 Taylor R. Church
Taylor R. Church Seth S. Margolis
Seth S. Margolis- 1Department of Biological Chemistry, The Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 2Solomon H. Snyder Department of Neuroscience, The Johns Hopkins University School of Medicine, Baltimore, MD, United States
Neurodegenerative diseases are characterized by the progressive breakdown of neuronal structure and function and the pathological accumulation of misfolded protein aggregates and toxic protein oligomers. A major contributor to the deterioration of neuronal physiology is the disruption of protein catabolic pathways mediated by the proteasome, a large protease complex responsible for most cellular protein degradation. Previously, it was believed that proteolysis by the proteasome required tagging of protein targets with polyubiquitin chains, a pathway called the ubiquitin-proteasome system (UPS). Because of this, most research on proteasomal roles in neurodegeneration has historically focused on the UPS. However, additional ubiquitin-independent pathways and their importance in neurodegeneration are increasingly recognized. In this review, we discuss the range of ubiquitin-independent proteasome pathways, focusing on substrate identification and targeting, regulatory molecules and adaptors, proteasome activators and alternative caps, and diverse proteasome complexes including the 20S proteasome, the neuronal membrane proteasome, the immunoproteasome, extracellular proteasomes, and hybrid proteasomes. These pathways are further discussed in the context of aging, oxidative stress, protein aggregation, and age-associated neurodegenerative diseases, with a special focus on Alzheimer’s Disease, Huntington’s Disease, and Parkinson’s Disease. A mechanistic understanding of ubiquitin-independent proteasome function and regulation in neurodegeneration is critical for the development of therapies to treat these devastating conditions. This review summarizes the current state of ubiquitin-independent proteasome research in neurodegeneration.
Introduction
Neurodegeneration causes an irreversible decline in cognition and motor coordination due to the progressive breakdown of neuronal structure and function. A defining feature of neurodegeneration is the accumulation of misfolded protein aggregates, which are toxic to the cell and cause neuronal damage by disrupting essential cellular processes. A fundamental mechanism in the formation of these aggregates is disruption of neuronal proteostasis, the balance of protein synthesis and degradation. This is mediated in part through the proteasome, a multi-subunit protease complex responsible for the majority of protein degradation, including the misfolded and damaged proteins implicated in neurodegenerative diseases (Zheng et al., 2016; Türker et al., 2021; Cuanalo-Contreras et al., 2023; Davidson and Pickering, 2023). The proteasome functions through multiple proteolytic mechanisms based on its composition and interactors.
The cell’s main degradative machinery, called the 26S proteasome, consists of a cylindrical 20S core particle (20S) that contains catalytic sites for proteolysis and a 19S regulatory cap (19S) that acts as a proteasome activator (PA) to facilitate recognition, unfolding, and rapid degradation of substrates. The 26S proteasome is the central hub for the ubiquitin-proteasome system (UPS), a catabolic pathway that targets proteins for destruction through 1) covalent attachment of polyubiquitin chains by a series of ubiquitin ligases, 2) recognition and de-ubiquitination by the 19S cap, and 3) ATPase-dependent unfolding and translocation of substrate proteins to the interior of the 20S core for degradation (Hershko et al., 1981; Hershko and Ciechanover, 1992; Ciechanover and Schwartz, 1998). The catalytic subunits of the 20S core include β5 (PSMB5; chymotrypsin-like activity), β2 (PSMB7; trypsin-like activity), and β1 (PSMB6; caspase-like activity), which cleave peptide bonds with different specificities and are responsible for the breakdown of proteins into short peptides (Baumeister et al., 1997; Kisselev et al., 1999; Unno et al., 2002). These peptide products are then used as a source of amino acids for biosynthesis or for other cell type-specific functions including antigen recognition, modulation of neuronal signaling, and intercellular communication (Vabulas and Hartl, 2005; Basler et al., 2013; Ramachandran and Margolis, 2017; Limanaqi et al., 2019; Türker et al., 2024). While the UPS is the best-characterized mechanism of proteasome activity (Bingol and Schuman, 2005; Patrick, 2006; Yi and Ehlers, 2007) and extensive reviews have been written on its role in neurodegenerative diseases (Ciechanover and Brundin, 2003; Dantuma and Bott, 2014; Zheng et al., 2016; Watanabe et al., 2020; Schmidt et al., 2021; Davidson and Pickering, 2023), many questions remain which are being actively explored and which will not be the focus of this review.
In addition to the UPS, the 20S proteasome is highly abundant and found with a broad set of activators and associated proteins important for various proteolytic functions, especially those affected by neurodegenerative disease (Fabre et al., 2014; Opoku-Nsiah and Gestwicki, 2018; Türker et al., 2023). While 20S proteasomes were previously believed to be non-functional without a regulatory 19S cap, increasing evidence has indicated unique, ubiquitin-independent roles of the 20S core particle and its interacting partners, particularly in degradation of intrinsically disordered, oxidized, or misfolded proteins (Jariel-Encontre et al., 2008; Baugh et al., 2009; Ben-Nissan and Sharon, 2014; Erales and Coffino, 2014; Opoku-Nsiah and Gestwicki, 2018; Davidson and Pickering, 2023), important hallmarks of neurodegeneration. Because a large portion of proteins in the human genome contain intrinsically disordered regions under physiological conditions, and because 20% of proteins may be degraded through ubiquitin-independent proteasome pathways under normal or stress conditions, it is likely that these pathways are more important for quotidian function than previously appreciated (Baugh et al., 2009; Ben-Nissan and Sharon, 2014; Pepelnjak et al., 2024). In this review, we focus on ubiquitin-independent proteasomal mechanisms and the emerging role these mechanisms plays in neurodegeneration.
Search scheme and article selection
PubMed and Google Scholar search engines were first used to identify research articles using search terms including ubiquitin-independent proteasome, neurodegeneration, 20S proteasome, Alzheimer’s Disease, Parkinson’s Disease, Huntington’s Disease, protein aggregation, oxidative stress, PA200, PA28, and aging, among others. However, results from these keyword search terms did not distinguish well between the UPS and ubiquitin-independent mechanisms, so alternative tools employing artificial intelligence (AI) were used. These included Consensus and Semantic Scholar–the primary tools used–as well as Elicit and Research Rabbit. Consensus, Elicit, and Semantic Scholar were leveraged to search databases of >200 million peer-reviewed scientific papers using natural language processing to interpret questions about research topics rather than keywords (e.g., “What roles do ubiquitin-independent proteasome mechanisms play in neurodegenerative disease?” or “Can the 20S proteasome degrade tau without a proteasome activator?”), using machine learning to process the content and context of literature, and using large language models to suggest relevant articles or provide a summary of conflicts and consensus in the literature with references, reducing bias in the search results. After curating a collection of the most relevant articles based on these searches, Research Rabbit was used to visualize connected papers, identifying additional article suggestions. Aside from AI tools, other papers were identified by scanning the references of pertinent articles. After identification, full-text articles published in or before October 2024 were reviewed for ubiquitin-independent proteasome mechanistic relevance. Original articles and reviews were included, and retracted papers were excluded. All articles were peer-reviewed except for one pre-print (indicated in the text). The writing of this review was not AI-generated, and AI tools were used for article identification only.
Proteasome substrate identification
For decades, evidence has demonstrated that the proteasome has diverse mechanisms of substrate recognition and degradation beyond the canonical UPS pathway (Jariel-Encontre et al., 2008; Baugh et al., 2009; Ben-Nissan and Sharon, 2014; Erales and Coffino, 2014), and a growing number of protein substrates targeted to the proteasome without ubiquitination have been discovered (Rosenberg-Hasson et al., 1989; Bercovich and Kahana, 1993; Coffino, 1998; Sheaff et al., 2000; Bossis et al., 2003; Myers et al., 2018; Makaros et al., 2023). Recent advances include a systematic analysis of human 20S proteasome substrates using a method called proteasomal-induced proteolysis mass spectrometry, developed by Pepelnjak et al. to identify a range of proteins degraded by the ubiquitin-independent 20S (Pepelnjak et al., 2024). Another technique, Global Protein Stability peptidome screening, was developed by Koren et al. (2018) and subsequently applied to identify ubiquitin-independent proteasome substrates (Makaros et al., 2023). These papers and others have demonstrated that proteins central to neurodegeneration, including tau (important in AD and other tauopathies) (David et al., 2002; Grune et al., 2010; Ukmar-Godec et al., 2020), α-synuclein (important in PD and other synucleinopathies) (Tofaris et al., 2001; Nakajima et al., 2005; Alvarez-Castelao et al., 2014; Makaros et al., 2023), huntingtin (important in HD) (Juenemann et al., 2013), as well as many proteins important in stress, transcriptional regulation (like RNA-binding partners and transcription factors), phase granule separation (Myers et al., 2018), and cell cycle regulation (Sheaff et al., 2000; Touitou et al., 2001; Asher et al., 2005; Wiggins et al., 2011), can be degraded through ubiquitin-independent mechanisms (Pepelnjak et al., 2024). These techniques suggest that ubiquitin-independent degradation is far more prevalent than previously believed, although in some cases, more orthogonal approaches or in vivo data approximating normal physiology may be needed to definitively support this claim. Although significant advances are rapidly emerging, the exact targeting mechanisms of ubiquitin-independent degradation are still under investigation. However, the 20S proteasome is known to degrade intrinsically disordered proteins (IDPs) like α-synuclein (α-syn) and tau more efficiently than structured proteins (Grune et al., 2010; Alvarez-Castelao et al., 2014; Myers et al., 2018; Ukmar-Godec et al., 2020), and there may be additional specific motifs (Touitou et al., 2001; Bossis et al., 2003; Makaros et al., 2023), including C-terminal degrons (Touitou et al., 2001; Makaros et al., 2023), and structural features including exposed hydrophobic residues (Kisselev et al., 2002), that are recognized for targeted degradation. It has also recently been demonstrated that the 20S can degrade ubiquitinated substrates, degrading the ubiquitin tag along with the protein more quickly than the 26S can deubiquitinate and digest, a mechanism increased during hypoxic stress conditions to clear misfolded/damaged proteins rapidly (Sahu et al., 2021).
To protect the cell from excessive proteolysis, entry into the catalytic chamber of the 20S core is tightly regulated, with its external α-rings partially obstructing the protease active sites (Wenzel and Baumeister, 1995; Groll et al., 2000). To allow substrate entry, 20S proteasomes may interact with pore-opening proteasome activators (PAs) (Knowlton et al., 1997; Hendil et al., 1998; Ortega et al., 2005), or they may allow direct substrate access without a PA (Kisselev et al., 2002). As a standalone molecule, the 20S can recognize and interact with hydrophobic regions of misfolded proteins, which act as degradation signals, as well as IDPs and oxidized proteins (Kisselev et al., 2002; Förster et al., 2003; Raynes et al., 2016; Deshmukh et al., 2023). Because IDPs lack a rigid, well-defined structure, they are more flexible and can more easily enter the narrow entry channel of the 20S proteasome (Suskiewicz et al., 2011), whereas structured proteins require unfolding or linearization by the 19S cap ATPases (Wenzel and Baumeister, 1995; Dong et al., 2019). The independent 20S can undergo conformational changes in its α-rings without ATP hydrolysis that permit self-gated entry of unstructured, oxidized, or misfolded proteins through the narrow entry pore (Kisselev et al., 2002; Förster et al., 2003). This capacity for protein degradation without ubiquitination or energy consumption makes the 20S proteasome uniquely suited to remediate the accumulation of toxic protein aggregates in neurodegenerative diseases, which often cause mitochondrial damage, oxidative stress, and further impairment of the UPS (Ding et al., 2006; Pickering et al., 2010; Li et al., 2011; Huang et al., 2013; Höhn et al., 2020). Importantly, substrate degradation by ubiquitin-independent proteasomal mechanisms, the UPS, or non-proteasomal pathways like autophagy are not necessarily mutually exclusive in the cell, and some substrates may be degraded by one mechanism in some conditions and another mechanism in other conditions, such as oxidative stress (Grune et al., 2010; Suskiewicz et al., 2011; Ben-Nissan and Sharon, 2014; Manfredonia and Kraut, 2022). It is also well-documented that autophagic pathways may be used to clear certain isoforms of these proteins, hypermodified forms, or aggregates, which will not be discussed here (Lee et al., 2013; Watanabe et al., 2020). Increasing research has shed light on how these substrates are targeted and the variety of mechanisms used to facilitate or regulate their degradation through the proteasome.
Proteasome activators (PAS)
The best-understood mechanisms of substrate targeting are through PAs. Prior research has demonstrated that interaction with 20S molecules require many PAs to use a C-terminal tri-peptide HbYX motif (hydrophobic residue, followed by tyrosine, then any amino acid) that docks into the spaces between ⍺-ring subunits, called 20S ⍺-pockets (Smith et al., 2007; Rabl et al., 2008; Sadre-Bazzaz et al., 2010). In contrast to research on the HbYX motif in archaea models (Smith et al., 2007; Rabl et al., 2008; Yu et al., 2010), recent research shows that human 20S proteasomes, which are hetero-oligomers with seven distinct ⍺-subunits in the outer ⍺-ring rather than the homo-oligomers formed by archaea, may have more heterologous signals, dubbed YΦ motifs by the Gestwicki group in 2022 (Opoku-Nsiah et al., 2022). While HbYX motifs are tripeptides, the YΦ motifs tested were hexapeptide sequences, showing an effect on degradation for each of the last 6 residues of the C-terminus. YΦ refers to Y-F/Y residues at the antepenultimate and penultimate positions at the C-terminus. In addition, as opposed to monovalent PAs like PA200 that require adherence to HbYX/YΦ rules (Sadre-Bazzaz et al., 2010), hetero-oligomeric PAs, which have increased valency due to interactions with multiple ⍺-subunit pockets, allowed some flexibility in C-terminal gating association outside of HbYX/YΦ rules (Opoku-Nsiah et al., 2022). Further research must be performed to catalog the full range of interaction sequences present in human 20S proteasomes, and this may provide insight into regulation of PAs, the importance of any post-translational modifications, and their role in neurodegenerative diseases.
An important implication of these recognition sequences is that they may be useful as drug targets. It has been demonstrated that synthesized HbYX-like peptide mimetics can open the 20S pore and stimulate degradation of unstructured protein substrates, posing a possible therapeutic option for neurodegenerative disease (Chuah et al., 2023). In addition to increasing degradation of tau by the 20S, HbYX mimetics also completely block 20S inhibition by amyloid-β, α-syn, and huntingtin oligomers, further demonstrating the potential for small molecule treatments to restore 20S proteasome activity and increase degradation of disordered substrates prone to aggregation (Chuah et al., 2023).
The most famous PA is the 19S regulatory cap (also called PA700), which in addition to being the canonical activator complex in the UPS has some capacity for facilitating ubiquitin-independent degradation through the 26S proteasome, generating a different set of peptides than 20S alone (Kisselev et al., 1999; Asher et al., 2005; Baugh et al., 2009; Winkler et al., 2013; Ben-Nissan and Sharon, 2014; Tsvetkov et al., 2020). In addition, Tsvetkov et al. showed in vitro that when the assembled 26S is stabilized by binding of NADH, an important molecule in aging and metabolism that is sensitive to cellular redox state, it can facilitate degradation of IDPs even in the absence of ATP. However, 20S proteasome catalytic activity was not affected by NADH or NAD+ in vitro (Tsvetkov et al., 2020). Other PAs that can bind to the exterior of the 20S core and enhance its activity by inducing central pore opening include PA28 (also called 11S or PSME1) and PA200 (or Blm10 in yeast), both of which use ubiquitin-independent mechanisms to facilitate substrate entry and often target oxidized, unstructured/intrinsically-disordered, or misfolded proteins in specific subcellular locations (Dubiel et al., 1992; Ma et al., 1992; Ustrell et al., 2002; Ortega et al., 2005; Cascio, 2021). In addition to PAs, other major proteasome interactors include PI31 (also called PSMF1), an adaptor protein for proteasome transport in neurons (Liu et al., 2019); midnolin, a regulator of immediate early gene protein and transcription factor degradation (Gu et al., 2023); ECPAS (“Ecm29 proteasome adaptor and scaffold”; also called PSMG1, or Ecm29), a modulator of 26S assembly and disassembly participating in stress responses (Wang et al., 2017; Lee et al., 2020), and catalytic core regulators (CCRs), which allosterically modulate uncapped 20S activity (Deshmukh et al., 2023). These will be explored in greater detail in the following subsections. See Figure 1 and Table 1.
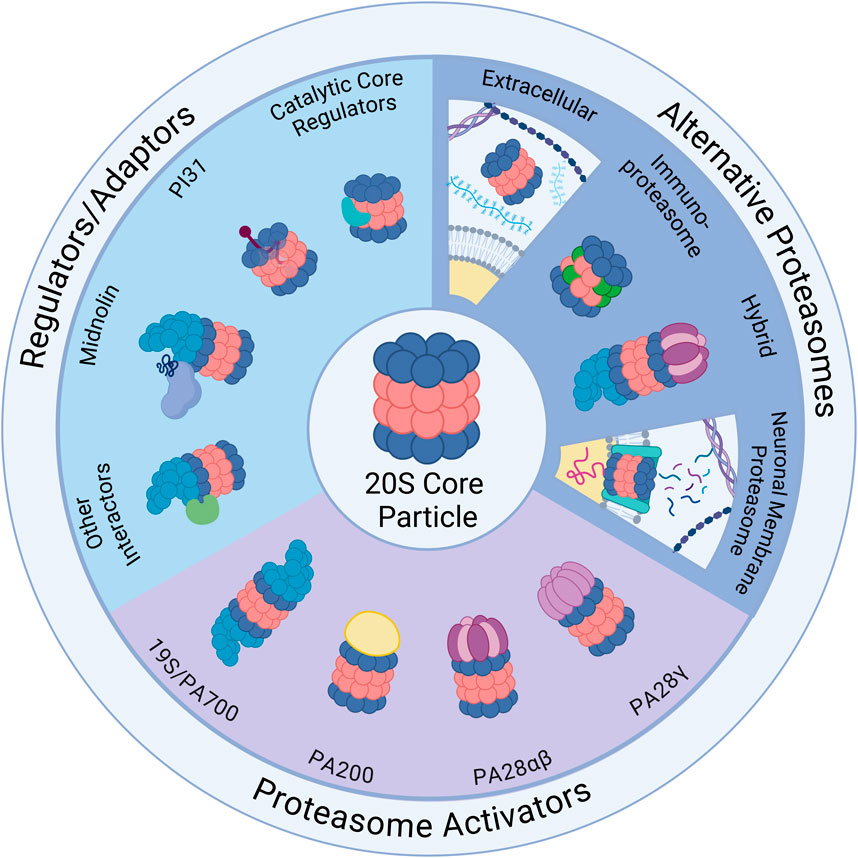
Figure 1. Ubiquitin-Independent Proteasome Complexes and Mechanisms. Illustrated above are the diverse complexes that can form in combination with the 20S core particle (20S) (center illustration). The 20S has 28 subunits that form a barrel structure with 14 α-subunits (blue) and 14 β-subunits (red). Alternative Proteasomes: The 20S can be found in various forms including the immunoproteasome, which has alternative β catalytic subunits (green) and can be induced as part of the immune response, and the neuronal membrane proteasome (NMP), which is a neuron-specific proteasome complex localized to the plasma membrane that is used for signaling. The 20S can also be found in the extracellular space and can exist in hybrid forms which have two distinct cap structures on each side of the 20S (shown here with 19S and PA28αβ). Regulators/Adaptors: The 20S interacts with several important regulators/adaptors including catalytic core regulators (CCRs), midnolin, PI31, and a broad category encompassing other interactors. Aside from PI31, which interacts with 20S subunits from inside the barrel, most of these regulators associate with the 20S exterior, and in some cases (midnolin and several other interactors) with the 19S-capped 20S. Proteasome Activators (PAs): The 20S interacts with a variety of PAs that increase 20S activity by opening the gate formed by α-subunits and permitting substrate entry for degradation. Those pictured include: the 19S cap, which combines with the 20S to form the 26S; PA200, which is monomeric and mainly found in the nucleus; PA28αβ, which is typically cytoplasmic and plays an important role in the immune response; and PA28γ, which has high expression in the brain and serves an important role in cell cycle regulation. Created in BioRender. Church, T. (2025) https://BioRender.com/o73i883.
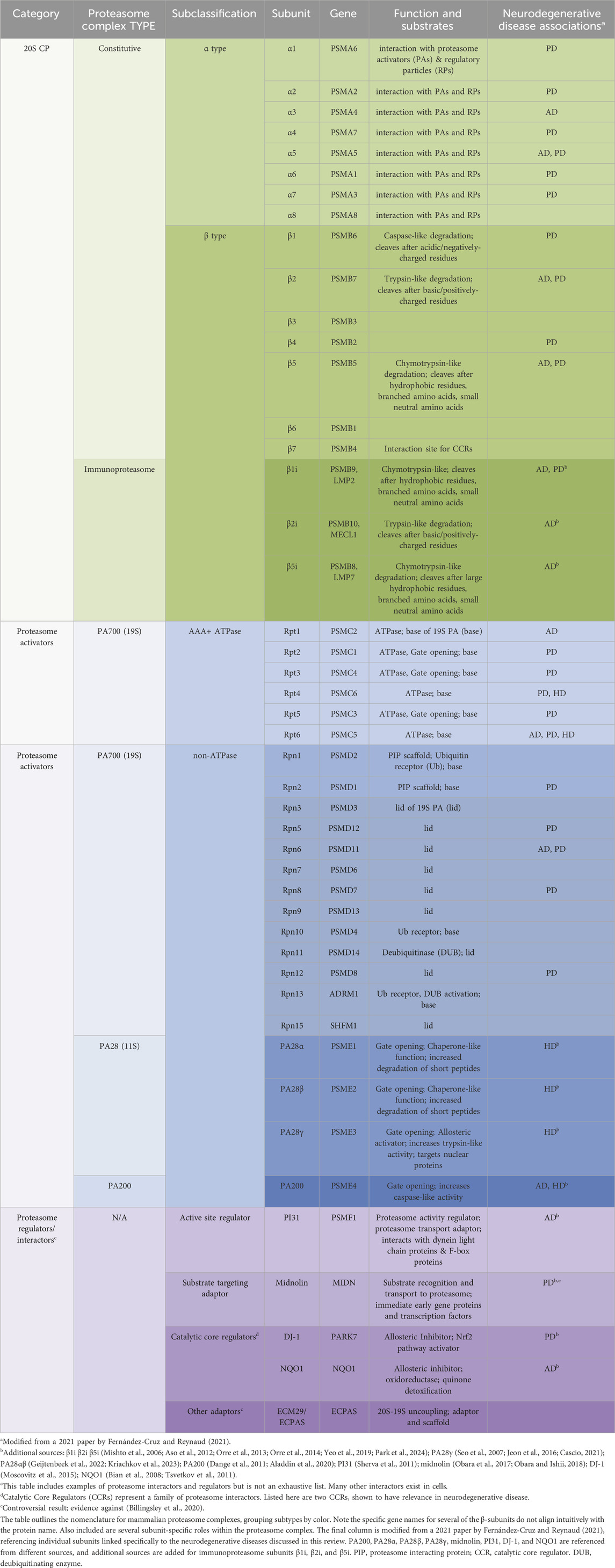
Table 1. Proteasome nomenclature and subunit-specific links to neurodegenerative disease.
PA28
PA28 is a heptameric, ring-shaped, and ATP- and ubiquitin-independent 20S PA that promotes rapid degradation of small, unstructured protein fragments, short peptides, and oxidized or misfolded proteins in the nucleus and cytoplasm by binding to the ends of the 20S core and inducing conformational changes that widen the 20S pore (Ma et al., 1992; Knowlton et al., 1997; Zhang et al., 1999; Thomas and Smith, 2022). Importantly, there are multiple isoforms of PA28, including PA28α (also called REGα and PSME1), PA28β (also called REGβ and PSME2), and PA28γ (also called REGγ and PSME3), which have distinct sets of functions and substrates (Knowlton et al., 1997; Thomas and Smith, 2022). PA28α and PA28β are mainly cytoplasmic and typically combine to form heteroheptamers, but PA28α is expressed at higher levels in the brain than PA28β and can form a homoheptamer (Knowlton et al., 1997; Zhang et al., 1999; Noda et al., 2000). PA28γ also forms homoheptamers and is primarily nuclear, ubiquitously expressed in all organ systems, with particularly high expression in the brain (Noda et al., 2000; Cascio, 2021; Frayssinhes et al., 2021). PA28γ is an interferon-γ- (IFNγ) and ubiquitin-independent PA which serves as a regulator of DNA replication, DNA repair, transcription, cell cycle control, and p53 tumor suppressor stability (Zhang and Zhang, 2008).
Because it is not an ATPase, PA28 cannot unfold proteins and has a preference for disordered or partially unfolded proteins, which can enter the 20S catalytic chamber without additional unfolding (Frayssinhes et al., 2021). It can interact with both the typical 20S core (constitutive 20S) and the immunoproteasome (a modified, inducible complex described in the “Immunoproteasome” section of this review), inducing different allosteric effects on each (Lesne et al., 2020). Indeed, PA28 becomes increasingly important in aging and neurodegenerative disease, when the UPS is compromised and damaged proteins accumulate (Seo et al., 2007), through its regulation of 20S activity, its ability to activate 26S as a hybrid proteasome (19S-20S-11S/PA28) (Tanahashi et al., 2000), and as a standalone chaperone-like molecule and chaperone regulator (Minami et al., 2000; Adelöf et al., 2018; Adelöf et al., 2021). PA28 expression is also upregulated under conditions of high protein damage including oxidative stress, indicating an important role in maintaining proteostasis by mitigating oxidative damage, and it often accompanies upregulation of the immunoproteasome (Pickering et al., 2010; Pickering et al., 2012).
While best characterized in other cell types, studies in neurons have shown that PA28 promotes ubiquitin-independent proteasomal degradation of oxidized and misfolded proteins and protects against oxidative stress (Li et al., 2011; Pickering and Davies, 2012), increasing evidence for its role as a 20S regulator in oxidatively-burdened neurodegenerative disease states. In addition, PA28⍺β overexpression showed sex-specific benefits for female mice in preventing age-related protein aggregation, hypothesized by the authors to be a novel, proteasome-independent, chaperone-like function (Adelöf et al., 2018). Moreover, PA28⍺β, plays major roles in the immune system through regulation of the immunoproteasome, described in the “Immunoproteasome” section below. In neurons and microglia, exposure to cytokine IFNγ or other pro-inflammatory factors during an immune or inflammatory response increases PA28αβ expression and its association with the immunoproteasome (Rivett et al., 2001; Pickering et al., 2010; Pintado et al., 2012). Because neuroinflammation is increasingly recognized as a contributor to the development of neurodegenerative diseases, dysregulation of PA28αβ - and therefore the immunoproteasome - can contribute to neuroinflammatory processes through neurons and glia (Leng and Edison, 2021; Malek et al., 2024). Notably, studies have also reported altered PA28 expression in Alzheimer’s disease brains (Krzyzanowska et al., 2015) and PA28γ may play a complex role in the etiology of HD, which will be described in the “Huntington’s Disease” section below (Cascio, 2021). Expansion on the significance of PA28 in specific neurodegenerative disease will be included in sections below.
PA200
PA200 is a large, monomeric, and ATP- and ubiquitin-independent 20S proteasome activator found predominantly in the nucleus which associates with the 20S core and regulates DNA repair mechanisms, transcription, and the cell cycle through targeted, acetylation-dependent degradation of histones and other protein targets (Ustrell et al., 2002). Its structure has two apertures for substrate entry and forms a dome-like cap on the 20S to open it (Guan et al., 2020). Some evidence suggests PA200 alters the relative activity of the 20S β catalytic subunits, increasing β1 (Ustrell et al., 2002) or β2 (Toste Rêgo and Da Fonseca, 2019) activity compared to the uncapped 20S.
Because PA200 does not have ATPase activity, it primarily acts on peptides and disordered and partially unfolded proteins, although there is a possibility it has some intrinsic unfolding ability through recruitment of other factors or conformational changes of substrates. In addition to possible regulatory roles in proteasome stability or maturation (VerPlank et al., 2024), PA200 is upregulated in response to DNA damage and induces opening of the α-ring substrate entry channel of the 20S, allowing for rapid clearance of oxidized, aggregated, and misfolded substrates (Ortega et al., 2005). These substrates include tau (Dange et al., 2011) and N-terminal huntingtin protein fragments (Aladdin et al., 2020), the proteins responsible for the pathogenic aggregates in AD and HD, respectively. Because research on PA200 function in the nervous system is still limited, its role in neurodegenerative diseases, its regulation and interactions with other PAs, and its cell type-specific characteristics in neurons remain mostly unknown. In fact, depending on the disease state, PA200 may ameliorate or worsen neurodegeneration in in vivo disease models (Aladdin et al., 2020; VerPlank et al., 2024).
Additional cap conformations and hybrid proteasomes
Further research is being performed to investigate the existence and roles of additional alternative caps, including hybrid proteasomes with different caps (e.g., one 19S and one alternative cap associated with a 20S core) (Hendil et al., 1998; Tanahashi et al., 2000; Cascio et al., 2002). While initially thought to be absent from the brain (Noda et al., 2000), along with PA28β, more recent data have demonstrated the presence of hybrid proteasomes and PA28β (Adelöf et al., 2018; Kriachkov et al., 2023). The significance of hybrid proteasomes in neurons is not yet well understood, but they may provide finely calibrated regulation of substrates or alter proteasome catalytic activity to generate a different set of peptides, as has been seen in the immune system to modify peptide products for antigen presentation (Hendil et al., 1998; Cascio et al., 2002). Hybrid proteasomes have also been proposed to use the 19S for protein unfolding and entry into the 20S chamber and PA28 for the rapid release of digestion products (Pratt and Rechsteiner, 2008; Kriachkov et al., 2023). It is unknown if PI31 or other adaptors are involved in hybrid proteasomes.
In addition, there may be tissue-specific, ubiquitin-independent alternative caps, as has been noted for ubiquitin-dependent PAs (Goldberg et al., 2021), or transient caps that do not assemble into stable structures like PA28 or PA200 but which interact briefly with the 20S core to modulate its activity or substrate channel access (Clemen et al., 2015; Esaki et al., 2018; Goldberg et al., 2021). These phenomena may be especially likely in neurons, which have specialized proteasomes, unique activators, and additional proteasomal physiological functions (Bingol and Schuman, 2005; Ramachandran and Margolis, 2017; Goldberg et al., 2021). An example of this is the characterization of neurodegenerative disease associated protein valosin-containing protein (also called Cdc48, TER94, and p97), a HbYX motif-containing PA which plays a role in ubiquitin-dependent degradation (Johnson et al., 2010; Esaki et al., 2018). In addition, some evidence suggests that 19S caps may not require ubiquitin for degradation of some substrates (Kisselev et al., 1999; Winkler et al., 2013; Tsvetkov et al., 2020). The full range of endogenous proteasome interactors and alternative caps are still being explored, and more molecules are likely to emerge.
Proteasome regulators
In addition to PAs, other proteasome adaptor and interactor proteins can regulate proteasome assembly and disassembly, link the proteasome to signaling pathways, regulate substrate specificity, and direct intracellular trafficking of proteasomes. The functions of these adaptors vary by cellular conditions, cell type, and activation of intersecting regulatory pathways (Arkinson et al., 2024). See Figure 1 and Table 1.
PI31
While not a PA, PI31 has been proposed as an endogenous 20S proteasome regulator and is targeted to the 20S via a HbYX motif. In vitro, it interacts with both 20S and 26S constitutive proteasomes and has been found to inhibit the 20S and to prevent binding of the 19S cap and PA28 (McCutchen-Maloney et al., 2000; Li et al., 2014; Liu et al., 2019; Wang et al., 2024). In contrast to the constitutive 20S and 26S, immunoproteasomes are capable of cleaving the PI31 C-terminus, preventing its binding and its inhibition of the core catalytic subunits (Wang et al., 2024). Separately, another study suggests PI31 affects immunoproteasome assembly (Zaiss et al., 2002). Although much of the research regarding PI31 has been performed in vitro (Li et al., 2014; Wang et al., 2024), in vivo experiments have contributed to a complex picture of PI31-mediated proteasome regulation. In a more physiological context, PI31 may in fact activate proteasome degradation through the 26S, and in addition, both knockout and overexpression of PI31 are lethal, indicating that cells are sensitive to PI31 amount (Bader et al., 2011). In vivo experiments in mouse motor neurons have demonstrated that PI31 acts as a proteasome regulator and adaptor protein that connects the proteasome to transport machinery for translocation down neuronal projections including axons, an essential function for maintaining a healthy proteasome supply to diverse cellular locations for their various functions (Bader et al., 2011; Liu et al., 2019). Loss of PI31 contributes to neurodegeneration, as its regulatory activity is required for normal proteasome function, maintenance of synapses, and neuronal survival (Bader et al., 2011; Liu et al., 2019; Minis et al., 2019). Genome-wide association studies have linked PI31 to AD risk (Sherva et al., 2011), and a direct antagonist of PI31, called valosin-containing protein (VCP), causes a familial type of the neurodegenerative disorder amyotrophic lateral sclerosis (Johnson et al., 2010; Clemen et al., 2015).
Catalytic core regulators (CCRs)
As the importance of non-UPS proteasome activity is becoming more apparent, it is increasingly critical to study regulators of these mechanisms. A newly discovered family of multi-functional regulatory proteins that directly interact with the 20S core to closely modulate its cap-independent degradation of IDPs and damaged, partially-unfolded proteins are the Catalytic Core Regulators (CCRs) (Olshina et al., 2020; Deshmukh et al., 2023). These CCRs are allosteric regulators with shared structural features including a common N-terminal sequence motif and a Rossman fold, providing further evidence that HbYX motifs represent only a portion of the structural features characterizing proteasome regulators. CCRs bind to the external surface of the 20S β7 (PSMB4) subunit and induce a conformational change that inhibits all three catalytic mechanisms of degradation without plugging the substrate entry gate and can protect substrates from degradation, including ⍺-syn, which forms toxic oligomers in PD and other synucleinopathies (Deshmukh et al., 2023). CCRs are critical for coordinating the oxidative stress response through interaction with transcription factor Nrf2 and activation of a range of response factors including upregulation of 20S subunits (Olshina et al., 2020; Deshmukh et al., 2023). The identification of the structural features underlying allosteric regulation of degradation by the 20S also provides insight relevant to the development of selective, synthesized inhibitors of 20S proteasomes and possible therapeutic options for neurodegenerative diseases like PD, which is directly affected by a CCR called DJ-1 and is discussed later in this review (Moscovitz et al., 2015).
Midnolin
Midnolin is an inducible, chaperone-like protein that associates with the 26S proteasome to promote selective, ubiquitin-independent degradation of transcription factors and other nuclear proteins (Gu et al., 2023). Recent studies indicate its targeting mechanism uses an internally symmetrical “catch” domain that induces a conformational change in unstructured regions of protein substrates to capture them for destruction by the proteasome. Midnolin associates with the proteasome using a C-terminal ⍺ helix, which does not contain a HbYX motif or YΦ motif, through an unknown mechanism, and it facilitates degradation of targets bound to the catch domain using an N-terminal ubiquitin-like domain. While the reasons for its preference for the 26S over the 20S are not well understood, midnolin co-immunoprecipitates with both 19S and 20S subunits (Gu et al., 2023).
Substrates of midnolin include transcriptional regulators and immediate early gene products, which are rapidly induced upon stimulation by a variety of stimuli and regulate transcription of longer-term sets of proteins in response to a particular stimulus (Chiba et al., 2024; Gu et al., 2023). Notably, midnolin and some of its substrates have been identified as PD risk genes, with deletion of midnolin resulting in loss of parkin expression, increased expression of ⍺-syn, and induction of PD phenotypes including loss of neurite outgrowth (Obara et al., 2017; Obara and Ishii, 2018). Obara et al. used microarray analysis to show that 10.5% of sporadic PD patients and 0% of healthy controls lack one copy of midnolin, positing a role for midnolin loss in development of PD (Obara et al., 2017). These data were supported by another large cohort study by the same research team in 2019, which showed a significant odds ratio of 4.35 with midnolin copy number loss for development of PD, with the odds ratio increasing to 22.3 when copy number loss is defined by large deletions (Obara et al., 2019). However, using whole genome sequencing and analysis of a public database of structural variants, Billingsley et al. and the International Parkinson’s Genomics Consortium did not identify PD-associated midnolin deletions and disputed the determination of midnolin as a PD risk gene, indicating that further study with orthogonal methods is required to investigate midnolin association with PD and resolve controversy (Billingsley et al., 2020).
Additional regulators/adaptors
Beyond the above regulators, there are other critical 20S interactors and adaptors that affect 20S activity and assembly briefly described here. Chaperones PAC1-PAC4 and POMP are crucial for the proper formation and maturation of the proteasome from its constituent monomeric subunits (Hirano et al., 2005; Hirano et al., 2006; Fricke et al., 2007; Le Tallec et al., 2007), and adaptors including ECPAS contribute to 26S assembly and disassembly in vivo (Wang et al., 2017; Choi et al., 2023). The ECPAS-20S interaction regulates the 20S:26S ratio and modulates the balance between ubiquitin-dependent and -independent mechanisms for adaptation to conditions like glucose deprivation or oxidative stress, in which it facilitates disassembly of 26S to 20S to support degradation of oxidized and misfolded substrates (Leggett et al., 2002; Wang et al., 2017; Choi et al., 2023). Interactions among the proteasome, ECPAS, and ankyrin G also regulate critical remodeling of the axon initial segment of neurons, found to have significant structural abnormalities in AD-affected neurons (Lee et al., 2020).
In addition to proteins directly bound to the 20S, there are also substrate-bound proteins critical to its regulation, deemed “nanny” proteins, that protect newly synthesized intrinsically disordered 20S substrates from degradation and allow new intrinsically disordered proteins (IDPs) to mature (Tsvetkov et al., 2009). Potential nanny proteins suggested by Tsvetkov et al., have been linked to a variety of nervous system disorders (Enokido et al., 2010; Kamińska et al., 2024; Yuhan et al., 2024). Conversely, chaperones like Hsp70 and Hsp110, dysfunction of which has been linked to neurodegenerative diseases, facilitate the targeting of substrates to the proteasome for ubiquitin-dependent and ubiquitin-independent degradation (Eroglu et al., 2010; Turturici et al., 2011; Hjerpe et al., 2016; Kandasamy and Andréasson, 2018; Taguchi et al., 2019; Vinokurov et al., 2024). Hsp70 has further been demonstrated to interact with ubiquilin2, a shuttling factor that brings substrates to the proteasome and plays a role in neurodegeneration (Wang et al., 2006; Zhang et al., 2014; Hjerpe et al., 2016; Ma et al., 2023). While members of the ubiquilin family typically require ubiquitin for trafficking proteasome degradative targets (Zhang et al., 2014; Itakura et al., 2016), Makaros et al. demonstrated that ubiquilins may also mediate ubiquitin-independent proteasome substrate identification (Makaros et al., 2023).
There are likely many additional yet-uncharacterized proteins that regulate 20S proteasome activity in neurons. In addition, post-translational modifications like phosphorylation, oxidation, or acetylation can also alter proteasome activity and 20S interaction with regulators in various cell types, especially as cells age (Bulteau et al., 2000; Bulteau et al., 2001; Ishii et al., 2005; Kors et al., 2019).
Specialized proteasomes
Immunoproteasome
When stimulated by interferon-γ (IFNγ) or oxidative stress, immune cells and some other cell types (e.g., microglia in the nervous system (Orre et al., 2013; Malek et al., 2024)) can produce a modified proteasome, the immunoproteasome, which can act through ubiquitin independent or ubiquitin dependent mechanisms and replaces the three catalytic β-subunits (β1, β2, β5) in the constitutive proteasome core with three unique catalytic subunits (β1i, β2i, β5i, also called PSMB9/LMP2, PSMB10/MECL-1, PSMB8/LMP7) (Noda et al., 2000; Basler et al., 2013; Freudenburg et al., 2013; Rock et al., 2014; Johnston-Carey et al., 2015; Ettari et al., 2017; Winter et al., 2017; Abi Habib et al., 2022). This subunit replacement allows for the generation of longer peptides which can be further processed and presented as antigens that allow cells to determine self vs. non-self, an important component of immune responses, although an increasing number of roles for the immunoproteasome are being recognized (Pickering et al., 2010; Abi Habib et al., 2020; Tundo et al., 2023). It appears to be particularly important for the clearance of oxidized and misfolded proteins in response to oxidative stress (Pickering et al., 2010; Pickering and Davies, 2012). Immunoproteasomes are upregulated in reactive glia in AD mouse models (Orre et al., 2013; Orre et al., 2014), which may be critical for elimination of misfolded or damaged proteins that could spread between cells and cause disease progression. Studies using immunoproteasome-specific inhibitors demonstrate improvements in cognitive decline in AD mice, showing that the immunoproteasome may contribute to pathology in association with neurodegeneration (Yeo et al., 2019; Park et al., 2024). The ability to generate highly selective inhibitors for modified immunoproteasome subunits provides a pathway to evaluate the effects of these complexes without affecting constitutive proteasome activity, a therapy that could have benefits in neurodegenerative diseases impacted by neuroinflammation (Johnston-Carey et al., 2015; Malek et al., 2024).
While the expression of immunoproteasome in the young and healthy brain is very low or negligible, and for a long time it was believed that the immunoproteasome was not expressed in the brain at all (Noda et al., 2000), immunoproteasome has been detected in both neurons and glia in aged healthy brains and brains with neurodegeneration (Diaz-Hernandez et al., 2003; Mishto et al., 2006; Aso et al., 2012; Ugras et al., 2018), suggesting that the induction of immunoproteasome in the brain may be a result of aging, neurodegeneration, or neuroinflammation. Neuroinflammation can exacerbate neurodegeneration but requires additional impaired proteasomal degradation to induce disease phenotypes (Pintado et al., 2012; Malek et al., 2024). Indeed, proteasome research in AD brains has demonstrated a notable induction of immunoproteasome subunits and changes in proteasome activity and composition, although the specific changes observed have varied substantially across studies (e.g., a decrease of only trypsin-like activity vs decreases in both chymotrypsin-like and caspase-like activity; a decrease in β1 expression and a proportional increase in β1i/LMP2 expression vs little change in expression levels) (Mishto et al., 2006; Aso et al., 2012; Keller et al., 2000a; Davidson and Pickering, 2023). In contrast to findings showing decreased activity, a recent study using advanced activity-based probes to detect global proteasome activity in human AD brain tissue detected elevated activity (Türker et al., 2023).
An induction of immunoproteasome has also been detected in HD brains and PD brains (Diaz-Hernandez et al., 2003; Ugras et al., 2018). Concurrent with an increase in LMP2 and LMP7 expression, an increase in trypsin- and chymotrypsin-like activity was observed in HD brains in the areas most affected (Diaz-Hernandez et al., 2003), and in PD brains, an increase in expression of immunoproteasome subunit LMP7 (β5i) was observed (Ugras et al., 2018). A recent study in mice found that knocking out immunoproteasome in brain can also cause seizures, tau hyperphosphorylation, increased polyubiquitination, and neurodegeneration, and the authors suggest that immunoproteasome has a role in healthy brain aging (Leister et al., 2024). In-depth descriptions of the immunoproteasome in neurodegeneration can be found in other recent reviews (Zerfas et al., 2020; Tundo et al., 2023).
Neuronal membrane proteasome
A specialized proteasome found in the plasma membrane of neurons, called the neuronal membrane proteasome (NMP), is another form of 20S proteasome that functions through ubiquitin-independent mechanisms (Ramachandran and Margolis, 2017). The NMP degrades nascent polypeptide chains from ribosomes closely associated with the membrane to form its signaling molecules (Ramachandran et al., 2018), but it is not yet known how substrate selection or recognition motifs to the NMP may differ from other 20S proteasomes in neurons. Unlike proteasomes whose primary role is in protein turnover, the NMP degrades intracellular proteins into peptides expelled into the extracellular space, creating small, specific peptide signaling molecules that serve additional functions in neurons that are important in synaptic regulation, including NMDA receptor activity modulation, and in pain sensation modulation (Ramachandran and Margolis, 2017; Ramachandran et al., 2018; Türker et al., 2024; Villalón Landeros et al., 2024). Because the NMP regulates neuronal circuits and is essential in learning-induced behavioral plasticity (He et al., 2023), it is possible that dysfunction of the 20S proteasome induced by aging and proteotoxic aggregates in neurodegeneration may also disrupt NMP function, further contributing to declining cognition in neurodegenerative diseases. Supporting this hypothesis, a preprint in bioRxiv by Paradise et al. showed an association between NMP and ApoE, a critical AD risk gene (Paradise et al., 2023). In their experiments, NMP co-purified with ApoE, suggesting a physical interaction, and inhibition of the NMP was sufficient to cause aggregation of newly-synthesized tau. Future studies will reveal the full details of this novel proteasome complex and its function in brain health and disease.
Extracellular proteasome
While proteasomes are typically thought of as intracellular, increasing evidence has shown 20S proteasomes in extracellular vesicles (EVs) and free-floating in a variety of body fluids, including the interstitial fluid and the cerebrospinal fluid of the brain (Mueller et al., 2012; Ben-Nissan et al., 2022). It has been reported that extracellular proteasomes rarely contain 19S or PA200 PAs (Kulichkova et al., 2017; Tsimokha et al., 2020), although this is debated and may be body fluid/tissue specific (Ben-Nissan et al., 2022). However, they are often free floating as 20S or accompanied by PA28αβ or PI31, and they have significant levels of post-translational modifications, including several unique from other proteasome complexes (Tsimokha et al., 2020; Ben-Nissan et al., 2022), which may affect their function or localization to the extracellular space. The mechanisms of extracellular release and the source of these proteasomes are not yet fully understood, but data have shown release of 20S core particles and PA28 molecules by immune cells in microparticles subject to later dissolution (Bochmann et al., 2014; Bonhoure et al., 2022). It is possible that neurons or glia could release proteasomes as part of normal physiology or as part of a stress response (Ben-Nissan et al., 2022), or that cell damage or cell death causes intracellular proteasomes to leak into the extracellular space. As extracellular proteasomes have been shown to participate in a variety of cellular functions across cell types and disease states (Ben-Nissan et al., 2022), they could serve important roles in regulating protein clearance, especially of IDPs and damaged or oxidized proteins (Bonhoure et al., 2022), or in reducing neuroinflammation (Dianzani et al., 2019) by degrading pro-inflammatory cytokines in the extracellular milieu in the nervous system. Indeed, Dianzani et al. demonstrated that extracellular proteasomes can generate functional peptides and play a role in regulating cell migration and inflammation through cleavage of extracellular osteopontin, a cytokine implicated in diseases like multiple sclerosis (Dianzani et al., 2017; Dianzani et al., 2019).
Notably, early data has suggested differential regulation and expression of extracellular proteasomes and proteasome regulators found in EVs in neurodegenerative diseases including PD, raising the possibility of using proteasomal changes in EVs as a biomarker (Wang et al., 2019; Thompson et al., 2020). These results are encouraging for development of screening tools for neurogenerative disease and require more follow-up study. Critical questions about their functions remain, from regulation of their release to their role in healthy versus diseased brains (Dwivedi et al., 2021). While still in early stages, study of extracellular proteasomes in neurodegeneration has significant promise for understanding disease etiology and identifying novel therapies, including possibly for degradation of extracellular aggregates like amyloid-β plaques. The development of standardized, reliable tools for measuring their activity in the brain or spinal cord are needed. This recent review by Ben-Nissan et al. (2022) discusses the current understanding of the extracellular proteasome in depth, as well as experimental strategies to study this increasingly-recognized molecule.
Aging, oxidative stress, and protein aggregation
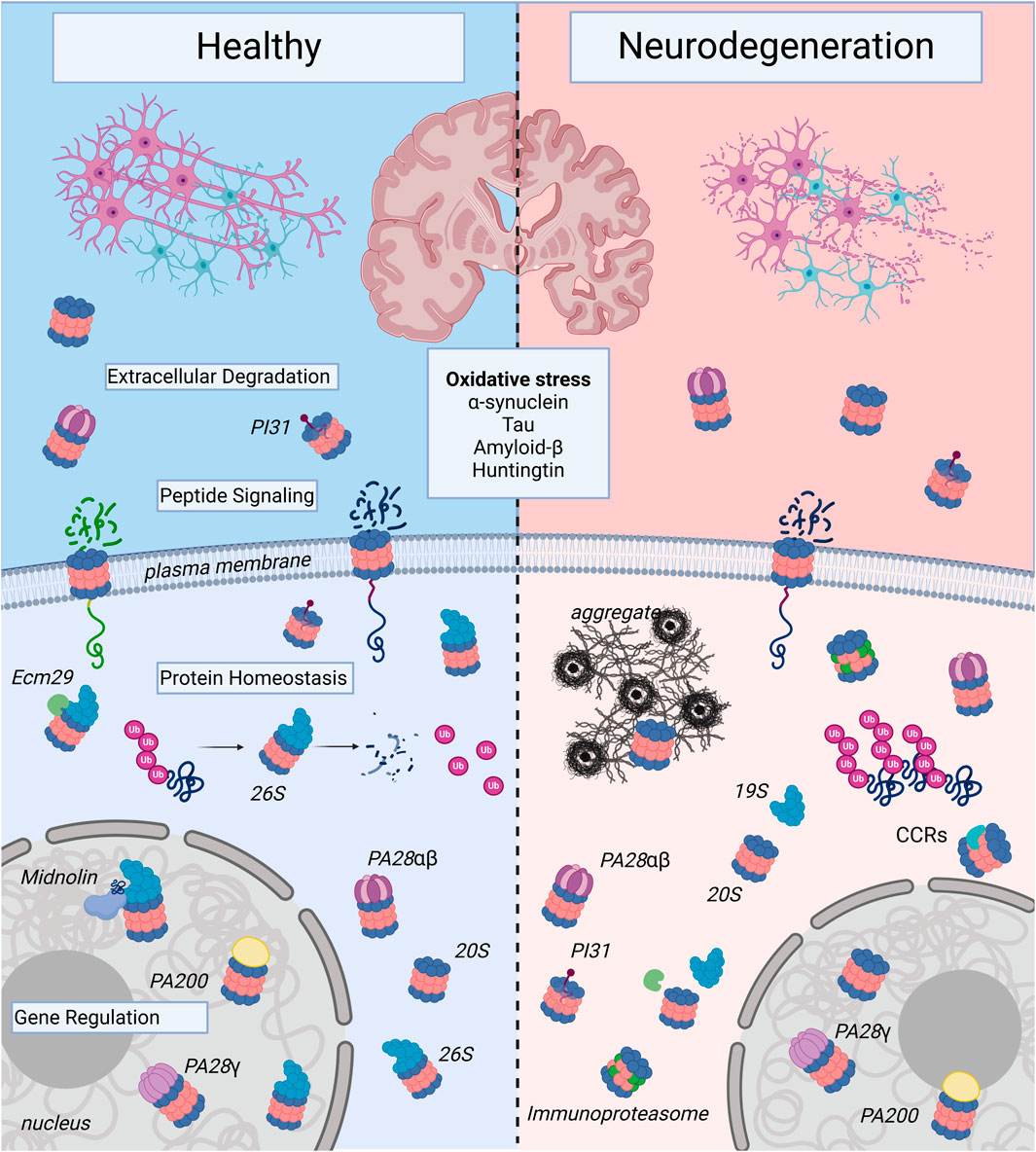
Figure 2. Proteasome Complexes in Health and Neurodegenerative Disease: The illustration shows healthy neurons and neurons affected by neurodegenerative disease. Note, the neurons undergoing neurodegeneration show fragmentation to illustrate decreased survival, which contributes to the decline in memory, cognition, and motor control seen in age-associated neurodegenerative illnesses such as Alzheimer’s Disease, Huntington’s Disease, and Parkinson’s Disease. At the molecular level, this inevitable cell death occurs in part due to proteasome dysregulation and is triggered by oxidative stress and toxic forms of aggregate-forming proteins such as α-synuclein, tau, amyloid-β, and huntingtin. In healthy neurons, there are a variety of proteasome complexes that exist under physiological conditions. In the nucleus, proteasomes with different proteasome activators (PAs; PA200, PA28γ, etc.) or uncapped 20S core particles (20S) have been demonstrated to regulate gene transcription. Cytosolic 20S proteasomes can also be activator-associated (e.g., 26S, PA28αβ) or uncapped. 26S proteasomes can mediate degradation of ubiquitinated proteins and produce small peptide fragments and free ubiquitin (Ub). In the extracellular space, there can be free as well as PA-capped 20S proteasomes, and in neurons, the 20S can be localized to the plasma membrane, where it serves a signaling function. In neurodegenerative diseases, proteasomes shift from predominantly ubiquitin-dependent degradation through the 26S to ubiquitin-independent degradation through alternatively-capped or uncapped 20S complexes. In addition, expression of immunoproteasome subunits is induced, particularly in the setting of chronic neuroinflammation. Proteasome complexes are regulated by adaptors like Ecm29, which mediates assembly and disassembly of the 26S proteasome from the 19S and 20S); PI31, a regulatory molecule that modulates proteasome activity from inside the 20S core; and catalytic core regulators (CCRs), which allosterically regulate the 20S to protect vital intrinsically-disordered proteins from degradation and serve critical roles in the oxidative stress response. Proteasomes are complex and heterogeneous molecules, and targeting different forms of the proteasome may prove useful for the development of preventative and disease-modifying therapies in neurodegeneration, an active area of research. Created in BioRender. Church, T. (2025) https://BioRender.com/k79b247.
Aging
As neurons age, damage to the proteome increases and overall neuronal proteasome activity decreases (Davidson and Pickering, 2023). Both the UPS and ubiquitin-independent proteasome function decline, further contributing to the aggregation of neurodegeneration-associated proteins such as tau, amyloid-β (Aβ), and α-syn (Bulteau et al., 2000; Keller et al., 2000b; Ding et al., 2006; Kelmer Sacramento et al., 2020; Cuanalo-Contreras et al., 2023; Davidson and Pickering, 2023). In addition, while glia normally secrete chaperones that assist neurons in maintaining proper protein folding, age-related 20S dysfunction could disrupt this process and further contribute to a decline in neuronal proteostasis (Chaplot et al., 2020; Leng and Edison, 2021).
With age, there is a shift from 26S to 20S proteasome activity concurrent with the decline in proteasome function, causing a relative increase in 20S activity (Keller et al., 2000b; Ding et al., 2006; Tonoki et al., 2009; Choi et al., 2023; Davidson and Pickering, 2023; Türker et al., 2023). Factors that contribute to this change include: oxidative stress, calpain activation, impaired assembly and recycling, and increased demand for IDP degradation (Huang et al., 2013; Coskuner-Weber et al., 2022; Davidson and Pickering, 2023). As aging is associated with increased oxidative stress and a decline in mitochondrial function, oxidation and damage to proteasome subunits can occur, especially subunits of the 19S cap (Reinheckel et al., 1998; Huang et al., 2013). Because 26S activity, assembly, and recycling are ATP-dependent, dysfunction of mitochondria also limits energy availability, further impairing 26S function and turnover. Furthermore, as oxidative damage accumulates, NADH, which stabilizes the 26S, oxidizes to its NAD+ form, compounding 19S dissociation from the 20S (Tsvetkov et al., 2014). As 26S activity declines, the 20S, which is more resilient to oxidative damage (Reinheckel et al., 1998), becomes more active in neurons by comparison, and its relative levels increase as association with the 19S cap decreases (Huang et al., 2013). Calpains, which are calcium-dependent proteases activated in aging and neurodegenerative diseases, cleave and inactive Rpn10, a 19S subunit, further decreasing 26S activity (Huang et al., 2013). Because aggregation-prone IDPs and damaged proteins with intrinsically disordered regions (IDRs) are preferentially degraded by the 20S rather than 26S, this shift to 20S activity may help to counteract the buildup of toxic protein aggregates over time in aging neurons (Opoku-Nsiah and Gestwicki, 2018; Davidson and Pickering, 2023).
Oxidative stress
In aging and neurodegenerative disease, a snowball effect of increasing oxidative stress, production of damaged and oxidized proteins, and inhibited proteasome activity can contribute to a progressively worsening cycle leading to neuronal dysfunction and cytotoxicity (Davies, 2001; Raynes et al., 2016). Sulfhydryl groups of the 19S PA are especially vulnerable to oxidation, causing 19S caps to lose their capacity to facilitate proteolysis and resulting in 26S disassembly by Ecm29/ECPAS as an adaptive response to oxidative stress (Shringarpure et al., 2001; Choi et al., 2023; Arkinson et al., 2024). 19S caps are sequestered by Hsp70 during the oxidative insult (Grune et al., 2011), and 20S subunits, immunoproteasome, and PA28αβ are upregulated (Pickering et al., 2010; Moscovitz et al., 2015; Davidson and Pickering, 2023). Because of the shift to favor 20S-mediated protein degradation during oxidative stress, 20S proteasomes are responsible for the majority of proteasome-mediated degradation in these conditions and do not require ubiquitin (Shringarpure et al., 2001; Ding et al., 2006; Raynes et al., 2016). Additionally, while transient mild oxidative stress increases ubiquitin activating/conjugating activity, this activity decreases during sustained oxidative stress (Shang and Taylor, 2011). Because the 20S proteasome does not require a ubiquitin tag to identify substrates, its degradation of substrates must be regulated to destroy harmful proteins while protecting IDPs and IDRs important for the oxidative stress response and normal cellular functions like cell cycle regulators, tumor suppressors, and signaling proteins. There are diverse posttranslational modifications that can regulate the relative activities of different proteasome species, including under oxidative stress conditions, but additional robust protective pathways are required. Several of these regulatory mechanisms (Bi et al., 2021), including catalytic core regulators (CCRs; see “Catalytic Core Regulators” section above) and PI31-mediated activity modulation, have only recently been uncovered, and investigation continues (Liu et al., 2019; Minis et al., 2019; Olshina et al., 2020; Deshmukh et al., 2023).
A necessary antioxidant pathway that emphasizes the role of ubiquitin-independent proteasome degradation involves the 20S proteasome, CCRs DJ-1 and NQO1, and transcription factor Nrf2 (nuclear factor E2-related factor 2) (Pickering and Davies, 2012). DJ-1 (also called PARK7) is a critical regulatory protein that stabilizes Nrf2 under oxidative stress conditions. Nrf2 translocates to the nucleus, where it upregulates a variety of proteins important in antioxidant defense, including subunits of the 20S proteasome and PA28⍺β, but not the immunoproteasome (which is induced by a different mechanism) or 19S subunit (Clements et al., 2006; Pickering and Davies, 2012). Nrf2 also induces the expression of NQO1 (NAD(P)H quinone oxidoreductase 1), an enzyme which prevents reactive oxygen species formation and acts as a sensor for cellular redox state, avoiding degradation during oxidative stress but being rapidly degraded as cellular conditions normalize (Moscovitz et al., 2012; Yuhan et al., 2024). In addition, both DJ-1 and NQO1 are CCRs and act allosterically to inhibit the 20S, thus rescuing partially unfolded proteins from degradation, an important protective mechanism as the 20S proteasome becomes the predominant proteasomal pathway in oxidative stress (Olshina et al., 2020; Deshmukh et al., 2023). This allows for a rapid, fine-tuned response in which damaged proteins are degraded but critical proteins are preserved as the redox-sensing mechanism facilitates fast termination of the oxidative stress response. Induction of this Nrf2 pathway in Drosophila increases proteasome subunit expression and decreases age-associated phenotypes (Tsakiri et al., 2013), while in human fibroblasts, this activation increases proteasome activity and delays cellular senescence (Kapeta et al., 2010). As cellular redox state normalizes to basal conditions, Nrf2 can be degraded by the 26S proteasome, terminating the oxidative stress response. This pathway is important in aging and neurodegenerative disease, and mutations in DJ-1 and NQO1 are associated with increased risk of developing PD and AD, respectively (Bian et al., 2008; Tsvetkov et al., 2011; Moscovitz et al., 2015).
Protein aggregate formation in neurodegenerative diseases
Protein aggregates, a hallmark of neurodegenerative diseases, form through a complex interplay of factors that disrupt proteostasis including aging, chronic oxidative stress, mutations, breakdown of degradative pathways and chaperones, and errors during protein synthesis. Aggregation-prone proteins undergo structural changes in response to stress that increase disorder, form incorrect intramolecular bonds, and expose hydrophobic residues, making them targets for ubiquitin-independent degradation by the 20S proteasome (Kisselev et al., 2002; Saez and Vilchez, 2014; Nago et al., 2024). As aggregates form, they may sequester and deplete functional proteins, trigger inflammatory responses, and disrupt cellular membranes and signaling pathways, leading to a toxic cascade of neurodegenerative damage, cell death, and disease progression (Nago et al., 2024).
Aggregates are characteristic of neurodegenerative diseases, but they are not the most cytotoxic species of their constituent neurodegeneration-associated proteins, which include amyloid-β (Aβ) and tau in AD, mutant huntingtin (mHTT) in HD, and α-synuclein (α-syn) in PD. Instead, decades of data have indicated that the most damage is caused by misfolded soluble oligomers that interrupt cellular functions including proteasome degradation pathways (Takahashi et al., 2008; Tai et al., 2012; Usenovic et al., 2015; Fiolek T. et al., 2021). While early studies posited that oligomers inhibit proteasome activity by directly blocking the 20S pore or by acting as competitive substrates (Gregori et al., 1997; Zhao and Yang, 2010), more recent data show that at least three of these disease-associated oligomers (Aβ, α-syn, and HTT) act as allosteric inhibitors, forming a common three-dimensional conformation that allows them to bind and disrupt the 20S proteasome via stabilization of its closed state, thus preventing opening of its substrate entry gate and blocking access to HbYX motif-containing PAs and regulators like the 19S cap, PA200, and PI31 (Thibaudeau et al., 2018). This interaction further reduces the proteasome’s ability to degrade misfolded and aggregated proteins. As these oligomers accumulate and interact, formation of the large, insoluble aggregates may actually be a protective mechanism mitigating the effects of the toxic oligomers (Arrasate et al., 2004; Carrell et al., 2008; Boulos et al., 2024), although this is debated. Finally, the oligomers may interact with other regulators of the proteasome, impairing their activity (Olshina et al., 2020; Deshmukh et al., 2023).
Therapeutic strategies to target ubiquitin-independent proteasome activity
Development of therapies for neurodegenerative disease have focused on enhancing clearance mechanisms, reducing and preventing misfolding, and eliminating toxic oligomers and aggregates. Most studies targeting the 20S proteasome for the treatment of neurodegenerative diseases have demonstrated that increasing 20S activation can reduce toxic protein aggregate levels in vitro, in cell culture, and in animal models of neurodegeneration (Pickhardt et al., 2005; Zhou et al., 2019; Cekala et al., 2022; Staerz et al., 2022; Sadahiro et al., 2024; Staerz et al., 2024), although at least one study has demonstrated that increases in certain PAs can increase neurodegenerative pathology (VerPlank et al., 2024). Activators that have been tested include activated endogenous PAs (as described in prior sections), peptidomimetics of PAs (Cekala et al., 2022; Cekala et al., 2024), as well as small molecule activators of 20S, such as fluspirilene analogs and dihydroquinazolines, which have been shown to rescue impaired proteasome activity and prevent pathological IDP aggregation of Aβ and α-syn (Fiolek T. et al., 2021; Fiolek T. J. et al., 2021). Drawbacks to using small molecule activators or activated endogenous PAs include a lack of specificity and the possibility of off-target effects in other essential cellular functions. Previously, there were no methods for targeting specific proteins to ubiquitin-independent proteasome pathways as there are for the UPS - called Proteolysis Target Chimeras (PROTAC) and molecular glues (Hyun and Shin, 2021) – meaning many activators could have off-target effects on essential proteins and cause more neuronal damage (Gao et al., 2020). However, several labs have recently published techniques, including chemical inducers of degradation (CIDEs) and direct-to-proteasome degraders (DPDs), to bypass the requirement for substrate polyubiquitination using chimera molecules or chemical dimerizers that directly target the desired substrate to the proteasome (Wilmington and Matouschek, 2016; Bashore et al., 2023; Balzarini et al., 2024). It is important to note that the value of 20S activators as disease-modifying therapies might also vary among different neurodegenerative diseases or at different stages of disease progression. Further research, especially in vivo, is necessary. Comprehensive reviews of recent therapeutic research on ubiquitin-independent proteasome mechanisms in age-related neurodegenerative disease can be found elsewhere (Harding and Tong, 2018; Opoku-Nsiah and Gestwicki, 2018; Hyun and Shin, 2021; Schmidt et al., 2021; Rawat et al., 2022; Tundo et al., 2023).
Neurodegenerative disease-specific changes in ubiquitin-independent proteasome degradation
Alzheimer’s disease
Alzheimer’s Disease (AD) is a progressive neurodegenerative disorder characterized by cognitive decline, memory loss, and neuropathological changes including aggregation of extracellular amyloid-β (Aβ) plaques and intracellular tau neurofibrillary tangles. While there is still debate about the mechanism by which each of these proteins contributes to neuronal deterioration, recent research has emphasized the role of synergistic crosstalk between the two proteins in producing AD pathology (Busche et al., 2019; Rawat et al., 2022; Roda et al., 2022). As in other age-related neurodegenerative diseases, oxidative stress, neuroinflammation, and disrupted proteostasis play important roles in the etiology of AD. Misfolding and posttranslational modifications induced by oxidation and inflammation interrupt the physiological functions of Aβ and tau in cytoskeletal support, recovery from injury, stabilization of microtubules, and synaptic plasticity (Haass and Selkoe, 2007; Orre et al., 2013; Bonet-Costa et al., 2016; Brothers et al., 2018), and this misfolding can cause toxic gain-of-function effects including disruption of normal protein degradation (Poppek et al., 2006; Opoku-Nsiah and Gestwicki, 2018; Davidson and Pickering, 2023), leading to cytotoxicity and cell death.
In AD, proteasome dysfunction is more severe than the decline associated with normal aging (Bonet-Costa et al., 2016). This compromised clearance pathway contributes to the formation of toxic protein oligomers that accumulate as aggregates. According to several studies, both tau and Aβ are IDPs that can be degraded by the proteasome through ubiquitin-independent mechanisms during physiological conditions, although alternative complementary degradative pathways may participate in degradation depending on cellular context or if the proteasome is inhibited (Grune et al., 2010; Zhao and Yang, 2010; Watanabe et al., 2020). In fact, based on mouse model data, impaired proteasome activity may induce AD pathology in individuals with an underlying diathesis. Prior to the development of pathology in AD model mice (3xTg-AD), impaired or inhibited proteasome activity can increase tau and Aβ accumulation, a process which can be rescued with Aβ immunotherapy against Aβ oligomers, reducing protein accumulation and restoring proteasome activity (Oddo et al., 2004; Oh et al., 2005; Tseng et al., 2008). Notably, proteasome activity can be inhibited during oxidative stress and neuroinflammation by CCRs, and mutations of CCRs that regulate cellular defense against oxidative stress, including NQO1, are associated with increased risk of developing AD (Bian et al., 2008; Tsvetkov et al., 2011; Moscovitz et al., 2015).
A study by Poppek et al. showed that tau is more resistant to oxidative stress than many other proteins, and oxidized tau is degraded equally as well as native, non-oxidized tau through the 20S proteasome in vitro. However, tau degradation in cells showed a different pattern. It was enhanced in cellular models (Poppek et al., 2006) of acute oxidative stress in which tau was oxidized and not phosphorylated but was strongly inhibited in models of chronic inflammation-induced oxidative stress, which caused hyper phosphorylation of tau, making it resistant to 20S proteasomal degradation. These data suggest an indirect mechanism affecting ubiquitin-independent proteasomal degradation wherein oxidized tau is rapidly degraded, but as the cell activates response pathways in chronic inflammation, tau is phosphorylated and the resulting hyperphosphorylated forms resist proteasomal degradation. Both ubiquitin-independent mechanisms and the UPS are impaired in AD, and hyperphosphorylated tau forms paired helical filaments that can directly bind to 20S proteasome complexes, affecting both 26S and 20S degradation mechanisms, and can associate into large neurofibrillary tangles (Keck et al., 2003; Poppek et al., 2006; Rankin et al., 2007; Min et al., 2010; Rawat et al., 2022).
Tau contributes to UPS dysfunction (Poppek et al., 2006; Tai et al., 2012; Thibaudeau et al., 2018), with higher levels of oligomeric and aggregated tau associated with a decrease in 26S activity without a decrease in subunit expression (Myeku et al., 2016). As discussed in the “Protein Aggregation” and “Oxidative Stress” sections of this review, soluble oligomers are the most toxic species of both tau and Aβ (Usenovic et al., 2015; Thibaudeau et al., 2018). Mouse models of AD show a physical association between tau and the 26S molecule that impairs degradation of ubiquitinated substrates and small peptides by the 26S and results in an increase in ubiquitinated protein burden. Treatment of healthy mice with tau oligomers also show decreasing degradative capacity, supporting the hypothesis that tau is proteotoxic, and this effect was rescued with activation of cAMP-protein kinase A (Myeku et al., 2016). It is worth noting, however, that the authors did not detect 20S activity, which has been shown in other studies to be active in AD brains (Gillardon et al., 2007; Türker et al., 2023) and to be directly inhibited by paired helical filament binding (Keck et al., 2003), so there may be underlying differences in experimental approaches that are important to revisit.
The other protein capable of forming aggregates characteristic of AD is Aβ, which has a direct inhibitory effect on proteasomal proteolytic pathways through allosteric stabilization of the 20S core particle in its closed state (Thibaudeau et al., 2018). Aged mouse models of AD overexpressing Aβ show a decrease in proteasome function that correlates with Aβ level, an effect which was reproduced in cultured neurons through extracellular Aβ application on cells (Favit et al., 2000; Oh et al., 2005) and in vitro (Gregori et al., 1995). Extracellular application of Aβ was specifically shown to affect chymotrypsin-like activity without affecting ubiquitination or deubiquitination levels, implying that the changes occurring in proteasomal pathways are due to direct effects on the proteasome rather than other UPS components (Gregori et al., 1995). Proteasome impairment correlates with Aβ oligomer levels but not aggregate levels (Tseng et al., 2008). Notably, although these and many other studies on Aβ and proteasomes are meant to describe the UPS, many do not distinguish between ubiquitin-dependent and ubiquitin-independent proteasome activity, so it is likely that at least some of the effects on proteasome activity are attributable to ubiquitin-independent mechanisms, especially as cellular stress increases (Zheng et al., 2016; Davidson and Pickering, 2023) and induces protein misfolding and the formation of toxic protein species.
In a comparative study of the inhibitory effects of Aβ monomers, oligomers, or fibrils on 20S activity, it was demonstrated that oligomers inhibit proteasomal degradation more than monomers or fibrils, supporting evidence that it is toxic oligomers rather than macroscopic aggregates that induce proteasome dysfunction (Tseng et al., 2008; Zhao and Yang, 2010). However, in another study, the opposite was found, with oligomeric and fibrillar Aβ increasing proteasome activity, and fibrillar species showing a much greater effect. This difference is likely due to a difference in experimental methods, with many groups using AMC peptide hydrolysis assays to measure activity and others using orthogonal approaches like activity ELISAs and activity-based probes (Orre et al., 2013; Türker et al., 2023).
Increasing data have shown that tau and Aβ each amplify the pathology of the other, driving an accelerated cycle of cellular dysfunction, resistance to and inhibition of proteasomal degradation, and neuronal deterioration. Aβ can trigger neuroinflammation and induce hyperphosphorylation of tau (Amadoro et al., 2011; Ising et al., 2019; Roda et al., 2022), paired helical filament formation, and tau aggregation. Conversely, excess misfolded tau causes Aβ aggregation, abnormal trafficking of the precursor protein for Aβ (APP), and increased prion-like propagation of both Aβ and tau pathology to other cells via exosomes and extracellular secretion of excess protein (Frost et al., 2009; Amadoro et al., 2011; Guo and Lee, 2011). As AD progresses, propelled by the damaging bidirectional pathway between tau and Aβ, changes occur to the composition of the proteasome, including significant upregulation of immunoproteasome subunits (Orre et al., 2013), and normal proteasomal degradation pathways are altered.
Huntington’s disease
Huntington’s Disease (HD) is a progressive, autosomal dominant neurodegenerative disorder that causes motor dysfunction, mental health symptoms, cognitive decline, and eventually death. It is caused by a mutation in the N-terminus of the huntingtin (HTT) gene that leads to a large chain (>35–40) of glutamine amino acids, called a polyglutamine (polyQ) expansion, which is prone to misfolding, aggregation, and the formation of toxic peptide fragments and interferes with normal HTT roles in cellular trafficking, endocytosis, and transcription regulation, among others (Miller et al., 2011; El-Daher et al., 2015; Saudou and Humbert, 2016; Guo et al., 2018). Misfolded aggregates of mutant huntingtin (mHTT) form inclusion bodies, which are proposed to have both protective effects in reducing toxic peptide fragments and oligomers (Arrasate et al., 2004; Takahashi et al., 2008) as well as harmful effects through pathophysiological interactions and disruption of cellular processes, based on disease progression and cellular context (Waelter et al., 2001; Arrasate et al., 2004; Takahashi et al., 2008; Saudou and Humbert, 2016; Riguet et al., 2021).
The degradation pathways used for HTT breakdown likely vary by the species of HTT protein present (e.g., fragments, monomers, oligomers, aggregates). There is clear involvement of both autophagy and the UPS in HD, the roles of which are reviewed elsewhere (Martin et al., 2015; Sap et al., 2023), and the third degradative mechanism relevant to HD is ubiquitin-independent proteasomal degradation. mHTT has significant IDRs, especially in its polyQ region, which could make it a good substrate for the 20S proteasome and non-ATPase PAs (Juenemann et al., 2013). One in vitro study found that mammalian 20S proteasomes do not completely degrade polyQ repeats (Venkatraman et al., 2004), but many others have found the opposite, showing that the 20S can degrade wildtype HTT and mHTT (Rousseau et al., 2009; Juenemann et al., 2013) and that this effect is modulated with the addition of PA28αβ (Geijtenbeek et al., 2022). The 20S and ubiquitin-independent PAs may be especially critical in breaking down toxic mHTT fragments (Juenemann et al., 2013; Geijtenbeek et al., 2022), particularly as HD advances and the UPS is overwhelmed or compromised (Geijtenbeek et al., 2022). Indirectly, the 20S proteasome may also play a role in HD by maintaining overall proteostasis and mitigating the damage caused by oxidative and proteotoxic stress in neurons (Pickering et al., 2010; Höhn et al., 2020). More research is needed to directly show the extent to which ubiquitin-independent 20S proteasome activity drives degradation of mHTT and what regulatory and targeting mechanisms may exist.
While it was previously thought that HTT aggregates impair the UPS and proteasomal degradation by sequestering complexes within mHTT inclusion bodies (Holmberg et al., 2004), it is more likely that the co-localization of proteasome complexes and inclusion bodies reflects dynamic, targeted recruitment of ubiquitin and catalytically-active proteasomes that can facilitate both ubiquitin-dependent and ubiquitin-independent degradation (Schipper-Krom et al., 2014; Juenemann et al., 2018). In fact, 20S, 26S, PA28, PA200, and 19S all colocalize with perinuclear inclusions (Waelter et al., 2001; Aladdin et al., 2020), supporting evidence that multiple proteasomal pathways are involved in HTT clearance mechanisms.
Another component of the interaction between HD and ubiquitin-independent proteasomal degradation is the disruption of proteostasis pathways including the UPS. This could either increase ubiquitin-independent activity through the 20S or accompany disruption in ubiquitin-independent degradation depending on which proteasome molecules or regulators are most affected by the pathology of HD. Some studies do show an increase in proteasome activity in HD in early disease and in postmortem brains, likely providing a compensatory mechanism to account for proteostatic deficits elsewhere (Diaz-Hernandez et al., 2003; Thompson et al., 2009). Other studies demonstrate no deficits in 20S activity in proportion to protein aggregation, suggesting that aggregates do not directly impair proteasome activity (Diaz-Hernandez et al., 2003). Compatible with this research, data has suggested indirect mechanisms through which oligomers and aggregates impair proteasome activity, including mitochondrial dysfunction and disruption of overall proteostasis (Jana et al., 2001; Hipp et al., 2012). Another study using mHTT species derived from cells suggests that instead of aggregates, it is mHTT filaments, modified by posttranslational modifications, that impair proteasome function, and that these disruptions are especially harmful to 26S proteasomes rather than 20S, favoring a shift to 20S activity (Diaz-Hernandez et al., 2006). It is important to note that many in vitro studies use synthesized polyQ tracts, which do not have physiological posttranslational modifications and could diverge from native conditions. There is significant heterogeneity in results and conclusions drawn from the available literature regarding the effects of mHTT in its various forms on proteasome activity, and differences in experimental design are likely to explain some of the discrepancies.
Investigating interactions between mHTT and proteasomes can grant additional mechanistic insight. Some HTT found in HD cells is ubiquitinated, and data shows that ubiquitinated mHTT does not directly clog the 20S catalytic chamber in aggregate or soluble form (Hipp et al., 2012). However, when the concentration of mutant fragments reaches a certain threshold, cytoplasmic inclusions accumulate and deficits are observed in both the UPS and ubiquitin-independent mechanisms, likely demonstrating that it is an overall deficit in proteostasis rather than a dose-dependent effect of impaired ubiquitin conjugation mediating dysfunction (Hipp et al., 2012). Importantly, the authors note that in cells, most mHTT they found was not ubiquitin-conjugated, and that degradation of these fragments was not fully captured in their study.
There are various forms of ubiquitin-independent proteasomal degradation mechanisms which could have relevance in HD. One mechanism which has emerged is through alternative proteasome activator, PA28. In vitro studies have shown that a PA28γ mutant increases 20S catalytic activity and can promote complete degradation of polyQ peptides, suggesting that PAs may promote significant degradation of polyQ tracts synergistically with the 20S (Pratt and Rechsteiner, 2008). Wildtype PA28γ overexpression improved cell survival in excitotoxic and proteasome-inhibited states in a neuronal model of HD (Seo et al., 2007), and lentiviral-delivered gene therapy increasing PA28γ expression improved motor coordination in mouse models of HD and reduced ubiquitin-positive inclusion body expression, although the decrease in mHTT in inclusion bodies was not significant (Jeon et al., 2016). However, in a separate HD mouse model, it was noted that knockdown of PA28γ did not worsen polyQ-related pathology (Bett et al., 2006), so the exact effects of this PA in HD are not yet fully understood. In many cases, it is unclear what proportion of these effects is due to PA28γ interaction with the proteasome through ubiquitin-independent mechanisms versus its chaperone-like function or crosstalk with the UPS, and this will be an important area of future mechanistic study (Yersak et al., 2017). For example, in another polyQ-expansion disease, spinal and bulbar muscular atrophy, PA28γ had two opposing effects, increasing cell viability in association with its proteasome binding activity and conversely increasing aggregate formation and oligomer toxicity independently of its proteasome binding activity (Yersak et al., 2017). The effect of PA28γ is likely dependent on its cellular context.
In a study by Geijtenbeek et al., reduction in PAαβ activation in HD-model mice (R6/2) increased mHTT aggregation in the brain, and as the disease progressed, PA28αβ increasingly dissociated from the 20S proteasomes. This disassembly was specific to brain areas particularly affected by HD. This study also noted that in vitro, PA28αβ can enhance polyQ degradation through 20S proteasomes, but decreases overall mHTT degradation, implying that the regulatory effect of PA28αβ may be indirect (Geijtenbeek et al., 2022). Another recent study also found that PA28αβ can increase polyQ breakdown and suggested hybrid proteasomes may have a role (Kriachkov et al., 2023).
Another proteasome regulator and PA with relevance to HD is PA200, which is mainly found in the nucleus and recognizes short peptides and unstructured protein regions. Aladdin et al. (2020) recently demonstrated that human PA200 can bind to mHTT fragments and that the loss of PA200 in human cells contributed to aggregate formation and increased cytotoxicity. Additionally, the yeast ortholog of PA200 increased 20S degradation of soluble mHTT fragments in vitro, identifying that PA200-bound proteasomes may contribute to mHTT degradation, particularly in the nucleus (Aladdin et al., 2020). PA200 may also form hybrid proteasomes (Schmidt et al., 2005; Blickwedehl et al., 2008) and function in parallel to UPS-mediated degradation of ubiquitinated HTT to enhance digestion of disordered proteins, including aberrant species of HTT like soluble non-ubiquitinated polyQ sequences and oligomers.
In addition to the effects of PAs on proteasome activity, it is possible that HD pathology can induce an effect on proteasome composition. Diaz-Hernandez et al. found that the catalytic activity of the 20S core was preserved in HD mouse model brain extracts, and there was induction of immunoproteasome subunits LMP2 and LMP7 in the cortex and striatum of human brains and mouse models, with immunohistochemistry showing the highest expression in neurons. This induction only occurred after the development of significant HD pathology, and it was accompanied by reactive gliosis, with LMP2 and LMP7 also induced in nearby glia (Diaz-Hernandez et al., 2003). A follow-up study showed that expression of mHTT alone was insufficient to induce the observed changes in 20S activity but that mHTT works synergistically with IFNγ, causing an increase in immunoproteasome proportional to the severity of neuroinflammation-associated neurodegeneration (Diaz-Hernandez et al., 2004). Because immunoproteasomes can be beneficial in oxidative stress responses (Pickering et al., 2010), they likely serve a protective effect against damage from disrupted proteolysis and mitochondrial dysfunction.
There are many debates surrounding HD etiology, progression, and proteostasis. One possible contributor to these questions is that many studies in the literature presumed to describe the UPS ignore or do not control for ubiquitin-independent activity, and so it is possible that some of the complexity and contradictions in the literature about proteasomal pathways in HD are due to these diverging mechanisms. Further complicating the study of many neurodegenerative diseases, including HD, are the variations and limitations of model systems in reproducing critical aspects of the diseases (Bett et al., 2006; Seo et al., 2007; Ortega and Lucas, 2014). Additionally, some mechanisms may behave differently between in vitro experiments and cellular models, even within the same study (Geijtenbeek et al., 2022). The development of additional tools to study HD in physiological models will grant a better understanding of the biology of HD and inform the design of future studies and possible treatment avenues. Because the UPS becomes dysfunctional in late-stage HD (Jana et al., 2001), increasing degradation of mHTT by ubiquitin-independent 20S mechanisms could have therapeutic benefit. A recent review discusses therapeutic strategies in targeting different proteostasis pathways in HD (Harding and Tong, 2018).
Parkinson’s disease
Parkinson’s Disease (PD) is a progressive, age-associated movement disorder caused by neurodegenerative changes primarily in dopaminergic neurons of the substantia nigra region of the brain. It is characterized by protein inclusions predominantly composed of aggregated α-synuclein (α-syn), called Lewy bodies, as well as failures of proteostasis, neuroinflammation, and oxidative and mitochondrial damage. In its physiological role, neuronal α-syn regulates neurotransmitter release through interaction with pre-synaptic membranes and synaptic vesicle release machinery (Calabresi et al., 2023). Different forms of α-syn are degraded through multiple proteasomal and lysosomal pathways, and impairment of one or more of these pathways can contribute to development of PD pathology and affect the proteolytic activity of other pathways (Ancolio et al., 2000; Webb et al., 2003; Arawaka et al., 2017; Stefanis et al., 2019).
In studying which pathways are used to degrade α-syn in normal and pathophysiological states, evidence has been mixed, and results conflict across experimental models and conditions, creating a complicated picture of intersecting proteolytic pathways, substrate degradation regulation through posttranslational modifications, and brain area-specific variations and vulnerabilities to neurodegenerative damage (Vogiatzi et al., 2008; Stefanis et al., 2019). For these reasons, data must be carefully compared across in vitro, cellular, and in vivo models when considering the broader PD proteostasis field. However, evidence in normal and PD-affected brains has consistently supported a role for ubiquitin-independent 20S proteasome degradation of α-syn, an intrinsically disordered protein (IDP) and known target of the 20S, especially in the context of oxidative stress (Tofaris et al., 2001; Machiya et al., 2010; Höhn et al., 2020; Coskuner-Weber et al., 2022). Like in other neurodegenerative diseases, mitochondrial dysfunction and oxidative stress are significant features of PD (Calabresi et al., 2023), and ubiquitin-independent proteasomal degradation is critical to clear accumulated damaged and oxidized proteins (Tofaris et al., 2001; Opoku-Nsiah and Gestwicki, 2018).
In PD, oxidative stress induces posttranslational modifications of α-syn including oxidation of methionines, which affect its degradation by the proteasome. Because cleavage of α-syn by the 20S requires first that the α-syn N-terminus binds to the 20S α7 subunit C-terminus, oxidation of α-syn N-terminal methionines during oxidative stress directly inhibits this degradation, slowing down clearance of α-syn and allowing accumulation within the cell (Alvarez-Castelao et al., 2014). Oxidized α-syn then continues to aggregate (forming non-fibrillar oligomers, then protofibrils, and finally fibrillar aggregates), becoming insoluble, and its degradation is significantly impaired relative to non-oxidized α-syn (Alvarez-Castelao et al., 2014). Certain modifications to α-syn that occur during oxidative stress are irreversible through normal pathways (Binolfi et al., 2016), and some of these modifications both inhibit protective modifications and facilitate harmful ones like phosphorylation of α-syn Serine-129 (pS129) (Anderson et al., 2006; Schildknecht et al., 2013). pS129 is an especially pathological modification present in over 90% of the α-syn in Lewy bodies, but under 4% of α-syn found in normal brains (Fujiwara et al., 2002; Anderson et al., 2006). Data in rat primary cortical cultures and SH-SY5Y neuroblastoma cells demonstrate that ubiquitin-independent proteasomal degradation is the primary mechanism through which pS129 α-syn in soluble monomeric form is degraded (Machiya et al., 2010), emphasizing the mechanism’s importance in mitigating pathology in PD. In insoluble form, pS129 α-syn is degraded both by ubiquitin-independent proteasome mechanisms and by the lysosome. However, after extensive aggregation, pS129 α-syn can no longer be degraded by the proteasome and collects within Lewy bodies (Arawaka et al., 2017). To relieve intracellular protein overload, α-syn can be exported into the extracellular space through exosomes, which can transfer α-syn between cells and may contribute to the spread of toxic α-syn species from cell-to-cell, nucleating pathological aggregation in those cells (Danzer et al., 2012; Lee et al., 2014; Stefanis et al., 2019). Extracellular α-syn can also induce neuroinflammation through activation of microglia, further compounding neurodegenerative changes in cellular stress (Lee et al., 2014; Calabresi et al., 2023).
As described in the “Catalytic Core Regulators” (CCRs) and “Oxidative Stress” sections, allosteric regulation of the 20S by CCRs is especially important during oxidative stress, and failure of the pathways regulating the oxidative stress response is central to PD pathology. Mutations in PARK7, the gene encoding PD-associated protein deglycase DJ-1 (also called Parkinson disease protein 7) increase vulnerability of cells to oxidative damage due in part to defects in regulation of ubiquitin-independent 20S proteasome degradation by DJ-1, which is upregulated in response to oxidative stress, coordinates the critically important Nrf2 antioxidant response pathway (see “Oxidative Stress” section), and allosterically inhibits ubiquitin-independent degradation by the 20S, protecting important physiological IDPs from degradation while allowing rapid destruction of damaged and oxidized proteins (Clements et al., 2006; Moscovitz et al., 2015; Deshmukh et al., 2023). The oxidation state of DJ-1 also affects the propensity of α-syn to form fibrils and affects its chaperone-like activity (Zhou et al., 2006).
Beyond changes to α-syn, as PD progresses, there are also alterations to proteasome expression and composition. These changes are most pronounced in areas most affected by PD pathology, like the substantia nigra pars compacta. McNaught et al. showed that in brains affected by sporadic PD, there are deficits in proteostasis associated with selective loss of 20S ⍺-subunits in dopaminergic neurons of the substantia nigra pars compacta but not in other areas (McNaught et al., 2002). Α-subunits, specifically α4, interact directly with parkin, an E3 ligase and one of the most important components of Parkinson’s disease.
The loss of these subunits causes structural instability of the proteasome and can prevent its coordinated assembly, contributing to the breakdown in proteostasis observed in PD and possibly the accumulation of Lewy bodies and dopaminergic cell death (McNaught et al., 2002). In addition, all three types of proteolytic activity in the 20S (chymotrypsin-like, trypsin-like, and caspase-like) are impaired in the substantia nigra of brains from patients who died of sporadic PD by up to 42% (McNaught and Jenner, 2001). Other studies have found that in neuronal cell models, the substantia nigra pars compacta may have lower baseline expression of PAs including 19S and PA28 and that dopaminergic neurons may not effectively upregulate PAs in response to stress as well as other cell types, making them increasingly vulnerable to damage by defective proteostasis (McNaught et al., 2010).
Mutations in parkin, an E3 ligase, can induce proteasome dysfunction. Parkin activates 26S in a ubiquitin-ligase-independent manner and enhances interactions between 19S subunits, playing a role in proteasome assembly that is disrupted by parkin mutations (Um et al., 2010). It also interacts directly with the α4 subunit of the 20S (Dachsel et al., 2005) and is proposed to have some function in substrate identification by the 20S/ubiquitin-independent pathways (Sanchez-Lanzas and Castano, 2014). A review by Sanchez-Lanzas and Castano (2014) describes several other 20S proteasome interactors and their relationships to ubiquitin-independent pathways.
Conclusion
The proteasome is a cylindrical degradation complex composed of four stacked heptameric rings of α/β subunits (α7β7β7α7) around a central proteolytic chamber. Proteasomes catalyze the majority of protein degradation in mammalian cells through multiple mechanisms, the best characterized of which is the ubiquitin-proteasome system (UPS), a pathway employing ubiquitin protein tags, a 19S cap, and ATP hydrolysis to recognize, unfold, and degrade protein targets (Coux et al., 1996; Ciechanover, 1998; Ciechanover and Schwartz, 1998; Ben-Nissan and Sharon, 2014). Ubiquitin-independent proteasomal degradation is more recently studied and can be mediated by the 20S proteasome core or by a variety of proteasome activator complexes (e.g., PA28, PA200, and 19S) which facilitate degradation of unfolded and intrinsically disordered proteins without ATP (Opoku-Nsiah and Gestwicki, 2018). Increasing evidence supports a central role for ubiquitin-independent proteasome degradation in the oxidative stress response and clearance of protein aggregates in age-related neurodegenerative diseases (Kazee and Han, 1995; Lopez Salon et al., 2000; Bence et al., 2001; Iwata et al., 2005; Bennett et al., 2007; Wilson et al., 2011; Hipp et al., 2012; Ben Yehuda et al., 2017).
Literature on aging, neurodegenerative disease, and ubiquitin-independent 20S proteasome activity in different cellular contexts is ongoing and has had some conflicting results. Conflicts in the literature around the ubiquitin-independent proteasome are highly dependent on model system (species, in vitro vs. cultures vs. in vivo, etc.) and experimental design differences, and orthogonal approaches are necessary to definitively determine the behavior of the physiological system (Oh et al., 2005; Türker et al., 2023). In fact, it is possible some research describing the UPS in neurodegenerative disease reflects effects of both the UPS and ubiquitin-independent degradation because many studies rely on measures of global proteasome activity and pan-proteasome inhibitors, without isolating the ubiquitin-independent proteasome activity for assessment separately from the UPS. An additional barrier is the multifactorial changes associated with aging that complicate mechanistic investigations in vivo. As tools differentiating between proteasomal mechanisms are developed, the respective contributions of each mechanism to neurodegenerative disease may be better clarified.
Looking forward, many questions remain about how alternative mechanisms of protein degradation impact and are impacted by neurodegenerative processes. These include identifying how degradative pathways cooperate, determining how changes in proteasome activity and composition vary by cell type and cell compartment specificity in different diseases, further investigating the roles of alternative proteasome activator and regulators, investigating regulation of ubiquitin-independent proteasomal mechanisms by posttranslational modifications and interaction with binding partners, and confirming how these changes occur in vivo. Additionally, there have been recent developments in diverse neurodegenerative disease and proteasome research topics including liquid-liquid phase separation (Myers et al., 2018; Cohen-Kaplan et al., 2020; Yasuda et al., 2020; Zbinden et al., 2020; Hayashi et al., 2021; Mee Hayes et al., 2022; Hurtle et al., 2023), diagnostic and molecular tools (Devitt et al., 2018; Barthel et al., 2022), posttranslational 20S proteasomal processing of substrates for unique functions (Moorthy et al., 2006; Solomon et al., 2017), endoproteolytic proteasomal cleavage of disordered residues (Liu et al., 2003), proteasome-catalyzed peptide splicing (Liepe et al., 2016; Soh et al., 2024), ATP-independent 26S proteasomal degradation (Tsvetkov et al., 2020), which may further propel and nuance the understanding of proteasomal roles in neurodegenerative disease. Several of these topics are controversial and need more validation, but they reflect an appreciation of proteasome roles and regulation beyond the canonical UPS. Overall, this review examines the regulators, 20S-associated proteasome activators, and complexes involved in ubiquitin-independent proteasomal degradation, focusing on their bidirectional impact on age-associated neurodegenerative diseases.
Author contributions
TC: Conceptualization, Funding acquisition, Project administration, Writing–review and editing, Data curation, Formal Analysis, Investigation, Methodology, Resources, Software, Supervision, Writing–original draft. SM: Conceptualization, Funding acquisition, Project administration, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. TC was supported by NIH-NINDS NRSA F31NS134239. This work was funded by institutional funding and NIH grant R01 NS110754 (SM).
Acknowledgments
We thank the funding agencies for supporting our study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abi Habib, J., De Plaen, E., Stroobant, V., Zivkovic, D., Bousquet, M. P., Guillaume, B., et al. (2020). Efficiency of the four proteasome subtypes to degrade ubiquitinated or oxidized proteins. Sci. Rep. 10, 15765. doi:10.1038/s41598-020-71550-5
Abi Habib, J., Lesenfants, J., Vigneron, N., and Van Den Eynde, B. J. (2022). Functional differences between proteasome subtypes. Cells 11, 421. doi:10.3390/cells11030421
Adelöf, J., Andersson, M., Porritt, M., Petersen, A., Zetterberg, M., Wiseman, J., et al. (2018). PA28αβ overexpression enhances learning and memory of female mice without inducing 20S proteasome activity. BMC Neurosci. 19, 70. doi:10.1186/s12868-018-0468-2
Adelöf, J., Wiseman, J., Zetterberg, M., and Hernebring, M. (2021). PA28α overexpressing female mice maintain exploratory behavior and capacity to prevent protein aggregation in hippocampus as they age. Aging Cell. 20, e13336. doi:10.1111/acel.13336
Aladdin, A., Yao, Y., Yang, C., Kahlert, G., Ghani, M., Király, N., et al. (2020). The proteasome activators blm10/PA200 enhance the proteasomal degradation of N-terminal huntingtin. Biomolecules 10, 1581. doi:10.3390/biom10111581
Alvarez-Castelao, B., Goethals, M., Vandekerckhove, J., and Castaño, J. G. (2014). Mechanism of cleavage of alpha-synuclein by the 20S proteasome and modulation of its degradation by the RedOx state of the N-terminal methionines. Biochimica Biophysica Acta (BBA) - Mol. Cell. Res. 1843, 352–365. doi:10.1016/j.bbamcr.2013.11.018
Amadoro, G., Corsetti, V., Ciotti, M. T., Florenzano, F., Capsoni, S., Amato, G., et al. (2011). Endogenous Aβ causes cell death via early tau hyperphosphorylation. Neurobiol. Aging 32, 969–990. doi:10.1016/j.neurobiolaging.2009.06.005
Ancolio, K., Alves Da Costa, C., Uéda, K., and Checler, F. (2000). Alpha-synuclein and the Parkinson's disease-related mutant Ala53Thr-alpha-synuclein do not undergo proteasomal degradation in HEK293 and neuronal cells. Neurosci. Lett. 285, 79–82. doi:10.1016/s0304-3940(00)01049-1
Anderson, J. P., Walker, D. E., Goldstein, J. M., De Laat, R., Banducci, K., Caccavello, R. J., et al. (2006). Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 281, 29739–29752. doi:10.1074/jbc.M600933200
Arawaka, S., Sato, H., Sasaki, A., Koyama, S., and Kato, T. (2017). Mechanisms underlying extensive Ser129-phosphorylation in α-synuclein aggregates. Acta Neuropathol. Commun. 5, 48. doi:10.1186/s40478-017-0452-6
Arkinson, C., Dong, K. C., Gee, C. L., and Martin, A. (2024). Mechanisms and regulation of substrate degradation by the 26S proteasome. Nat. Rev. Mol. Cell. Biol. doi:10.1038/s41580-024-00778-0
Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R., and Finkbeiner, S. (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–810. doi:10.1038/nature02998
Asher, G., Tsvetkov, P., Kahana, C., and Shaul, Y. (2005). A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes and Dev. 19, 316–321. doi:10.1101/gad.319905
Aso, E., Lomoio, S., Lopez-Gonzalez, I., Joda, L., Carmona, M., Fernandez-Yague, N., et al. (2012). Amyloid generation and dysfunctional immunoproteasome activation with disease progression in animal model of familial Alzheimer's disease. Brain Pathol. 22, 636–653. doi:10.1111/j.1750-3639.2011.00560.x
Bader, M., Benjamin, S., Wapinski, O. L., Smith, D. M., Goldberg, A. L., and Steller, H. (2011). A conserved F box regulatory complex controls proteasome activity in Drosophila. Cell. 145, 371–382. doi:10.1016/j.cell.2011.03.021
Balzarini, M., Tong, J., Gui, W., Jayalath, I. M., Schell, B.-B., and Kodadek, T. (2024). Recruitment to the proteasome is necessary but not sufficient for chemically induced, ubiquitin-independent degradation of native proteins. ACS Chem. Biol. 19, 2323–2335. doi:10.1021/acschembio.4c00422
Barthel, H., Villemagne, V. L., and Drzezga, A. (2022). Future directions in molecular imaging of neurodegenerative disorders. J. Nucl. Med. 63, 68S–74S. doi:10.2967/jnumed.121.263202
Bashore, C., Prakash, S., Johnson, M. C., Conrad, R. J., Kekessie, I. A., Scales, S. J., et al. (2023). Targeted degradation via direct 26S proteasome recruitment. Nat. Chem. Biol. 19, 55–63. doi:10.1038/s41589-022-01218-w
Basler, M., Kirk, C. J., and Groettrup, M. (2013). The immunoproteasome in antigen processing and other immunological functions. Curr. Opin. Immunol. 25, 74–80. doi:10.1016/j.coi.2012.11.004
Baugh, J. M., Viktorova, E. G., and Pilipenko, E. V. (2009). Proteasomes can degrade a significant proportion of cellular proteins independent of ubiquitination. J. Mol. Biol. 386, 814–827. doi:10.1016/j.jmb.2008.12.081
Baumeister, W., Cejka, Z., Kania, M., and Seemüller, E. (1997). The proteasome: a macromolecular assembly designed to confine proteolysis to a nanocompartment. Biol. Chem. 378, 121–130. doi:10.1515/bchm.1997.378.3-4.121
Bence, N. F., Sampat, R. M., and Kopito, R. R. (2001). Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292, 1552–1555. doi:10.1126/science.292.5521.1552
Bennett, E. J., Shaler, T. A., Woodman, B., Ryu, K. Y., Zaitseva, T. S., Becker, C. H., et al. (2007). Global changes to the ubiquitin system in Huntington's disease. Nature 448, 704–708. doi:10.1038/nature06022
Ben-Nissan, G., Katzir, N., Fuzesi-Levi, M. G., and Sharon, M. (2022). Biology of the extracellular proteasome. Biomolecules 12, 619. doi:10.3390/biom12050619
Ben-Nissan, G., and Sharon, M. (2014). Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 4, 862–884. doi:10.3390/biom4030862
Ben Yehuda, A., Risheq, M., Novoplansky, O., Bersuker, K., Kopito, R. R., Goldberg, M., et al. (2017). Ubiquitin accumulation on disease associated protein aggregates is correlated with nuclear ubiquitin depletion, histone de-ubiquitination and impaired DNA damage response. PLoS One 12, e0169054. doi:10.1371/journal.pone.0169054
Bercovich, Z., and Kahana, C. (1993). Involvement of the 20S proteasome in the degradation of ornithine decarboxylase. Eur. J. Biochem. 213, 205–210. doi:10.1111/j.1432-1033.1993.tb17749.x
Bett, J. S., Goellner, G. M., Woodman, B., Pratt, G., Rechsteiner, M., and Bates, G. P. (2006). Proteasome impairment does not contribute to pathogenesis in R6/2 Huntington's disease mice: exclusion of proteasome activator REGgamma as a therapeutic target. Hum. Mol. Genet. 15, 33–44. doi:10.1093/hmg/ddi423
Bi, M., Du, X., Jiao, Q., Chen, X., and Jiang, H. (2021). Expanding the role of proteasome homeostasis in Parkinson's disease: beyond protein breakdown. Cell. Death Dis. 12, 154. doi:10.1038/s41419-021-03441-0
Bian, J.-T., Zhao, H.-L., Zhang, Z.-X., Bi, X.-H., and Zhang, J.-W. (2008). Association of NAD(P)H:quinone oxidoreductase 1 polymorphism and Alzheimer's disease in Chinese. J. Mol. Neurosci. MN 34, 235–240. doi:10.1007/s12031-008-9036-z
Billingsley, K. J., Bandres-Ciga, S., Ding, J., Hernandez, D., Gibbs, J. R., Blauwendraat, C., et al. (2020). MIDN locus structural variants and Parkinson's Disease risk. Ann. Clin. Transl. Neurology 7, 602–603. doi:10.1002/acn3.51012
Bingol, B., and Schuman, E. M. (2005). Synaptic protein degradation by the ubiquitin proteasome system. Curr. Opin. Neurobiol. 15, 536–541. doi:10.1016/j.conb.2005.08.016
Binolfi, A., Limatola, A., Verzini, S., Kosten, J., Theillet, F. X., Rose, H. M., et al. (2016). Intracellular repair of oxidation-damaged α-synuclein fails to target C-terminal modification sites. Nat. Commun. 7, 10251. doi:10.1038/ncomms10251
Blickwedehl, J., Agarwal, M., Seong, C., Pandita, R. K., Melendy, T., Sung, P., et al. (2008). Role for proteasome activator PA200 and postglutamyl proteasome activity in genomic stability. Proc. Natl. Acad. Sci. U. S. A. 105, 16165–16170. doi:10.1073/pnas.0803145105
Bochmann, I., Ebstein, F., Lehmann, A., Wohlschlaeger, J., Sixt, S. U., Kloetzel, P. M., et al. (2014). T lymphocytes export proteasomes by way of microparticles: a possible mechanism for generation of extracellular proteasomes. J. Cell. Mol. Med. 18, 59–68. doi:10.1111/jcmm.12160
Bonet-Costa, V., Pomatto, L. C., and Davies, K. J. (2016). The proteasome and oxidative stress in alzheimer's disease. Antioxid. Redox Signal 25, 886–901. doi:10.1089/ars.2016.6802
Bonhoure, A., Henry, L., Bich, C., Blanc, L., Bergeret, B., Bousquet, M. P., et al. (2022). Extracellular 20S proteasome secreted via microvesicles can degrade poorly folded proteins and inhibit Galectin-3 agglutination activity. Traffic 23, 287–304. doi:10.1111/tra.12840
Bossis, G., Ferrara, P., Acquaviva, C., Jariel-Encontre, I., and Piechaczyk, M. (2003). c-Fos proto-oncoprotein is degraded by the proteasome independently of its own ubiquitinylation in vivo. Mol. Cell. Biol. 23, 7425–7436. doi:10.1128/MCB.23.20.7425-7436.2003
Boulos, A., Maroun, D., Ciechanover, A., and Ziv, N. E. (2024). Peripheral sequestration of huntingtin delays neuronal death and depends on N-terminal ubiquitination. Commun. Biol. 7, 1014. doi:10.1038/s42003-024-06733-1
Brothers, H. M., Gosztyla, M. L., and Robinson, S. R. (2018). The physiological roles of amyloid-β peptide hint at new ways to treat alzheimer's disease. Front. Aging Neurosci. 10, 118. doi:10.3389/fnagi.2018.00118
Bulteau, A. L., Lundberg, K. C., Humphries, K. M., Sadek, H. A., Szweda, P. A., Friguet, B., et al. (2001). Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J. Biol. Chem. 276, 30057–30063. doi:10.1074/jbc.M100142200
Bulteau, A. L., Petropoulos, I., and Friguet, B. (2000). Age-related alterations of proteasome structure and function in aging epidermis. Exp. Gerontol. 35, 767–777. doi:10.1016/S0531-5565(00)00136-4
Busche, M. A., Wegmann, S., Dujardin, S., Commins, C., Schiantarelli, J., Klickstein, N., et al. (2019). Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat. Neurosci. 22, 57–64. doi:10.1038/s41593-018-0289-8
Calabresi, P., Mechelli, A., Natale, G., Volpicelli-Daley, L., Di Lazzaro, G., and Ghiglieri, V. (2023). Alpha-synuclein in Parkinson's disease and other synucleinopathies: from overt neurodegeneration back to early synaptic dysfunction. Cell. Death Dis. 14, 176. doi:10.1038/s41419-023-05672-9
Carrell, R. W., Mushunje, A., and Zhou, A. (2008). Serpins show structural basis for oligomer toxicity and amyloid ubiquity. FEBS Lett. 582, 2537–2541. doi:10.1016/j.febslet.2008.06.021
Cascio, P. (2021). PA28γ: new insights on an ancient proteasome activator. Biomolecules 11, 228. doi:10.3390/biom11020228
Cascio, P., Call, M., Petre, B. M., Walz, T., and Goldberg, A. L. (2002). Properties of the hybrid form of the 26S proteasome containing both 19S and PA28 complexes. EMBO J. 21, 2636–2645. doi:10.1093/emboj/21.11.2636
Cekala, K., Trepczyk, K., Sowik, D., Karpowicz, P., Gieldon, A., Witkowska, J., et al. (2022). Peptidomimetics based on C-terminus of Blm10 stimulate human 20S proteasome activity and promote degradation of proteins. Biomolecules 12, 777. doi:10.3390/biom12060777
Cekala, K., Trepczyk, K., Witkowska, J., Jankowska, E., and Wieczerzak, E. (2024). Rpt5-Derived analogs stimulate human proteasome activity in cells and degrade proteins forming toxic aggregates in age-related diseases. Int. J. Mol. Sci. 25, 4663. doi:10.3390/ijms25094663
Chaplot, K., Jarvela, T. S., and Lindberg, I. (2020). Secreted chaperones in neurodegeneration. Front. Aging Neurosci. 12, 268. doi:10.3389/fnagi.2020.00268
Chiba, A., Kato, C., Nakagawa, T., Osaki, T., Nakamura, K., Norota, I., et al. (2024). Midnolin, a genetic risk factor for Parkinson’s disease, promotes neurite outgrowth accompanied by early growth response 1 activation in PC12 cells. Mol. Cell. Biol. 0, 516–527. doi:10.1080/10985549.2024.2399358
Choi, W. H., Yun, Y., Byun, I., Kim, S., Lee, S., Sim, J., et al. (2023). ECPAS/Ecm29-mediated 26S proteasome disassembly is an adaptive response to glucose starvation. Cell. Rep. 42, 112701. doi:10.1016/j.celrep.2023.112701
Chuah, J. J. Y., Thibaudeau, T. A., and Smith, D. M. (2023). Minimal mechanistic component of HbYX-dependent proteasome activation that reverses impairment by neurodegenerative-associated oligomers. Commun. Biol. 6, 725. doi:10.1038/s42003-023-05082-9
Ciechanover, A. (1998). The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 17, 7151–7160. doi:10.1093/emboj/17.24.7151
Ciechanover, A., and Brundin, P. (2003). The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40, 427–446. doi:10.1016/S0896-6273(03)00606-8
Ciechanover, A., and Schwartz, A. L. (1998). The ubiquitin-proteasome pathway: the complexity and myriad functions of proteins death. Proc. Natl. Acad. Sci. U. S. A. 95, 2727–2730. doi:10.1073/pnas.95.6.2727
Clemen, C. S., Marko, M., Strucksberg, K.-H., Behrens, J., Wittig, I., Gärtner, L., et al. (2015). VCP and PSMF1: antagonistic regulators of proteasome activity. Biochem. Biophysical Res. Commun. 463, 1210–1217. doi:10.1016/j.bbrc.2015.06.086
Clements, C. M., Mcnally, R. S., Conti, B. J., Mak, T. W., and Ting, J. P. (2006). DJ-1, a cancer- and Parkinson's disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. U. S. A. 103, 15091–15096. doi:10.1073/pnas.0607260103
Coffino, P. (1998). “Degradation of ornithine decarboxylase,” in Ubiquitin and the biology of the cell. Editors J.-M. Peters, J. R. Harris, and D. Finley (Boston, MA: Springer US), 411–428.
Cohen-Kaplan, V., Livneh, I., and Ciechanover, A. (2020). Proteasome phase separation: a novel layer of quality control. Cell. Res. 30, 374–375. doi:10.1038/s41422-020-0306-9
Coskuner-Weber, O., Mirzanli, O., and Uversky, V. N. (2022). Intrinsically disordered proteins and proteins with intrinsically disordered regions in neurodegenerative diseases. Biophys. Rev. 14, 679–707. doi:10.1007/s12551-022-00968-0
Coux, O., Tanaka, K., and Goldberg, A. L. (1996). Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 65, 801–847. doi:10.1146/annurev.bi.65.070196.004101
Cuanalo-Contreras, K., Schulz, J., Mukherjee, A., Park, K.-W., Armijo, E., and Soto, C. (2023). Extensive accumulation of misfolded protein aggregates during natural aging and senescence. Front. Aging Neurosci. 14, 1090109. doi:10.3389/fnagi.2022.1090109
Dachsel, J. C., Lucking, C. B., Deeg, S., Schultz, E., Lalowski, M., Casademunt, E., et al. (2005). Parkin interacts with the proteasome subunit alpha4. FEBS Lett. 579, 3913–3919. doi:10.1016/j.febslet.2005.06.003
Dange, T., Smith, D., Noy, T., Rommel, P. C., Jurzitza, L., Cordero, R. J. B., et al. (2011). Blm10 protein promotes proteasomal substrate turnover by an active gating mechanism. J. Biol. Chem. 286, 42830–42839. doi:10.1074/jbc.M111.300178
Dantuma, N. P., and Bott, L. C. (2014). The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front. Mol. Neurosci. 7, 70. doi:10.3389/fnmol.2014.00070
Danzer, K. M., Kranich, L. R., Ruf, W. P., Cagsal-Getkin, O., Winslow, A. R., Zhu, L., et al. (2012). Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 7, 42. doi:10.1186/1750-1326-7-42
David, D. C., Layfield, R., Serpell, L., Narain, Y., Goedert, M., and Spillantini, M. G. (2002). Proteasomal degradation of tau protein. J. Neurochem. 83, 176–185. doi:10.1046/j.1471-4159.2002.01137.x
Davidson, K., and Pickering, A. M. (2023). The proteasome: a key modulator of nervous system function, brain aging, and neurodegenerative disease. Front. Cell. Dev. Biol. 11, 1124907. doi:10.3389/fcell.2023.1124907
Davies, K. J. (2001). Degradation of oxidized proteins by the 20S proteasome. Biochimie 83, 301–310. doi:10.1016/s0300-9084(01)01250-0
Deshmukh, F. K., Ben-Nissan, G., Olshina, M. A., Füzesi-Levi, M. G., Polkinghorn, C., Arkind, G., et al. (2023). Allosteric regulation of the 20S proteasome by the catalytic core regulators (CCRs) family. Nat. Commun. 14, 3126. doi:10.1038/s41467-023-38404-w
Devitt, G., Howard, K., Mudher, A., and Mahajan, S. (2018). Raman spectroscopy: an emerging tool in neurodegenerative disease research and diagnosis. ACS Chem. Neurosci. 9, 404–420. doi:10.1021/acschemneuro.7b00413
Dianzani, C., Bellavista, E., Liepe, J., Verderio, C., Martucci, M., Santoro, A., et al. (2017). Extracellular proteasome-osteopontin circuit regulates cell migration with implications in multiple sclerosis. Sci. Rep. 7, 43718. doi:10.1038/srep43718
Dianzani, C., Vecchio, D., Clemente, N., Chiocchetti, A., Martinelli Boneschi, F., Galimberti, D., et al. (2019). Untangling extracellular proteasome-osteopontin circuit dynamics in multiple sclerosis. Cells 8, 262. doi:10.3390/cells8030262
Diaz-Hernandez, M., Hernandez, F., Martin-Aparicio, E., Gomez-Ramos, P., Moran, M. A., Castano, J. G., et al. (2003). Neuronal induction of the immunoproteasome in Huntington's disease. J. Neurosci. 23, 11653–11661. doi:10.1523/JNEUROSCI.23-37-11653.2003
Diaz-Hernandez, M., Martin-Aparicio, E., Avila, J., Hernandez, F., and Lucas, J. J. (2004). Enhanced induction of the immunoproteasome by interferon gamma in neurons expressing mutant Huntingtin. Neurotox. Res. 6, 463–468. doi:10.1007/BF03033282
Diaz-Hernandez, M., Valera, A. G., Moran, M. A., Gomez-Ramos, P., Alvarez-Castelao, B., Castano, J. G., et al. (2006). Inhibition of 26S proteasome activity by huntingtin filaments but not inclusion bodies isolated from mouse and human brain. J. Neurochem. 98, 1585–1596. doi:10.1111/j.1471-4159.2006.03968.x
Ding, Q., Dimayuga, E., and Keller, J. N. (2006). Proteasome regulation of oxidative stress in aging and age-related diseases of the CNS. Antioxid. Redox Signal 8, 163–172. doi:10.1089/ars.2006.8.163
Dong, Y., Zhang, S., Wu, Z., Li, X., Wang, W. L., Zhu, Y., et al. (2019). Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 565, 49–55. doi:10.1038/s41586-018-0736-4
Dubiel, W., Pratt, G., Ferrell, K., and Rechsteiner, M. (1992). Purification of an 11 S regulator of the multicatalytic protease. J. Biol. Chem. 267, 22369–22377. doi:10.1016/s0021-9258(18)41681-x
Dwivedi, V., Yaniv, K., and Sharon, M. (2021). Beyond cells: the extracellular circulating 20S proteasomes. Biochim. Biophys. Acta Mol. Basis Dis. 1867, 166041. doi:10.1016/j.bbadis.2020.166041
El-Daher, M. T., Hangen, E., Bruyère, J., Poizat, G., Al-Ramahi, I., Pardo, R., et al. (2015). Huntingtin proteolysis releases non-polyQ fragments that cause toxicity through dynamin 1 dysregulation. EMBO J. 34, 2255–2271. doi:10.15252/embj.201490808
Enokido, Y., Tamura, T., Ito, H., Arumughan, A., Komuro, A., Shiwaku, H., et al. (2010). Mutant huntingtin impairs Ku70-mediated DNA repair. J. Cell. Biol. 189, 425–443. doi:10.1083/jcb.200905138
Erales, J., and Coffino, P. (2014). Ubiquitin-independent proteasomal degradation. Biochimica biophysica acta 1843, 216–221. doi:10.1016/j.bbamcr.2013.05.008
Eroglu, B., Moskophidis, D., and Mivechi, N. F. (2010). Loss of Hsp110 leads to age-dependent tau hyperphosphorylation and early accumulation of insoluble amyloid beta. Mol. Cell. Biol. 30, 4626–4643. doi:10.1128/mcb.01493-09
Esaki, M., Johjima-Murata, A., Islam, M. T., and Ogura, T. (2018). Biological and pathological implications of an alternative ATP-powered proteasomal assembly with Cdc48 and the 20S Peptidase. Front. Mol. Biosci. 5, 56. doi:10.3389/fmolb.2018.00056
Ettari, R., Zappala, M., Grasso, S., Musolino, C., Innao, V., and Allegra, A. (2017). Immunoproteasome-selective and non-selective inhibitors: a promising approach for the treatment of multiple myeloma. Pharmacol. Ther. 182, 176–192. doi:10.1016/j.pharmthera.2017.09.001
Fabre, B., Lambour, T., Garrigues, L., Ducoux-Petit, M., Amalric, F., Monsarrat, B., et al. (2014). Label-free quantitative proteomics reveals the dynamics of proteasome complexes composition and stoichiometry in a wide range of human cell lines. J. Proteome Res. 13, 3027–3037. doi:10.1021/pr500193k
Favit, A., Grimaldi, M., and Alkon, D. L. (2000). Prevention of beta-amyloid neurotoxicity by blockade of the ubiquitin-proteasome proteolytic pathway. J. Neurochem. 75, 1258–1263. doi:10.1046/j.1471-4159.2000.0751258.x
Fernández-Cruz, I., and Reynaud, E. (2021). Proteasome subunits involved in neurodegenerative diseases. Archives Med. Res. 52, 1–14. doi:10.1016/j.arcmed.2020.09.007
Fiolek, T., Magyar, C. L., Wall, T. J., Davie, S. B., Campbell, M. V., Savich, C. J., et al. (2021a). Dihydroquinazolines enhance 20S proteasome activity and induce degradation of α-synuclein, an intrinsically disordered protein associated with neurodegeneration. Bioorg. and Med. Chem. Lett. 36, 127821. doi:10.1016/j.bmcl.2021.127821
Fiolek, T. J., Keel, K. L., and Tepe, J. J. (2021b). Fluspirilene analogs activate the 20S proteasome and overcome proteasome impairment by intrinsically disordered protein oligomers. ACS Chem. Neurosci. 12, 1438–1448. doi:10.1021/acschemneuro.1c00099
Förster, A., Whitby, F. G., and Hill, C. P. (2003). The pore of activated 20S proteasomes has an ordered 7-fold symmetric conformation. EMBO J. 22, 4356–4364. doi:10.1093/emboj/cdg436
Frayssinhes, J.-Y. A., Cerruti, F., Laulin, J., Cattaneo, A., Bachi, A., Apcher, S., et al. (2021). PA28γ–20S proteasome is a proteolytic complex committed to degrade unfolded proteins. Cell. Mol. Life Sci. 79, 45. doi:10.1007/s00018-021-04045-9
Freudenburg, W., Gautam, M., Chakraborty, P., James, J., Richards, J., Salvatori, A. S., et al. (2013). Immunoproteasome activation during early antiviral response in mouse pancreatic beta-cells: new insights into auto-antigen generation in type I diabetes? J. Clin. Cell. Immunol. 4, 141. doi:10.4172/2155-9899.1000141
Fricke, B., Heink, S., Steffen, J., Kloetzel, P. M., and Krüger, E. (2007). The proteasome maturation protein POMP facilitates major steps of 20S proteasome formation at the endoplasmic reticulum. EMBO Rep. 8, 1170–1175. doi:10.1038/sj.embor.7401091
Frost, B., Jacks, R. L., and Diamond, M. I. (2009). Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 284, 12845–12852. doi:10.1074/jbc.M808759200
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M. S., et al. (2002). alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell. Biol. 4, 160–164. doi:10.1038/ncb748
Gao, H., Sun, X., and Rao, Y. (2020). PROTAC Technology: opportunities and challenges. ACS Med. Chem. Lett. 11, 237–240. doi:10.1021/acsmedchemlett.9b00597
Geijtenbeek, K. W., Janzen, J., Bury, A. E., Sanz-Sanz, A., Hoebe, R. A., Bondulich, M. K., et al. (2022). Reduction in PA28αβ activation in HD mouse brain correlates to increased mHTT aggregation in cell models. PLOS ONE 17, e0278130. doi:10.1371/journal.pone.0278130
Gillardon, F., Kloß, A., Berg, M., Neumann, M., Mechtler, K., Hengerer, B., et al. (2007). The 20S proteasome isolated from Alzheimer’s disease brain shows post-translational modifications but unchanged proteolytic activity. J. Neurochem. 101, 1483–1490. doi:10.1111/j.1471-4159.2006.04438.x
Goldberg, A. L., Kim, H. T., Lee, D., and Collins, G. A. (2021). Mechanisms that activate 26S proteasomes and enhance protein degradation. Biomolecules 11, 779. doi:10.3390/biom11060779
Gregori, L., Fuchs, C., Figueiredo-Pereira, M. E., Van Nostrand, W. E., and Goldgaber, D. (1995). Amyloid beta-protein inhibits ubiquitin-dependent protein degradation in vitro. J. Biol. Chem. 270, 19702–19708. doi:10.1074/jbc.270.34.19702
Gregori, L., Hainfeld, J. F., Simon, M. N., and Goldgaber, D. (1997). Binding of amyloid beta protein to the 20 S proteasome. J. Biol. Chem. 272, 58–62. doi:10.1074/jbc.272.1.58
Groll, M., Bajorek, M., Köhler, A., Moroder, L., Rubin, D. M., Huber, R., et al. (2000). A gated channel into the proteasome core particle. Nat. Struct. Biol. 7, 1062–1067. doi:10.1038/80992
Grune, T., Botzen, D., Engels, M., Voss, P., Kaiser, B., Jung, T., et al. (2010). Tau protein degradation is catalyzed by the ATP/ubiquitin-independent 20S proteasome under normal cell conditions. Archives Biochem. Biophysics 500, 181–188. doi:10.1016/j.abb.2010.05.008
Grune, T., Catalgol, B., Licht, A., Ermak, G., Pickering, A. M., Ngo, J. K., et al. (2011). HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free Radic. Biol. Med. 51, 1355–1364. doi:10.1016/j.freeradbiomed.2011.06.015
Gu, X., Nardone, C., Kamitaki, N., Mao, A., Elledge, S. J., and Greenberg, M. E. (2023). The midnolin-proteasome pathway catches proteins for ubiquitination-independent degradation. Science 381, eadh5021. doi:10.1126/science.adh5021
Guan, H., Wang, Y., Yu, T., Huang, Y., Li, M., Saeed, A. F. U. H., et al. (2020). Cryo-EM structures of the human PA200 and PA200-20S complex reveal regulation of proteasome gate opening and two PA200 apertures. PLoS Biol. 18, e3000654. doi:10.1371/journal.pbio.3000654
Guo, J. L., and Lee, V. M. (2011). Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 286, 15317–15331. doi:10.1074/jbc.M110.209296
Guo, Q., Bin, H., Cheng, J., Seefelder, M., Engler, T., Pfeifer, G., et al. (2018). The cryo-electron microscopy structure of huntingtin. Nature 555, 117–120. doi:10.1038/nature25502
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat. Rev. Mol. Cell. Biol. 8, 101–112. doi:10.1038/nrm2101
Harding, R. J., and Tong, Y. F. (2018). Proteostasis in Huntington's disease: disease mechanisms and therapeutic opportunities. Acta Pharmacol. Sin. 39, 754–769. doi:10.1038/aps.2018.11
Hayashi, Y., Ford, L. K., Fioriti, L., Mcgurk, L., and Zhang, M. (2021). Liquid-liquid phase separation in physiology and pathophysiology of the nervous system. J. Neurosci. 41, 834–844. doi:10.1523/jneurosci.1656-20.2020
He, H.-Y., Ahsan, A., Bera, R., Mclain, N., Faulkner, R., Ramachandran, K. V., et al. (2023). Neuronal membrane proteasomes regulate neuronal circuit activity in vivo and are required for learning-induced behavioral plasticity. Proc. Natl. Acad. Sci. 120, e2216537120. doi:10.1073/pnas.2216537120
Hendil, K. B., Khan, S., and Tanaka, K. (1998). Simultaneous binding of PA28 and PA700 activators to 20 S proteasomes. Biochem. J. 332 (Pt 3), 749–754. doi:10.1042/bj3320749
Hershko, A., and Ciechanover, A. (1992). The ubiquitin system for protein degradation. Annu. Rev. Biochem. 61, 761–807. doi:10.1146/annurev.bi.61.070192.003553
Hershko, A., Ciechanover, A., and Rose, I. A. (1981). Identification of the active amino acid residue of the polypeptide of ATP-dependent protein breakdown. J. Biol. Chem. 256, 1525–1528. doi:10.1016/S0021-9258(19)69833-9
Hipp, M. S., Patel, C. N., Bersuker, K., Riley, B. E., Kaiser, S. E., Shaler, T. A., et al. (2012). Indirect inhibition of 26S proteasome activity in a cellular model of Huntington's disease. J. Cell. Biol. 196, 573–587. doi:10.1083/jcb.201110093
Hirano, Y., Hayashi, H., Iemura, S.-I., Hendil, K. B., Niwa, S.-I., Kishimoto, T., et al. (2006). Cooperation of multiple chaperones required for the assembly of mammalian 20S proteasomes. Mol. Cell. 24, 977–984. doi:10.1016/j.molcel.2006.11.015
Hirano, Y., Hendil, K. B., Yashiroda, H., Iemura, S.-I., Nagane, R., Hioki, Y., et al. (2005). A heterodimeric complex that promotes the assembly of mammalian 20S proteasomes. Nature 437, 1381–1385. doi:10.1038/nature04106
Hjerpe, R., Bett, J. S., Keuss, M. J., Solovyova, A., Mcwilliams, T. G., Johnson, C., et al. (2016). UBQLN2 mediates autophagy-independent protein aggregate clearance by the proteasome. Cell. 166, 935–949. doi:10.1016/j.cell.2016.07.001
Höhn, A., Tramutola, A., and Cascella, R. (2020). Proteostasis failure in neurodegenerative diseases: focus on oxidative stress. Oxidative Med. Cell. Longev. 2020, 5497046. doi:10.1155/2020/5497046
Holmberg, C. I., Staniszewski, K. E., Mensah, K. N., Matouschek, A., and Morimoto, R. I. (2004). Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J. 23, 4307–4318. doi:10.1038/sj.emboj.7600426
Huang, Q., Wang, H., Perry, S. W., and Figueiredo-Pereira, M. E. (2013). Negative regulation of 26S proteasome stability via calpain-mediated cleavage of Rpn10 subunit upon mitochondrial dysfunction in neurons. J. Biol. Chem. 288, 12161–12174. doi:10.1074/jbc.M113.464552
Hurtle, B. T., Xie, L., and Donnelly, C. J. (2023). Disrupting pathologic phase transitions in neurodegeneration. J. Clin. Invest 133, e168549. doi:10.1172/JCI168549
Hyun, S., and Shin, D. (2021). Chemical-mediated targeted protein degradation in neurodegenerative diseases. Life (Basel) 11, 607. doi:10.3390/life11070607
Ishii, T., Sakurai, T., Usami, H., and Uchida, K. (2005). Oxidative modification of proteasome: identification of an oxidation-sensitive subunit in 26 S proteasome. Biochemistry 44, 13893–13901. doi:10.1021/bi051336u
Ising, C., Venegas, C., Zhang, S., Scheiblich, H., Schmidt, S. V., Vieira-Saecker, A., et al. (2019). NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673. doi:10.1038/s41586-019-1769-z
Itakura, E., Zavodszky, E., Shao, S., Wohlever, M. l., Keenan, R. j., and Hegde, R. s. (2016). Ubiquilins chaperone and triage mitochondrial membrane proteins for degradation. Mol. Cell. 63, 21–33. doi:10.1016/j.molcel.2016.05.020
Iwata, A., Christianson, J. C., Bucci, M., Ellerby, L. M., Nukina, N., Forno, L. S., et al. (2005). Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc. Natl. Acad. Sci. U. S. A. 102, 13135–13140. doi:10.1073/pnas.0505801102
Jana, N. R., Zemskov, E. A., Wang, G., and Nukina, N. (2001). Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum. Mol. Genet. 10, 1049–1059. doi:10.1093/hmg/10.10.1049
Jariel-Encontre, I., Bossis, G., and Piechaczyk, M. (2008). Ubiquitin-independent degradation of proteins by the proteasome. Biochimica Biophysica Acta (BBA) - Rev. Cancer 1786, 153–177. doi:10.1016/j.bbcan.2008.05.004
Jeon, J., Kim, W., Jang, J., Isacson, O., and Seo, H. (2016). Gene therapy by proteasome activator, PA28γ, improves motor coordination and proteasome function in Huntington’s disease YAC128 mice. Neuroscience 324, 20–28. doi:10.1016/j.neuroscience.2016.02.054
Johnson, J. O., Mandrioli, J., Benatar, M., Abramzon, Y., Van Deerlin, V. M., Trojanowski, J. Q., et al. (2010). Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864. doi:10.1016/j.neuron.2010.11.036
Johnston-Carey, H. K., Pomatto, L. C., and Davies, K. J. (2015). The Immunoproteasome in oxidative stress, aging, and disease. Crit. Rev. Biochem. Mol. Biol. 51, 268–281. doi:10.3109/10409238.2016.1172554
Juenemann, K., Jansen, A. H. P., Van Riel, L., Merkx, R., Mulder, M. P. C., An, H., et al. (2018). Dynamic recruitment of ubiquitin to mutant huntingtin inclusion bodies. Sci. Rep. 8, 1405. doi:10.1038/s41598-018-19538-0
Juenemann, K., Schipper-Krom, S., Wiemhoefer, A., Kloss, A., Sanz Sanz, A., and Reits, E. a.J. (2013). Expanded polyglutamine-containing N-terminal huntingtin fragments are entirely degraded by mammalian proteasomes. J. Biol. Chem. 288, 27068–27084. doi:10.1074/jbc.M113.486076
Kamińska, J., Tylicka, M., Sutkowska, K., Gacuta, K. M., Sawicka, M. M., Kowalewska, E., et al. (2024). The preliminary study suggests an association between NF-ĸB pathway activation and increased plasma 20S proteasome activity in intracranial aneurysm patients. Sci. Rep. 14, 3941. doi:10.1038/s41598-024-54692-8
Kandasamy, G., and Andréasson, C. (2018). Hsp70–Hsp110 chaperones deliver ubiquitin-dependent and -independent substrates to the 26S proteasome for proteolysis in yeast. J. Cell. Sci. 131, jcs210948. doi:10.1242/jcs.210948
Kapeta, S., Chondrogianni, N., and Gonos, E. S. (2010). Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts. J. Biol. Chem. 285, 8171–8184. doi:10.1074/jbc.M109.031575
Kazee, A. M., and Han, L. Y. (1995). Cortical Lewy bodies in Alzheimer's disease. Arch. Pathol. Lab. Med. 119, 448–453.
Keck, S., Nitsch, R., Grune, T., and Ullrich, O. (2003). Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer's disease. J. Neurochem. 85, 115–122. doi:10.1046/j.1471-4159.2003.01642.x
Keller, J. N., Hanni, K. B., and Markesbery, W. R. (2000a). Impaired proteasome function in Alzheimer's disease. J. Neurochem. 75, 436–439. doi:10.1046/j.1471-4159.2000.0750436.x
Keller, J. N., Huang, F. F., and Markesbery, W. R. (2000b). Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience 98, 149–156. doi:10.1016/s0306-4522(00)00067-1
Kelmer Sacramento, E., Kirkpatrick, J. M., Mazzetto, M., Baumgart, M., Bartolome, A., Di Sanzo, S., et al. (2020). Reduced proteasome activity in the aging brain results in ribosome stoichiometry loss and aggregation. Mol. Syst. Biol. 16, e9596. doi:10.15252/msb.20209596
Kisselev, A. F., Akopian, T. N., Woo, K. M., and Goldberg, A. L. (1999). The sizes of peptides generated from protein by mammalian 26 and 20 S proteasomes. Implications for understanding the degradative mechanism and antigen presentation. J. Biol. Chem. 274, 3363–3371. doi:10.1074/jbc.274.6.3363
Kisselev, A. F., Kaganovich, D., and Goldberg, A. L. (2002). Binding of hydrophobic peptides to several non-catalytic sites promotes peptide hydrolysis by all active sites of 20 S proteasomes: evidence for peptide-induced channel opening in the α-rings. J. Biol. Chem. 277, 22260–22270. doi:10.1074/jbc.M112360200
Knowlton, J. R., Johnston, S. C., Whitby, F. G., Realini, C., Zhang, Z., Rechsteiner, M., et al. (1997). Structure of the proteasome activator REGalpha (PA28alpha). Nature 390, 639–643. doi:10.1038/37670
Koren, I., Timms, R. T., Kula, T., Xu, Q., Li, M. Z., and Elledge, S. J. (2018). The eukaryotic proteome is shaped by E3 ubiquitin ligases targeting C-terminal degrons. Cell. 173, 1622–1635. doi:10.1016/j.cell.2018.04.028
Kors, S., Geijtenbeek, K., Reits, E., and Schipper-Krom, S. (2019). Regulation of proteasome activity by (Post-)transcriptional mechanisms. Front. Mol. Biosci. 6, 48. doi:10.3389/fmolb.2019.00048
Kriachkov, V. A., Gotmanova, N. N., Tashlitsky, V. N., and Bacheva, A. V. (2023). Brain-derived 11S regulator (PA28αβ) promotes proteasomal hydrolysis of elongated oligoglutamine-containing peptides. Int. J. Mol. Sci. 24, 13275. doi:10.3390/ijms241713275
Krzyzanowska, A., García-Consuegra, I., Pascual, C., Antequera, D., Ferrer, I., and Carro, E. (2015). Expression of regulatory proteins in choroid plexus changes in early stages of alzheimer disease. J. Neuropathology and Exp. Neurology 74, 359–369. doi:10.1097/NEN.0000000000000181
Kulichkova, V. A., Artamonova, T. O., Lyublinskaya, O. G., Khodorkovskii, M. A., Tomilin, A. N., and Tsimokha, A. S. (2017). Proteomic analysis of affinity-purified extracellular proteasomes reveals exclusively 20S complexes. Oncotarget 8, 102134–102149. doi:10.18632/oncotarget.22230
Lee, H. J., Bae, E. J., and Lee, S. J. (2014). Extracellular α--synuclein-a novel and crucial factor in Lewy body diseases. Nat. Rev. Neurol. 10, 92–98. doi:10.1038/nrneurol.2013.275
Lee, M., Liu, Y.-C., Chen, C., Lu, C.-H., Lu, S.-T., Huang, T.-N., et al. (2020). Ecm29-mediated proteasomal distribution modulates excitatory GABA responses in the developing brain. J. Cell. Biol. 219, e201903033. doi:10.1083/jcb.201903033
Lee, M. J., Lee, J. H., and Rubinsztein, D. C. (2013). Tau degradation: the ubiquitin–proteasome system versus the autophagy-lysosome system. Prog. Neurobiol. 105, 49–59. doi:10.1016/j.pneurobio.2013.03.001
Leggett, D. S., Hanna, J., Borodovsky, A., Crosas, B., Schmidt, M., Baker, R. T., et al. (2002). Multiple associated proteins regulate proteasome structure and function. Mol. Cell. 10, 495–507. doi:10.1016/s1097-2765(02)00638-x
Leister, H., Krause, F. F., Gil, B., Prus, R., Prus, I., Hellhund-Zingel, A., et al. (2024). Immunoproteasome deficiency results in age-dependent development of epilepsy. Brain Commun. 6, fcae017. doi:10.1093/braincomms/fcae017
Leng, F., and Edison, P. (2021). Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat. Rev. Neurol. 17, 157–172. doi:10.1038/s41582-020-00435-y
Lesne, J., Locard-Paulet, M., Parra, J., Zivković, D., Menneteau, T., Bousquet, M.-P., et al. (2020). Conformational maps of human 20S proteasomes reveal PA28-and immuno-dependent inter-ring crosstalks. Nat. Commun. 11, 6140. doi:10.1038/s41467-020-19934-z
Le Tallec, B., Barrault, M.-B., Courbeyrette, R., Guérois, R., Marsolier-Kergoat, M.-C., and Peyroche, A. (2007). 20S proteasome assembly is orchestrated by two distinct pairs of chaperones in yeast and in mammals. Mol. Cell. 27, 660–674. doi:10.1016/j.molcel.2007.06.025
Li, J., Powell, S. R., and Wang, X. (2011). Enhancement of proteasome function by PA28α overexpression protects against oxidative stress. FASEB J. 25, 883. doi:10.1096/fj.10-160895
Li, X., Thompson, D., Kumar, B., and Demartino, G. N. (2014). Molecular and cellular roles of PI31 (PSMF1) protein in regulation of proteasome function. J. Biol. Chem. 289, 17392–17405. doi:10.1074/jbc.M114.561183
Liepe, J., Marino, F., Sidney, J., Jeko, A., Bunting, D. E., Sette, A., et al. (2016). A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science 354, 354–358. doi:10.1126/science.aaf4384
Limanaqi, F., Biagioni, F., Gaglione, A., Busceti, C. L., and Fornai, F. (2019). A sentinel in the crosstalk between the nervous and immune system: the (Immuno)-Proteasome. Front. Immunol. 10, 628. doi:10.3389/fimmu.2019.00628
Liu, C. W., Corboy, M. J., Demartino, G. N., and Thomas, P. J. (2003). Endoproteolytic activity of the proteasome. Science 299, 408–411. doi:10.1126/science.1079293
Liu, K., Jones, S., Minis, A., Rodriguez, J., Molina, H., and Steller, H. (2019). PI31 is an adaptor protein for proteasome transport in axons and required for synaptic development. Dev. Cell. 50, 509–524. doi:10.1016/j.devcel.2019.06.009
Lopez Salon, M., Morelli, L., Castano, E. M., Soto, E. F., and Pasquini, J. M. (2000). Defective ubiquitination of cerebral proteins in Alzheimer's disease. J. Neurosci. Res. 62, 302–310. doi:10.1002/1097-4547(20001015)62:2<302::AID-JNR15>3.0.CO;2-L
Ma, C. P., Slaughter, C. A., and Demartino, G. N. (1992). Identification, purification, and characterization of a protein activator (PA28) of the 20 S proteasome (macropain). J. Biol. Chem. 267, 10515–10523. doi:10.1016/s0021-9258(19)50047-3
Ma, Q., Xin, J., Peng, Q., Li, N., Sun, S., Hou, H., et al. (2023). UBQLN2 and HSP70 participate in Parkin-mediated mitophagy by facilitating outer mitochondrial membrane rupture. EMBO Rep. 24, e55859. doi:10.15252/embr.202255859
Machiya, Y., Hara, S., Arawaka, S., Fukushima, S., Sato, H., Sakamoto, M., et al. (2010). Phosphorylated alpha-synuclein at Ser-129 is targeted to the proteasome pathway in a ubiquitin-independent manner. J. Biol. Chem. 285, 40732–40744. doi:10.1074/jbc.M110.141952
Makaros, Y., Raiff, A., Timms, R. T., Wagh, A. R., Gueta, M. I., Bekturova, A., et al. (2023). Ubiquitin-independent proteasomal degradation driven by C-degron pathways. Mol. Cell. 83, 1921–1935.e7. doi:10.1016/j.molcel.2023.04.023
Malek, N., Gladysz, R., Stelmach, N., and Drag, M. (2024). Targeting microglial immunoproteasome: a novel approach in neuroinflammatory-related disorders. ACS Chem. Neurosci. 15, 2532–2544. doi:10.1021/acschemneuro.4c00099
Manfredonia, A. J., and Kraut, D. A. (2022). The 26S proteasome switches between ATP-dependent and -independent mechanisms in response to substrate ubiquitination. Biomolecules 12, 750. doi:10.3390/biom12060750
Martin, D. D., Ladha, S., Ehrnhoefer, D. E., and Hayden, M. R. (2015). Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 38, 26–35. doi:10.1016/j.tins.2014.09.003
Mccutchen-Maloney, S. L., Matsuda, K., Shimbara, N., Binns, D. D., Tanaka, K., Slaughter, C. A., et al. (2000). cDNA cloning, expression, and functional characterization of PI31, a proline-rich inhibitor of the proteasome. J. Biol. Chem. 275, 18557–18565. doi:10.1074/jbc.M001697200
Mcnaught, K. S. P., Belizaire, R., Jenner, P., Olanow, C. W., and Isacson, O. (2002). Selective loss of 20S proteasome alpha-subunits in the substantia nigra pars compacta in Parkinson's disease. Neurosci. Lett. 326, 155–158. doi:10.1016/s0304-3940(02)00296-3
Mcnaught, K. S. P., and Jenner, P. (2001). Proteasomal function is impaired in substantia nigra in Parkinson's disease. Neurosci. Lett. 297, 191–194. doi:10.1016/S0304-3940(00)01701-8
Mcnaught, K. S. P., Jnobaptiste, R., Jackson, T., and Jengelley, T.-A. (2010). The pattern of neuronal loss and survival may reflect differential expression of proteasome activators in Parkinson's disease. Synapse 64, 241–250. doi:10.1002/syn.20719
Mee Hayes, E., Sirvio, L., and Ye, Y. (2022). A potential mechanism for targeting aggregates with proteasomes and disaggregases in liquid droplets. Front. Aging Neurosci. 14, 854380. doi:10.3389/fnagi.2022.854380
Miller, J., Arrasate, M., Brooks, E., Libeu, C. P., Legleiter, J., Hatters, D., et al. (2011). Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat. Chem. Biol. 7, 925–934. doi:10.1038/nchembio.694
Min, S. W., Cho, S. H., Zhou, Y., Schroeder, S., Haroutunian, V., Seeley, W. W., et al. (2010). Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966. doi:10.1016/j.neuron.2010.08.044
Minami, Y., Kawasaki, H., Minami, M., Tanahashi, N., Tanaka, K., and Yahara, I. (2000). A critical role for the proteasome activator PA28 in the Hsp90-dependent protein refolding. J. Biol. Chem. 275, 9055–9061. doi:10.1074/jbc.275.12.9055
Minis, A., Rodriguez, J. A., Levin, A., Liu, K., Govek, E.-E., Hatten, M. E., et al. (2019). The proteasome regulator PI31 is required for protein homeostasis, synapse maintenance, and neuronal survival in mice. Proc. Natl. Acad. Sci. 116, 24639–24650. doi:10.1073/pnas.1911921116
Mishto, M., Bellavista, E., Santoro, A., Stolzing, A., Ligorio, C., Nacmias, B., et al. (2006). Immunoproteasome and LMP2 polymorphism in aged and Alzheimer's disease brains. Neurobiol. Aging 27, 54–66. doi:10.1016/j.neurobiolaging.2004.12.004
Moorthy, A. K., Savinova, O. V., Ho, J. Q., Wang, V. Y., Vu, D., and Ghosh, G. (2006). The 20S proteasome processes NF-kappaB1 p105 into p50 in a translation-independent manner. EMBO J. 25, 1945–1956. doi:10.1038/sj.emboj.7601081
Moscovitz, O., Ben-Nissan, G., Fainer, I., Pollack, D., Mizrachi, L., and Sharon, M. (2015). The Parkinson’s-associated protein DJ-1 regulates the 20S proteasome. Nat. Commun. 6, 6609. doi:10.1038/ncomms7609
Moscovitz, O., Tsvetkov, P., Hazan, N., Michaelevski, I., Keisar, H., Ben-Nissan, G., et al. (2012). A mutually inhibitory feedback loop between the 20S proteasome and its regulator, NQO1. Mol. Cell. 47, 76–86. doi:10.1016/j.molcel.2012.05.049
Mueller, O., Anlasik, T., Wiedemann, J., Thomassen, J., Wohlschlaeger, J., Hagel, V., et al. (2012). Circulating extracellular proteasome in the cerebrospinal fluid: a study on concentration and proteolytic activity. J. Mol. Neurosci. 46, 509–515. doi:10.1007/s12031-011-9631-2
Myeku, N., Clelland, C. L., Emrani, S., Kukushkin, N. V., Yu, W. H., Goldberg, A. L., et al. (2016). Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 22, 46–53. doi:10.1038/nm.4011
Myers, N., Olender, T., Savidor, A., Levin, Y., Reuven, N., and Shaul, Y. (2018). The disordered landscape of the 20S proteasome substrates reveals tight association with phase separated granules. PROTEOMICS 18, doi:10.1002/pmic.201800076
Nago, N., Murata, S., Tanaka, K., and Tanahashi, N. (2024). Changes in brain proteasome dynamics associated with aging. Genes cells. 29, 438–445. doi:10.1111/gtc.13113
Nakajima, T., Takauchi, S., Ohara, K., Kokai, M., Nishii, R., Maeda, S., et al. (2005). Alpha-synuclein-positive structures induced in leupeptin-infused rats. Brain Res. 1040, 73–80. doi:10.1016/j.brainres.2005.01.099
Noda, C., Tanahashi, N., Shimbara, N., Hendil, K. B., and Tanaka, K. (2000). Tissue distribution of constitutive proteasomes, immunoproteasomes, and PA28 in rats. Biochem. Biophysical Res. Commun. 277, 348–354. doi:10.1006/bbrc.2000.3676
Obara, Y., Imai, T., Sato, H., Takeda, Y., Kato, T., and Ishii, K. (2017). Midnolin is a novel regulator of parkin expression and is associated with Parkinson’s Disease. Sci. Rep. 7, 5885. doi:10.1038/s41598-017-05456-0
Obara, Y., and Ishii, K. (2018). Transcriptome analysis reveals that midnolin regulates mRNA expression levels of multiple Parkinson’s disease causative genes. Biol. Pharm. Bull. 41, 20–23. doi:10.1248/bpb.b17-00663
Obara, Y., Sato, H., Nakayama, T., Kato, T., and Ishii, K. (2019). Midnolin is a confirmed genetic risk factor for Parkinson’s disease. Ann. Clin. Transl. Neurology 6, 2205–2211. doi:10.1002/acn3.50914
Oddo, S., Billings, L., Kesslak, J. P., Cribbs, D. H., and Laferla, F. M. (2004). Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 43, 321–332. doi:10.1016/j.neuron.2004.07.003
Oh, S., Hong, H. S., Hwang, E., Sim, H. J., Lee, W., Shin, S. J., et al. (2005). Amyloid peptide attenuates the proteasome activity in neuronal cells. Mech. Ageing Dev. 126, 1292–1299. doi:10.1016/j.mad.2005.07.006
Olshina, M. A., Arkind, G., Kumar Deshmukh, F., Fainer, I., Taranavsky, M., Hayat, D., et al. (2020). Regulation of the 20S proteasome by a novel family of inhibitory proteins. Antioxidants and Redox Signal. 32, 636–655. doi:10.1089/ars.2019.7816
Opoku-Nsiah, K. A., De La Pena, A. H., Williams, S. K., Chopra, N., Sali, A., Lander, G. C., et al. (2022). The YΦ motif defines the structure-activity relationships of human 20S proteasome activators. Nat. Commun. 13, 1226. doi:10.1038/s41467-022-28864-x
Opoku-Nsiah, K. A., and Gestwicki, J. E. (2018). Aim for the core: suitability of the ubiquitin-independent 20S proteasome as a drug target in neurodegeneration. Transl. Res. 198, 48–57. doi:10.1016/j.trsl.2018.05.002
Orre, M., Kamphuis, W., Dooves, S., Kooijman, L., Chan, E. T., Kirk, C. J., et al. (2013). Reactive glia show increased immunoproteasome activity in Alzheimer's disease. Brain 136, 1415–1431. doi:10.1093/brain/awt083
Orre, M., Kamphuis, W., Osborn, L. M., Jansen, A. H. P., Kooijman, L., Bossers, K., et al. (2014). Isolation of glia from Alzheimer's mice reveals inflammation and dysfunction. Neurobiol. Aging 35, 2746–2760. doi:10.1016/j.neurobiolaging.2014.06.004
Ortega, J., Heymann, J. B., Kajava, A. V., Ustrell, V., Rechsteiner, M., and Steven, A. C. (2005). The axial channel of the 20S proteasome opens upon binding of the PA200 activator. J. Mol. Biol. 346, 1221–1227. doi:10.1016/j.jmb.2004.12.049
Ortega, Z., and Lucas, J. J. (2014). Ubiquitin-proteasome system involvement in Huntington's disease. Front. Mol. Neurosci. 7, 77. doi:10.3389/fnmol.2014.00077
Paradise, V., Sabu, M., Bafia, J., Sharif, N. A., Nguyen, C., Konrad-Vicario, K. D., et al. (2023). Dysregulation of neuroproteasomes by ApoE isoforms drives endogenous Tau aggregation. doi:10.1101/2022.11.29.518293
Park, J. E., Chaudhary, C. L., Bhattarai, D., and Kim, K. B. (2024). Brain-permeable immunoproteasome-targeting macrocyclic peptide epoxyketones for alzheimer's disease. J. Med. Chem. 67, 7146–7157. doi:10.1021/acs.jmedchem.3c02488
Patrick, G. N. (2006). Synapse formation and plasticity: recent insights from the perspective of the ubiquitin proteasome system. Curr. Opin. Neurobiol. 16, 90–94. doi:10.1016/j.conb.2006.01.007
Pepelnjak, M., Rogawski, R., Arkind, G., Leushkin, Y., Fainer, I., Ben-Nissan, G., et al. (2024). Systematic identification of 20S proteasome substrates. Mol. Syst. Biol. 20, 403–427. doi:10.1038/s44320-024-00015-y
Pickering, A. M., and Davies, K. J. A. (2012). “Chapter 6 - degradation of damaged proteins: the main function of the 20S proteasome,” in Progress in molecular biology and translational science. Editor T. Grune (Academic Press), 227–248.
Pickering, A. m., Koop, A. l., Teoh, C. y., Ermak, G., Grune, T., and Davies, K. j.A. (2010). The immunoproteasome, the 20S proteasome and the PA28αβ proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem. J. 432, 585–594. doi:10.1042/BJ20100878
Pickering, A. M., Linder, R. A., Zhang, H., Forman, H. J., and Davies, K. J. A. (2012). Nrf2-dependent induction of proteasome and Pa28αβ regulator are required for adaptation to oxidative stress. J. Biol. Chem. 287, 10021–10031. doi:10.1074/jbc.M111.277145
Pickhardt, M., Gazova, Z., Von Bergen, M., Khlistunova, I., Wang, Y., Hascher, A., et al. (2005). Anthraquinones inhibit tau aggregation and dissolve Alzheimer's paired helical filaments in vitro and in cells. J. Biol. Chem. 280, 3628–3635. doi:10.1074/jbc.M410984200
Pintado, C., Gavilan, M. P., Gavilan, E., Garcia-Cuervo, L., Gutierrez, A., Vitorica, J., et al. (2012). Lipopolysaccharide-induced neuroinflammation leads to the accumulation of ubiquitinated proteins and increases susceptibility to neurodegeneration induced by proteasome inhibition in rat hippocampus. J. Neuroinflammation 9, 87. doi:10.1186/1742-2094-9-87
Poppek, D., Keck, S., Ermak, G., Jung, T., Stolzing, A., Ullrich, O., et al. (2006). Phosphorylation inhibits turnover of the tau protein by the proteasome: influence of RCAN1 and oxidative stress. Biochem. J. 400, 511–520. doi:10.1042/BJ20060463
Pratt, G., and Rechsteiner, M. (2008). Proteasomes cleave at multiple sites within polyglutamine tracts: activation by PA28gamma(K188E). J. Biol. Chem. 283, 12919–12925. doi:10.1074/jbc.M709347200
Rabl, J., Smith, D. M., Yu, Y., Chang, S.-C., Goldberg, A. L., and Cheng, Y. (2008). Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol. Cell. 30, 360–368. doi:10.1016/j.molcel.2008.03.004
Ramachandran, K. V., Fu, J. M., Schaffer, T. B., Na, C. H., Delannoy, M., and Margolis, S. S. (2018). Activity-dependent degradation of the nascentome by the neuronal membrane proteasome. Mol. Cell. 71, 169–177. doi:10.1016/j.molcel.2018.06.013
Ramachandran, K. V., and Margolis, S. S. (2017). A mammalian nervous-system-specific plasma membrane proteasome complex that modulates neuronal function. Nat. Struct. Mol. Biol. 24, 419–430. doi:10.1038/nsmb.3389
Rankin, C. A., Sun, Q., and Gamblin, T. C. (2007). Tau phosphorylation by GSK-3beta promotes tangle-like filament morphology. Mol. Neurodegener. 2, 12. doi:10.1186/1750-1326-2-12
Rawat, P., Sehar, U., Bisht, J., Selman, A., Culberson, J., and Reddy, P. H. (2022). Phosphorylated tau in alzheimer's disease and other tauopathies. Int. J. Mol. Sci. 23, 12841. doi:10.3390/ijms232112841
Raynes, R., Pomatto, L. C. D., and Davies, K. J. A. (2016). Degradation of oxidized proteins by the proteasome: distinguishing between the 20S, 26S, and immunoproteasome proteolytic pathways. Mol. Aspects Med. 50, 41–55. doi:10.1016/j.mam.2016.05.001
Reinheckel, T., Sitte, N., Ullrich, O., Kuckelkorn, U., Davies, K. J. A., and Grune, T. (1998). Comparative resistance of the 20S and 26S proteasome to oxidative stress. Biochem. J. 335, 637–642. doi:10.1042/bj3350637
Riguet, N., Mahul-Mellier, A.-L., Maharjan, N., Burtscher, J., Croisier, M., Knott, G., et al. (2021). Nuclear and cytoplasmic huntingtin inclusions exhibit distinct biochemical composition, interactome and ultrastructural properties. Nat. Commun. 12, 6579. doi:10.1038/s41467-021-26684-z
Rivett, A. J., Bose, S., Brooks, P., and Broadfoot, K. I. (2001). Regulation of proteasome complexes by gamma-interferon and phosphorylation. Biochimie 83, 363–366. doi:10.1016/s0300-9084(01)01249-4
Rock, K. L., Farfan-Arribas, D. J., Colbert, J. D., and Goldberg, A. L. (2014). Re-examining class-I presentation and the DRiP hypothesis. Trends Immunol. 35, 144–152. doi:10.1016/j.it.2014.01.002
Roda, A. R., Serra-Mir, G., Montoliu-Gaya, L., Tiessler, L., and Villegas, S. (2022). Amyloid-beta peptide and tau protein crosstalk in Alzheimer's disease. Neural Regen. Res. 17, 1666–1674. doi:10.4103/1673-5374.332127
Rosenberg-Hasson, Y., Bercovich, Z., Ciechanover, A., and Kahana, C. (1989). Degradation of ornithine decarboxylase in mammalian cells is ATP dependent but ubiquitin independent. Eur. J. Biochem. 185, 469–474. doi:10.1111/j.1432-1033.1989.tb15138.x
Rousseau, E., Kojima, R., Hoffner, G., Djian, P., and Bertolotti, A. (2009). Misfolding of proteins with a polyglutamine expansion is facilitated by proteasomal chaperones. J. Biol. Chem. 284, 1917–1929. doi:10.1074/jbc.M806256200
Sadahiro, Y., Nishimura, S., Hitora, Y., and Tsukamoto, S. (2024). Syrosingopine enhances 20S proteasome activity and degradation of α-synuclein. J. Nat. Prod. 87, 554–559. doi:10.1021/acs.jnatprod.3c00661
Sadre-Bazzaz, K., Whitby, F. G., Robinson, H., Formosa, T., and Hill, C. P. (2010). Structure of a Blm10 complex reveals common mechanisms for proteasome binding and gate opening. Mol. Cell. 37, 728–735. doi:10.1016/j.molcel.2010.02.002
Saez, I., and Vilchez, D. (2014). The mechanistic links between proteasome activity, aging and age-related diseases. Curr. Genomics 15, 38–51. doi:10.2174/138920291501140306113344
Sahu, I., Mali, S. M., Sulkshane, P., Xu, C., Rozenberg, A., Morag, R., et al. (2021). The 20S as a stand-alone proteasome in cells can degrade the ubiquitin tag. Nat. Commun. 12, 6173. doi:10.1038/s41467-021-26427-0
Sanchez-Lanzas, R., and Castano, J. G. (2014). Proteins directly interacting with mammalian 20S proteasomal subunits and ubiquitin-independent proteasomal degradation. Biomolecules 4, 1140–1154. doi:10.3390/biom4041140
Sap, K. A., Geijtenbeek, K. W., Schipper-Krom, S., Guler, A. T., and Reits, E. A. (2023). Ubiquitin-modifying enzymes in Huntington's disease. Front. Mol. Biosci. 10, 1107323. doi:10.3389/fmolb.2023.1107323
Saudou, F., and Humbert, S. (2016). The biology of huntingtin. Neuron 89, 910–926. doi:10.1016/j.neuron.2016.02.003
Schildknecht, S., Gerding, H. R., Karreman, C., Drescher, M., Lashuel, H. A., Outeiro, T. F., et al. (2013). Oxidative and nitrative alpha-synuclein modifications and proteostatic stress: implications for disease mechanisms and interventions in synucleinopathies. J. Neurochem. 125, 491–511. doi:10.1111/jnc.12226
Schipper-Krom, S., Juenemann, K., Jansen, A. H., Wiemhoefer, A., Van Den Nieuwendijk, R., Smith, D. L., et al. (2014). Dynamic recruitment of active proteasomes into polyglutamine initiated inclusion bodies. FEBS Lett. 588, 151–159. doi:10.1016/j.febslet.2013.11.023
Schmidt, M., Haas, W., Crosas, B., Santamaria, P. G., Gygi, S. P., Walz, T., et al. (2005). The HEAT repeat protein Blm10 regulates the yeast proteasome by capping the core particle. Nat. Struct. and Mol. Biol. 12, 294–303. doi:10.1038/nsmb914
Schmidt, M. F., Gan, Z. Y., Komander, D., and Dewson, G. (2021). Ubiquitin signalling in neurodegeneration: mechanisms and therapeutic opportunities. Cell. Death and Differ. 28, 570–590. doi:10.1038/s41418-020-00706-7
Seo, H., Sonntag, K.-C., Kim, W., Cattaneo, E., and Isacson, O. (2007). Proteasome activator enhances survival of huntington's disease neuronal model cells. PLoS ONE 2, e238. doi:10.1371/journal.pone.0000238
Shang, F., and Taylor, A. (2011). Ubiquitin-proteasome pathway and cellular responses to oxidative stress. Free Radic. Biol. Med. 51, 5–16. doi:10.1016/j.freeradbiomed.2011.03.031
Sheaff, R. J., Singer, J. D., Swanger, J., Smitherman, M., Roberts, J. M., and Clurman, B. E. (2000). Proteasomal turnover of p21Cip1 does not require p21Cip1 ubiquitination. Mol. Cell. 5, 403–410. doi:10.1016/S1097-2765(00)80435-9
Sherva, R., Baldwin, C. T., Inzelberg, R., Vardarajan, B., Cupples, L. A., Lunetta, K., et al. (2011). Identification of novel candidate genes for Alzheimer's disease by autozygosity mapping using genome wide SNP data. J. Alzheimer's Dis. JAD 23, 349–359. doi:10.3233/JAD-2010-100714
Shringarpure, R., Grune, T., and Davies, K. J. (2001). Protein oxidation and 20S proteasome-dependent proteolysis in mammalian cells. Cell. Mol. life Sci. CMLS 58, 1442–1450. doi:10.1007/PL00000787
Smith, D. M., Chang, S.-C., Park, S., Finley, D., Cheng, Y., and Goldberg, A. L. (2007). Docking of the proteasomal ATPases' carboxyl termini in the 20S proteasome's alpha ring opens the gate for substrate entry. Mol. Cell. 27, 731–744. doi:10.1016/j.molcel.2007.06.033
Soh, W. T., Roetschke, H. P., Cormican, J. A., Teo, B. F., Chiam, N. C., Raabe, M., et al. (2024). Protein degradation by human 20S proteasomes elucidates the interplay between peptide hydrolysis and splicing. Nat. Commun. 15, 1147. doi:10.1038/s41467-024-45339-3
Solomon, H., Brauning, B., Fainer, I., Ben-Nissan, G., Rabani, S., Goldfinger, N., et al. (2017). Post-translational regulation of p53 function through 20S proteasome-mediated cleavage. Cell. Death Differ. 24, 2187–2198. doi:10.1038/cdd.2017.139
Staerz, S. D., Anamoah, C., and Tepe, J. J. (2024). 20S proteasome enhancers prevent cytotoxic tubulin polymerization-promoting protein induced α-synuclein aggregation. iScience 27, 110166. doi:10.1016/j.isci.2024.110166
Staerz, S. D., Jones, C. L., and Tepe, J. J. (2022). Design, synthesis, and biological evaluation of potent 20S proteasome activators for the potential treatment of α-synucleinopathies. J. Med. Chem. 65, 6631–6642. doi:10.1021/acs.jmedchem.1c02158
Stefanis, L., Emmanouilidou, E., Pantazopoulou, M., Kirik, D., Vekrellis, K., and Tofaris, G. K. (2019). How is alpha-synuclein cleared from the cell? J. Neurochem. 150, 577–590. doi:10.1111/jnc.14704
Suskiewicz, M. J., Sussman, J. L., Silman, I., and Shaul, Y. (2011). Context-dependent resistance to proteolysis of intrinsically disordered proteins. Protein Sci. 20, 1285–1297. doi:10.1002/pro.657
Taguchi, Y. V., Gorenberg, E. L., Nagy, M., Thrasher, D., Fenton, W. A., Volpicelli-Daley, L., et al. (2019). Hsp110 mitigates α-synuclein pathology in vivo. Proc. Natl. Acad. Sci. 116, 24310–24316. doi:10.1073/pnas.1903268116
Tai, H. C., Serrano-Pozo, A., Hashimoto, T., Frosch, M. P., Spires-Jones, T. L., and Hyman, B. T. (2012). The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 181, 1426–1435. doi:10.1016/j.ajpath.2012.06.033
Takahashi, T., Kikuchi, S., Katada, S., Nagai, Y., Nishizawa, M., and Onodera, O. (2008). Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum. Mol. Genet. 17, 345–356. doi:10.1093/hmg/ddm311
Tanahashi, N., Murakami, Y., Minami, Y., Shimbara, N., Hendil, K. B., and Tanaka, K. (2000). Hybrid Proteasomes: induction by interferon-γ and contribution to atp-dependent proteolysis. J. Biol. Chem. 275, 14336–14345. doi:10.1074/jbc.275.19.14336
Thibaudeau, T. A., Anderson, R. T., and Smith, D. M. (2018). A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 9, 1097. doi:10.1038/s41467-018-03509-0
Thomas, T. A., and Smith, D. M. (2022). Proteasome activator 28γ (PA28γ) allosterically activates trypsin-like proteolysis by binding to the α-ring of the 20S proteasome. J. Biol. Chem. 298, 102140. doi:10.1016/j.jbc.2022.102140
Thompson, A. G., Gray, E., Mager, I., Thezenas, M. L., Charles, P. D., Talbot, K., et al. (2020). CSF extracellular vesicle proteomics demonstrates altered protein homeostasis in amyotrophic lateral sclerosis. Clin. Proteomics 17, 31. doi:10.1186/s12014-020-09294-7
Thompson, L. M., Aiken, C. T., Kaltenbach, L. S., Agrawal, N., Illes, K., Khoshnan, A., et al. (2009). IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell. Biol. 187, 1083–1099. doi:10.1083/jcb.200909067
Tofaris, G. K., Layfield, R., and Spillantini, M. G. (2001). alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 509, 22–26. doi:10.1016/S0014-5793(01)03115-5
Tonoki, A., Kuranaga, E., Tomioka, T., Hamazaki, J., Murata, S., Tanaka, K., et al. (2009). Genetic evidence linking age-dependent attenuation of the 26S proteasome with the aging process. Mol. Cell. Biol. 29, 1095–1106. doi:10.1128/MCB.01227-08
Toste Rêgo, A., and Da Fonseca, P. C. A. (2019). Characterization of fully recombinant human 20S and 20S-PA200 proteasome complexes. Mol. Cell. 76, 138–147. doi:10.1016/j.molcel.2019.07.014
Touitou, R., Richardson, J., Bose, S., Nakanishi, M., Rivett, J., and Allday, M. J. (2001). A degradation signal located in the C-terminus of p21WAF1/CIP1 is a binding site for the C8 alpha-subunit of the 20S proteasome. EMBO J. 20, 2367–2375. doi:10.1093/emboj/20.10.2367
Tsakiri, E. N., Sykiotis, G. P., Papassideri, I. S., Gorgoulis, V. G., Bohmann, D., and Trougakos, I. P. (2013). Differential regulation of proteasome functionality in reproductive vs. somatic tissues of Drosophila during aging or oxidative stress. FASEB J. 27, 2407–2420. doi:10.1096/fj.12-221408
Tseng, B. P., Green, K. N., Chan, J. L., Blurton-Jones, M., and Laferla, F. M. (2008). Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol. Aging 29, 1607–1618. doi:10.1016/j.neurobiolaging.2007.04.014
Tsimokha, A. S., Artamonova, T. O., Diakonov, E. E., Khodorkovskii, M. A., and Tomilin, A. N. (2020). Post-translational modifications of extracellular proteasome. Molecules 25, 3504. doi:10.3390/molecules25153504
Tsvetkov, P., Adamovich, Y., Elliott, E., and Shaul, Y. (2011). E3 ligase STUB1/CHIP regulates NAD(P)H:quinone oxidoreductase 1 (NQO1) accumulation in aged brain, a process impaired in certain Alzheimer disease patients. J. Biol. Chem. 286, 8839–8845. doi:10.1074/jbc.M110.193276
Tsvetkov, P., Myers, N., Adler, J., and Shaul, Y. (2020). Degradation of intrinsically disordered proteins by the NADH 26S proteasome. Biomolecules 10, 1642. doi:10.3390/biom10121642
Tsvetkov, P., Myers, N., Eliav, R., Adamovich, Y., Hagai, T., Adler, J., et al. (2014). NADH binds and stabilizes the 26S proteasomes independent of ATP. J. Biol. Chem. 289, 11272–11281. doi:10.1074/jbc.M113.537175
Tsvetkov, P., Reuven, N., and Shaul, Y. (2009). The nanny model for IDPs. Nat. Chem. Biol. 5, 778–781. doi:10.1038/nchembio.233
Tundo, G. R., Cascio, P., Milardi, D., Santoro, A. M., Graziani, G., Lacal, P. M., et al. (2023). Targeting immunoproteasome in neurodegeneration: a glance to the future. Pharmacol. Ther. 241, 108329. doi:10.1016/j.pharmthera.2022.108329
Türker, F., Bharadwaj, R. A., Kleinman, J. E., Weinberger, D. R., Hyde, T. M., White, C. J., et al. (2023). Orthogonal approaches required to measure proteasome composition and activity in mammalian brain tissue. J. Biol. Chem. 299, 104811. doi:10.1016/j.jbc.2023.104811
Türker, F., Brennan, A., and Margolis, S. S. (2024). Neuronal membrane proteasome-derived peptides modulate NMDAR-dependent neuronal signaling to promote changes in gene expression. Mol. Biol. Cell. 35, ar6. doi:10.1091/mbc.E23-06-0218
Türker, F., Cook, E. K., and Margolis, S. S. (2021). The proteasome and its role in the nervous system. Cell. Chem. Biol. 28, 903–917. doi:10.1016/j.chembiol.2021.04.003
Turturici, G., Sconzo, G., and Geraci, F. (2011). Hsp70 and its molecular role in nervous system diseases. Biochem. Res. Int. 2011, 618127. doi:10.1155/2011/618127
Ugras, S., Daniels, M. J., Fazelinia, H., Gould, N. S., Yocum, A. K., Luk, K. C., et al. (2018). Induction of the immunoproteasome subunit Lmp7 links proteostasis and immunity in α-synuclein aggregation disorders. EBioMedicine 31, 307–319. doi:10.1016/j.ebiom.2018.05.007
Ukmar-Godec, T., Fang, P., Ibáñez De Opakua, A., Henneberg, F., Godec, A., Pan, K. T., et al. (2020). Proteasomal degradation of the intrinsically disordered protein tau at single-residue resolution. Sci. Adv. 6, eaba3916. doi:10.1126/sciadv.aba3916
Um, J. W., Im, E., Lee, H. J., Min, B., Yoo, L., Yoo, J., et al. (2010). Parkin directly modulates 26S proteasome activity. J. Neurosci. 30, 11805–11814. doi:10.1523/JNEUROSCI.2862-09.2010
Unno, M., Mizushima, T., Morimoto, Y., Tomisugi, Y., Tanaka, K., Yasuoka, N., et al. (2002). The structure of the mammalian 20S proteasome at 2.75 Å resolution. Structure 10, 609–618. doi:10.1016/S0969-2126(02)00748-7
Usenovic, M., Niroomand, S., Drolet, R. E., Yao, L., Gaspar, R. C., Hatcher, N. G., et al. (2015). Internalized tau oligomers cause neurodegeneration by inducing accumulation of pathogenic tau in human neurons derived from induced pluripotent stem cells. J. Neurosci. 35, 14234–14250. doi:10.1523/JNEUROSCI.1523-15.2015
Ustrell, V., Hoffman, L., Pratt, G., and Rechsteiner, M. (2002). PA200, a nuclear proteasome activator involved in DNA repair. EMBO J. 21, 3516–3525. doi:10.1093/emboj/cdf333
Vabulas, R. M., and Hartl, F. U. (2005). Protein synthesis upon acute nutrient restriction relies on proteasome function. Science 310, 1960–1963. doi:10.1126/science.1121925
Venkatraman, P., Wetzel, R., Tanaka, M., Nukina, N., and Goldberg, A. L. (2004). Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell. 14, 95–104. doi:10.1016/s1097-2765(04)00151-0
Verplank, J. J. S., Gawron, J. M., Silvestri, N. J., Wrabetz, L., and Feltri, M. L. (2024). Knockout of PA200 improves proteasomal degradation and myelination in a proteotoxic neuropathy. Life Sci. Alliance 7, e202302349. doi:10.26508/lsa.202302349
Villalón Landeros, E., Kho, S. C., Church, T. R., Brennan, A., Türker, F., Delannoy, M., et al. (2024). The nociceptive activity of peripheral sensory neurons is modulated by the neuronal membrane proteasome. Cell. Rep. 43, 114058. doi:10.1016/j.celrep.2024.114058
Vinokurov, A. Y., Palalov, A. A., Kritskaya, K. A., Demyanenko, S. V., Garbuz, D. G., Evgen'ev, M. B., et al. (2024). Cell-permeable HSP70 protects neurons and astrocytes against cell death in the rotenone-induced and familial models of Parkinson's disease. Mol. Neurobiol. 61, 7785–7795. doi:10.1007/s12035-024-04077-9
Vogiatzi, T., Xilouri, M., Vekrellis, K., and Stefanis, L. (2008). Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 283, 23542–23556. doi:10.1074/jbc.M801992200
Waelter, S., Boeddrich, A., Lurz, R., Scherzinger, E., Lueder, G., Lehrach, H., et al. (2001). Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol. Biol. Cell. 12, 1393–1407. doi:10.1091/mbc.12.5.1393
Wang, H., Lim, P. J., Yin, C., Rieckher, M., Vogel, B. E., and Monteiro, M. J. (2006). Suppression of polyglutamine-induced toxicity in cell and animal models of Huntington's disease by ubiquilin. Hum. Mol. Genet. 15, 1025–1041. doi:10.1093/hmg/ddl017
Wang, J., Kjellgren, A., and Demartino, G. N. (2024). Differential interactions of the proteasome inhibitor PI31 with constitutive and immuno-20S proteasomes. Biochemistry 63, 1000–1015. doi:10.1021/acs.biochem.3c00707
Wang, S., Kojima, K., Mobley, J. A., and West, A. B. (2019). Proteomic analysis of urinary extracellular vesicles reveal biomarkers for neurologic disease. EBioMedicine 45, 351–361. doi:10.1016/j.ebiom.2019.06.021
Wang, X., Chemmama, I. E., Yu, C., Huszagh, A., Xu, Y., Viner, R., et al. (2017). The proteasome-interacting Ecm29 protein disassembles the 26S proteasome in response to oxidative stress. J. Biol. Chem. 292, 16310–16320. doi:10.1074/jbc.M117.803619
Watanabe, Y., Taguchi, K., and Tanaka, M. (2020). Ubiquitin, autophagy and neurodegenerative diseases. Cells 9, 2022. doi:10.3390/cells9092022
Webb, J. L., Ravikumar, B., Atkins, J., Skepper, J. N., and Rubinsztein, D. C. (2003). Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278, 25009–25013. doi:10.1074/jbc.M300227200
Wenzel, T., and Baumeister, W. (1995). Conformational constraints in protein degradation by the 20S proteasome. Nat. Struct. Biol. 2, 199–204. doi:10.1038/nsb0395-199
Wiggins, C. M., Tsvetkov, P., Johnson, M., Joyce, C. L., Lamb, C. A., Bryant, N. J., et al. (2011). BIM(EL), an intrinsically disordered protein, is degraded by 20S proteasomes in the absence of poly-ubiquitylation. J. Cell. Sci. 124, 969–977. doi:10.1242/jcs.058438
Wilmington, S. R., and Matouschek, A. (2016). An inducible system for rapid degradation of specific cellular proteins using proteasome adaptors. PLoS One 11, e0152679. doi:10.1371/journal.pone.0152679
Wilson, A. C., Dugger, B. N., Dickson, D. W., and Wang, D. S. (2011). TDP-43 in aging and Alzheimer's disease - a review. Int. J. Clin. Exp. Pathol. 4, 147–155.
Winkler, L. L., Hwang, J., and Kalejta, R. F. (2013). Ubiquitin-independent proteasomal degradation of tumor suppressors by human cytomegalovirus pp71 requires the 19S regulatory particle. J. Virol. 87, 4665–4671. doi:10.1128/JVI.03301-12
Winter, M. B., La Greca, F., Arastu-Kapur, S., Caiazza, F., Cimermancic, P., Buchholz, T. J., et al. (2017). Immunoproteasome functions explained by divergence in cleavage specificity and regulation. Elife 6, e27364. doi:10.7554/eLife.27364
Yasuda, S., Tsuchiya, H., Kaiho, A., Guo, Q., Ikeuchi, K., Endo, A., et al. (2020). Stress- and ubiquitylation-dependent phase separation of the proteasome. Nature 578, 296–300. doi:10.1038/s41586-020-1982-9
Yeo, I. J., Lee, M. J., Baek, A., Miller, Z., Bhattarai, D., Baek, Y. M., et al. (2019). A dual inhibitor of the proteasome catalytic subunits LMP2 and Y attenuates disease progression in mouse models of Alzheimer's disease. Sci. Rep. 9, 18393. doi:10.1038/s41598-019-54846-z
Yersak, J. M., Montie, H. L., Chevalier-Larsen, E. S., Liu, Y., Huang, L., Rechsteiner, M., et al. (2017). The 11S proteasomal activator REGγ impacts polyglutamine-expanded androgen receptor aggregation and motor neuron viability through distinct mechanisms. Front. Mol. Neurosci. 10, 159. doi:10.3389/fnmol.2017.00159
Yi, J. J., and Ehlers, M. D. (2007). Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacol. Rev. 59, 14–39. doi:10.1124/pr.59.1.4
Yu, Y., Smith, D. M., Kim, H. M., Rodriguez, V., Goldberg, A. L., and Cheng, Y. (2010). Interactions of PAN's C-termini with archaeal 20S proteasome and implications for the eukaryotic proteasome-ATPase interactions. EMBO J. 29, 692–702. doi:10.1038/emboj.2009.382
Yuhan, L., Khaleghi Ghadiri, M., and Gorji, A. (2024). Impact of NQO1 dysregulation in CNS disorders. J. Transl. Med. 22, 4. doi:10.1186/s12967-023-04802-3
Zaiss, D. M., Standera, S., Kloetzel, P. M., and Sijts, A. J. (2002). PI31 is a modulator of proteasome formation and antigen processing. Proc. Natl. Acad. Sci. U. S. A. 99, 14344–14349. doi:10.1073/pnas.212257299
Zbinden, A., Pérez-Berlanga, M., De Rossi, P., and Polymenidou, M. (2020). Phase separation and neurodegenerative diseases: a disturbance in the force. Dev. Cell. 55, 45–68. doi:10.1016/j.devcel.2020.09.014
Zerfas, B. L., Maresh, M. E., and Trader, D. J. (2020). The immunoproteasome: an emerging target in cancer and autoimmune and neurological disorders. J. Med. Chem. 63, 1841–1858. doi:10.1021/acs.jmedchem.9b01226
Zhang, K. Y., Yang, S., Warraich, S. T., and Blair, I. P. (2014). Ubiquilin 2: a component of the ubiquitin–proteasome system with an emerging role in neurodegeneration. Int. J. Biochem. and Cell. Biol. 50, 123–126. doi:10.1016/j.biocel.2014.02.018
Zhang, Z., Krutchinsky, A., Endicott, S., Realini, C., Rechsteiner, M., and Standing, K. G. (1999). Proteasome activator 11S REG or PA28: recombinant REG alpha/REG beta hetero-oligomers are heptamers. Biochemistry 38, 5651–5658. doi:10.1021/bi990056+
Zhang, Z., and Zhang, R. (2008). Proteasome activator PA28γ regulates p53 by enhancing its MDM2-mediated degradation. EMBO J. 27, 852–864. doi:10.1038/emboj.2008.25
Zhao, X., and Yang, J. (2010). Amyloid-β peptide is a substrate of the human 20S proteasome. ACS Chem. Neurosci. 1, 655–660. doi:10.1021/cn100067e
Zheng, Q., Huang, T., Zhang, L., Zhou, Y., Luo, H., Xu, H., et al. (2016). Dysregulation of ubiquitin-proteasome system in neurodegenerative diseases. Front. Aging Neurosci. 8, 303. doi:10.3389/fnagi.2016.00303
Zhou, H., Shao, M., Guo, B., Li, C., Lu, Y., Yang, X., et al. (2019). Tetramethylpyrazine analogue T-006 promotes the clearance of alpha-synuclein by enhancing proteasome activity in Parkinson's disease models. Neurotherapeutics 16, 1225–1236. doi:10.1007/s13311-019-00759-8
Keywords: neurodegenerative disease, ubiquitin independent, protein degradation, proteasome, Alzheimer's disease, Huntington's disease, Parkinson's disease, oxidative stress
Citation: Church TR and Margolis SS (2025) Mechanisms of ubiquitin-independent proteasomal degradation and their roles in age-related neurodegenerative disease. Front. Cell Dev. Biol. 12:1531797. doi: 10.3389/fcell.2024.1531797
Received: 20 November 2024; Accepted: 23 December 2024;
Published: 07 February 2025.
Edited by:
Ralf Stohwasser, Brandenburg University of Technology Cottbus-Senftenberg, GermanyReviewed by:
Elena Panizza, Cornell University, United StatesHeidi Olzscha, Martin Luther University of Halle-Wittenberg, Germany
Copyright © 2025 Church and Margolis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Taylor R. Church, dGNodXJjaDlAamhtaS5lZHU=; Seth S. Margolis, c21hcmdvbDdAamhtaS5lZHU=