoeassin C (3), which was cleaved with DDQ to form the hydroxyl group in the C-5 position of the aglycone moiety. Ipomoeassin F (6) differs from ipomoeassin A (1) only by the presence of two additional methylene units in the tail of aglycone, and no synthetic strategy was changed for the ipomoeassin F (6), except for the substitute of heptenol with nonenol to form the building block 28 (Figure 3) instead of 14.
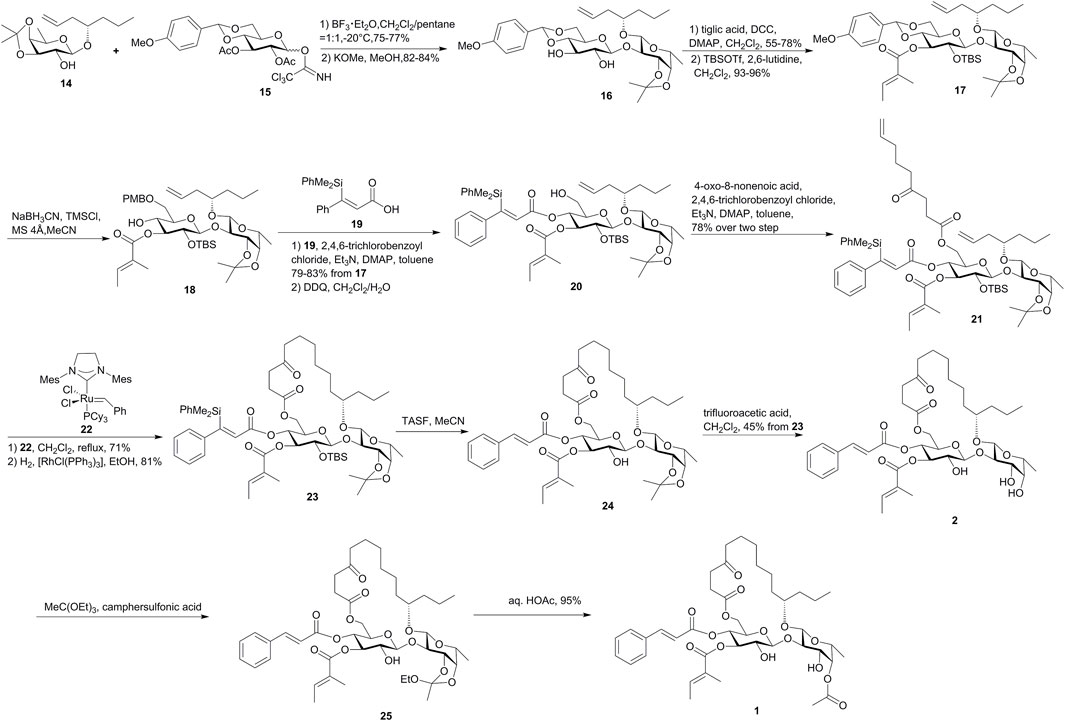
FIGURE 2. Fürstner’s approach toward the synthesis of ipomoeassins A–F (1–6). TASF, tris(dimethylamino)sulfonium difluorotrimethylsilicate.
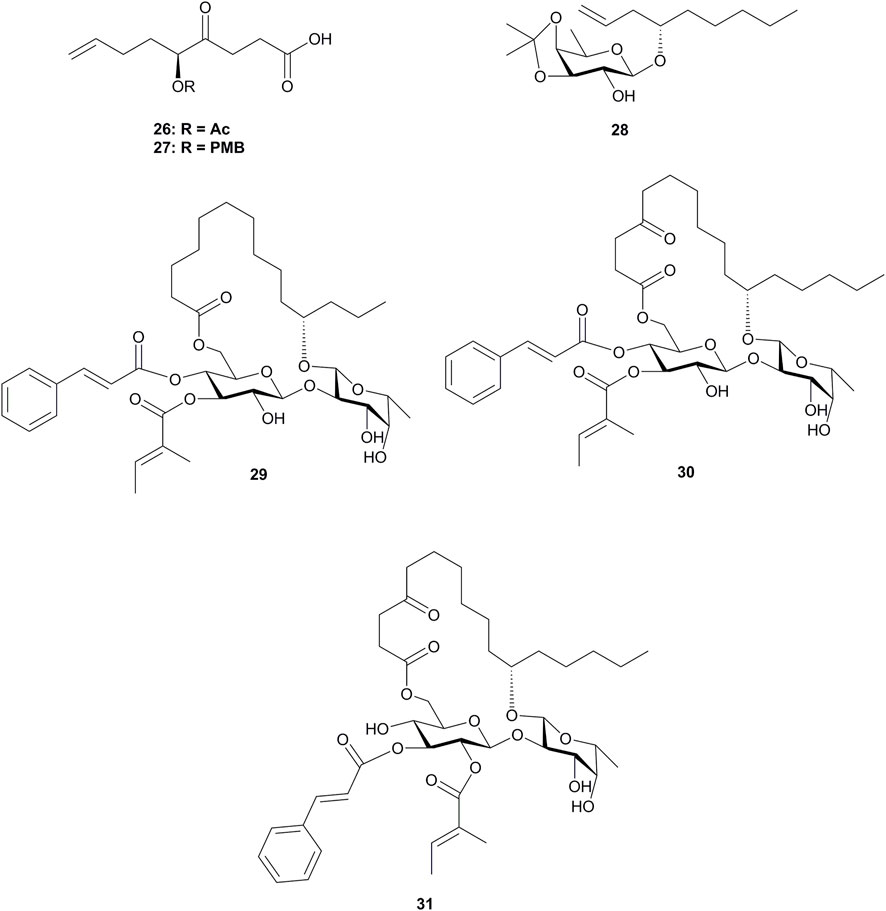
FIGURE 3. Structures of 26–31.
Apart from ipomoeassins A–F (1–6), three analogues 29–31 (Figure 3) were also synthesized by Fürstner’s group. By comparing the cytotoxic activity of these compounds, it was observed that the acetylation of the axial 4-OH group of fucopyranose and hydroxylation of the C-5 position in the lipophilic tether are detrimental. In addition, the ketone in the tether, the exact location of the two unsaturated esters, and the lipophilicity/hydrophilicity balance also showed strong influences on the bioactivity. A cell cycle analysis showed that those ipomoeassins could cause G0/G1 arrest in L-929 cells, followed by the apoptotic induction (Nagano et al., 2009).
Approach by Postema’s group
The synthetic approach for ipomoeassin F (6) proposed by Postema’s group also used RCM as the macrolactonization method and differed with that of Fürstner by the introduction of the unsaturated acid in the late stage (Postema et al., 2009, Figure 4).
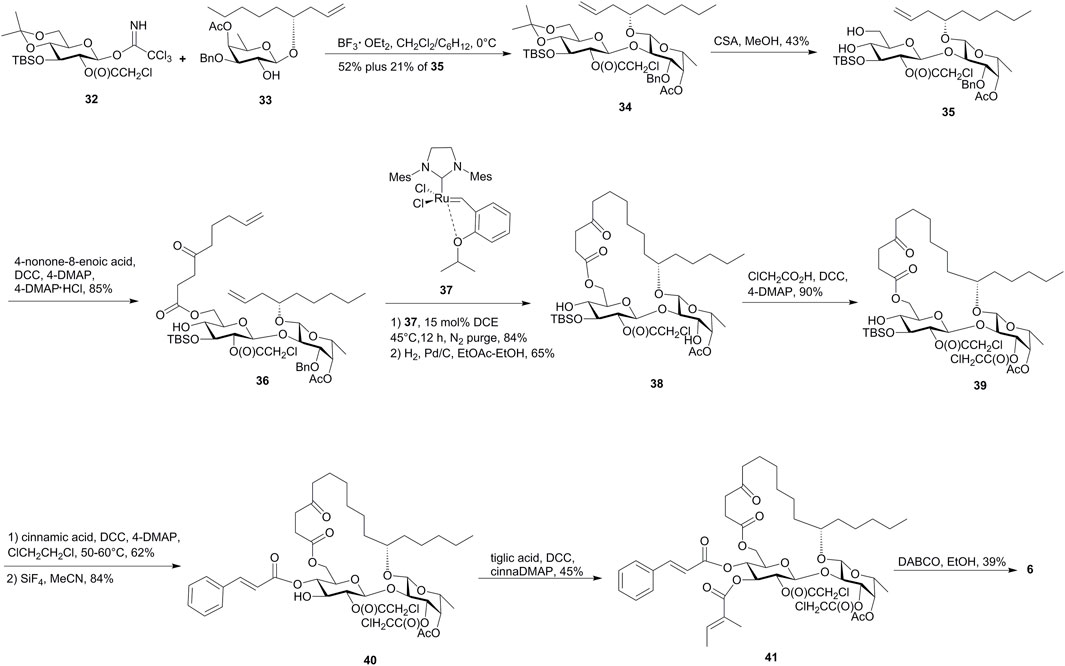
FIGURE 4. Postema’s approach toward the synthesis of ipomoeassin F (6). CSA, camphor-10-sulfonic acid.
In this approach, the obtained diene (36) cleanly cyclized to the desired 20-membered ring on reaction with ruthenium carbine (37), which was directly subjected to hydrogenation and cleavage of the O-3 benzyl group of fucopyranose to deliver 38. The subsequent selective chloroacetylation on the O-3 hydroxyl group of fucopyranose afforded 39. After the installment of the cinnamoyl group at the O-4 hydroxyl of the glucopyranose, the TBS group was effectively removed and following tigloylation gave the fully blocked precursor 41. Finally, the removal of R-chloroacetates by employing an excess of DABCO furnished ipomoeassin F (6) in 39% yield after purification by flash chromatography and HPLC.
Approaches by Shi’s group
Two strategies for the construction of ipomoeassin F (6) were described by Shi and colleagues and were both based on the RCM in constructing ring structures, with the difference on the introduction stage of the tiglate moiety (Zong et al., 2015; Zong et al., 2017b).
In the first strategy shown in Figure 5, the tiglate was introduced into the glucosyl donor 42 in an early stage, and the fucosyl acceptor 43 was designed with minimal protection steps from the fucosyl donor and chiral alcohol. The regioselective Schmidt glycosylation of 43 and 42, followed by the acetylation of the remaining hydroxyl group, gave disaccharide 44 in 71% yield. The resulting alcohol obtained by the removal of the Alloc group was treated with TBSOTf and 2,6-lutidine to afford the silyl ether 45 as the major product (66%). The removal of the isopropylidene group followed by the esterification of 6-OH-Glcp with 4-oxo-8-nonenoic acid gave diene 47 as the RCM precursor. Treatment of diene 47 with the Hoveyda–Grubbs catalyst and the subsequent hydrogenation over Wilkinson’s catalyst gave 48. After the introduction of the cinnamate moiety and the removal of the TBS groups, ipomoeassin F (6) was finally obtained. In this route, ipomoeassin F was synthesized in 3.8% overall yield over 17 steps of the longest linear sequence from commercially available starting materials, and thus the synthesis can be easily scaled up to gram-scale (Zong et al., 2015).
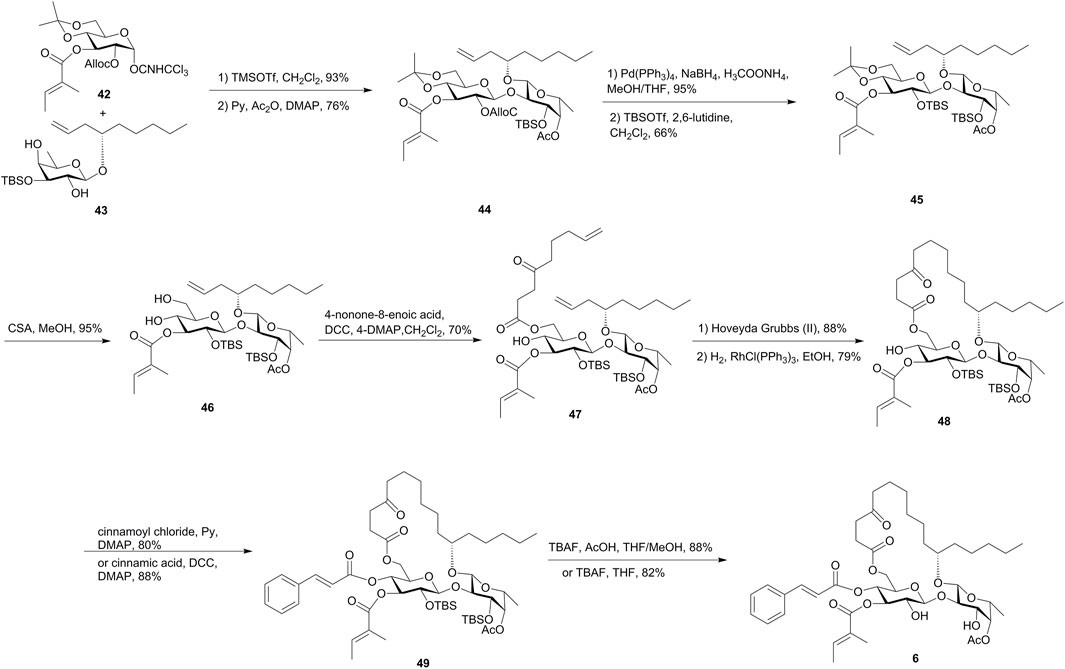
FIGURE 5. First Shi’s approach toward the synthesis of ipomoeassin F (6). Alloc, allyloxycarbonyl; CSA, camphor-10-sulfonic acid.
In the second strategy, Shi’s group tried to install two acyl groups in the late stage by utilizing the highly regioselective esterification of the glucose diol (Figure 6). The disaccharide diol 52 was obtained by the removal of the isopropylidene group of 51, which was built through regioselective glycosylation of the glucosyl donor 50 and fucoside acceptor 43, followed by acetylation of the remaining 4-OH-Glcp. The chemoselective esterification of 6-OH-Glcp in diol 52 with carboxylic acid 53 gave diene 54 as the RCM precursor. The treatment of diene 54 with the Hoveyda–Grubbs catalyst and the subsequent hydrogenation reaction gave the ring structure 55. The cinnamate moiety was then introduced using Steglich esterification to furnish disaccharide 56. The removal of Lev groups followed by the cleavage of ketal in the presence of CSA gave the key intermediate 57. Steglich or Mukaiyama esterification of 57 with tiglic acid yielded the desired 3-O-tigloyl product 58, the TBS group of which was removed to afford ipomoeassin F (6) (Zong et al., 2017b). This route was developed with an overall yield of 4.0% over 17 steps of the longest linear sequence from commercially available D-glucose penta-acetate and was particularly suitable for the efficient preparation of ipomoeassin F analogues with modifications at 3-OH-Glcp.
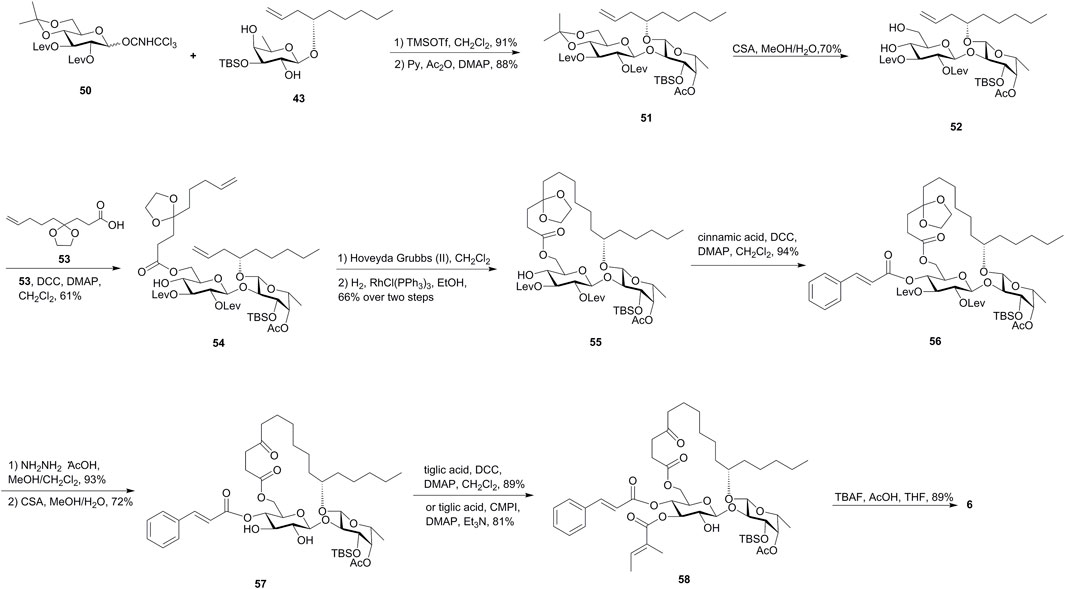
FIGURE 6. Second Shi’s approach toward the synthesis of ipomoeassin F (6). Lev, levulinoyl; CSA, camphor-10-sulfonic acid; CMPI, 2-chloro-1-methylpyridinium iodide.
Using these two strategies, Shi’s group also synthesized a series of ipomoeassin F analogues, including the 11R-epimer (59), acyl group-removed or -modified analogues (30, 60–71), chain-modified or open-chain analogues (72–83), and monosaccharide analogues (84–86) (Zong et al., 2015; Zong et al. 2016; Zong et al. 2017a; Zong et al. 2017b; Zong et al. 2018; Zong et al. 2020, Figure 7). These analogues were evaluated for the cytotoxic activity, followed by the structure–activity relationship study. Based on the evaluation result, two α, β-unsaturated esters, the natural 11S configuration of aglycone, the size and flexibility of the ring, and the 6-membered ring of the fucoside moiety are critical for the activity, whereas, the cyclic skeleton, ketone in the aglycone, and 3-OH of the Fuc unit showed less influences on the activities of ipomoeassin F (6).
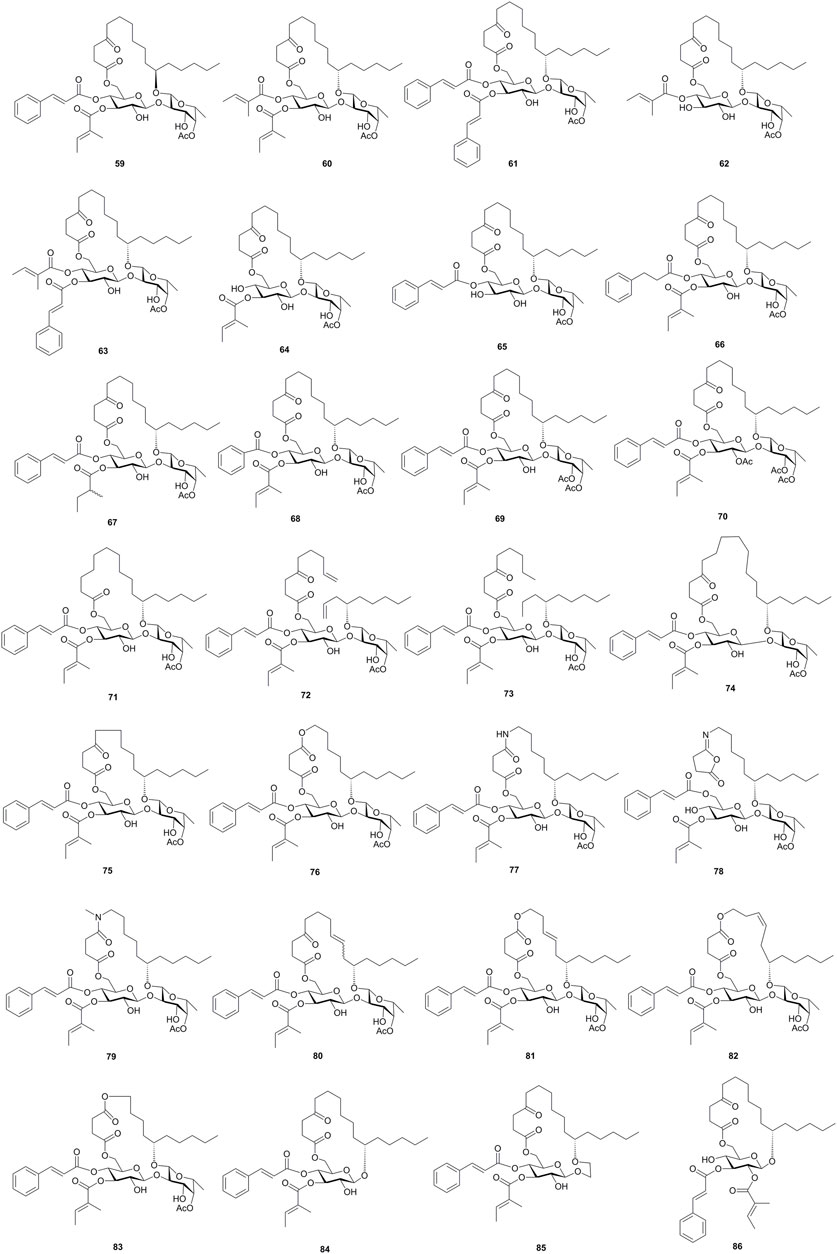
FIGURE 7. Structures of synthesized ipomoeassins F analogues 59–86.
In addition, the biotin analogue of ipomoeassin F was synthesized by Shi’s group and was used to identify Sec61α, the pore-forming subunit of the Sec61protein translocon, as a direct binding partner of ipomoeassin F (6) (Zong et al., 2019). A ring-expanded ipomoeassin F analogue (74, Figure 7) exhibited improved cytotoxicity and in vitro protein translocation inhibition, implying that the binding pocket of Sec61α can accommodate further structural modifications, likely in the fatty acid portion (Zong et al., 2020). Due to the Sec61 inhibitory potential, ipomoeassin F (6) was developed as a valuable tool to research the protein biogenesis at the endoplasmic reticulum (Leznicki et al., 2022; O'Keefe et al., 2021a; 2021b; Roboti et al., 2021).
Total synthesis of batatoside L (7)
Approach by Yang’s group
Batatoside L (7) is a cytotoxic resin glycoside isolated from Ipomoea batatas, a plant with the common name of sweet potato (Yin et al., 2009). Due to the appealing structural and bioactive features, it was synthesized by Yang’s group. The crucial step was the construction of the 18-membered macrolactone framework through the Corey–Nicolaou macrolactonization approach (Xie et al., 2010, Figure 8).
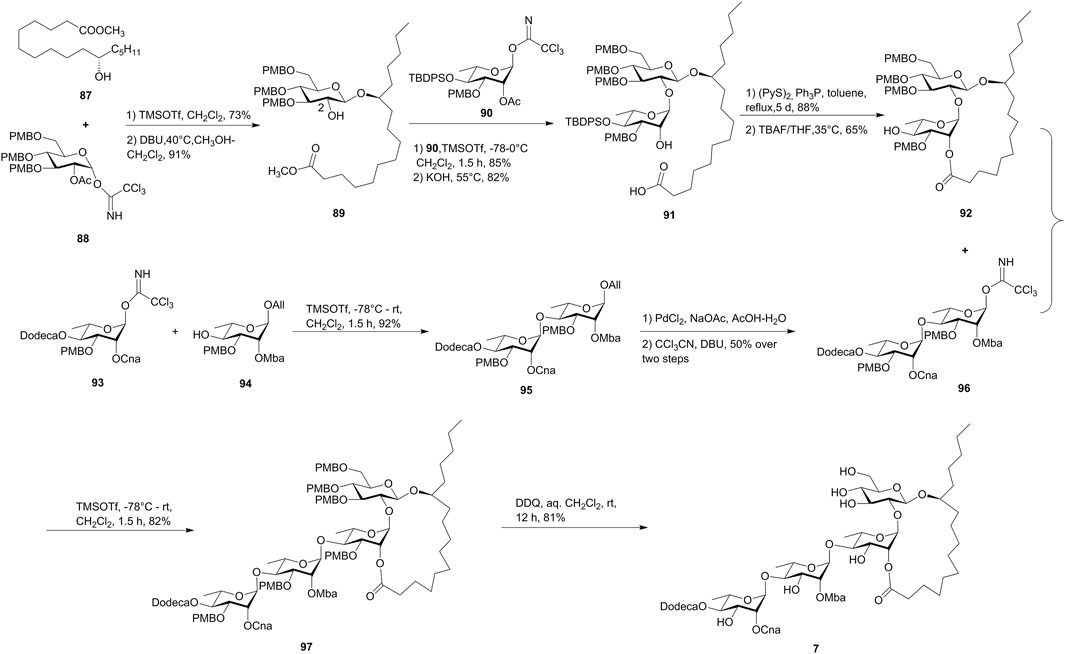
FIGURE 8. Yang’s approach toward the synthesis of batatoside L (7). Dodeca, n-dodecanoyl; Cna, trans-cinnamyl; Mba, 2S-methylbutyryl; All, allyl.
The synthetic approach started with the utilization of the synthesized chiral methyl 11(S)-jalapinolate (87) using commercially available (R)-glycidol as a starting material. The chiral hydroxyl ester (87) was glycosylated with D-glucosyl donors 88, followed by the selective cleavage of C-2 ester functionality, to deliver β-D-glucopyranoside 89. The subsequent coupling of 89 and R-L-rhamnopyranosyl trichloroacetimidate 90, followed by the saponification with KOH, afforded the disaccharide acid 91. The Corey–Nicolaou method was used for the macrolactonization, which yielded the desired lactone alcohol 92 by the removal of the silyl moiety. The exocyclic dirhamnopyranose fragment 95 was prepared by the glycosylation of monosaccharide modules 93 and 94 and was transformed to the trichloroacetimidate derivative 96. Finally, the “inverse glycosylation” procedure developed by Schmidt et al. was used to form the tetrasaccharide 97 in 82% yield, which produced target batatoside L (7) by the cleavage of PMB-ether functions. The synthetic batatoside L (7) was found to be identical with the natural isolate on the basis of 1H and 13C NMR spectra and specific rotation comparisons (Xie et al., 2010).
Total synthesis of murucoidins IV (8) and V (9)
Approach by Wan’s group
Murucoidins IV (8) and V (9) were isolated from the flowers of Ipomoea murucoides, a plant with a vernacular name of “cazahuate” in Mexico (Chérigo and Pereda-Miranda. 2006). They present very similar structures, containing the same oligosaccharide chain and an 11S-hydroxyhexadecanoic acid aglycone. Their only difference lies in the macrolactone rings, which is an 18-membered ring and a 19-membered ring in murucoidins IV (8) and V (9), respectively. Different from the classic glycosylation reactions utilizing the trichloroacetimidate glycosyl donors or thioglycosyl donors, the interrupted Pummerer reaction-mediated (IPRm) glycosylation reactions were developed by Wan’s group and were used for the total synthesis of murucoidins IV (8) and V (9) (Fang et al., 2019, Figure 9).
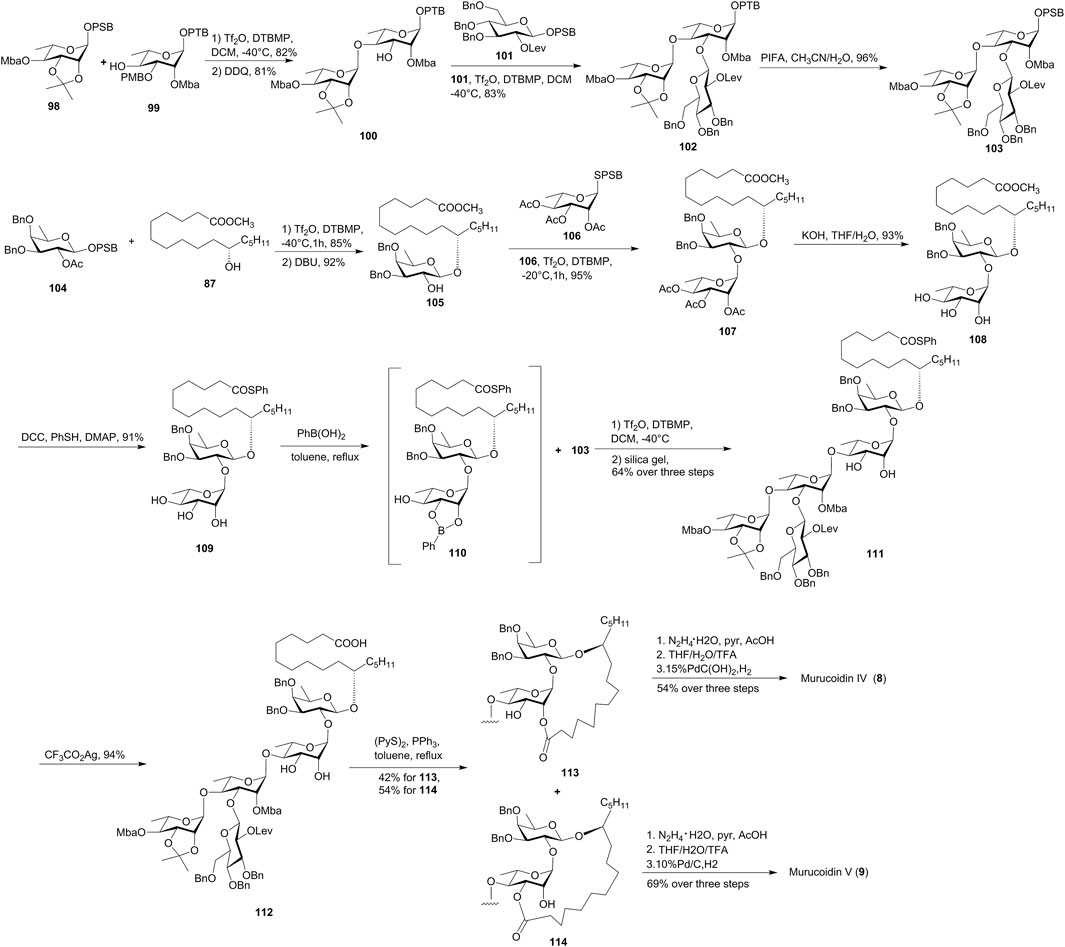
FIGURE 9. Wan’s approach toward the synthesis of murucoidins IV (8) and V (9). Mba, 2S-methylbutyryl; OPSB: O-2-[(propan-2-yl)sulfinyl]benzyl; OPTB, O-2-[(propan-2-yl)thio]benzyl; Lev, levulinoyl; PIFA, bis(trifluoro-acetoxyiodo)benzene; SPSB, S-2-[(propan-2-yl)sulfinyl] benzyl.
The coupling of latent OPTB glycoside 99 with active OPSB glycosyl donor 98, followed by the removal of the PMB-protecting group, furnished latent disaccharide 100. The glycosylation of 100 with OPSB glycoside 101 produced trisaccharide 102, which was subsequently oxidized to yield exocyclic glycosyl donor 103 with the active OPSB group.
The synthesis of glycosyl acceptor fragment 108 was started with the glycosylation of glycosyl donor 104 with methyl 11(S)-jalapinolate (87). Release of the C2-hydroxyl group of the product gave acceptor 105, which was coupled with the disarmed SPSB glycosyl donor 106 to provide disaccharide 107. The global saponification of all the ester groups under basic conditions furnished the key intermediate 108. Thioester was introduced for the protection of fatty acid to form 109, and cis-dihydroxyl groups were protected by forming a cyclic boronic acid ester 110. The site-selective [3 + 2] glycosidation of 110 and 103, followed by the cleavage of the cyclic boronic ester using silica gel column chromatography, afforded pentasaccharide 111 in 64% total yield over three steps. The macrolactonization of 112 under Corey–Nicolaou conditions proceeded smoothly to furnish macrolactones 113 and 114 with 18- and 19-membered rings. Although there was no selectivity obtained for this macrolactonization, both skeletons belong to murucoidins IV (8) and V (9), respectively. Finally, global removal of the protecting groups of 113 and 114 accomplished the divergent synthesis of murucoidins IV (8) and V (9), respectively. The NMR data exactly matched with the natural isolate, which endorsed their real structures of murucoidins IV (8) and V (9) (Fang et al., 2019).
Total synthesis of merremoside D (10)
Merremoside resin glycosides were isolated from Merremia mammosa (Bidara upas) and exhibited ionophoretic activity (Kitagawa et al., 1996a; Kitagawa et al., 1996b). Among them, merremoside D (10) was synthesized by two different strategies (Sharif et al., 2014; Xiao et al., 2020).
Approach by O’Doherty’s group
A de novo asymmetric synthesis of merremoside D (10) based on pyranone glycosyl donor 115 and Corey–Nicolaou macrolactonization was proposed by O’Doherty and the co-workers (Sharif et al., 2014, Figure 10).
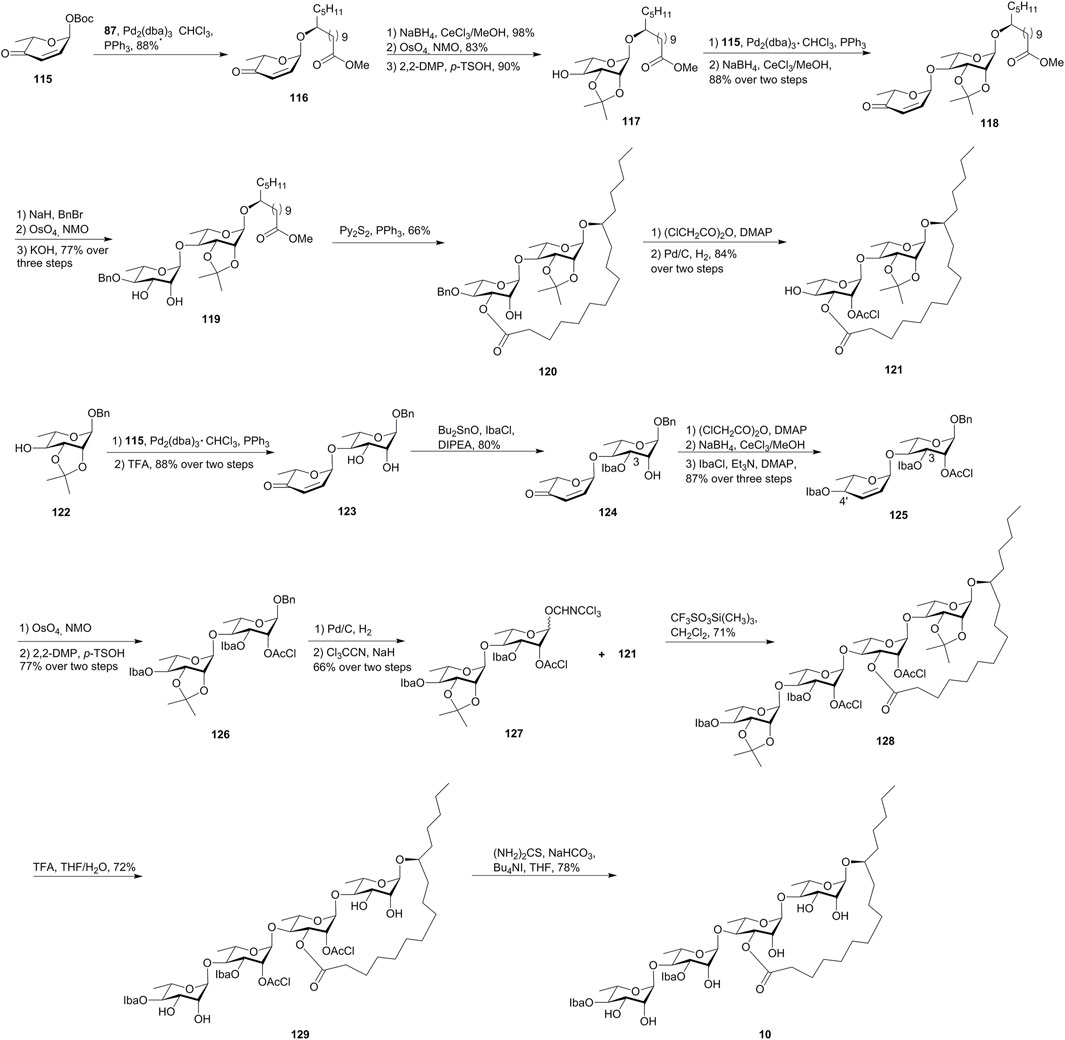
FIGURE 10. O’Doherty’s approach toward the synthesis of merremoside D (10). Iba, isobutyryl; DIPEA, N, N-diisopropylethylamine.
The synthesis of the macrolactone disaccharide began with a Pd-catalyzed stereoselective glycosylation of 115 with methyl 11(S)-jalapinolate (87) giving the enone 116, which was converted to the glycosyl acceptor 117 via the Luche reduction, the Upjohn dihydroxylation and syn-diol protection. Then, a second Pd-catalyzed glycosylation of 117 with pyranone 115, followed by enone reduction, gave allylic alcohol 118. The benzylation of allylic alcohol 118, followed by alkene dihydroxylation and methyl ester saponification, yielded diol-acid 119. The macrolactonization of 119 under Corey–Nicolaou conditions gave macrolactone 120 as the major product (66% yield). The remaining hydroxyl group on macrolactone 120 was protected as chloroacetic ester, and benzylether was removed to form the glycosyl acceptor 121.
The donor disaccharide was also prepared via the de novo asymmetric strategy. The protected rhamnopyranoside 122 was converted from pyranone 115 and then was linked by 115 via Pd-catalyzed glycosylation followed by acetonide removal to afford 123. The introduction of the C-3 Iba-ester was accomplished via a tin-mediated esterification with isobutyryl chloride (IbaCl), and the C-2 hydroxyl group was then protected as chloroacetic ester, followed by the Luche reduction/esterification to yield allylic ester 125. The glycosyl donor 127 was synthesized from 125 by functionalizing the double bond and anomeric activation and then reacted with the glycosyl acceptor 121 under Schmidt’s glycosylation conditions to afford 128 in 71% yield. Finally, the removal of acetonide groups and chloroacetates provided merremoside D (10). The comparison of the assignments of the synthetic product with the limited NMR data reported for the natural isolate allowed for the tentative confirmation of the structure of merremoside D (10). The total synthesis of merremoside D was achieved in 22 longest linear steps with a 3% overall yield and demonstrated the power of a de novo asymmetric approach to a stereochemically complex oligosaccharide natural product.
Approach by Wan’s group
A novel one-pot relay glycosylation was established by Wan’s group and was used for the synthesis of merremoside D (10). The method capitalizes on the in situ generated cyclic-thiosulfonium ion as the relay activator, which directly activates the newly formed thioglycoside in one pot and thus constructs two glycosidic bonds with only one equivalent of triflic anhydride (Xiao et al., 2020, Figure 11).
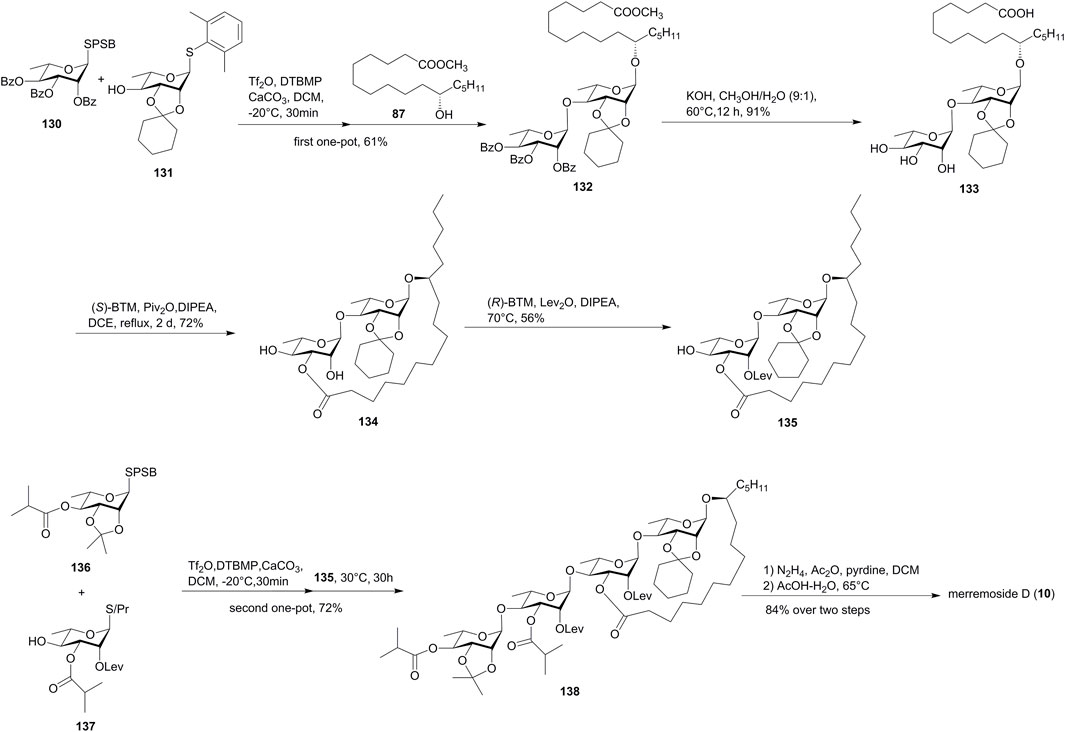
FIGURE 11. Wan’s approach toward the synthesis of merremoside D (10). SPSB, S-2-[(propan-2-yl)sulfinyl] benzyl; BTM, benzotetramisole; DIPEA, N, N-diisopropylethylamine; Lev: levulinoyl.
Merremoside D (10) was synthesized with two key one-pot relay glycosylations. The first one-pot reaction produced disaccharide 132 by the coupling of SPSB glycoside 130, thioglycoside 131, and methyl 11(S)-jalapinolate (87). The hydrolyzation of the ester groups in 132 afforded acid 133 with three continuous free hydroxy groups. The cation–n interaction-mediated site-selective acylation reaction was used for the construction of the macrolide structure. The site-selective macrolactonization of 133 with 1.5 equiv of (S)-BTM (benzotetramisole) along with 1.0 equiv of Piv2O as esterification reagents gave 134 in 72% yield. The following site-selective installation of the levulinoyl (Lev) group on the C2′ position produced 135. The second one-pot glycosylation of 135 with monosaccharide modules 136 and 137 produced the desired tetrasccharide 138 in 72% yield, which eventually furnished merremoside D (10) by global deprotection.
Total synthesis of tricolorin A (11)
Approach by Wan’s group
Tricolorin A (11) is a resin glycoside isolated from Ipomoea tricolor (heavenly blue morning glory) (Pereda-Miranda et al., 1993; Bah and Pereda-Miranda. 1996). The structure of tricolorin A (11) consists of a 19-membered macrolactone and a tetrasaccharide backbone. Tricolorin A (11) had been synthesized by several groups before 2009 (Pereda-Miranda et al., 2010). Herein, we introduced the recently reported synthetic approach by Wan’s group via the IPRm glycosylation and one-pot relay glycosylation (Sun et al., 2020, Figure 12).
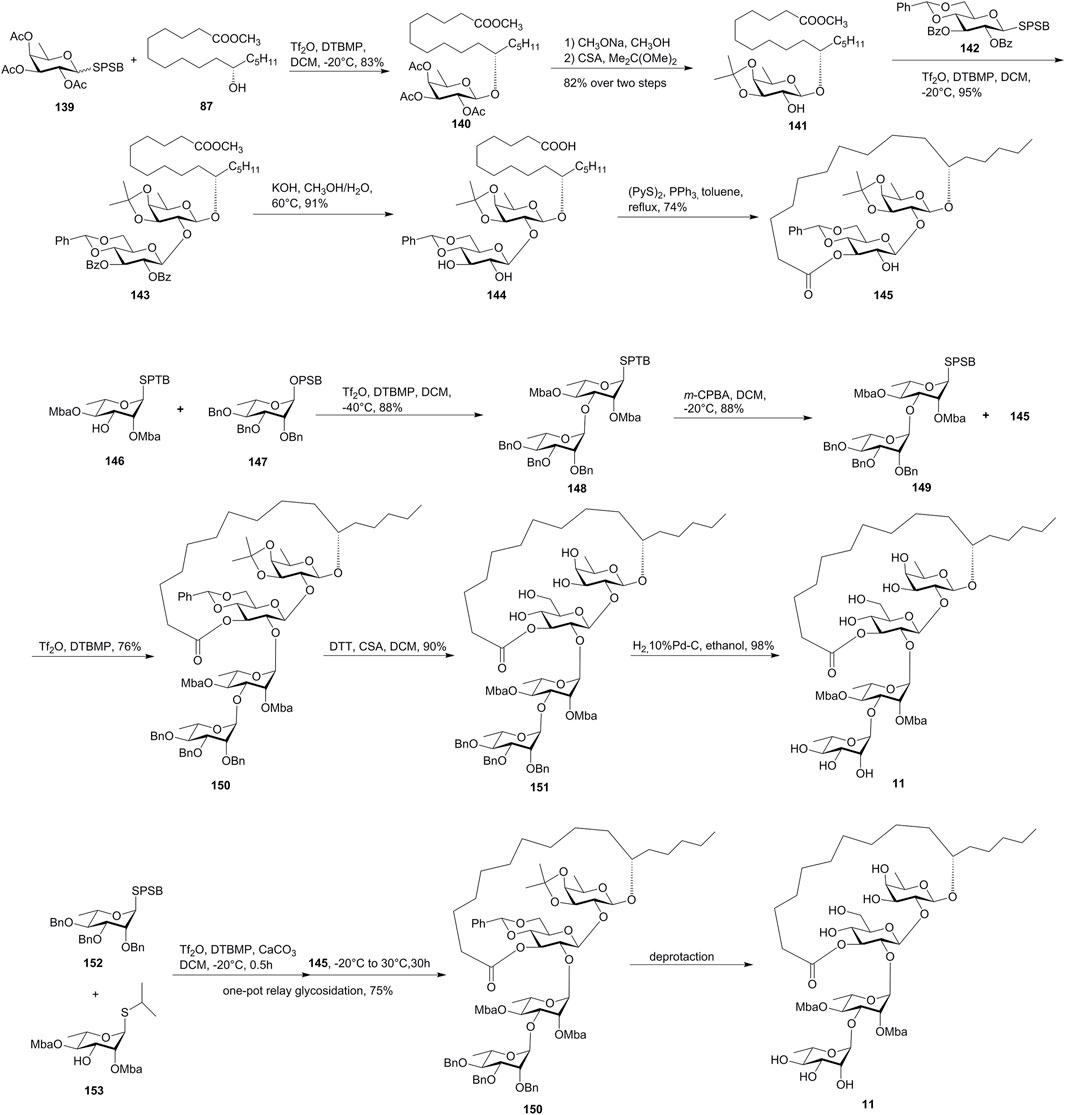
FIGURE 12. Wan’s approaches toward the synthesis of tricolorin A (11). SPSB, S-2-[(propan-2-yl)sulfinyl]benzyl; CSA, camphor-10-sulfonic acid; Mba, 2S-methylbutyryl; SPTB: S-2-[(propan-2-yl)sulfanyl]benzyl; OPSB, O-2-[(propan-2-yl) sulfinyl]benzyl.
The macrolactone fragment was prepared using IPRm glycosylation, which was started with the glycosylation of active SPSB donor 139 with methyl 11(S)-jalapinolate (87). The subsequent deacetylation and isopropylidenization of 140 afforded glycosyl acceptor 141, which was coupled with the SPSB donor 142 under the IPRm glycosylation conditions to furnish disaccharide 143. The saponification of 143 released the free dihydroxy groups and the aglycon carboxyl group, which formed the desired 19-membered macrolactone 145 under the Corey–Nicolaou condition in 74% yield.
The installation of the donor disaccharide fragment could be both achieved by IPRm glycosylation or one-pot relay glycosylation.
In the IPRm glycosylation approach, coupling of SPTB glycosyl acceptor 146 with the OPSB glycosyl donor 147 under IPRm glycosylation conditions produced disaccharide 148, which was then selectively oxidized to the SPSB donor 149. Coupling of the two key building blocks 149 and 145 under the standard IPRm glycosylation conditions furnished the desired tetrasaccharide 150 in moderate 1,2-trans selectivity (76% yield), while using the corresponding OPSB donor of 149 produced 150 in 92% yield. The removal of isopropylidene and benzylidene acetal groups of tetrasaccharide 150, followed by the final debenzylation, yielded tricolorin A (11) (Figure 12). The NMR data exactly matched with the natural and the reported synthetic tricolorin A (11). The longest linear sequence in this route is 16 steps with 12.5% overall yield.
In the one-pot relay glycosylation approach, monosaccharide unit 152 was first coupled with 153 in the presence of Tf2O, DTBMP, and CaCO3, and then macrolactone 145 was added to yield precursor tetrasaccharide 150 in 75% yield in one-pot glycosylation (Figure 12). Compared to the stepwise synthesis, the one-pot procedure offered a slightly higher overall yield and great convenience.
Total synthesis of tricolorin F (12)
Approach by Sakairi’s group
Tricolorin F (12) is another resin glycoside isolated from I. tricolor. It is a linear hetero-trisaccharide of jalapinolic acid, with the macrolactone bond linked at the C-2 position to the third sugar unit to form a 21-membered ring (Bah and Pereda-Miranda. 1996).
Sakairi’s group provided an intramolecular glycosylation for the construction of the macrolide structures of resin glycosides, which was applied for the total synthesis of tricolorin F (12) (Son et al., 2009, Figure 13). The D-fucosyl donor 154 was first coupled with aglycone methyl 11(S)-jalapinolate (87), followed by Zemplén de-O-benzoylation, giving the D-fucosyl acceptor 141. The glycosylation of 141 with the donor 155 under the MeOTf-promoted glycosylation, followed by hydrolyzation with aqueous NaOH, afforded the disaccharide 156. The coupling of 156 with excess thioquinovoside 157 gave the trisaccharide 158. Then, the intramolecular glycosylation of 158 using the MeOTf method gave the fully protected derivative 159 in 70% yield, which afforded tricolorin F (12) by global deprotection. In addition to macrolactonization and RCM, this new methodology would provide various synthetic analogues useful for the systematic survey of the biological functions of resin glycosides.
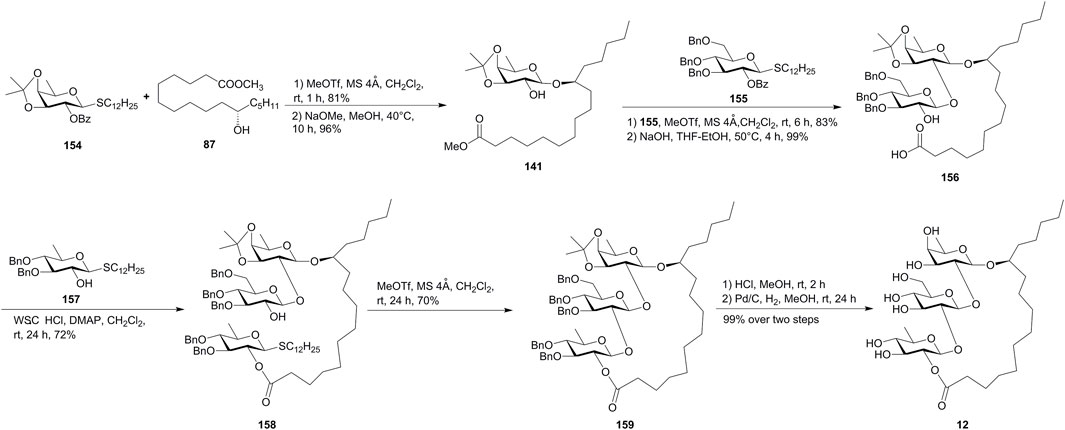
FIGURE 13. Sakairi’s approach toward the synthesis of tricolorin F (12). WSCHCl, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride.
Total synthesis of batatin VI (13)
Approach by Yang’s group
Batatin VI (13) is an ester-type resin glycoside dimer isolated from Ipomoea batatas (Rosas-Ramírez et al., 2011) and was synthesized by Yang’s group via a convergent [5 + 3] glycosidic coupling approach. An effective Keck macrolactonization was utilized for the formation of the 18-membered ring (Zhu et al., 2013, Figure 14).
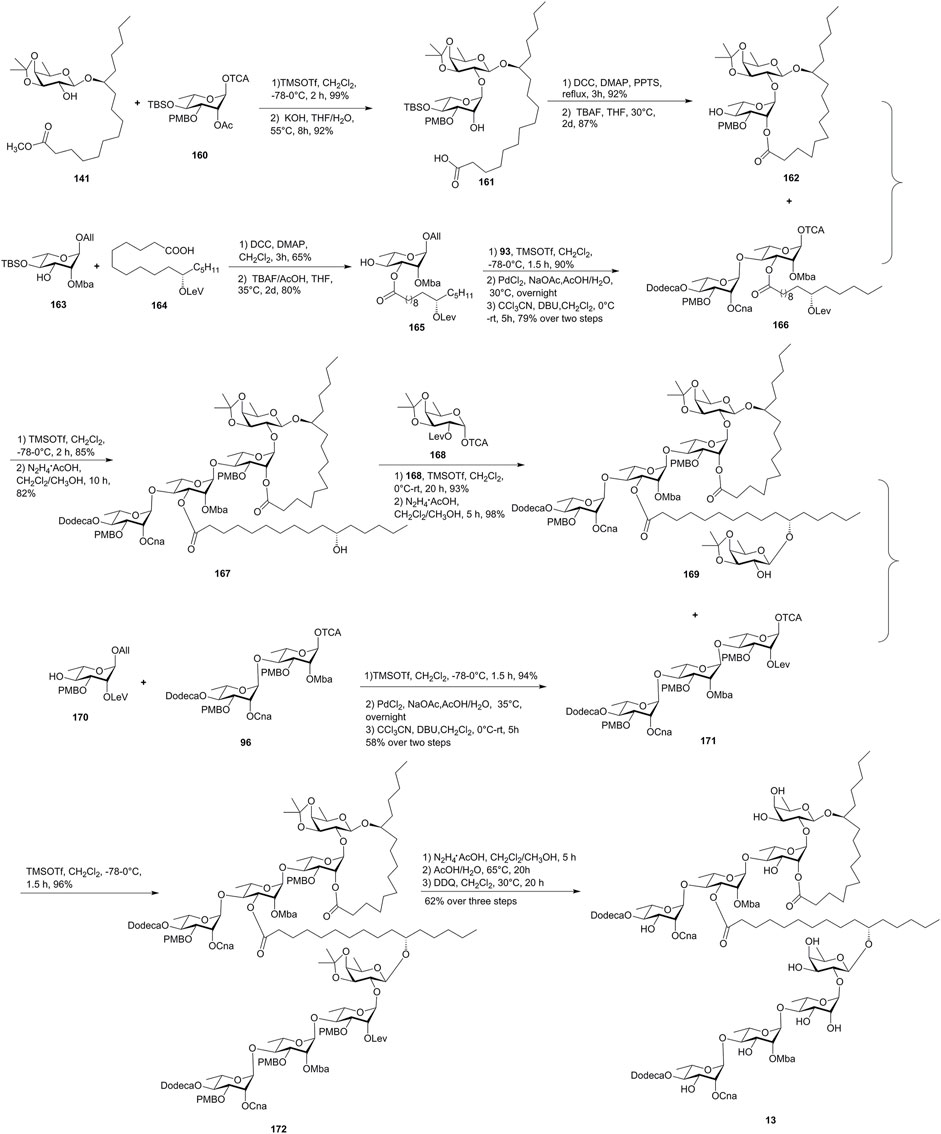
FIGURE 14. Yang’s approach toward the synthesis of batatin VI (13). All, allyl; Mba, 2S-methylbutyryl; Lev, levulinoyl; Dodeca, n-dodecanoyl; Cna, trans-cinnamyl.
The macrolactone precursor 161 was obtained by the Schmidt glycosylation of the known 141 with monosaccharide 160, followed by the saponification with KOH. Then, the Keck macrolactonization followed by the subsequent desilylation gave the required lactone alcohol 162 in 87% yield. The disaccharide donor 166 was obtained by the coupling of 165 with the chiral rhamnosyl intermediate 93, followed by the deallylation and activation of the resulting crude hemiacetal. The macrocycle acceptor 162 was coupled with 166 under TMSOTf-activated inverse glycosylation conditions in 85% yield, and then the levulinate was selectively hydrolyzed to afford tetrasaccharide 167. The glycosylation of 167 with monosaccharide 168 and the removal of the Lev group afforded crucial the pentasaccharide intermediate 169. Meanwhile, the trisaccharide intermediate 171 was obtained by the glycosidic coupling of 170 with 96. Finally, the fragments 169 and 171 were also linked together under TMSOTf-activated inverse glycosylation conditions to furnish protected octasaccharide 172 in 96% yield. The following global deprotection of 172 afforded the target batatin VI (13). Unfortunately, the NMR data of the synthetic product were not in full agreement with those of the naturally occurring batatin VI (13), suggesting that a structural revision to batatin VI (13) may be required (Zhu et al., 2013).
Synthesis of resin glycoside intermediates
There are also some research studies on the efficient synthesis of resin glycoside intermediates, which are summarized in Figure 15 and were introduced here briefly. Zhang’s group tried the Keck macrolactonization method to synthesize the macrolactone structures 159, 173, and 174 (Huang et al., 2015). The intramolecular glycosylation method was used for the construction of the macrolide cores 175 and 176, which were the intermediates for tricolorin A (11) and calysolin IX (177) (Son et al., 2009; Nawój et al., 2020). In addition, a suitably protected trichloroacetimidate (178) was synthesized by a block synthesis approach, which could serve as an exocyclic intermediate in the total synthesis of resin glycoside merremoside H2 (179) (Shen et al., 2009).
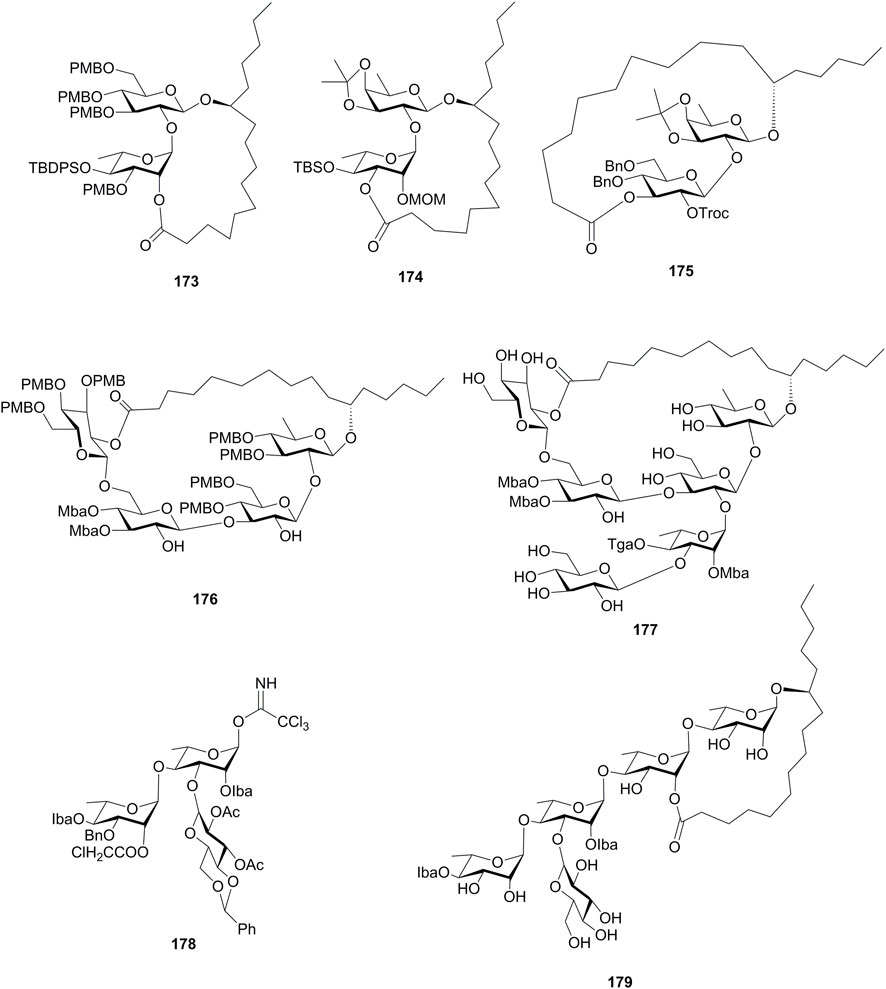
FIGURE 15. Structures of 173–179. TBDPS, tert-butyldiphenylsilyl; Troc, trichloroethoxycarbonyl; Mba, 2S-methylbutyryl; Tga, tiglyl; Iba, isobutyryl.
Conclusion
Given the structural diversification and promising bioactivities, considerable progress has been made in the total synthesis of resin glycosides. In this review, we highlighted reports on various total syntheses of resin glycosides in the last decade.
The construction of the macrolide structure is the most crucial step. The ring-closing metathesis (RCM) method was the major approach for the synthesis of resin glycoside ipomoeassins A–F (1–6), especially Shi’s group developed two strategies for the synthesis of ipomoeassin F (6) and its analogues, which were used for comprehensive structure–activity relationship studies on the cytotoxic activity. Several macrolactonization approaches were also used for the construction of the macrolide structure, including the Corey–Nicolaou macrolactonization reaction for batatoside L (7), murucoidins IV (8) and V (9), merremoside D (10), and tricolorin A (11), the cation–n interaction-mediated site-selective acylation reaction for merremoside D (10), and the Keck macrolactonization reaction for batatin VI (13) and the macrolactone intermediates 159, 173, and 174. In addition, the intramolecular glycosylation method was used for the total synthesis of tricolorin F (12), as well as the macrolide cores 175 and 176.
The construction of the glycosidic bond is another important step. Although the common activation of trichloroacetimidate glycosyl donors or thioglycosyl donors were the mostly used glycosylation methods, Wan’s group have developed the interrupted Pummerer reaction-mediated (IPRm) glycosylation and one-pot relay glycosylation methods, which were successfully utilized for the synthesis of murucoidins IV (8) and V (9), merremoside D (10), and tricolorin A (11). A Pd-catalyzed stereoselective glycosylation was used for the synthesis of merremoside D (10).
In general, the synthesized resin glycosides and their analogues helped in structural verification, structural modification, and further biological activity exploration, as well as provided enlightenment for the synthesis of glycoside compounds. However, compared with the vigorous structure and bioactivity studies, only a limited number of bioactive resin glycosides have been synthesized. The development of novel methods for the synthesis of resin glycosides and their analogues with better biological outcomes would be of strategic importance for further drug discovery.
Author contributions
BF and J-LL conceived the concept of review. WW, YL, and YH wrote the manuscript. All authors discussed, reviewed, and commented on the manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (82070600 and 32100317), Natural Science Project of Nantong Science and Technology Board (JC2021094), Large-Scale Instrument and Equipment Open Fund of Nantong University (KFJN2273), Postgraduate Research and Practice Innovation Program of Jiangsu Province (KYCX22-3381), and Students’ Platform for Innovation and Entrepreneurship Training Program of Nantong University (2022238).
Conflict of interest
Author YH is employed by Jinghua Pharmaceutical Group Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bah, M., and Pereda-Miranda, R. (1996). Bioactive natural products from traditionally used Mexican plants. Part IV. Detailed FAB-mass spectrometry and high resolution NMR investigations of tricolorins A-E, individual oligosaccharides from the resins of Ipomoea tricolor (Convolvulaceae). Tetrahedron 52, 13063–13080. doi:10.1016/0040-4020(96)00789-2
Cao, S., Guza, R. C., Wisse, J. H., Miller, J. S., Evans, R., and Kingston, D. G. (2005). Ipomoeassins A–E, cytotoxic macrocyclic glycoresins from the leaves of Ipomoea squamosa from the Suriname rainforest. J. Nat. Prod. (Gorakhpur). 68, 487–492. doi:10.1021/np049629w
Cao, S., Norris, A., Wisse, J. H., Miller, J. S., Evans, R., and Kingston, D. G. (2007). Ipomoeassin F, a new cytotoxic macrocyclic glycoresin from the leaves of Ipomoea squamosa from the Suriname rainforest. Nat. Prod. Res. 21, 872–876. doi:10.1080/14786410600929576
Castro-García, J. M., León-Rivera, I., and Gutiérrez, M. D. C. (2018). Resin glycosides evoke the Gaba release by sodium- and/or calcium-dependent mechanism. Biomed. Pharmacother. 97, 496–502. doi:10.1016/j.biopha.2017.10.089
Chen, C., Ma, T., Zhang, C., Zhang, H., Bai, L., Kong, L., et al. (2017). Down-regulation of aquaporin 5-mediated epithelial-mesenchymal transition and anti-metastatic effect by natural product Cairicoside E in colorectal cancer. Mol. Carcinog. 56, 2692–2705. doi:10.1002/mc.22712
Chérigo, L., and Pereda-Miranda, R. (2006). Resin glycosides from the flowers of Ipomoea murucoides. J. Nat. Prod. (Gorakhpur). 69, 595–599. doi:10.1021/np0504457
Fan, B. Y., Gu, Y. C., He, Y., Li, Z. R., Luo, J. G., and Kong, L. Y. (2014). Cytotoxic resin glycosides from Ipomoea aquatica and their effects on intracellular Ca2+ concentrations. J. Nat. Prod. (Gorakhpur). 77, 2264–2272. doi:10.1021/np5005246
Fan, B. Y., Jiang, X., Li, Y. X., Wang, W. L., Yang, M., Li, J. L., et al. (2022). Chemistry and biological activity of resin glycosides from Convolvulaceae species. Med. Res. Rev. 42, 2025–2066. doi:10.1002/med.21916
Fang, J., Zeng, J., Sun, J., Zhang, S., Xiao, X., Lu, Z., et al. (2019). Total syntheses of resin glycosides murucoidins IV and V. Org. Lett. 21, 6213–6216. doi:10.1021/acs.orglett.9b02004
Figueroa-González, G., Jacobo-Herrera, N., Zentella-Dehesa, A., and Pereda-Miranda, R. (2012). Reversal of multidrug resistance by morning glory resin glycosides in human breast cancer cells. J. Nat. Prod. (Gorakhpur). 75, 93–97. doi:10.1021/np200864m
Fürstner, A., and Nagano, T. (2007). Total syntheses of ipomoeassin B and E. J. Am. Chem. Soc. 129, 1906–1907. doi:10.1021/ja068901g
Huang, H., Yang, C. S., Zeng, L. N., and Zhang, L. L. (2015). Synthesis of key macrolactone structure of resin glycosides using a Keck macrolactonization method. J. Asian Nat. Prod. Res. 17, 289–298. doi:10.1080/10286020.2014.998653
Kitagawa, I., Baek, N. I., Kawashima, K., Yokokawa, Y., Yoshikawa, M., Ohashi, K., et al. (1996a). Indonesian medicinal plants. XV. Chemical structures of five new resin-glycosides, merremosides a, b, c, d, and e, from the tuber of Merremia mammosa (Convolvulaceae). Chem. Pharm. Bull. 44, 1680–1692. doi:10.1248/cpb.44.1680
Kitagawa, I., Baek, N. I., Yokokawa, Y., Yoshikawa, M., Ohashi, K., and Shibuya, H. (1996b). Indonesian medicinal plants. XVI. Chemical structures of four new resin-glycosides, merremosides f, g, h1, and h2, from the tuber of Merremia mammosa (Convolvulaceae). Chem. Pharm. Bull. 44, 1693–1699. doi:10.1248/cpb.44.1693
León-Rivera, I., Castro, J. M., Mirón-López, G., del Río-Portilla, F., Enríquez, R. G., Reynolds, W. F., et al. (2014). Resin glycosides from Ipomoea tyrianthina and their sedative and vasorelaxant effects. J. Nat. Med. 68, 655–667. doi:10.1007/s11418-014-0844-x
Leznicki, P., Schneider, H. O., Harvey, J. V., Shi, W. Q., and High, S. (2022). Co-translational biogenesis of lipid droplet integral membrane proteins. J. Cell Sci. 135, jcs259220. doi:10.1242/jcs.259220
Lira-Ricárdez, J., and Pereda-Miranda, R. (2020). Reversal of multidrug resistance by amphiphilic morning glory resin glycosides in bacterial pathogens and human cancer cells. Phytochem. Rev. 19, 1211–1229. doi:10.1007/s11101-019-09631-1
Lu, S. F., O’yang, Q., Guo, Z. W., Yu, B., and Hui, Y. Z. (1997). Total synthesis of tricolorin A. J. Org. Chem. 62, 8400–8405. doi:10.1021/jo9711450
Nagano, T., Pospísil, J., Chollet, G., Schulthoff, S., Hickmann, V., Moulin, E., et al. (2009). Total synthesis and biological evaluation of the cytotoxic resin glycosides ipomoeassin A-F and analogues. Chem. Eur. J. 15, 9697–9706. doi:10.1002/chem.200901449
Nawój, M., Grobelny, A., and Mlynarski, J. (2020). Macrolide core synthesis of Calysolin IX using an intramolecular glycosylation approach. Eur. J. Org. Chem. 2020, 47–51. doi:10.1002/ejoc.201901480
Newman, D. J., and Cragg, G. M. (2020). Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. (Gorakhpur). 83, 770–803. doi:10.1021/acs.jnatprod.9b01285
O'Keefe, S., Roboti, P., Duah, K. B., Zong, G., Schneider, H., Shi, W. Q., et al. (2021a). Ipomoeassin-F inhibits the in vitro biogenesis of the SARS-CoV-2 spike protein and its host cell membrane receptor. J. Cell Sci. 134, jcs257758. doi:10.1242/jcs.257758
O'Keefe, S., Zong, G., Duah, K. B., Andrews, L. E., Shi, W. Q., and High, S. (2021b). An alternative pathway for membrane protein biogenesis at the endoplasmic reticulum. Commun. Biol. 4, 828. doi:10.1038/s42003-021-02363-z
Ono, M., Azuchi, M., Ichio, M., Jiyoubi, Y., Tsutsumi, S., Yasuda, S., et al. (2019). Seven new resin glycosides from the seeds of Quamoclit × multifida. J. Nat. Med. 73, 11–22. doi:10.1007/s11418-018-1236-4
Ono, M., Kawakami, G., Takigawa, A., Kabata, K., Okawa, M., Kinjo, J., et al. (2014). Calysolins X-XIII, resin glycosides from Calystegia soldanella, and their antiviral activity toward herpes simplex virus. Chem. Pharm. Bull. 62, 839–844. doi:10.1248/cpb.c14-00356
Pan, J. T., Yu, B. W., Yin, Y. Q., Li, J. H., Wang, L., Guo, L. B., et al. (2015). Four new pentasaccharide resin glycosides from Ipomoea cairica with strong α-glucosidase inhibitory activity. Molecules 20, 6601–6610. doi:10.3390/molecules20046601
Pereda-Miranda, R., Mata, R., Anaya, A. L., Wickramaratne, D. B., Pezzuto, J. M., and Kinghorn, A. D. (1993). Tricolorin A, major phytogrowth inhibitor from Ipomoea tricolor. J. Nat. Prod. (Gorakhpur). 56, 571–582. doi:10.1021/np50094a018
Pereda-Miranda, R., Rosas-Ramírez, D., and Castańeda-Gómez, J. (2010). Resin glycosides from the morning glory family. Prog. Chem. Org. Nat. Prod. 92, 77–153. doi:10.1007/978-3-211-99661-4_2
Postema, M. H., TenDyke, K., Cutter, J., Kuznetsov, G., and Xu, Q. (2009). Total synthesis of ipomoeassin F. Org. Lett. 11, 1417–1420. doi:10.1021/ol900086b
Roboti, P., O'Keefe, S., Duah, K. B., Shi, W. Q., and High, S. (2021). Ipomoeassin-F disrupts multiple aspects of secretory protein biogenesis. Sci. Rep. 11, 11562. doi:10.1038/s41598-021-91107-4
Rosas-Ramírez, D., Escalante-Sánchez, E., and Pereda-Miranda, R. (2011). Batatins III-VI, glycolipid ester-type dimers from Ipomoea batatas. Phytochemistry 72, 773–780. doi:10.1016/j.phytochem.2011.03.002
Sharif, E. U., Wang, H. Y., Akhmedov, N. G., and O'Doherty, G. A. (2014). Merremoside D: De novo synthesis of the purported structure, NMR analysis, and comparison of spectral data. Org. Lett. 16, 492–495. doi:10.1021/ol403369h
Shen, X. Q., Xie, L., Gao, L., He, L. L., Yang, Q., and Yang, J. S. (2009). Synthetic studies on the trisaccharide intermediate of resin glycoside merremoside H2. Carbohydr. Res. 344, 2063–2068. doi:10.1016/j.carres.2009.08.005
Son, S. H., Yanagiya, N., Furukawa, J. I., and Sakairi, N. (2009). Intramolecular glycosylation approach toward constructing the macrocyclic structure of resin glycosides. Synlett 2009, 2957–2960. doi:10.1055/s-0029-1218272
Sun, J., Fang, J., Xiao, X., Cai, L., Zhao, X., Zeng, J., et al. (2020). Total synthesis of tricolorin A via interrupted Pummerer reaction-mediated glycosylation and one-pot relay glycosylation. Org. Biomol. Chem. 18, 3818–3822. doi:10.1039/d0ob00513d
Wang, W. Q., Liu, S. S., Song, W. B., Li, J., and Xuan, L. J. (2018). Resin glycosides from the seeds of Ipomoea muricata and their multidrug resistance reversal activities. Phytochemistry 149, 24–30. doi:10.1016/j.phytochem.2018.01.020
Xiao, X., Zeng, J., Fang, J., Sun, J., Li, T., Song, Z., et al. (2020). One-pot relay glycosylation. J. Am. Chem. Soc. 142, 5498–5503. doi:10.1021/jacs.0c00447
Xie, L., Zhu, S. Y., Shen, X. Q., He, L. L., and Yang, J. S. (2010). Total synthesis of batatoside L. J. Org. Chem. 75, 5764–5767. doi:10.1021/jo101231r
Yin, Y. Q., Wang, J. S., Luo, J. G., and Kong, L. Y. (2009). Novel acylated lipo-oligosaccharides from the tubers of Ipomoea batatas. Carbohydr. Res. 344, 466–473. doi:10.1016/j.carres.2008.12.022
Zhu, S. Y., Huang, J. S., Zheng, S. S., Zhu, K., and Yang, J. S. (2013). First total synthesis of the proposed structure of batatin VI. Org. Lett. 15, 4154–4157. doi:10.1021/ol4020255
Zong, G., Aljewari, H., Hu, Z., and Shi, W. Q. (2016). Revealing the pharmacophore of ipomoeassin F through molecular editing. Org. Lett. 18, 1674–1677. doi:10.1021/acs.orglett.6b00555
Zong, G., Barber, E., Aljewari, H., Zhou, J., Hu, Z., Du, Y., et al. (2015). Total synthesis and biological evaluation of ipomoeassin F and its unnatural 11R-epimer. J. Org. Chem. 80, 9279–9291. doi:10.1021/acs.joc.5b01765
Zong, G., Hirsch, M., Mondrik, C., Hu, Z., and Shi, W. Q. (2017a). Design, synthesis and biological evaluation of fucose-truncated monosaccharide analogues of ipomoeassin F. Bioorg. Med. Chem. Lett. 27, 2752–2756. doi:10.1016/j.bmcl.2017.04.065
Zong, G., Hu, Z., Duah, K. B., Andrews, L. E., Zhou, J., O'Keefe, S., et al. (2020). Ring expansion leads to a more potent analogue of ipomoeassin F. J. Org. Chem. 85, 16226–16235. doi:10.1021/acs.joc.0c01659
Zong, G., Hu, Z., O'Keefe, S., Tranter, D., Iannotti, M. J., Baron, L., et al. (2019). Ipomoeassin F binds sec61α to inhibit protein translocation. J. Am. Chem. Soc. 141, 8450–8461. doi:10.1021/jacs.8b13506
Zong, G., Sun, X., Bhakta, R., Whisenhunt, L., Hu, Z., Wang, F., et al. (2018). New insights into structure-activity relationship of ipomoeassin F from its bioisosteric 5-oxa/aza analogues. Eur. J. Med. Chem. 144, 751–757. doi:10.1016/j.ejmech.2017.11.022
Keywords: resin glycosides, total synthesis, glycolipids, macrolactonization, glycosylation
Citation: Wang W, Li Y, He Y, Jiang X, Yi Y, Zhang X, Zhang S, Chen G, Yang M, Luo J-L and Fan B (2022) Progress in the total synthesis of resin glycosides. Front. Chem. 10:1036954. doi: 10.3389/fchem.2022.1036954
Received: 05 September 2022; Accepted: 31 October 2022;
Published: 11 November 2022.
Edited by:
M. Carmen Galan, University of Bristol, United KingdomReviewed by:
John F. Trant, University of Windsor, CanadaJing Zeng, Huazhong University of Science and Technology, China
Copyright © 2022 Wang, Li, He, Jiang, Yi, Zhang, Zhang, Chen, Yang, Luo and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Boyi Fan, ZmFuYm95aUBudHUuZWR1LmNu; Jia-Lie Luo, bHVvamlhbGllQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship