 Zhong-Hui Shen
Zhong-Hui Shen Si-Yuan Lu
Si-Yuan Lu Jin-Bao Peng
Jin-Bao Peng Ai-Jun Ma
Ai-Jun Ma
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 05 October 2022
Sec. Organic Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.1022533
This article is part of the Research Topic Bioactive Natural Products-Oriented Synthetic and Functional Studies View all 24 articles
Bioassay-guided isolation of spiroaspertrione A from cultures of Aspergillus sp. TJ23 in 2017 demonstrated potent resensitization of oxacillin against methicillin-resistant Staphylococcus aureus by lowering the oxacillin minimal inhibitory concentration up to 32-fold. To construct this unique spiro[bicyclo[3.2.2]nonane-2,1′-cyclohexane] system, a protocol for ceric ammonium nitrate-induced intramolecular cross-coupling of silyl enolate is disclosed.
The expansion of multidrug-resistant pathogens is a threat to human health that can effectively take us back to the pre-antibiotic era for many infectious diseases (Walsh et al., 2011). Considering its grave roles in hospital and community-acquired infections, methicillin-resistant Staphylococcus aureus (MRSA) is a “superbug” with an extreme array of resistance and virulence factors (Gonzales, et al., 2015). Drug-resistance gene mutations of MRSA are exemplified by mecA, the disruption of which can produce inducible resistance to β-lactam antibiotics because it encodes penicillin-binding protein 2a (Fuda et al., 2005). With the rapid acquisition of resistance restricting therapeutic options for MRSA, many scientists have explored treatment methods combining the use of small molecules to render MRSA sensitive to the effects of conventional β-lactam antibiotics (Van Hal et al., 2011; Long et al., 2014; Bush, 2015; Gonzales et al., 2015).
In 2017, Zhang group used a bioassay-guided approach to isolate a novel terpene-polyketide hybrid spiromero-terpenoid from a culture of Aspergillus sp. TJ23, spiroaspertrione A (1), which bears a unique spiro[bicyclo[3.2.2]nonane-2,1′- cyclohexane] carbocyclic skeleton (Figure 1) (He et al., 2017). Spiroaspertrione A demonstrated potent resensitization of oxacillin against MRSA by lowering the oxacillin minimal inhibitory concentration (MIC) up to 32-fold from 32 μg/mL to 1 μg/mL (He et al., 2017). This promising bioactivity together with a unique spiro[bicyclo[3.2.2]nonane-2,1-cyclohexane] carbocyclic skeleton renders spiroaspertrione A an interesting and challenging target for total synthesis. To date, no synthesis method for spiroaspertrione A has been reported.
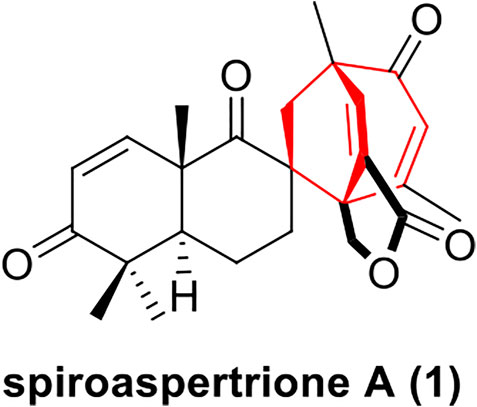
FIGURE 1. Structure of spiroaspertrione A (1).
Here, we analyzed and constructed the core skeleton of this spiromeroterpenoid, culminating in a strategy of intramolecular enol oxidative coupling (Figure 2). When designing this synthetic strategy for spiroaspertrione A, we noticed that the E lactone ring can be obtained by simple lactonization during later synthesis. Construction of the spiro[bicyclo[3.2.2]nonane] system of the ABCD rings exhibits a high degree of ring tension and rigidity–the most interesting and challenging feature of spiroaspertrione A synthesis. The core skeleton of spiroaspertrione A could be constructed through Birch reduction followed by methylation of the naphthene compound 2. This synthetically significant and more tractable spiro-ring system can then be built by an intramolecular enol oxidative coupling (EOC) reaction of precursor 3, which can then be traced back to a 1,4-conjugate addition of western fragment 5 and eastern fragment 4.
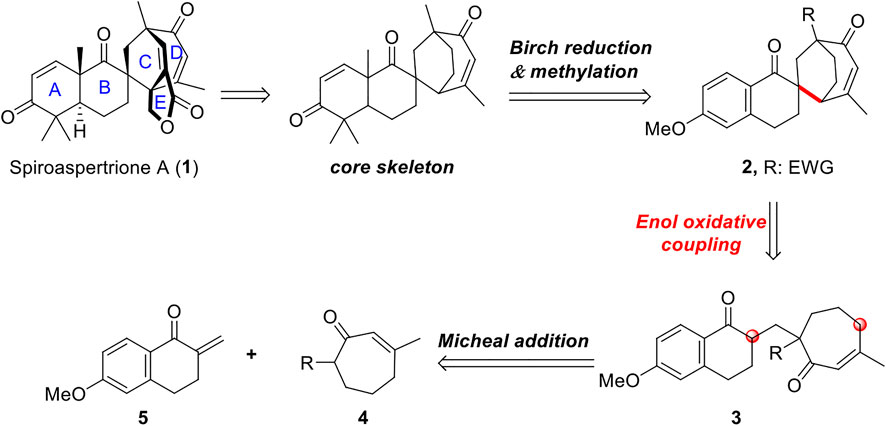
FIGURE 2. Retrosynthetic analysis.
As an efficient synthetic method to directly construct C-C bonds, the oxidative coupling reaction of enol derivatives has been applied in the syntheses of polyketides, alkaloids, and other natural products. (Murarka and Antonchick 2018). Although the first oxidative coupling reaction of enol derivatives dates back to 1935, it did not receive widespread attention from chemists until the 1970s because the efficiency and practicality of this reaction were less than satisfactory (Fujii et al., 1992; Kohno and Narasaka 1995; Ryter and Livinghouse 1998; Ekebergh et al., 2011; Rathke and Lindert 1971; Dessau and Heiba 1974; Xie and Huang 2010; Renaud and Fox 1998). In 2005, Baran group began to conduct in-depth research on the oxidative coupling reaction of enolates and successfully applied their findings to the total synthesis of multiple complex natural products (Baran et al., 2005; Richter et al., 2007; DeMartino et al., 2008). To date, the oxidative coupling reaction of enol derivates remains under constant development and optimization. The EOC reaction can be broadly divided into two categories: direct oxidation, involving the construction of C-C bonds under single-electron oxidants (e.g., ketones, carboxylic acids, esters, and amides) bound to the corresponding enols or enolates; and indirect oxidation, in which single-electron oxidants are converted to the corresponding enol (e.g., silanes and enamines) prior to construction of the C-C bonds (Figure 3). The EOC reaction has been reviewed by Plumet (Csákÿ and Plumet 2001), Baran (Baran 2006), Dong (Yeung Dong 2011), Thomson (Guo et al., 2012), Ma (Nagaraju and Ma 2018), Chen (Chen and Liu 2021), and others.
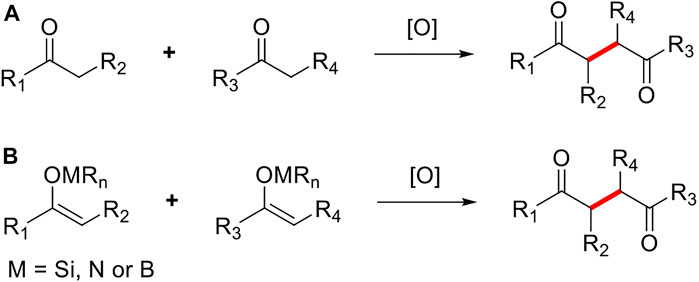
FIGURE 3. Types of enol oxidative coupling reaction.
In recent years, intermolecular and intramolecular EOC reactions have been applied to many natural products as an efficient method of constructing C-C bonds (Figure 4). Baran group completed the construction of the core skeleton of the natural product maoecrystal V using intermolecular EOC reactions (Krawczuk et al., 2009). Furthermore, Yang group (You et al., 2015) used enol silyl ethers substrates to realize the enantioselective synthesis of propindilactone G from the Schisandra family by cross-oxidative coupling reaction. Moreover, Thomson groups disclosed a method of self-intermolecular EOC reactions applied to the synthesis of dimerized natural product bis-murrayaquinone A (Konkol et al., 2011).
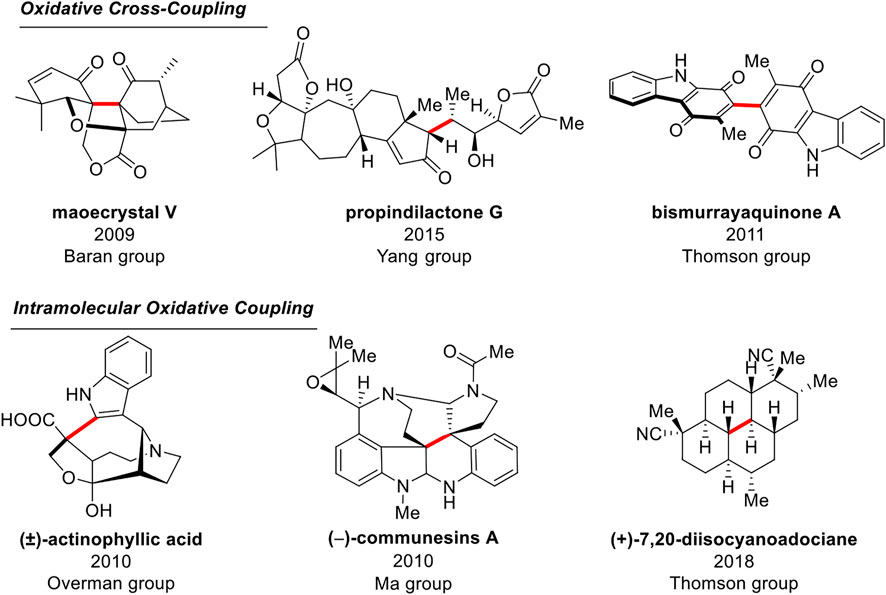
FIGURE 4. Application of EOC reaction in the synthesis of natural products.
Using intramolecular EOC reactions, Overman group (Martin et al., 2008; Martin et al., 2010) made important progress toward the total synthesis of the indole alkaloids (±)-actinophyllic acid. From 2010 to 2014, Ma group (Zuo et al., 2010; Zuo and Ma 2011; Zi et al., 2012; Wei et al., 2013; Teng et al., 2014) realized the efficient construction of the core skeleton of indole alkaloids and synthesized several indole alkaloids such as (+)-communesins A. In 2018, Thomson group (Guo et al., 2012; Jones et al., 2014; Robinson and Thomson, 2018) reported the strategy of intra-EOC reaction using enol di-silyl ether to realize the formal synthesis of natural products (+)-7,20-diisocyanoadociane and other derived products.
Unless otherwise noted, all reactions were carried out under N2 atmosphere. All reagents were from commercial sources and used as received without further purification. All solvents were dried by standard techniques and distilled prior to use. Column chromatography was performed on silica gel (200–300 meshes) using petrol etherand ethyl acetate as eluent. NMR spectra were recorded on a Bruker Avance operating at for 1H NMR at 500 MHz, 13C NMR at 126 MHz and spectral data were reported in ppm relative to tetramethylsilane (TMS) as internal standard and CDCl3 (1H NMR δ 7.26, 13C NMR δ 77.0) as solvent. All high-resolution mass spectra (HRMS) were obtained by Thermo Scientific's UltiMate 3,000 Series liquid system and Thermo Scientific Q-Exactive combined quadrupole Orbitrap mass spectrometer.
According to our retrosynthetic analysis, we chose known compound 5 (Li et al., 2019) and 4 self-prepared from 3-methylcyclohept-2-en-1-one as substrates to form the important precursor 3. Following screening of various conditions, we obtained compound 3 as a minor product with a yield of 30% under NaH and MeOH, and compound 5’s O-1,4-addition byproduct as the major product. Subsequent screening of several Lewis acids, such as BF3·OEt and TiCl4, yielded substrate 5’s O-DA reaction byproduct as the major product. To our delight, conducting the reaction in acetone at 55°C in the presence of K2CO3(2 eq) afforded the desired 1,4-addition product 3 with 60%–68% yield (Figure 5).
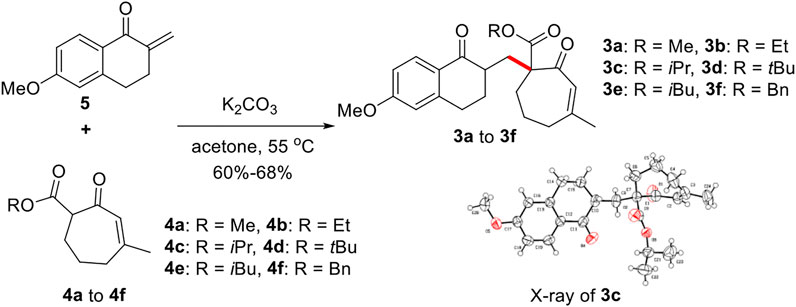
FIGURE 5. Synthesis of precursor 3.
With precursor 3a in hand, we intended to construct the desired C-C bond by single-electron oxidation under conditions including a metal base (Table 1). In the presence of LHMDS and cupric chloride (CuCl2) or ferric chloride (FeCl3), we obtained a very small amount of the EOC product 2a and 2a′, although the recovery yield was 60% (Table 1, entries 1 and 2). We assumed that LHMDS conditions were not conducive to the formation of stable enolates, thus, screened various metal bases. When using LDA as the base, the reaction only provided a complex mixture and trace amount of product with the oxidant CuCl2 (Table 1, entry 3). The substrate was completely consumed under conditions including KHMDS or NaHMDS (Table 1, entries 4 and 5). Although we tried numerous oxidative conditions, the yield of oxidative coupling products was not significantly improved (Table 1, entries 6 to 11).
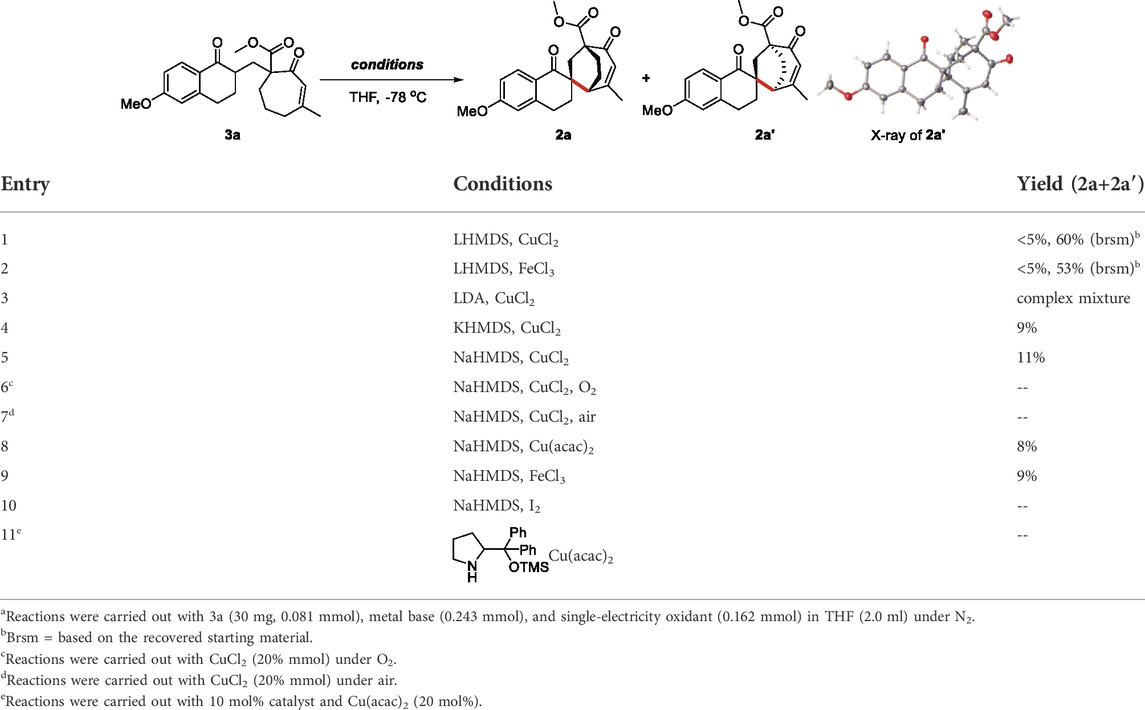
TABLE 1. Optimization of the reaction conditionsa.
According to the unsatisfactory experimental results described above, we assumed that substrate 3a may form more stable metal complexes with Cu(II) or Fe(III) ions under the alkaline system, thereby inhibiting the process of oxidative coupling. Therefore, we envisaged the replacement of this stable complex by enol silyl ether (Table 2). We chose compound 3a as a substrate to first optimize the silyl bis-enol etherification condition. The desired silyl bis-enol ether product 6a was obtained with 21% yield in THF (2.0 ml) at -78°C under N2 in the presence of LHMDS (0.243 mmol) and TBSOTf (0.162 mmol) (Table 2, entry 2). As previously mentioned, it was not conducive to obtain silyl enol ethers and could be broken down using LDA as the base (Table 2, entry 1). Encouraged by this result, we surveyed other bases including NaHMDS, KHMDS and Et3N, and found that NaHMDS generated the best yield (58%) while Et3N only generated monosilyl product (Table 2, entries 3–6).
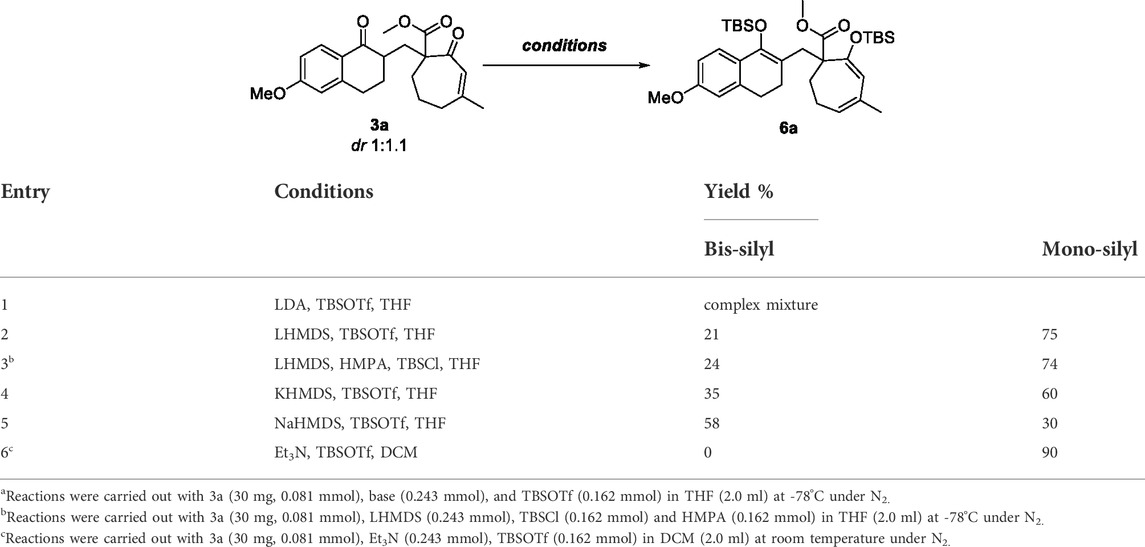
TABLE 2. Optimization of silyl bis-enol etherification conditionsa.
With the enol bis-silyl ether 6a in hand, we intended to optimize the intramolecular enol oxidative coupling reaction (Table 3). Conducting the reaction in CH3CN/THF at 0°C in the presence of CAN (0.24 mmol) and NaHCO3 (0.48 mmol) exclusively afforded the coupling products in 77% isolated yield after 0.5 h (Table 3, entry 1). However, the main product 4a′ identified by X-ray analyses was an undesired stereoisomer. We intended to optimize the diastereomeric ratio (dr) by changing the ester group of 6 (Table 3, entries 2–5) and found that the isopropyl dr of ester substrate (3c) reached 4.9:1. We also explored the effect of different silicon groups (Table 3, entries 6–7). Unfortunately, changing the silicon groups did not decisively progress the EOC reaction. Finally, we evaluated different oxidants to optimize the dr of products (Table 3, entries 8–12). Cu(II) chloride and Fe(III) chloride produced the coupling products with 21% and 23% yields, but even more of the desilylation product 3 (Table 3, entries 8 and 10). Other metal oxidants, including Cu(acac)2 and AgF, provided a complex mixture of product and raw product 6c (Table 3, entries 9 and 11), and the result of Koser’s reagent (PhI(OH)OTs) was also unsatisfactory (Table 3, entry 12).
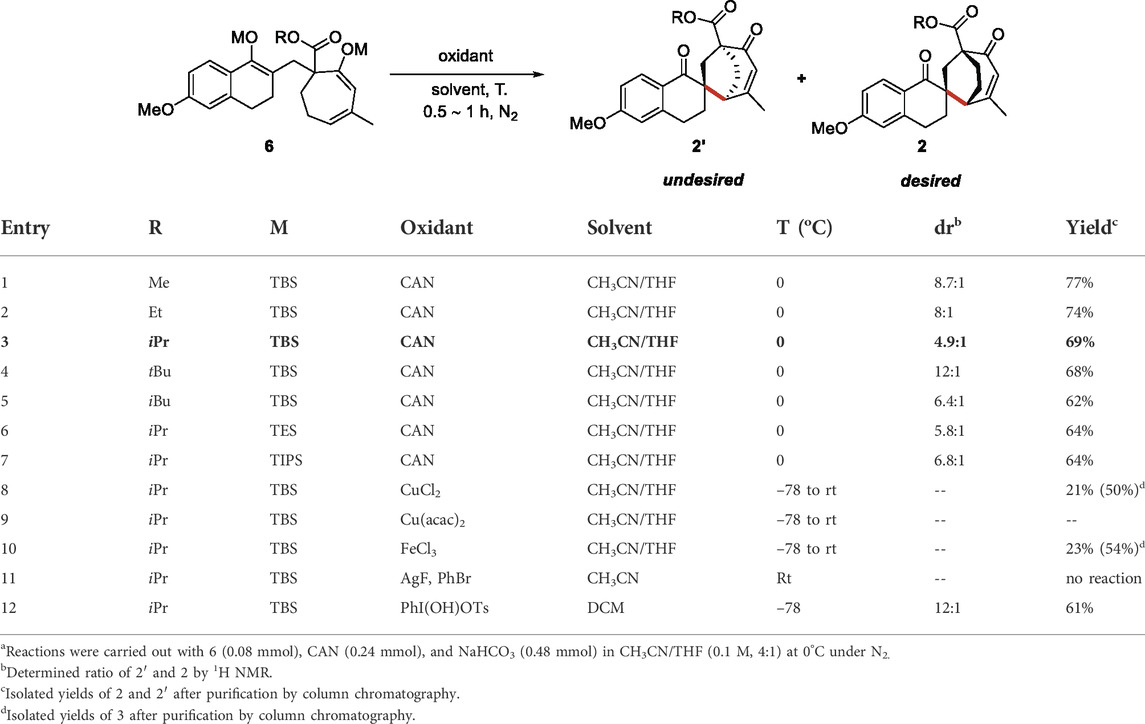
TABLE 3. Optimization of intramolecular EOC reaction conditionsa.
In conclusion, we developed an efficient method of constructing the spiro[bicyclo[3.2.2]nonane] system by intramolecular enol oxidative coupling reaction. Although the diastereomeric ratio of products is embarrassing, the high yield of this remote oxidative coupling reaction to build rigid spiro[bicyclo[3.2.2]nonane] structures is encouraging. Our findings once again confirm the practicality of enol oxidative coupling reactions in natural products and provide a new strategy for the synthesis of spiroaspertrione A. Further study for the total synthesis of spiroaspertrione A is underway in our laboratory.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
AM and ZS designed the project and wrote the manuscript. ZS, SL, JZ, XZ, JP, and JZ performed the experiments. All authors contributed to the article and approved the submitted version.
This work was supported by the Guangdong Province Rural Revitalization Strategy Special Fund (No. Jiangke [2021] 183), the Department of Education of Guangdong Province (2020KCXTD036 and 2021KQNCX101), the Hong Kong–Macao Joint Research and Development Fund of Wuyi University (2019WGALH12 and 2021WGALH12), the Science Foundation for Young Teachers (2019td02) of Wuyi University, the Jiangmen City Science and Technology Basic Research Project (2021030102630004945), and the Innovations in Graduate Education Program (YJS-SFJD-21–01).
We thank Liwen Bianji (Edanz) (www.liwenbianji.cn/) for basic language editing of a draft of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.1022533/full#supplementary-material
Baran, P. S., Richter, J. M., and Lin, D. W. (2005). Direct coupling of pyrroles with carbonyl compounds: Short enantioselective synthesis of (S)‐Ketorolac. Angew. Chem. 117, 615–618. doi:10.1002/ange.200462048
Baron, P. S. (2006). Enantioselective total synthesis of avrainvillamide and the stephacidins. J. Am. Chem. Soc. 128, 8678–8693. doi:10.1021/ja061660s
Bush, K. (2015). Synergistic MRSA combinations. Nat. Chem. Biol. 11, 832–833. doi:10.1038/nchembio.1935
Chen, W., and Liu, Q. (2021). Recent advances in the oxidative coupling reaction of enol derivatives. Chin. J. Org. Chem. 41, 3414–3430. doi:10.6023/cjoc202104058
Csákÿ, A. G., and Plumet, J. (2001). Stereoselective coupling of ketone and carboxylate enolates. Chem. Soc. Rev. 30, 313–320. doi:10.1039/B104000F
DeMartino, M. P., Chen, K., and Baran, P. S. (2008). Intermolecular enolate heterocoupling: Scope, mechanism, and application. J. Am. Chem. Soc. 130, 11546–11560. doi:10.1021/ja804159y
Dessau, R. M., and Heiba, E. A. I. (1974). Oxidation by metal salts. XII. Novel one-step synthesis of 1, 4-diketones. J. Org. Chem. 93, 3457–3459. doi:10.1021/jo00937a053
Ekebergh, A., Karlsson, I., Mete, R., Pan, Y., Börje, A., and Mårtensson, J. (2011). Oxidative coupling as a biomimetic approach to the synthesis of scytonemin. Org. Lett. 13, 4458–4461. doi:10.1021/ol201812n
Fuda, C., Hesek, D., Lee, M., Morio, K. I., Nowak, T., and Mobashery, S. (2005). Activation for catalysis of penicillin-binding protein 2a from methicillin-resistant Staphylococcus a ureus by bacterial cell wall. J. Am. Chem. Soc. 127, 2056–2057. doi:10.1021/ja0434376
Fujii, T., Hirao, T., and Ohshiro, Y. (1992). Oxovanadium-induced oxidative desilylation for the selective synthesis of 1, 4-diketones. Tetrahedron Lett. 33, 5823–5826. doi:10.1016/0040-4039(92)89041-A
Gonzales, P. R., Pesesky, M. W., Bouley, R., Ballard, A., Biddy, B. A., Suckow, M. A., et al. (2015). Synergistic, collaterally sensitive β-lactam combinations suppress resistance in MRSA. Nat. Chem. Biol. 11, 855–861. doi:10.1038/nchembio.1911
Guo, F., Clift, M. D., and Thomson, R. J. (2012). Oxidative coupling of enolates, enol silanes, and enamines: Methods and natural product synthesis. Eur. J. Org. Chem. 2012, 4881–4896. doi:10.1002/ejoc.201200665
He, Y., Hu, Z., Sun, W., Li, Q., Li, X. N., Zhu, H., et al. (2017). Spiroaspertrione A, a bridged spirocyclic meroterpenoid, as a potent potentiator of oxacillin against methicillin-resistant Staphylococcus aureus from Aspergillus sp. TJ23. J. Org. Chem. 82, 3125–3131. doi:10.1021/acs.joc.7b00056
Jones, B. T., Avetta, C. T., and Thomson, R. J. (2014). Total synthesis of propolisbenzofuran B. Chem. Sci. 5, 1794–1798. doi:10.1039/C4SC00356J
Kise, N., Tokioka, K., Aoyama, Y., and Matsumura, Y. (1995). Enantioselective synthesis of 2, 3-disubstituted succinic acids by oxidative homocoupling of optically active 3-acyl-2-oxazolidones. J. Org. Chem. 60, 1100–1101. doi:10.1021/jo00110a003
Kohno, Y., and Narasaka, K. (1995). Oxidative generation of α-radicals of carbonyl compounds from the α-stannyl derivatives and their reactions with electron-rich olefins. Bull. Chem. Soc. Jpn. 68, 322–329. doi:10.1246/bcsj.68.322
Konkol, L. C., Guo, F., Sarjeant, A. A., and Thomson, R. J. (2011). Enantioselective total synthesis and studies into the configurational stability of bismurrayaquinone A. Angew. Chem. Int. Ed. 50, 9931–9934. doi:10.1002/anie.201104726
Krawczuk, P. J., Schöne, N., and Baran, P. S. (2009). A synthesis of the carbon skeleton of maoecrystal V. Org. Lett. 11, 4774–4776. doi:10.1021/ol901963v
Li, Y. P., Li, Z. Q., Zhou, B., Li, M. L., Xue, X. S., Zhu, S. F., et al. (2019). Chiral spiro phosphoric acid-catalyzed friedel–crafts conjugate addition/enantioselective protonation reactions. ACS Catal. 9, 6522–6529. doi:10.1021/acscatal.9b01502
Long, S. W., Olsen, R. J., Mehta, S. C., Palzkill, T., Cernoch, P. L., Perez, K. K., et al. (2014). PBP2a mutations causing high-level ceftaroline resistance in clinical methicillin-resistant Staphylococcus aureus isolates. Antimicrob. Agents Chemother. 58, 6668–6674. doi:10.1128/AAC.03622-14
Martin, C. L., Overman, L. E., and Rohde, J. M. (2008). Total synthesis of (±)-actinophyllic acid. J. Am. Chem. Soc. 130, 7568–7569. doi:10.1021/ja803158y
Martin, C. L., Overman, L. E., and Rohde, J. M. (2010). Total synthesis of (±)-and (−)-actinophyllic acid. J. Am. Chem. Soc. 132, 4894–4906. doi:10.1021/ja100178u
Murarka, S., and Antonchick, A. P. (2018). Metal-catalyzed oxidative coupling of ketones and ketone enolates. Synthesis 50, 2150–2162. doi:10.1055/s-0037-1609715
Nagaraju, K., and Ma, D. (2018). Oxidative coupling strategies for the synthesis of indole alkaloids. Chem. Soc. Rev. 47, 8018–8029. doi:10.1039/C8CS00305J
Rathke, M. W., and Lindert, A. (1971). Reaction of ester enolates with copper (II) salts. Synthesis of substituted succinate esters. J. Am. Chem. Soc. 93, 4605–4606. doi:10.1021/ja00747a051
Renaud, P., and Fox, M. A. (1988). Reaction of dilithiated carboxylic acids with iodine: Evidence for the formation of a radical anion intermediate. J. Org. Chem. 53, 3745–3752. doi:10.1021/jo00251a015
Richter, J. M., Whitefield, B. W., Maimone, T. J., Lin, D. W., Castroviejo, M. P., and Baran, P. S. (2007). Scope and mechanism of direct indole and pyrrole couplings adjacent to carbonyl compounds: Total synthesis of acremoauxin A and oxazinin 3. J. Am. Chem. Soc. 129, 12857–12869. doi:10.1021/ja074392m
Robinson, E. E., and Thomson, R. J. (2018). A strategy for the convergent and stereoselective assembly of polycyclic molecules. J. Am. Chem. Soc. 140, 1956–1965. doi:10.1021/jacs.7b13234
Ryter, K., and Livinghouse, T. (1998). Dichloro (2, 2, 2-trifluoroethoxy) oxovanadium (V). A remarkably effective reagent for promoting one-electron oxidative cyclization and unsymmetrical coupling of silyl enol ethers. J. Am. Chem. Soc. 120, 2658–2659. doi:10.1021/ja973585e
Teng, M., Zi, W., and Ma, D. (2014). Total synthesis of the monoterpenoid indole alkaloid (±)-Aspidophylline A. Angew. Chem. Int. Ed. 53, 1814–1817. doi:10.1002/anie.201310928
Van Hal, S. J., Paterson, D. L., and Gosbell, I. B. (2011). Emergence of daptomycin resistance following vancomycin-unresponsive Staphylococcus aureus bacteraemia in a daptomycin-naïve patient—A review of the literature. Eur. J. Clin. Microbiol. Infect. Dis. 30, 603–610. doi:10.1007/s10096-010-1128-3
Walsh, T. R., Weeks, J., Livermore, D. M., and Toleman, M. A. (2011). Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: An environmental point prevalence study. Lancet Infect. Dis. 11, 355–362. doi:10.1016/S1473-3099(11)70059-7
Wei, Y., Zhao, D., and Ma, D. (2013). Total synthesis of the indole alkaloid (±)-and (+)-Methyl N-decarbomethoxychanofruticosinate. Angew. Chem. 125, 13226–13229. doi:10.1002/ange.201307788
Xie, J., and Huang, Z. Z. (2010). The cascade carbo-carbonylation of unactivated alkenes catalyzed by an organocatalyst and a transition metal catalyst: A facile approach to γ-diketones and γ-carbonyl aldehydes from arylalkenes under air. Chem. Commun. 46, 1947–1949. doi:10.1039/B921310D
Yeung, C. S., and Dong, V. M. (2011). Catalytic dehydrogenative cross-coupling: Forming carbon− carbon bonds by oxidizing two carbon-hydrogen bonds. Chem. Rev. 111, 1215–1292. doi:10.1021/cr100280d
You, L., Liang, X. T., Xu, L. M., Wang, Y. F., Zhang, J. J., Su, Q., et al. (2015). Asymmetric total synthesis of propindilactone G. J. Am. Chem. Soc. 137, 10120–10123. doi:10.1021/jacs.5b06480
Zi, W., Xie, W., and Ma, D. (2012). Total synthesis of akuammiline alkaloid (−)-vincorine via intramolecular oxidative coupling. J. Am. Chem. Soc. 134, 9126–9129. doi:10.1021/ja303602f
Zuo, Z., and Ma, D. (2011). Enantioselective total syntheses of communesins A and B. Angew. Chem. Int. Ed. 50, 12008–12011. doi:10.1002/anie.201106205
Keywords: studies, synthesis, core skeleton, spiroaspertrione A, natural product
Citation: Shen Z-H, Lu S-Y, Zheng J-Y, Zhang X-Z, Peng J-B and Ma A-J (2022) Studies toward synthesis of the core skeleton of spiroaspertrione A. Front. Chem. 10:1022533. doi: 10.3389/fchem.2022.1022533
Received: 18 August 2022; Accepted: 30 August 2022;
Published: 05 October 2022.
Edited by:
Xi Zheng, The State University of New Jersey, United StatesCopyright © 2022 Shen, Lu, Zheng, Zhang, Peng and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ai-Jun Ma, d3l1Y2hlbW1hakAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.