
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 29 March 2021
Sec. Organic Chemistry
Volume 9 - 2021 | https://doi.org/10.3389/fchem.2021.657028
 Zhenhua Liu1,2,3Yun Niu1,4Li Zhou1,4Lijun Meng1,4Sitan Chen1,4Mengke Wang1,4
Zhenhua Liu1,2,3Yun Niu1,4Li Zhou1,4Lijun Meng1,4Sitan Chen1,4Mengke Wang1,4 Jiangmiao Hu2,3*
Jiangmiao Hu2,3* Wenyi Kang1,4*
Wenyi Kang1,4*Patrinia scabiosaefolia is a medical and edible Chinese herb with high nutritional and medicinal value. The continuing study of its chemical constituents led to the discovery of nine unique iridoids and iridoid glycosides, including three new iridoids (1-3) and six previously unknown irioid glycosides (5-10), and one known compound (4). Among them, compound 1 was a deformed iridoid, while compounds 3, 5-7, and 10 formed a new ring in their skeletons which was uncommon in this genus. For compound 3, the new ring existed between C-3 and C-10, while a 1,3-dioxane appeared between C-7 and C-10 in compounds 5-7 and 10. Moreover, compound 10 was a bis-iridoid glycoside, which was the first reported in P. scabiosaefolia. And the sugar of irioid glycosides (5-10) was glucose at C-11, except in 9 which had a 5-deoxyglucose moiety. All their structures were confirmed based on the extensive spectroscopic analysis, including IR, UV, HR-ESI-MS, ECD, and 1D- and 2D-NMR experiments. Their cytotoxic activities against HL-60, A-549, SMMC-7721, MCF-7, SW480 were also tested.
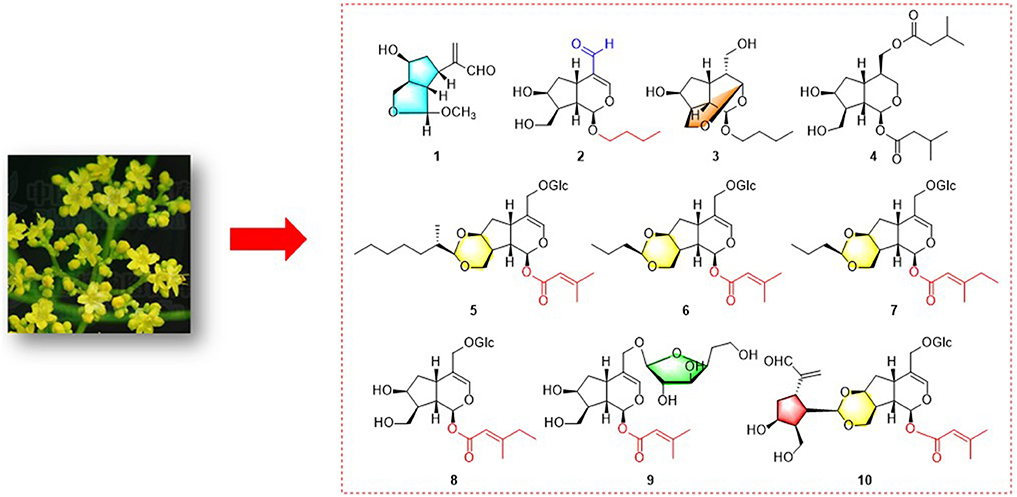
Graphical Abstract. Nine unique iridoids and iridoid glycosides from Patrinia scabiosaefolia.
There are about 400 species in 13 genera of Valerianaceae, mostly distributed in northern temperate zones and the subtropics or frigid zones. In China, this family comprises three genera, Nardostachys, Valeriana, and Patrinia, which can be found all over the country (Delectis Flora Reipublicae Popularis Sinicae Agendae Academiae Sinicae Edita., 1986). Plants from the genus Patrinia, such as P. scabiosaefolia, P. villosa, and P. heterophylla, have a long history of use as a traditional Chinese medicine for detoxification, swelling, empyema, liver protection, and cholagogue,. Some species are also edible as wild herbs, for example P. scabiosaefolia, P. villosa, P. punctiflora, and P. angustifolia (Xiao et al., 2007).
Among them, P. scabiosaefolia is a medical and edible Chinese herb, first recorded in “Sheng Nong's Herbal Classic.” It is of great nutritional value and is enriched with amino acids, vitamins, β-carotene, and trace elements. The content of vitamin C per 100 grams is 42.65 mg, which is higher than that in some vegetables and fruits. Also, there is 14995.69 mg of amino acids in it, including eight kinds of essential amino acids which account for 36.06% of the total amount of amino acids (Zhong et al., 2001). It was also verified to have effects on the initial stages of edema, appendicitis, endometriosis, and inflammation (Delectis Flora Reipublicae Popularis Sinicae Agendae Academiae Sinicae Edita., 1986).
Phytochemical research showed that different kinds of compounds existed in the Patrinia genus, including triterpenes, iridoids, flavones, lignans, and their glycosides (Kim and Kang, 2013). Among these, iridoids are considered as the main components with diverse structures and various activities, which attract our attention to further explore the plant P. scabiosaefolia. As a result, we found a series of iridoids from ethyl acetate extract of P. scabiosaefolia, including three bis-iridoids which were first reported (Liu et al., 2017a,b; Liu et al., 2019).
In the continuing study, chemical constituents on n-butanol extract of P. scabiosaefolia led to the discovery of nine unique iridoids and iridoid glycosides, including three new iridoids (1-3) and six novel irioid glycosides (5-10), and one known compound (4) (Figure 1, Graphical Abstract). Among them, compound 1 was a deformed iridoid, compound 3 formed a cycle between C-3 and C-10, compounds 5-7 with a 1,3-dioxane between C-7 and C-10, and compound 10 was a bis-iridoid glycoside, which was the first reported in P. scabiosaefolia. Herein, we discussed their isolation, structure elucidation, and their cytotoxic activities.
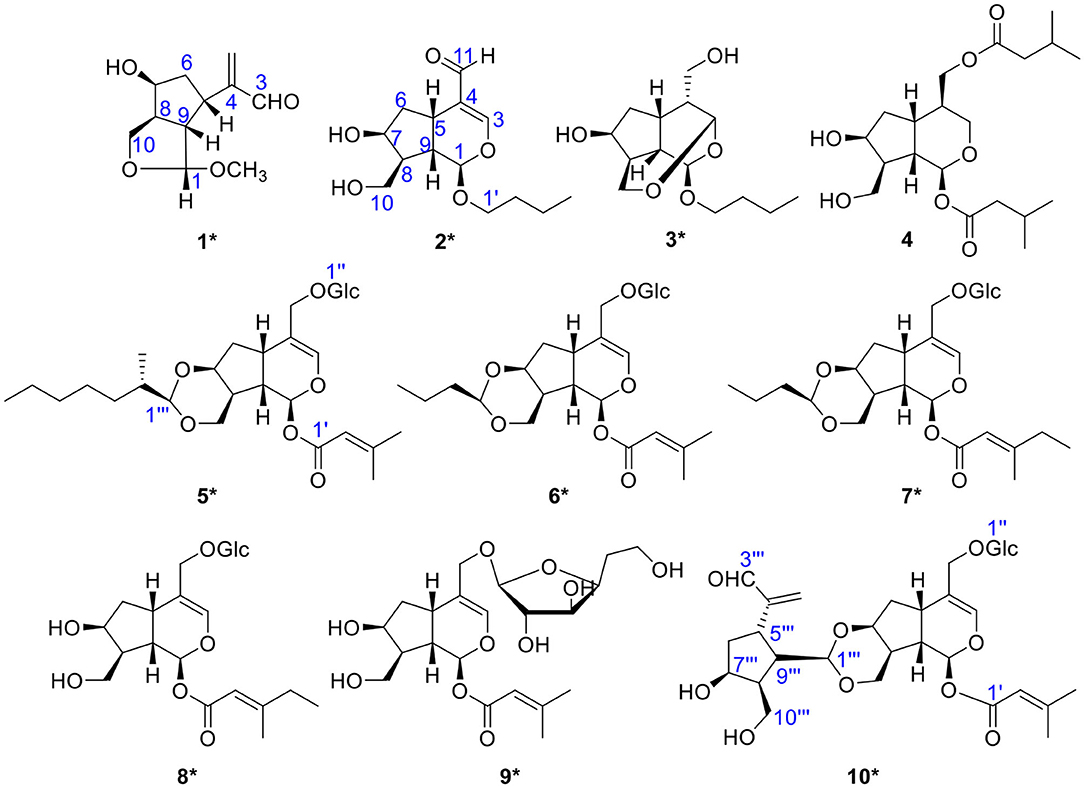
Figure 1. The chemical structures of compounds 1-10 (*new compounds).
Optical rotation was obtained on a JASCO P-1020 digital polarimeter (Horiba, Tokyo, Japan). UV spectra were measured by a Shimadzu UV-2401 PC spectrophotometer (Shimadzu, Kyoto, Japan). IR spectra were obtained on a Bruker Tensor 27 infrared spectrophotometer (Bruker Optics GmbH, Ettlingen, Germany) with KBr pellets. Mass spectra were performed on an API QSTAR time-of-flight spectrometer (MDS Sciqaszex, Concord, Ontario, Canada) and LC/MS-IT-TOF (Shimadzu, Kyoto, Japan) spectrometer. NMR spectra were recorded on Bruker DRX-500 and Av III-800 instruments with TMS as the internal standard (Bruker, Bremerhaven, Germany). The chemical shifts were given in δ (ppm) with reference to the solvent signal. Column chromatography was performed on silica gel (200–300 and 300–400 mesh, Qingdao Marine Chemical Inc., Qingdao, China), Lichroprep Rp-18 gel (40–63 μm, Merck, Darmstadt, Germany), MCI gel CHP-20P (75–150 μm, Mitsubishi Chemical Corp., Tokyo, Japan), Sephadex LH-20 (20–150 μm, Amersham Biosciences, Uppsala, Sweden), and YMC*GEL ODS-A-HG (50 μm, YMC Co. Ltd. Japan). Fractions were monitored by TLC, and spots were visualized by UV light and sprayed with 10% H2SO4 in EtOH, followed by heating.
The whole plants of Patrinia scabiosaefolia were collected in October 2010 from Shucheng county, Anhui Province, People's Republic of China, and were stored in a cool and dry place at room temperature. The material was identified by Prof. Shou-Jin Liu in Anhui University of Chinese Medicine and a voucher specimen (Wan1295) was deposited in Anhui University of Chinese Medicine. The plants of P. scabiosaefolia are common in the local area and the collection was permitted. We also ensured that the local population of P. scabiosaefolia was not destroyed through the means of collection at different locations.
The air-dried and powdered whole plants (29 kg) of P. scabiosaefolia were extracted with 95% ethanol (3 × 75 L) under room temperature and concentrated under reduced pressure. Then the residue (3 kg) was dissolved in water and partitioned successively with n-butanol to yield n-butanol extract (0.85 kg) after concentration. The n-butanol extract was subjected to silica gel column chromatography eluted with a gradient of CHCl3-MeOH (8:1→ 0:1, v/v) to obtain five fractions 1-5 by TLC plate analysis.
Fraction 2 (22 g) was separated by silica gel column chromatography eluted with a gradient system of CHCl3-MeOH (6:1→ 1:1, v/v) to afford 4 subfractions (Fr.2-1 to Fr.2-4). Fr.2-1 (5.9 g) was performed by Rp-18 column chromatography (MeOH-H2O, 50:50→ 100:0, v/v) to afford 6 subfractions (Fr.2-1-1 to Fr.2-1-6). Fraction 2-1-2 (1.0 g) was applied to Sephadex LH-20 column chromatography (MeOH-H2O, 90:10, v/v) and then purified by silica gel column chromatography repeatedly to obtain 1 (1.0 mg). Fraction 2-2-3 (1.3 g) was submitted to silica gel column chromatography eluted with a gradient of EtOAc-MeOH (4:1→ 1:1, v/v) to afford A (180 mg) and B (240 mg), and then purified by semi-prep. HPLC (MeOH-H2O, 65:35, v/v) to obtain 3 (7.0 mg) and 4 (16.0 mg) respectively. Fraction 2-2-4 (2.1 g) was separated by the same methods as Fraction 2-2-3 and purified by semi-prep. HPLC (MeOH-H2O, 55:45, v/v) to afford 5 (1.0 mg) and 10 (16.0 mg).
Fraction 3 (15 g) was subjected to silica gel column chromatography eluted with a gradient system of CHCl3-MeOH (5:1→ 1:1, v/v) to afford 6 subfractions (Fr.3-1 to Fr.3-6). Fr.3-3 (4.3 g) was separated by Rp-18 column chromatography (MeOH-H2O, 50:50→ 100:0, v/v) to get 5 subfractions (Fr.3-3-1 to Fr.3-3-5). Fraction 3-3-3 (560 mg) was applied to Sephadex LH-20 column chromatography (MeOH-H2O, 90:10, v/v), and silica gel column chromatography eluted with CHCl3- MeOH (4:1, v/v) to obtain 2 (3.0 mg).
Fraction 4 (28 g) was submitted to Rp-18 column chromatography (MeOH-H2O, 30:70→ 100:0, v/v) to obtain 6 subfractions (Fr.4-1 to Fr.4-6). Fr.4-2 (3.4 g) was applied to silica gel column chromatography eluted with a gradient system of EtOAc-MeOH (4:1→ 1:1, v/v), then isolated by Sephadex LH-20 column chromatography (MeOH-H2O, 90:10, v/v), and further purified by semi-prep. HPLC (MeOH-H2O, 55:45, v/v) to afford 6 (3.0 mg) and 7 (10 mg). Fr.4-4 (5.6 g) was repeatedly separated by silica gel column chromatography and Sephadex LH-20 column chromatography (MeOH-H2O, 90:10, v/v), and finally purified by semi-prep. HPLC (MeOH-H2O, 55:45, v/v) to afford 8 (3.0 mg) and 9 (16.0 mg).
Patrirscabioin M (1): colorless oil; [α]23 D-32.2 (c0.03, MeOH); UV (MeOH) λmax (log ε): 204 (3.42) nm; IR (KBr) νmax 3,428, 2,925, 2,855, 1,632, 1,384, 1,101 cm−1; 1H and 13C NMR data, see Table 1; positive ESIMS m/z 235 [M + Na]+; HREIMS m/z 235.0943 [M + Na]+ (calcd for C11H16O4Na, 235.0941).
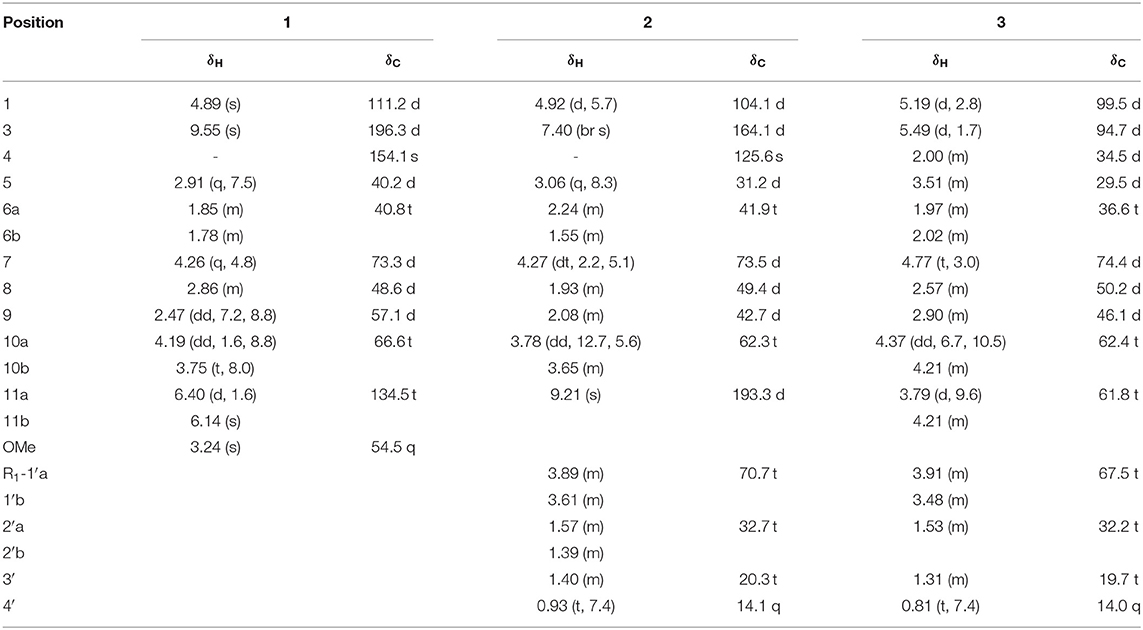
Table 1. The 1H and 13C NMR data of 1-4 (CD3OD, δ in ppm, J in Hz).
Patrirscabioin N (2): light yellow oil; [α]24 D-42.6 (c0.05, MeOH); UV (MeOH) λmax (log ε): 248 (3.72) nm; 203 (3.69) nm; IR (KBr) νmax 3,425, 2,927, 2,874, 1,727, 1,625, 1,384, 1,161, 1,088 cm−1; 1H and 13C NMR data, see Table 1; positive ESIMS m/z 293 [M + Na]+; HREIMS m/z 293.1354 [M + Na]+ (calcd for C14H22O5Na, 293.1359).
Patrirscabioin O (3): light yellow oil; [α]23 D-11.3 (c0.05, MeOH); UV (MeOH) λmax (log ε): 203 (3.03) nm; IR (KBr) νmax 3,444, 2,926, 2,870, 1,725, 1,634, 1,383, 1,088 cm−1; 1H and 13C NMR data, see Table 1; positive ESIMS m/z 295 [M + Na]+; HREIMS m/z 295.1515 [M + Na]+ (calcd for C14H24O5Na, 295.1516).
Patrinoside B (5): light yellow oil; [α]23 D-54.6 (c0.02, MeOH); UV (MeOH) λmax (log ε): 218 (4.15) nm; IR (KBr) νmax 3,426, 2,929, 2,873, 1,727, 1,642, 1,076 cm−1; 1H and 13C NMR data, see Table 2; positive ESIMS m/z 593 [M + Na]+; HREIMS m/z 593.2931 [M + Na]+ (calcd for C29H46O11Na, 593.2932).
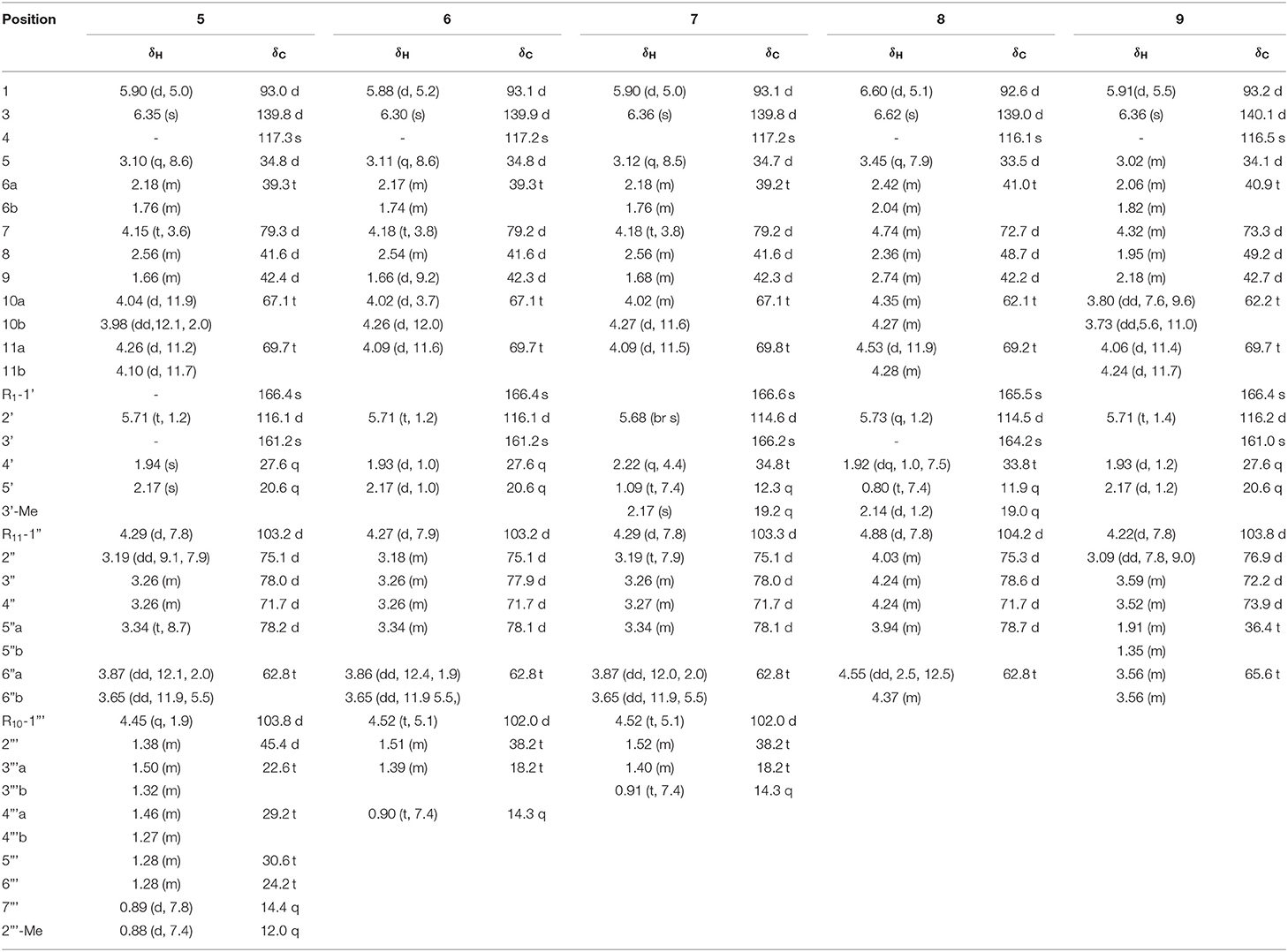
Table 2. The 1H and 13C NMR data of 5-9 (CD3OD, δ in ppm, J in Hz).
Patrinoside C (6): light yellow oil; [α]24 D-61.4 (c0.14, MeOH); UV (MeOH) λmax (log ε): 209 (4.16) nm, 217 (4.18) nm; IR (KBr) νmax 3,431, 2,926, 2,876, 1,727, 1,637, 1,384, 1,075 cm−1; 1H and 13C NMR data, see Table 2; positive ESIMS m/z 537 [M + Na]+; HREIMS m/z 537.2302 [M + Na]+ (calcd for C25H38O11Na, 537.2306).
Patrinoside D (7): light yellow oil; [α]24 D-54.2 (c0.05, MeOH); UV (MeOH) λmax (log ε): 209 (4.06) nm, 217 (4.06) nm; IR (KBr) νmax 3,429, 2,924, 2,855, 1,724, 1,635, 1,384, 1,075 cm−1; 1H and 13C NMR data, see Table 2; positive ESIMS m/z 551 [M + Na]+; HREIMS m/z 551.2467 [M + Na]+ (calcd for C26H40O11Na, 551.2463).
Patrinoside E (8): light yellow oil; [α]23 D-23.7 (c0.08, MeOH); UV (MeOH) λmax (log ε): 205 (3.95) nm, 214 (3.94) nm; IR (KBr) νmax 3,429, 2,925, 2,857, 1,725, 1,635, 1,384, 1,077 cm−1; 1H and 13C NMR data, see Table 2; positive ESIMS m/z 497 [M + Na]+; HREIMS m/z 497.1988 [M + Na]+ (calcd for C22H34O11Na, 497.1993).
Patrinoside F (9): light yellow oil; [α]24 D-74.0 (c0.32, MeOH); UV (MeOH) λmax (log ε): 207 (4.18) nm; IR (KBr) νmax 3,441, 2,925, 2,860, 1,727, 1,639, 1,121, 1,065 cm−1; 1H and 13C NMR data, see Table 2; positive ESIMS m/z 467 [M + Na]+; HREIMS m/z 467.1889 [M + Na]+ (calcd for C26H40O11Na, 467.1888).
Patrirscabiobisin D (10): light yellow oil; [α]23 D-34.1 (c0.17, MeOH); UV (MeOH) λmax (log ε): 209 (4.24) nm, 217 (4.25) nm; IR (KBr) νmax 3,426, 2,926, 1,724, 1641, 1,077 cm−1; 1H and 13C NMR data, see Table 3; positive ESIMS m/z 663 [M + Na]+; HREIMS m/z 663.2627 [M + Na]+ (calcd for C31H44O14Na, 663.2623).
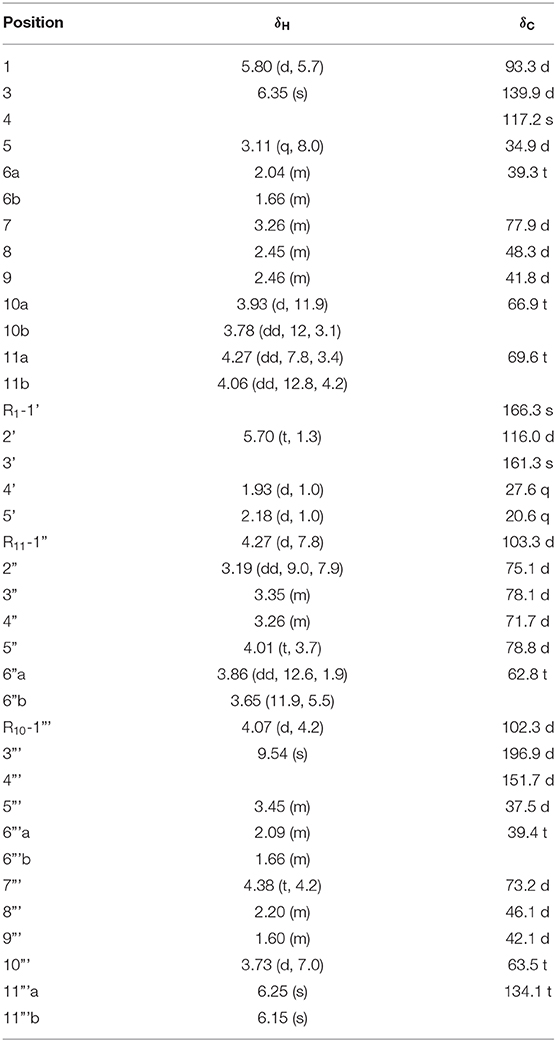
Table 3. The 1H and 13C NMR data of 10 (CD3OD δ in ppm, J in Hz).
The following human tumor cell lines were used: HL-60, SMMC-7721, A-549, MCF-7, and SW-480. These were obtained from ATCC (Manassas, VA, USA). All the cells were cultured in RPMI-1640 or DMEM medium (Hyclone, Logan, UT, USA), supplemented with 10% fetal bovine serum (Hyclone) at 37°C in a humidified atmosphere with 5% CO2. Cell viability was assessed by conducting colorimetric measurements of the amount of insoluble formazan formed in living cells based on the reduction of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2- (4-sulfopheny)-2H-tetrazolium (MTS) (Sigma, St. Louis, MO, USA) (Monks et al., 1991). In brief, 100 μL of adherent cells were seeded into each well of a 96-well cell culture plate and allowed to adhere for 12 h before drug addition, while suspended cells were seeded just before drug addition, both with an initial density of 1 × 105 cells/mL in 100 μL medium. Each tumor cell line was exposed to the test compound at various concentrations in triplicate for 48 h, with cisplatin and paclitaxel (Sigma) as positive controls. After the incubation, MTS (100 μg) was added to each well, and the incubation continued for 4 h at 37 °C. The cells were lysed with 100 μL of 20% SDS-50% DMF after removal of 100 μL medium. The optical density of the lysate was measured at 490 nm in a 96-well microtiter plate reader (Bio-Rad 680). The IC50 value of each compound was calculated by the Reed and Muench method (1938).
The CHARMM force field and DFT/TDDFT calculations were performed with Discovery Studio 4.0 and Gaussian09 program package, respectively. Conflex conformational search generated low-energy conformers within a 20 kcal/mol energy window and were subjected to geometry optimization using DFT method without imposing any symmetry constraints at the B3LYP/6-31G(d) level. Frequency calculations were carried out using the same level to verify that the molecular structures were true minimum. The calculated ECD spectra were generated by the program SpecDis2 using a Gaussian band shape with 0.3eV exponential half-width from dipole-length dipolar and rotational strengths (Bruhn et al., 2013).
Compound 1 was indicated as C11H16O4 by HR-ESI-MS [m/z 235.0943 [M + Na]+, calcd. for 235.0941]. Preliminary inspection of the 1H and 13C NMR (Table 1) spectroscopic data of compound 1 revealed the presence of one conjugated aldehyde group [δH 9.55 (s), δC 196.3 (d)] and a pair of double bond [δC 154.1 (s), δC 134.5 (t)], two oxygenated methine groups [δH 4.89 (s), δC 111.2 (d); δH 4.26 (q), δC 73.3 (d)], and one oxygenated methylene at δC 66.6 (t). These data were similar to those of 8,9-didehydro-7-hydroxydolichodial (Veith et al., 1986), except for the disappearance of conjugated aldehyde and double bonds, and the appearance of two methines and one oxygenated methane, especially the change of methyl at C-10 to oxygenated methylene. Therefore, we speculated that the methyl at C-10 of 8,9-didehydro-7-hydroxydolichodial was oxidized to oxygenated methylene, and then underwent a hemiacetal formation with the aldehyde group at C-1 to form the deformed iridoid—compound 1 with 5/5 rings, which occurred simultaneously to the hydrogenation at C-8 and C-9, and finally the C-1 was methylated (Figure 2). This conclusion was confirmed by HMBC spectrum with the key correlations from H-1 [δH 4.89 (s)] to C-10 (δC 66.6), C-8 (δC 48.6), and C-9 (δC 57.1), and OMe (δC 54.5) were observed, and the correlations of H-1/H-9 and H-9/H-5 in the ROESY spectrum (Figure 3).
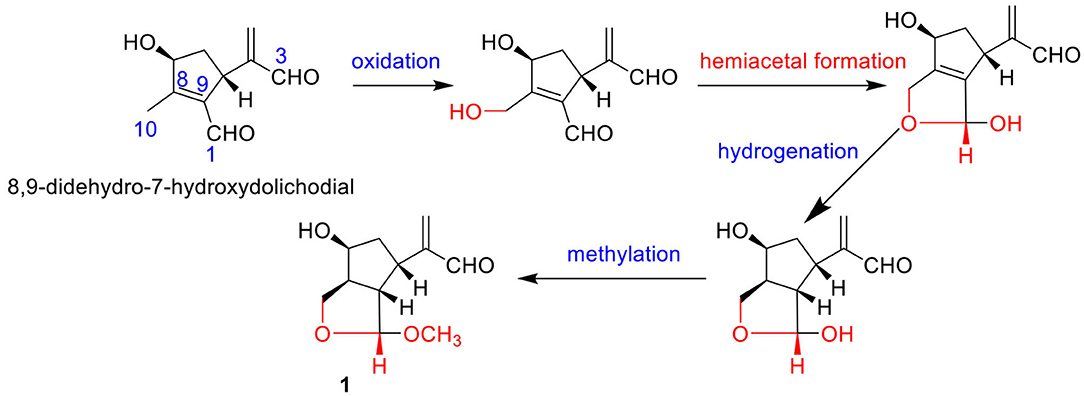
Figure 2. The possible pathway for the transformation of compound 1.
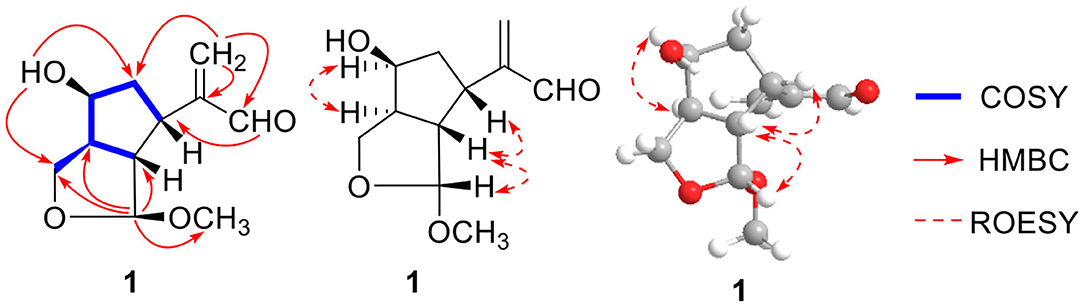
Figure 3. Key 1H-1H COSY, HMBC, and ROESY correlations of compound 1.
The absolute configurations of H-5 and H-7 were both S deduced from the comparison of chemical shift value to the known compound 8,9-didehydro-7-hydroxydolichodial (Veith et al., 1986). The α-orientation of H-8 was elucidated by the correlation of H-7 with H-8 in ROESY (Figure 3). The β-orientations of H-1 and H-9 were confirmed by the ROESY correlations from H-5 to H-1 and H-9 (Figure 2). And the R-configuration of the new formed acetal methine at C-1 was further confirmed by the comparison of experimental and calculated ECD spectra, which the positive Cotton effect near 200 nm in calculated ECD spectra of R-configuration at C-1 agreed with the experimental ECD spectra (Figure 4). Hence, the structure of compound 1 was defined as (1R,5S,7S,8S,9S)-1,10-epoxy-7,10-dihydroxy-dolichodial, named Patriscabioin M.
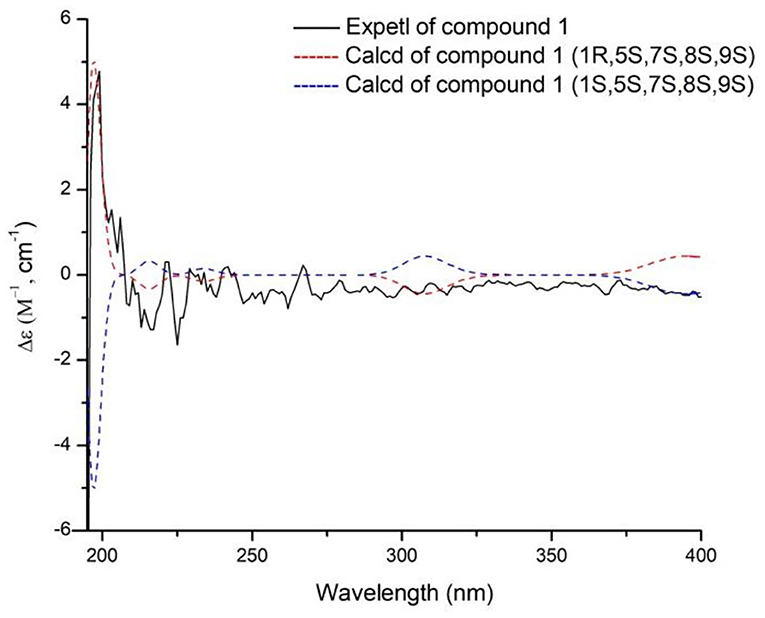
Figure 4. Calculated and experimental ECD spectra of compound 1 at TDDFT/B3LYP/6-31G(d) level.
Compound 2, a light yellow oil, had a molecular formula of C14H22O5 based on HRESIMS ([M + Na]+, m/z 293.1354, calcd. for 293.1359). Detailed comparisons of 1H-NMR and 13C-NMR (Table 1) suggested it was a typical iridoid and included a hemiketal methine at δH 4.92 (d, J = 5.7 Hz, H-1) and δC 104.1 (d, C-1), a conjugated olefinic bond at δC 193.3 (d, C-11); δC 164.1 (d, C-3), δC 125.6 (s, C-4), and one oxygenated methylene at δC 62.3 (t, C-10). These data were similar to patriscabioin I (Liu et al., 2017), except for the substituent at C-1 (Figure 5). In compound 2 it was n-butanol group [δC 70.7 (t), 32.7 (t), 20.3 (t), 14.1 (q)], while it was methyl in patriscabioin I. The correlation from H-1 [4.92 (d, 5.7)] to C-1' [δC 70.7 (t)] in HMBC implied the location of n-butanol group at C-1 (Figure 5). The configuration of compound 2 was the same as patriscabioin I. Therefore, the structure of compound 2 was defined as (1R,5S,7S,8S,9S)-1-n-butoxy-7,10-dihydroxy-11-aldehyde-5,6-dihydrovaltrate hydrin, named patriscabioin N.
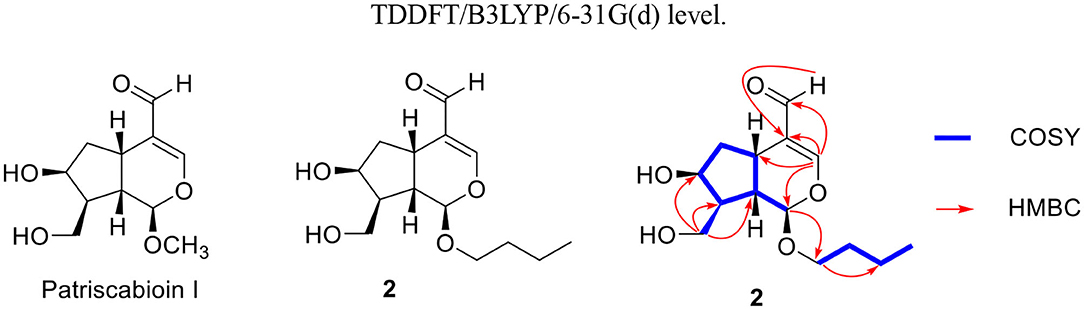
Figure 5. Key 1H-1H COSY and HMBC correlations of compound 2 and the structure of patriscabioin I.
Compound 3 was found to have a molecular formula of C14H24O5, determined by its positive HRESIMS data ([M + Na]+, m/z 295.1515, calcd. for 295.1516) and 13C-NMR spectrum (Table 1), with 3 degrees of unsaturation. From the NMR data, compound 3 showed spectral characteristics of iridoid at δH 5.19 (d, J = 2.8 Hz, H-1) and δC 99.5 (d, C-1), with a n-butanol group [δC 67.5 (t), 32.2 (t), 19.7 (t), 14.0 (q)]) located in C-1, which was confirmed by the correlation from H-1 to C-1' in HMBC (Figure 6). However, the biggest difference from other iridoids' skeletons (such as compounds 2, 5-9) was the added 1 unsaturation, of which the former's degree was 2, and the latter was 3. Meanwhile, the NMR spectra exhibited the disappeared double bonds and the appearance of two methines, among which the chemical shift was δC 94.7 (d, C-3), but there was no other unsaturated group. Therefore, it was presumed that there was a ring that existed in the skeleton of compound 3. The new ring was formed by the ether bond between C-3 and C-10, which was further verified by the fragment of H-3—H-4—H-5—H-9—H-8—H-10 in COSY, and the correlation from H-3 to C-10 in HMBC (Figure 7). According to the S-configuration of H-5 and H-9 and R-configuration of H-1, the configuration of H-4 was determined as R-configuration and the configurations of H-3 and H-8 were determined as S-configuration by the correlations of H-4/H-5, H-3/H-11, and H-7/H-8 in ROESY (Figure 6). There was good agreement between experimental and calculated ECD spectra, which was further verified these configurations (Figure 7). Hence, the structure of 3 was established as (1R,3S,4R,5S,7S,8S,9S)-1-n-butoxy-3,10-epoxy-7-hydroxy-3,4,5,6-tetrahydrovaltrate hydrinm, named patriscabioin O.
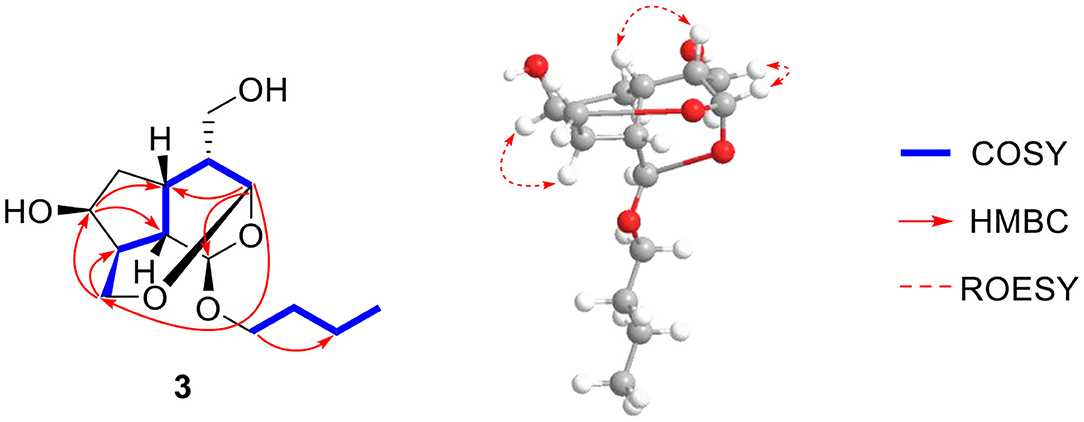
Figure 6. Key 1H-1H COSY, HMBC, and ROESY correlations of compound 3.
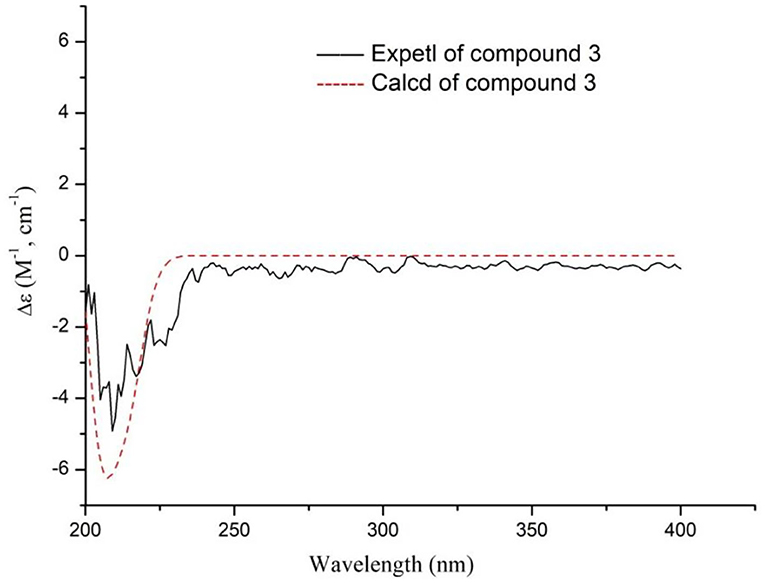
Figure 7. Calculated and experimental ECD spectra of compound 3 at TDDFT/B3LYP/6-31G(d) level.
Compound 5 was obtained as a slight yellow oil. Its molecular formula was assigned as C29H46O11 on the basis of positive-ion HR-ESI-MS at m/z 593.2931 [M + Na]+ (calcd for 593.2932) and 13C-NMR data (Table 2). Carefully analysis of 1H-NMR and 13C-NMR led to the discovery of a hemiketal methine at δH 5.90 (1H, d, J = 5.0 Hz, H-1) and δC 93.0 (d, C-1), a trisubstituted olefinic bond at δH 6.35 (1H, s, H-3), δC 139.8 (d, C-3), and δC117.3 (s, C-4), an oxymethine at δC 79.3 (C-7) and two oxygenated methylenes at δC 67.1 (t, C-10) and δC 69.7 (t, C-11). The above data indicated the existence of 7,10,11-trihydroxy-3-en valtratehydrin. It was found that 5 had a 3-methylcrotonyl group at δC 166.4 (s), δC 116.1 (d), δC 161.2 (s), δC 27.6 (q), δC 20.6 (q) and a glucopyranose unit at δC 103.2 (d), δC 78.2 (d), δC 78.0 (d), δC 75.1(d), δC 71.7 (d), δC 62.8 (t) through further detailed analysis of its 1D-NMR and 2D-NMR. The 3-methylcrotonyl group and glucopyranose were arranged at C-1 and C-11 by the correlations from δH 5.90 (H-1) to δC 166.4 (C-1'), and δH 4.29 (H-1”) to δC 69.7 (C-11”) in HMBC spectrum (Figure 8). The typical signal for β-configuration (δH 4.29, d, J = 7.8 Hz) of H-1” could also be observed. Thus, eight carbons remained, including two methyl groups, four methylene groups, and two methine groups. The 1H-1H COSY correlations (Figure 8) showed the fragments: CH(H-1”')—CH(H-2”')—CH2 (H-3”')—CH2(H-4”')—CH (H-5”', H-6”')—CH3 (H-7”') and CH(H-2”')—CH3(2”'-Me). Combined with the correlations of H-2”'/2”'-Me, and H-1”'/ C-10, C-7 in HMBC, these revealed the presence of a 1,3-dioxane through acetalation between OH-7, OH-10 and C-1.”' It meant that 1,3-dioxane was formed by hemiacetal formation from hydroxyl groups of C-7 and C-10 and aldehyde of 2-methyl-heptaldehyde.
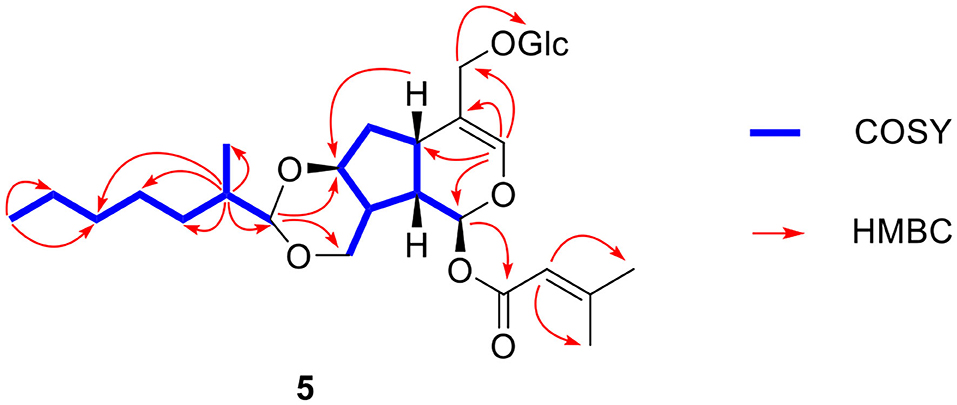
Figure 8. Key 1H-1H COSY and HMBC correlations of compound 5.
The relative configuration of 5 was similar to patriscabioins A-L and patriscabiobisins A and B (Liu et al., 2017a,b) from P. scabiosaefolia through comparison with their ROESY and CD spectrum (Figure 11). The ROESY cross peaks of H-1”'/H-8 and 2”'-Me/H-1”' suggested the α-orientation of H-1”' and 2”'-Me. Thus, the structure of 5 was identified as shown and named patrinoside B.
Compound 6 had the molecular formula C25H38O11 based on its quasimolecular ion peak at m/z 537.2302 [M + Na]+ (calcd for 537.2306) in HR-ESI-MS spectrum and 13C-NMR data (Table 2). Comparison of the 13C NMR and DEPT spectra of 6 with those of 5 revealed that 6 shared the same aglycone, a glucose at C-11, and the 3-methylcrotonyl group at C-1 as compound 5, and the significant difference between them was the disappearance of two methylenes, one methine and one methyl in the high field region, and the molecular weight of 6 was 56 less than that of 5. This indicated that the fragment of 2-methyl-heptaldehyde in compound 5 was changed to n-butyraldehyde in compound 6, which was further verified by the correlations from H-1”' (δH 4.52) to C-2”' (δC 38.2), and H-4”' (δH 0.90) to C-2”' (δC 38.2), C-3”' (δC 18.2) in HMBC. Therefore, the structure of 6 was deduced as shown and named patrinoside C.
The molecular formula of 7 was identified as C26H40O11 from HR-ESI-MS at m/z 551.2467 [M + Na]+ (calcd for 551.2463). Comparing 1H-NMR and 13C-NMR data of 7 with those of 6, it was found that they shared similar NMR data, and the obvious distinction between them was the appearance of a methylene (δC 34.8) and the change in chemical shift of methyl signal of C-4' (δC 27.6→ 12.3). It indicated that a 3-methylcrotonyl group at C-1 in 6 was replaced by the 3,4-dimethylcrotonyl group in 7. This was ultimately confirmed by 1D and 2D NMR spectra. In HMBC spectrum, the correlations between H-2' (δH 5.68) and C-4' (δC 34.8) and C-5' (δC 12.3) provided solid evidence for the existence of the 3,4-dimethylcrotonyl group. Finally, the structure of 7 was elucidated as shown and named patrinoside D.
Compound 8 was analyzed to have a molecular formula of C22H34O11 by HR-ESI-MS at m/z 497.1988 [M + Na]+ (calcd for 497.1993), whose molecular weight is 54 less than that of 7. Careful analysis of its NMR data found that it had the same aglycone as compound 7, as well as a glucose at C-11 and a 3,4-dimethylcrotonyl group at C-1, but lacked an oxymethine at δC 102.0, two methylenes and a methyl, which suggested that it did not have a 1,3-dioxaneformed at C-7 and C-10. This could be further verified by the chemical shifts at C-7 and C-10 which were shifted up field from δC 79.2 to 72.7 and form δC 67.1 to 62.1, respectively, compared with compound 7. Finally, the structure of compound 8 was characterized as (1S,5S,7S,8S,9S)-1-O-(3,4-dimethylcrotonyl)-7,10-dihydroxy-11-β-D-glucose-5,6-dihydrovaltrate hydrin and named patrinoside E.
Compound 9 was found to have a molecular formula of C21H32O10 by HR-ESI-MS at m/z 467.1889 [M + Na]+ (calcd for 467.1888). Careful analysis of its 1D and 2D NMR data led to finding that it was a 7,10,11-trihydroxy-3-envaltratehydrin iridoid with a 3-methylcrotonyl group at C-1 and a sugar. The 1H-1H COSY correlations (Figure 9) showed a fragment: CH (H-1”, δH 4.22)—CH (H-2”, δH 3.09)—CH (H-3”, δH 3.59)—CH (H-4”, δH 3.52)—CH2 (H-5”, δH 1.35)—CH2 (H-6”, δH 3.56). Combined with the correlations between H-5” (δH 1.35) and C-2” (δC 76.9) and C-3” (δC 72.2) in the HMBC, all these signals hinted the sugar was 5-deoxyglucose, which was linked to C-11 from the correlation of H-11 (δH 4.06, 4.24)/C-1” (δC 103.8). The β-configuration was determined by the coupled constant of H-1” (δH 4.22, d, J = 7.8 Hz). Ultimately, the structure of compound 9 was confirmed as (1S,5S,7S,8S,9S)-1-O-(3-methylcrotonyl)-7,10-dihydroxy-11-(5-deoxy-β-D-glucofuranose)-5,6-dihydrovaltrate hydrin and named patrinoside F.
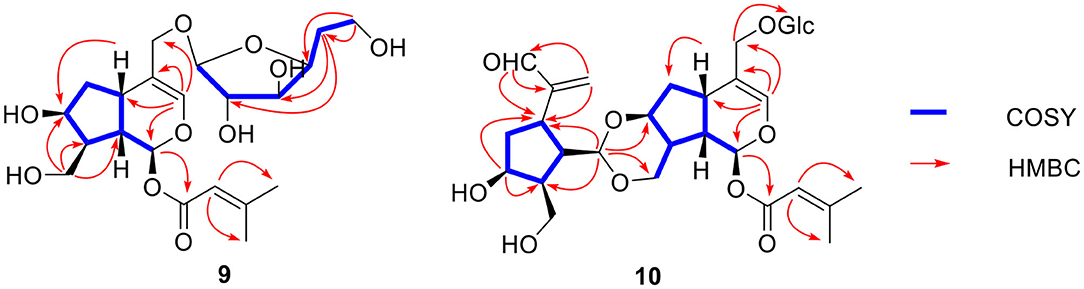
Figure 9. Key 1H-1H COSY and HMBC correlations of compounds 9-10.
Compound 10 was formulated as C31H44O14 from HR-ESI-MS at m/z 663.2627 [M + Na]+ (calcd for 663.2623) and 13C-NMR data (Table 3). The carbon resonances at δC 103.3 (d), 75.1 (d), 78.1 (d), 71.7 (d), 78.8 (d), 62.8 (t) and the anomeric proton signals at δH 4.27 (d, J = 7.8 Hz) suggested that compound 10 contained a glucose at C-11 by the correlation from the anomeric proton (δH 4.27) to C-11 (δC 69.6) in HMBC (Figure 9). The remaining carbon signals showed two iridoid units, which means it was a bis-iridoidglycoside. Detailed analysis of the NMR data at δC 93.3 (d), δC 139.9 (d), δC 117.2 (s), δC 77.9 (d), δC 66.9 (t), δC 69.6 (t), and δH 5.80 (d, 5.7), combined with the 3-methylcrotonyl [δC 166.3 (s), δC 161.3 (s), δC 116.0 (d), δC 27.6 (q), δC 20.6 (q)] at C-1, found one iridoid was patrinoside A (Liu et al., 2019). The other iridoid was similar to 8,9-didehydro-7-hydroxydolichodial (Georg and Joerg, 1985) through the conjugated aldehyde and double bond at δC 196.9 (d), δC 151.7 (s), δC 134.1 (t), two methylene at δC 39.4 (t), and δC 63.5 (t), and four methynes at δC 37.5 (t), δC 73.2 (d), δC 46.1 (d), δC 42.1 (d) in 13C NMR, except for the absence of one methyl, one pair of double bonds, and an aldehyde group. However, it did include an oxygenated methylene, two methines, and an acetal at δC 102.3 (d). This suggested that the other iridoid was 7,10-dihydroxydolichodial. And two units were linked through the aldehyde group between the aldehyde group at C-1' of 7,10 -dihydroxydolichodial and hydroxyl groups at C-7 and C-10 of patrinoside A, which was further verified by the correlations of H-1'/C-7, C-10 in the HMBC spectrum. The α-orientations of H-1”', H-8”', and H-9”' were determined by the ROESY correlations of H-7”' with H-1”', H-8”,' and H-9”'. Additionally, these were further proven by the good agreement of ECD spectra between compound 10 and patriscabiobisin A and B which were similar to compound 10. Thus, the structure of compound 10 was characterized as shown, namely patriscabiobisin C.
In conclusion, compounds 1-10 were a series of 5,-dihydrovaltrate hydrins with characteristic substituents, such as 3-methylcrotonyl group or 3,4-dimethylcrotonyl group in the Valerianaceae family, belonging to iridoids derived from iridodial. Iridoial was enoxylated and then subjected to intramolecular hemiacetal formation to form iridoid. And iridoid suffered a series of chemical changes to achieve diverse compounds (1-10) (Figure 10). Iridodial was oxidized and then underwent the hemiacetal formation between C-1 and C-10 to form the deformed iridoid—compound 1 with 5/5 rings. Iridoid was oxidized and combined with different substituents to constitute compounds 2, 4, 8-9, and when it was dehydration condensed between C-10 and C-3 to generate a new ring, like compound 3. However, when the hydroxyl of C-7 and C-10 of iridoid combined with the aldehyde compounds, a 1,3-dioxane group would be formed, such as compounds 5-7. Furthermore, if the aldehyde group came from an iridoid, then it would generate a bis-iridoid, like compound 10. And the configurations of compounds 1-10 were further confirmed by ECD (Figure 11).
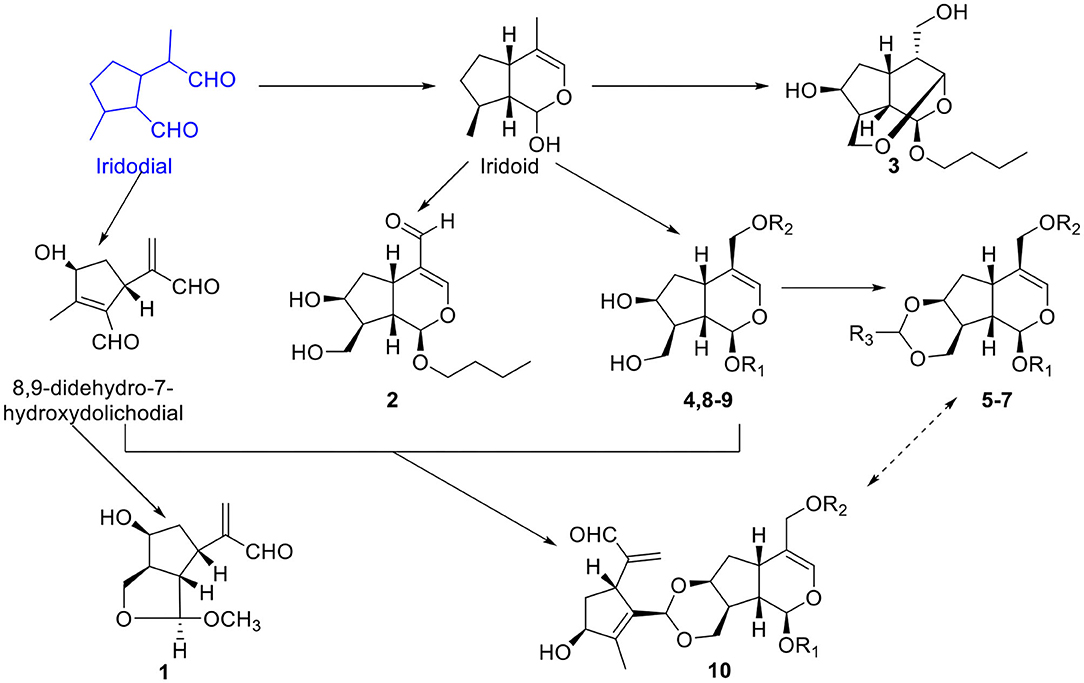
Figure 10. The possible pathway for the transformation of compounds 1-10.
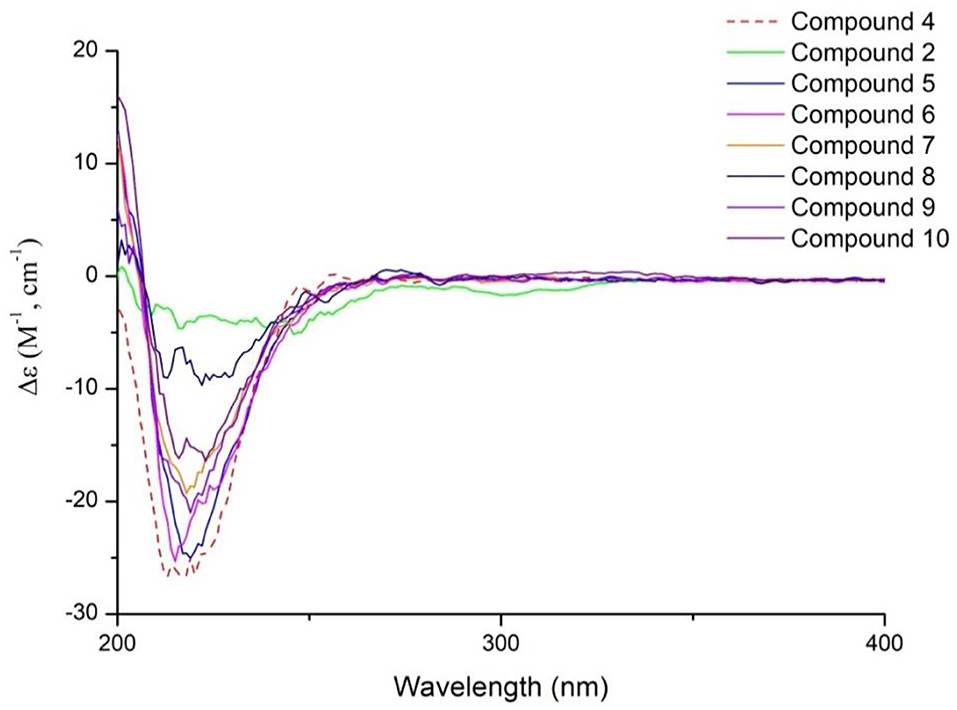
Figure 11. Experimental ECD spectra of compounds 2, 4-10 at TDDFT/B3LYP/6-31G(d) level.
Finally, the cytotoxic activities of these compounds were evaluated against five human cancer cell lines (HL-60, A-549, SMMC-7721, MCF-7, and SW-480). Unfortunately, none of the compounds showed significant cytotoxicities at 40 μM, except for compound 5 which showed the cell inhibition of 102.42, 95.13, 73.07, and 80.93% against HL-60, SMMC-7721, MCF-7, and SW-480 respectively (Figure 12).
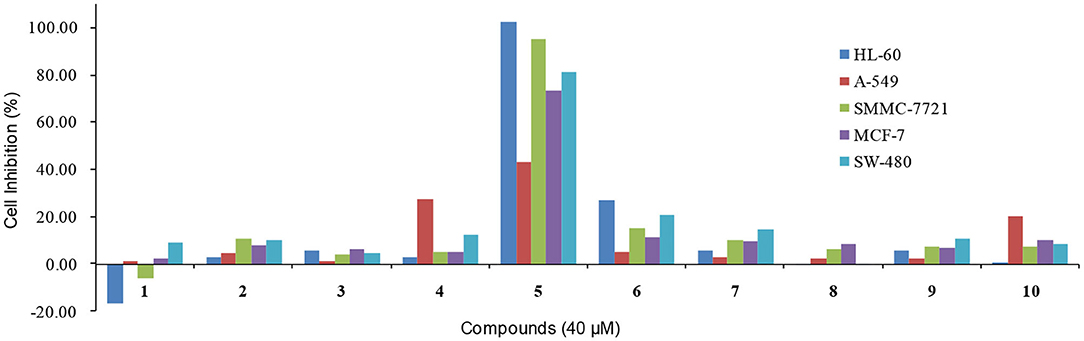
Figure 12. The cytotoxic activities against five human cancer cell lines of compounds 1-10.
P. scabiosaefolia is a medical and edible Chinese herb with high nutritional value and a wide range of biological activities. Research showed plants in the Patrinia genus are rich in iridoids and terpenoids. In our ongoing study, we found a series of iridoids and iridoid glycosides, including three new iridoids (1-3) and six novel irioid glycosides (5-10), and one known compound (4). Among them, compound 1 was a deformed iridoid, compound 3 formed a cycle between C-3 and C-10, compounds 5-7 with a 1,3-dioxane between C-7 and C-10, and compound 10 was a bis-iridoid glycoside, which was the first reported in P. scabiosaefolia. Cytotoxicity assays found compound 5 showed good cell inhibition against HL-60, SMMC-7721, MCF-7, and SW-480. All these results enriched the study on the chemical constituents and activities of Patrinia genus. However, the activities of these compounds were not thorough, just the evaluation of the cytotoxicity assays, and the other activities and their mechanisms are worth further exploration.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
ZL, JH, and WK conceived the research subject. ZL, YN, LZ, LM, SC, and MW contributed to the development and writing of the manuscript. WK and ZL contributed in validating, reviewing, and supervising the project. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (31900292), Science and Technology Development Program of Henan Province (202102110149, 212102110469), and the Science and Technology Project of Kaifeng (1908005).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.657028/full#supplementary-material
Bruhn, T., Schaumlöffel, A., Hemberger, Y., and Bringmann, G. (2013). Specdis: quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 25, 25243–25249. doi: 10.1002/chir.22138
Delectis Flora Reipublicae Popularis Sinicae Agendae Academiae Sinicae Edita. (1986). Flora of China. Beijing: Science Press. 73:006.
Georg, S., and Joerg, V. (1985). Valepotriat-artefakte aus centranthus ruber (L.) Dc. Arch. Pharm. 318, 515–519. doi: 10.1002/ardp.19853180607
Kim, J. S., and Kang, S. S. (2013). Chemical constituents of plants from the genus Patrinia. Nat. Prod. Sci. 19, 77–119. Available online at: http://www.e-nps.or.kr/.
Liu, Z. H., Hou, B., Yang, L., Ma, R. J., Li, J. Y., Hu, et al. (2017a). Iridoids and bis-iridoids from Patrinia scabiosaefolia. RSC Adv. 7, 24940–24949. doi: 10.1039/C7RA03345A
Liu, Z. H., Ma, R. J., Yang, L., Li, J. Y., Hu, J. M., and Zhou, J. (2017b). Triterpenoids and iridoids from Patrinia scabiosaefolia. Fitoterapia 119, 130–135. doi: 10.1016/j.fitote.2017.04.011
Liu, Z. H., Xu, L. T., Xu, X. Q., Niu, Y., Saadeldeen, F. S. A., and Kang, W. Y. (2019). Effects and mechanisms of iridoid glycosides from Patrinia scabiosaefolia. Food Chem. Toxicol. 134, 110806–110811. doi: 10.1016/j.fct.2019.110806
Monks, A., Scudiero, D., Skehan, P., Shoemaker, R., Paull, K., Vistica, et al. (1991). Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Insit. 83, 757–766. doi: 10.1093/jnci/83.11.757
Reed, L. J., and Muench, H. (1938). A simple method of estimating fifty percent endo-points. Amer. J. Hygiene. 27, 493–797. doi: 10.1093/oxfordjournals.aje.a118408
Veith, J., Schneider, G., and Lemmer, B. (1986). The influence of some degradation products of valepotriates on the motor activity of light-dark synchronized mice. Planta Med. 3, 179–183. doi: 10.1243/PIME_PROC_1954_168_069_02
Xiao, M., Zhu, S., and Zhang, A. (2007). Research advance in medicinal and edible utilization of plants of Parinia. J. Jinling Inst. Technol. 23, 83–86. doi: 10.3969/j.issn.1672-755X.2007.03.021
Keywords: Patrinia scabiosaefolia, iridoid, iridoid glycoside, bis-iridoid glycoside, cytotoxic activity
Citation: Liu Z, Niu Y, Zhou L, Meng L, Chen S, Wang M, Hu J and Kang W (2021) Nine Unique Iridoids and Iridoid Glycosides From Patrinia scabiosaefolia. Front. Chem. 9:657028. doi: 10.3389/fchem.2021.657028
Received: 22 January 2021; Accepted: 04 March 2021;
Published: 29 March 2021.
Edited by:
Thierry Brigaud, Université de Cergy-Pontoise, FranceReviewed by:
Ya-nan Yang, Institute of Materia Medica, ChinaCopyright © 2021 Liu, Niu, Zhou, Meng, Chen, Wang, Hu and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenyi Kang, a2FuZ3dlbnlAaG90bWFpbC5jb20=; Jiangmiao Hu, aHVqaWFuZ21pYW9AbWFpbC5raWIuYWMuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.