 Wen Zhu
Wen Zhu Alanya J. Nardone
Alanya J. Nardone Lucciano A. Pearce
Lucciano A. Pearce
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Chem. Biol. , 06 June 2024
Sec. Molecular Sciences
Volume 3 - 2024 | https://doi.org/10.3389/fchbi.2024.1410435
This article is part of the Research Topic Structure-function relationship of enzymes through a chemical lens View all 6 articles
Bifunctional enzymes, characterized by their dual active sites, enable efficient chemical conversion and substrate channeling using elegant coupling mechanisms to coordinate the two active sites. In humans, several bifunctional enzymes synthesize de novo carbon-nitrogen bonds by hydrolyzing glutamine and ATP in distinct active sites. Notable examples include guanosine monophosphate synthetase, cytidine triphosphate synthetase, phosphoribosylformyl-glycinamidine synthase, asparagine synthetase, and nicotinamide adenine dinucleotide synthetase. A more complex example of multifunctional glutamine-hydrolyzing synthetases in humans is carbamoyl phosphate synthetase. These enzymes are crucial for the biosynthesis of amino acids, nucleic acids, and co-factors, thereby playing pivotal roles in human health. This review delineates recent progress in understanding the structural characteristics, regulatory mechanisms, and disease relevance of glutamine-hydrolyzing synthetases in humans. Insights into their catalysis and activity regulation offer potential pathways for developing novel therapeutics.
Bifunctional enzymes typically contain multiple domains with separate active sites, facilitating efficient substrate conversions (Moore, 2004). Linking two domains that possess distinct catalytic functions allows the committing steps in one biological process to produce localized substrates to feed into the next step of the reaction, leading to tunable cooperativity within metabolic pathways (Pareek et al., 2021). Colocalization of catalytic domains also prevents the potential leakage of toxic intermediate metabolites, causing unwanted damage (Knudsen et al., 2018). Free ammonia is a toxic metabolite, even though it is a useful nitrogen donor for amination reactions (Warren, 1962). Members of the glutamine amidotransferase (GATase) and glutamine-dependent nitrilase families can produce ammonia via glutamine hydrolysis. When the glutaminase domain (GD) is fused with an additional domain, the ammonia can be transported across the domain interface and utilized to introduce new carbon-nitrogen (C-N) bonds in other substrates that are activated in the second active site (Figure 1A). This strategy effectively overcomes the low availability of unprotonated ammonia at physiological pH and prevents cytotoxicity arising from ammonia leakage from the enzyme. As the substrate for one active site relies on the product of another active site, precise coordination and regulation of catalytic activities are widely observed in these bifunctional enzymes.
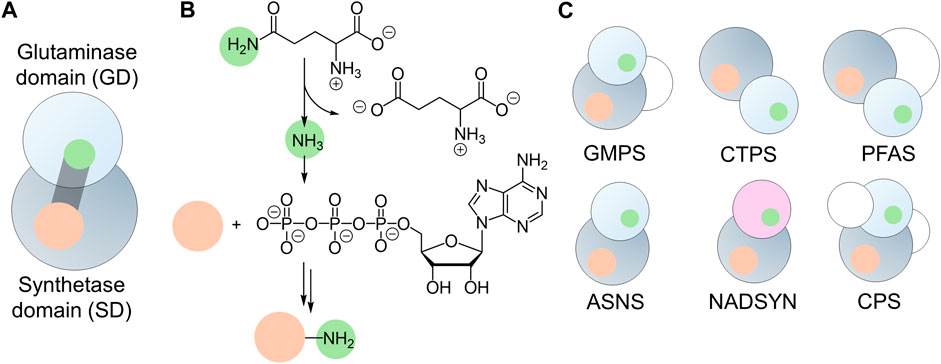
Figure 1. Architecture of Glutamine-hydrolyzing synthetases. (A) Cartoon illustration of the two prominent domains: the glutaminase domain (GD, light gray sphere) and the synthetase domain (SD, dark gray sphere), connected by an ammonia tunnel (gray cylinder). This tunnel links the GD active site (green) with the SD active site (orange). (B) General reaction catalyzed by glutamine-hydrolyzing synthetases, showcasing the substrate (orange sphere) activated by ATP in the synthetase domain and ligated to ammonia (green sphere) produced through glutamine hydrolysis. (C) Domain organization of six glutamine-hydrolyzing synthetases found in humans, featuring GATase (light gray spheres) or nitrilase (light purple sphere) as the glutaminase domain, synthetase domain (dark gray spheres), and additional domains (white spheres).
GATases are widely distributed in all kingdoms of life and are used to assimilate nitrogen in biological systems by participating in the biosynthesis of amino acids, nucleic acids, and co-factors (Massière and Badet-Denisot, 1998). GATases can be heteromeric complexes, in which the assembly of multiple subunits containing independent active sites relies on protein-protein interactions (Semmelmann et al., 2019). Examples of such heteromeric GATases include histidine amidotransferase (histidine biosynthesis) and anthranilate synthase (tryptophan biosynthesis) (Crawford, 1989; Alifano et al., 1996). Members of the GATase family also include multi-domain enzymes, in which the GD and additional domain(s) are fused into a single polypeptide chain. An example of such an enzyme is glutamine-fructose-6-phosphate aminotransferase. This enzyme contains a sugar isomerase domain, allowing the transformation of D-fructose-6-phosphate into D-glucosamine-6-phosphate (Nakaishi et al., 2009). GATases have been generally classified by the GD architecture into two evolutionary independent classes (Massière and Badet-Denisot, 1998). Class I (or trpG-type) GATase contains a cysteine-histidine-glutamate catalytic triad within a three-layer αβα architecture, in which cysteine serves as the nucleophile during the C-N bond cleavage, and histidine and glutamate modulate the nucleophilicity of cysteine. Class II (or purF-type) GATases, on the other hand, have a GD that adopts a four-layer αββα fold and lacks a catalytic triad. Instead, the active site of Class II GATase is defined by an N-terminal cysteine, together with asparagine and glycine residues forming an oxyanion hole to stabilize the tetrahedral intermediate after the nucleophilic attack of the cysteine on the substrate glutamine. Given that the catalytic cysteine is at the N-terminus, Class II GATases are also called N-terminal nucleophile (Ntn)-type GATases. In addition, glutaminase activity has also been found in enzymes that belong to the nitrilase superfamily, which is evolutionary unrelated to the GATases. The nitrilase superfamily can be divided into thirteen branches, and only two of them are glutamine-hydrolyzing enzymes that share the same glutaminase activity as GATases, both of which are NAD+ synthetases (Pace and Brenner, 2001).
A subgroup of bifunctional ammonia-assimilating enzymes introduces de novo C-N bonds into molecules that are activated by ATP hydrolysis in a synthetase domain (SD). Synthetases are ligases, in which the bond formation is usually driven by ATP hydrolysis (Frey and Hegeman, 2007). The use of ATP to activate inert functional groups provides a significant energetic driving force for ammonia incorporation (Figure 1B). Structurally, most SDs share a core αβα sandwich-fold, adopting either a canonical phosphate-binding loop (P-loop) or pyrophosphate binding loop (PP-loop) for ATP hydrolysis (Walker et al., 1982; Bork and Koonin, 1994). They are denoted as ATP phosphatases and ATP pyrophosphatases, respectively. To transport ammonia from the GD active site to the SD active site, these bifunctional enzymes also require a tunnel within the interior of the protein to avoid the ammonia being released into the solution.
While the enzymes featuring both glutaminase and synthetase domains are more functionally diverse in microorganisms, humans possess only a limited selection of glutamine-hydrolyzing synthetases. These include guanosine monophosphate (GMP) synthetase (GMPS, EC 6.3.5.2), cytidine triphosphate (CTP) synthetase (CTPS, EC 6.3.4.2), phosphoribosylformylglycinamidine (PFA) synthase (PFAS, EC 6.3.5.3), asparagine synthetase (ASNS, EC 6.3.5.4), and nicotinamide adenine dinucleotide (NAD+) synthetase (NADSYN, EC 6.3.5.1). In addition to these five enzymes, a multifunctional enzyme, carbamoyl phosphate synthetase (CPS, EC 6.3.5.5), is also present in humans (Figure 1C). As their name suggests, these bi- and multifunctional enzymes are involved in the central biological pathways in humans. Genetic mutations and up- or down-regulation of the expression levels can lead to pathological outcomes, highlighting the importance of understanding the structure-function relationship at the molecular level for a targeted drug discovery. The mechanistic and structural characterizations of bacterial and mammalian homologs of these enzymes that began decades ago have uncovered the chemical insights into their catalytic steps. Only recently has the complexity of the activity regulation by dynamics of the human enzymes and their involvement in numerous diseases started to be understood. Several recent reviews have discussed each individual enzyme from different organisms (Bearne et al., 2022; Ballut et al., 2023). We aim to provide a comparative analysis of recent advances in a few examples of human enzymes that couple glutamine and ATP hydrolysis. Understanding the reaction mechanisms, structural characteristics, and dynamical regulation of these examples, together with the diseases associated with their dysfunction will offer insights into novel rational ligand designs targeting the disease associated with the malfunction of these enzymes.
Human GMPS catalyzes the final step in the de novo biosynthesis of GMP from xanthosine 5′-monophosphate (XMP) (Figure 2A) (Ballut et al., 2023). The crystal structure of human GMPS homodimer has been resolved in the presence of XMP to 2.5 Å resolution (Figure 2B) (Welin et al., 2013). It features an N-terminal Class I GATase domain with a Cys104-His190-Glu192 catalytic triad and a C-terminal ATP pyrophosphatase domain in which amination of XMP occurs (Figures 2C, D) (Nakamura et al., 1995; Tesmer et al., 1996). The catalytic triad at the GD is approximately 40 Å away from the PP-loop, SGGVDS, of the SD within the same monomer (Figure 2B). The GD active site is widely exposed to the solvent and no ammonia tunnel is present in the structure. At the C-terminus of the SD, GMPS also contains two dimerization subdomains at the C-terminus of the SD, named D1 and D2, both of which contribute to dimerization and XMP binding. In this structure, XMP is anchored by salt bridges and hydrogen bonds with residues in D1 and D2, positioning the carbonyl at the C2 position of purine so that it directly points towards the PP-loop in the SD active site (Figure 2D) (Welin et al., 2013).
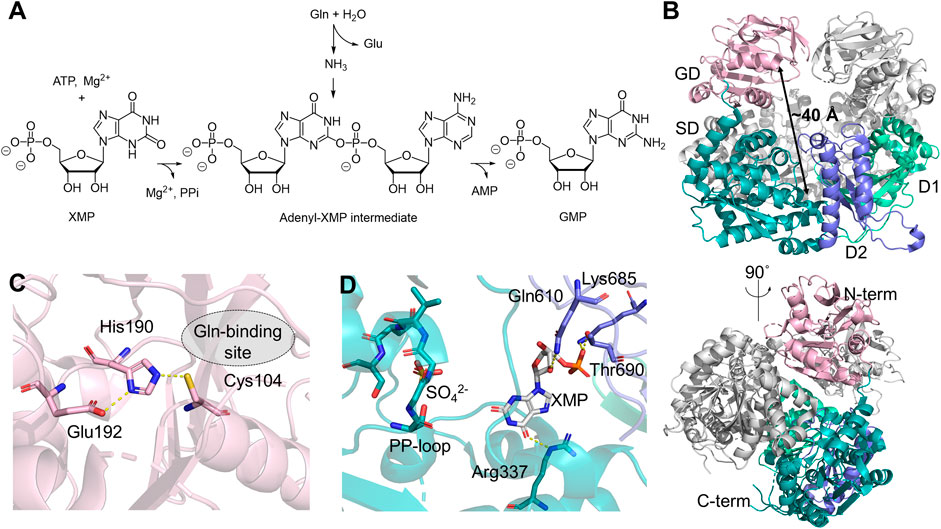
Figure 2. Human GMPS Structure and Function. (A) Reaction catalyzed by GMPS involves converting glutamine (Gln), ATP, and XMP into glutamate (Glu), GMP, adenosine monophosphate (AMP), and pyrophosphate (PPi). (B) Structure of the GMPS homodimer (PDB:2VXO), with the glutaminase domain (GD, light pink) and the synthetase domain (SD, teal) in one monomer. The subdomains, D1 and D2, are shown in light green and purple, respectively. (C) Catalytic triad in the GD, residues in 3 Å are depicted as sticks. (D) Key residues involved in the binding of XMP (white stick) and the orientation of the PP-loop (stick) in the SD.
Steady-state kinetics have been determined by following 14C incorporation into GMP from isotopically labeled substrates (Nakamura and Lou, 1995). ATP and glutamine follow classical Michaelis-Menten behavior, while the XMP-dependent kinetics is sigmoidal, suggesting an allosterically controlled activity (Nakamura and Lou, 1995). It has been shown that GMPS largely exists as a monomer in solution, regardless of the substrate binding in the SD, while the glutamine analog, 6-diazo-5-oxo-l-norleucine (DON), can induce dimerization, which is speculated to be the source of positivity cooperativity observed in the XMP-dependent kinetic measurements (Welin et al., 2013). The stoichiometric production of glutamate and GMP suggests that the activities in the two domains are highly synchronized (Nakamura and Lou, 1995). Although mounting evidence supports the ammonia channeling between two active sites based on enzyme kinetics (Nakamura and Lou, 1995; Oliver et al., 2013; Shivakumaraswamy et al., 2022), analytical gel filtration (Bhat et al., 2008; Welin et al., 2013), and cross-link mass spectrometry (Welin et al., 2013) using GMPS from several model organisms, no ammonia tunnels have been observed in any crystal structures, including that of the human homolog. Transient substrate-induced conformational changes, therefore, have been proposed to explain the formation of the ammonia tunnel (Welin et al., 2013). Molecular dynamics (MD) simulation of Escherichia coli GMPS predicts that rotation of the GD is necessary to allow direct contact between two domains. A disordered region located near the PP-loop in the SD may become organized upon nucleotide binding, thereby inducing conformational changes that assemble the ammonia tunnel (Tesmer et al., 1996). On the other hand, since domain rotation could adversely perturb enzyme dimerization, it remains unclear whether a similar domain rotation occurs in human GMPS, given that the E. coli enzyme lacks the D1 dimerization subdomain.
GMPS bridges amino acid pool and nucleotide biosynthesis, directly impacting the supply of building blocks for DNA. As one of the key enzymes in the purine biosynthesis pathway, altered GMPS expression levels have been found in many cancer types. High GMPS expression is linked to decreased survival rates and disease progression in prostate cancer (Wang Q. et al., 2021) and esophageal squamous cell carcinoma (Wang et al., 2021). Since GMPS expression is repressed by p53, GMPS could serve as a biomarker for p53-related cancer types (Holzer et al., 2017). Besides its biochemical role in cellular metabolism, GMPS also functions as a ubiquitin-specific protease activator via protein-protein interaction (Van Der Knaap et al., 2010). The ubiquitin-specific protease USP7 (or HAUSP) regulates pathways related to two major tumor suppressors, p53/MDM2 and FOXO4/PTEN, both of which are central to apoptosis (Wang et al., 2021). GMPS enhances the activity of USP7 by interacting with its C-terminal HUBL domains, promoting deubiquitinase activity towards substrates, such as p53 (Wang et al., 2021), MDM2, histone H2B. In Epstein-Barr virus (EBV) infected cells, the USP7-GMPS interaction also impacts the chromatin structure at a region of the EBV genome that controls EBV persistence (Wang et al., 2021). It remains unclear whether the USP7-GMPS interaction alters the dimer structure of GMPS or is associated with GMPS enzymatic activity in vivo.
CTPS is another member of the Class I GATase family involved in de novo nucleotide biosynthesis. It exists as a homotetramer in solution, with each monomer containing an N-terminal SD and the C-terminal GD, which converts uridine 5′-triphosphate (UTP) into CTP (Figures 3A, B) (Lieberman, 1956). In the GD, glutamine is hydrolyzed into glutamate and ammonia by the catalytic triad Cys399-His526-Glu528 (Figure 3C). The SD utilizes ATP to phosphorylate the C4 carbonyl of pyrimidine, generating ADP and a 4-phospho-UTP intermediate, which then reacts with ammonia. ATP hydrolysis is mediated by a conserved P-loop that features a GX3GXGK motif for coordinating the triphosphate group in ATP and magnesium (Figure 3D) (Kursula et al., 2006). In humans, there are two CTPS isozymes, encoded by the CTPS1 and CTPS2 genes located in Chromosome 1 and X, respectively, which share 74% sequence identity (van Kuilenburg et al., 2000). Structures of CTPS1 (Kursula et al., 2006; Lynch et al., 2021) and CTPS2 (Lynch et al., 2021) have both been resolved up to 2.8 Å resolution in the presence of substrates and/or products (Figure 3E). Both isoforms exist as a homotetramer, which stacks into filamentous structures (Figure 3F), although the condition for filament formation varies between the two isoforms. The polymerization of CTPS1 is triggered only by UTP binding, while CTPS2 can also polymerize in the presence of CTP and ADP. The substrate-induced CTPS1 polymerization results in a domain rotation that aligns the two active sites, creating openings at both active sites that can potentially facilitate ammonia relocation, although the complete tunnel architecture remains to be identified. In CTPS2, an ammonia tunnel is only present in UTP-bound CTPS2 filaments (Figure 3G) (Lynch et al., 2017), but not in the ADP/CTP-bound structure, in which a “gating” loop containing Pro52-His55-Val58 blocks the connection between two active sites (Lynch and Kollman, 2020). In contrast, an ammonia tunnel is readily present in the E. coli CTPS tetramer, and a hydrophobic gating residue, Val60, near the highly conserved PX2HX2V motif, has been proposed to regulate the tunnel access (McCluskey and Bearne, 2018; Bearne et al., 2022).
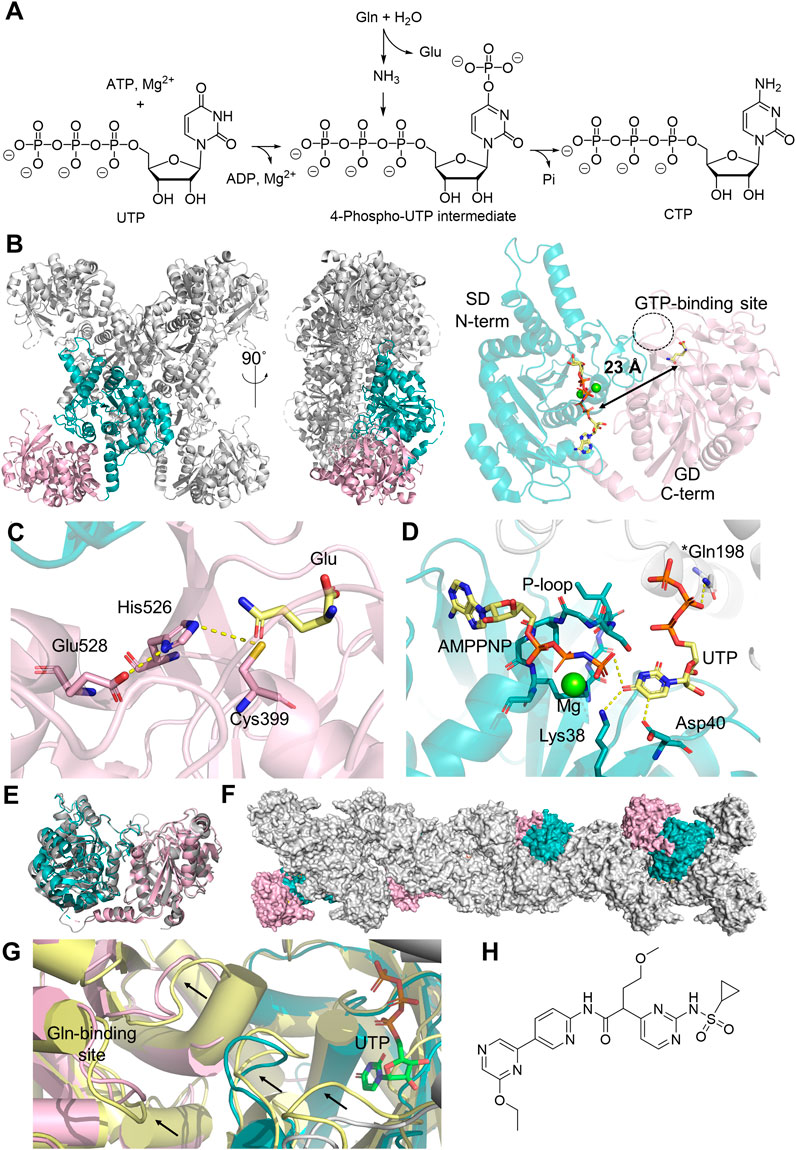
Figure 3. Human CTPS Structure and Function. (A) Reaction catalyzed by CTPS1 involves converting glutamine (Gln), ATP and UMP into glutamate (Glu), CTP, adenosine diphosphate (ADP) and phosphate (Pi). (B) Homotetramer structure of CTPS1 (PDB:7MGZ, gray), with the glutaminase domain (GD, light pink) and the synthetase domain (SD, teal) highlighted in one monomer. A 90-degree rotation illustrates the orientation from the N-terminal to the C-terminal domain. The monomer structure on the right displays the distance (black arrow) from the catalytic triad in the GD to the active site in the SD, where XMP (yellow sticks) and magnesium ions (green sphere) are located. The GTP binding site is shown in a dotted circle. (C) Zoomed-in glutaminase active site and the catalytic triad (stick) models and the binding of glutamate (yellow sticks). (D) SD active site highlighting UTP (yellow sticks) binding and its interactions with amino acids (sticks) within 3 Å (yellow dash line), alongside the ATP analog ATPPNP (yellow sticks) and a magnesium ion (green sphere). An asterisk on Gln198 indicates this residue is located at a different monomer. (E) Aligned monomer structures of CTPS1 (PDB:7MGZ, teal and light pink) and CTPS2 (PDB:7MH1, gray). (F) Polymerized CTPS1 (PDB:5U03, gray), with monomer highlighted in color. (G) Structural comparison of CTPS1 under non-filament forming condition (PDB:7MH0, yellow) and the filament-forming condition (PDB:5U03, color code for teal/pink/gray is the same as panel (G)), black arrows indicating the conformation changes seen in two different structures. UTP in the SD is shown in green sticks. (H) Chemical structure of sulfonaminopyrimidine inhibitor of CTPS1.
Detailed kinetic characterizations on CTPS from several model organisms have illustrated that CTPS activity is tightly regulated (Kassel et al., 2010). First, CTP inhibits CTPS by competing with the UTP binding in the SD. The physiological concentrations of UTP and CTP are 253 μM and 91 μM, respectively, and the IC50 of CTP for human CTPS1 is ∼40 μM when UTP is held at 100 μM (Kassel et al., 2010). This suggests that the rate of CTPS-catalyzed reaction is sensitive to fluctuations of the substrate-product ratio in the cell. Second, GTP acts as an allosteric activator at low concentrations, promoting glutamine hydrolysis in the GD but inhibiting synthetase activity when GTP exceeds the 10 mM threshold due to its competition with substrates in the SD (Kassel et al., 2010). The GTP binding site has been identified in a cryogenic electron microscopy (cryo-EM) structure of Drosophila melanogaster CTPS. This allosteric pocket is in direct contact with the glutaminase active site via a mobile loop, which is also close to the domain interface within the same monomer, further organizing the ATP/UTP binding at the N-terminal active site (Zhou et al., 2021; Bearne et al., 2022). The GTP binding site also bridges the contact with the N-terminal SD of the adjacent monomer in the tetrameric structure, supporting the functional importance of ternary complex formation in activity regulation (Zhou et al., 2021). Even though the structure of the human CTPS1 or CTPS2 has not been resolved in its GTP-bound form, the sequence alignments of CTPS homologs from D. melanogaster and humans indicate that the GTP-binding pocket is highly conserved (Bearne et al., 2022). Third, and remarkably, the CTPS filamentous structures formed in the cell, named cytoophidia also contribute to the activity regulation (Figure 3F). This phenomenon has been observed in CTPS from all kingdoms of life (Ingerson-Mahar et al., 2010; Noree et al., 2010; Chen et al., 2011), and the impact of filament formation on enzyme activity has been investigated for several model organisms. For example, E. coli CTPS polymer exhibits reduced enzyme activity due to the stabilization of an inactive conformation by the adjacent monomers, while polymerization promotes human CTPS1 activity and improves the cooperative regulation in CTPS2 (Lynch et al., 2017). Studies have also shown that CTPS cytoophidia assist in the maintenance of cell morphology and mediate lipid homeostasis (Liu et al., 2023). Additionally, the activity of human CTPSs is known to be negatively regulated by phosphorylation at serine residues located in the C-terminal tail region (Higgins et al., 2007). Protein kinase A (Choi and Carman, 2007) and glycogen synthetase kinase 3 (Higgins et al., 2007) have been proposed to be responsible for CTPS1 phosphorylation, while CTPS2 being phosphorylated by casein kinase 1 (Kassel et al., 2010). The mechanism behind the negative regulation observed in the phosphorylated enzyme is hindered by the absence of structural information for the C-terminal tail, likely a result of its flexibility in the unphosphorylated state.
CTPS plays a significant role in the homeostasis of the nucleotide pool due to its relevance to four nucleotides, including ATP, GTP, UTP, and CTP. CTPS1 exhibits higher enzymatic activity and is considered the major contributor to T-cell proliferation; CTPS2, on the other hand, meets the CTP demand for other tissues and is required for cell growth if CTPS1 function is impaired (Minet et al., 2023). Even though the CTP salvage pathways can generate CTP from cytidine, CTPS remains central to the lymphocyte proliferation in the immune response because rapidly dividing cells require nucleotides for DNA synthesis (Chaplin, 2010; Martin et al., 2014). CTPS1 deficiency (CTPSD) is a rare immune deficiency disorder caused by genetic mutations in CTPS1 (Martin et al., 2014). Individuals with CTPSD experience recurrent bacterial and viral infections but present no additional phenotype outside of the hemopoietic system. All identified cases, to date, carry a homozygous mutation near the acceptor splice site at introns 17–18, leading to abnormal gene splicing and the expression of a truncated protein missing residues at the C-terminal domain (Martin et al., 2020). The disease-linked variants exhibit reduced expression levels and decreased protein stability in the cell, but enzyme activity is preserved. The highly specific link between CTPS1 and lymphocyte proliferation also suggests that CTPS1 could be a potential target for immunosuppressive agents to treat autoimmune disease and inflammation. Several CTPS inhibitors have been shown to improve disease phenotypes in models of delayed-type hypersensitivity, collagen-induced arthritis, and experimental autoimmune encephalomyelitis (Nademi et al., 2019; McElwee et al., 2023).
In normal T cells, increased CTPS1 levels are only seen during the S phase of the activated lymphocytes, but the expression of CTPS1 is elevated in tumor cells at all stages of the cell cycle, providing a plausible basis for CTPS1-targeted therapy in oncology (Asnagli et al., 2023). Studies have shown that CTPS1 expression is elevated in acute lymphocytic leukemia (ALL), acute myeloid leukemia (AML), hepatoma, subtypes of non-Hodgkin’s lymphoma, multiple myeloma, triple-negative breast cancer, colon cancer, renal carcinoma (Verschuur et al., 2000; Verschuur, A.C., 2007; Pfeiffer et al., 2024). Although CTPS2 does not show a significant correlation with advanced disease stage and poor prognosis in these cancer types, CTPS2 expression levels have been linked to fluorouracil-resistant colorectal cancer (Tan et al., 2011). The pan-CTPS inhibitors that exhibit low selectivity between CTPS1 and CTPS2 lead to toxicity and efficacy issues in clinical trials (Quddus, 2020). Sulfonaminopyrimidine derivatives that selectively targets CTPS1 can induce apoptosis at nanomolar concentration in cell based assay (Figure 3H) (Quddus, 2020). These compounds inhibit hematological malignancies in preclinical studies and have shown promise as ‘first-in-class’ drugs targeting CTPS1 due to their high selectivity against two isoforms and are currently being tested in clinical trials.
Human phosphoribosylformylglycinamide synthetase (PFAS), also known as phosphoformylglycinamidine ribonucleotide (FGAM) synthetase (FGAMS or FGAS) or PurL, is a 144 kDa multidomain enzyme that mediates the fourth step of de novo inosine 5′-monophosphate (IMP) biosynthesis (Zhang et al., 2008). PFAS catalyzes the glutamine-dependent amination of phosphoformylglycinamide ribonucleotide (FGAR) to FGAM through activation of the amide oxygen of FGAR by the γ-phosphate of ATP, leading to the formation of a monophosphate intermediate, followed by nucleophilic attack of ammonia generated from glutamine (Zhao et al., 2013). The end products of this process are FGAM, ADP, phosphate, and glutamate (Figure 4A). Eukaryotic PFAS, sometimes designated large-PurL, is a single peptide chain that carries functional domains equivalent to a multienzyme cascade consisting of small-PurL, PurQ, and PurS in Gram-positive bacteria (Schendel et al., 1989; Patterson et al., 1999; Saxild and Nygaard, 2000; Anand et al., 2004). There is no structural information available for human PFAS. AlphaFold structural prediction shows that more than 90% of protein exhibits high or very high prediction confidence (pLDDTs > 70). The remaining regions, which display medium to low prediction confidence, are all likely to be solvent-exposed surface areas (Varadi et al., 2022).
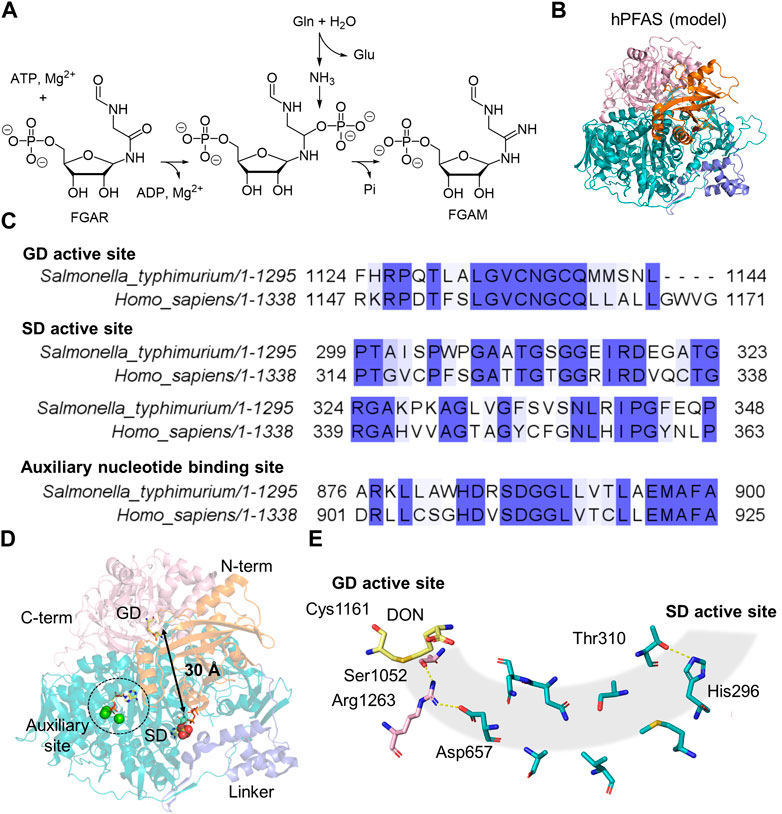
Figure 4. PFAS and PurL Structure and Function. (A) Amination reaction catalyzed by PFAS involves converting glutamine (Gln), ATP, and FGAR into glutamate (Glu), ADP, FGAM, and phosphate (Pi). (B) Predicted structure of human PFAS (AlphaFold: AF-O15067-F1), containing four domains: an N-terminal domain (orange), a linker domain (blue), a synthetase domain (SD, teal), and a glutaminase C-terminal domain (light pink). (C) Sequence alignment of StPurL and human PFAS, displaying the conserved regions in the two species within the GD, SD, and auxiliary regions. (D) Structure of StPurL (PDB:3UMM), highlighting the distance between the catalytic active sites (black arrow), the auxiliary site (dotted circle) and DON (yellow sticks) in GD, ATP (yellow sticks) and sulfate (spheres) in SD active site. (E) Key amino acid residues (sticks) necessary for the ammonia tunnel (gray shaded cylinder) formation and passage of ammonia from DON (yellow sticks) in the GD active site to the SD active site. The resides belong to the GD (light pink) and the SD (teal), which are either polar or charged residues.
One of the best-characterized homologs is Salmonella typhimurium PurL, which shares 37% sequence identity with the human enzyme (Figures 4B, C) (Anand et al., 2004; Tanwar et al., 2012). StPurL contains four domains: an N-terminal domain that is similar to the PurS dimer, a linker domain made of the tri-alpha helical bundle, an SD containing two nucleotide-binding sites in a pseudo-two-fold axis conformation, and a Class I GATase GD at the C-terminus. In the SD, only one nucleotide binding site that is in direct contact with the linker domain is catalytically active (Anand et al., 2004). This active site structurally does not share similarities with either the Rossman-fold or P-loop types as seen in many ATP phosphatases (Longo et al., 2020). The auxiliary nucleotide binding site is buried deep within the SD and is inaccessible to the solvent; it also lacks the positively charged residue, such as lysine, that could facilitate ATP hydrolysis (Figure 4D). Either ATP or ADP has been observed tightly bound in the auxiliary site of PurL isolated from different organisms. The functional importance of this auxiliary site remains to be investigated. In the C-terminal GD, the glutaminase active site is approximately 30 Å away from the synthetase active site (Anand et al., 2004). Although no readily formed ammonia tunnel has been identified in any of the StPurL structures, the N-terminal domain and a flexible loop region in the SD are thought to participate in the transient formation of the tunnel via conformational changes as seen in computational simulations (Sharma et al., 2020; 2022). Thus, when glutamine is bound in the active site, the tunnel can transiently open through altered interactions between Arg1263, Ser1052, and Asp657, along with a sidechain rotation to break a hydrogen bond between His296 and Thr310 (Figure 4E). Point mutations in residues lining the tunnel have a drastic impact on FGAM synthesis due to the leakage of ammonia from the StPurL variants (Sharma et al., 2022). Mutations in the SD catalytic loop, as well as residues in the N-terminal domain, compromise glutamine binding, suggesting an intimate interaction between each domain in StPurL.
PFAS has been shown to cluster into a higher-order assembly with other enzymes in purine biosynthesis, named purinosome, under purine-depleted conditions (An et al., 2008; An et al., 2010; Field et al., 2011; Pedley et al., 2022). The presence of purinosome potentially improves regulation of the metabolic flux and substrate channeling among enzymes in the pathway (An et al., 2008), although detailed protein-protein interactions within the purinosome remain to be explored structurally. Additionally, the PFAS-containing purinosome has been observed moving along microtubules in cells and eventually localizing with mitochondria (Chan et al., 2018). This interaction between the purinosome, mitochondria, and microtubules highlights a dynamic role in purine biosynthesis and suggests their potential regulatory influence on enzymatic functions.
Extensive studies have been conducted on PurL from bacterial pathogens because it plays a vital role in supplying precursors for GMP and adenosine monophosphate (AMP). Most viruses do not encode enzymes necessary for completing purine biosynthesis, even though viral replication is coupled with a high demand for nucleic acids in the host cells. For some viruses, such as γ-herpesviruses, which contain genes encoding putative PurL homologs, it is unclear whether these putative enzymes are catalytically competent (Ling et al., 2008; Tsai et al., 2015). Instead, these PurL homologs are known to be viral factors triggering proteasomal degradation, which is important for disarming the intrinsic antiviral machinery of the host. Human PFAS is located at Chromosome 17p13 (Patterson et al., 1999). No genetic diseases have been reported to be associated with PFAS variants. It has been shown that PFAS activity is related to cell proliferation of hepatomas, thymus, spleen, and testis in mouse models (Elliott and Weber, 1984). Under an oncogenic regime, phosphorylated ERK2 directly phosphorylates a threonine residue (Thr619) in PFAS, which, in turn, increases de novo nucleic acid biosynthesis. PFAS has also been found to be expressed in neurons (Mangold et al., 2018) and colocalized with mitochondria in the rat hippocampus (Williamson et al., 2017; Mangold et al., 2018; Ali et al., 2020).
As a part of human amino acid metabolism, ASNS converts aspartate into asparagine coupled with hydrolysis of glutamine and ATP (Figure 5A) (Richards and Schuster, 1998; Zhu et al., 2019). The two half-reactions are carried out in two domains: the N-terminal GD and the C-terminal SD. ASNS belongs to the Class II GATase, which requires the presence of cysteine at the N-terminus (Cys1) for glutamine hydrolysis (Zhu et al., 2019). Therefore, ASNS, like many other Class II GATase, needs to be post-translationally processed by methionine aminopeptidase to become catalytically competent in the glutamine-dependent reaction (Xiao et al., 2010). For this reason, attempts to express human ASNS using E. coli yield little active enzyme in the absence of ammonia (Van Heeke and Schuster, 1989). In the SD, ATP activates the carboxylate side chain of aspartate, forming pyrophosphate and a β-aspartyl-AMP intermediate that eventually reacts with ammonia. The first crystal structure of human ASNS was obtained in 1.85 Å resolution (Figure 5B) (Larsen et al., 1999; Zhu et al., 2019). In this structure, ASNS forms a head-to-head homodimer, in which Cys41 of two N-terminal domains are covalently linked. Size-exclusion chromatography has shown that ASNS exists as a stable dimer in solution (Li, 2007), although no cooperativity has been reported for ASNS. Particularly in the structure recently determined by cryo-EM, it was shown that dimerized ASNS exists in the absence of a disulfide bond (Coricello et al., 2023). It remains uncertain whether the covalent linkage in the homodimer structure is functionally essential. In the crystal structure, the catalytically active Cys1, which is covalently modified by DON is located between two layers of antiparallel β-sheets and is close to the domain interface between the GD and SD (Figure 5C). The C-terminal SD contains a PP-loop sharing a classical SGGLDS motif (Figure 5D). Two domains are connected by a flexible loop that is absent in the ASNS crystal structure, and the intramolecular tunnel connecting two active sites for ammonia transfer is also blocked (Zhu et al., 2019). This ammonia tunnel, however, is clearly visible in the crystal structure of E. coli homolog, AsnB (Larsen et al., 1999). Besides the clear variation in tunnel-lining residues between the two enzymes, one possible explanation is that the AsnB structure is solved in the presence of AMP using an inactive variant of the enzyme, in which the catalytic cysteine is replaced by alanine (Larsen et al., 1999). The MD simulations on human ASNS further suggest that the presence of β-aspartyl-AMP intermediate in the SD active site promotes tunnel formation by stabilizing the open conformation via residue networks between two active sites (Coricello et al., 2023). Therefore, the tunnel opening of the human ASNS is likely to be transient and controlled by conformational changes at the tunnel upon substrate binding or intermediate formation.
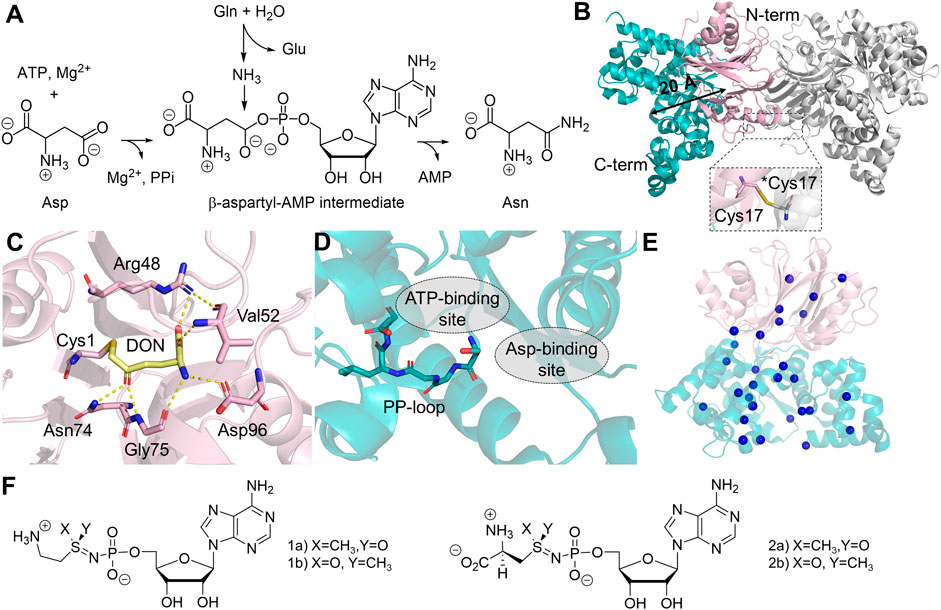
Figure 5. Structure and Reaction of Human ASNS. (A) Reaction catalyzed by ASNS involves converting glutamine (Gln), ATP, and aspartate (Asp) into glutamate (Glu), asparagine (Asn), AMP, and pyrophosphate (PPi). (B) The homodimer structure of ASNS (PDB:6GQ3, gray), featuring head-to-head dimerization and a monomer containing an N-terminal glutaminase domain (GD, light pink) and a C-terminal synthetase domain (SD, teal), with a distance of 20 Å (black arrow) between the two catalytic active sites. A close-up of Cys17 disulfide bond between two monomers at the N-terminal interfaces is shown. An asterisk indicates Cys17 in the adjacent monomer. (C) Catalytic active site in the GD, showing the interaction with DON (yellow sticks) and the catalytic residue (sticks) within 3 Å distance to DON. (D) The SD active site, featuring a stick model of the PP-loop, and the substrate binding sites for both ATP and aspartate (gray dotted circle). (E) Location of ASNSD-causing mutants in the ASNS structure, the mutations are represented as blue spheres on the α-carbon of the affected amino acids, with specific amino acid mutations listed in Table 1. (F) Chemical structures of two ASNS inhibitors.
The kinetic characterization of human ASNS, using either glutamine or ammonia as nitrogen sources, confirms that the apparent KM and kcat for each substrate are comparable under these conditions. Similar values were also obtained from the ASNS homologs purified from rat liver, mice pancreas, and bovine (Hongo et al., 1978; Milman et al., 1980; Markin et al., 1981). Step-wise kinetic mechanism using bacterial homologs from E. coli and Vibrio cholerae reveals that the glutaminase active site exhibits high basal activity in the absence of substrate at the SD, suggesting relatively high independence between two active sites (Boehlein et al., 1998; Tesson et al., 2003; Fresquet et al., 2004). In E. coli AsnB, ATP and aspartate sequentially bind to the SD prior to the glutamine binding, allowing the formation of a complex containing enzyme and all three substrates (Boehlein et al., 1998). Another common characteristic of ASNS across different organisms is the substantial inhibition by asparagine, which is likely due to the structural similarity among the four amino acids involved in the reaction. Ammonia can substitute glutamine in the in vitro assays, although the binding mode of aspartate and ATP only becomes ordered sequential in bovine ASNS when glutamine is used as a nitrogen source (Markin et al., 1981).
Human ASNS is encoded by the ASNS gene, located at Chromosome 7q21.3. Several isoforms with unknown biological functions containing a truncated N-terminal or C-terminal domain have also been detected. In the cells, ASNS exists in both cytosol and nucleus, and in both locations, ASNS has been shown to form higher-order complexes associated with mitotic spindles during cell division (Noree et al., 2018). In yeast, the high-order assembly occurs during nutrient limitation, although the molecular basis of this high-order assembly is unclear (Noree et al., 2010; Zhang et al., 2018). In humans, ASNS dysfunction seems to be extremely detrimental to brain development, leading to a severe neurometabolic disorder named asparagine synthetase deficiency (ASNSD). ASNSD is an autosomal recessive disease presented as cognitive delay, axial hypotonia, and early death in children (Ruzzo et al., 2013; Alfadhel et al., 2015; Ben-Salem et al., 2015). Diagnosis of ASNSD mainly relies on genomic sequencing, coupled with brain CT imaging and measurements of asparagine levels in cerebrospinal fluid and blood. Individuals with ASNSD harbor non-silent mutations in the ASNS gene. Even though dietary asparagine can be transported into cells by amino acid transporters such as LAT1 (Gauthier-Coles et al., 2021), asparagine supplements do not always lead to symptom relief in clinical settings for ASNSD, and sometimes, seizures worsen after the asparagine treatment (Alrifai and Alfadhel, 2016). Studies have shown that these disease-linked mutations usually impact the protein stability in cells or compromise enzyme activity. Among known cases, besides a few frameshifting (Yamamoto et al., 2017; Schleinitz et al., 2018; Chen et al., 2020) and truncating (Staklinski et al., 2023) mutations, ASNSD variants are mostly seen to be missense mutations (Table 1), resulting in amino acid changes spread across the protein (Figure 5E). Several residues are located in the C-terminal tail which are not resolved in the crystal structure. Studies have shown that the disordered C-terminal tail is functional and essential for synthetase activity in AsnB (Meyer, 2010), consistent with clinical observations of the ASNSD-linked C-terminal tail variants. Several ASNSD variants have been characterized using the purified protein and/or cell-based assays (Zhu et al., 2019; Matsumoto et al., 2021; Staklinski et al., 2022; 2023; Chang et al., 2023). In some cases, decreased stability and enzyme activity are attributed to ASNS dysfunction. Some variants, however, do not show a clear activity impairment in the enzyme assay. These findings suggest that the disease phenotype might involve a more complex mechanism of action beyond just the reduction of enzyme activity.
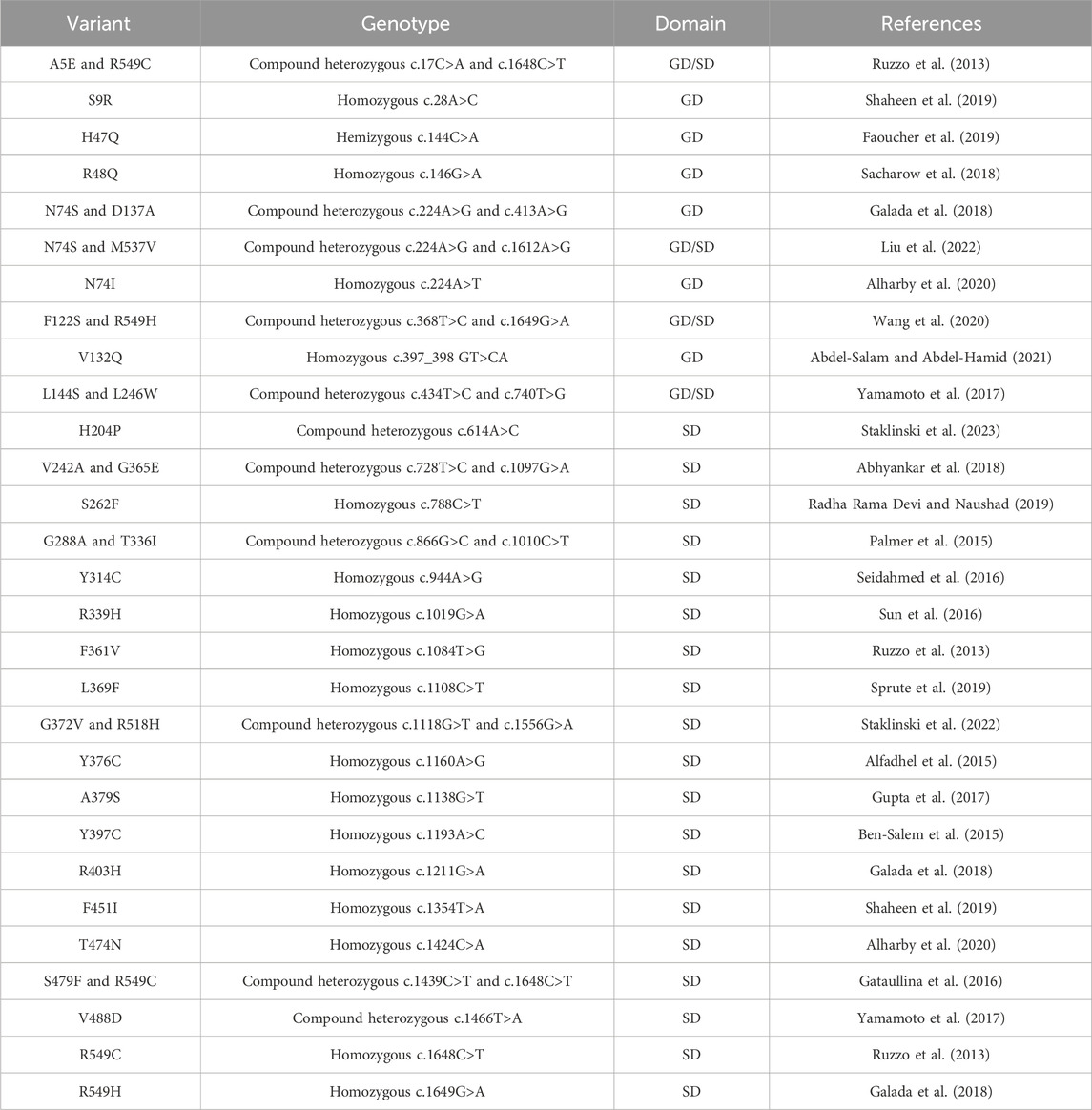
Table 1. Clinically identified missense mutations in individuals diagnosed with ASNSD. A list of missense mutations associated with ASNSD, detailing residue variants, genotypes, nucleotide mutations, and their locations in the glutaminase domain (GD) or synthetase domain (SD). For heterozygous genotypes, locations in both domains are specified.
Additionally, upregulation of ASNS has been observed in many cancer types, including colorectal (Du et al., 2019), breast (Yang et al., 2014; Knott et al., 2018), and prostate cancers (Sircar et al., 2012), and asparagine levels have long been linked to tumorigenesis and metastasis. Besides its obvious role in protein synthesis, asparagine also seems to be involved in electron transport chain inhibition, metastasis formation and indirectly participating in nucleotide synthesis via mammalian target of rapamycin complex 1 (mTORC1) signaling pathway (Yuan et al., 2024). In solid tumors, ASNS expression level is positively correlated to cell proliferation in ovarian carcinomas (Lorenzi and Weinstein, 2009) and gastric cancer (Yu et al., 2016). Knocking down ASNS in breast cancer cells also inhibits metastatic progression. In fact, triple-negative breast cancer has the highest ASNS expression level among others (Yang et al., 2014). The transcription of ASNS is induced by transcription factor 4 (Su and Kilberg, 2008; Balasubramanian et al., 2013) and C/EBP-β (Su and Kilberg, 2008), which is modulated via the P13K/AKT and KRAS-mTOR pathways (Toda et al., 2016). In ALL, ASNS is silenced, resulting in asparagine acquisition from the extracellular environment so that proliferation can take place (Radadiya et al., 2020). Therefore, asparaginase, which removes asparagine extracellularly, is an effective chemotherapy drug for treating ALL (Cooney and Handschumacher, 1970; Van Trimpont et al., 2022). Yet in drug-resistant ALL, ASNS is upregulated, leading to an intracellular asparagine supply in the tumor cells; therefore, inhibiting ASNS is widely thought to be a strategy to treat drug-resistant ALL. A transition state analog has been tested in vitro, which exhibits nano-molar affinity to ASNS and has relatively high selectivity against other SD-containing enzymes in the HCT-116 cell line (Figure 5F) (Zhu et al., 2019). Recently, however, the mechanism of action for asparaginase has been challenged because the expression level of ASNS did not dictate B cell lymphoma proliferation in mice (Grima-Reyes et al., 2022). Instead, the glutaminase activity, a side reaction of asparaginase, was proposed to maximize the anticancer effect. ASNS is at the intersection of balancing glutamate and asparagine concentration in the cell as glutamate concentration is also correlated to tumorigenesis and cell signaling (Yi et al., 2020). Nevertheless, modulating ASNS activity, either by increasing its glutaminase activity or decreasing asparagine production, could lead to the discovery of anticancer drugs with a new mechanism of action.
NAD+ is a redox cofactor that cycles between its oxidized and reduced forms in over 1,500 redox-active enzymatic reactions (Cantó et al., 2015). The last step of human NAD+ biosynthesis is mediated by NADSYN, an enzyme that utilizes ammonia generated in the GD to produce NAD+ from deamido-NAD+ (NaAD+) and ATP in the SD (Figure 6A). The structure of human NADSYN has been resolved to 2.84 Å resolution (Chuenchor et al., 2020). A homodimer is present in one asymmetric unit, and four of the asymmetric units assemble into an octamer, which is believed to be the biologically competent form of the enzyme (Figure 6B). The N-terminal GD of NADSYN belongs to neither the Class I nor Class II GATase families but is structurally similar to members of the nitrilase superfamily. It possesses a Lys114-Cys175-Glu45 catalytic triad in order to modulate the pKa and reactivity of the catalytic cysteine residue (Figure 6C) (Chuenchor et al., 2020). The C-terminal synthetase active site contains a substrate binding cleft, in which the SGGVDS motif of the PP-loop is buried in the SD, and the NaAD+ binding site is located at the homodimer interface (Chuenchor et al., 2020). Even though the SD of NADSYN structure was resolved in the presence of NaAD+, AMP, pyrophosphate, and magnesium, the ammonia channel that must connect the two active sites was sealed off at both ends (Figure 6D). Ammonia channeling, therefore, be coupled with conformational changes that take place during the catalytic cycle.
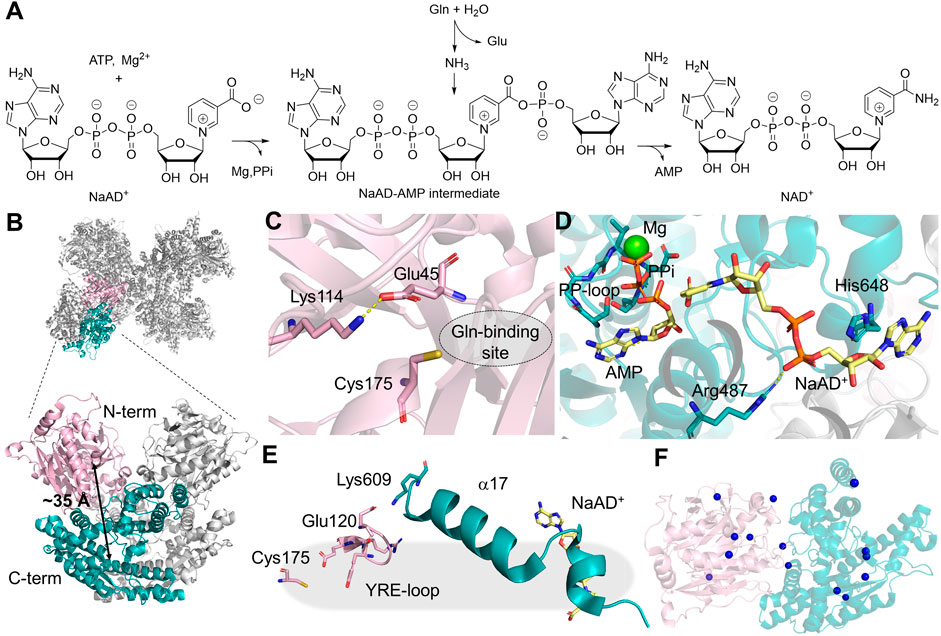
Figure 6. NADSYN Structure and Function. (A) mination reaction catalyzed by NADSYN involves converting glutamine (Gln), ATP, and NaAD+ into glutamate (Glu), NAD+, AMP, and pyrophosphate (PPi) (B) Octamer structure of NADSYN (PDB:6OFB), glutaminase domain (GD, light pink), and synthetase domain (SD, teal) with a distance of ∼35 Å (black arrow) between the two active sites, an ammonia tunnel will form to transfer ammonia from one site to the next. (C) Representation of the GD active site, with the catalytic triad as pink stick as well as the Gln binding site (dotted circle) which is located next to the triad; (D) Binding site in the SD, bindings of the by-product AMP (yellow sticks) with a coordinating magnesium (green sphere) and substrate NaAD+(yellow sticks), near the PP-Loop (teal sticks) necessary for ATP binding; (E) YRE loop (light pink) present in the GD, utilized in regulation, and interacts with the SD (teal); (F) The locations of the missense mutations, denoted as blue spheres on the α-carbon of the affected amino acids, in NADSYN1 causing CNDD, located across both domains with specific residues listed in Table 2.
Activity coupling between two active sites in NADSYN has been confirmed through kinetic assays and computational simulation. Glutaminase activity is enhanced 31-fold when an adenylated reaction intermediate is present in the SD, suggesting an interaction network that is present in NADSYN to prevent unproductive glutamine hydrolysis. The glutaminase activity regulation involves the movement of a loop containing Tyr123-Arg124-Glu125 (YRE). The YRE loop not only assists in orientating the geometry of the catalytic triad in the GD, but also gates the glutaminase active site, shifting the residue interaction via a helical segment connecting to the SD (Figure 6E). The strength of such regulation, however, varies from species to species, providing an opportunity for targeting NADSYN for antibiotic design. (Chuenchor et al., 2020). Additionally, it appears that the human NADSYN does not exhibit different catalytic efficiency when ammonia is directly supplied as a nitrogen source, suggesting that the activity of the SD can be decoupled from the GD.
The NAD+ level in the cell is essential for maintaining redox balance and energy supplies, which also significantly impacts other metabolic pathways. Genetic mutations in NADSYN that result in decreased NAD+ have been linked to congenital NAD+ deficiency disorder (CNDD) (Lin et al., 2021). Mutations in multiple enzymes in the NAD biosynthetic pathway have been linked to CNDD. Pathological phenotypes associated with biallelic variants of NADSYN lead to defects in multiple organs and limbs, and affected individuals do not usually survive for a few months after birth (Szot et al., 2020). The 23 discovered variants throughout both the N- and C-terminal domains arise from missense (Table 2), nonsense (Kortbawi et al., 2022; Szot et al., 2024), deletion (Lin et al., 2021), truncation (Szot et al., 2024), and frameshifts (Szot et al., 2020; 2024) in the NADSYN gene (Figure 6F). The human NADSYN variants all seem to have decreased activity compared to that of the wild-type enzyme and/or low expression (Lin et al., 2021) suggesting that solely restoring enzyme activity may not be enough for treating disease-linked NADSYN variants.
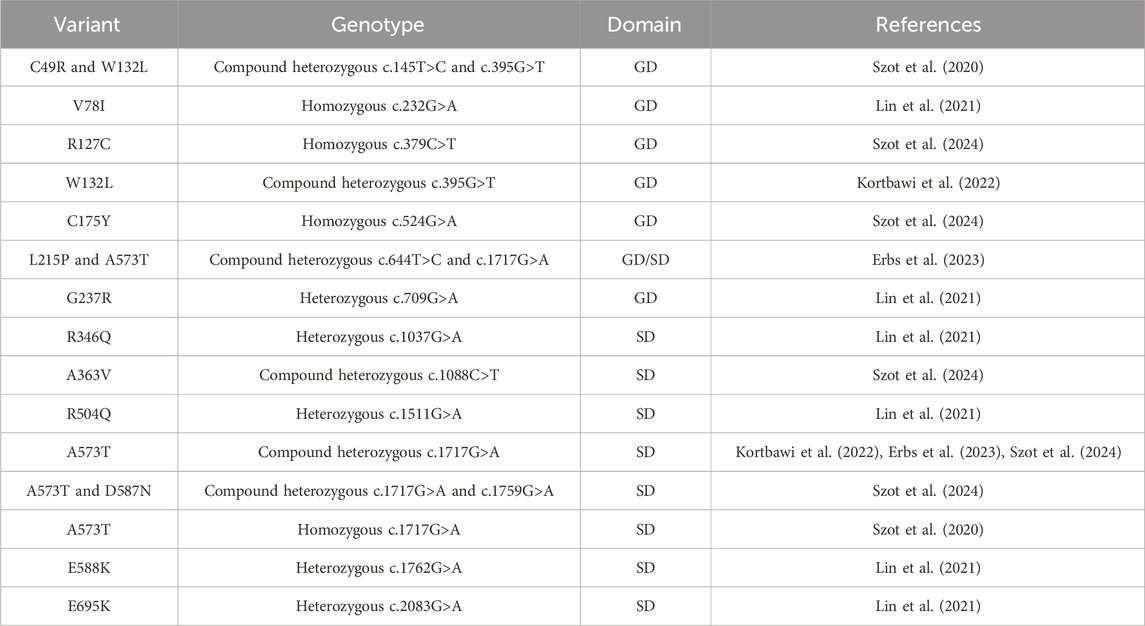
Table 2. Clinically identified missense NADSYN mutations in individuals diagnosed with CNDD. A list of missense mutations associated with CNDD, detailing residue variants, genotypes, nucleotide mutations, and their locations in the glutaminase domain (GD) or synthetase domain (SD). For heterozygous genotypes, locations in both domains are specified.
In addition to the five bifunctional enzymes discussed above, in humans, carbamoyl phosphate synthetase 2 (CPS2) is also capable of hydrolyzing ATP and glutamine for amination reactions in the cytosol (Grande-García et al., 2014). Human CPS2 is part of a multifunctional enzyme named CAD, which contains additional domains that are homologs to aspartate transcarbamylase (ATCase) and dihydroorotase (DHOase). CAD catalyzes the first three steps of pyrimidine de novo biosynthesis to generate carbamoyl phosphate (Figure 7A). Ammonia generated by glutamine hydrolysis in the N-terminal GD is combined with bicarbonate, mediated by ATP hydrolysis at the first SD active site; the product carbamate is subsequently phosphorylated in the second active site of SD to produce carbamoyl phosphate. CPS2 is a member of the Class I GATase, sharing the common Cys252-His336-Glu338 triad as GMPS, CTPS, and PFAS. Carbamoyl phosphate, the product of CPS2, is further processed by two additional domains that share homology with DHOase and ATCase to generate dihydroorotate as the final product, both of which have been structurally characterized separately (Grande-García et al., 2014; Ruiz-Ramos et al., 2016; Del Caño-Ochoa et al., 2018). Kinetic and structural characterization has been performed on the enzyme complex of E. coli homologs, which share high degrees of sequence conservation with human enzyme (Del Caño-Ochoa and Ramón-Maiques, 2021), although no structural information or kinetic characterization is available for the human full-length CAD protein.
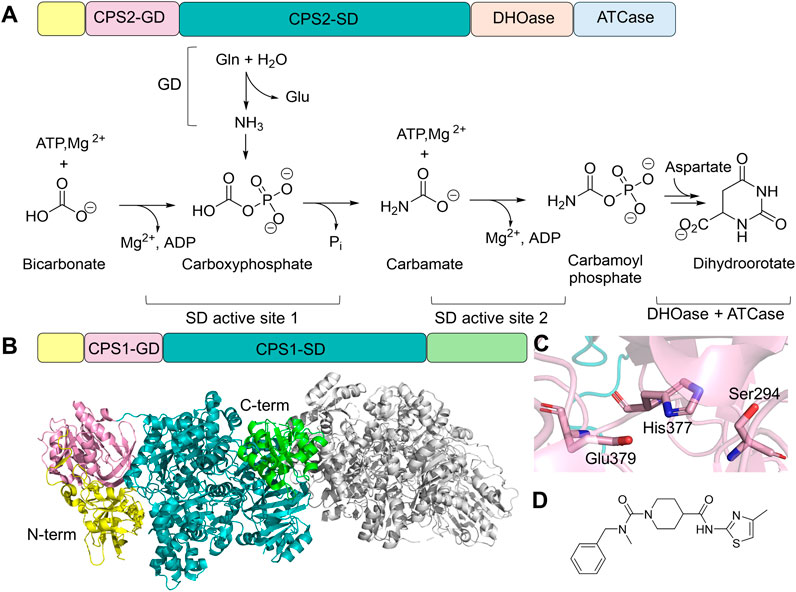
Figure 7. CPS Structure and Function. (A) Protein domain architecture and chemical reaction catalyzed by CAD. CAD contains an N-terminal small domain (yellow), GD (pink), SD (teal), DHOase domain (light orange), and ATCase domain (light blue). Two active sites in the SD domain each carry a half-reaction. (B) Domain architecture of CPS1 and the dimer structure of CPS1 (gray, PDB: 5DOU), highlighting N-terminal (yellow), Non-functional glutaminase domain (GD, light pink), synthetase domain (SD, teal), and allosteric domain (green) of one of the monomers. (C) Residue triad within CPS1-GD, where cysteine is replaced by a serine (light pink sticks). (D) Chemical structure of an allosteric inhibitor of CPS1, H30B-120, which binds to the C-terminal allosteric domain.
Humans also possess another enzyme called CPS1, located in the mitochondria, which contains the GD, SD, and an allosteric N-acetyl-L-glutamate-binding domain (Figure 7B). Despite sharing a similar SD architecture with CPS2, CPS1 has lost its glutaminase activity due to the replacement of cysteine by serine (Ser294) in the catalytic triad of the glutaminase active site (Holden et al., 1999; De Cima et al., 2015). It is, therefore, part of the urea cycle, facilitating ammonia removal (Summar et al., 2003). Structural characterization confirmed the presence of a functionally impaired Ser294-His377-Glu379 triad (Figure 7C) (De Cima et al., 2015). Although CPS1 does not require an ammonia tunnel, a carbamate tunnel is present in the enzyme, allowing the reactive intermediate generated in the first SD active site to travel to the second SD active site. Furthermore, the formation of the carbamate tunnel is triggered by the binding of N-acetyl-L-glutamate in the allosteric regulatory domain. Loop rearrangement and domain-domain interactions facilitate the expansion of the intramolecular tunnel.
Loss-of-function CAD variants, primarily located at the DHOase and ATCase region, have been found in genetic disorders characterized by global development delay (Li et al., 2021). The therapeutic potential of CAD is also frequently discussed in oncology, as pyrimidine biosynthesis is upregulated in cancer cells to meet their nucleotide demands (Christopherson et al., 2002). Inhibitors targeting ATCase and DHOase have been tested in clinical trials, yet none of them moved beyond Phase II evaluation (Li et al., 2021). Due to the essential role of CPS1 in ammonia detoxification for urea synthesis, loss-of-function mutations in the gene encoding CPS1 lead to hyperammonemia. CPS1 deficiency is an autosomal recessive inborn error characterized by protein intolerance, seizures in late-onset types, and prenatal/neonatal death in early-onset types (Yan et al., 2019; Wang et al., 2023). Treatment of hyperammonemia ranges from ammonia removal, such as hemodialysis (Ames et al., 2020), dietary management, and/or interfering ammonia production-absorbing process in the intestine. CPS1 is also suggested to play a role in cardiovascular disease and obesity (Nitzahn and Lipshutz, 2020). Even though CPS1 does not use glutamine to generate ammonia, carbamoyl phosphate produced by CPS1 can feed into the pyrimidine biosynthesis; therefore, inhibition of CPS1 has also been proposed as an anticancer strategy (Yao et al., 2020). Multiple chemical probes have been designed to further explore CSP1’s therapeutic potential for novel anticancer reagent development (Figure 7D) (Rolfe et al., 2020).
Domains in bifunctional enzymes typically evolve separately before genetic fusion brings the two segments together. These bifunctional enzymes are widely present in organisms from other kingdoms, such as fungi (Nguyen et al., 2022), archaea (Zhou et al., 2020), and plants (Qu et al., 2019); the human glutamine-hydrolyzing synthetases discussed here all have their own evolutionary ancestors. In fact, the same biological function can also be carried out by other single-domain synthetases, such as ammonia-dependent NAD+ synthetase and ammonia-dependent asparagine synthetase, in other organisms, which may or may not be structurally similar to bifunctional enzymes (Roy et al., 2003; De Ingeniis et al., 2012). The ‘mosaic distribution’ of these enzymes in single-domain and bifunctional forms across the tree of different species indicates a rather complicated picture of evolution (De Ingeniis et al., 2012). Therefore, the activity regulation, active site coupling, and domain architecture of these bifunctional enzymes exhibit their unique features.
Despite not being closely related evolutionarily, the glutamine-hydrolyzing synthetases discussed here clearly benefit from the structural complexity to adapt sophisticated regulatory mechanisms. One remarkable feature is the regulation of ammonia tunnels. In all cases, the opening of the ammonia tunnel is regulated by the ligand binding in the SD via long-distance interaction networks. The gating residues are located at the GD, SD, or both. Prior to full activation, ammonia transport is restricted by protein motions at either end of the active site or both. The reciprocal regulation, in which the glutaminase activity facilitates ATP hydrolysis, seems relatively rare, although this observation might be biased since glutaminase activity is not universally reported for all enzymes. This is also supported by the absence of a tunnel observed in the crystal structures containing DON-modified GD. In Table 3, we have summarized the ammonia- and glutamine-dependent activity reported for four of the six human enzyme. Each enzyme has a different response to the change of nitrogen source from glutamine to ammonia. Assuming that the ammonia-dependent activity does not require tunnel formation, i.e., enzyme access the ammonia from the bulk solvent, the changes in apparent KM should report the cooperativity of the glutaminase activity and the tunnel formation. Since the rate-limiting step, up to and including ammonia trafficking, remains unclear for many of the human enzymes discussed here, the kinetic coupling between reactivity and the tunnel opening frequency remains to be explored. To further address the time scale of conformational dynamics during tunnel formation in relation to the rate of the catalysis, MD simulation has provided insights into possible gating mechanisms that lead to transient tunnel formation. The high-energy state of the transient tunnel opening triggered by reaction-induced conformational changes may exist as part of the conformational sampling in the absence of ligands but exists in a much lower population that cannot be easily captured by X-ray crystallography. Given that the size of these multi-domain enzymes limits dynamical analysis by NMR, techniques such as cryo-EM and/or room-temperature X-ray crystallography may provide important insights into tunnel-forming mechanisms.
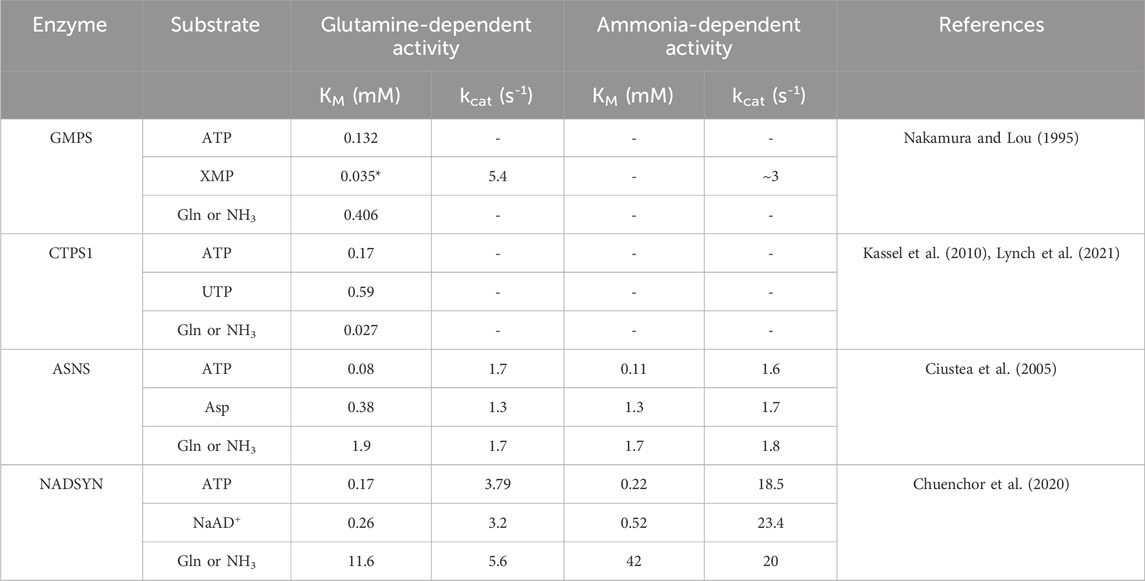
Table 3. Steady-state kinetics of human bifunctional glutamine-hydrolyzing synthetases. Comparison of the steady-state kinetics parameter of four human bifunctional glutamine-hydrolyzing synthetases using glutamine or ammonia as nitrogen sources. Dashes indicate that the data is not available. References for each enzyme detail the individual assay conditions. An asterisk indicates the half-saturation value under the reported condition in the reference.
Besides the domain-domain interaction, another interesting feature is that several enzymes discussed here all require the assembly of monomers into oligomers. CPS1 takes a step further on the complex assembly by fusing two additional downstream enzymes directly in a single polypeptide chain. CTPS, ASNS, and PFAS are all found to polymerize or be associated with other enzymes in the cell, suggesting that the quaternary structure may contribute to additional activity regulation. This higher-order assembly seems to be reversible and often occurs under specific conditions. Only CTPS polymers have been structurally characterized in vitro. Further structural insights into purinosome and ASNS-spindle association would be beneficial for better understanding the role of these glutamine-hydrolyzing synthetases beyond their catalytic function. Additionally, the difference seen in bacterial and human homologs regarding how the polymer formation impacts enzyme activity further suggests that the activity regulation in human enzymes could be drastically different from the bacterial homologs despite sharing similarities in catalysis. Further investigation of the activity regulation of human enzymes will prompt valuable insights for targeted therapeutic development.
Ammonia assimilation is fundamental to the biosynthesis and regulation of critical biological pathways in all forms of life. This is accomplished by structurally complex enzymes that are involved in the biosynthesis of essential metabolites. The activity regulation in the glutamine-hydrolyzing synthetases discussed here, partially arising from ammonia transfer between active sites, underscores the complexity and evolutionary significance of multifunctional enzymes. Knowledge of the regulatory mechanism not only enhances our understanding of fundamental biochemical processes in the cell cycle, which is valuable for treating oncologic diseases but also brings possibilities for targeted therapeutic interventions in genetic diseases associated with the loss-of-function variants. In addition to developing inhibitors suitable for oncology, small molecule activators that can restore function in disease-linked enzyme variants also have the potential to expand the toolbox of therapeutic strategy. Harnessing the regulatory mechanisms in these bifunctional enzymes will potentially translate the fundamental understanding of the enzyme structure-function relationship to innovative drug discovery.
WZ: Visualization, Writing–original draft, Writing–review and editing. AN: Writing–original draft, Writing–review and editing, Visualization. LP: Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article.
We acknowledge the start-up funding provided by FSU (WZ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdel-Salam, G. M. H., and Abdel-Hamid, M. S. (2021). Asparagine synthetase deficiency with intracranial hemorrhage can mimic molybdenum cofactor deficiency. Neuropediatrics 52 (03), 201–207. doi:10.1055/s-0040-1718917
Abhyankar, A., Lamendola-Essel, M., Brennan, K., Giordano, J. L., Esteves, C., Felice, V., et al. (2018). Clinical whole exome sequencing from dried blood spot identifies novel genetic defect underlying asparagine synthetase deficiency. Clin. Case Rep. 6 (1), 200–205. doi:10.1002/ccr3.1284
Alfadhel, M., Alrifai, M. T., Trujillano, D., Alshaalan, H., Al Othaim, A., Al Rasheed, S., et al. (2015). Asparagine synthetase deficiency: new inborn errors of metabolism. JIMD Rep. 22, 11–16. doi:10.1007/8904_2014_405
Alharby, E., Faqeih, E. A., Saleh, M., Alameer, S., Almuntashri, M., Pastore, A., et al. (2020). Clinical, molecular, and biochemical delineation of asparagine synthetase deficiency in Saudi cohort. Genet. Med. 22 (12), 2071–2080. doi:10.1038/s41436-020-0919-x
Ali, E. S., Sahu, U., Villa, E., O’Hara, B. P., Gao, P., Beaudet, C., et al. (2020). ERK2 phosphorylates PFAS to mediate posttranslational control of de novo purine synthesis. Mol. Cell 78 (6), 1178–1191.e6. doi:10.1016/j.molcel.2020.05.001
Alifano, P., Fani, R., Liò, P., Lazcano, A., Bazzicalupo, M., Carlomagno, M. S., et al. (1996). Histidine biosynthetic pathway and genes: structure, regulation, and evolution. Microbiol. Rev. 60 (1), 44–69. doi:10.1128/mr.60.1.44-69.1996
Alrifai, M. T., and Alfadhel, M. (2016). Worsening of seizures after asparagine supplementation in a child with asparagine synthetase deficiency. Pediatr. Neurol. 58, 98–100. doi:10.1016/j.pediatrneurol.2016.01.024
Ames, E. G., Luckritz, K. E., and Ahmad, A. (2020). A retrospective review of outcomes in the treatment of hyperammonemia with renal replacement therapy due to inborn errors of metabolism. Pediatr. Nephrol. 35 (9), 1761–1769. doi:10.1007/s00467-020-04533-3
An, S., Deng, Y., Tomsho, J. W., Kyoung, M., and Benkovic, S. J. (2010). Microtubule-assisted mechanism for functional metabolic macromolecular complex formation. Proc. Natl. Acad. Sci. 107 (29), 12872–12876. doi:10.1073/pnas.1008451107
An, S., Kumar, R., Sheets, E. D., and Benkovic, S. J. (2008). Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 320 (5872), 103–106. doi:10.1126/science.1152241
Anand, R., Hoskins, A. A., Stubbe, J., and Ealick, S. E. (2004). Domain organization of Salmonella typhimurium formylglycinamide ribonucleotide amidotransferase revealed by X-ray crystallography. Biochemistry 43 (32), 10328–10342. doi:10.1021/bi0491301
Asnagli, H., Minet, N., Pfeiffer, C., Hoeben, E., Lane, R., Laughton, D., et al. (2023). CTP synthase 1 is a novel therapeutic target in lymphoma. HemaSphere 7 (4), e864. doi:10.1097/HS9.0000000000000864
Balasubramanian, M. N., Butterworth, E. A., and Kilberg, M. S. (2013). Asparagine synthetase: regulation by cell stress and involvement in tumor biology. Am. J. Physiology-Endocrinology Metabolism 304 (8), E789–E799. doi:10.1152/ajpendo.00015.2013
Ballut, L., Violot, S., Kumar, S., Aghajari, N., and Balaram, H. (2023). GMP synthetase: allostery, structure, and function. Biomolecules 13 (9), 1379. doi:10.3390/biom13091379
Bearne, S. L., Guo, C.-J., and Liu, J.-L. (2022). GTP-dependent regulation of CTP synthase: evolving insights into allosteric activation and NH3 translocation. Biomolecules 12 (5), 647. doi:10.3390/biom12050647
Ben-Salem, S., Gleeson, J. G., Al-Shamsi, A. M., Islam, B., Hertecant, J., Ali, B. R., et al. (2015). Asparagine synthetase deficiency detected by whole exome sequencing causes congenital microcephaly, epileptic encephalopathy and psychomotor delay. Metab. Brain Dis. 30 (3), 687–694. doi:10.1007/s11011-014-9618-0
Bhat, J. Y., Shastri, B. G., and Balaram, H. (2008). Kinetic and biochemical characterization of Plasmodium falciparum GMP synthetase. Biochem. J. 409 (1), 263–273. doi:10.1042/BJ20070996
Boehlein, S. K., Stewart, J. D., Walworth, E. S., Thirumoorthy, R., Richards, N. G. J., and Schuster, S. M. (1998). Kinetic mechanism of Escherichia coli asparagine synthetase B. Biochemistry 37 (38), 13230–13238. doi:10.1021/bi981058h
Bork, P., and Koonin, E. V. (1994). A P-loop-like motif in a widespread ATP pyrophosphatase domain: implications for the evolution of sequence motifs and enzyme activity. Proteins Struct. Funct. Bioinforma. 20 (4), 347–355. doi:10.1002/prot.340200407
Cantó, C., Menzies, K. J., and Auwerx, J. (2015). NAD+ metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 22 (1), 31–53. doi:10.1016/j.cmet.2015.05.023
Chan, C. Y., Pedley, A. M., Kim, D., Xia, C., Zhuang, X., and Benkovic, S. J. (2018). Microtubule-directed transport of purine metabolons drives their cytosolic transit to mitochondria. Proc. Natl. Acad. Sci. 115 (51), 13009–13014. doi:10.1073/pnas.1814042115
Chang, M. C., Staklinski, S. J., Merritt, M. E., and Kilberg, M. S. (2023). A method for measurement of human asparagine synthetase (ASNS) activity and application to ASNS protein variants associated with ASNS deficiency. Biol. methods and Protoc. 8 (1), bpad026. doi:10.1093/biomethods/bpad026
Chaplin, D. D. (2010). Overview of the immune response. J. Allergy Clin. Immunol. 125 (2), S3–S23. doi:10.1016/j.jaci.2009.12.980
Chen, C., Hao, Y., Liang, J., and Liu, X. (2020). Congenital microcephaly with early onset epileptic encephalopathy caused by ASNS gene mutation: a case report. Medicine 99 (22), e20507. doi:10.1097/MD.0000000000020507
Chen, K., Zhang, J., Tastan, Ö. Y., Deussen, Z. A., Siswick, M. Y. Y., and Liu, J. L. (2011). Glutamine analogs promote cytoophidium assembly in human and Drosophila cells. J. Genet. Genomics 38 (9), 391–402. doi:10.1016/j.jgg.2011.08.004
Choi, M.-G., and Carman, G. M. (2007). Phosphorylation of human CTP synthetase 1 by protein kinase A. J. Biol. Chem. 282 (8), 5367–5377. doi:10.1074/jbc.M610993200
Christopherson, R. I., Lyons, S. D., and Wilson, P. K. (2002). Inhibitors of de novo nucleotide biosynthesis as drugs. Accounts Chem. Res. 35 (11), 961–971. doi:10.1021/ar0000509
Chuenchor, W., Doukov, T. I., Chang, K. T., Resto, M., Yun, C. S., and Gerratana, B. (2020). Different ways to transport ammonia in human and Mycobacterium tuberculosis NAD+ synthetases. Nat. Commun. 11 (1), 16. doi:10.1038/s41467-019-13845-4
Ciustea, M., Gutierrez, J. A., Abbatiello, S. E., Eyler, J. R., and Richards, N. G. (2005). Efficient expression, purification, and characterization of C-terminally tagged, recombinant human asparagine synthetase. Archives Biochem. Biophysics 440 (1), 18–27. doi:10.1016/j.abb.2005.05.023
Cooney, D. A., and Handschumacher, R. E. (1970). L-asparaginase and L-asparagine metabolism. Annu. Rev. Pharmacol. 10 (1), 421–440. doi:10.1146/annurev.pa.10.040170.002225
Coricello, A., Zhu, W., Lupia, A., Gratteri, C., Vos, M., Chaptal, V., et al. (2023). Cryo-EM and molecular dynamics simulations reveal hidden conformational dynamics controlling ammonia transport in human asparagine synthetase. bioRxiv: the preprint server for biology. doi:10.1101/2023.05.16.541009
Crawford, I. P. (1989). Evolution of A biosynthetic pathway: the tryptophan paradigm. Annu. Rev. Microbiol. 43 (1), 567–600. doi:10.1146/annurev.mi.43.100189.003031
De Cima, S., Polo, L. M., Díez-Fernández, C., Martínez, A. I., Cervera, J., Fita, I., et al. (2015). Structure of human carbamoyl phosphate synthetase: deciphering the on/off switch of human ureagenesis. Sci. Rep. 5 (1), 16950. doi:10.1038/srep16950
De Ingeniis, J., Kazanov, M. D., Shatalin, K., Gelfand, M. S., Osterman, A. L., and Sorci, L. (2012). Glutamine versus ammonia utilization in the NAD synthetase family. PLoS ONE 7. doi:10.1371/journal.pone.0039115
Del Caño-Ochoa, F., Grande-García, A., Reverte-López, M., D’Abramo, M., and Ramón-Maiques, S. (2018). Characterization of the catalytic flexible loop in the dihydroorotase domain of the human multi-enzymatic protein CAD. J. Biol. Chem. 293 (49), 18903–18913. doi:10.1074/jbc.RA118.005494
Del Caño-Ochoa, F., and Ramón-Maiques, S. (2021). Deciphering CAD: structure and function of a mega-enzymatic pyrimidine factory in health and disease. Protein Sci. 30 (10), 1995–2008. doi:10.1002/pro.4158
Du, F., Chen, J., Liu, H., Cai, Y., Cao, T., Han, W., et al. (2019). SOX12 promotes colorectal cancer cell proliferation and metastasis by regulating asparagine synthesis. Cell Death Dis. 10 (3), 239. doi:10.1038/s41419-019-1481-9
Elliott, W. L., and Weber, G. (1984). Proliferation-linked increase in phosphoribosylformylglycinamidine synthetase activity (EC 6.3.5.3). Cancer Res. 44 (6), 2430–2434.
Erbs, E., Brasen, C. L., Lund, A. M., and Rasmussen, M. (2023). Adult patient diagnosed with NADSYN1 associated congenital NAD deficiency and analysis of NAD levels to be published in: European Journal of Medical Genetics. Eur. J. Med. Genet. 66 (3), 104698. doi:10.1016/j.ejmg.2023.104698
Faoucher, M., Poulat, A. L., Chatron, N., Labalme, A., Schluth-Bolard, C., Till, M., et al. (2019). Asparagine synthetase deficiency: a novel case with an unusual molecular mechanism. Mol. Genet. Metabolism Rep. 21, 100509. doi:10.1016/j.ymgmr.2019.100509
Field, M. S., Anderson, D. D., and Stover, P. J. (2011). Mthfs is an essential gene in mice and a component of the purinosome. Front. Genet. 2, 36. doi:10.3389/fgene.2011.00036
Fresquet, V., Thoden, J. B., Holden, H. M., and Raushel, F. M. (2004). Kinetic mechanism of asparagine synthetase from Vibrio cholerae. Bioorg. Chem. 32 (2), 63–75. doi:10.1016/j.bioorg.2003.10.002
Frey, P. A., and Hegeman, A. D. (2007). “ATP-dependent synthetases and ligases,” in Enzymatic reaction mechanisms. Editors P. A. Frey,, and A. D. Hegeman (Oxford: Oxford University Press). doi:10.1093/oso/9780195122589.003.0015
Galada, C., Hebbar, M., Lewis, L., Soans, S., Kadavigere, R., Srivastava, A., et al. (2018). Report of four novel variants in ASNS causing asparagine synthetase deficiency and review of literature. Congenit. Anomalies 58 (5), 181–182. doi:10.1111/cga.12275
Gauthier-Coles, G., Vennitti, J., Zhang, Z., Comb, W. C., Xing, S., Javed, K., et al. (2021). Quantitative modelling of amino acid transport and homeostasis in mammalian cells. Nat. Commun. 12 (1), 5282. doi:10.1038/s41467-021-25563-x
Grande-García, A., Lallous, N., Díaz-Tejada, C., and Ramón-Maiques, S. (2014). Structure, functional characterization, and evolution of the dihydroorotase domain of human CAD. Structure 22 (2), 185–198. doi:10.1016/j.str.2013.10.016
Grima-Reyes, M., Vandenberghe, A., Nemazanyy, I., Meola, P., Paul, R., Reverso-Meinietti, J., et al. (2022). Tumoral microenvironment prevents de novo asparagine biosynthesis in B cell lymphoma, regardless of ASNS expression. Sci. Adv. 8 (27), eabn6491. doi:10.1126/sciadv.abn6491
Gupta, N., Tewari, V. V., Kumar, M., Langeh, N., Gupta, A., Mishra, P., et al. (2017). Asparagine Synthetase deficiency-report of a novel mutation and review of literature. Metab. Brain Dis. 32 (6), 1889–1900. doi:10.1007/s11011-017-0073-6
Higgins, M. J., Graves, P. R., and Graves, L. M. (2007). Regulation of human cytidine triphosphate synthetase 1 by glycogen synthase kinase 3. J. Biol. Chem. 282 (40), 29493–29503. doi:10.1074/jbc.M703948200
Holden, H. M., Thoden, J. B., and Raushel, F. M. (1999). Carbamoyl phosphate synthetase: an amazing biochemical odyssey from substrate to product. Cell. Mol. Life Sci. (CMLS) 56 (5–6), 507–522. doi:10.1007/s000180050448
Holzer, K., Drucker, E., Roessler, S., Dauch, D., Heinzmann, F., Waldburger, N., et al. (2017). Proteomic analysis reveals GMP synthetase as p53 repression target in liver cancer. Am. J. Pathology 187 (2), 228–235. doi:10.1016/j.ajpath.2016.09.022
Hongo, S., Matsumoto, T., and Sato, T. (1978). Purification and properties of asparagine synthetase from rat liver. Biochimica Biophysica Acta (BBA) - Enzym. 522 (1), 258–266. doi:10.1016/0005-2744(78)90342-X
Ingerson-Mahar, M., Briegel, A., Werner, J. N., Jensen, G. J., and Gitai, Z. (2010). The metabolic enzyme CTP synthase forms cytoskeletal filaments. Nat. Cell Biol. 12 (8), 739–746. doi:10.1038/ncb2087
Kassel, K. M., Au, D. R., Higgins, M. J., Hines, M., and Graves, L. M. (2010). Regulation of human cytidine triphosphate synthetase 2 by phosphorylation. J. Biol. Chem. 285 (44), 33727–33736. doi:10.1074/jbc.M110.178566
Knott, S. R. V., Wagenblast, E., Khan, S., Kim, S. Y., Soto, M., Wagner, M., et al. (2018). Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 554 (7692), 378–381. doi:10.1038/nature25465
Knudsen, C., Janeshawari Gallage, N., Cetti Hansen, C., Lindberg Møller, B., and Laursen, T. (2018). Dynamic metabolic solutions to the sessile life style of plants. Nat. Prod. Rep. 35 (11), 1140–1155. doi:10.1039/C8NP00037A
Kortbawi, H., Ames, E., Pritchard, A., Devine, P., van Ziffle, J., and Slavotinek, A. (2022). Further description of two patients with biallelic variants in NADSYN1 in association with cardiac and vertebral anomalies. Am. J. Med. Genet. Part A 188 (8), 2479–2484. doi:10.1002/ajmg.a.62765
Kursula, P., Flodin, S., Ehn, M., Hammarström, M., Schüler, H., Nordlund, P., et al. (2006). Structure of the synthetase domain of human CTP synthetase, a target for anticancer therapy. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 62 (7), 613–617. doi:10.1107/S1744309106018136
Larsen, T. M., Boehlein, S. K., Schuster, S. M., Richards, N. G. J., Thoden, J. B., Holden, H. M., et al. (1999). Three-dimensional structure of Escherichia coli asparagine synthetase B: a short journey from substrate to product. Biochemistry 38 (49), 16146–16157. doi:10.1021/bi9915768
Lauer-Zillhardt, J., Kaminska, A., Galmiche-Rolland, L., Bahi-Buisson, N., Pontoizeau, C., Ottolenghi, C., et al. (2016). Epileptic phenotype of two siblings with asparagine synthesis deficiency mimics neonatal pyridoxine-dependent epilepsy. Neuropediatrics 47 (06), 399–403. doi:10.1055/s-0036-1586222
Li, G., Li, D., Wang, T., and He, S. (2021). Pyrimidine biosynthetic enzyme CAD: its function, regulation, and diagnostic potential. Int. J. Mol. Sci. 22 (19), 10253. doi:10.3390/ijms221910253
Li, K. (2007). Intramolecular tunnel and regulatory mechanisms of asparagine synthetase (ASNS). Univ. Fla. ProQuest Diss. Publ. 2007, 3300760.
Lieberman, I. (1956). Enzymatic amination of uridine triphosphate to cytidine triphosphate. J. Biol. Chem. 222 (2), 765–775. doi:10.1016/S0021-9258(20)89934-7
Lin, J., Zhao, L., Zhao, S., Li, S., Zhao, Z., Chen, Z., et al. (2021). Disruptive NADSYN1 variants implicated in congenital vertebral malformations. Genes 12 (10), 1615. doi:10.3390/genes12101615
Ling, P. D., Tan, J., Sewatanon, J., and Peng, R. (2008). Murine gammaherpesvirus 68 open reading frame 75c tegument protein induces the degradation of PML and is essential for production of infectious virus. J. Virology 82 (16), 8000–8012. doi:10.1128/JVI.02752-07
Liu, J., Zhang, Y., Wang, Q. Q., Zhou, Y., and Liu, J. L. (2023). Fat body-specific reduction of CTPS alleviates HFD-induced obesity. eLife 12, e85293. doi:10.7554/eLife.85293
Liu, L., Wang, J., Li, H., Dong, Y., Li, Y., Xia, L., et al. (2022). An intractable epilepsy phenotype of ASNS novel mutation in two patients with asparagine synthetase deficiency. Clin. Chim. Acta 531, 331–336. doi:10.1016/j.cca.2022.04.989
Longo, L. M., Jabłońska, J., Vyas, P., Kanade, M., Kolodny, R., Ben-Tal, N., et al. (2020). On the emergence of P-Loop NTPase and Rossmann enzymes from a Beta-Alpha-Beta ancestral fragment. eLife 9, e64415. doi:10.7554/eLife.64415
Lorenzi, P. L., and Weinstein, J. N. (2009). Asparagine synthetase: a new potential biomarker in ovarian cancer. Drug News Perspect. 22 (1), 61. doi:10.1358/dnp.2009.22.1.1303820
Lynch, E. M., DiMattia, M. A., Albanese, S., van Zundert, G. C. P., Hansen, J. M., Quispe, J. D., et al. (2021). Structural basis for isoform-specific inhibition of human CTPS1. Proc. Natl. Acad. Sci. U. S. A. 118 (40), e2107968118. doi:10.1073/pnas.2107968118
Lynch, E. M., Hicks, D. R., Shepherd, M., Endrizzi, J. A., Maker, A., Hansen, J. M., et al. (2017). Human CTP synthase filament structure reveals the active enzyme conformation. Nat. Struct. Mol. Biol. 24 (6), 507–514. doi:10.1038/nsmb.3407
Lynch, E. M., and Kollman, J. M. (2020). Coupled structural transitions enable highly cooperative regulation of human CTPS2 filaments. Nat. Struct. Mol. Biol. 27 (1), 42–48. doi:10.1038/s41594-019-0352-5
Mangold, C. A., Yao, P. J., Du, M., Freeman, W. M., Benkovic, S. J., and Szpara, M. L. (2018). Expression of the purine biosynthetic enzyme phosphoribosyl formylglycinamidine synthase in neurons. J. Neurochem. 144 (6), 723–735. doi:10.1111/jnc.14304
Markin, R. S., Luehr, C. A., and Schuster, S. M. (1981). Kinetic mechanism of beef pancreatic L-asparagine synthetase. Biochemistry 20 (25), 7226–7232. doi:10.1021/bi00528a027
Martin, E., Latour, S., Minet, N., Boschat, A. C., Sanquer, S., Sobrino, S., et al. (2020). Impaired lymphocyte function and differentiation in CTPS1-deficient patients result from a hypomorphic homozygous mutation. JCI Insight 5 (5), e133880. doi:10.1172/jci.insight.133880
Martin, E., Palmic, N., Sanquer, S., Lenoir, C., Hauck, F., Mongellaz, C., et al. (2014). CTP synthase 1 deficiency in humans reveals its central role in lymphocyte proliferation. Nature 510 (7504), 288–292. doi:10.1038/nature13386
Massière, F., and Badet-Denisot, M.-A. (1998). The mechanism of glutamine-dependent amidotransferases. Cell. Mol. Life Sci. CMLS 54 (3), 205–222. doi:10.1007/s000180050145
Matsumoto, H., Kawashima, N., Yamamoto, T., Nakama, M., Otsuka, H., Ago, Y., et al. (2021). In vitro functional analysis of four variants of human asparagine synthetase. J. Inherit. Metabolic Dis. 44 (5), 1226–1234. doi:10.1002/jimd.12408
McCluskey, G. D., and Bearne, S. L. (2018). “Pinching” the ammonia tunnel of CTP synthase unveils coordinated catalytic and allosteric-dependent control of ammonia passage. Biochimica Biophysica Acta (BBA) - General Subj. 1862 (12), 2714–2727. doi:10.1016/j.bbagen.2018.08.008
McElwee, J. J., Kaila, N., Carreiro, S., Kumar, S., Toms, A. V., Phadke, A., et al. (2023). Discovery and characterization of novel inhibitors of CTP synthase 1 (CTPS1) for the treatment of autoimmune and inflammatory disease. J. Immunol. 210 (1_Suppl. ment), 165.16. doi:10.4049/jimmunol.210.Supp.165.16
Meyer, M. E. (2010). Inhibition, characterization, and crystallization of glutamine-dependent asparagine synthetase. Univ. Fla. ProQuest Diss. Publ. 2010, 3416706.
Milman, H. A., Cooney, D. A., and Huang, C. Y. (1980). Studies on the mechanism of the glutamine-dependent reaction catalyzed by asparagine synthetase from mouse pancreas. J. Biol. Chem. 255 (5), 1862–1866. doi:10.1016/S0021-9258(19)85961-6
Minet, N., Boschat, A. C., Lane, R., Laughton, D., Beer, P., Asnagli, H., et al. (2023). Differential roles of CTP synthetases CTPS1 and CTPS2 in cell proliferation. Life Sci. Alliance 6 (9), e202302066. doi:10.26508/lsa.202302066
Moore, B. D. (2004). Bifunctional and moonlighting enzymes: lighting the way to regulatory control. Trends Plant Sci. 9 (5), 221–228. doi:10.1016/j.tplants.2004.03.005
Nademi, Z., Wynn, R. F., Slatter, M., Hughes, S. M., Bonney, D., Qasim, W., et al. (2019). Hematopoietic stem cell transplantation for cytidine triphosphate synthase 1 (CTPS1) deficiency. Bone Marrow Transplant. 54 (1), 130–133. doi:10.1038/s41409-018-0246-x
Nakaishi, Y., Bando, M., Shimizu, H., Watanabe, K., Goto, F., Tsuge, H., et al. (2009). Structural analysis of human glutamine:fructose-6-phosphate amidotransferase, a key regulator in type 2 diabetes. FEBS Lett. 583 (1), 163–167. doi:10.1016/j.febslet.2008.11.041
Nakamura, J., and Lou, L. (1995). Biochemical characterization of human GMP synthetase. J. Biol. Chem. 270 (13), 7347–7353. doi:10.1074/jbc.270.13.7347
Nakamura, J., Straub, K., Wu, J., and Lou, L. (1995). The glutamine hydrolysis function of human GMP synthetase: Identification of an essential active site cysteine. J. Biol. Chem. 270 (40), 23450–23455. doi:10.1074/jbc.270.40.23450
Nguyen, S., Jovcevski, B., Pukala, T. L., Bruning, J. B., and Laursen, T. (2022). Structural insights into the antifungal drug target guanosine monophosphate synthase from Aspergillus fumigatus. Acta Crystallogr. Sect. D Struct. Biol. 78 (2), 248–259. doi:10.1107/S2059798321012031
Nitzahn, M., and Lipshutz, G. S. (2020). CPS1: looking at an ancient enzyme in a modern light. Mol. Genet. Metabolism 131 (3), 289–298. doi:10.1016/j.ymgme.2020.10.003
Noree, C., Monfort, E., and Shotelersuk, V. (2018). Human asparagine synthetase associates with the mitotic spindle. Biol. Open 7, 038307. doi:10.1242/bio.038307
Noree, C., Sato, B. K., Broyer, R. M., and Wilhelm, J. E. (2010). Identification of novel filament-forming proteins in Saccharomyces cerevisiae and Drosophila melanogaster. J. Cell Biol. 190 (4), 541–551. doi:10.1083/jcb.201003001
Oliver, J. C., Linger, R. S., Chittur, S. V., and Davisson, V. J. (2013). Substrate activation and conformational dynamics of guanosine 5′-monophosphate synthetase, Biochemistry 52, 5225–5235. doi:10.1021/bi3017075
Pace, H. C., and Brenner, C. (2001). The nitrilase superfamily: classification, structure and function. Genome Biol. 2 (1), reviews0001–1. doi:10.1186/gb-2001-2-1-reviews0001
Palmer, E. E., Hayner, J., Sachdev, R., Cardamone, M., Kandula, T., Morris, P., et al. (2015). Asparagine Synthetase Deficiency causes reduced proliferation of cells under conditions of limited asparagine. Mol. Genet. Metabolism 116 (3), 178–186. doi:10.1016/j.ymgme.2015.08.007
Pareek, V., Sha, Z., He, J., Wingreen, N. S., and Benkovic, S. J. (2021). Metabolic channeling: predictions, deductions, and evidence. Mol. Cell 81 (18), 3775–3785. doi:10.1016/j.molcel.2021.08.030
Patterson, D., Bleskan, J., Gardiner, K., and Bowersox, J. (1999). Human phosphoribosylformylglycineamide amidotransferase (FGARAT): regional mapping, complete coding sequence, isolation of a functional genomic clone, and DNA sequence analysis. Gene 239 (2), 381–391. doi:10.1016/S0378-1119(99)00378-9
Pedley, A. M., Pareek, V., and Benkovic, S. J. (2022). The purinosome: a case study for a mammalian metabolon. Annu. Rev. Biochem. 91 (1), 89–106. doi:10.1146/annurev-biochem-032620-105728
Pfeiffer, C., Grandits, A. M., Asnagli, H., Schneller, A., Huber, J., Zojer, N., et al. (2024). CTPS1 is a novel therapeutic target in multiple myeloma which synergizes with inhibition of CHEK1, ATR or WEE1. Leukemia 38 (1), 181–192. doi:10.1038/s41375-023-02071-z
Qu, C., Hao, B., Xu, X., Wang, Y., Yang, C., Xu, Z., et al. (2019). Functional research on three presumed asparagine synthetase family members in poplar. Genes 10 (5), 326. doi:10.3390/genes10050326
Quddus, A. (2020). Step Pharma SAS. AUSTRALIAN PATENT OFFICE Aminopyrimidine/pyrazine derivatives as ctps1 inhibitors. Aust. Pat. Off. WO2020083975A1.
Radadiya, A., Zhu, W., Coricello, A., Alcaro, S., and Richards, N. G. J. (2020). Improving the treatment of acute lymphoblastic leukemia. Biochemistry 59 (35), 3193–3200. doi:10.1021/acs.biochem.0c00354
Radha Rama Devi, A., and Naushad, S. M. (2019). Molecular diagnosis of asparagine synthetase (ASNS) deficiency in two Indian families and literature review of 29 ASNS deficient cases. Gene 704, 97–102. doi:10.1016/j.gene.2019.04.024
Richards, N. G. J., and Schuster, S. M. (1998). “Mechanistic issues in asparagine synthetase catalysis,” in Advances in enzymology - and related areas of molecular Biology. 1st edn (United States: Wiley), 145–198. doi:10.1002/9780470123188.ch5
Rolfe, A., Yao, S., Nguyen, T. V., Omoto, K., Colombo, F., Virrankoski, M., et al. (2020). Discovery of 2,6-dimethylpiperazines as allosteric inhibitors of CPS1. ACS Med. Chem. Lett. 11 (6), 1305–1309. doi:10.1021/acsmedchemlett.0c00145
Roy, H., Becker, H. D., Reinbolt, J., and Kern, D. (2003). When contemporary aminoacyl-tRNA synthetases invent their cognate amino acid metabolism. Proc. Natl. Acad. Sci. 100 (17), 9837–9842. doi:10.1073/pnas.1632156100
Ruiz-Ramos, A., Velázquez-Campoy, A., Grande-García, A., Moreno-Morcillo, M., and Ramón-Maiques, S. (2016). Structure and functional characterization of human aspartate transcarbamoylase, the target of the anti-tumoral drug PALA. Structure 24 (7), 1081–1094. doi:10.1016/j.str.2016.05.001
Ruzzo, E. K., Capo-Chichi, J. M., Ben-Zeev, B., Chitayat, D., Mao, H., Pappas, A., et al. (2013). Deficiency of asparagine synthetase causes congenital microcephaly and a progressive form of encephalopathy. Neuron 80 (2), 429–441. doi:10.1016/j.neuron.2013.08.013
Sacharow, S. J., Dudenhausen, E. E., Lomelino, C. L., Rodan, L., El Achkar, C. M., Olson, H. E., et al. (2018). Characterization of a novel variant in siblings with asparagine synthetase deficiency. Mol. Genet. Metabolism 123 (3), 317–325. doi:10.1016/j.ymgme.2017.12.433
Saxild, H. H., and Nygaard, P. (2000). The yexA gene product is required for phosphoribosylformylglycinamidine synthetase activity in Bacillus subtilis. Microbiology 146 (4), 807–814. doi:10.1099/00221287-146-4-807
Schendel, F. J., Mueller, E., Stubbe, J., Shiau, A., and Smith, J. M. (1989). Formylglycinamide ribonucleotide synthetase from Escherichia coli: cloning, sequencing, overproduction, isolation, and characterization. Biochemistry 28 (6), 2459–2471. doi:10.1021/bi00432a017
Schleinitz, D., Seidel, A., Stassart, R., Klammt, J., Hirrlinger, P. G., Winkler, U., et al. (2018). Novel mutations in the asparagine synthetase gene (ASNS) associated with microcephaly. Front. Genet. 9, 245. doi:10.3389/fgene.2018.00245
Seidahmed, M. Z., Salih, M. A., Abdulbasit, O. B., Samadi, A., Al Hussien, K., Miqdad, A. M., et al. (2016). Hyperekplexia, microcephaly and simplified gyral pattern caused by novel ASNS mutations, case report. BMC Neurol. 16 (1), 105. doi:10.1186/s12883-016-0633-0
Semmelmann, F., Hupfeld, E., Heizinger, L., Merkl, R., and Sterner, R. (2019). A fold-independent interface residue is crucial for complex formation and allosteric signaling in class I glutamine amidotransferases. Biochemistry 58 (22), 2584–2588. doi:10.1021/acs.biochem.9b00286
Shaheen, R., Maddirevula, S., Ewida, N., Alsahli, S., Abdel-Salam, G. M., Zaki, M. S., et al. (2019). Genomic and phenotypic delineation of congenital microcephaly. Genet. Med. 21 (3), 545–552. doi:10.1038/s41436-018-0140-3
Sharma, N., Ahalawat, N., Sandhu, P., Strauss, E., Mondal, J., and Anand, R. (2020). Role of allosteric switches and adaptor domains in long-distance cross-talk and transient tunnel formation. Sci. Adv. 6 (14), eaay7919. doi:10.1126/sciadv.aay7919
Sharma, N., Singh, S., Tanwar, A. S., Mondal, J., and Anand, R. (2022). Mechanism of coordinated gating and signal transduction in purine biosynthetic enzyme formylglycinamidine synthetase. ACS Catal. 12 (3), 1930–1944. doi:10.1021/acscatal.1c05521
Shivakumaraswamy, S., Kumar, S., Bellur, A., Polisetty, S. D., and Balaram, H. (2022). Mechanistic insights into the functioning of a two-subunit GMP synthetase, an allosterically regulated, ammonia channeling enzyme. Biochemistry 61 (18), 1988–2006. doi:10.1021/acs.biochem.2c00151
Sircar, K., Huang, H., Hu, L., Cogdell, D., Dhillon, J., Tzelepi, V., et al. (2012). Integrative molecular profiling reveals asparagine synthetase is a target in castration-resistant prostate cancer. Am. J. Pathology 180 (3), 895–903. doi:10.1016/j.ajpath.2011.11.030
Sprute, R., Ardicli, D., Oguz, K. K., Malenica-Mandel, A., Daimagüler, H. S., Koy, A., et al. (2019). Clinical outcomes of two patients with a novel pathogenic variant in ASNS: response to asparagine supplementation and review of the literature. Hum. Genome Var. 6 (1), 24–10. doi:10.1038/s41439-019-0055-9
Staklinski, S. J., Chang, M. C., Ahrens-Nicklas, R. C., Kaur, S., Stefanatos, A. K., Dudenhausen, E. E., et al. (2023). Characterizing asparagine synthetase deficiency variants in lymphoblastoid cell lines. JIMD Rep. 64 (2), 167–179. doi:10.1002/jmd2.12356
Staklinski, S. J., Chang, M. C., Yu, F., Collins Ruff, K., Franz, D. N., Qian, Z., et al. (2022). Cellular and molecular characterization of two novel asparagine synthetase gene mutations linked to asparagine synthetase deficiency. J. Biol. Chem. 298 (9), 102385. doi:10.1016/j.jbc.2022.102385
Su, N., and Kilberg, M. S. (2008). C/EBP homology protein (CHOP) interacts with activating transcription factor 4 (ATF4) and negatively regulates the stress-dependent induction of the asparagine synthetase gene. J. Biol. Chem. 283 (50), 35106–35117. doi:10.1074/jbc.M806874200
Summar, M. L., Hall, L., Eeds, A., Hutcheson, H., Kuo, A., Willis, A., et al. (2003). Characterization of genomic structure and polymorphisms in the human carbamyl phosphate synthetase I gene. Gene 311, 51–57. doi:10.1016/S0378-1119(03)00528-6
Sun, J., McGillivray, A. J., Pinner, J., Yan, Z., Liu, F., Bratkovic, D., et al. (2017). Diaphragmatic eventration in sisters with asparagine synthetase deficiency: a novel homozygous ASNS mutation and expanded phenotype. JIMD Rep. 34, 1–9. doi:10.1007/8904_2016_3
Szot, J. O., Campagnolo, C., Cao, Y., Iyer, K. R., Cuny, H., Drysdale, T., et al. (2020). Bi-Allelic mutations in NADSYN1 cause multiple organ defects and expand the genotypic spectrum of congenital NAD deficiency disorders. Am. J. Hum. Genet. 106 (1), 129–136. doi:10.1016/j.ajhg.2019.12.006
Szot, J. O., Cuny, H., Martin, E. M., Sheng, D. Z., Iyer, K., Portelli, S., et al. (2024). A metabolic signature for NADSYN1-dependent congenital NAD deficiency disorder. J. Clin. investigation 134 (4), e174824. doi:10.1172/JCI174824
Tan, W. L., Bhattacharya, B., Loh, M., Balasubramanian, I., Akram, M., Dong, D., et al. (2011). Low cytosine triphosphate synthase 2 expression renders resistance to 5-fluorouracil in colorectal cancer. Cancer Biol. Ther. 11 (6), 599–608. doi:10.4161/cbt.11.6.14670
Tanwar, A. S., Morar, M., Panjikar, S., and Anand, R. (2012). Formylglycinamide ribonucleotide amidotransferase from Salmonella typhimurium: role of ATP complexation and the glutaminase domain in catalytic coupling. Acta Crystallogr. Sect. D. Biol. Crystallogr. 68 (6), 627–636. doi:10.1107/S0907444912006543
Tesmer, J. J. G., Klem, T. J., Deras, M. L., Davisson, V. J., and Smith, J. L. (1996). The crystal structure of GMP synthetase reveals a novel catalytic triad and is a structural paradigm for two enzyme families. Nat. Struct. Biol. 3 (1), 74–86. doi:10.1038/nsb0196-74
Tesson, A. R., Soper, T. S., Ciustea, M., and Richards, N. (2003). Revisiting the steady state kinetic mechanism of glutamine-dependent asparagine synthetase from Escherichia coli. Archives Biochem. Biophysics 413 (1), 23–31. doi:10.1016/S0003-9861(03)00118-8
Toda, K., Kawada, K., Iwamoto, M., Inamoto, S., Sasazuki, T., Shirasawa, S., et al. (2016). Metabolic alterations caused by KRAS mutations in colorectal cancer contribute to cell adaptation to glutamine depletion by upregulation of asparagine synthetase. Neoplasia 18 (11), 654–665. doi:10.1016/j.neo.2016.09.004
Tsai, K., Messick, T. E., and Lieberman, P. M. (2015). Disruption of host antiviral resistances by gammaherpesvirus tegument proteins with homology to the FGARAT purine biosynthesis enzyme. Curr. Opin. Virology 14, 30–40. doi:10.1016/j.coviro.2015.07.008
Van Der Knaap, J. A., Kozhevnikova, E., Langenberg, K., Moshkin, Y. M., and Verrijzer, C. P. (2010). Biosynthetic enzyme GMP synthetase cooperates with ubiquitin-specific protease 7 in transcriptional regulation of ecdysteroid target genes. Mol. Cell. Biol. 30 (3), 736–744. doi:10.1128/MCB.01121-09
Van Heeke, G., and Schuster, S. M. (1989). The N-terminal cysteine of human asparagine synthetase is essential for glutamine-dependent activity. J. Biol. Chem. 264 (33), 19475–19477. doi:10.1016/S0021-9258(19)47138-X
van Kuilenburg, A. B. P., Meinsma, R., Vreken, P., Waterham, H. R., and van Gennip, A. H. (2000). Identification of a cDNA encoding an isoform of human CTP synthetase. Biochimica Biophysica Acta (BBA) - Gene Struct. Expr. 1492 (2), 548–552. doi:10.1016/S0167-4781(00)00141-X
Van Trimpont, M., Peeters, E., De Visser, Y., Schalk, A. M., Mondelaers, V., De Moerloose, B., et al. (2022). Novel insights on the use of L-asparaginase as an efficient and safe anti-cancer therapy. Cancers 14 (4), 902. doi:10.3390/cancers14040902
Varadi, M., Anyango, S., Deshpande, M., Nair, S., Natassia, C., Yordanova, G., et al. (2022). AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50 (D1), D439–D444. doi:10.1093/nar/gkab1061
Verschuur, A. C. (2007). Cytidine triphosphate synthetase (CTP synthetase) as a druggable target in cancer. Drugs Future 32 (12), 1071. doi:10.1358/dof.2007.032.12.1155384
Verschuur, A. C., Van Gennip, A., Leen, R., Muller, E., Elzinga, L., Voûte, P., et al. (2000). Cyclopentenyl cytosine inhibits cytidine triphosphate synthetase in paediatric acute non-lymphocytic leukaemia. Eur. J. Cancer 36 (5), 627–635. doi:10.1016/S0959-8049(00)00021-6
Walker, J. E., Saraste, M., Runswick, M., and Gay, N. (1982). Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1 (8), 945–951. doi:10.1002/j.1460-2075.1982.tb01276.x
Wang, C., He, G., Ge, Y., Li, R., Li, Z., and Lin, Y. (2020). A novel compound heterozygous missense mutation in ASNS broadens the spectrum of asparagine synthetase deficiency. Mol. Genet. Genomic Med. 8 (6), e1235. doi:10.1002/mgg3.1235
Wang, J., Luo, F. F., Huang, T. J., Mei, Y., Peng, L. X., Qian, C. N., et al. (2021). The upregulated expression of RFC4 and GMPS mediated by DNA copy number alteration is associated with the early diagnosis and immune escape of ESCC based on a bioinformatic analysis. Aging 13 (17), 21758–21777. doi:10.18632/aging.203520
Wang, J., Wu, Y., Li, Y., Wang, Y., Shen, F., Zhou, J., et al. (2021). Guanosine monophosphate synthase upregulation mediates cervical cancer progression by inhibiting the apoptosis of cervical cancer cells via the Stat3/P53 pathway. Int. J. Oncol. 58 (4), 3. doi:10.3892/ijo.2021.5183
Wang, Q., Guan, Y. F., Hancock, S. E., Wahi, K., van Geldermalsen, M., Zhang, B. K., et al. (2021). Inhibition of guanosine monophosphate synthetase (GMPS) blocks glutamine metabolism and prostate cancer growth. J. Pathology 254 (2), 135–146. doi:10.1002/path.5665
Wang, S., Chen, J., Zhu, X., Huang, T., Xu, H., Ying, G., et al. (2023). Clinical and genetic analysis of a case of late onset carbamoyl phosphate synthase I deficiency caused by CPS1 mutation and literature review. BMC Med. Genomics 16 (1), 145. doi:10.1186/s12920-023-01569-w
Welin, M., Lehtiö, L., Johansson, A., Flodin, S., Nyman, T., Trésaugues, L., et al. (2013). Substrate specificity and oligomerization of human GMP synthetase. J. Mol. Biol. 425 (22), 4323–4333. doi:10.1016/j.jmb.2013.06.032
Williamson, J., Petralia, R. S., Wang, Y. X., Mattson, M. P., and Yao, P. J. (2017). Purine biosynthesis enzymes in hippocampal neurons. NeuroMolecular Med. 19 (4), 518–524. doi:10.1007/s12017-017-8466-6
Xiao, Q., Zhang, F., Nacev, B. A., Liu, J. O., and Pei, D. (2010). Protein N-terminal processing: substrate specificity of Escherichia coli and human methionine aminopeptidases. Biochemistry 49 (26), 5588–5599. doi:10.1021/bi1005464
Yamamoto, T., Endo, W., Ohnishi, H., Kubota, K., Kawamoto, N., Inui, T., et al. (2017). The first report of Japanese patients with asparagine synthetase deficiency. Brain Dev. 39 (3), 236–242. doi:10.1016/j.braindev.2016.09.010
Yan, B., Wang, C., Zhang, K., Zhang, H., Gao, M., Lv, Y., et al. (2019). Novel neonatal variants of the carbamoyl phosphate synthetase 1 deficiency: two case reports and review of literature. Front. Genet. 10, 718. doi:10.3389/fgene.2019.00718
Yang, H., He, X., Zheng, Y., Feng, W., Xia, X., Yu, X., et al. (2014). Down-regulation of asparagine synthetase induces cell cycle arrest and inhibits cell proliferation of breast cancer. Chem. Biol. Drug Des. 84 (5), 578–584. doi:10.1111/cbdd.12348
Yao, S., Nguyen, T. V., Rolfe, A., Agrawal, A. A., Ke, J., Peng, S., et al. (2020). Small molecule inhibition of CPS1 activity through an allosteric pocket. Cell Chem. Biol. 27 (3), 259–268.e5. doi:10.1016/j.chembiol.2020.01.009
Yi, H., Talmon, G., and Wang, J. (2020). Glutamate in cancers: from metabolism to signaling. J. Biomed. Res. 34 (4), 260. doi:10.7555/JBR.34.20190037
Yu, Q., Wang, X., Wang, L., Zheng, J., Wang, J., and Wang, B. (2016). Knockdown of asparagine synthetase (ASNS) suppresses cell proliferation and inhibits tumor growth in gastric cancer cells. Scand. J. Gastroenterology 51 (10), 1220–1226. doi:10.1080/00365521.2016.1190399
Yuan, Q., Yin, L., He, J., Zeng, Q., Liang, Y., Shen, Y., et al. (2024). Metabolism of asparagine in the physiological state and cancer. Cell Commun. Signal. 22 (1), 163. doi:10.1186/s12964-024-01540-x
Zhang, S., Ding, K., Shen, Q. J., Zhao, S., and Liu, J. L. (2018). Filamentation of asparagine synthetase in Saccharomyces cerevisiae. PLOS Genet. 14 (10), e1007737. doi:10.1371/journal.pgen.1007737
Zhang, Y., Morar, M., and Ealick, S. E. (2008). Structural biology of the purine biosynthetic pathway. Cell. Mol. Life Sci. 65 (23), 3699–3724. doi:10.1007/s00018-008-8295-8
Zhao, H., French, J. B., Fang, Y., and Benkovic, S. J. (2013). The purinosome, a multi-protein complex involved in the de novo biosynthesis of purines in humans, Chem. Commun. Camb. Engl. 49, 4444. doi:10.1039/c3cc41437j
Zhou, X., Guo, C. J., Chang, C. C., Zhong, J., Hu, H. H., Lu, G. M., et al. (2021). Structural basis for ligand binding modes of CTP synthase. Proc. Natl. Acad. Sci. U. S. A. 118 (30), e2026621118. doi:10.1073/pnas.2026621118
Zhou, S., Xiang, H., and Liu, J.-L. (2020). CTP synthase forms cytoophidia in archaea. J. Genet. Genomics. 47 (4), 213–223. doi:10.1016/j.jgg.2020.03.004
Keywords: GMP synthetase, CTP synthetase, asparagine synthetase, NAD+ synthetase, FGAS, carbamoyl phosphatase synthetase, activity regulation, ammonia tunnel
Citation: Zhu W, Nardone AJ and Pearce LA (2024) Advances in human glutamine-hydrolyzing synthetases and their therapeutic potential. Front. Chem. Biol 3:1410435. doi: 10.3389/fchbi.2024.1410435
Received: 01 April 2024; Accepted: 09 May 2024;
Published: 06 June 2024.
Edited by:
Adam Offenbacher, East Carolina University, United StatesReviewed by:
Ales Hnizda, Charles University, CzechiaCopyright © 2024 Zhu, Nardone and Pearce. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen Zhu, d3podUBjaGVtLmZzdS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.