 Mansour Akbari
Mansour Akbari- 1Department of Medical Biology, Faculty of Health Sciences, UiT-The Arctic University of Norway, Tromsø, Norway
- 2Department of Clinical Molecular Biology, Institute of Clinical Medicine, University of Oslo, Oslo, Norway
- 3Unit for precision medicine, Akershus University Hospital, Nordbyhagen, Norway
- 4Department of Microbiology, Oslo University Hospital, Oslo, Norway
Mitochondria are the primary sites for cellular energy production and are required for many essential cellular processes. Mitochondrial DNA (mtDNA) is a 16.6 kb circular DNA molecule that encodes only 13 gene products of the approximately 90 different proteins of the respiratory chain complexes and an estimated 1,200 mitochondrial proteins. MtDNA is, however, crucial for organismal development, normal function, and survival. MtDNA maintenance requires mitochondrially targeted nuclear DNA repair enzymes, a mtDNA replisome that is unique to mitochondria, and systems that control mitochondrial morphology and quality control. Here, we provide an overview of the current literature on mtDNA repair and transcription machineries and discuss how dynamic functional interactions between the components of these systems regulate mtDNA maintenance and transcription. A profound understanding of the molecular mechanisms that control mtDNA maintenance and transcription is important as loss of mtDNA integrity is implicated in normal process of aging, inflammation, and the etiology and pathogenesis of a number of diseases.
1 Introduction
Research on mitochondria and their gene architecture has led to the hypothesis that this organelle has evolved from an endosymbiotic event 1.5 billion years ago, when an archaea-like host engulfed a proteobacterium-like ancestor (Lang et al., 1999; Dyall et al., 2004). Thus, mitochondria have their own genome and mitochondrial respiratory chain complexes have high homology with those found in bacteria. Further, to some extent mitochondria rely on their own ribosomes, tRNAs and associated protein factors as protein translation machinery resembling the bacterial ancestors. Mitochondria have an outer and an inner membrane which separate the innermost compartment, the mitochondrial matrix, from the cytosol (Figure 1). Ions and small uncharged molecules can freely pass through the outer membrane through porins, the pore-forming outer membrane proteins. Larger molecules and proteins, require specific translocases for import. The inner membrane is a tight and selective barrier that forms extensive invaginations into the matrix called cristae (Figure 1). Most complexes of the electron transport chain (ETC) are embedded into or are tightly associated with the inner membrane ETC complexes establish the electrochemical gradient across the inner membrane that is then utilized by the ATP synthase to generate ATP, the “energy currency” of the cells. This process is called oxidative phosphorylation (OXPHOS).
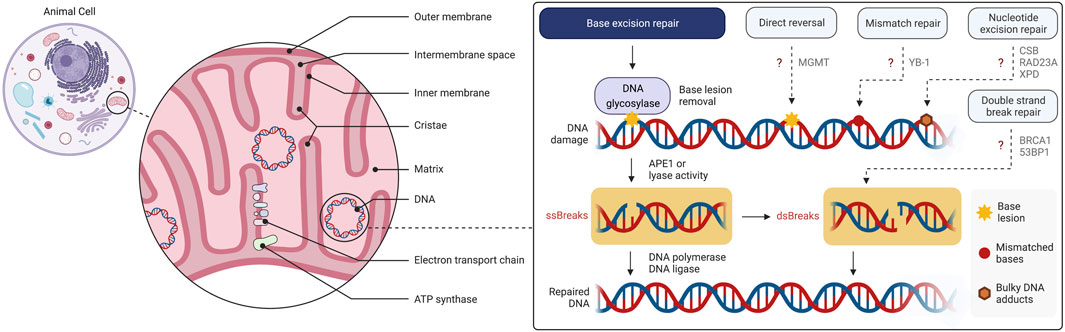
FIGURE 1. Overview of the mitochondrial structure and mtDNA damage and repair. Base excision repair (BER) is the primary mtDNA repair pathway. Canonical forms of the nuclear DNA repair pathways, mismatch repair (MMR), nucleotide excision repair (NER), direct reversal (DR) and double strand break repair (DSBR), have not been demonstrated to operate in mitochondria although individual enzymes of these pathways may be present. The figure has been generated using Biorender.com.
Mitochondria also play a central role in several other key biological processes such as regulation of apoptosis (Gahl et al., 2016), cytoplasmic calcium buffering (Drago et al., 2012), and reactive oxygen species (ROS) mediated signaling pathways (Shadel and Horvath, 2015). The intracellular localization, biogenesis, and bioenergetics of mitochondria are largely determined through interplay between these processes and are tightly connected to mitochondrial function and cellular need for energy (Fernandez-Vizarra et al., 2011).
Our genome resides in the nucleus and in the mitochondria. The mitochondrial genome is a 16.6 kb, maternally inherited, intron-less, circular DNA. Mitochondrial DNA (mtDNA) is packaged with mitochondrial transcription factor A (TFAM) and other proteins into structures called nucleoids tightly associated with the inner mitochondrial membrane (Kukat et al., 2011).
Most of the mtDNA has been transferred to the nuclear genome during the evolution (Lopez et al., 1994; Mourier et al., 2001). Mitochondria contain approximately 1,200 proteins (Rath et al., 2021); nearly all of them are encoded by nuclear DNA and transported into the mitochondria. Human mtDNA has retained 13 protein coding genes encoding subunits of electron transport chain complexes CI, CIII, CIV, and CV or ATP synthase, which are all essential for OXPHOS (Anderson et al., 1981). Each mitochondrion contains multiple copies of mtDNA. The mtDNA copy number varies significantly among different tissues (D'Erchia et al., 2015). Altered mtDNA copy number (Grady et al., 2018; Filograna et al., 2021) and mtDNA depletions caused by disturbed mitochondrial dNTP pool (Viscomi and Zeviani, 2017) are associated with human disease. Many pathogenic mtDNA mutations have been reported (MITO-MAP). Single nucleotide mutations (Goto et al., 1990) and large scale deletions (Moraes et al., 1989) can cause diseases with a wide variety of clinical manifestations ranging from myopathy to neurological disorders (Gorman et al., 2016). MtDNA mutations can be present in a fraction of total mtDNA or in all mtDNA molecules referred to as mtDNA heteroplasmy and homoplasmy, respectively.
Accumulation of various forms of mtDNA damage correlates with aging (DeBalsi et al., 2017), indicating that diminished mtDNA integrity is tightly connected to normal process of tissue aging. Replication errors followed by clonal expansion of mutated mtDNA driven by positive selection and random genetic drift, are commonly thought prominent mechanisms driving the accumulation of mtDNA mutation in various tissues with age (Elson et al., 2001; Ameur et al., 2011; DeBalsi et al., 2017).
The importance of mtDNA for normal cellular and organismal function is illustrated by the observation that defect in the core components of the mtDNA replication and repair machineries cause diseases with a range of clinical presentations from severe infantile multi-tissue diseases to adult-onset progressive degeneration of muscles and neurons (Takashima et al., 2002; Das et al., 2010; Gao et al., 2011; Sykora et al., 2011; DeBalsi et al., 2017; Viscomi and Zeviani, 2017).
The maintenance of mtDNA involves mechanisms distinct from nuclear DNA repair and maintenance systems. Mitochondria lack the full repertoire of DNA repair pathways present in the nucleus, and mtDNA homeostasis involves processes that control mitochondrial morphology and dynamics (Sabouny and Shutt, 2021).
2 Mitochondrial maintenance: Mitochondrial dynamics, clearance, and repair
2.1 Mitochondrial DNA repair
In the nuclear genome, lesions are repaired by different DNA repair pathways depending on the nature of the lesion, phases of the cell cycle, and posttranslational modifications of the repair proteins (Akbari et al., 2009; Mjelle et al., 2015; Carter and Parsons, 2016; Fowler et al., 2022). The main pathways include mismatch repair (MMR), nucleotide excision repair (NER), base excision repair (BER) and the double strand break (DSB) repair (DSBR) pathways homologous recombination (HR) or non-homologous end joining (NHEJ). Some enzymes involved in these nuclear pathways have been reported to localize in mitochondria. It is not known if the presence of these enzymes represents non-canonical forms of the nuclear pathways or different mechanisms which might be exclusive to mitochondria. For instance, low-level repair activity on nicked heteroduplex substrate with a GT or a GG mismatch was identified in rat liver mitochondrial lysate. Similar to the nuclear MMR, the activity was mismatch-selective, bi-directional, ATP dependent and EDTA-sensitive (Mason et al., 2003). However, later it was found that this MMR activity was dependent on the Y-box binding protein (YB-1) as its depletion led to a decrease of MMR correction activity in the mitochondrial extracts and an increase in mtDNA mutations (de Souza-Pinto et al., 2009), thus suggesting an atypical mechanism.
Some enzymes acting in nuclear NER have been reported to localize in the mitochondria. Cockayne syndrome group B (CSB) localizes in the mitochondria, where it stimulates mtDNA stability likely by facilitating BER (Aamann et al., 2010). RAD23 homolog A (RAD23A) can be recruited to mitochondria following induction of oxidative mtDNA damage and upon mt-Ox treatment. Accordingly, Rad23a−/− mouse embryonic fibroblasts (MEFs) showed elevated 8-oxoG levels in mtDNA compared to Rad23a+/+ cells (Wisnovsky et al., 2016). Xeroderma pigmentosus group D (XPD) localizes in mitochondria and protects the mitochondrial genome from oxidative DNA damage (Liu et al., 2015). Thus, a small selection of NER proteins appears to be present in the mitochondria, but several enzymes and activities required for NER are lacking.
A DNA end-joining activity for repair of DSBs have been reported in mitochondrial extracts from mammalian cells (Lakshmipathy and Campbell, 1999) and Breast cancer type 1 susceptibility protein (BRCA1) and p53-binding protein 1 (53BP1) have been reported to be in the mammalian mitochondria (Coene et al., 2005; Youn et al., 2017).
Some biochemical evidence suggests O6-methylguanine (O6-MeG) lesions removal in mitochondria (Myers et al., 1988). However, O6-methylguanine DNA methyltransferase (MGMT), the enzyme responsible for O6MeG direct reversal (DR) in the nucleus, has not been verified in the mitochondrial compartment, and the existence of DR in mammalian mitochondria remains uncertain (Satoh et al., 1988; LeDoux et al., 1992). In conclusion, the presence of NER, MMR, and DSBR in mitochondria and how mitochondrial deal with lesions typically removed by these pathways need further investigation. There is, however, substantial evidence for the presence of BER in mitochondria (Akbari et al., 2008; Kazak et al., 2012).
BER is the key pathway for repair of a number of different types of DNA base lesions, as well as apurinic-apyrimidinic sites (AP sites), and single-strand DNA breaks (Krokan and Bjoras, 2013) (Figure 1). BER is initiated by removal of damaged base by a DNA glycosylase. After removal of the substrate base, apurinic endonuclease 1 (APE1) incises immediately 5′ to the AP site leaving a gap in DNA that is filled by DNA polymerase. The gap filling can be in the form of single nucleotide insertion, or through incorporation of several nucleotides called long-patch BER (Akbari et al., 2008). The final step in BER is the end-joining or ligation of the DNA phosphodiester backbone by DNA ligase (Krokan and Bjoras, 2013).
Mechanistically, nuclear and mitochondrial BER are identical, and they share most of their proteins. However, mitochondrial BER seems to be somewhat less robust than the nuclear BER (Yakes and Van Houten, 1997; Akbari et al., 2014; Akbari et al., 2015).
In an early study, the repair capacity for lesions generated by treatment with 200 μM hydrogen peroxide on mtDNA was investigated (Yakes and Van Houten, 1997). Following the treatment, a 16.2 kb mtDNA fragment and a 17.7 kb fragment flanking the β-globin gene were PCR amplified and the amplicons were used to measure the level of damage and repair in the target DNA regions. When the treatments were limited to 15 min, mtDNA damage was repaired with similar kinetics as the nuclear fragment. However, following a 60 min treatment, damage to the nuclear fragment was completely repaired within 90 min, whereas no DNA repair in the mitochondrion was detected (Yakes and Van Houten, 1997), suggesting that prolonged DNA damage may exhaust the mitochondrial BER capacity.
In another study, mitochondrial BER capacity was investigated using a circular DNA substrate containing uracil at a specific position in highly purified mitochondrial extracts from human U2OS and HeLa cell lines (Akbari et al., 2014). DNA ligation was significantly slower than the preceding mitochondrial BER steps. Moreover, overexpression of DNA ligase III in mitochondria improved the rate of overall BER, and increased cell survival after extraneously induced oxidative stress (Akbari et al., 2014). An efficient ligation step in BER is particularly important to prevent the accumulation of nicks in mtDNA that can give rise to DSBs and block DNA replication and transcription.
Mammalian cells contain at least 11 different DNA glycosylases. Nuclear and mitochondrial isoforms of DNA glycosylases are encoded by the same gene; however, mitochondrial DNA glycosylases are usually generated by alternative splicing and alternative transcription initiation (Nilsen et al., 1997; Takao et al., 1999; Ohtsubo et al., 2000). Mitochondrial DNA glycosylases include DNA glycosylases 8-oxoguanin DNA glycosylase (OGG1), MutY homologue (MYH), Endonuclease III-like protein 1 (NTHL1), Endonuclease VIII-like protein 1 (NEIL1), Endonuclease VIII-like protein 2 (NEIL2), that initiate the repair of oxidative DNA base lesions; uracil-DNA glycosylase 1 (UNG1) that initiate the repair of uracil; Alkyladenine DNA glycosylase (AAG) which repairs alkylated DNA lesions.
APE1 is the prominent endonuclease responsible for the processing of the AP sites generated following the removal of damaged base by DNA glycosylase. APE1 contains a nuclear localization signal within its N-terminal domain that directs the protein within the nucleus (Jackson et al., 2005). APE1 is imported into mitochondria through the translocase of the outer membrane (TOM) pore complex (Li et al., 2010). APE1 may contain a putative mitochondrial localization signal within amino acids 289–318 in its C- terminus (Li et al., 2010).
Abortive DNA-ligase activity during replication and BER could result in 5′-AMP–DNA overhang. Aprataxin (APTX), a member of the histidine triad superfamily, removes 5′-AMP from DNA. Defect in APTX causes the ataxia oculomotor apraxia (AOA1) an autosomal recessive spinocerebellar ataxia syndrome (Date et al., 2001). APTX is present in both the nucleus and mitochondria (Sykora et al., 2011). Biochemical experiments suggest that contrary to nuclear DNA repair where long-patch BER can remove 5′-AMP from DNA as part of the displaced 5′ strand (5′flap), mitochondrial BER is not able to compensate for APTX deficiency resulting in the persistent 5′-AMP lesions in mtDNA and may thus contribute to AOA1 pathology (Akbari et al., 2015).
Polymerase gamma (Pol γ) is a Family A replicative DNA polymerase and functions in mtDNA replication and repair. Pol γ is a heterotrimer composed of a 140 kDa catalytic subunit and a homodimer 110 kDa accessory subunit (POLG2) that increases processivity of DNA synthesis (Krasich and Copeland, 2017). Pol γ contains a C-terminus polymerase domain and an N-terminus proofreading 3′–5′ exonuclease domain. The catalytic subunit has an associated lyase activity that catalyzes the removal of 5′-deoxyribose phosphate residue from incised AP site during BER (Longley et al., 1998).
DNA polymerases other than Pol γ might exist in mitochondria in specific tissues or transiently in response to stress (Krasich and Copeland, 2017; Sykora et al., 2017; Zheng et al., 2020). Functional Pol γ is, however, indispensable for normal mitochondrial and organismal function indicating a prominent role of Pol γ in mtDNA maintenance and integrity (DeBalsi et al., 2017).
In higher eukaryotes, there are three distinct DNA ligases: DNA ligase I is required for the ligation of Okazaki fragments during lagging strand DNA synthesis and is involved in several DNA repair pathways; DNA ligase III has been associated with BER and NER; DNA ligase IV is mainly involved in NHEJ. A mitochondrial isoform of DNA ligase III is responsible for DNA ligation during mtDNA replication and repair (Lakshmipathy and Campbell, 2001; Gao et al., 2011; Simsek et al., 2011).
2.2 Disconnection of mtDNA damage and mtDNA mutation load
In nuclear DNA, unrepaired lesions can be fixed as mutations following DNA replication and somewhat by the process of DNA repair itself. In mtDNA this link between DNA lesion and the generation of mutation appears to be weak despite less robust DNA repair activity in mitochondria compared with the nucleus (Itsara et al., 2014; Valente et al., 2016; Kauppila et al., 2018).
Mitochondrial dysfunction is a pathological hallmark of several neurodegenerative diseases and is often associated with apoptosis and neuronal loss. The effect of mtDNA instability in mitochondrial dysfunction and neurodegeneration was investigated in mice engineered to generate high levels of AP sites in mtDNA in forebrain neurons by tissue-specific expression of a mutated version of the mitochondrial uracil-DNA glycosylase that removes thymine instead of uracil from mtDNA (Lauritzen et al., 2010). High levels of AP sites in mtDNA from the transgenic mice was confirmed by a probe that reacts with the exposed aldehyde group at AP sites. These mice showed aberrant mitochondrial function, neuronal death, and behavioral deficits consistent with the crucial role of mitochondria in neuronal function and survival. MtDNA mutation load was tested by PCR amplification of cytochrome B gene followed by cloning, and sequencing of the amplicons. The results failed to detect any differences in the mutation frequency between mtDNA from transgenic mice compared with the wild-type littermates (Lauritzen et al., 2010). The authors speculated that the lack of mutation elevation could be due to efficient repair of AP sites, though this conclusion was not supported by mtDNA copy number analysis to rule out degradation of damaged mtDNA.
The inner mitochondrial membrane is commonly thought to be the primary site of ROS production. ROS can generate a plethora of pre-mutagenic DNA lesions (Akbari and Krokan, 2008; Murphy, 2009), including 8-oxodG, a common oxidative DNA base lesion which can mispair with adenine during DNA replication giving rise to G to T transversion mutations (Bjelland and Seeberg, 2003). Consistent with the relatively high number of oxidative base damage events in mtDNA, and the central function of OGG1 in 8-oxodG repair, mtDNA from OGG1-deficient mice contained 20-fold more 8-oxodG than mtDNA from wild-type animals (de Souza-Pinto et al., 2001).
DNA glycosylases OGG1 and MYH functionally cooperate to prevent G to T mutations arising from 8-oxoG lesions. Mice deficient in both MYH and OGG1 DNA glycosylases display strong tumorigenesis and severely reduced lifespan (Xie et al., 2004). Most of the tumors showed G to T transversion mutations in nuclear DNA determined by PCR amplification of the nuclear k-RAS gene followed by nucleotide sequencing analysis of cloned PCR products (Xie et al., 2004). A separate study, using PCR-based methods, failed to detect elevated mtDNA mutations in myh and ogg1 double knockout mice. However, biochemical analysis of the mitochondrial ETC complex I activity and mitochondrial respiration measurements demonstrated a significant reduction in respiratory capacity in brain mitochondria from the adult double knockout mice (Halsne et al., 2012).
In a recent study, CRISPR-Cas9 based gene editing was used to generate cells lacking DNA glycosylase OGG1 that removes 8-oxoG from DNA, or MTH1 that hydrolyses 8-oxo-dGTP to 8-oxo-dGMP to prevent its insertion into DNA. In vivo mitochondrial BER activity was measured in OGG1-and MTH1-deficient cells using a cell-and mitochondrial-permeable fluorescent probe that reacts with AP sites in mtDNA (Jun et al., 2022). Following exposure of the cells to an exogenous oxidating compound, mitochondrial BER was greatly elevated in the MTH1 KO cell line demonstrating the importance of sanitizing 8-oxoG precursors in mitochondria. The OGG1 KO cells showed lower BER activity than both MTH1 KO cells and the wild-type control cells resulting in the accumulation of 8-oxoG lesions in mtDNA (Jun et al., 2022).
Thus, despite recent reports showing that BER deficiency does not lead to increased mutation load in mtDNA or mtRNA in mice (Kauppila et al., 2018), the fact that BER, as well as the nucleotide pool sanitizing enzymes MTH1 and deoxyuridine nucleotidohydrolase (dUTPase) that eliminates dUTP, exist in mitochondria indicate the importance of preventing and repairing base lesions in mtDNA (Kang et al., 1995; Ladner and Caradonna, 1997; Ichikawa et al., 2008; Jun et al., 2022).
Several mechanisms may account for the disconnection between mtDNA damage and mutation (Figure 2). Pol γ is a high fidelity DNA polymerase showing an error rate of ∼3 × 10–5 (Johnson and Johnson, 2001) comparable with the replicative DNA polymerases Polε and Polδ each displaying an error rate of 10−5–10–6 and 10−4–10–6, respectively (Burgers and Kunkel, 2017). Because 8-oxoG can mispair with adenine during DNA synthesis and 8-oxoG is highly relevant mtDNA lesion, biochemical experiments have been caried out to understand the efficiency and fidelity of 8-oxoG bypass. Pol γ has been found to display moderate DNA translesion-like synthesis activity and is able to insert the correct dCMP (≈ dAMP ≫ dGMP) opposite 8-oxo-dG, thus counteracting the pre-mutagenic capacity of 8-oxoG in mtDNA (Figure 2). No dTMP incorporation was observed (Graziewicz et al., 2007).
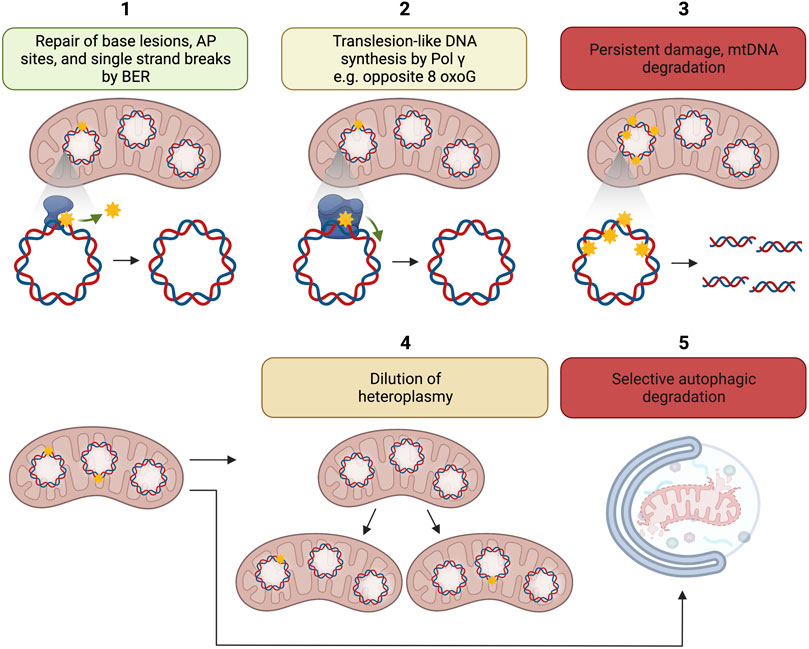
FIGURE 2. Putative mechanisms for disconnection between mtDNA damage and mtDNA mutation load. 1. Repair of base lesions, AP sites, and single strand breaks by BER; 2. Translesion like DNA synthesis by Pol γ e.g., opposite 8-oxoG; 3. Persistent damage may lead to the degradation of damaged mtDNA; 4. Dilution of heteroplasmy, i.e., ratio of mutant to wild-type mtDNA by mitochondrial dynamics; 5. Possible selective autophagic degradation of mitochondria with high levels of mtDNA damage. The figure has been generated using Biorender. com.
MtDNA degradation is another potential mechanism disconnecting mtDNA damage to mtDNA mutation accumulation (Figure 2).
It has long been known that mtDNA has a higher turnover than nuclear DNA (Gross et al., 1969). In an early study, high-performance liquid chromatography was used to measure the amount of 8-oxoG in mtDNA isolated from rat liver mitochondria. The results showed that long and intact circular mtDNA contained much lower amounts of 8-oxoG than shorter fragmented mtDNA molecules (Suter and Richter, 1999) suggesting elevated turnover of highly damaged mtDNA molecules. Evidence now supports the idea that degradation of highly damaged mtDNA is an important mechanism for the preservation of mtDNA integrity (Shokolenko et al., 2009; Shokolenko et al., 2013).
Several mechanisms have been shown to facilitate the degradation of damaged mtDNA. DNA double-strand breaks (DSBs) occur in mtDNA because of defective replication as well as through ROS generated DNA damage. Mitochondria lack the robust DSB repair that is present in the nucleus. Generation of target specific mtDNA DSBs resulted in the loss of the damaged mtDNA molecules because of extensive mtDNA degradation (Moretton et al., 2017). Components of the mtDNA replisome i.e., Pol γ, DNA helicase TWINKLE, and the 5′-3′exonuclease MGME1, all seem to contribute to the degradation of linear mtDNA generated following DSBs (Nissanka et al., 2018; Peeva et al., 2018).
TFAM is an abundant mitochondrial protein that coats the entire mtDNA (Kukat et al., 2011), and regulates mtDNA copy number (Ekstrand et al., 2004). TFAM may promote the degradation of mtDNA containing AP sites. Purified TFAM complexed with AP site containing oligodeoxynucleotides, reduced the half-life of AP sites by DNA strand incision at AP sites. Chemical trapping of the reaction by sodium cyanoborohydride suggested that the AP site incision reaction occurred via Schiff base intermediate which is a necessary step in the β-elimination cleavage by AP lyases (Xu et al., 2019). Considering the central role of TFAM in mtDNA metabolism and the emerging importance of mtDNA degradation as an active mechanism to counteract mtDNA damage accumulation, more research is warranted to determine the biological significance of this finding.
2.3 Mitochondrial dynamics in mtDNA maintenance
Mitochondria fuse together to form larger mitochondria and build tubular networks. They also undergo fission to form smaller mitochondria. These events, together with the mechanisms that control mitochondrial quality are collectively referred to as mitochondrial dynamics and are controlled by a number of intra- and extracellular cues (Giacomello et al., 2020). Defect in the regulators of mitochondrial dynamics cause disease, often in the form of severe neuromuscular and neurodegenerative disorders (Dorn and Dang, 2022; Romanello and Sandri, 2022).
Mitochondrial fusion and fission involve a group of dynamin-like proteins and GTPases. The key proteins in fusion include the outer mitochondrial membrane proteins mitofusin 1 (MFN1) and mitofusin 2 (MFN2), and the inner mitochondrial membrane protein optic atrophy type 1 (OPA1) (Koshiba et al., 2004). The cytosolic dynamin-related protein 1 (DRP1), and several mitochondrial outer membrane DRP1-binding proteins or receptors are key regulators of mitochondrial fission (Loson et al., 2013).
Mitochondrial dynamics exert a profound effect on mtDNA homeostasis manifested by mtDNA copy number and integrity. Mitochondrial dynamics also controls the distribution of normal and mutant mtDNA in the cellular mitochondrial population, i.e., heteroplasmy, to prevent that the number of mutant mtDNA molecules in each mitochondrion exceed a threshold that could impair mitochondrial function (Figure 2 box 4) (Amati-Bonneau et al., 2008; Hudson et al., 2008; Chen et al., 2010; Yu-Wai-Man et al., 2010; Elachouri et al., 2011; Peralta et al., 2018; Sabouny and Shutt, 2021; Sidarala et al., 2022).
Mutations in OPA1 cause dominant optic atrophy a disease affecting retinal ganglion cells. Clinical presentation of the disease includes optic atrophy, progressive external ophthalmoplegia, ataxia, deafness, and sensory-motor neuropathy (Alexander et al., 2000; Delettre et al., 2000). Mitochondria in skeletal muscle from patients with OPA1 mutation show mosaic cytochrome c oxidase deficiency and contain mtDNA with multiple deletions (Amati-Bonneau et al., 2008; Hudson et al., 2008). Fibroblasts from patients with OPA1 mutation display mitochondrial network fragmentation (Bonifert et al., 2014; Liao et al., 2017).
Homozygous Opa1 mutant mice die in utero during embryogenesis, but heterozygous Opa1 mutants display the main features of human dominant optic atrophy including abnormal mitochondrial morphology, disorganized cristae structure, mitochondrial dysfunction and mtDNA instability (Alavi et al., 2007; Davies et al., 2007; Chen et al., 2012).
Complete depletion of MFN1 and MFN2 in mice cause embryonic lethality (Chen et al., 2003). Target specific deletion of Mfn1 and Mfn2 in skeletal muscle during development resulted in severe mtDNA depletion in muscle that preceded physiological abnormalities (Chen et al., 2010). In another study, β-cells specific deletion of Mfn1 and Mfn2 in mice showed that MFN1 and MFN2 control glucose homeostasis in a manner that was connected to their role in the preservation of mtDNA number by regulating the expression of Tfam (Sidarala et al., 2022). Thus, MFN1 and MFN2 appear to control mitochondrial homeostasis through regulation of mitochondrial structure as well as mtDNA copy number maintenance.
FBXL4 is a member of F-box protein family. Mutations in FBXL4 were identified in patients with lactic acidosis and severe encephalopathic syndrome (Bonnen et al., 2013; Gai et al., 2013). Analysis of patient tissue samples identified various mitochondrial abnormalities including reduced mtDNA content and impaired mitochondrial morphology (Bonnen et al., 2013; Gai et al., 2013; Antoun et al., 2016). However, it is possible that mitochondrial phenotypes in FBXL4 deficient cells might be related to a putative role of FBXL4 in controlling mitochondrial turnover by autophagy rather than a direct role of FBXL4 in mitochondrial biogenesis and mtDNA copy number regulation (Alsina et al., 2020).
2.4 Mitochondrial clearance
Dysfunctional or superfluous mitochondria can be degraded via bulk autophagy or mitochondria-specific autophagy (mitophagy) (Figure 2 box 5) (Li et al., 2022). Mutations in mitophagy regulators, such as PTEN-induced putative kinase 1 (PINK1) and the cytosolic E3 ligase Parkin, are known as causal genes for familiar Parkinson disease (Kitada et al., 1998; Valente et al., 2004; Narendra et al., 2010). Mitochondria harboring mtDNA damage seem to be able to escape mitophagy and propagate in the cell. The missed clearance of these mutation-bearing mitochondria causes somatically mutated mtDNA to undergo clonal expansion during aging and thereby cause mosaic respiratory chain deficiency in various tissues (Larsson, 2010). Patients with inherited mtDNA depletion syndrome suffer from strong tissue-specific reductions in mtDNA levels (Viscomi and Zeviani, 2017). Thus, whether, and to what extent, mtDNA damage could activate mitophagy needs further investigation.
2.5 MtDNA maintenance in disease
Mitochondrial DNA maintenance defects (MDMDs) are a class of diseases caused by pathogenic mutations in nuclear-encoded mtDNA maintenance proteins. These diseases are characterized by substantial reduction in mtDNA copy number, accumulation of mutations, and large-scale deletions. MDMDs can be autosomal recessive or dominantly inherited, and they can cause a wide range of symptoms. MDMDs have been related to pathogenic variants in 20 nuclear genes that are important for mtDNA maintenance. These mutations have been found in three gene categories: genes for mtDNA replication machinery enzymes (POLG, POLG2, TWNK, TFAM, RNASEH1, MGME1, and DNA2); genes encoding proteins involved in mitochondrial nucleotide pool synthesis (TK2, DGUOK, SUCLG1, SUCLA2, ABAT, RRM2B TYMP, SLC25A4, AGK, MPV17); and genes encoding proteins involved in mitochondrial dynamics (OPA1, MFN2, FBXL4) (reviewed in (El-Hattab et al., 2017)).
New evidence suggests that BER causes accumulation of single-stranded DNA breaks in an age-dependent manner and contributes to Parkinson disease–like pathology in a C. elegans model. NTH-1 DNA glycosylase mediated mitochondrial and nuclear genomic instability promoting degeneration of dopaminergic neurons in old nematodes. Conversely, NTH-1 deficiency protects against α-synuclein-induced neurotoxicity, maintaining neuronal function with age (SenGupta et al., 2021). In humans, whole-exome sequencing analysis from two independent cohorts of clinically validated idiopathic PD and controls, the Norwegian ParkWest cohort (n = 411) and the North American Parkinson’s Progression Markers Initiative (n = 640) showed that, compared to controls, individuals with idiopathic PD had a significant polygenic enrichment of rare, nonsynonymous variants in the genes encoding enzymes involved in mtDNA maintenance (Gaare et al., 2018). This work suggests that the results found in C. elegans might be relevant in human settings. However, more studies in human cells are required to confirm such putative mechanisms.
2.6 Methods for measuring mtDNA topology, damage, and repair
To interpret the functional implications of mtDNA maintenance mechanisms and their relevance to disease, it is important to be aware of both deliverables and limitations of the various assays used to detect mtDNA topology and damage. Here we review the assays used for measuring mtDNA topology, copy number. and mtDNA damage.
2.6.1 Assays for measuring mtDNA topology and copy number
In 1963, transmission electron microscopy studies of chicken embryo mitochondria revealed that mitochondria have their own DNA (Nass and Nass, 1963a; Nass and Nass, 1963b). Transmission electron microscopy in combination with other techniques and enzymatic assays has been used in mtDNA topology and replication studies (Kasamatsu et al., 1971; Kosar et al., 2021).
Southern blot is a technique where DNA fragments of different lengths are separated by size via gel electrophoresis, transferred to a membrane and visualized by hybridization with radioactive or non-radioactive labeled sequence-specific DNA probes. One dimensional agarose gel (1D gel) electrophoresis technique allows separation of molecules roughly according to their mass. 1D gel electrophoresis of mtDNA provides a qualitative distribution of total mtDNA topoisomers, catenated circles, nicked circular mtDNAs, linear and supercoiled circular DNA molecules (Pohjoismaki et al., 2006). Gel electrophoresis of mtDNA linearized with restriction enzymes provides information about mtDNA content (Wheeler et al., 2019) and large deletions (Kaukonen et al., 2000).
Initiation, elongation, and termination of DNA replication are each associated with distinct, nonlinear DNA structures. The two-dimensional agarose gel (2D gel) electrophoresis technique, developed by Brewer and Fangman in 1987 (Brewer and Fangman, 1987) allows separation of these nonlinear structures based on two dimensions: The first dimension gel is run at low voltage in low percentage agarose (0.4%) to separate DNA molecules in proportion to their mass; the second dimension is run at high voltage in a higher agarose concentration (≥1%) gel in the presence of ethidium bromide (or a similar intercalating agent), so that the mobility of a non-linear molecule is greatly influenced by its shape (Brewer and Fangman, 1987). This technique allows the resolution of nonlinear replication intermediates in specific and predictable ways and was adapted to study mtDNA replication mechanisms (Han and Stachow, 1994; Holt et al., 2000). The simplest type of replication intermediate is the standard replication fork, and investigations of these structures led to the identification of two distinct replication origins in mtDNA (Han and Stachow, 1994; Holt et al., 2000).
Gel electrophoresis based mtDNA analyses do not require a prior mtDNA purification step. However, they might require relatively large amounts of DNA which may pose a limitation for small tissue samples.
PCR-based techniques enable very sensitive and precise measurement of mtDNA damage and content allowing for rapid examination of large sample number starting from nanogram of template. Real-time quantitative PCR (qPCR) assay: In qPCR, a short DNA fragment, typically shorter than 200 nucleotides, is amplified in a mixture containing dNTPs, Taq DNA polymerase, and a DNA-intercalating dye. Primer design is of extreme importance for this technique. This is especially true for mtDNA studies given the presence of inactive copies of mitochondrial genes in the nuclear DNA called nuclear mitochondrial pseudogenes (NUMTs). The presence of mtDNA-like DNA segments in the nuclear genome of eukaryote cells was first suggested in 1967 (du Buy and Riley, 1967) and has subsequently been confirmed in human (Tsuzuki et al., 1983).
Quantification of the number of mtDNA molecules which correlates with the cellular energy demand of the cell and ATP production has been used to assess the state of mitochondrial function (Clay Montier et al., 2009; Fuke et al., 2011). Relative mtDNA copy numbers can be determined by qPCR using singleplex Taqman assays designed to target the mitochondrial MT-ND1 gene and normalize it to a nuclear gene such as B2M (Venegas and Halberg, 2012). 18S ribosomal RNA (18s rDNA) is also frequently used as a control for mtDNA copy number normalizations (Filograna et al., 2019). mtDNA copy numbers can be also quantified using droplet digital PCR. In this technique, the sample is partitioned in single droplets in a water-oil emulsion to the extent that single template DNA molecules can be amplified individually. This technique provides increased precision and sensitivity compared to qPCR. Importantly, it allows the quantitation of absolute DNA copy number (Hindson et al., 2011). These assays do not provide any information about topology, deletions, or lesions in the mtDNA.
The multiplex MT-ND1/MT-ND4 Taqman qPCR assay determines the proportion of deleted mtDNA. The assay is based on the observations that the positions of mtDNA deletions are non-random (Gokey et al., 2004; Taylor and Turnbull, 2005). Of the mtDNA deletions (reported in the MitoBreak database (Damas et al., 2014a)), loss of the MT-ND4 sequence occurs in approximately 90% of all reported break points (Damas et al., 2014b). The common deletion encompassing MT-ND4 spans 4,977 bp (8,483–13459) (Figure 3). Therefore, amplification of this region provides a direct evaluation of deletions. MT-ND1 gene is used for normalization as it is normally not affected by the deletion. This assay does not provide any information about mtDNA copy number or mtDNA lesions (Phillips et al., 2014).
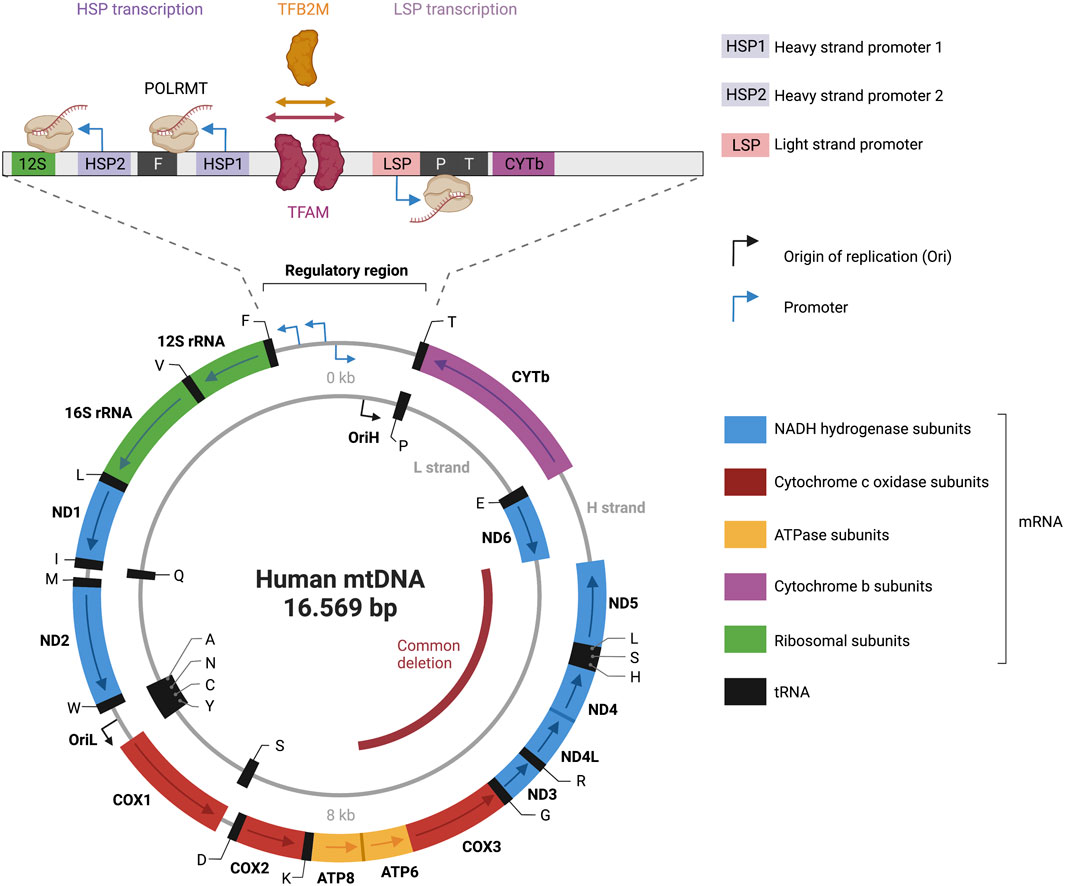
FIGURE 3. Representation of the human mitochondrial DNA transcription. Depicted is a map of the 16.569 bp circular, double-stranded mitochondrial genome. The heavy strand is depicted as outer circle and the light strand as inner circle. mRNA genes coding for NADH hydrogenase subunits are labeled in blue; mRNA genes coding for Cytochrome c oxidase subunits are labeled in red; mRNA genes coding for ATPase subunits are labeled in yellow; mRNA genes coding for cytochrome b subunits are labelled in purple; rRNA genes are labelled in green; tRNAs genes are labelled in black. Transcriptional directionality is specified for mRNAs and rRNAs. The displacement loop (D-loop) noncoding control region is depicted as expanded). D-loop encompasses three promoters: heavy-strand promoter 1 and 2 (HSP1, HSP2), and light-strand promoter (LSP). Transcription of the heavy strand initiated from HSP1 generates a short transcript terminating at the 16S rRNA. Transcription of the heavy strand initiated from HSP2, generates a polycistronic mRNA including 12 mRNA genes, 2 rRNAs and 14 tRNA genes. Light-strand transcription from the light-strand promoter (LSP) generates ND6 mRNA and 8 tRNAs. The transcription start site (TSS) of LSP, HSP1 and HSP2 has been established to be located position 407/408, 561, 643/644 respectively (Montoya et al., 1983; Chang and Clayton, 1984; Zollo et al., 2012). A comparison of the three promoter sequences revealed a mild consensus in the base pairs around the TSS with the -7A,-3C,+1 to +3AAA, and +5A being conserved positions in all three promoters (Basu et al., 2020). The transcription activator TFAM (transcription factor A, mitochondrial) and dimethyladenosine transferase 2, mitochondrial (TFB2M) are required to bind mtDNA upstream of all three promoters, recruiting a single subunit RNA polymerase (POLRMT). The figure has been generated using Biorender. com.
2.6.2 Assays for measuring mtDNA damage
Long-PCR assay: This assay is built on the principle that DNA lesions prevent the DNA polymerase from progressing along a long DNA template, resulting in reduced amplification of the target DNA (Santos et al., 2006). This test monitors the integrity of mtDNA directly from total cellular DNA, eliminating the requirement for a mitochondria separation step. Another significant advantage of this approach is its sensitivity, which allows for the identification of about one lesion per 100,000 nucleotides. A major limitation is that it likely identifies only single- and double-strand DNA breaks, whereas lesions such as 8-oxoG, which do not significantly impede PCR DNA polymerase progression, will not be identified with high efficiency (Santos et al., 2006). Lesions which can block replicative polymerases, such as the oxidized pyrimidine 5,6 dihydroxy-5,6 dihydrothimine (Thymine glycol, Tg) (Clark and Beardsley, 1987) have also the capacity to reduce the amplification efficiency when are present in the template strand.
Quantification of nDNA or mtDNA damage can be accomplished by Real-time qPCR Analysis of Damage Frequency (RADF) (Wang et al., 2016). This method is based on the ability of a lesion in one strand to inhibit restriction enzyme TaqI digestion of double-stranded DNA. Subsequent amplification of the complementary strand after restriction cleavage gives a quantitative measure of the damage content in that site. Since Taql needs to detect double stranded TCGA sequences, DNA lesions of any of the four bases -A, T, G, or C- have the same ability to affect the cut efficiency. For this reason, this assay is informative about general DNA lesions levels while it is not informative about specific lesions.
Over the last few decades, several analytical methods for measuring DNA adducts have been developed allowing for measurement of oxidative DNA lesions by mass spectrometric techniques. Chromatographic techniques including high performance liquid chromatography with electrochemical detection (HPLC-ECD) and gas chromatography-mass spectroscopy (GC-MS) are among the analytical methods that have been developed to measure oxidative base and sugar lesions in DNA (Maynard et al., 2010; Dizdaroglu et al., 2015). In these methods, preparation of highly purified mitochondria devoid of contaminating nuclear DNA is needed. Moreover, care should be taking to avoid introducing oxidative damage to mtDNA during DNA sample purification from intact mitochondria, for instance, by including antioxidants during sample preparation. In general, although these methods can accurately measure mtDNA lesions, they need special equipment and expertise and are therefore not applicable in most clinical and research laboratories.
3 Mitochondrial transcription
The mitochondrial genome is transcribed in a strand-specific, bacterial-like polycistronic manner (Aloni and Attardi, 1971). The precursor polycistronic transcript is then processed to generate mature mRNAs, tRNAs and rRNAs. In a cesium chloride density gradient, the mtDNA molecule can be dissociated into two strands: a guanine-rich heavy strand and a cytosine-rich light strand (Borst, 1972) (Figure 3). The mtDNA codes for 13 messenger RNAs (mRNAs), two ribosomal RNAs (12S and 16S rRNAs), and 22 transfer RNAs (tRNAs). The mRNAs encode for NADH ubiquinone oxidoreductase subunits ND1–ND6, ND4L, cytochrome B CYTB, cytochrome oxidase subunits CO1–CO3, and ATP synthase subunits ATP6 and ATP8, which are all involved in the OXPHOS.
3.1 Mitochondrial transcription initiation
Sequencing results in the early 1980s revealed that the mitochondrial genome is incredibly compact. MtDNA is mostly coding (>90%) and both strands harbor different amounts of genetic information (Anderson et al., 1981). The only noncoding region in mtDNA is a 1.1-kb region called displacement loop (D-loop), that features most of the known transcription and replication regulatory elements (Nicholls and Minczuk, 2014). Three promoters are located within a 250-bp section of the D-loop: two heavy-strand promoters (HSP1 and HSP2), and a light-strand promoter (LSP) (Figure 3). The closely spaced HSP1 and HSP2 promoters in the heavy strand drive the expression of 12 mRNAs, two rRNAs, and 14 tRNAs. The LSP drives the expression of one mRNA and eight tRNAs. HSP and LSP drive transcriptions in opposite directions.
DNA-directed RNA polymerase (POLRMT) catalyzes the transcription of a polycistronic RNA transcript, which is then processed to yield mature RNAs. POLRMT shows a significant structural and sequence similarity to the RNA polymerases found in T3 and T7 bacteriophages (Cermakian et al., 1997; Tiranti et al., 1997; Ringel et al., 2011).
The C-terminal domain (CTD) structure of POLRMT constitute the catalytic core and resembles the classic “right hand” shape composed of the thumb, palm, and fingers domains (Jeruzalmi and Steitz, 1998; Cheetham et al., 1999; Ringel et al., 2011). The palm and fingers domains contain the active site responsible for catalyzing nucleotide incorporation, while the thumb domain is essential for DNA binding (Sousa et al., 1993). The N-terminal domain (NTD) contains two promoter-recognizing structural elements, the AT-rich recognition loop and the intercalating hairpin (ICH); the third element, the specificity loop, is in the CTD. The NTD also contains two pentatricopeptide repeat (PPR) typical of RNA-associated proteins and necessary for site-specific interactions (Ringel et al., 2011; Barkan et al., 2012).
The promoter-binding elements in POLRMT have diverged from T7 RNAP causing reduced promoter sequence recognition capacity and increasing reliance on transcription factors. Dimethyladenosine transferase 2, mitochondrial (TFB2M), is required for transcription from all three promoters (Morozov et al., 2014). There are significant differences in the transcription initiation mechanism at the three promoters that likely arise from the differences in promoter sequences and their requirement for TFAM. TFAM molecules can attach to the region between the HSP1 and LSP and regulate the two promoters (Uchida et al., 2017). In vitro studies indicate that LSP and HSP1 require TFAM for optimal transcription (Litonin et al., 2010; Ramachandran et al., 2017), whereas TFAM inhibits transcription from HSP2 (Lodeiro et al., 2012; Zollo et al., 2012).
Once POLRMT and the transcription factors bind at the promoter regions they form a transcriptional complex and catalyze the RNA synthesis. TFAM can specifically bind to LSP (Fisher and Clayton, 1985; Uchida et al., 2017), and can form a complex with POLRMT on the DNA (Morozov et al., 2014). Mechanistically it is proposed that TFAM recruits POLRMT to LSP promoter forming a pre-initiation complex. POLRMT and TFB2M alone can bind with high efficiency and catalyze promoter proximal transcription (Basu et al., 2020). However, it is not clear how this model differs for the HSP1 and HSP2 (Basu et al., 2020). The initiation complex structure with POLRMT, TFAM, and TFB2M shows that the three proteins cover the promoter site (Hillen et al., 2017a).
3.2 Mitochondrial transcription elongation and termination
For the elongation stage, POLRMT requires transcription elongation factor (TEFM) (Minczuk et al., 2011; Hillen et al., 2017b) which promotes processivity stimulating the longer transcript formation in vitro (Posse et al., 2015) as its depletion leads to a reduction in promoter–distal transcription elongation products (Minczuk et al., 2011). Transcriptional termination has been proposed to be dependent on MTERF1 which binds within the tRNA encoding leucine downstream the rRNAs genes downstream HSP promoters (Kruse et al., 1989; Asin-Cayuela et al., 2005; Guja and Garcia-Diaz, 2012).
3.3 Transcription-associated BER: From nuclear to mitochondrial gene expression regulation
Unrepaired DNA damage can influence cells via mutagenesis and beyond. Base damage, for example, can influence the affinities of DNA binding proteins, such as transcription factors, and interfere with transcription fidelity, resulting in modified transcripts when damage is bypassed by the transcription machinery. While complex DNA lesions can induce cytotoxic effects by blocking polymerases (Bjelland and Seeberg, 2003), base damage, albeit with some important exceptions, does not generally cause DNA helix distortions and in most instances does not result in RNA polymerase stalling during transcription elongation in the nucleus (Lans et al., 2019). Base lesions can however cause polymerase pausing or alter fidelity (Charlet-Berguerand et al., 2006; Agapov et al., 2022). It is safe to assume that, similarly, base damage does not block POLRMT during mitochondrial transcription elongation but might cause pausing or fidelity alterations. On the other hand, unrepaired base lesions which are recognized and excised during transcription, and interference between DNA repair and transcription, may lead to the exposure of transcription-blocking DNA repair intermediates such as AP sites or single strand breaks (SSB). Although little is known about the impact of a single DNA base damage on transcription efficiency, current research supports the idea of a nuclear transcription associated-BER, given preferential base repair on actively transcribed regions. Here we discuss the plausibility of a transcription associated-BER in mitochondria (Figure 4).
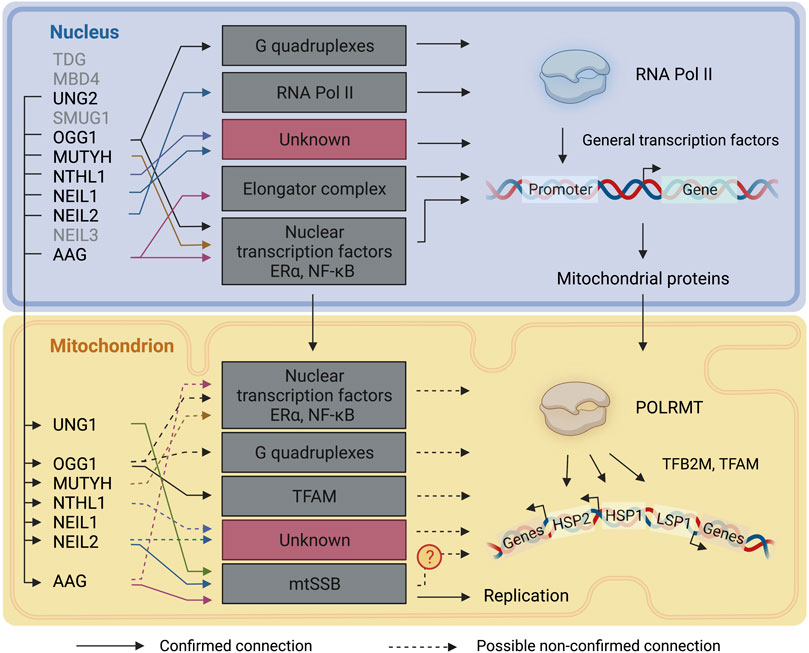
FIGURE 4. DNA glycosylases involvement in transcriptional regulation. A representation of the connections between DNA glycosylases and the transcription mechanisms in the nucleus and mitochondria. Continuous lines represent connections that have been described. Dashed lines represent connections that are likely to take place but have not been directly described. TDG: Thymine-DNA glycosylase; MBD4: Methyl-CpG-domain protein 4; UNG1/2: Uracil-DNA glycosylase; SMUG1: Single-strand selective monofunctional uracil DNA glycosylase 1; OGG1:8-Oxoguanine-DNA glycosylase 1; MUTYH: Adenine-DNA glycosylase; NTHL1: Endonuclease III-like protein 1; NEIL1: Nei endonuclease VIII-like 1; NEIL2: Nei endonuclease VIII-like 2; NEIL3: Nei endonuclease VIII-like 3; AAG: Alkyladenine-DNA glycosylase. RNA Pol II: RNA polymerase II: POLRMT: DNA-dependent RNA polymerase; TFAM: mitochondrial transcription factor A; TFB2M: Dimethyladenosine transferase 2, mitochondrial; HSP: heavy-strand promoter; LSP: light-strand promoter. The figure has been generated using Biorender. com.
Canugovi et al. showed that TFAM preferentially binds DNA containing 8-oxoG substrate over normal DNA (Canugovi et al., 2010), but did not show any preference for uracil, AP sites or gap-containing substrates. Furthermore, when tested for its ability to modulate in vitro BER enzymatic activities, TFAM inhibited OGG1, UDG, APE1, pol γ and, additionally, OGG1 in a cell-based assay (Canugovi et al., 2010). The same work showed that TFAM’s interaction with p53 modifies TFAM DNA binding and has the capacity to promote BER in mtDNA. In other words, TFAM could be an important regulator of BER supporting the model where association of TFAM with DNA can regulate BER proteins access to the DNA. As the major functions of TFAM are to compact mtDNA and regulate transcriptional initiation, this work highlights the possibility of a link between BER and transcription in mtDNA.
3.3.1 8-Oxoguanine-DNA glycosylase 1 (OGG1) and adenine-DNA glycosylase (MYH)
8-oxoguanine-DNA glycosylase 1 (OGG1) repairs oxidized pyrimidines, with 8-oxoG being the classical substrate. Some reports suggest that OGG1 is also able to excise ring-opened formamidopyrimidines FapyG and FapyA from DNA (Krishnamurthy et al., 2008; Ba and Boldogh, 2018). The mitochondrial isoform, OGG1-β, differs from the nuclear isoform OGG1-α, as it lacks the nuclear localization signal near the C- terminus encoded by exon 7. Exon 8 replaces exon 7 in the OGG1-β mRNA (Nishioka et al., 1999). The importance of a mitochondrial OGG1 is illustrated by the fact that mtDNA accumulates high levels of 8-oxoG (Mecocci et al., 1993; Hudson et al., 1998). Furthermore, 8-oxoG levels in mtDNA isolated from liver, heart and brain were 6-, 16- and 23-fold higher than nDNA, suggesting that tissues with high oxidative load accumulate more oxidative DNA damage in mtDNA than in nuclear DNA (Hamilton et al., 2001). 8-oxoG accumulation in mtDNA is increased in Ogg1−/− mice compared to WT thus suggesting that repair depended on OGG1 (de Souza-Pinto et al., 2001). Correspondingly, mitochondrial targeted human OGG1 was shown to enhance mitochondrial DNA repair, increase mitochondrial function, and protect different cell types from oxidant mediated death (Rachek et al., 2002; Yuzefovych et al., 2012; Yuzefovych et al., 2013).
In the nucleus, recruitment of OGG1 on 8-oxoG lesions in DNA can alter gene expression through three different mechanisms (reviewed in (Wang et al., 2018; Bordin et al., 2021)):
(1) The first mechanism involves secondary DNA structures. Depending on certain sequence motifs and interactions with other proteins, DNA can adopt a variety of alternative conformations in addition to the right-handed double helix shape. G-quadruplexes (G4) are among these secondary structures and are formed when G-quartet - four guanine molecules form a square planar arrangement in which each guanine is hydrogen bonded to the two adjacent guanines are stacked. Recent evidence suggests involvement of G4 structures in transcription, replication, genome stability, and epigenetic regulation. 8-oxoG accumulation in G4 forming sequences can influence transcription depending in which strand the lesion occurs. In the coding strand, after OGG1 recognition, APE1 recruits transcription factors to the site, enhancing transcription (Fleming et al., 2017a). In the template strand, the 8-oxoG processing through OGG1 and APE1 leads to transcription inhibition (Fleming et al., 2017b; Zhu et al., 2018). APE1 modulates G4-mediated expression of genes as its binding to AP sites promotes G4 folding (Roychoudhury et al., 2020). G-quadruplexes are not exclusive structures of nuclear DNA but can occur also in mtDNA (Dong et al., 2014) and thus influence the regulation of mitochondrial gene expression (Falabella et al., 2019). Thus, it is possible that OGG1 can interact with these structures in mtDNA as well, but data on the subject is lacking.
(2) A second mechanism that suggests OGG1 influences transcription is the recognition of 8-oxoG at the promoters of NF-κB target genes. OGG1 binding facilitates DNA bending at the site, enhancing NF-κB occupancy and recruitment of specific transcription factors (Ba et al., 2014; Ba and Boldogh, 2018). Mitochondrial localization was recently described for the proteins p50/NF-kB1 and RelA (p65), two subunits of the NF-kB family of transcription factors (Barshad et al., 2018). Furthermore, in mouse keratinocytes, STAT3 bound mtDNA and co-immunoprecipitated with TFAM. STAT3 knockout increased the transcription of several mitochondrial genes, pointing to a potential inhibitory role of STAT3 in mtDNA transcription (Macias et al., 2014).
(3) In a third mechanism, 8-oxoG regulates chromatin relaxation and remodeling in estrogen- and retinoic acid-induced genes (Perillo et al., 2008; Zuchegna et al., 2014). Estrogen receptors class α and β (ERα and ERβ, respectively) are transcriptional activators which are primed by estradiol (estrogen hormone family) binding. In the nucleus, ER targeted demethylation of histone 3 lysine 9 (H3K9) via local activation of LSD1 (Lysine demethylase 1A) causes local formation of H2O2 followed by a rapid highly controlled increment of 8-oxoG in discrete foci in proximal promoters and enhancers. OGG1 together with topoisomerase IIβ initiates the BER pathway, that generates the required nicks used by topoisomerases to relax and open the chromatin for estrogen mediated transcription (Perillo et al., 2008). ERs have been shown to localize to mitochondria and bind putative estrogen-response elements within mtDNA, and there is evidence that ERs may directly regulate the expression of both mitochondrial- and nuclear encoded OXPHOS subunit genes (Klinge, 2020). ERs have been shown to indirectly up-regulate mtDNA-encoded OXPHOS subunits through the NRF/TFAM pathway (Pronsato et al., 2020). Although indirectly these findings suggest that OGG1 could regulate effectors of mitochondrial transcription through already described nuclear mechanisms more studies are necessary to confirm these putative mechanisms.
More direct evidence of OGG1 involvement in mitochondria showed that in OGG1Tg mice constitutively overexpressing human OGG1 in the mitochondria, the expression of Tfam as well as several mitochondrial transcripts were significantly upregulated, indicating increased transcription of mitochondrially-encoded genes in these animals compared to the WT mice (Komakula et al., 2018). mtDNA 8-oxoG immunoreactivities colocalize with TFAM upon exposure to menadione in MEFs lacking mitochondrial Ogg1 (Oka et al., 2008).
MYH recognizes A:G, A:C and A:8-oxoG mispairs. While the effect of MYH alone on transcription has not been described, transcriptome analysis from Ogg1−/− Myh−/− mice hippocampi showed upregulation of ERα target genes involved in anxiety and cognition regulation, thus suggesting that the two DNA glycosylases might act in concert to regulate ERα signaling (Bjorge et al., 2015). It is not known if a similar mechanism is present in mitochondria.
3.3.2 Endonuclease III-like protein 1 (NTHL1) and endonuclease 8-like 1–2 (NEIL1-2)
Endonuclease III-like protein 1 (NTHL1) has glycosylase activity on oxidized pyrimidines 5,6 dihydroxy-5,6dihydrothimine (Thymine glycol, Tg), 2,6-diamino-4-oxo-5-formamidopyrimidine (FapyG), 4,6-diamino-5-formamidopyrimidine (FapyA), 5-hydroxyuracil (5-ohU), 5-hydroxycytosine (5-ohC) as well as 8-oxoG (Michaels et al., 1992; Aspinwall et al., 1997; Hilbert et al., 1997; Ikeda et al., 1998). The mitochondrial NTHL1 activity is higher than their nuclear isoforms (Karahalil et al., 2002). C. elegans, is equipped with only two DNA glycosylases: endonuclease III-1 (NTH-1) ortholog of human NTHL1 and uracil DNA glycosylase-1 (UNG-1). NTH-1 has been shown to be a key mediator of age-dependent genomic instability and compromised NTH-1 activity promotes neuroprotection in a Parkinson disease model in nematodes as a direct effect of defective BER in mitochondria. Importantly, loss of NTH-1 activity altered mitochondrial transcription output inducing general overexpression of mitochondrial genes compared to control in an age-dependent manner (SenGupta et al., 2021). This evidence suggests that NTH-1 can regulate mitochondrial transcription in C. elegans and hints the existing of a similar mechanism in humans. However, NTH-1 is the only DNA glycosylase involved in the repair of oxidative DNA damage in C. elegans (Elsakrmy et al., 2020), while other DNA glycosylases with comparable substrate selectivity to NTHL1 are present in humans (comparison between eukaryotic organisms reviewed in (Hindi et al., 2021)), suggesting a more complex control, dependent on several DNA glycosylase.
Endonuclease 8-like 1 and 2 (NEIL1 and NEIL2) have similar substrate specificity. NEIL1 was originally shown to recognize Tg, 5,6-dihydrothymine (DHT), 5,6-dihydrothymine (DHU), 5-ohC, FapyG and FapyA (Hazra et al., 2002a; Bandaru et al., 2002; Jaruga et al., 2004). NEIL2 is active on 5-ohU, 5-ohC as well as Tg, DHT and DHU (Hazra et al., 2002b; Wallace et al., 2003).
NEIL1 and NEIL2 have activity on bubble structured or ssDNA in vitro, suggesting preferential repair activity in actively transcribed DNA (Dou et al., 2003). NEIL2 in complex with the transcription machinery repairs lesions preferentially in the transcribed genes and not on transcriptionally silent genes in mammalian cells (Englander and Ma, 2006; Banerjee et al., 2011). Consistently, in shRNA-NEIL2 stable cell lines, transcribed genes showed significant increase in DNA lesion accumulation in comparison to non-transcribed genes (Banerjee et al., 2011).
NEIL2 was shown to localize and to be active in mitochondria (Mandal et al., 2012) and chromatin immunoprecipitation analysis showed NEIL2 binding to the mitochondrial genes MT-CO2 and MT-CO3 216. Neil2-morphant whole embryos of Xenopus laevis showed upregulation of mitochondrial (mt) gene expression (namely mt-Nd1,4 and 5, mt-co3) as response to mitochondrial dysfunction (Han et al., 2019). Further, NEIL1 interacts with mitochondrial single-stranded binding protein (mtSSB1), an essential protein for mtDNA replication (Jiang et al., 2021). MtSSB1 defects cause transcriptional alterations including hypoxia response in C. elegans (Sugimoto et al., 2008) highlighting that this protein might be involved in mitochondrial transcription. MtSSB1 interacts with NEIL1 to inhibit its AP-lyase activity (Sharma et al., 2018).
3.3.3 Alkyladenine DNA glycosylase (AAG)
Alkyladenine DNA glycosylase (AAG), also called N-methylpurine DNA glycosylase (MPG) recognizes and excises alkylated bases 7-methylguanine (7-meG), 3-methyladenine (3-meA) and 3-methylguanine (3-meG). These adducts can be generated using the alkylating agent methyl methane sulfonate (MMS) (Chakravarti et al., 1991; Samson et al., 1991; Alseth et al., 2006). AAG is also able to remove hypoxanthine (Hx) from DNA. In the nucleus, AAG interacts with several transcription factors. One interactor is estrogen receptor alpha (ERα). AAG modulates ERα capacity to bind Estrogen response elements (ERE) containing genes, resulting in the repression of transcription, (Likhite et al., 2004). ERα influences AAG capacity to repair Hx containing DNA. Another AAG interactor is methylated DNA-binding domain 1 (MBD1), a repressor of transcription able to recognize methylated regions. Once an AAG-MBD1 complex is established they repress methylated promoters and facilitate AAG mediated repair of MMS induced lesions (Watanabe et al., 2003). AAG has been shown to interact with RNA pol II through the interaction with the Elongator complex. This interaction allows the recruitment of BER components to the chromatin to complete the AAG-initiated repair in a transcription coordinated manner (Montaldo et al., 2019). This suggests that AAG influences nuclear transcription to repair aberrantly methylated bases.
In the mitochondria, AAG interacts with mtSSB which inhibits AAG repair activity in single-stranded, but not in double-stranded DNA (van Loon and Samson, 2013). The consequences of AAG-mtSSB interaction have not been explored further and it is not clear whether AAG is able to interact with the mitochondrial replication or transcription machinery. Loss of AAG does not seem to have any effect on mitochondrial dysfunction caused by alkylative DNA damage (Alhumaydhi et al., 2020). Further studies are required to address the impact of this glycosylase on mitochondrial transcription.
3.3.4 Uracil-DNA glycosylase (UNG)
Uracil-DNA glycosylase (UNG) removes uracil from U:A pairs that arise from incorporation of dUMP in the place of dTMP during DNA replication (nuclear and mitochondrial), premutagenic U:G mismatches that are formed from spontaneous or enzymatic deamination of cytosine, and uracil in single-stranded DNA (Sekiguchi et al., 1976; Olsen et al., 1989; Liu et al., 2002; Krokan and Bjoras, 2013).
The human UNG gene encodes both nuclear (UNG2) and mitochondrial (UNG1) isoforms of the enzyme by a mechanism involving two different promoters and alternative splicing. The mRNAs for UNG2 and UNG1 encode 44 and 35 unique N-terminal residues that are required for mitochondrial and nuclear translocation, respectively, whereas the downstream 269 residues encompassing the catalytic region of the enzyme are common. Therefore, the two isoforms have the same catalytic activity (Slupphaug et al., 1993; Nilsen et al., 1997; Otterlei et al., 1998). Mitochondrial UNG1 activity has been reported (Karahalil et al., 2002). Interestingly, mtSSB completely inhibits uracil-excision by UNG from single-stranded DNA (Wollen Steen et al., 2012).
Patients carrying inactivating mutations in the UNG gene show skewed somatic hypermutation and reduced class-switch recombination mechanism (Imai et al., 2003). However, whether these patients show symptoms of mitochondrial dysfunction has not been reported.
3.3.5 Apurinic/apyrimidinic endonuclease (APE1)
APE1 acts downstream of DNA glycosylases and incises at the 5′-end of AP sites. APE1 AP-site cleavage is inhibited by mtSSB (Wollen Steen et al., 2012). Photodynamic therapy (PDT), a promising cancer treatment strategy, generates ROS using a laser in the presence of photosensitizers. Mitochondrial dysfunction is thought to be one of the major causes of cell death upon PDT. Protein–protein interaction experiments showed that, upon PDT, APE1 localizes to the mitochondria where it interacts with TFAM and TFB2M and regulates TFAM transcriptional activity (Li et al., 2012). APE1 was independently identified as a reductive function as a redox effector factor (Ref-1) for several transcription factors including AP-1 (c-Jun/Fos heterodimer), hypoxia-inducible factor HIF1-α, and p53 (Xanthoudakis and Curran, 1992; Tell et al., 2005; Bhakat et al., 2009). Consequently, APE1 could operate as a redox factor enzyme in regulating transcription the mitochondria.
4 Conclusion
The importance of mitochondrial functions is highlighted by their multiple cellular roles, complex relationship with the nucleus, and the wide spectrum of illnesses linked with their malfunction. Beyond its crucial function in the preservation of nuclear and mtDNA, BER has been demonstrated to contribute to the regulation of nuclear transcription (Figure 4). While some data suggests that this might also be true in mitochondria, the importance of BER in mitochondrial transcription is still elusive (Figure 4). Two essential mitochondrial proteins involved in mtDNA metabolism appear to interact with the BER pathway. TFAM, an important factor for packaging mtDNA and for transcriptional initiation, influences BER enzymatic activity in a replication- and transcription-dependent manner. A second factor, mtSSB, important for mtDNA replication and perhaps also transcription, interacts with several DNA glycosylases. Defect in mtssb1-causes transcriptional alterations in C. elegans (Sugimoto et al., 2008), hinting at the possibility of a tight regulation of transcription, replication, and DNA repair in mitochondria. Thus, more research is warranted to investigate the possible role of BER in mitochondrial transcription regulation, which could aid to develop therapeutic avenues for mitochondrial diseases caused by loss of mtDNA and transcription integrity.
Author contributions
Conceptualization, MA, HN, and NM; Writing-original draft, validation, and editing MA, HN, and NM; Visual graphics, NM; All listed authors have read, made intellectual contribution, and approved the published version of the manuscript.
Funding
HN has been granted South East Regional Health Authority (Project no. 279922 and 2017029) and Norwegian Research Council (Grant no. 302483) NM has been granted Anders Jahre’s fond for the promotion of science and medicine (Project no. 352999).
Acknowledgments
The authors thank the excellent contributions of numerous researchers whose published works we were unable to cite due to word limits.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aamann, M. D., Sorensen, M. M., Hvitby, C., Berquist, B. R., Muftuoglu, M., Tian, J., et al. (2010). Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. FASEB J. 24, 2334–2346. doi:10.1096/fj.09-147991
Agapov, A., Olina, A., and Kulbachinskiy, A. (2022). RNA polymerase pausing, stalling and bypass during transcription of damaged DNA: From molecular basis to functional consequences. Nucleic Acids Res. 50, 3018–3041. doi:10.1093/nar/gkac174
Akbari, M., Keijzers, G., Maynard, S., Scheibye-Knudsen, M., Desler, C., Hickson, I. D., et al. (2014). Overexpression of DNA ligase III in mitochondria protects cells against oxidative stress and improves mitochondrial DNA base excision repair. DNA Repair (Amst) 16, 44–53. doi:10.1016/j.dnarep.2014.01.015
Akbari, M., and Krokan, H. E. (2008). Cytotoxicity and mutagenicity of endogenous DNA base lesions as potential cause of human aging. Mech. Ageing Dev. 129, 353–365. doi:10.1016/j.mad.2008.01.007
Akbari, M., Pena-Diaz, J., Andersen, S., Liabakk, N. B., Otterlei, M., and Krokan, H. E. (2009). Extracts of proliferating and non-proliferating human cells display different base excision pathways and repair fidelity. DNA Repair (Amst) 8, 834–843. doi:10.1016/j.dnarep.2009.04.002
Akbari, M., Sykora, P., and Bohr, V. A. (2015). Slow mitochondrial repair of 5'-AMP renders mtDNA susceptible to damage in APTX deficient cells. Sci. Rep. 5, 12876. doi:10.1038/srep12876
Akbari, M., Visnes, T., Krokan, H. E., and Otterlei, M. (2008). Mitochondrial base excision repair of uracil and AP sites takes place by single-nucleotide insertion and long-patch DNA synthesis. DNA Repair (Amst) 7, 605–616. doi:10.1016/j.dnarep.2008.01.002
Alavi, M. V., Bette, S., Schimpf, S., Schuettauf, F., Schraermeyer, U., Wehrl, H. F., et al. (2007). A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain 130, 1029–1042. doi:10.1093/brain/awm005
Alexander, C., VotrubaM., , Pesch, U. E., Thiselton, D. L., Mayer, S., Moore, A., et al. (2000). OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 26, 211–215. doi:10.1038/79944
Alhumaydhi, F. A., de O Lopes, D., Bordin, D. L., Aljohani, A. S. M., Lloyd, C. B., McNicholas, M. D., et al. (2020). Alkyladenine DNA glycosylase deficiency uncouples alkylation-induced strand break generation from PARP-1 activation and glycolysis inhibition. Sci. Rep. 10, 2209. doi:10.1038/s41598-020-59072-6
Aloni, Y., and Attardi, G. (1971). Symmetrical in vivo transcription of mitochondrial DNA in HeLa cells. Proc. Natl. Acad. Sci. U. S. A. 68, 1757–1761. doi:10.1073/pnas.68.8.1757
Alseth, I., Rognes, T., Lindback, T., Solberg, I., Robertsen, K., Kristiansen, K. I., et al. (2006). A new protein superfamily includes two novel 3-methyladenine DNA glycosylases from Bacillus cereus, AlkC and AlkD. Mol. Microbiol. 59, 1602–1609. doi:10.1111/j.1365-2958.2006.05044.x
Alsina, D., Lytovchenko, O., Schab, A., Atanassov, I., Schober, F. A., Jiang, M., et al. (2020). FBXL4 deficiency increases mitochondrial removal by autophagy. EMBO Mol. Med. 12, e11659. doi:10.15252/emmm.201911659
Amati-Bonneau, P., Valentino, M. L., Reynier, P., Gallardo, M. E., Bornstein, B., Boissiere, A., et al. (2008). OPA1 mutations induce mitochondrial DNA instability and optic atrophy 'plus' phenotypes. Brain 131, 338–351. doi:10.1093/brain/awm298
Ameur, A., Stewart, J. B., Freyer, C., Hagstrom, E., Ingman, M., Larsson, N. G., et al. (2011). Ultra-deep sequencing of mouse mitochondrial DNA: Mutational patterns and their origins. PLoS Genet. 7, e1002028. doi:10.1371/journal.pgen.1002028
Anderson, S., Bankier, A. T., Barrell, B. G., de Bruijn, M. H., Coulson, A. R., Drouin, J., et al. (1981). Sequence and organization of the human mitochondrial genome. Nature 290, 457–465. doi:10.1038/290457a0
Antoun, G., McBride, S., Vanstone, J. R., Naas, T., Michaud, J., Redpath, S., et al. (2016). Detailed biochemical and bioenergetic characterization of FBXL4-related encephalomyopathic mitochondrial DNA depletion. JIMD Rep. 27, 1–9. doi:10.1007/8904_2015_491
Asin-Cayuela, J., Schwend, T., Farge, G., and Gustafsson, C. M. (2005). The human mitochondrial transcription termination factor (mTERF) is fully active in vitro in the non-phosphorylated form. J. Biol. Chem. 280, 25499–25505. doi:10.1074/jbc.M501145200
Aspinwall, R., Rothwell, D. G., Roldan-Arjona, T., Anselmino, C., Ward, C. J., Cheadle, J. P., et al. (1997). Cloning and characterization of a functional human homolog of Escherichia coli endonuclease III. Proc. Natl. Acad. Sci. U. S. A. 94, 109–114. doi:10.1073/pnas.94.1.109
Ba, X., Bacsi, A., Luo, J., Aguilera-Aguirre, L., Zeng, X., Radak, Z., et al. (2014). 8-oxoguanine DNA glycosylase-1 augments proinflammatory gene expression by facilitating the recruitment of site-specific transcription factors. J. Immunol. 192, 2384–2394. doi:10.4049/jimmunol.1302472
Ba, X., and Boldogh, I. (2018). 8-Oxoguanine DNA glycosylase 1: Beyond repair of the oxidatively modified base lesions. Redox Biol. 14, 669–678. doi:10.1016/j.redox.2017.11.008
Bandaru, V., Sunkara, S., Wallace, S. S., and Bond, J. P. (2002). A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair (Amst) 1, 517–529. doi:10.1016/s1568-7864(02)00036-8
Banerjee, D., Mandal, S. M., Das, A., Hegde, M. L., Das, S., Bhakat, K. K., et al. (2011). Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J. Biol. Chem. 286, 6006–6016. doi:10.1074/jbc.M110.198796
Barkan, A., Rojas, M., Fujii, S., Yap, A., Chong, Y. S., Bond, C. S., et al. (2012). A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLoS Genet. 8, e1002910. doi:10.1371/journal.pgen.1002910
Barshad, G., Marom, S., Cohen, T., and Mishmar, D. (2018). Mitochondrial DNA transcription and its regulation: An evolutionary perspective. Trends Genet. 34, 682–692. doi:10.1016/j.tig.2018.05.009
Basu, U., Bostwick, A. M., Das, K., Dittenhafer-Reed, K. E., and Patel, S. S. (2020). Structure, mechanism, and regulation of mitochondrial DNA transcription initiation. J. Biol. Chem. 295, 18406–18425. doi:10.1074/jbc.REV120.011202
Bhakat, K. K., Mantha, A. K., and Mitra, S. (2009). Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxid. Redox Signal. 11, 621–638. doi:10.1089/ARS.2008.2198
Bjelland, S., and Seeberg, E. (2003). Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat. Res. 531, 37–80. doi:10.1016/j.mrfmmm.2003.07.002
Bjorge, M. D., Hildrestrand, G. A., Scheffler, K., Suganthan, R., Rolseth, V., Kusnierczyk, A., et al. (2015). Synergistic actions of Ogg1 and mutyh DNA glycosylases modulate anxiety-like behavior in mice. Cell Rep. 13, 2671–2678. doi:10.1016/j.celrep.2015.12.001
Bonifert, T., Karle, K. N., Tonagel, F., Batra, M., Wilhelm, C., Theurer, Y., et al. (2014). Pure and syndromic optic atrophy explained by deep intronic OPA1 mutations and an intralocus modifier. Brain 137, 2164–2177. doi:10.1093/brain/awu165
Bonnen, P. E., Yarham, J. W., Besse, A., Wu, P., Faqeih, E. A., Al-Asmari, A. M., et al. (2013). Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am. J. Hum. Genet. 93, 471–481. doi:10.1016/j.ajhg.2013.07.017
Bordin, D. L., Lirussi, L., and Nilsen, H. (2021). Cellular response to endogenous DNA damage: DNA base modifications in gene expression regulation. DNA Repair (Amst) 99, 103051. doi:10.1016/j.dnarep.2021.103051
Borst, P. (1972). Mitochondrial nucleic acids. Annu. Rev. Biochem. 41, 333–376. doi:10.1146/annurev.bi.41.070172.002001
Brewer, B. J., and Fangman, W. L. (1987). The localization of replication origins on ARS plasmids in S. cerevisiae. Cell 51, 463–471. doi:10.1016/0092-8674(87)90642-8
Burgers, P. M. J., and Kunkel, T. A. (2017). Eukaryotic DNA replication fork. Annu. Rev. Biochem. 86, 417–438. doi:10.1146/annurev-biochem-061516-044709
Canugovi, C., Maynard, S., Bayne, A. C. V., Sykora, P., Tian, J., de Souza-Pinto, N. C., et al. (2010). The mitochondrial transcription factor A functions in mitochondrial base excision repair. DNA Repair (Amst) 9, 1080–1089. doi:10.1016/j.dnarep.2010.07.009
Carter, R. J., and Parsons, J. L. (2016). Base excision repair, a pathway regulated by posttranslational modifications. Mol. Cell. Biol. 36, 1426–1437. doi:10.1128/MCB.00030-16
Cermakian, N., Ikeda, T. M., Miramontes, P., Lang, B. F., Gray, M. W., and CedeRgRen, R. (1997). On the evolution of the single-subunit RNA polymerases. J. Mol. Evol. 45, 671–681. doi:10.1007/pl00006271
Chakravarti, D., Ibeanu, G. C., Tano, K., and Mitra, S. (1991). Cloning and expression in Escherichia coli of a human cDNA encoding the DNA repair protein N-methylpurine-DNA glycosylase. J. Biol. Chem. 266, 15710–15715. doi:10.1016/s0021-9258(18)98467-x
Chang, D. D., and Clayton, D. A. (1984). Precise identification of individual promoters for transcription of each strand of human mitochondrial DNA. Cell 36, 635–643. doi:10.1016/0092-8674(84)90343-x
Charlet-Berguerand, N., Feuerhahn, S., Kong, S. E., Ziserman, H., Conaway, J. W., Conaway, R., et al. (2006). RNA polymerase II bypass of oxidative DNA damage is regulated by transcription elongation factors. EMBO J. 25, 5481–5491. doi:10.1038/sj.emboj.7601403
Cheetham, G. M., Jeruzalmi, D., and Steitz, T. A. (1999). Structural basis for initiation of transcription from an RNA polymerase-promoter complex. Nature 399, 80–83. doi:10.1038/19999
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200. doi:10.1083/jcb.200211046
Chen, H., Vermulst, M., Wang, Y. E., Chomyn, A., Prolla, T. A., McCaffery, J. M., et al. (2010). Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289. doi:10.1016/j.cell.2010.02.026
Chen, L., Liu, T., Tran, A., Lu, X., Tomilov, A. A., Davies, V., et al. (2012). OPA1 mutation and late-onset cardiomyopathy: Mitochondrial dysfunction and mtDNA instability. J. Am. Heart Assoc. 1, e003012. doi:10.1161/JAHA.112.003012
Clark, J. M., and Beardsley, G. P. (1987). Functional effects of cis-thymine glycol lesions on DNA synthesis in vitro. Biochemistry 26, 5398–5403. doi:10.1021/bi00391a027
Clay Montier, L. L., Deng, J. J., and Bai, Y. (2009). Number matters: Control of mammalian mitochondrial DNA copy number. J. Genet. Genomics 36, 125–131. doi:10.1016/S1673-8527(08)60099-5
Coene, E. D., Hollinshead, M. S., Waeytens, A. A. T., Schelfhout, V. R. J., Eechaute, W. P., Shaw, M. K., et al. (2005). Phosphorylated BRCA1 is predominantly located in the nucleus and mitochondria. Mol. Biol. Cell 16, 997–1010. doi:10.1091/mbc.e04-10-0895
D'Erchia, A. M., Gadaleta, G., Pavesi, G., Chiara, M., De Virgilio, C., et al. (2015). Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion 20, 13–21. doi:10.1016/j.mito.2014.10.005
Damas, J., Carneiro, J., Amorim, A., and Pereira, F. (2014). MitoBreak: The mitochondrial DNA breakpoints database. Nucleic Acids Res. 42, D1261–D1268. doi:10.1093/nar/gkt982
Damas, J., Samuels, D. C., Carneiro, J., Amorim, A., and Pereira, F. (2014). Mitochondrial DNA rearrangements in health and disease--a comprehensive study. Hum. Mutat. 35, 1–14. doi:10.1002/humu.22452
Das, B. B., Dexheimer, T. S., Maddali, K., and Pommier, Y. (2010). Role of tyrosyl-DNA phosphodiesterase (TDP1) in mitochondria. Proc. Natl. Acad. Sci. U. S. A. 107, 19790–19795. doi:10.1073/pnas.1009814107
Date, H., OnOdera, O., Tanaka, H., Iwabuchi, K., UeKawa, K., IgaraShi, S., et al. (2001). Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat. Genet. 29, 184–188. doi:10.1038/ng1001-184
Davies, V. J., Hollins, A. J., Piechota, M. J., Yip, W., Davies, J. R., White, K. E., et al. (2007). Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum. Mol. Genet. 16, 1307–1318. doi:10.1093/hmg/ddm079
de Souza-Pinto, N. C., Eide, L., Hogue, B. A., Thybo, T., STevnsner, T., SEEbErg, E., et al. (2001). Repair of 8-oxodeoxyguanosine lesions in mitochondrial dna depends on the oxoguanine dna glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial dna of OGG1-defective mice. Cancer Res. 61, 5378–5381.
de Souza-Pinto, N. C., Mason, P. A., Hashiguchi, K., Weissman, L., Tian, J., Guay, D., et al. (2009). Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair (Amst) 8, 704–719. doi:10.1016/j.dnarep.2009.01.021
DeBalsi, K. L., Hoff, K. E., and Copeland, W. C. (2017). Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Ageing Res. Rev. 33, 89–104. doi:10.1016/j.arr.2016.04.006
Delettre, C., Lenaers, G., Griffoin, J. M., GigarelN., , Lorenzo, C., Belenguer, P., et al. (2000). Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 26, 207–210. doi:10.1038/79936
Dizdaroglu, M., Coskun, E., and Jaruga, P. (2015). Measurement of oxidatively induced DNA damage and its repair, by mass spectrometric techniques. Free Radic. Res. 49, 525–548. doi:10.3109/10715762.2015.1014814
Dong, D. W., Pereira, F., Barrett, S. P., Kolesar, J. E., Cao, K., Damas, J., et al. (2014). Association of G-quadruplex forming sequences with human mtDNA deletion breakpoints. BMC Genomics 15, 677. doi:10.1186/1471-2164-15-677
Dorn, G. W., and Dang, X. (2022). Predicting mitochondrial dynamic behavior in genetically defined neurodegenerative diseases. Cells 11, 1049. doi:10.3390/cells11061049
Dou, H., Mitra, S., and Hazra, T. K. (2003). Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J. Biol. Chem. 278, 49679–49684. doi:10.1074/jbc.M308658200
Drago, I., De Stefani, D., Rizzuto, R., and Pozzan, T. (2012). Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. U. S. A. 109, 12986–12991. doi:10.1073/pnas.1210718109
du Buy, H. G., and Riley, F. L. (1967). Hybridization between the nuclear and kinetoplast dna's of leishmania enriettii and between nuclear and mitochondrial dna's of mouse liver. Proc. Natl. Acad. Sci. U. S. A. 57, 790–797. doi:10.1073/pnas.57.3.790
Dyall, S. D., Brown, M. T., and Johnson, P. J. (2004). Ancient invasions: From endosymbionts to organelles. Science 304, 253–257. doi:10.1126/science.1094884
Ekstrand, M. I., Falkenberg, M., Rantanen, A., Park, C. B., Gaspari, M., Hultenby, K., et al. (2004). Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 13, 935–944. doi:10.1093/hmg/ddh109
El-Hattab, A. W., Craigen, W. J., and Scaglia, F. (2017). Mitochondrial DNA maintenance defects. Biochim. Biophys. Acta. Mol. Basis Dis. 1863, 1539–1555. doi:10.1016/j.bbadis.2017.02.017
Elachouri, G., Vidoni, S., Zanna, C., Pattyn, A., Boukhaddaoui, H., Gaget, K., et al. (2011). OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution. Genome Res. 21, 12–20. doi:10.1101/gr.108696.110
Elsakrmy, N., Zhang-Akiyama, Q. M., and Ramotar, D. (2020). The base excision repair pathway in the nematode Caenorhabditis elegans. Front. Cell Dev. Biol. 8, 598860. doi:10.3389/fcell.2020.598860
Elson, J. L., Samuels, D. C., Turnbull, D. M., and Chinnery, P. F. (2001). Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am. J. Hum. Genet. 68, 802–806. doi:10.1086/318801
Englander, E. W., and Ma, H. (2006). Differential modulation of base excision repair activities during brain ontogeny: Implications for repair of transcribed DNA. Mech. Ageing Dev. 127, 64–69. doi:10.1016/j.mad.2005.09.008
Falabella, M., Kolesar, J. E., WallaCe, C., De Jesus, D., Sun, L., Taguchi, Y. V., et al. (2019). G-quadruplex dynamics contribute to regulation of mitochondrial gene expression. Sci. Rep. 9, 5605. doi:10.1038/s41598-019-41464-y
Fernandez-Vizarra, E., Enriquez, J. A., Perez-Martos, A., Montoya, J., and Fernandez-Silva, P. (2011). Tissue-specific differences in mitochondrial activity and biogenesis. Mitochondrion 11, 207–213. doi:10.1016/j.mito.2010.09.011
Filograna, R., Koolmeister, C., UpadhyayM., , PAjAk, A., Clemente, P., Wibom, R., et al. (2019). Modulation of mtDNA copy number ameliorates the pathological consequences of a heteroplasmic mtDNA mutation in the mouse. Sci. Adv. 5, eaav9824. doi:10.1126/sciadv.aav9824
Filograna, R., Mennuni, M., Alsina, D., and Larsson, N. G. (2021). Mitochondrial DNA copy number in human disease: The more the better? FEBS Lett. 595, 976–1002. doi:10.1002/1873-3468.14021
Fisher, R. P., and Clayton, D. A. (1985). A transcription factor required for promoter recognition by human mitochondrial RNA polymerase. Accurate initiation at the heavy- and light-strand promoters dissected and reconstituted in vitro. J. Biol. Chem. 260, 11330–11338. doi:10.1016/s0021-9258(17)39184-6
Fleming, A. M., Ding, Y., and Burrows, C. J. (2017). Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc. Natl. Acad. Sci. U. S. A. 114, 2604–2609. doi:10.1073/pnas.1619809114
Fleming, A. M., Zhu, J., Ding, Y., and Burrows, C. J. (2017). 8-Oxo-7, 8-dihydroguanine in the context of a gene promoter G-quadruplex is an on-off switch for transcription. ACS Chem. Biol. 12, 2417–2426. doi:10.1021/acschembio.7b00636
Fowler, F. C., Chen, B. R., Zolnerowich, N., Wu, W., Pavani, R., Paiano, J., et al. (2022). DNA-PK promotes DNA end resection at DNA double strand breaks in G0 cells. Elife 11, e74700. doi:10.7554/eLife.74700
Fuke, S., Kubota-Sakashita, M., Kasahara, T., Shigeyoshi, Y., and Kato, T. (2011). Regional variation in mitochondrial DNA copy number in mouse brain. Biochim. Biophys. Acta 1807, 270–274. doi:10.1016/j.bbabio.2010.11.016
Gaare, J. J., Nido, G. S., Sztromwasser, P., Knappskog, P. M., Dahl, O., Lund-Johansen, M., et al. (2018). Rare genetic variation in mitochondrial pathways influences the risk for Parkinson's disease. Mov. Disord. 33, 1591–1600. doi:10.1002/mds.64
Gahl, R. F., Dwivedi, P., and Tjandra, N. (2016). Bcl-2 proteins bid and bax form a network to permeabilize the mitochondria at the onset of apoptosis. Cell Death Dis. 7, e2424. doi:10.1038/cddis.2016.320
Gai, X., Ghezzi, D., Johnson, M. A., Biagosch, C. A., Shamseldin, H. E., Haack, T. B., et al. (2013). Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am. J. Hum. Genet. 93, 482–495. doi:10.1016/j.ajhg.2013.07.016
Gao, Y., Katyal, S., Lee, Y., Zhao, J., Rehg, J. E., Russell, H. R., et al. (2011). DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature 471, 240–244. doi:10.1038/nature09773
Giacomello, M., Pyakurel, A., Glytsou, C., and Scorrano, L. (2020). The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 21, 204–224. doi:10.1038/s41580-020-0210-7
Gokey, N. G., Cao, Z., Pak, J. W., Lee, D., McKiernan, S. H., McKenzie, D., et al. (2004). Molecular analyses of mtDNA deletion mutations in microdissected skeletal muscle fibers from aged rhesus monkeys. Aging Cell 3, 319–326. doi:10.1111/j.1474-9728.2004.00122.x
Gorman, G. S., Chinnery, P. F., DiMauro, S., Hirano, M., Koga, Y., McFarland, R., et al. (2016). Mitochondrial diseases. Nat. Rev. Dis. Prim. 2, 16080. doi:10.1038/nrdp.2016.80
Goto, Y., Nonaka, I., and Horai, S. (1990). A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 348, 651–653. doi:10.1038/348651a0
Grady, J. P., Pickett, S. J., Ng, Y. S., Alston, C. L., Blakely, E. L., Hardy, S. A., et al. (2018). mtDNA heteroplasmy level and copy number indicate disease burden in m.3243A>G mitochondrial disease. EMBO Mol. Med. 10, e8262. doi:10.15252/emmm.201708262
Graziewicz, M. A., Bienstock, R. J., and Copeland, W. C. (2007). The DNA polymerase gamma Y955C disease variant associated with PEO and parkinsonism mediates the incorporation and translesion synthesis opposite 7, 8-dihydro-8-oxo-2'-deoxyguanosine. Hum. Mol. Genet. 16, 2729–2739. doi:10.1093/hmg/ddm227
Gross, N. J., Getz, G. S., and Rabinowitz, M. (1969). Apparent turnover of mitochondrial deoxyribonucleic acid and mitochondrial phospholipids in the tissues of the rat. J. Biol. Chem. 244, 1552–1562. doi:10.1016/s0021-9258(18)91795-3
Guja, K. E., and Garcia-Diaz, M. (2012). Hitting the brakes: Termination of mitochondrial transcription. Biochim. Biophys. Acta 1819, 939–947. doi:10.1016/j.bbagrm.2011.11.004
Halsne, R., Esbensen, Y., Wang, W., Scheffler, K., Suganthan, R., Bjoras, M., et al. (2012). Lack of the DNA glycosylases MYH and OGG1 in the cancer prone double mutant mouse does not increase mitochondrial DNA mutagenesis. DNA Repair (Amst) 11, 278–285. doi:10.1016/j.dnarep.2011.12.001
Hamilton, M. L., Guo, Z., Fuller, C. D., Van Remmen, H., Ward, W. F., Austad, S. N., et al. (2001). A reliable assessment of 8-oxo-2-deoxyguanosine levels in nuclear and mitochondrial DNA using the sodium iodide method to isolate DNA. Nucleic Acids Res. 29, 2117–2126. doi:10.1093/nar/29.10.2117
Han, D., Schomacher, L., Schule, K. M., Mallick, M., Musheev, M. U., Karaulanov, E., et al. (2019). NEIL1 and NEIL2 DNA glycosylases protect neural crest development against mitochondrial oxidative stress. Elife 8, e49044. doi:10.7554/eLife.49044
Han, Z., and Stachow, C. (1994). Analysis of Schizosaccharomyces pombe mitochondrial DNA replication by two dimensional gel electrophoresis. Chromosoma 103, 162–170. doi:10.1007/BF00368008
Hazra, T. K., Izumi, T., Boldogh, I., Imhoff, B., Kow, Y. W., Jaruga, P., et al. (2002). Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. U. S. A. 99, 3523–3528. doi:10.1073/pnas.062053799
Hazra, T. K., Kow, Y. W., Hatahet, Z., Imhoff, B., Boldogh, I., Mokkapati, S. K., et al. (2002). Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J. Biol. Chem. 277, 30417–30420. doi:10.1074/jbc.C200355200
Hilbert, T. P., Chaung, W., Boorstein, R. J., Cunningham, R. P., and Teebor, G. W. (1997). Cloning and expression of the cDNA encoding the human homologue of the DNA repair enzyme, Escherichia coli endonuclease III. J. Biol. Chem. 272, 6733–6740. doi:10.1074/jbc.272.10.6733
Hillen, H. S., Morozov, Y. I., Sarfallah, A., Temiakov, D., and Cramer, P. (2017). Structural basis of mitochondrial transcription initiation. Cell 171, 1072–1081. doi:10.1016/j.cell.2017.10.036
Hillen, H. S., Parshin, A. V., Agaronyan, K., Morozov, Y. I., Graber, J. J., Chernev, A., et al. (2017). Mechanism of transcription anti-termination in human mitochondria. Cell 171, 1082–1093. e1013. doi:10.1016/j.cell.2017.09.035
Hindi, N. N., Elsakrmy, N., and Ramotar, D. (2021). The base excision repair process: Comparison between higher and lower eukaryotes. Cell. Mol. Life Sci. 78, 7943–7965. doi:10.1007/s00018-021-03990-9
Hindson, B. J., Ness, K. D., Masquelier, D. A., Belgrader, P., Heredia, N. J., Makarewicz, A. J., et al. (2011). High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 83, 8604–8610. doi:10.1021/ac202028g
Holt, I. J., Lorimer, H. E., and Jacobs, H. T. (2000). Coupled leading- and lagging-strand synthesis of mammalian mitochondrial DNA. Cell 100, 515–524. doi:10.1016/s0092-8674(00)80688-1
Hudson, E. K., Hogue, B. A., Souza-Pinto, N. C., Croteau, D. L., Anson, R. M., Bohr, V. A., et al. (1998). Age-associated change in mitochondrial DNA damage. Free Radic. Res. 29, 573–579. doi:10.1080/10715769800300611
Hudson, G., Amati-Bonneau, P., Blakely, E. L., Stewart, J. D., He, L., Schaefer, A. M., et al. (2008). Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: A novel disorder of mtDNA maintenance. Brain 131, 329–337. doi:10.1093/brain/awm272
Ichikawa, J., Tsuchimoto, D., Oka, S., Ohno, M., Furuichi, M., Sakumi, K., et al. (2008). Oxidation of mitochondrial deoxynucleotide pools by exposure to sodium nitroprusside induces cell death. DNA Repair (Amst) 7, 418–430. doi:10.1016/j.dnarep.2007.11.007
Ikeda, S., Biswas, T., Roy, R., Izumi, T., Boldogh, I., Kurosky, A., et al. (1998). Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. J. Biol. Chem. 273, 21585–21593. doi:10.1074/jbc.273.34.21585
Imai, K., Slupphaug, G., Lee, W. I., Revy, P., Nonoyama, S., Catalan, N., et al. (2003). Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat. Immunol. 4, 1023–1028. doi:10.1038/ni974
Itsara, L. S., Kennedy, S. R., Fox, E. J., Yu, S., Hewitt, J. J., Sanchez-Contreras, M., et al. (2014). Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PLoS Genet. 10, e1003974. doi:10.1371/journal.pgen.1003974
Jackson, E. B., Theriot, C. A., Chattopadhyay, R., Mitra, S., and Izumi, T. (2005). Analysis of nuclear transport signals in the human apurinic/apyrimidinic endonuclease (APE1/Ref1). Nucleic Acids Res. 33, 3303–3312. doi:10.1093/nar/gki641
Jaruga, P., Birincioglu, M., Rosenquist, T. A., and Dizdaroglu, M. (2004). Mouse NEIL1 protein is specific for excision of 2, 6-diamino-4-hydroxy-5-formamidopyrimidine and 4, 6-diamino-5-formamidopyrimidine from oxidatively damaged DNA. Biochemistry 43, 15909–15914. doi:10.1021/bi048162l
Jeruzalmi, D., and Steitz, T. A. (1998). Structure of T7 RNA polymerase complexed to the transcriptional inhibitor T7 lysozyme. EMBO J. 17, 4101–4113. doi:10.1093/emboj/17.14.4101
Jiang, M., Xie, X., Zhu, X., Jiang, S., Milenkovic, D., Misic, J., et al. (2021). The mitochondrial single-stranded DNA binding protein is essential for initiation of mtDNA replication. Sci. Adv. 7, eabf8631. doi:10.1126/sciadv.abf8631
Johnson, A. A., and Johnson, K. A. (2001). Fidelity of nucleotide incorporation by human mitochondrial DNA polymerase. J. Biol. Chem. 276, 38090–38096. doi:10.1074/jbc.M106045200
Jun, Y. W., Albarran, E., Wilson, D. L., Ding, J., and Kool, E. T. (2022). Fluorescence imaging of mitochondrial DNA base excision repair reveals dynamics of oxidative stress responses. Angew. Chem. Int. Ed. Engl. 61, e202111829. doi:10.1002/anie.202111829
Kang, D., Nishida, J., IyAmA, A., Nakabeppu, Y., FuruichiM., , Fujiwara, T., et al. (1995). Intracellular localization of 8-oxo-dGTPase in human cells, with special reference to the role of the enzyme in mitochondria. J. Biol. Chem. 270, 14659–14665. doi:10.1074/jbc.270.24.14659
Karahalil, B., Hogue, B. A., de Souza-Pinto, N. C., and Bohr, V. A. (2002). Base excision repair capacity in mitochondria and nuclei: Tissue-specific variations. FASEB J. 16, 1895–1902. doi:10.1096/fj.02-0463com
Kasamatsu, H., Robberson, D. L., and Vinograd, J. (1971). A novel closed-circular mitochondrial DNA with properties of a replicating intermediate. Proc. Natl. Acad. Sci. U. S. A. 68, 2252–2257. doi:10.1073/pnas.68.9.2252
Kaukonen, J., Juselius, J. K., Tiranti, V., Kyttala, A., ZevianiM., , Comi, G. P., et al. (2000). Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science 289, 782–785. doi:10.1126/science.289.5480.782
Kauppila, J. H. K., Bonekamp, N. A., Mourier, A., Isokallio, M. A., Just, A., Kauppila, T. E. S., et al. (2018). Base-excision repair deficiency alone or combined with increased oxidative stress does not increase mtDNA point mutations in mice. Nucleic Acids Res. 46, 6642–6669. doi:10.1093/nar/gky456
Kazak, L., Reyes, A., and Holt, I. J. (2012). Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 13, 659–671. doi:10.1038/nrm3439
Kitada, T., ASakawa, S., HattoriN., , Matsumine, H., Yamamura, Y., MinoShima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi:10.1038/33416
Klinge, C. M. (2020). Estrogenic control of mitochondrial function. Redox Biol. 31, 101435. doi:10.1016/j.redox.2020.101435
Komakula, S. S. B., Tumova, J., Kumaraswamy, D., Burchat, N., Vartanian, V., Ye, H., et al. (2018). The DNA repair protein OGG1 protects against obesity by altering mitochondrial energetics in white adipose tissue. Sci. Rep. 8, 14886. doi:10.1038/s41598-018-33151-1
Kosar, M., Piccini, D., Foiani, M., and Giannattasio, M. (2021). A rapid method to visualize human mitochondrial DNA replication through rotary shadowing and transmission electron microscopy. Nucleic Acids Res. 49, e121. doi:10.1093/nar/gkab770
Koshiba, T., Detmer, S. A., Kaiser, J. T., Chen, H., McCaffery, J. M., and Chan, D. C. (2004). Structural basis of mitochondrial tethering by mitofusin complexes. Science 305, 858–862. doi:10.1126/science.1099793
Krasich, R., and Copeland, W. C. (2017). DNA polymerases in the mitochondria: A critical review of the evidence. Front. Biosci. 22, 692–709. doi:10.2741/4510
Krishnamurthy, N., Haraguchi, K., Greenberg, M. M., and David, S. S. (2008). Efficient removal of formamidopyrimidines by 8-oxoguanine glycosylases. Biochemistry 47, 1043–1050. doi:10.1021/bi701619u
Krokan, H. E., and Bjoras, M. (2013). Base excision repair. Cold Spring Harb. Perspect. Biol. 5, a012583. doi:10.1101/cshperspect.a012583
Kruse, B., Narasimhan, N., and Attardi, G. (1989). Termination of transcription in human mitochondria: Identification and purification of a DNA binding protein factor that promotes termination. Cell 58, 391–397. doi:10.1016/0092-8674(89)90853-2
Kukat, C., Wurm, C. A., Spahr, H., Falkenberg, M., Larsson, N. G., and Jakobs, S. (2011). Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Natl. Acad. Sci. U. S. A. 108, 13534–13539. doi:10.1073/pnas.1109263108
Ladner, R. D., and Caradonna, S. J. (1997). The human dUTPase gene encodes both nuclear and mitochondrial isoforms. Differential expression of the isoforms and characterization of a cDNA encoding the mitochondrial species. J. Biol. Chem. 272, 19072–19080. doi:10.1074/jbc.272.30.19072
Lakshmipathy, U., and Campbell, C. (2001). Antisense-mediated decrease in DNA ligase III expression results in reduced mitochondrial DNA integrity. Nucleic Acids Res. 29, 668–676. doi:10.1093/nar/29.3.668
Lakshmipathy, U., and Campbell, C. (1999). Double strand break rejoining by mammalian mitochondrial extracts. Nucleic Acids Res. 27, 1198–1204. doi:10.1093/nar/27.4.1198
Lang, B. F., Gray, M. W., and Burger, G. (1999). Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 33, 351–397. doi:10.1146/annurev.genet.33.1.351
Lans, H., Hoeijmakers, J. H. J., Vermeulen, W., and Marteijn, J. A. (2019). The DNA damage response to transcription stress. Nat. Rev. Mol. Cell Biol. 20, 766–784. doi:10.1038/s41580-019-0169-4
Larsson, N. G. (2010). Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 79, 683–706. doi:10.1146/annurev-biochem-060408-093701
Lauritzen, K. H., Moldestad, O., Eide, L., Carlsen, H., Nesse, G., Storm, J. F., et al. (2010). Mitochondrial DNA toxicity in forebrain neurons causes apoptosis, neurodegeneration, and impaired behavior. Mol. Cell. Biol. 30, 1357–1367. doi:10.1128/MCB.01149-09
LeDoux, S. P., Wilson, G. L., Beecham, E. J., STevnsner, T., Wassermann, K., and Bohr, V. A. (1992). Repair of mitochondrial DNA after various types of DNA damage in Chinese hamster ovary cells. Carcinogenesis 13, 1967–1973. doi:10.1093/carcin/13.11.1967
Li, A., Gao, M., Liu, B., Qin, Y., Chen, L., Liu, H., et al. (2022). Mitochondrial autophagy: Molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. 13, 444. doi:10.1038/s41419-022-04906-6
Li, M. X., Shan, J. L., Wang, D., He, Y., Zhou, Q., Xia, L., et al. (2012). Human apurinic/apyrimidinic endonuclease 1 translocalizes to mitochondria after photodynamic therapy and protects cells from apoptosis. Cancer Sci. 103, 882–888. doi:10.1111/j.1349-7006.2012.02239.x
Li, M., Zhong, Z., Zhu, J., Xiang, D., Dai, N., Cao, X., et al. (2010). Identification and characterization of mitochondrial targeting sequence of human apurinic/apyrimidinic endonuclease 1. J. Biol. Chem. 285, 14871–14881. doi:10.1074/jbc.M109.069591
Liao, C., Ashley, N., Diot, A., Morten, K., Phadwal, K., Williams, A., et al. (2017). Dysregulated mitophagy and mitochondrial organization in optic atrophy due to OPA1 mutations. Neurology 88, 131–142. doi:10.1212/WNL.0000000000003491
Likhite, V. S., Cass, E. I., Anderson, S. D., Yates, J. R., and Nardulli, A. M. (2004). Interaction of estrogen receptor alpha with 3-methyladenine DNA glycosylase modulates transcription and DNA repair. J. Biol. Chem. 279, 16875–16882. doi:10.1074/jbc.M313155200
Litonin, D., Sologub, M., Shi, Y., Savkina, M., Anikin, M., Falkenberg, M., et al. (2010). Human mitochondrial transcription revisited: Only TFAM and TFB2M are required for transcription of the mitochondrial genes in vitro. J. Biol. Chem. 285, 18129–18133. doi:10.1074/jbc.C110.128918
Liu, J., Fang, H., Chi, Z., Wu, Z., Wei, D., Mo, D., et al. (2015). XPD localizes in mitochondria and protects the mitochondrial genome from oxidative DNA damage. Nucleic Acids Res. 43, 5476–5488. doi:10.1093/nar/gkv472
Liu, P., Burdzy, A., and Sowers, L. C. (2002). Substrate recognition by a family of uracil-DNA glycosylases: UNG, MUG, and TDG. Chem. Res. Toxicol. 15, 1001–1009. doi:10.1021/tx020030a
Lodeiro, M. F., Uchida, A., Bestwick, M., Moustafa, I. M., Arnold, J. J., Shadel, G. S., et al. (2012). Transcription from the second heavy-strand promoter of human mtDNA is repressed by transcription factor A in vitro. Proc. Natl. Acad. Sci. U. S. A. 109, 6513–6518. doi:10.1073/pnas.1118710109
Longley, M. J., Prasad, R., Srivastava, D. K., Wilson, S. H., and Copeland, W. C. (1998). Identification of 5'-deoxyribose phosphate lyase activity in human DNA polymerase gamma and its role in mitochondrial base excision repair in vitro. Proc. Natl. Acad. Sci. U. S. A. 95, 12244–12248. doi:10.1073/pnas.95.21.12244
Lopez, J. V., Yuhki, N., Masuda, R., Modi, W., O'Brien, S. J., and Numt, S. J. (1994). Numt, a recent transfer and tandem amplification of mitochondrial DNA to the nuclear genome of the domestic cat. J. Mol. Evol. 39, 174–190. doi:10.1007/BF00163806
Loson, O. C., Song, Z., Chen, H., Chan, D. C., and Fis1, Mff (2013). Fis1, mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 24, 659–667. doi:10.1091/mbc.E12-10-0721
Macias, E., Rao, D., Carbajal, S., Kiguchi, K., and DiGiovanni, J. (2014). Stat3 binds to mtDNA and regulates mitochondrial gene expression in keratinocytes. J. Invest. Dermatol. 134, 1971–1980. doi:10.1038/jid.2014.68
Mandal, S. M., Hegde, M. L., Chatterjee, A., Hegde, P. M., Szczesny, B., Banerjee, D., et al. (2012). Role of human DNA glycosylase Nei-like 2 (NEIL2) and single strand break repair protein polynucleotide kinase 3'-phosphatase in maintenance of mitochondrial genome. J. Biol. Chem. 287, 2819–2829. doi:10.1074/jbc.M111.272179
Mason, P. A., Matheson, E. C., Hall, A. G., and Lightowlers, R. N. (2003). Mismatch repair activity in mammalian mitochondria. Nucleic Acids Res. 31, 1052–1058. doi:10.1093/nar/gkg167
Maynard, S., de Souza-Pinto, N. C., Scheibye-Knudsen, M., and Bohr, V. A. (2010). Mitochondrial base excision repair assays. Methods 51, 416–425. doi:10.1016/j.ymeth.2010.02.020
Mecocci, P., MacGarvey, U., Kaufman, A. E., Koontz, D., Shoffner, J. M., Wallace, D. C., et al. (1993). Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann. Neurol. 34, 609–616. doi:10.1002/ana.410340416
Michaels, M. L., Cruz, C., Grollman, A. P., and Miller, J. H. (1992). Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc. Natl. Acad. Sci. U. S. A. 89, 7022–7025. doi:10.1073/pnas.89.15.7022
Minczuk, M., He, J., Duch, A. M., Ettema, T. J., Chlebowski, A., Dzionek, K., et al. (2011). TEFM (c17orf42) is necessary for transcription of human mtDNA. Nucleic Acids Res. 39, 4284–4299. doi:10.1093/nar/gkq1224
Mjelle, R., Hegre, S. A., Aas, P. A., Slupphaug, G., Drablos, F., Saetrom, P., et al. (2015). Cell cycle regulation of human DNA repair and chromatin remodeling genes. DNA Repair (Amst) 30, 53–67. doi:10.1016/j.dnarep.2015.03.007
Montaldo, N. P., Bordin, D. L., Brambilla, A., Rosinger, M., Fordyce Martin, S. L., Bjoras, K. O., et al. (2019). Alkyladenine DNA glycosylase associates with transcription elongation to coordinate DNA repair with gene expression. Nat. Commun. 10, 5460. doi:10.1038/s41467-019-13394-w
Montoya, J., Gaines, G. L., and Attardi, G. (1983). The pattern of transcription of the human mitochondrial rRNA genes reveals two overlapping transcription units. Cell 34, 151–159. doi:10.1016/0092-8674(83)90145-9
Moraes, C. T., DiMauro, S., ZevianiM., , Lombes, A., ShanSke, S., Miranda, A. F., et al. (1989). Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. N. Engl. J. Med. 320, 1293–1299. doi:10.1056/NEJM198905183202001
Moretton, A., Morel, F., Macao, B., Lachaume, P., Ishak, L., Lefebvre, M., et al. (2017). Selective mitochondrial DNA degradation following double-strand breaks. PLoS One 12, e0176795. doi:10.1371/journal.pone.0176795
Morozov, Y. I., Agaronyan, K., Cheung, A. C. M., Anikin, M., Cramer, P., and Temiakov, D. (2014). A novel intermediate in transcription initiation by human mitochondrial RNA polymerase. Nucleic Acids Res. 42, 3884–3893. doi:10.1093/nar/gkt1356
Mourier, T., Hansen, A. J., Willerslev, E., and Arctander, P. (2001). The Human Genome Project reveals a continuous transfer of large mitochondrial fragments to the nucleus. Mol. Biol. Evol. 18, 1833–1837. doi:10.1093/oxfordjournals.molbev.a003971
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi:10.1042/BJ20081386
Myers, K. A., Saffhill, R., and O'Connor, P. J. (1988). Repair of alkylated purines in the hepatic DNA of mitochondria and nuclei in the rat. Carcinogenesis 9, 285–292. doi:10.1093/carcin/9.2.285
Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D. F., Gautier, C. A., Shen, J., et al. (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298. doi:10.1371/journal.pbio.1000298
Nass, M. M., and Nass, S. (1963). Intramitochondrial fibers with DNA characteristics. I. Fixation and electron staining reactions. J. Cell Biol. 19, 593–611. doi:10.1083/jcb.19.3.593
Nass, S., and Nass, M. M. (1963). Intramitochondrial fibers with DNA characteristics. Ii. Enzymatic and other hydrolytic treatments. J. Cell Biol. 19, 613–629. doi:10.1083/jcb.19.3.613
Nicholls, T. J., and Minczuk, M. (2014). In D-loop: 40 years of mitochondrial 7S DNA. Exp. Gerontol. 56, 175–181. doi:10.1016/j.exger.2014.03.027
Nilsen, H., OtterleiM., , Haug, T., Solum, K., Nagelhus, T. A., SkorpenF., , et al. (1997). Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res. 25, 750–755. doi:10.1093/nar/25.4.750
Nishioka, K., OhTsubo, T., Oda, H., Fujiwara, T., Kang, D., Sugimachi, K., et al. (1999). Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell 10, 1637–1652. doi:10.1091/mbc.10.5.1637
Nissanka, N., Bacman, S. R., Plastini, M. J., and Moraes, C. T. (2018). The mitochondrial DNA polymerase gamma degrades linear DNA fragments precluding the formation of deletions. Nat. Commun. 9, 2491. doi:10.1038/s41467-018-04895-1
Ohtsubo, T., NishioKa, K., Imaiso, Y., Iwai, S., SHimokawa, H., Oda, H., et al. (2000). Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res. 28, 1355–1364. doi:10.1093/nar/28.6.1355
Oka, S., Ohno, M., Tsuchimoto, D., Sakumi, K., Furuichi, M., and Nakabeppu, Y. (2008). Two distinct pathways of cell death triggered by oxidative damage to nuclear and mitochondrial DNAs. EMBO J. 27, 421–432. doi:10.1038/sj.emboj.7601975
Olsen, L. C., Aasland, R., Wittwer, C. U., Krokan, H. E., and Helland, D. E. (1989). Molecular cloning of human uracil-DNA glycosylase, a highly conserved DNA repair enzyme. EMBO J. 8, 3121–3125. doi:10.1002/j.1460-2075.1989.tb08464.x
Otterlei, M., Haug, T., Nagelhus, T. A., SlupphauG, G., Lindmo, T., and Krokan, H. E. (1998). Nuclear and mitochondrial splice forms of human uracil-DNA glycosylase contain a complex nuclear localisation signal and a strong classical mitochondrial localisation signal, respectively. Nucleic Acids Res. 26, 4611–4617. doi:10.1093/nar/26.20.4611
Peeva, V., Blei, D., Trombly, G., Corsi, S., Szukszto, M. J., Rebelo-Guiomar, P., et al. (2018). Linear mitochondrial DNA is rapidly degraded by components of the replication machinery. Nat. Commun. 9, 1727. doi:10.1038/s41467-018-04131-w
Peralta, S., Goffart, S., Williams, S. L., Diaz, F., Garcia, S., Nissanka, N., et al. (2018). ATAD3 controls mitochondrial cristae structure in mouse muscle, influencing mtDNA replication and cholesterol levels. J. Cell Sci. 131, jcs217075. doi:10.1242/jcs.217075
Perillo, B., Ombra, M. N., Bertoni, A., Cuozzo, C., Sacchetti, S., Sasso, A., et al. (2008). DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 319, 202–206. doi:10.1126/science.1147674
Phillips, N. R., Sprouse, M. L., and Roby, R. K. (2014). Simultaneous quantification of mitochondrial DNA copy number and deletion ratio: A multiplex real-time PCR assay. Sci. Rep. 4, 3887. doi:10.1038/srep03887
Pohjoismaki, J. L., Wanrooij, S., Hyvarinen, A. K., Goffart, S., Holt, I. J., Spelbrink, J. N., et al. (2006). Alterations to the expression level of mitochondrial transcription factor A, TFAM, modify the mode of mitochondrial DNA replication in cultured human cells. Nucleic Acids Res. 34, 5815–5828. doi:10.1093/nar/gkl703
Posse, V., Shahzad, S., Falkenberg, M., Hallberg, B. M., and Gustafsson, C. M. (2015). TEFM is a potent stimulator of mitochondrial transcription elongation in vitro. Nucleic Acids Res. 43, 2615–2624. doi:10.1093/nar/gkv105
Pronsato, L., Milanesi, L., and Vasconsuelo, A. (2020). Testosterone induces up-regulation of mitochondrial gene expression in murine C2C12 skeletal muscle cells accompanied by an increase of nuclear respiratory factor-1 and its downstream effectors. Mol. Cell. Endocrinol. 500, 110631. doi:10.1016/j.mce.2019.110631
Rachek, L. I., Grishko, V. I., Musiyenko, S. I., Kelley, M. R., LeDoux, S. P., and Wilson, G. L. (2002). Conditional targeting of the DNA repair enzyme hOGG1 into mitochondria. J. Biol. Chem. 277, 44932–44937. doi:10.1074/jbc.M208770200
Ramachandran, A., Basu, U., Sultana, S., Nandakumar, D., and Patel, S. S. (2017). Human mitochondrial transcription factors TFAM and TFB2M work synergistically in promoter melting during transcription initiation. Nucleic Acids Res. 45, 861–874. doi:10.1093/nar/gkw1157
Rath, S., Sharma, R., Gupta, R., Ast, T., Chan, C., Durham, T. J., et al. (2021). MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49, D1541–D1547. doi:10.1093/nar/gkaa1011
Ringel, R., Sologub, M., Morozov, Y. I., Litonin, D., Cramer, P., and Temiakov, D. (2011). Structure of human mitochondrial RNA polymerase. Nature 478, 269–273. doi:10.1038/nature10435
Romanello, V., and Sandri, M. (2022). Implications of mitochondrial fusion and fission in skeletal muscle mass and health. Semin. Cell Dev. Biol. S1084-9521 (22), 00050–00057. doi:10.1016/j.semcdb.2022.02.011
Roychoudhury, S., Pramanik, S., Harris, H. L., Tarpley, M., Sarkar, A., Spagnol, G., et al. (2020). Endogenous oxidized DNA bases and APE1 regulate the formation of G-quadruplex structures in the genome. Proc. Natl. Acad. Sci. U. S. A. 117, 11409–11420. doi:10.1073/pnas.1912355117
Sabouny, R., and Shutt, T. E. (2021). The role of mitochondrial dynamics in mtDNA maintenance. J. Cell Sci. 134, jcs258944. doi:10.1242/jcs.258944
Samson, L., Derfler, B., Boosalis, M., and Call, K. (1991). Cloning and characterization of a 3-methyladenine DNA glycosylase cDNA from human cells whose gene maps to chromosome 16. Proc. Natl. Acad. Sci. U. S. A. 88, 9127–9131. doi:10.1073/pnas.88.20.9127
Santos, J. H., Meyer, J. N., Mandavilli, B. S., and Van Houten, B. (2006). Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol. 314, 183–199. doi:10.1385/1-59259-973-7:183
Satoh, M. S., Huh, N., Rajewsky, M. F., and Kuroki, T. (1988). Enzymatic removal of O6-ethylguanine from mitochondrial DNA in rat tissues exposed to N-ethyl-N-nitrosourea in vivo. J. Biol. Chem. 263, 6854–6856. doi:10.1016/s0021-9258(18)68722-8
Sekiguchi, M., Hayakawa, H., Makino, F., Tanaka, K., and Okada, Y. (1976). A human enzyme that liberates uracil from DNA. Biochem. Biophys. Res. Commun. 73, 293–299. doi:10.1016/0006-291x(76)90706-3
SenGupta, T., Palikaras, K., Esbensen, Y. Q., Konstantinidis, G., Galindo, F. J. N., Achanta, K., et al. (2021). Base excision repair causes age-dependent accumulation of single-stranded DNA breaks that contribute to Parkinson disease pathology. Cell Rep. 36, 109668. doi:10.1016/j.celrep.2021.109668
Shadel, G. S., and Horvath, T. L. (2015). Mitochondrial ROS signaling in organismal homeostasis. Cell 163, 560–569. doi:10.1016/j.cell.2015.10.001
Sharma, N., Chakravarthy, S., Longley, M. J., Copeland, W. C., and Prakash, A. (2018). The C-terminal tail of the NEIL1 DNA glycosylase interacts with the human mitochondrial single-stranded DNA binding protein. DNA Repair (Amst) 65, 11–19. doi:10.1016/j.dnarep.2018.02.012
Shokolenko, I. N., Wilson, G. L., and Alexeyev, M. F. (2013). Persistent damage induces mitochondrial DNA degradation. DNA Repair (Amst) 12, 488–499. doi:10.1016/j.dnarep.2013.04.023
Shokolenko, I., Venediktova, N., Bochkareva, A., Wilson, G. L., and Alexeyev, M. F. (2009). Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 37, 2539–2548. doi:10.1093/nar/gkp100
Sidarala, V., Zhu, J., Levi-D'Ancona, E., Pearson, G. L., Reck, E. C., Walker, E. M., et al. (2022). Mitofusin 1 and 2 regulation of mitochondrial DNA content is a critical determinant of glucose homeostasis. Nat. Commun. 13, 2340. doi:10.1038/s41467-022-29945-7
Simsek, D., Furda, A., Gao, Y., Artus, J., Brunet, E., Hadjantonakis, A. K., et al. (2011). Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature 471, 245–248. doi:10.1038/nature09794
Slupphaug, G., Markussen, F. H., Olsen, L. C., Aasland, R., AarsaetherN., , Bakke, O., et al. (1993). Nuclear and mitochondrial forms of human uracil-DNA glycosylase are encoded by the same gene. Nucleic Acids Res. 21, 2579–2584. doi:10.1093/nar/21.11.2579
Sousa, R., Chung, Y. J., Rose, J. P., and Wang, B. C. (1993). Crystal structure of bacteriophage T7 RNA polymerase at 3.3 A resolution. Nature 364, 593–599. doi:10.1038/364593a0
Sugimoto, T., Mori, C., Takanami, T., Sasagawa, Y., Saito, R., Ichiishi, E., et al. (2008). Caenorhabditis elegans par2.1/mtssb-1 is essential for mitochondrial DNA replication and its defect causes comprehensive transcriptional alterations including a hypoxia response. Exp. Cell Res. 314, 103–114. doi:10.1016/j.yexcr.2007.08.015
Suter, M., and Richter, C. (1999). Fragmented mitochondrial DNA is the predominant carrier of oxidized DNA bases. Biochemistry 38, 459–464. doi:10.1021/bi9811922
Sykora, P., Croteau, D. L., Bohr, V. A., and Wilson, D. M. (2011). Aprataxin localizes to mitochondria and preserves mitochondrial function. Proc. Natl. Acad. Sci. U. S. A. 108, 7437–7442. doi:10.1073/pnas.1100084108
Sykora, P., Kanno, S., AkbariM., , Kulikowicz, T., Baptiste, B. A., Leandro, G. S., et al. (2017). DNA polymerase beta participates in mitochondrial DNA repair. Mol. Cell. Biol. 37, e00237–17. doi:10.1128/MCB.00237-17
Takao, M., Zhang, Q. M., Yonei, S., and Yasui, A. (1999). Differential subcellular localization of human MutY homolog (hMYH) and the functional activity of adenine:8-oxoguanine DNA glycosylase. Nucleic Acids Res. 27, 3638–3644. doi:10.1093/nar/27.18.3638
Takashima, H., Boerkoel, C. F., John, J., Saifi, G. M., Salih, M. A. M., Armstrong, D., et al. (2002). Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat. Genet. 32, 267–272. doi:10.1038/ng987
Taylor, R. W., and Turnbull, D. M. (2005). Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 6, 389–402. doi:10.1038/nrg1606
Tell, G., Damante, G., Caldwell, D., and Kelley, M. R. (2005). The intracellular localization of APE1/ref-1: More than a passive phenomenon? Antioxid. Redox Signal. 7, 367–384. doi:10.1089/ars.2005.7.367
Tiranti, V., SAvoiA, A., FortiF., , D'Apolito, M. F., CentraM., , RocchiM., , et al. (1997). Identification of the gene encoding the human mitochondrial RNA polymerase (h-mtRPOL) by cyberscreening of the Expressed Sequence Tags database. Hum. Mol. Genet. 6, 615–625. doi:10.1093/hmg/6.4.615
Tsuzuki, T., Nomiyama, H., Setoyama, C., Maeda, S., and Shimada, K. (1983). Presence of mitochondrial-DNA-like sequences in the human nuclear DNA. Gene 25, 223–229. doi:10.1016/0378-1119(83)90226-3
Uchida, A., Murugesapillai, D., Kastner, M., Wang, Y., Lodeiro, M. F., Prabhakar, S., et al. (2017). Unexpected sequences and structures of mtDNA required for efficient transcription from the first heavy-strand promoter. Elife 6, e27283. doi:10.7554/eLife.27283
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M. K., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304, 1158–1160. doi:10.1126/science.1096284
Valente, W. J., Ericson, N. G., Long, A. S., White, P. A., Marchetti, F., and Bielas, J. H. (2016). Mitochondrial DNA exhibits resistance to induced point and deletion mutations. Nucleic Acids Res. 44, 8513–8524. doi:10.1093/nar/gkw716
van Loon, B., and Samson, L. D. (2013). Alkyladenine DNA glycosylase (AAG) localizes to mitochondria and interacts with mitochondrial single-stranded binding protein (mtSSB). DNA Repair (Amst) 12, 177–187. doi:10.1016/j.dnarep.2012.11.009
Venegas, V., and Halberg, M. C. (2012). Measurement of mitochondrial DNA copy number. Methods Mol. Biol. 837, 327–335. doi:10.1007/978-1-61779-504-6_22
Viscomi, C., and Zeviani, M. (2017). MtDNA-maintenance defects: Syndromes and genes. J. Inherit. Metab. Dis. 40, 587–599. doi:10.1007/s10545-017-0027-5
Wallace, S. S., Bandaru, V., Kathe, S. D., and Bond, J. P. (2003). The enigma of endonuclease VIII. DNA Repair (Amst) 2, 441–453. doi:10.1016/s1568-7864(02)00182-9
Wang, R., Hao, W., Pan, L., Boldogh, I., and Ba, X. (2018). The roles of base excision repair enzyme OGG1 in gene expression. Cell. Mol. Life Sci. 75, 3741–3750. doi:10.1007/s00018-018-2887-8
Wang, W., Scheffler, K., Esbensen, Y., and Eide, L. (2016). Quantification of DNA damage by real-time qPCR. Methods Mol. Biol. 1351, 27–32. doi:10.1007/978-1-4939-3040-1_3
Watanabe, S., Ichimura, T., Fujita, N., Tsuruzoe, S., Ohki, I., Shirakawa, M., et al. (2003). Methylated DNA-binding domain 1 and methylpurine-DNA glycosylase link transcriptional repression and DNA repair in chromatin. Proc. Natl. Acad. Sci. U. S. A. 100, 12859–12864. doi:10.1073/pnas.2131819100
Wheeler, J. H., Young, C. K. J., and Young, M. J. (2019). Analysis of human mitochondrial DNA content by southern blotting and nonradioactive probe hybridization. Curr. Protoc. Toxicol. 80, e75. doi:10.1002/cptx.75
Wisnovsky, S., Jean, S. R., and Kelley, S. O. (2016). Mitochondrial DNA repair and replication proteins revealed by targeted chemical probes. Nat. Chem. Biol. 12, 567–573. doi:10.1038/nchembio.2102
Wollen Steen, K., Doseth, B., P Westbye, M., Akbari, M., Kang, D., Falkenberg, M., et al. (2012). mtSSB may sequester UNG1 at mitochondrial ssDNA and delay uracil processing until the dsDNA conformation is restored. DNA Repair (Amst) 11, 82–91. doi:10.1016/j.dnarep.2011.10.026
Xanthoudakis, S., and Curran, T. (1992). Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 11, 653–665. doi:10.1002/j.1460-2075.1992.tb05097.x
Xie, Y., Yang, H., Cunanan, C., Okamoto, K., Shibata, D., Pan, J., et al. (2004). Deficiencies in mouse Myh and Ogg1 result in tumor predisposition and G to T mutations in codon 12 of the K-ras oncogene in lung tumors. Cancer Res. 64, 3096–3102. doi:10.1158/0008-5472.can-03-3834
Xu, W., Boyd, R. M., Tree, M. O., Samkari, F., and Zhao, L. (2019). Mitochondrial transcription factor A promotes DNA strand cleavage at abasic sites. Proc. Natl. Acad. Sci. U. S. A. 116, 17792–17799. doi:10.1073/pnas.1911252116
Yakes, F. M., and Van Houten, B. (1997). Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. U. S. A. 94, 514–519. doi:10.1073/pnas.94.2.514
Youn, C. K., Kim, H. B., Wu, T. T., Park, S., Cho, S. I., and Lee, J. H. (2017). 53BP1 contributes to regulation of autophagic clearance of mitochondria. Sci. Rep. 7, 45290. doi:10.1038/srep45290
Yu-Wai-Man, P., Sitarz, K. S., Samuels, D. C., Griffiths, P. G., Reeve, A. K., Bindoff, L. A., et al. (2010). OPA1 mutations cause cytochrome c oxidase deficiency due to loss of wild-type mtDNA molecules. Hum. Mol. Genet. 19, 3043–3052. doi:10.1093/hmg/ddq209
Yuzefovych, L. V., Schuler, A. M., Chen, J., Alvarez, D. F., Eide, L., Ledoux, S. P., et al. (2013). Alteration of mitochondrial function and insulin sensitivity in primary mouse skeletal muscle cells isolated from transgenic and knockout mice: Role of ogg1. Endocrinology 154, 2640–2649. doi:10.1210/en.2013-1076
Yuzefovych, L. V., Solodushko, V. A., Wilson, G. L., and Rachek, L. I. (2012). Protection from palmitate-induced mitochondrial DNA damage prevents from mitochondrial oxidative stress, mitochondrial dysfunction, apoptosis, and impaired insulin signaling in rat L6 skeletal muscle cells. Endocrinology 153, 92–100. doi:10.1210/en.2011-1442
Zheng, J., Akbari, M., Schirmer, C., Reynaert, M. L., Loyens, A., Lefebvre, B., et al. (2020). Hippocampal tau oligomerization early in tau pathology coincides with a transient alteration of mitochondrial homeostasis and DNA repair in a mouse model of tauopathy. Acta Neuropathol. Commun. 8, 25. doi:10.1186/s40478-020-00896-8
Zhu, J., Fleming, A. M., and Burrows, C. J. (2018). The RAD17 promoter sequence contains a potential tail-dependent G-quadruplex that downregulates gene expression upon oxidative modification. ACS Chem. Biol. 13, 2577–2584. doi:10.1021/acschembio.8b00522
Zollo, O., Tiranti, V., and Sondheimer, N. (2012). Transcriptional requirements of the distal heavy-strand promoter of mtDNA. Proc. Natl. Acad. Sci. U. S. A. 109, 6508–6512. doi:10.1073/pnas.1118594109
Keywords: mitochondria, DNA repair, base excision repair (BER), base excision repair (BER)glycosylases, transcription, mitochdrial damage
Citation: Akbari M, Nilsen HL and Montaldo NP (2022) Dynamic features of human mitochondrial DNA maintenance and transcription. Front. Cell Dev. Biol. 10:984245. doi: 10.3389/fcell.2022.984245
Received: 01 July 2022; Accepted: 02 August 2022;
Published: 07 September 2022.
Edited by:
Konstantinos Palikaras, National and Kapodistrian University of Athens, GreeceReviewed by:
Maria Markaki, Foundation for Research and Technology Hellas (FORTH), GreeceMara Doimo, Umeå University, Sweden
Copyright © 2022 Akbari, Nilsen and Montaldo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola Pietro Montaldo, n.p.montaldo@medisin.uio.no