 Joshua D. Hartman
Joshua D. Hartman Amanda Mathews1
Amanda Mathews1- 1Department of Chemistry, Mt. San Jacinto College, Menifee, CA, United States
- 2Department of Chemistry and Biochemistry, Brigham Young University, Provo, UT, United States
Modern approaches for calculating electric field gradient (EFF) tensors in molecular solids rely upon plane-wave calculations employing periodic boundary conditions (PBC). In practice, models employing PBCs are limited to generalized gradient approximation (GGA) density functionals. Hybrid density functionals applied in the context of gauge-including atomic orbital (GIAO) calculations have been shown to substantially improve the accuracy of predicted NMR parameters. Here we propose an efficient method that effectively combines the benefits of both periodic calculations and single-molecule techniques for predicting electric field gradient tensors in molecular solids. Periodic calculations using plane-wave basis sets were used to model the crystalline environment. We then introduce a molecular correction to the periodic result obtained from a single-molecule calculation performed with a hybrid density functional. Single-molecule calculations performed using hybrid density functionals were found to significantly improve the agreement of predicted 17O quadrupolar coupling constants (Cq) with experiment. We demonstrate a 31% reduction in the RMS error for the predicted 17O Cq values relative to standard plane-wave methods using a carefully constructed test set comprised of 22 oxygen-containing molecular crystals. We show comparable improvements in accuracy using five different hybrid density functionals and find predicted Cq values to be relatively insensitive to the choice of basis set used in the single molecule calculation. Finally, the utility of high-accuracy 17O Cq predictions is demonstrated by examining the disordered 4-Nitrobenzaldehyde crystal structure.
1 Introduction
Solid-state nuclear magnetic resonance (SSNMR) spectroscopy has proven highly effective at characterizing molecular crystals. Advances in NMR hardware and the development of novel pulse sequences continue to improve the accuracy and availability of experimental data. However, mapping relationships between NMR observables and structural features remains a formidable challenge. SSNMR investigations are often coupled with X-ray diffraction and first-principals calculations to form the interdisciplinary field of NMR crystallography. The success of NMR crystallography has been greatly facilitated by the availability of accurate computational methods for predicting NMR parameters which typically employ density functional theory (DFT) (Gervais et al., 2005; Wu, 2008; Kong et al., 2013; Yang et al., 2016; Kong et al., 2017; Soss et al., 2017; Holmes and Schurko, 2018; Yamada et al., 2020; Chalek et al., 2021; Wang et al., 2021).
There are two broad classifications for DFT-based methods commonly applied to molecular crystals. First, the gauge-including projected augmented wave (GIPAW) method (Pickard and Mauri, 2001) employs plane-wave basis sets to naturally capture the periodic nature of the crystalline lattice. Alternatively, the gauge-including atomic orbital (GIAO) approach (Ditchfield, 1974; Wolinski et al., 1990) relies on fragments or clusters of molecules constructed to mimic the crystalline environment (Hartman and Beran, 2014; Holmes et al., 2014; Hartman et al., 2015). Plane-wave and GIAO-based calculations have both proven highly effective in modeling a range of NMR parameters derived from the chemical shielding (CS) tensor and the electric field gradient (EFG) (Nakajima, 2017; Holmes and Schurko, 2018; Dračínský et al., 2019; Gregorovič, 2020).
Plane-wave methods have a natural advantage over fragment and cluster-based calculations when predicting NMR parameters for molecular crystals due to the explicit quantum mechanical treatment of crystal lattice effects. However, plane-wave methods are limited in practice to density functionals based on the generalized gradient approximation (GGA). Numerous studies have shown that using hybrid density functionals improves the accuracy of predicted NMR parameters (Hartman and Beran, 2014; Holmes et al., 2014; Hartman et al., 2015; Hartman et al., 2017). Previous benchmark studies involving 1H, 13C, 15N and 51V nuclei have shown fragment methods with hybrid density functionals improve the accuracy of predicted isotropic chemical shifts (Hartman et al., 2016; Hartman et al., 2017; Mathews and Hartman, 2021). Fragment methods employing hybrid density functionals coupled with electrostatic embedding techniques have demonstrated improved accuracy in the prediction of CS tensor principal components (Hartman and Beran, 2018) and predicted Cq values for 14N (Gregorovič, 2020).
Recently, a novel approach involving GIPAW with a molecular correction (GIPAW + MC) was put forward for modeling the chemical shift tensor (Nakajima, 2017; Dračínský et al., 2019). This scheme combines the strengths of plane-wave and molecular calculations. The GIPAW + MC approach relies upon GGA-based GIPAW calculations to capture long-range effects and then introduces a correction obtained from a molecular calculation performed on an isolated gas-phase molecule. The geometry of the isolated molecule is taken from the optimized crystal structure. In this way, hybrid density functionals or even post-Hartree-Fock wave function methods can be used in the molecular calculation to more accurately model the intramolecular effects on the CS tensor. A more detailed description of the GIPAW + MC method applied to CS tensor predictions can be found in the literature (Nakajima, 2017; Dračínský et al., 2019; Bártová et al., 2020).
Applying the GIPAW + MC approach (vide infra) to CS tensors has proven particularly effective in modeling NMR parameters for the quadrupolar 17O nucleus (Dračínský et al., 2019). Specifically, GIPAW + MC calculations using the PBE0 hybrid density functional and a 6-311+G (2d,p) basis reduces the mean absolute error (MAE) for predicted 17O chemical shifts by 17% relative to GIPAW (Dračínský et al., 2019). Improving the accuracy of predicted NMR parameters for quadrupolar nuclei is of particular interest given that quadrupolar nuclei account for approximately 73% of NMR-active nuclei (Hamaed et al., 2010).
In addition to improving the accuracy of CS tensor predictions, monomer correction methods have proven successful in modeling the energetics of conformational polymorphs (Greenwell and Beran, 2020). Here we extend the GIPAW + MC model to EFG tensor calculations and apply GIPAW + MC tensor calculations to a benchmark set of 22 molecular crystals with a total of 46 unique 17O environments. We demonstrate a 31% improvement in the accuracy of predicted Cq values relative to traditional plane-wave methods with a negligible increase in computational cost. These findings are particularly promising for NMR crystallography applications given the crucial role EFG tensor predictions have come to play in crystal structure refinement (Holmes and Schurko, 2018; Yamada et al., 2020), understanding hydrogen bond properties (Kazuhiko et al., 2000; Samadi et al., 2008) and investigating chemical reactivity and dynamics (Ashbrook and Sneddon, 2014; Bernard et al., 2018).
In the following, benchmark data are employed to examine basis set convergence and the accuracy of the predicted Cq values for Pople, core-valence Dunning-type basis sets, and the pcs-n (n = 1–4) series of basis sets. The performance of six commonly used density functionals are compared to demonstrate uniform improvement in accuracy through the application of a range of hybrid density functionals in the molecular calculation. We examine the relative improvement in EFG and CS tensor GIPAW calculations through the application of a molecular correction. Finally, GIPAW + MC EFG calculations are shown to accurately predict Cq values for the disordered oxygen atoms in the 4-Nitrobenzaldehyde crystal structure.
2 Theory and Methods
The EFG tensor is obtained from the second spatial derivative of the electrostatic potential V resulting from the charge distribution surrounding the nucleus.
Following Eq. 1, the EFG tensor is a symmetric 3 × 3 tensor with zero trace. Diagonalization of the EFG tensor yields three principal components defined such that |V33|≥|V22|≥|V11|. The principal components of the EFG tensor are used to derive two NMR observables. First, the nuclear quadrupolar coupling constant Cq is obtained from V33 according to,
where e is the elementary charge, Q = −25.58 mb (Pyykkö, 2001) is the nuclear quadrupole moment for 17O, and h is Plank’s constant. Second, the asymmetry parameter ηq is obtained from the ratio of the difference in V11 and V22 to V33.
Minor structural changes can have a pronounced impact on both Cq and ηq. However, previous studies suggest that the impact of structural changes on the EFG tensor is highly local (Gregorovič, 2020), and the GIPAW + MC approach to EFG tensor calculations is motivated by this assumption. The GIPAW + MC EFG tensor (Vcorr) is constructed from three separate calculations as follows,
First, the EFG tensor is computed using a full plane-wave GIPAW calculation at the lower level of theory (typically PBE) to obtain
According to Eq. 4, the GIPAW + MC model treats intermolecular interactions within the crystalline lattice using plane-wave methods. Critical intramolecular effects are then included in the form of a molecular correction. Molecular calculations performed within the GIAO framework can readily accommodate a higher level of theory since both Vmol terms rely on isolated gas-phase molecule calculations. Finally, the corrected EFG tensor is then subject to diagonalization to obtain the principal components which are used along with Eqs 2, 3 to predict the NMR observables.
3 Computational Methods
Molecular crystals were selected for inclusion in the benchmark study based on the availability of high-quality X-ray diffraction data and experimental NMR data providing high-accuracy Cq values (see Table 1). In all cases, both the experimental diffraction and NMR data were obtained at room temperature. Structural data from diffraction studies are obtained from the Cambridge Structure Database (CSD) maintained by the Cambridge Crystallographic Data Center. The CSD reference codes and the experimental references for NMR data are provided in Table 2. The crystal structure for 4-Nitrobenzaldehyde (KAYSUY) with a disordered oxygen is used as an application (Wu et al., 2008).
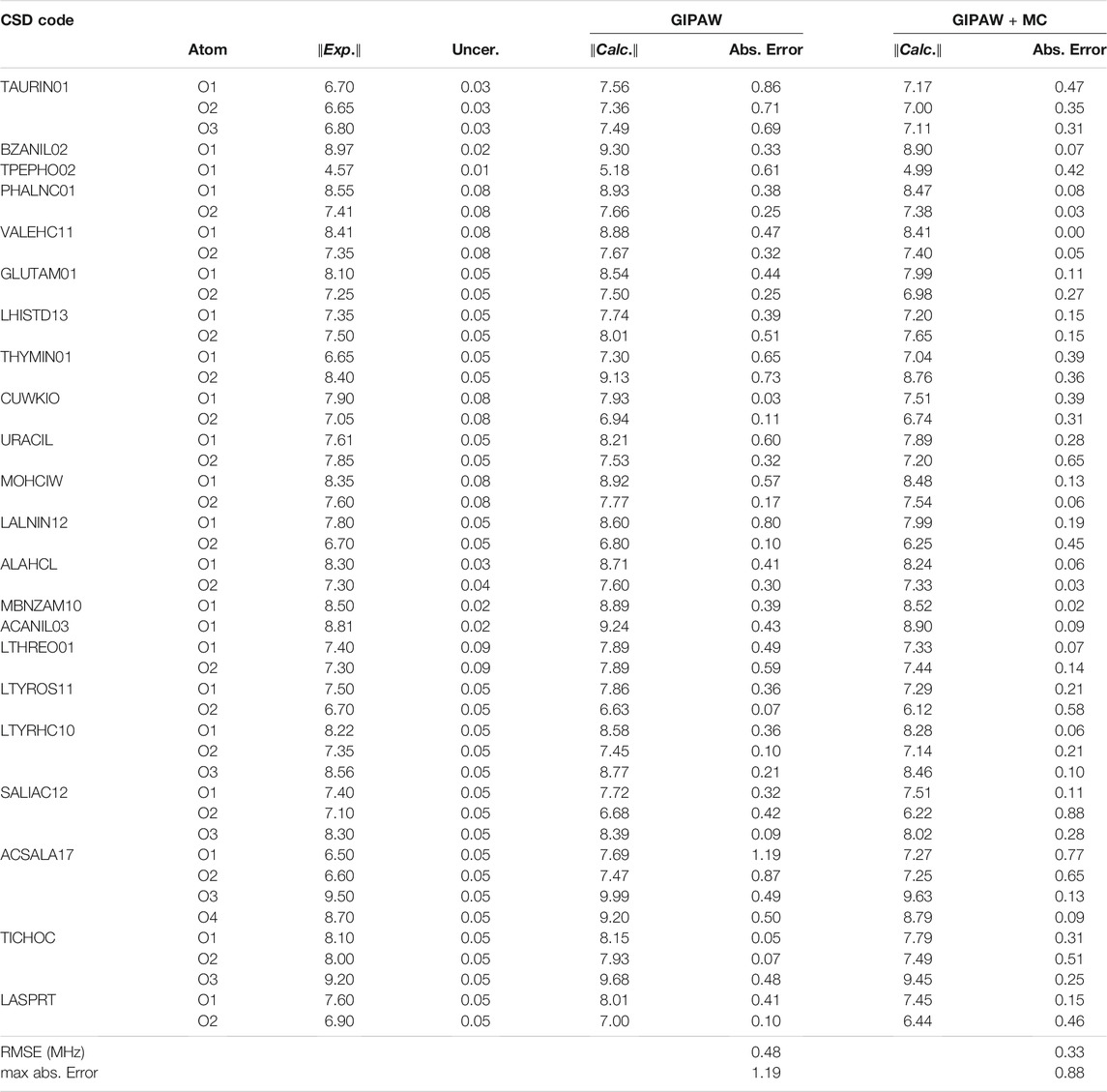
TABLE 1. Experimental17O Cq values with the reported uncertainty for each structure in the benchmark set. Calculated Cq for GIPAW and GIPAW + MC calculations provided along with the absolute errors. All values are reported in MHz and the GIPAW + MC calculations were performed with a PBE0/cc-pCVTZ correction.
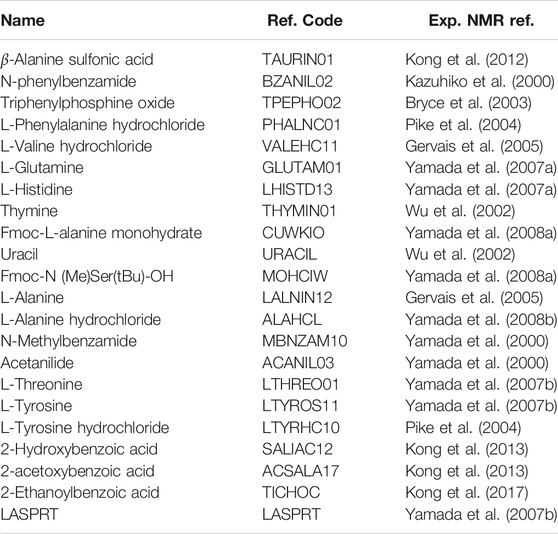
TABLE 2. Chemical name, CSD reference code, and experimental NMR reference for all crystal structures included in the17O benchmark study.
3.1 Crystal Structure Optimization
The experimental X-ray diffraction structures were used as a starting point for all-atom geometry optimizations subject to fixed experimental room-temperature lattice parameters. All-atom geometry optimization was carried out using dispersion-corrected DFT with the D3 dispersion correction (Grimme et al., 2010) and a maximum k-point spacing of 0.05 Å −1. The open-source Quantum Espresso (Giannozzi et al., 2009) software package, the PBE density functional, and an 80 Ry plane-wave cutoff were employed for the geometry optimizations. The following ultrasoft pseudopotentials were used: H.pbe-rrkjus.UPF, C.pbe-rrkjus.UPF, N.pbe-rrkjus.UPF, O.pbe-rrkjus.UPF, S.pbe-n-rrkjus_psl.0.1.UPF, Cl.pbe-n-rrkjus_psl.0.1.UPF, P.pbe-n-rrkjus_psl.0.1.UPF. All pseudopotentials used in the present work can be obtained from http://www.quantum-espresso.org.
3.2 EFG Tensor Calculations
Gauge-including projector augmented wave (GIPAW) chemical shielding calculations were performed using the optimized geometries. Calculations were performed using CASTEP (Clark et al., 2005) with the PBE density functional, ultrasoft pseudopotentials generated on-the-fly, and an 850 eV plane-wave basis set cut-off. Sampling for k-points was performed on a Monkhorst–Pack grid to give a maximum separation between k-points of 0.05 Å−1. These parameters were chosen based on previous benchmark studies involving quadrupolar nuclei (Hartman et al., 2016; Mathews and Hartman, 2021). Full space group symmetry was used in all GIPAW calculations.
EFG tensor calculations for the isolated molecules were carried out using Gaussian09 (Frisch et al., 1984) and the PBE0 (Adamo and Barone, 1999), PBE (Perdew et al., 1996), B3LYP (Stephens et al., 1994), TPSSh (Staroverov et al., 2003), ωB97XD (Chai and Head-Gordon, 2008), and mPW1PW91 (Adamo and Barone, 1998) density functionals. A large DFT integration grid consisting of 150 radial and 974 Lebedev angular points was selected on the basis of previous work (Hartman and Beran, 2014). The large integration grid approaches rotational invariance thereby reducing noise in the monomer calculations. To explore basis set dependence three classes of basis sets were employed. The Pople basis set 6-311+G (2d,p) (Frisch et al., 1984; Clark et al., 1983) was used to facilitate direct comparison with previous work (Hartman et al., 2016; Dračínský et al., 2019; Mathews and Hartman, 2021). The Dunning-type core-valance basis sets (Woon and Dunning, 1995), were used to examine the impact of tight higher angular momentum basis functions. Finally, the pcs-n (n = 1–4) basis sets were used to determine if the accuracy of predicted Cq values could be improved using basis sets optimized for predicting NMR parameters. The Dunning-type and pcs-n basis sets where obtained from the basis set exchange (https://bse.pnl.gov/bse/portal) (Pritchard et al., 2019).
4 Results and Discussion
4.1 17O Quadrupole Coupling Constants
We compare the accuracy of GIPAW + MC predicted 17O EFG tensor parameters with those obtained experimentally from NMR and NQR spectroscopy. We have selected the quadrupolar 17O nucleus for two reasons. First, 17O has tremendous biological and pharmaceutical importance. Second, previous GIPAW + MC studies involving the CS tensor showed the largest magnitude improvement in predicted isotropic shieldings for 17O relative to other second-row nuclei (Dračínský et al., 2019). These findings are consistent with previous fragment-based investigations which showed predicted isotropic shieldings for 17O to be more sensitive to long-range electrostatic effects relative to hydrogen and nitrogen (Hartman et al., 2016; Hartman et al., 2017).
We have selected 22 crystal structures with 46 unique 17O environments to benchmark the accuracy of GIPAW + MC EFG tensor predictions. The experimental Cq values in the benchmark set range from 4.57 to 9.50 MHz with an experimental uncertainty ≤0.09 MHz. Table 1 provides a complete list of all structures included in the benchmark analysis along with the experimental Cq values and the reported uncertainty. The crystal structures with labeled oxygen atoms are depicted in the SI. Table 1 also provides the predicted Cq values for traditional plane-wave calculations (GIPAW) and those obtained from the GIPAW + MC calculations. In most cases, the sign of Cq cannot be determined from the NMR experiment therefore we provide the magnitudes of the predicted Cq values and report the absolute error (in MHz) relative to experiment.
Table 1 establishes improved accuracy in the predicted Cq values obtained from GIPAW + MC calculations relative to traditional GIPAW. Specifically, introducing a molecular correction carried out at the PBE0/cc-pCVTZ level reduces the RMS error by 31% and reduced the maximum absolute error by 26%. The choice of density functional and basis set used in the preliminary analysis was motivated by previous studies (Harbison, 2015; Dračínský et al., 2019). In the following sections we thoroughly examine basis set convergence and the relative performance of different hybrid density functionals.
4.2 Basis Set Convergence
Previous studies applying the GIPAW + MC approach to CS tensor calculations on second-row nuclei found the method to be relatively insensitive to the choice of basis set used in the molecular correction (Dračínský et al., 2019). However, the regression model used to compare the absolute shieldings obtained from CS tensor calculations to the experimental chemical shifts partially corrects for systematic error. Unlike the CS tensor, comparing predicted EFG tensors (Eqs 2, 3) with experiment does not involve regression and therefore does not benefit from systematic error correction. In addition, previous studies involving GIAO-based EFG tensor predictions demonstrated improved accuracy in the predicted Cq values through the introduction of tight d functions (Harbison, 2015). Therefore, care must be taken to ensure the predicted EFG tensor components are well-converged with respect to basis set.
Figure 1 illustrates the error distributions for the predicted Cq values and the corresponding RMS error for GIPAW + MC calculations employing the PBE0 hybrid density functional in the molecular correction. Figure 1 provides error distributions for all three classes of basis functions and the corresponding data is provided for traditional plane-wave calculations (GIPAW) in purple. The pCVTZ basis results shown in orange correspond to the data presented in Table 1. The 17O Cq error distributions, RMSE, and maximum absolute errors yield comparable performance across the different classes of basis sets. The PBE0 molecular corrections provide ∼30% improvement in RMS error relative to GIPAW. In all cases, the GIPAW + MC results are well-converged at the double-ζ level.
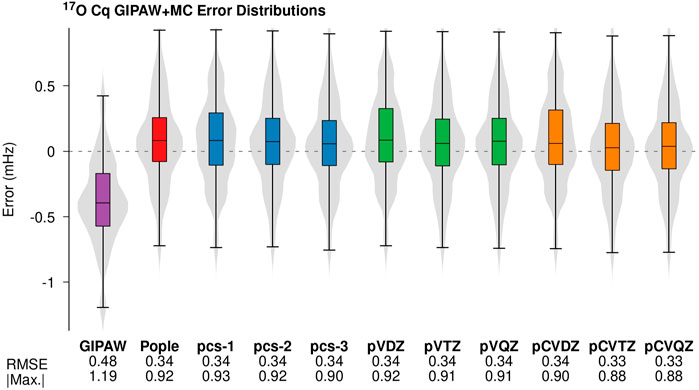
FIGURE 1. Errors in the 17O Cq predictions from PBE/GIPAW (purple) and GIPAW + MC calculations with PBE0 monomer corrections performed using different Gaussian basis sets. The PBE0/Pople basis (red) corresponds to the data in Table 1. The violin plots illustrate kernel density estimates for each error distribution. The box-plots within each violin provide the median (black horizontal line), middle two quartiles (colored box), and the whiskers represent the outer quartiles. The corresponding RMS error and maximum absolute errors are provided below each distribution. All values are reported in MHz.
Predicted Cq values rely on the largest magnitude principal component of EFG tensor (V33). To ensure convergence extends to all principal components we examine the deviation in all three principal components obtained from PBE0/pcs-n EFG tensor predictions for n = 1–3 relative to pcs-4. Figure 2 illustrates the RMS deviation in |V33| (red), |V22| (blue), and |V11| (green) relative to pcs-4 calculations applied to the benchmark set. The largest RMS deviations were observed for the largest EFG tensor component, V33, followed by V22, and V11 yields the smallest deviation. Together, Figures 1, 2 show EFG tensor predictions to be well-converged using standard triple-ζ basis sets. These findings are in agreement with previous work involving Dunning-type basis sets (Wu et al., 2008).
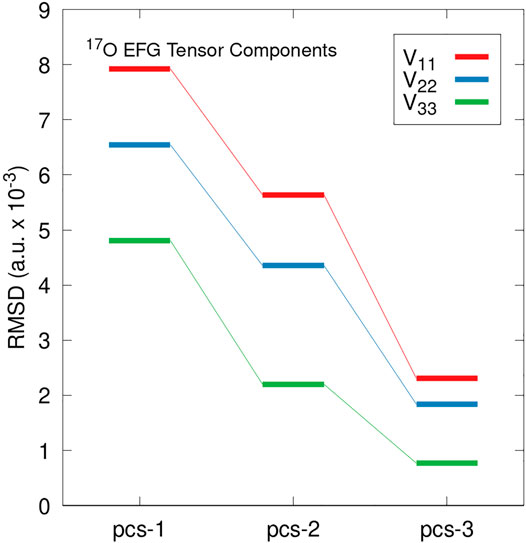
FIGURE 2. RMS deviations in predicted EFG tensor principal components relative to PBE0/pcs-4 calculations. Deviations in the |V33| component are shown in red, |V22| in blue, and |V11| in green. Values are reported in atomic units with a scaling factor of 10–3.
Figures 1, 2 suggest 17O Cq GIPAW + MC predictions employing DFT methods are relatively insensitive to the choice of basis set. The Dunning-type core-valance basis sets which include tight higher angular momentum basis functions (orange in Figure 1) do show a slight reduction (∼0.02 MHz) in the maximum absolute error. However, this improvement is equal to the average experimental uncertainty of the benchmark set. This result is surprising given previous results employing wave function-based correlation methods that showed improved accuracy in the predicted Cq values through the introduction of tight d functions (Harbison, 2015). Extending the GIPAW + MC model to wave function methods with custom Gaussian basis sets is a topic of an ongoing investigation.
4.3 Relative Performance of Density Functionals
The accuracy of predicted NMR parameters have been shown to vary substantially with different density functionals (Holmes et al., 2015). However, comparable performance is often observed within a given class of density functionals (Hartman et al., 2016). The results in the previous section establish the improved accuracy in Cq predictions through the introduction of a molecular correction based on the PBE0 hybrid density functional. In this section, we examine the relative performance of five other commonly used density functionals.
Figure 3 provides the error distributions associated with GIPAW + MC 17O Cq predictions using six common density functionals. GIPAW/PBE results are included in purple for comparison. A previous study identified the OPBE density functional as the best GGA-based density functional for predicting 13C, 15N, 17O, and 19F chemical shifts (Zhang et al., 2006). More recently, a benchmark study applying fragment methods to the prediction of isotropic 17O chemical shieldings in molecular crystals showed a slight improvement in the accuracy of OPBE relative to the PBE density functional (Hartman et al., 2016). Comparing the RMSE and maximum absolute error for GIPAW/PBE and GIPAW + MC/OPBE in Figure 3 suggests that the improved performance for OPBE in terms of predicting isotropic chemical shieldings does not translate to 17O Cq calculations.
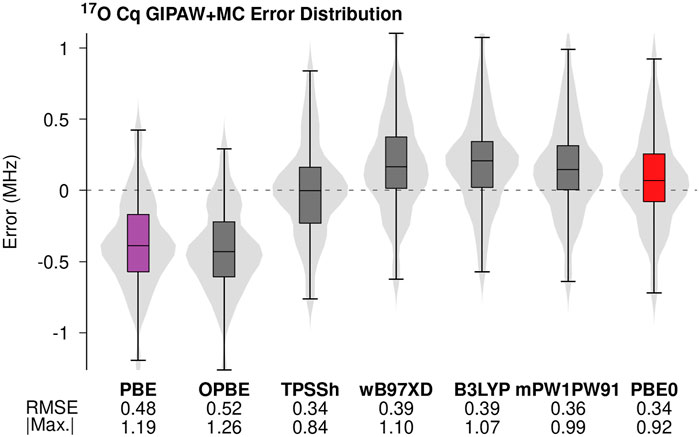
FIGURE 3. Errors in the predicted 17O Cq values for GIPAW and GIPAW + MC calculations were performed using a selection of commonly used density functionals. The molecular corrections were performed using the 6-311+G (2d,p) basis.
As expected, molecular corrections carried out using hybrid density functionals (TPSSh, wB97XD, mPW1PW91, B3LYP, and PBE0) uniformly improve the accuracy of predicted 17O Cq values relative to both GGA-based predictions. Recent work suggests double-hybrid density functionals further improves the accuracy of NMR parameter predictions (Stoychev et al., 2018). Extending the GIPAW + MC analysis to include double-hybrid density functionals is a topic of ongoing investigation. The RMS errors obtained for the different hybrid density functionals agree to within the maximum experimental uncertainty. However, the meta-hybrid TPSSh does show a small improvement in the maximum absolute error relative to the other density functionals. The hybrid density functional PBE0 (red in Figure 3) yields the second-lowest maximum absolute error. Once again, these findings represent a small deviation from previous studies involving 17O isotropic shielding calculations in which PBE0 and B3LYP predictions improved the accuracy relative to TPSSh (Hartman et al., 2016). Minor deviations aside, the trends in density functional choice illustrated in Figure 3 support the general consensus that hybrid density functionals improve the accuracy of predicted NMR parameters relative to GGA (Holmes et al., 2014; Holmes et al., 2015; Hartman et al., 2016; Hartman et al., 2017; Hartman and Beran, 2018).
4.4 Accuracy of Predicted 17O CS and EFG Tensors
Predicted 17O NMR parameters are highly sensitive to crystalline lattice effects (Hartman et al., 2016; Hartman et al., 2017). Previous work involving fragment-based methods coupled with electrostatic embedding have shown improved accuracy in the predicted isotropic shieldings for 1H, 13C, 15N, and 51V relative to GIPAW/PBE when hybrid density functionals are employed (Hartman et al., 2015; Hartman et al., 2016; Mathews and Hartman, 2021). However, GIPAW/PBE calculations yield more accurate 17O isotropic chemical shift predictions compared to fragment models with hybrid density functionals and self-consistent embedding (Hartman et al., 2017). Additionally, comparing predicted isotropic chemical shifts obtained from GIPAW + MC and GIPAW calculations shows a larger improvement in accuracy for 17O nuclei relative to both 13C and 15N (Dračínský et al., 2019). Specifically, GIPAW + MC improves the RMS error by 27% and reduces the maximum error by 26% relative to GIPAW/PBE (Dračínský et al., 2019). There findings suggest faithful reproduction of crystal lattice effects are essential for high-accuracy 17O NMR parameter prediction.
Based on the success in predicting 17O EFG parameters using GIPAW + MC, this approach was employed to also evaluate the accuracy of GIPAW + MC in predicting 17O CSA tensor components. This analysis represents the first benchmark study examining the accuracy of predicted 17O CSA tensor components obtained from GIPAW + MC calculations. We compare the accuracy of predicted 17O isotropic shieldings and CSA tensor elements with predicted Cq values for both GIPAW and GIPAW + MC. Because all three types of experimental data are not available for each benchmark compound, different numbers of compounds are included in each comparison. Specifically, the Cq error distribution and isotropic shielding data both include all 22 crystal structures in the benchmark set. The CSA tensor data includes 21 structures. To facilitate comparison with previous studies, the molecular correction was performed at the PBE0/6-311+G (2d,p) level and standard linear regression methods were used to map the predicted absolute shieldings to experiment. The details of the 17O regression models along with the experimental and calculated CS tensor data are provided in the supporting information.
Figure 4 illustrates the percent improvement in the RMS error (red) and maximum absolute error (blue) for GIPAW + MC calculations relative to GIPAW. We compare the percent improvement for the predicted 17O Cq values, isotropic shieldings (σiso) and the CSA tensor elements (σii). The RMS error for the 17O isotropic chemical shift predictions is 11.53 and 9.35 ppm for GIPAW and GIPAW + MC, respectively. This corresponds to a 19% relative improvement for GIPAW + MC which is in agreement with previous work (Dračínský et al., 2019). However, GIPAW + MC improves the maximum absolute error by only 2%. Similarly, GIPAW + MC improves the RMS error for the 17O CSA tensor elements by 4% (19.88 ppm for GIPAW compared to 18.99 ppm for GIPAW + MC). These results are influenced by the larger variation in experimental uncertainties for isotropic shifts and CSA tensor elements (see the Supplementary Material for details). Nevertheless, the roughly two-fold increase in RMS error for CSA tensor elements relative to isotropic shifts is in general agreement with previous results for 13C and 15N (Hartman et al., 2016; Hartman and Beran, 2018).
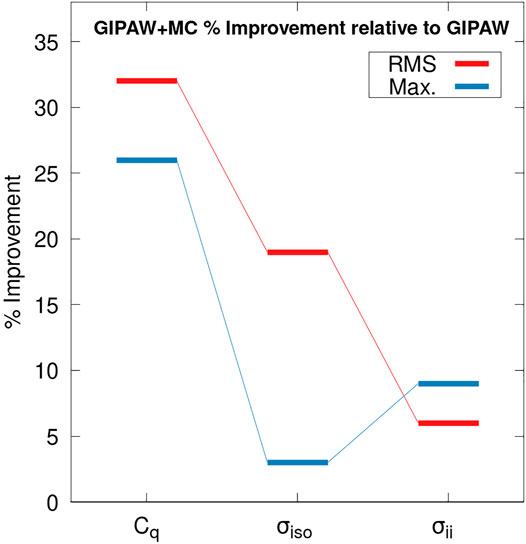
FIGURE 4. Percent improvement in the RMS error (red) and the maximum absolute error (blue) for GIPAW + MC calculations relative to GIPAW/PBE. Results are reported for the 17O quadrupole coupling constant (Cq), the isotropic chemical shift (σiso), and the CSA tensor components (σii). The molecular correction for the GIPAW + MC calculations were performed at the PBE0/6–311+G (2d,p) level.
Interestingly, the accuracy of GIPAW + MC 17O Cq predictions show a larger percent improvement relative to GIPAW compared with both σiso and σii. In other words, the molecular correction has a more pronounced impact on improving the accuracy of predicted EFG tensors compared to the CS tensor. These results support previous findings which suggest the EFG tensor is a highly local property (Michaelis et al., 2015; Gregorovič, 2020). Unlike the Cq values, the isotropic chemical shift and CSA tensor predictions benefit from a partial correction of systematic error through the application of a regression model discussed in the supporting information. Comparing the percent improvement between the different properties partially accounts for this difference. Nevertheless, care should be exercised when interpreting Figure 4.
4.5 Modeling Disorder in the 4-Nitrobenzaldehyde Crystal Structure
NMR parameters derived from the CS and EFG tensors have been used extensively in molecular structure refinement (Olsen et al., 2003; Witter et al., 2006; Harris et al., 2010; Apperley et al., 2012; Harper et al., 2013; Kalakewich et al., 2015; Yang et al., 2016; Soss et al., 2017; Chalek et al., 2021; Wang et al., 2021). Recently, plane-wave EFG tensor predictions involving 14N, 17O, and 35Cl nuclei were used to obtain optimized damping parameters for crystal geometry refinement using Grimme’s DFT-D2 scheme (Holmes and Schurko, 2018). These findings suggest that high-accuracy NMR calculations can be used to help design geometry optimization protocols. Here we examine the sensitivity of 17O NMR parameters to subtle changes in geometry using the disordered crystal structure of 4-Nitrobenzaldehyde.
Disorder is reported in the O3 position for the crystal structure of 4-Nitrobenzaldehyde. The two diffraction structures differ by a 180-degree rotation around the bond between the ring and carbonyl carbon. Figure 5 depicts the unit cell for both crystal structures along with the GIPAW + MC 17O Cq predictions. Experimental evidence suggests structure A in Figure 5 is the dominant form, with a 90% occupancy for O3. The oxygen labeled O3’ in structure B has a 10% occupancy (Jackisch et al., 1989). Table 3 provides the experimental 17O Cq value (Wu et al., 2008) and the predicted Cq values for both structures obtained using GIPAW and GIPAW + MC calculations. The final column in Table 3 provides the weighted average of the predicted Cq values based on the experimental occupancy.
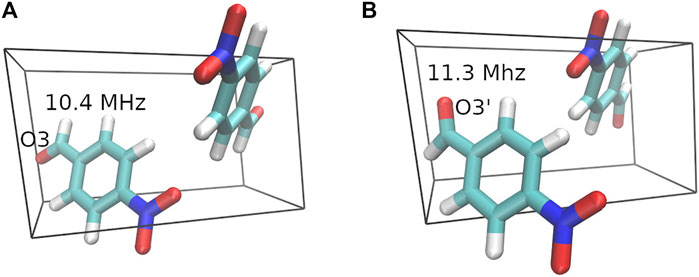
FIGURE 5. Crystal structures for the two 4-Nitrobenzaldehyde geometries. The dominant crystal structure (A) with a 90% occupancy is shown on the left and structure (B) with a 10% occupancy is shown on the right. The 17O Cq predictions obtained from GIPAW + MC calculations with a PBE0/6-311+G (2d,p) molecular correction are included in the figure.
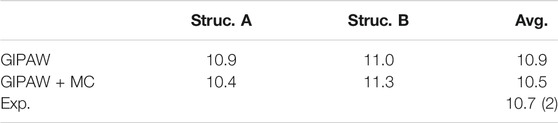
TABLE 3. Experimental and predicted17O Cq values for 4-Nitrobenzaldehyde in MHz. GIPAW + MC results obtained using a PBE0/6-311+G (2d,p) correction.
The predicted 17O quadrupole couplings for structure A from both GIPAW and GIPAW + MC are closer to the experimental value of 10.7 MHz, which agrees with the experimentally derived occupancies. The weighted average Cq predictions for both methods reproduce the experimental value to within the experimental uncertainty (0.2 MHz). However, the GIPAW Cq predictions in Table 3 show that both structures overshoot the experimental value. On the other hand, GIPAW + MC provides Cq predictions which bracket the experimental value. This results in a weighted average of the GIPAW + MC predicted Cq values which more closely matches experiment relative to either structure examined in isolation.
5 Conclusion
In summary, the present work establishes the GIPAW + MC method as a simple yet powerful approach for improving the accuracy of traditional plane-wave EFG tensor calculations. Introducing a correction based on the EFG tensor computed on an isolated monomer using a hybrid density functional substantially improves the accuracy of GIPAW calculations. The molecular correction is relatively insensitive to the choice of basis set, ensuring the cost of the molecular correction is a small fraction of the corresponding GIPAW calculation. In addition to improving the accuracy of predicted Cq values, we have shown the molecular correction improves the accuracy of predicted 17O CSA tensor elements. Comparing the relative improvement obtained through introducing a molecular correction to both EFG and CS tensor predictions we find a larger improvement in the accuracy of predicted EFG tensors. Finally, we apply GIPAW + MC EFG tensor calculations to the disordered crystal structure of 4-Nitrobenzaldehyde and show the molecular correction improves resolution between the different crystal geometries present in the disordered crystal (Stoychev et al., 2018).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
JoH developed the GIPAW + MC method and performed the computations. JoH prepared the figures and wrote the first draft of the manuscript. JH, AM, and JaH developed the benchmark set of 17-O-containing structures and contributed to the final version of the manuscript.
Funding
This work was supported by the National Science Foundation under CHE-2016185 to JaH. The Mt. San Jacinto College Foundation assisted with publication costs.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Prof. Gregory Beran at the University of California, Riverside for numerous helpful discussions and access to computational resources. We also thank the MSJC information technology department for its access to computational resources.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.751711/full#supplementary-material
References
Adamo, C., and Barone, V. (1998). Exchange Functionals with Improved Long-Range Behavior and Adiabatic Connection Methods without Adjustable Parameters: The mPW and mPW1PW Models. J. Chem. Phys. 108, 664–675. doi:10.1063/1.475428
Adamo, C., and Barone, V. (1999). Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 110, 6158–6170. doi:10.1063/1.478522
Apperley, D. C., Batsanov, A. S., Clark, S. J., Harris, R. K., Hodgkinson, P., and Jochym, D. B. (2012). Computation of Magnetic Shielding to Simultaneously Validate a crystal Structure and Assign a Solid-State NMR Spectrum. J. Mol. Struct. 1015, 192–201. doi:10.1016/j.molstruc.2011.10.024
Ashbrook, S. E., and Sneddon, S. (2014). New Methods and Applications in Solid-State NMR Spectroscopy of Quadrupolar Nuclei. J. Am. Chem. Soc. 136, 15440–15456. doi:10.1021/ja504734p
Bártová, K., Císařová, I., Lyčka, A., and Dračínský, M. (2020). Tautomerism of Azo Dyes in the Solid State Studied by 15N, 14N, 13C and 1H NMR Spectroscopy, X-ray Diffraction and Quantum-Chemical Calculations. Dyes Pigm. 178, 108342. doi:10.1016/j.dyepig.2020.108342
Bernard, G. M., Wasylishen, R. E., Ratcliffe, C. I., Terskikh, V., Wu, Q., Buriak, J. M., et al. (2018). Methylammonium Cation Dynamics in Methylammonium Lead Halide Perovskites: A Solid-State NMR Perspective. J. Phys. Chem. A. 122, 1560–1573. doi:10.1021/acs.jpca.7b11558
Bryce, D. L., Eichele, K., and Wasylishen, R. E. (2003). An 17O NMR and Quantum Chemical Study of Monoclinic and Orthorhombic Polymorphs of Triphenylphosphine Oxide. Inorg. Chem. 42, 5085–5096. doi:10.1021/ic020706p
Chai, J. D., and Head-Gordon, M. (2008). Long-range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 10, 6615–6620. doi:10.1039/b810189b
Chalek, K. R., Dong, X., Tong, F., Kudla, R. A., Zhu, L., Gill, A. D., et al. (2021). Bridging Photochemistry and Photomechanics with NMR Crystallography: the Molecular Basis for the Macroscopic Expansion of an Anthracene Ester Nanorod. Chem. Sci. 12, 453–463. doi:10.1039/d0sc05118g
Clark, S. J., Segall, M. D., Pickard, C. J., Hasnip, P. J., Probert, M. I., Refson, K., et al. (2005). First Principles Methods Using CASTEP. Z. Kristallogr, 220. 567–570. [Dataset]Frisch MJ. doi:10.1524/zkri.220.5.567.65075
Clark, T., Chandrasekhar, J., Spitznagel, G. W., and Schleyer, v. R. P. (1983). Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III.* the 3-21+G Basis Set for First-Row Elements. Li-f. J. Comp. Chem. 4, 294–301. doi:10.1002/jcc.540040303
Ditchfield, R. (1974). A Gauge-Invariant LCAO Method for N.M.R. Chemical Shifts. Mol. Phys. 27, 789. doi:10.1080/00268977400100711
Dračínský, M., Unzueta, P., and Beran, G. J. (2019). Improving the Accuracy of Solid-State Nuclear Magnetic Resonance Chemical Shift Prediction with a Simple Molecular Correction. Phys. Chem. Chem. Phys. 21, 14992–15000. doi:10.1039/c9cp01666j
Frisch, M. J., Pople, J. A., and Binkley, J. S. (1984). Self-consistent Molecular Orbital Methods 25. Supplementary Functions for Gaussian Basis Sets. J. Chem. Phys. 80, 3265–3269. doi:10.1063/1.447079
Gervais, C., Dupree, R., Pike, K. J., Bonhomme, C., Profeta, M., Pickard, C. J., et al. (2005). Combined First-Principles Computational and Experimental Multinuclear Solid-State NMR Investigation of Amino Acids. J. Phys. Chem. A. 109, 6960–6969. doi:10.1021/jp0513925
Giannozzi, P., Baroni, S., Bonini, N., Calandra, M., Car, R., Cavazzoni, C., et al. (2009). Quantum Espresso: a Modular and Open-Source Software Project for Quantum Simulations of Materials. J. Phys. Cond. Mat. 21, 395502. doi:10.1088/0953-8984/21/39/395502
Greenwell, C., and Beran, G. J. (2020). Inaccurate Conformational Energies Still Hinder Crystal Structure Prediction in Flexible Organic Molecules. Cryst. Growth Des. 20, 4875–4881. doi:10.1021/acs.cgd.0c00676
Gregorovič, A. (2020). The many-body Expansion Approach to Ab Initio Calculation of Electric Field Gradients in Molecular Crystals. J. Chem. Phys. 152. doi:10.1063/1.5144735
Grimme, S., Antony, J., Ehrlich, S., and Krieg, H. (2010). A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 132. doi:10.1063/1.3382344
Hamaed, H., Ye, E., Udachin, K., and Schurko, R. W. (2010). Solid-State 137Ba NMR Spectroscopy: An Experimental and Theoretical Investigation of 137Ba Electric Field Gradient Tensors and Their Relation to Structure and Symmetry. J. Phys. Chem. B 114, 6014–6022. doi:10.1021/jp102026m
Harbison, G. S. (2015). Polarization of Core Orbitals and Computation of Nuclear Quadrupole Coupling Constants Using Gaussian Basis Sets. J. Magn. Reson. 257, 24–31. doi:10.1016/j.jmr.2015.05.002
Harper, J. K., Iuliucci, R., Gruber, M., and Kalakewich, K. (2013). Refining crystal Structures with Experimental 13C NMR Shifts Tensors and Lattice-Including Electronic Structure Methods. Cryst. Eng. Comm. 15, 8693–8704. doi:10.1039/c3ce40108a
Harris, R. K., Hodgkinson, P., Zorin, V., Dumez, J. N., Herrmann, B. E., Emsley, L., et al. (2010). Computational NMR and Crystallography of Terbutaline Sulfate. Magn. Reason. Chem. 48, S103–S112. doi:10.1002/mrc.2636
Hartman, J., and Beran, G. (2018). Accurate 13-C and 15-N Molecular Cyrstal Chemical Shielding Tensors from Fragment-Based Electronic Sturcture Theory. Solid State. Nuc. Mang. Res. 96, 10–18. doi:10.1016/j.ssnmr.2018.09.003
Hartman, J. D., Balaji, A., and Beran, G. J. O. (2017). Improved Electrostatic Embedding for Fragment-Based Chemical Shift Calculations in Molecular Crystals. J. Chem. Theor. Comput. 13, 6043–6051. doi:10.1021/acs.jctc.7b00677
Hartman, J. D., and Beran, G. J. O. (2014). Fragment-based Electronic Structure Approach for Computing Nuclear Magnetic Resonance Chemical Shifts in Molecular Crystals. J. Chem. Theor. Comput. 10, 4862–4872. doi:10.1021/ct500749h
Hartman, J. D., Kudla, R. A., Day, G. M., Mueller, L. J., and Beran, G. J. O. (2016). Benchmark Fragment-Based 1H, 13C, 15N and 17O Chemical Shift Predictions in Molecular Crystals. Phys. Chem. Chem. Phys. 18, 21686–21709. doi:10.1039/C6CP01831A
Hartman, J. D., Monaco, S., Schatschneider, B., and Beran, G. J. O. (2015). Fragment-based 13C Nuclear Magnetic Resonance Chemical Shift Predictions in Molecular Crystals: An Alternative to Planewave Methods. J. Chem. Phys. 143, 102809. doi:10.1063/1.4922649
Holmes, S. T., Iuliucci, R. J., Mueller, K. T., and Dybowski, C. (2015). Critical Analysis of Cluster Models and Exchange-Correlation Functionals for Calculating Magnetic Shielding in Molecular Solids. J. Chem. Theor. Comput. 11, 5229–5241. doi:10.1021/acs.jctc.5b00752
Holmes, S. T., Iuliucci, R. J., Mueller, K. T., and Dybowski, C. (2014). Density Functional Investigation of Intermolecular Effects on 13C NMR Chemical-Shielding Tensors Modeled with Molecular Clusters. J. Chem. Phys. 141, 164121. doi:10.1063/1.4900158
Holmes, S. T., and Schurko, R. W. (2018). Refining Crystal Structures with Quadrupolar NMR and Dispersion-Corrected Density Functional Theory. J. Phys. Chem. C 122, 1809–1820. doi:10.1021/acs.jpcc.7b12314
Jackisch, M. A., Fronczek, F. R., and Butler, L. G. (1989). Structure of 4-nitrobenzaldehyde. Acta Crystallogr. Sect. C 45, 2016–2018. doi:10.1107/S0108270189008528
Kalakewich, K., Iuliucci, R., Mueller, K. T., Eloranta, H., and Harper, J. K. (2015). Monitoring the Refinement of crystal Structures with 15N Solid-State NMR Shift Tensor Data. J. Chem. Phys. 143, 1–10. doi:10.1063/1.4935367
Kazuhiko, Y., Shuan, D., and Wu, G. (2000). Solid-state 17O NMR Investigation of the Carbonyl Oxygen Electric-Field-Gradient Tensor and Chemical Shielding Tensor in Amides. J. Am. Chem. Soc. 122, 11602–11609. doi:10.1021/ja0008315
Kong, X., Dai, Y., and Wu, G. (2017). Solid-state 17O NMR Study of 2-acylbenzoic Acids and Warfarin. Solid State. Nucl. Magn. Reson. 84, 59–64. doi:10.1016/j.ssnmr.2016.12.011
Kong, X., O’Dell, L. A., Terskikh, V., Ye, E., Wang, R., and Wu, G. (2012). Variable-temperature 17O NMR Studies Allow Quantitative Evaluation of Molecular Dynamics in Organic Solids. J. Am. Chem. Soc. 134, 14609–14617. doi:10.1021/ja306227p
Kong, X., Shan, M., Terskikh, V., Hung, I., Gan, Z., and Wu, G. (2013). Solid-state 17O NMR of Pharmaceutical Compounds: Salicylic Acid and Aspirin. J. Phys. Chem. B 117, 9643–9654. doi:10.1021/jp405233f
Mathews, A., and Hartman, J. D. (2021). Accurate Fragment-Based 51-V Chemical Shift Predictions in Molecular Crystals. Solid State. Nucl. Magn. Reson. 114, 101733. doi:10.1016/j.ssnmr.2021.101733
Michaelis, V. K., Keeler, E. G., Ong, T. C., Craigen, K. N., Penzel, S., Wren, J. E., et al. (2015). Structural Insights into Bound Water in Crystalline Amino Acids: Experimental and Theoretical 17O NMR. J. Phys. Chem. B 119, 8024–8036. doi:10.1021/acs.jpcb.5b04647
Nakajima, T. (2017). An Extrapolation Scheme for Solid-State NMR Chemical Shift Calculations. Chem. Phys. Lett. 677, 99–106. doi:10.1016/j.cplett.2017.04.013
Olsen, R. A., Struppe, J., Elliott, D. W., Thomas, R. J., and Mueller, L. J. (2003). Through-Bond 13C–13C Correlation at the Natural Abundance Level: Refining Dynamic Regions in the Crystal Structure of Vitamin-D3 with Solid-State NMR. J. Am. Chem. Soc. 125, 11784–11785. doi:10.1021/ja036655s
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865. doi:10.1103/physrevlett.77.3865
Pickard, C. J., and Mauri, F. (2001). All-electron Magnetic Response with Pseudopotentials: NMR Chemical Shifts. Phys. Rev. B - Condensed Matter Mater. Phys. 63, 2451011–2451013. doi:10.1103/physrevb.63.245101
Pike, K. J., Lemaitre, V., Kukol, A., Anupõld, T., Samoson, A., Howes, A. P., et al. (2004). Solid-state 17 O NMR of Amino Acids. J. Phys. Chem. B 108, 9256–9263. doi:10.1021/jp049958x
Pritchard, B. P., Altarawy, D., Didier, B., Gibson, T. D., and Windus, T. L. (2019). New Basis Set Exchange: An Open, Up-To-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 59, 4814–4820. doi:10.1021/acs.jcim.9b00725
Pyykkö, P. (2001). Spectroscopic Nuclear Quadrupole Moments. Mol. Phys. 99, 1617–1629. doi:10.1080/00268970110069010
Samadi, Z., Mirzaei, M., Hadipour, N. L., and Abedini Khorami, S. (2008). Density Functional Calculations of Oxygen, Nitrogen and Hydrogen Electric Field Gradient and Chemical Shielding Tensors to Study Hydrogen Bonding Properties of Peptide Group (O{double Bond, long}C-NH) in Crystalline Acetamide. J. Mol. Graph. Model. 26, 977–981. doi:10.1016/j.jmgm.2007.08.003
Soss, S. E., Flynn, P. F., Iuliucci, R. J., Young, R. P., Mueller, L. J., Hartman, J., et al. (2017). Measuring and Modeling Highly Accurate 15N Chemical Shift Tensors in a Peptide. ChemPhysChem 18, 2225–2232. doi:10.1002/cphc.201700357
Staroverov, V. N., Scuseria, G. E., Tao, J., and Perdew, J. P. (2003). Comparative Assessment of a New Nonempirical Density Functional: Molecules and Hydrogen-Bonded Complexes. J. Chem. Phys. 119, 12129–12137. doi:10.1063/1.1626543
Stephens, P. J., Devlin, F. J., Chabalowski, C. F., and Frisch, M. J. (1994). Ab Initio calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force fields. J. Phys. Chem. 98, 11623–11627. doi:10.1021/j100096a001
Stoychev, G. L., Auer, A. A., and Neese, F. (2018). Efficient and Accurate Prediction of Nuclear Magnetic Resonance Shielding Tensors with Double-Hybrid Density Functional Theory. J. Chem. Theor. Comput. 14, 4756–4771. doi:10.1021/acs.jctc.8b00624
Wang, L., Elliott, A. B., Moore, S. D., Beran, G. J., Hartman, J. D., and Harper, J. K. (2021). Modeling Small Structural and Environmental Differences in Solids with 15N NMR Chemical Shift Tensors. ChemPhysChem 22, 1008–1017. doi:10.1002/cphc.202000985
Witter, R., Sternberg, U., Hesse, S., Kondo, T., Koch, F. T., and Ulrich, A. S. (2006). 13C Chemical Shift Constrained crystal Structure Refinement of Cellulose Iα and its Verification by NMR Anisotropy Experiments. Macromol 39, 6125–6132. doi:10.1021/ma052439n
Wolinski, K., Hinton, J. F., and Pulay, P. (1990). Efficient Implementation of the Gauge-independent Atomic Orbital Method for NMR Chemical Shift Calculations. J. Am. Chem. Soc. 112, 8251–8260. doi:10.1021/ja00179a005
Woon, D., and Dunning, T. (1995). Gaussian Basis Sets for Use in Correlated Molecular Calculations. V. Core-Valence Basis Sets for boron through Neon. J. Chem. Phys. 103, 4572. doi:10.1063/1.470645
Wu, G., Dong, S., Ida, R., and Reen, N. (2002). A Solid-State 17O Nuclear Magnetic Resonance Study of Nucleic Acid Bases. J. Am. Chem. Soc. 124, 1768–1777. doi:10.1021/ja011625f
Wu, G., Mason, P., Mo, X., and Terskikh, V. (2008). Experimental and Computational Characterization of the17O Quadrupole Coupling and Magnetic Shielding Tensors for P-Nitrobenzaldehyde and Formaldehyde. J. Phys. Chem. A. 112, 1024–1032. doi:10.1021/jp077558e
Wu, G. (2008). Solid-state 17O NMR Studies of Organic and Biological Molecules. Prog. Nucl. Magn. Reson. Spectrosc. 52, 118–169. doi:10.1016/j.pnmrs.2007.07.004
Yamada, K., Dong, S., and Wu, G. (2000). Solid-State 17O NMR Investigation of the Carbonyl Oxygen Electric-Filed-Gradient Tensor and Chemical Shielding Tensor in Amides. J. Am. Chem. Soc. 122, 11602–11609. doi:10.1021/ja0008315
Yamada, K., Hashizume, D., Shimizu, T., Ohki, S., and Yokoyama, S. (2008a). A Solid-State 17O NMR, X-ray, and Quantum Chemical Study of N-α-Fmoc-Protected Amino Acids. J. Mol. Struct. 888, 187–196. doi:10.1016/j.molstruc.2007.11.059
Yamada, K., Shimizu, T., Tansho, M., Nemoto, T., Asanuma, M., Yoshida, M., et al. (2007b). Solid-state 17O NMR Study of the Electric-Field-Gradient and Chemical Shielding Tensors in Polycrystalline Amino Acids. Magn. Reson. Chem. 45, 547–556. doi:10.1002/mrc.216710.1002/mrc.2002
Yamada, K., Shimizu, T., Yamazaki, T., and Ohki, S. (2008b). Determination of the Orientations for the 17O NMR Tensors in a Polycrystalline L-Alanine Hydrochloride. Solid State. Nucl. Magn. Reson. 33, 88–94. doi:10.1016/j.ssnmr.2008.04.001
Yamada, K., Shimizu, T., Yoshida, M., Asanuma, M., Tansho, M., Nemoto, T., et al. (2007a). Solid-state 17O NMR Study of Small Biological Compounds. Z. Naturforsch. - Sect. B J. Chem. Sci. 62, 1422–1432. doi:10.1515/znb-2007-1111
Yamada, K., Yamaguchi, Y., Uekusa, Y., Aoki, K., Shimada, I., Yamaguchi, T., et al. (2020). Solid-state 17O NMR Analysis of Synthetically 17O-Enriched D-Glucosamine. Chem. Phys. Lett. 749, 137455. doi:10.1016/j.cplett.2020.137455
Yang, C., Zhu, L., Kudla, R. A., Hartman, J. D., Al-Kaysi, R. O., Monaco, S., et al. (2016). Crystal Structure of the Meta-Stable Intermediate in the Photomechanical, crystal-to-crystal Reaction of 9-Tert-Butyl Anthracene Ester. CrystEngComm 18, 7319–7329. doi:10.1039/C6CE00742B
Keywords: nuclear magnetic resonance, electric field gradient, fragment methods, GIPAW, crystal structure, nuclear quadrupole coupling, 17-O
Citation: Hartman JD, Mathews A and Harper JK (2021) Fast and Accurate Electric Field Gradient Calculations in Molecular Solids With Density Functional Theory. Front. Chem. 9:751711. doi: 10.3389/fchem.2021.751711
Received: 01 August 2021; Accepted: 20 September 2021;
Published: 07 October 2021.
Edited by:
Oleksandr Loboda, AC2T Research, AustriaReviewed by:
Gerardo Andres Cisneros, University of North Texas, United StatesAmlan Kusum Roy, Indian Institute of Science Education and Research Kolkata, India
Ctirad Cervinka, University of Chemistry and Technology in Prague, Czechia
Copyright © 2021 Hartman, Mathews and Harper. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joshua D. Hartman, amhhcnRtYW5AbXNqYy5lZHU=