 Nicolai A. Aksenov1
Nicolai A. Aksenov1 Alexander V. Aksenov
Alexander V. Aksenov Dmitrii A. Aksenov
Dmitrii A. Aksenov
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Chem. , 11 February 2020
Sec. Organic Chemistry
Volume 8 - 2020 | https://doi.org/10.3389/fchem.2020.00077
This article is part of the Research Topic Nitro Compounds as Versatile Building Blocks for the Synthesis of Pharmaceutically Relevant Substances View all 7 articles
Unusual reactivity of nitroalkanes, involving formation of aci-forms (nitronic acids or nitronates) and their subsequent interaction with carbon-based nucleophiles, is surveyed in this review.
Aliphatic nitro compounds can be easily deprotonated at α-position in the presence of bases, and the formed anionic intermediates are normally employed in a variety of nucleophilic addition or substitution reactions. However, the same compounds can also serve as electrophilic synthons after Bronsted acid-assisted tautomerization into aci-forms. In these form nitro-compounds can be attacked, for example by electron rich arenes to form aryloximes, which can be further transformed into anilides (Yadav et al., 2010; Augustine et al., 2011), anilines (John and Bergens, 2011), ketones (Joseph et al., 1994), aldehydes (Ranu and Sarkar, 1988), or various products of their subsequent rearrangements (Sardarian and Shahsavari-Fard, 2008). It should be pointed out, that in-situ generated aci-form is only weakly nucleophilic and can efficiently interact with a limited number of relatively strong nucleophiles. This process could be severely impeded by unfavorable position of the tautomeric equilibrium, which is normally shifted toward nitro-form at low pH. This situation can be addressed by carrying out the reaction in the presence of excess acid at low temperature as in classical Nef reaction (Nef, 1894). To this end, strong Bronsted (Nakamura et al., 2007) or Lewis (Marce et al., 2016) acids could be employed. An alternative approach involves a tying the reactive aci-tautomer in acylated (Lehr et al., 1979), alkylated (Falck and Yu, 1992), or silated (Colvin et al., 1980) forms. Analysis of synthetic methods employing these unusual electrophilic synthons and their application in preparation of medicinally relevant scaffolds are analyzed in this review.
Processes of C-Aryl bonds formation with participation of nitroalkanes in a role of electrophilic synthon are pretty exotic. The reasons for this are summarized in our introduction and were very well-detailed in several reviews (Lalli, 2000; Smirnov et al., 2012; Tabolin et al., 2017, 2018). Arguably, one of the earliest contributions to this area was made by Fujisawa, who demonstrated interaction of nitroalkanes with organometallic reagents (Fujisawa et al., 1983). To this end, nitroethane 1.1 was treated with n-BuLi and then with Vilsmeier reagent (Scheme 1, Equation 1). It is known that lithium nitronate derivatives are not electrophilic, so even in the presence of excess n-BuLi they only afford products of 2-fold deprotonation. These dianions (1.7) can be employed as nucleophilic synthons in reactions with ketones (Seebach et al., 1982), as well as alkylating (Seebach et al., 1977) or acylating (Ram and Ehrenkaufer, 1986) reagents (Scheme 1, Equation 2). Interestingly, the generated aci-nitroiminium chloride species 1.9 could also be involved in the reaction with two equivalents of Grignard reagent to afford oximes 1.10 in good yields (Fujisawa et al., 1983). Catalytic version of this reaction was also investigated, to demonstrate that in the presence of copper(I) chloride the same oximes (1.10) were generated in nearly quantitative yields (Scheme 1, Equation 3) (Fujisawa et al., 1983). It should be pointed out that nitronates 1.12 could be generated in situ via addition of lithiumorganic compounds to trans-β-nitrostyrenes 1.11 and further reacted with Grignard reagents in the same manner (Scheme 1, Equation 4) (Fujisawa et al., 1983).
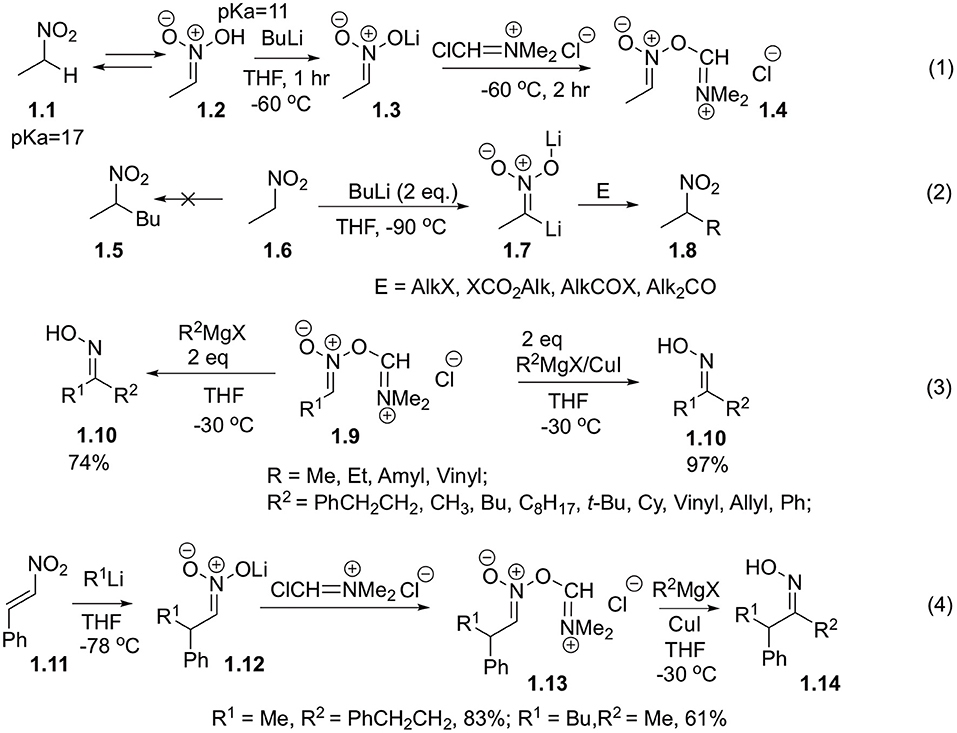
Scheme 1. Reactions of electrophilically activated nitroalkanes with Vilsmeier reagent.
Much more interesting are synthetic routes, involving direct interaction of nitroalkanes with nucleophilic arenes. Pioneering research in this area was performed by Ohwada et al. (1987a,b). He discovered that β-nitrostyrenes 2.1 in triflic acid form N, N-dihydroxyiminiumbenzyl dications 2.2, which was confirmed by a series of cryoscopic spectral methods, including 18O-labeling studies (Ohwada et al., 1986). These electrophiles are sufficiently reactive to be intercepted even by chlorobenzene. Nitronic acid formed in this process subsequently reacts with a second equivalent of arene affording 1,2,2-triarylethan-1-one oxime 2.3 (Scheme 2, Equation 1). Beckmann rearrangement of this oxime did not occur, probably, under highly acidic conditions it exists in diprotonated form. Influence of diprotonation on reaction yields was investigated in details. Thus, it was shown that upon employment of trifluoroacetic and sulfuric acids as well as in the presence of substoichiometric amounts of trific acid, the reaction afforded diarylation products in marginal yields (c.a. 30%), which was explained by low concentration of the diprotonated form in these media. Later, it was discovered that protonated nitronic acids could also be involved in reactions with arenes. To this end sodium nitronate 2.4, obtained from nitromethane or nitroethane was slowly added to a cold mixture of benzene and HF (Scheme 2, Equation 2). The authors pointed out that the reaction yields of oximes 2.7 were affected by a number of factors, and that the average reaction efficiency is related to competing non-productive C-protonation, which produce non-reactive nitroalkanes. This correlates well with trends observed in mechanistically related Nef reaction.
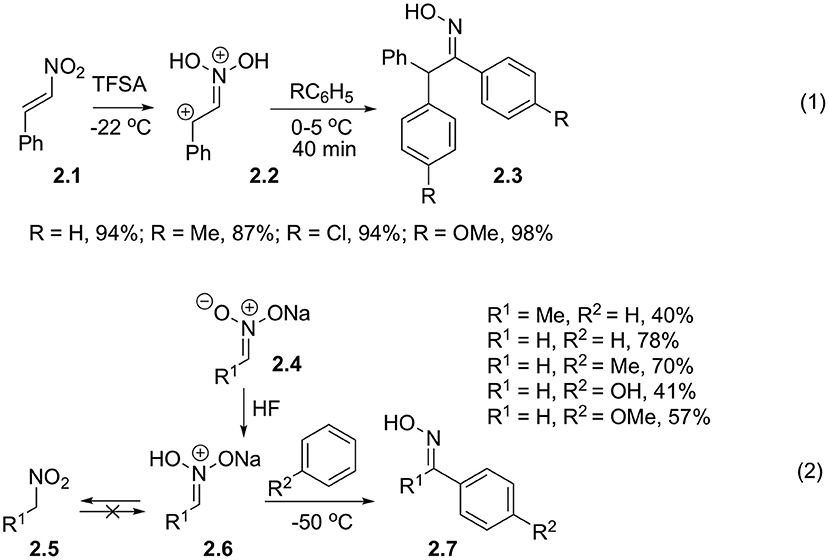
Scheme 2. Alpha-activation of nitroalkanes toward reaction with electron-rich arenes.
Ohwada with co-workers also investigated reactions of diprotonated 2-nitropropenes 3.1 with nucleophilic arenes. It was demonstrated, that in the presence of methanol aci-form 3.3 formed after Michael addition of arene to nitroalkene, could be efficiently involved in the Nef reaction, ultimately providing arylacetone 3.6 (Scheme 3, Equation 1) (Okabe et al., 1989; Ohwada et al., 1990). It should be pointed out, that this reaction might take a different route at elevated temperatures. Typically, in this case, nitronate 3.8 formed after addition of arene species to nitroalkene undergo rearrangement into unsaturated nitroso compound 3.9, which subsequently cyclizes into 1,2-benzoxazine 3.10 (Scheme 3, Equation 2) (Ohwada et al., 1990).
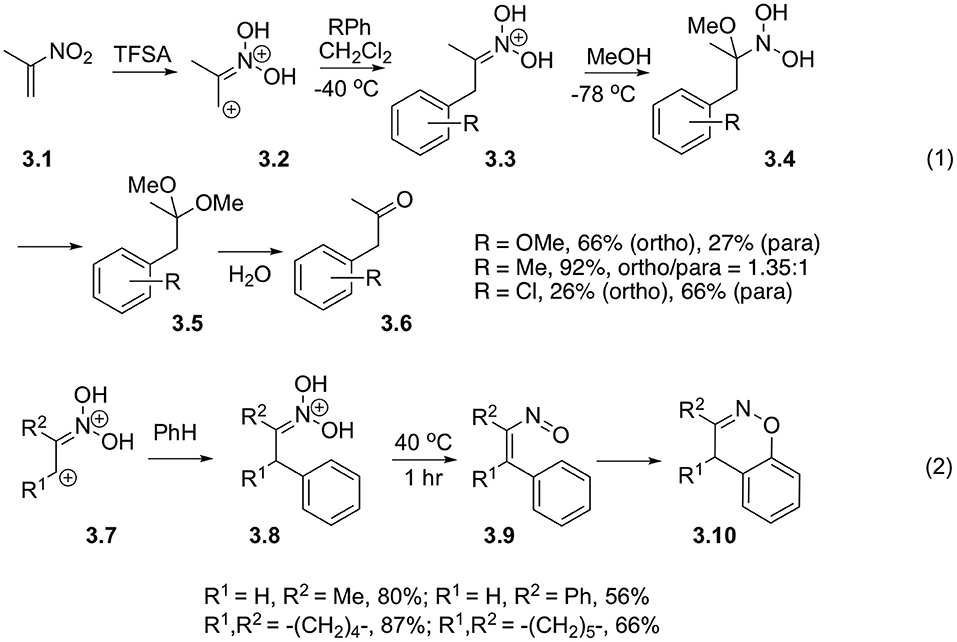
Scheme 3. Beta-activation of nitroalkanes toward reaction with electron-rich arenes.
Synthetic strategy capitalizing on double arylation of nitroalkenes was further developed in recent work by Golushko et al. (2018), describing reaction of 2,2,2-trihalogeno-1-nitroethylenes 4.1 with arenes in the presence of strong Brønsted and Lewis acids (Scheme 4, Equation 1). The best yields of bis-arylated products 4.3 were obtained in the presence of excess TfOH at room temperature. The reaction intermediates were studied by NMR spectroscopy and computer modeling. Regioselectivity of the reaction is not high and, typically, mixtures of regioisomers are formed. In reaction of sterically encumbered CBr3-derivatives high para-selectivity was achieved for the first (Michael reaction) step. Also, it should be pointed out that the products of ortho-substitution are forming mixtures of atropomers with axial chirality, which complicates analysis of the product mixture even further. An additional support for the reaction mechanism is obtained by intersection of the intermediate cations 4.6 with an appropriate nucleophile, for example chloride ion (Scheme 4, Equation 3). Under the same conditions, methanol only serves as a proton carrier and mediates isomerization of the aci- into nitro-form 4.5 (Scheme 4, Equation 2) (Golushko et al., 2018).
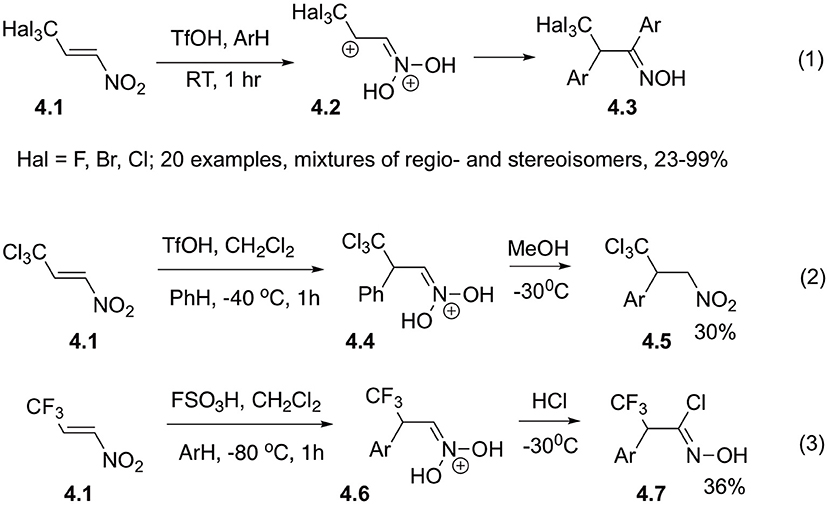
Scheme 4. Reaction of trihalogenmethylated nitro-olefins.
Reaction of nitroalkanes in aci-form (5.1) with non-activated arenes also occur in the presence of triflic acid and provides the corresponding oximes 5.3. In this case, participation of bis-protonated form of nitronic acid 5.2 was also postulated (Scheme 5, Equation 1). Utilization of 10% solution of triflic acid in TFA allows to improve the reaction efficiency (Ohwada et al., 1991). It should be pointed out, that benzoylnitromethanes 5.4 readily enolizable in the presence of strong Brønsted acid could participate in this type of transformations directly, without need for a separate conversion into nitronate form (Scheme 5, Equation 2). Computations suggest that reactive electrophilic species in this transformation is triple-protonated nitroketone species 5.5. This reaction was studies for a wide range of substrates, and typically provides good yields of products 5.6 derived from benzene, xylenes and anisole. In the latter case, however, para/ortho selectivity is quite low, providing mixtures of products c.a 2:1 (Scheme 5, Equation 2) (Ohwada et al., 1991; Takamoto et al., 2009).
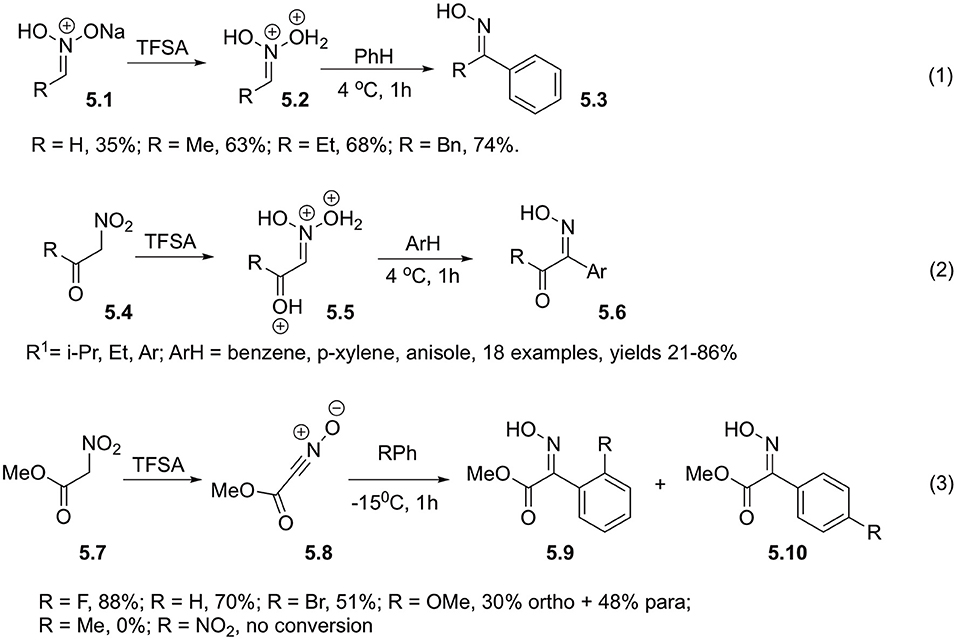
Scheme 5. Employment of TFSA to produce aci-form of nitroalkanes.
Nitroacetic esters 5.7 also react under the same conditions (Coustard, 1991; El Bahhaj et al., 2014). At 15°C they were shown to react with halogen- and alkoxy-substituted arenes affording oximes of α-ketoesters 5.9 and 5.10 in high yields (Scheme 5, Equation 3). These compounds were used as key intermediates in preparation of psammapline-like inhibitors of histone deacetylase. In contrast to Ohwada (Ohwada et al., 1991), advocating for formation of multi-protonated nitronates, Jacquesy in his work postulated formation of nitrile oxide 5.8 as a key electrophilic intermediate (Scheme 5, Equation 3) (Coustard et al., 1992); although this hypothesis was not experimentally verified.
It was shown that 1,1-bis(methylthio)-2-nitroethylene (6.1) could be transformed into the corresponding nitrile oxide 6.2 in superacidic medium (triflic acid or HF-SbF5). This nitrile oxide can be intercepted with various nucleophilic reagents, including electron rich arenes. Structures of products 6.3 and intermediates were investigated and the proposed mechanism was supported with NMR-spectroscopy and mass-spectrometry (Scheme 6, Equation 1). It should be pointed out, that high para-selectivity was observed in reactions involving anisole (Coustard and Jacquesy, 1993; Coustard, 1995). Similarly, at low temperature in superacidic medium proceeds diprotonation of 1-arylamino-1-methylthio-2-nitroethylenes 6.6. Subsequent elimination of water leads to the formation of nitriloxides 6.7, which could be intercepted with benzene to afford arylimino derivatives 6.8 (Scheme 6, Equation 2). Since the starting material 6.6 employed in this reaction contained aniline moiety, at higher temperature intramolecular cyclization occur to produce indole core 6.9 (Scheme 6, Equation 2) (Kearney et al., 1992b; Coustard, 1996; Cousson and Coustard, 1998). For better understanding of this process, reactivity of N-alkyl derivatives of 1-amino-2-nitroethylene 6.10 was also studied. It was confirmed, that in solution of triflic acid these substrates experience C,O-diprotonation to produce hydroxynitrilium ions 6.11, which could further react either with triflate anion or with benzene serving as a solvent. Reaction with triflate is a major detrimental factor affecting the efficiency of formation of the target oxime molecules 6.13 (Scheme 6, Equation 3) (Coustard, 1999).
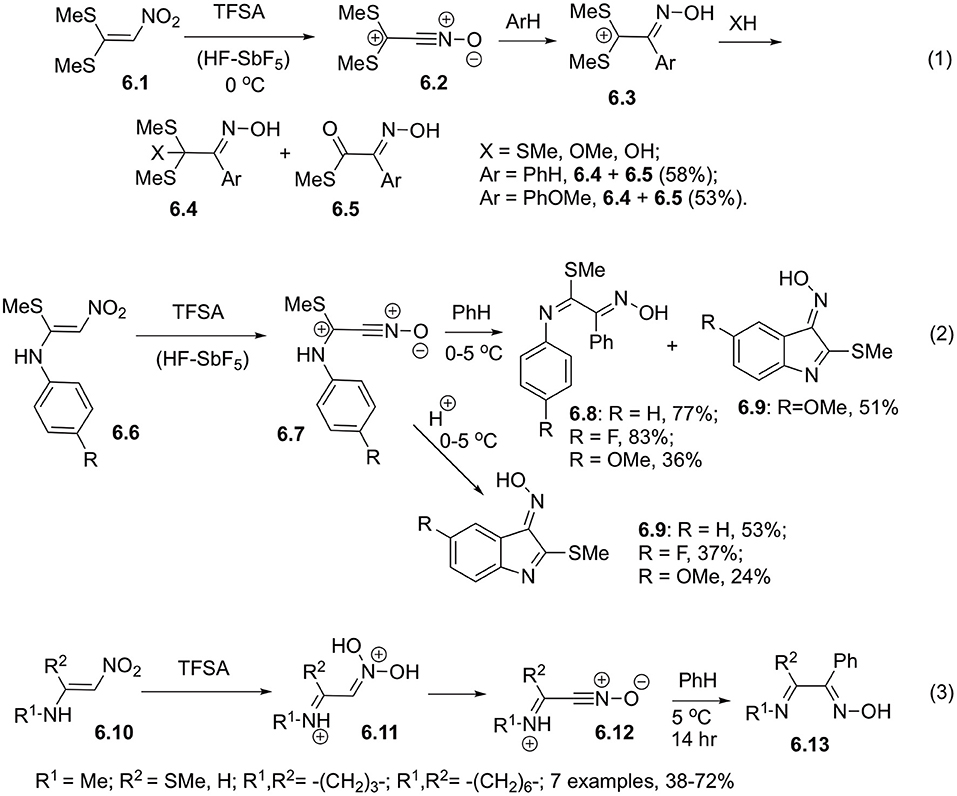
Scheme 6. Reactivity of 1,1-bis(methylthio)-2-nitroethylene.
Decarboxylation reaction could serve as a great alternative to α-deprotonation in the process of generation of aci-forms of nitroalkanes. Upon heating, dimethylnitromalonic ester 7.1 undergoes decarboxylation with subsequent elimination of water from nitro-functionality. The resulting methylcarbonocyanidate N-oxide 7.2 could undergo further dimerization into bis(carbomethoxy)furoxan or could be intercepted with dipolarophiles of electron rich aromatic compounds to afford oximes 7.3 in marginal yields. It should be mentioned that this reaction is limited to sterically hindered arene substrates, such as mesitylene, 1,2,3,4-tetramethylbenzene, pentamethylbenzene, or 3.3-dimethylanisole (Scheme 7, Equation 1) (Shimizu et al., 1985).
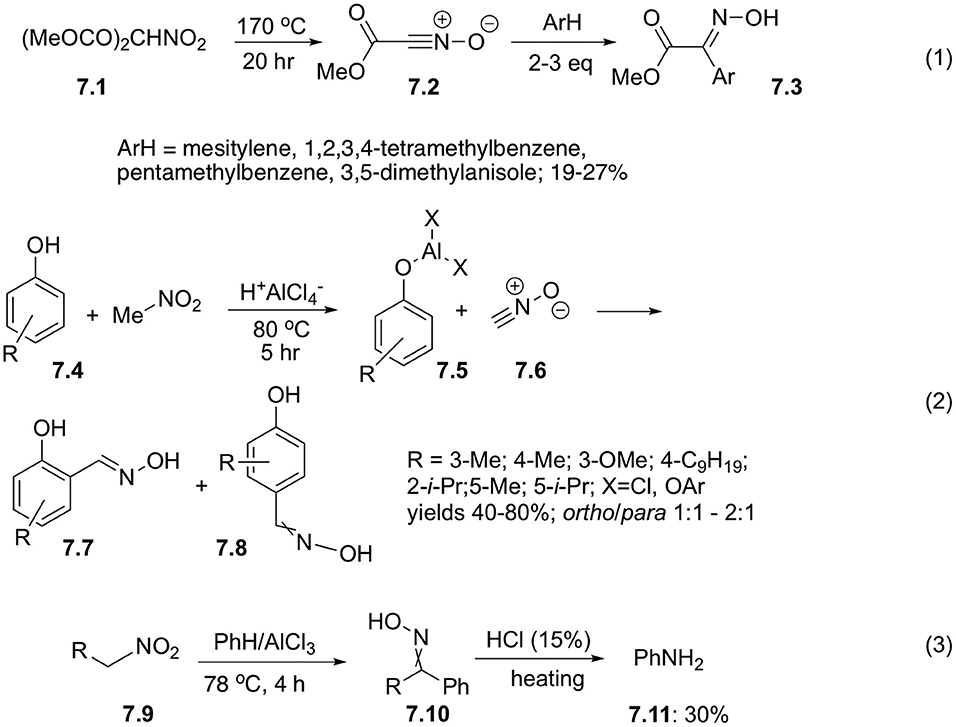
Scheme 7. Reactions of nitroalkanes with arenes under conditions of Frieden-Crafts reaction.
Satori and co-workers suggested that nitriloxides generated from nitroalkanes could react with phenoxides to afford salicylic oximes. Indeed, this reaction of phenols 7.4 involving nitromethane proceeds well in the presence of AlCl3, but higher nitroalkanes perform poorly (Sartori et al., 1994). Mild reaction conditions allow for preparation of wide variety of oximes 7.7 and 7.8 in reasonable yields, but regioselectivities are often marginal (Scheme 7, Equation 2). ortho-Oximes 7.7 were obtained as pure E-forms, which was explained by coordination with metal cation, while para-isomers 7.8 were isolated as mixtures of E- and Z-isomers (Sartori et al., 1994). It should be pointed out, that less reactive arenes, such as mesitylene, toluene, benzene, and even chlorobenzene, could also be involved into this reaction, but in this case the products were obtained as mixtures of stereo- and regioisomers in poor yields (Kim et al., 1995).
Sometimes, oximes 7.10 generated in situ in the reaction of arenes with electrophilically activated nitroalkanes 7.9 can be involved into subsequent Beckmann rearrangement. The very first example of such reactivity was reported by Lambert, who demonstrated that nitromethane could react with benzene in the presence of AlCl3 to afford benzaldehyde oxime, which after acidic quench afforded aniline (7.11) (Scheme 7, Equation 3) (Lambert et al., 1949).
Electrophilic activation of nitroalkanes in combination with Beckmann rearrangement could be used for designing of very powerful synthetic approaches employing C-H functionalization strategies. Remarkably, it was shown that the outcome of this transformation strongly depends on the nature of nitroalkane employed. Aksenov demonstrated, that nitroethane and other higher primary nitroalkanes 8.1 in polyphosphoric acid (PPA) media exist in the form of electrophilic nitronates, stabilized in phosphorylated form 8.2. These moieties could readily react with a variety of electron rich arenes and hetarenes to afford oxime derivatives 8.3, which upon heating in PPA undergo further Beckmann rearrangement to yield carboxamides 8.4 (Scheme 8). Overall, this sequence of step works as highly efficient direct acetamidation of aromatic substrates (Aksenov et al., 2010). Interestingly, that the same reaction can be carried out in the presence of additional nucleophilic substituents to design a one-pot cascade process toward more complex heterocyclic scaffolds. For example, reaction with para-substituted phenols 8.5 provided oximes 8.6, which after Beckmann rearrangement afforded carboxamide 8.7. Subsequent condensation, involving ortho-hydroxyl substituent resulted in formation of benzoxazoles 8.8 in good yields (Scheme 8) (Aksenov N. A. et al., 2015). Related processes, featuring nucleophilic secondary nucleophilic attack with nitrogen atom to form pyrimidine ring was also reported (Aksenov et al., 2016).
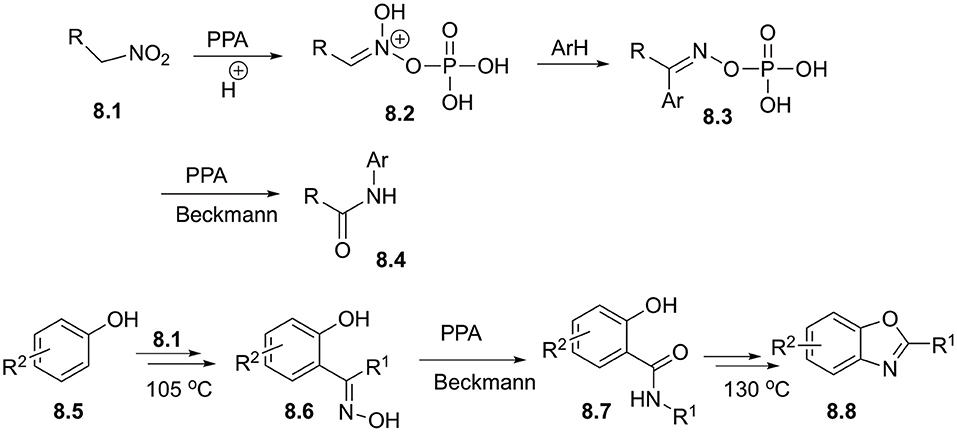
Scheme 8. Reactions of primary nitroalkanes with arenes in polyphosphoric acid.
Under the same reaction conditions process involving nitromethane 9.1 takes a different route (Scheme 1, Equation 1). Electrophilic nitronate 9.2 initially formed in PPA, first underwent similar reaction with electron-rich arenes to afford phosphorylated species 9.3, and following elimination of phosphoric acid leading to formation of aldoxime 9.4. The later species, however, does not undergo Beckmann rearrangement, losing a molecule of water instead and providing nitrile 9.5. Finally, upon hydrolysis, primary amides 9.6 or the corresponding carboxylic acids were isolated (Scheme 9, Equation 1). Overall, instead of a new C-N bond formed in the reaction of higher primary nitroalkane homologs, here new C-C bond was introduced (Aksenov et al., 2012).
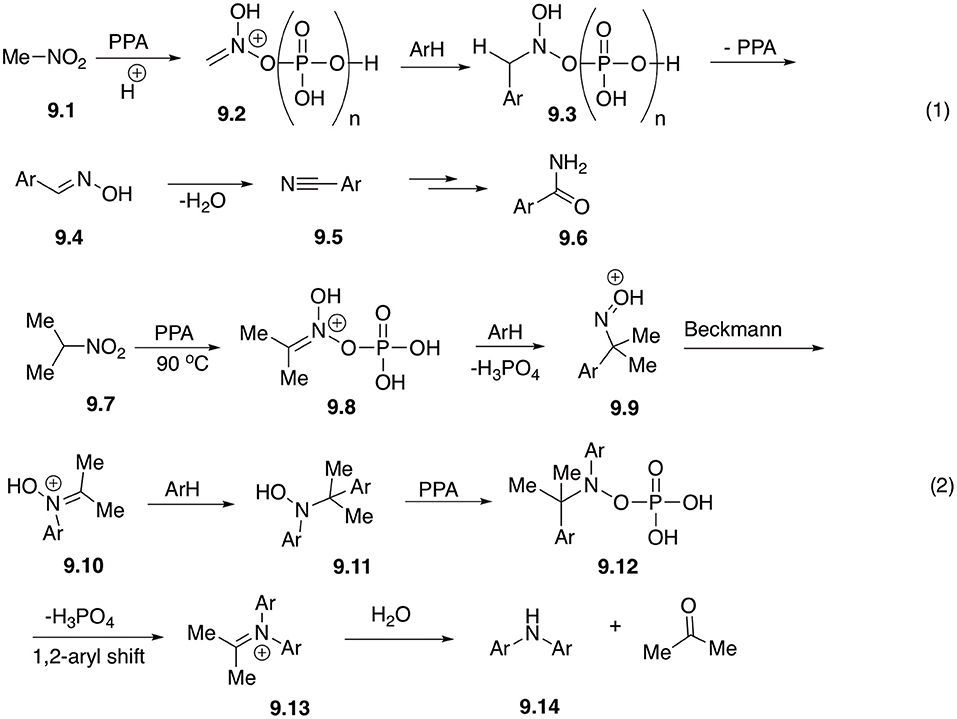
Scheme 9. Reactions of nitromethane and secondary nitroalkanes with arenes in polyphosphoric acid.
It was also shown that reactions involving secondary nitroalkanes, such as 2-nitropropane (9.7) proceed via a third alternative pathway (Scheme 9, Equation 2). Phosphorylated electrophilic nitronate species 9.8 first affords oxime 9.9, which undergoes Beckmann rearrangement. Produced nitrone 9.10, however, in this case lacks the α-CH bond and therefore cannot eliminate water to form a carboxamide. Instead, it experiences a second nucleophilic attack with arene to form hydroxylamine 9.11. Phosphorylated form of this hydroxylamine (9.12) experiences a subsequent 1,2-aryl shift (mechanistically related to pinacole rearrangement), to afford iminium species 9.13, which after hydrolytic quench produces diarylamine product 9.14 (Scheme 9, Equation 2) (Aksenov A. V. et al., 2015).
It is worth mentioning, that related intramolecular reactions involving electrophilically activated could also be very efficient. Thus, it was shown that cyclization of 2-nitroacetanylides 10.1 obtained via base-mediated hydrolysis of 1-arylamino-1-methylthio-2-nitroethylenes readily proceeded at room temperature in the presence of sulfuric or triflic acid to afford oximes of indoline-2,3-diones 10.3. It was stated, that protonated nitronates 10.2 serve as key intermediates in this transformation (Scheme 10, Equation 1) (Kearney et al., 1992a). It was also shown that intramolecular cyclizations of N, N-disubstituted nitroacetamides 10.6 bearing aryl substituent also occur in triflic acid. These starting materials can be generated in one-pot fashion in reaction between corresponding 3-(ω-arylalkylamino)propan-1-ols 10.4 and 1,1-bis(methylthio)-2-nitroethylene (10.5) (Scheme 10, Equation 2). To elucidate the mechanism of this transformation, the reaction was carried out at lowered temperature in sealed NMR tube. It was shown, that at 0°C nitroacetamide exists as triple-protonated species 10.7, but elevation of the temperature leads to the formation of nitrile oxide 10.8 and subsequent cyclization into lactones 10.9 (Scheme 10, Equation 2). It should be pointed out, that formation of six-membered ring is normally accompanied by loss of oxime functionality and aromatization, leading to the formation of isoquinoline-3-ones 10.10 in poor yields (Scheme 10, Equation 2) (Fante et al., 2014a).
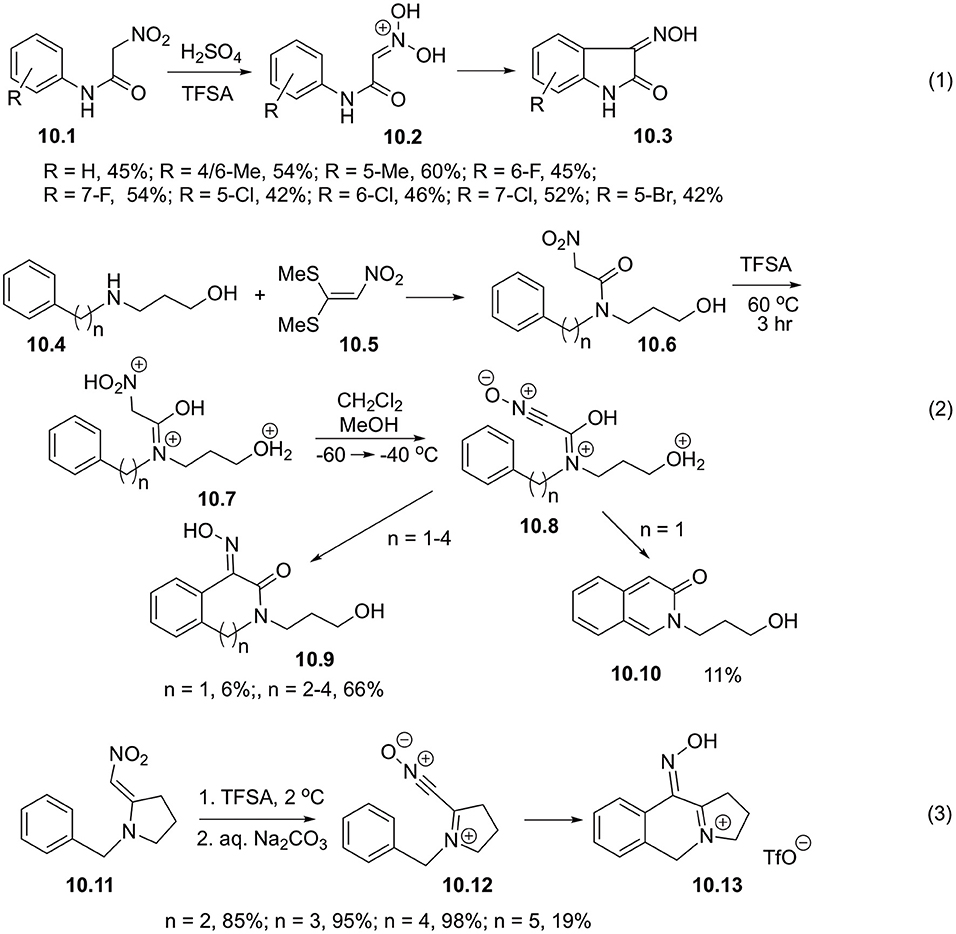
Scheme 10. Cyclizations of nitroalkanes involving intramolecular reactions with electron-rich arenes.
1-(ω-Phenylalkyl)-2-(nitromethylene)pyrrolidines 10.11 in triflic acid experience C,O-diprotonation with subsequent loss of water, leading to the formation of the corresponding nitrile oxides 10.12. The latter species can be readily involved into intramolecular SEAr reaction with phenyl substituent to afform tricyclic iminium species, isolated as triflic salts 10.13 in high yields (Scheme 10, Equation 3) (Fante et al., 2014b).
Expectedly, 2-nitromethylene-1-(ω-phenylalkyl)imidazolidines and 2-nitromethylene-1-(ω-phenylalkyl)hexahydropyrimidines (11.3) showcase very similar reactivity. It should be pointed out, that the precursors for these cyclizations could be obtained via reaction of readily available 1,2- or 1,3-diamines 11.1 with 1,1-bis(methylthio)-2-nitroethylene 11.2 in boiling acetonitrlile. Cyclization of intermediate 11.4 takes place at 60°C in triflic acid, and provide oximes 11.5 in good yields for 7-9-membered rings. More challenging closure of 10-membered rings is much less efficient (Scheme 11, Equation 1) (Coustard et al., 2006).
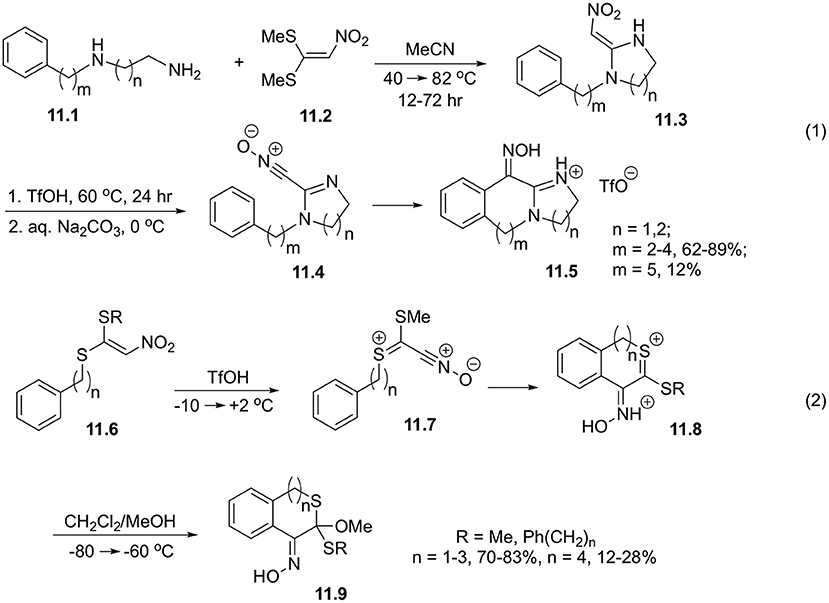
Scheme 11. Cyclization involving 1,1-bis(methylthio)-2-nitroethylene.
It was also shown that derivatives of 1-(methylthio)-2-nitro-1-(phenylalkylthio)ethylene 11.6 in triflic acid provided nitrile oxide 11.7, which underwent facile intramolecular cyclization affording intermediate dications 11.8, which were detected by NMR spectroscopy. Low temperature quenching with methanol afforded cyclic oximes of isothiochromanones 11.9, and their higher cyclic homologs. It was shown that ring sizes 6-8 could be assembled very efficiently, but larger rings were produced in marginal yields (Scheme 11, Equation 2) (Coustard, 2001).
Formally, vicarious substitution of hydrogen atom in hetarenes involving nitroalkanes in aci-form can also be regarded as related reaction. Thus, it was shown that anionic intermediate species 12.3 can be obtained in base-assisted interaction of nitroalkane 12.2 with triazines 12.1, further converting into corresponding oximes 12.4 in good yields (Scheme 12, Equation 1) (Rykowski and Makosza, 1984). 1,3-Dinitrobenzene 12.5 also showed similar reactivity in the presence of strong bases affording anion 12.6, which subsequently transformed into oxime 12.7 (Scheme 12, Equation 2) (Kawakami and Suzuki, 1999).
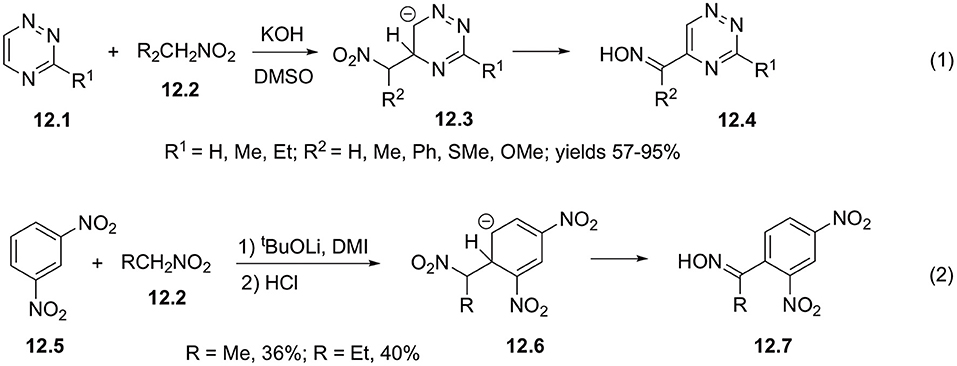
Scheme 12. Vicarious substitution off involving electrophilic mitroalkanes.
It should be pointed out that although cyclic nitronates are normally used as 1,3-dipoles, under certain conditions they could demonstrate electrophilic properties and can be readily engaged in the reactions with carbon-based nucleophiles. For example, cyclic species 13.1 was shown to react with TBSOTf to produce in situ highly reactive silated intermediate 13.2 susceptible for highly diastereoselective α-alkylation with ketene acetals, silylenol ethers, enamines, and allylstannanes (Scheme 13) (Smirnov et al., 2004). Reaction with silyl cyanides also was showcased to afford the corresponding α-cyanonitrozoacetal 13.7 in high yield (Scheme 13) (Mikhaylov et al., 2014).
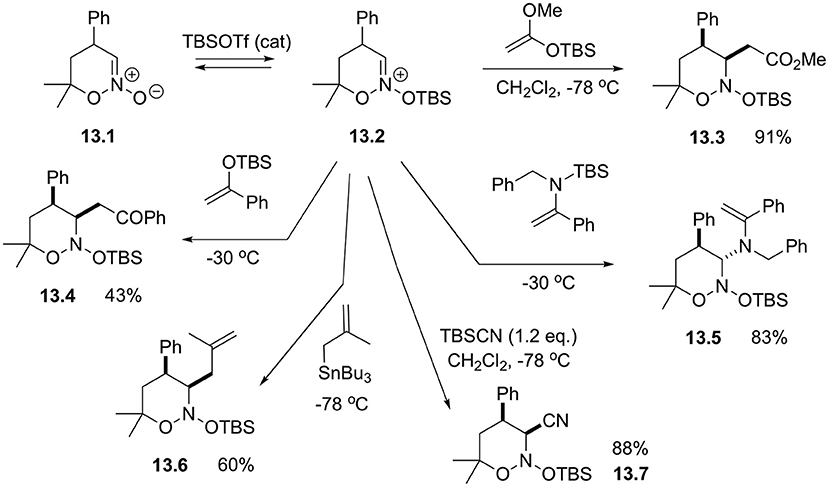
Scheme 13. Alpha-alkylations of cyclic nitronates.
It was also demonstrated that nitronates 14.1 bearing β-CH bonds afford N-silyloxyenamines 14.2 upon sylilation. Such species possess an electrophilic β-site and can react with alkali metal cyanide in the presence of Lewis acid. Desilation of the newly formed oxime function followed by nucleophilic cyclization to furnish isoxazole ring 14.5 in good yield (Scheme 14) (Ushakov et al., 2019).
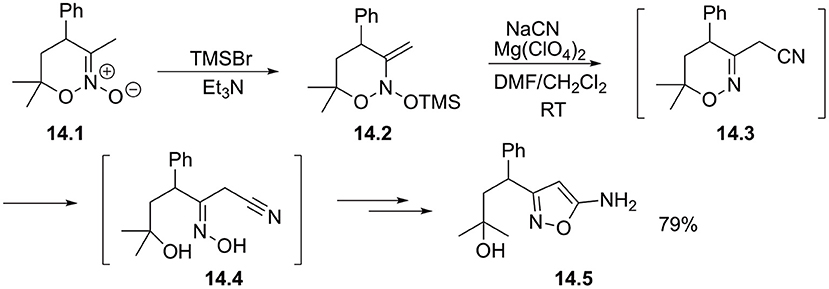
Scheme 14. Beta-cyanation of cyclic nitronates.
In conclusion, electrophilic activation of nitroalkane species in the presence of acidic and basic reagents is presented. Although these compounds are typically employed after deprotonation at α-position as nucleophilic synthons, there is a possibility to exploit the umpolung method of activation. Nitronic acids and nitronates (aka aci-forms of nitroalkanes) demonstrate electrophilic behavior and enable reactions with a variety of carbon-based nucleophiles. These unusual reactions allow to design novel processes, involving C-C bond forming steps, C-H-functionalizations, ring closures, providing highly versatile tool for organic synthesis and medicinal chemistry applications.
All authors were equally responsible for preparation of this review, which included literature search and analysis, and the preparation of the manuscript.
Preparation of this publication was supported by grant from the Russian Science Foundation (Grant number: 18-13-00238).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aksenov, A. V., Aksenov, N. A., Nadein, O. N., and Aksenova, I. V. (2010). Nitroethane in polyphosphoric acid: a new reagent for acetamidation and amination of aromatic compounds. Synlett 2628–2630. doi: 10.1055/s-0030-1258767
Aksenov, A. V., Aksenov, N. A., Nadein, O. N., and Aksenova, I. V. (2012). Nitromethane in polyphosphoric acid - a new reagent for carboxyamidation and carboxylation of activated aromatic compounds. Synth. Commun. 42, 541–547. doi: 10.1080/00397911.2010.526284
Aksenov, A. V., Aksenov, N. A., Orazova, N. A., Aksenov, D. A., Dmitriev, M. V., and Rubin, M. (2015). Direct metal-free synthesis of diarylamines from 2-nitropropane via the twofold C-H functionalization of arenes. RSC Adv. 5, 84849–84855. doi: 10.1039/C5RA17668A
Aksenov, A. V., Aksenov, N. A., Ovcharov, D. S., Aksenov, D. A., Griaznov, G., Voskressensky, L. G., et al. (2016). Rational design of an efficient one-pot synthesis of 6H-pyrrolo[2,3,4-gh] perimidines in polyphosphoric acid. RSC Adv. 6, 82425–82431. doi: 10.1039/C6RA17269E
Aksenov, N. A., Aksenov, A. V., Nadein, O. N., Aksenov, D. A., Smirnov, A. N., and Rubin, M. (2015). One-pot synthesis of benzoxazoles via the metal-free ortho-C-H functionalization of phenols with nitroalkanes. RSC Adv. 5, 71620–71626. doi: 10.1039/C5RA15128G
Augustine, J. K., Kumar, R., Bombrun, A., and Mandal, A. B. (2011). An efficient catalytic method for the Beckmann rearrangement of ketoximes to amides and aldoximes to nitriles mediated by propylphosphonic anhydride (T3P®). Tetrahedron Lett. 52, 1074–1077. doi: 10.1016/j.tetlet.2010.12.090
Colvin, E. W., Beck, A. K., Bastani, B., Seebach, D., Kaj, Y., and Dunitz, J. D. (1980). Synthesis, properties, and crystal structure of silyl nitronates (silyl esters of aci-nitroalkanes): towards the SN2 reaction path with retention of configuration at silicon. Helv. Chim. Acta 63, 697–710. doi: 10.1002/hlca.19800630320
Cousson, A., and Coustard, J.-M. (1998). X-ray structure determination of products resulting from trapping of hydroxynitrilium ions. Tetrahedron 54, 6523–6528. doi: 10.1016/S0040-4020(98)00325-1
Coustard, J.-M. (1995). 1,1-Bis(methylthio)-2-nitroethene in superacids: NMR study and reactivity of the formed hydroxynitrilium ion. Tetrahedron 51, 10929–10940. doi: 10.1016/0040-4020(95)00669-Y
Coustard, J.-M. (1996). 1-Arylamino-1-methylthio-2-nitroethene in superacids: NMR study and reactivity of the formed hydroxynitrilium ions. Tetrahedron 52, 9509–9520. doi: 10.1016/0040-4020(96)00485-1
Coustard, J.-M. (1999). 1-amino-2-Nitroethene derivatives in triflic acid: NMR study and triflates formation from their hydroxynitrilium ions. Tetrahedron 55, 5809–5820. doi: 10.1016/S0040-4020(99)00244-6
Coustard, J.-M. (2001). Synthesis of six- to nine-membered ring oximinoorthodithiolactones by cyclization of nitroketene S,S-acetals in trifluoromethanesulfonic acid. Eur. J. Org. Chem. 2001, 1525–1531. doi: 10.1002/1099-0690(200104)2001:8<1525::AID-EJOC1525>3.0.CO;2-O
Coustard, J.-M., Soro, Y., Siaka, S., Bamba, F., and Cousson, A. (2006). Synthesis of 6- to 10-membered ring (E)-hydroxyiminohydroazaazoniabenzocycloalkenes by cyclization of 2-nitromethylene-1-(ω-phenylalkyl)imidazolidine or 2-nitromethylene-1-(ω-phenylalkyl)hexahydropyrimidine in trifluoromethanesulfonic acid. Tetrahedron 62, 3320–3328. doi: 10.1016/j.tet.2006.01.057
Coustard, J. M., and Jacquesy, J. C. (1993). 1,1-Bis(methylthio)-2-nitroethene, a versatile synthon for the synthesis of oximinoorthothioesters. Phosphorus Sulfur Silicon Relat. Elem. 74, 427–428. doi: 10.1080/10426509308038148
Coustard, J. M., Jacquesy, J. C., and Violeau, B. (1991). Hydroxynitrilium ions as intermediates in the reaction of nitro derivatives with aromatics. Tetrahedron Lett. 32, 3075–3078. doi: 10.1016/0040-4039(91)80693-Z
Coustard, J. M., Jacquesy, J. C., and Violeau, B. (1992). Direct carbohydroximoylation of aromatics with primary nitroalkanes in triflic acid (TFSA). Tetrahedron Lett. 33, 8085–8086. doi: 10.1016/S0040-4039(00)74724-1
El Bahhaj, F., Desire, J., Blanquart, C., Martinet, N., Zwick, V., Simoes-Pires, C., et al. (2014). Superacid and thiol-ene reactions for access to psammaplin analogues with HDAC inhibition activities. Tetrahedron 70, 9702–9708. doi: 10.1016/j.tet.2014.10.053
Falck, J. R., and Yu, J. (1992). Synthesis of alkyl nitronates via Mitsunobu condensation. Tetrahedron Lett. 33, 6723–6726. doi: 10.1016/S0040-4039(00)61760-4
Fante, B., Soro, Y., Siaka, S., Marrot, J., and Coustard, J.-M. (2014a). Cyclization of nitroacetamide derivatives with a tethered phenyl ring in triflic acid. Synlett 25, 969–974. doi: 10.1055/s-0033-1340960
Fante, B., Soro, Y., Siaka, S., Marrot, J., and Coustard, J.-M. (2014b). Intramolecular Cyclization of 1-(ω-Phenylalkyl)-2-(nitromethylene)pyrrolidines in Triflic Acid. Synth. Commun. 44, 2377–2385. doi: 10.1080/00397911.2014.898307
Fujisawa, T., Kurita, Y., and Sato, T. (1983). A convenient synthesis of ketoximes from Grignard reagents and nitro compounds activated by N,N-dimethylchloromethyleniminium chloride. Chem. Lett. 12, 1537–1540. doi: 10.1246/cl.1983.1537
Golushko, A. A., Sandzhieva, M. A., Ivanov, A. Y., Boyarskaya, I. A., Khoroshilova, O. V., Barkov, A. Y., et al. (2018). Reactions of 3,3,3-trihalogeno-1-nitropropenes with arenes in the superacid CF3SO3H: synthesis of (Z)-3,3,3-trihalogeno-1,2-diarylpropan-1-one oximes and study on the reaction mechanism. J. Org. Chem. 83, 10142–10157. doi: 10.1021/acs.joc.8b01406
John, J. M., and Bergens, S. H. (2011). A highly active catalyst for the hydrogenation of amides to alcohols and amines. Angew. Chem. Int. Ed. 50, 10377–10380. doi: 10.1002/anie.201103137
Joseph, R., Sudalai, A., and Ravindranathan, T. (1994). Selective catalytic oxidative cleavage of oximes to carbonyl compounds with H2O2 over TS-1. Tetrahedron Lett. 35, 5493–5496. doi: 10.1016/S0040-4039(00)73533-7
Kawakami, T., and Suzuki, H. (1999). Masked acylation of m-dinitrobenzene and derivatives with nitroalkanes under basic conditions: nitromethylation and α-(hydroxyimino)alkylation. Tetrahedron Lett. 40, 1157–1160. doi: 10.1016/S0040-4039(98)02552-0
Kearney, T., Harris, P. A., Jackson, A., and Joule, J. A. (1992a). Synthesis of isatin 3-oximes from 2-nitroacetanilides. Synthesis 769–772. doi: 10.1134/s1068162011020105
Kearney, T., Joule, J. A., and Jackson, A. (1992b). Synthesis of 2-(methylthio)-3H-indol-3-one 3-oximes from 1-(arylamino)-1-(methylthio)-2-nitroethenes. Heterocycles 33, 757–762. doi: 10.3987/COM-91-S81
Kim, J. N., Song, J. H., and Ryu, E. K. (1995). Aluminum chloride-promoted carbohydroximoylation reaction of aromatic compounds with primary nitroalkanes. Synth. Commun. 25, 1711–1715. doi: 10.1080/00397919508015855
Lalli, K. K. (2000). Nitro and nitroso transformations in superacids. Coord. Chem. Rev. 210, 47–71. doi: 10.1016/S0010-8545(00)00316-7
Lambert, A., Rose, J. D., and Weedon, B. C. L. (1949). Aliphatic nitro compounds. XIX. Friedel-Crafts reactions with α- and β-nitro olefins. J. Chem. Soc. 42–46. doi: 10.1039/JR9490000042
Lehr, F., Gonnermann, J., and Seebach, D. (1979). Reactions of dilithionitroalkanes and -allylnitro derivatives with carbonyl compounds. Helv. Chim. Acta 62, 2258–2275. doi: 10.1002/hlca.19790620722
Marce, P., Lynch, J., Blacker, A. J., and Williams, J. M. J. (2016). Conversion of nitroalkanes into carboxylic acids via iodide catalysis in water. Chem. Commun. 52, 1013–1016. doi: 10.1039/C5CC08681G
Mikhaylov, A. A., Khomutova, Y. A., Arkhipov, D. E., Korlyukov, A. A., and Ioffe, S. L. (2014). ButMe2SiOTf-promoted cyanosilylation of six-membered cyclic nitronates with trialkylsilyl cyanides or tert-butyl isocyanide1. Mendeleev Commun. 24, 374–376. doi: 10.1016/j.mencom.2014.11.023
Nakamura, S., Sugimoto, H., and Ohwada, T. (2007). Formation of 4H-1,2-benzoxazines by intramolecular cyclization of nitroalkanes. scope of aromatic oxygen-functionalization reaction involving a nitro oxygen atom and mechanistic insights. J. Am. Chem. Soc. 129, 1724–1732. doi: 10.1021/ja067682w
Nef, J. U. (1894). Constitution of salts of the nitro-paraffins. Annalen 280, 263–291. doi: 10.1002/jlac.18942800209
Ohwada, T., Itai, A., Ohta, T., and Shudo, K. (1987a). Nitroethylene yields (N,N-dihydroxyiminium)methylium dication in trifluoromethanesulfonic acid. Dications stabilized by Y delocalization. J. Am. Chem. Soc. 109, 7036–7041. doi: 10.1021/ja00257a022
Ohwada, T., Ohta, T., and Shudo, K. (1986). Protonation of nitro groups. Diprotonation of β-nitrostyrenes in trifluoromethanesulfonic acid. J. Am. Chem. Soc. 108, 3029–3032. doi: 10.1021/ja00271a036
Ohwada, T., Ohta, T., and Shudo, K. (1987b). Reaction of β-nitrostyrenes with benzene catalyzed by trifluoromethanesulfonic acid. Formation and reaction of N,N-dihydroxyiminium-benzyl dications. Tetrahedron 43, 297–305. doi: 10.1016/S0040-4020(01)89957-9
Ohwada, T., Okabe, K., Ohta, T., and Shudo, K. (1990). Reactions of O,O-diprotonated nitro olefins with benzenes. Formation of phenylacetones, 4H-1,2-benzoxazines and biarylacetone oximes. Tetrahedron 46, 7539–7555. doi: 10.1016/S0040-4020(01)89066-9
Ohwada, T., Yamagata, N., and Shudo, K. (1991). Friedel-Crafts-type reactions involving di- and tricationic species. Onium-allyl dications and O,O-diprotonated aci-nitro species bearing a protonated carbonyl group. J. Am. Chem. Soc. 113, 1364–1373. doi: 10.1021/ja00004a044
Okabe, K., Ohwada, T., Ohta, T., and Shudo, K. (1989). Novel electrophilic species equivalent to α-keto cations. Reactions of 0,0-diprotonated nitro olefins with benzenes yield arylmethyl ketones. J. Org. Chem. 54, 733–734. doi: 10.1021/jo00265a001
Ram, S., and Ehrenkaufer, R. E. (1986). A facile synthesis of α-amino esters via reduction of α-nitro esters using ammonium formate as a catalytic hydrogen transfer agent. Synthesis. 133–135. doi: 10.1055/s-1986-31497
Ranu, B. C., and Sarkar, D. C. (1988). A simple, efficient, and highly selective method for the regeneration of carbonyl compounds from oximes and semicarbazones. J. Org. Chem. 53, 878–879. doi: 10.1021/jo00239a038
Rykowski, A., and Makosza, M. (1984). Reactions of organic anions. 126. Reaction of 1,2,4-triazines with nitronate anions, direct nucleophilic acylation of 1,2,4-triazines. Tetrahedron Lett. 25, 4795–4796. doi: 10.1016/S0040-4039(01)81522-7
Sardarian, A. R., and Shahsavari-Fard, Z. (2008). Efficient synthesis of 2-substituted benzoxazoles via Beckmann rearrangement of 2-hydroxyaryl ketoximes using diethyl chlorophosphate. Synlett 1391–1393. doi: 10.1055/s-2008-1072767
Sartori, G., Bigi, F., Maggi, R., and Tomasini, F. (1994). Reaction of nitromethane with aluminum phenolates: mild synthesis of salicylaldoximes. Tetrahedron Lett. 35, 2393–2396. doi: 10.1016/0040-4039(94)85228-6
Seebach, D., Beck, A. K., Mukhopadhyay, T., and Thomas, E. (1982). Diastereoselective synthesis of nitroaldol derivatives. Helv. Chim. Acta 65, 1101–1133. doi: 10.1002/hlca.19820650402
Seebach, D., Henning, R., Lehr, F., and Gonnermann, J. (1977). Carbon alkylations of α,α- and α,β-doubly deprotonated nitroalkanes. Tetrahedron Lett. 18, 1161–1164. doi: 10.1016/S0040-4039(01)92859-X
Shimizu, T., Hayashi, Y., and Teramura, K. (1985). A new synthetic method for alkyl carbonocyanidate N-oxides. Bull. Chem. Soc. Jpn. 58, 2519–2522. doi: 10.1246/bcsj.58.2519
Smirnov, V. O., Ioffe, S. L., Tishkov, A. A., Khomutova, Y. A., Nesterov, I. D., Antipin, M. Y., et al. (2004). New C-C coupling reaction of cyclic nitronates with carbon nucleophiles. Umpolung of the conventional reactivity of nitronates. J. Org. Chem. 69, 8485–8488. doi: 10.1021/jo048944k
Smirnov, V. O., Khomutova, Y. A., Tartakovsky, V. A., and Ioffe, S. L. (2012). C-C coupling of acyclic nitronates with silyl ketene acetals under silyl triflate catalysis: reactivity umpolung of aliphatic nitro compounds. Eur. J. Org. Chem. 2012, 3377–3384. doi: 10.1002/ejoc.201200239
Tabolin, A. A., Sukhorukov, A. Y., and Ioffe, S. L. (2018). α-Electrophilic reactivity of nitronates. Chem. Rec. 18, 1489–1500. doi: 10.1002/tcr.201800009
Tabolin, A. A., Sukhorukov, A. Y., Ioffe, S. L., and Dilman, A. D. (2017). Recent advances in the synthesis and chemistry of nitronates. Synthesis 49, 3255–3268. doi: 10.1055/s-0036-1589063
Takamoto, M., Kurouchi, H., Otani, Y., and Ohwada, T. (2009). Phenylation reaction of α-acylnitromethanes to give 1,2-diketone monooximes: involvement of carbon electrophile at the position α to the nitro group. Synthesis 4129–4136. doi: 10.1002/chin.201016076
Ushakov, P. Y., Tabolin, A. A., Ioffe, S. L., and Sukhorukov, A. Y. (2019). In situ generated magnesium cyanide as an efficient reagent for nucleophilic cyanation of nitrosoalkenes and parent nitronates. Eur. J. Org. Chem. 2019, 1888–1892. doi: 10.1002/ejoc.201801761
Keywords: nitroalkanes, electrophilic attack, synthetic methodology, C-H functionalization, oximes
Citation: Aksenov NA, Aksenov AV, Ovcharov SN, Aksenov DA and Rubin M (2020) Electrophilically Activated Nitroalkanes in Reactions With Carbon Based Nucleophiles. Front. Chem. 8:77. doi: 10.3389/fchem.2020.00077
Received: 23 November 2019; Accepted: 23 January 2020;
Published: 11 February 2020.
Edited by:
Alexey Sukhorukov, N.D. Zelinsky Institute of Organic Chemistry (RAS), RussiaReviewed by:
John Kallikat Augustine, Syngene International, IndiaCopyright © 2020 Aksenov, Aksenov, Ovcharov, Aksenov and Rubin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander V. Aksenov, YWxleGFrczA1QHJhbWJsZXIucnU=; Michael Rubin, bXJ1YmluQGt1LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.