
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Virol. , 12 January 2022
Sec. Fundamental Virology
Volume 1 - 2021 | https://doi.org/10.3389/fviro.2021.815388
 Massimo Pizzato*
Massimo Pizzato* Chiara Baraldi†
Chiara Baraldi† Giulia Boscato Sopetto†
Giulia Boscato Sopetto† Davide Finozzi†
Davide Finozzi† Carmelo Gentile†
Carmelo Gentile† Michele Domenico Gentile†
Michele Domenico Gentile† Roberta Marconi†
Roberta Marconi† Dalila Paladino†Alberto Raoss†
Dalila Paladino†Alberto Raoss† Ilary Riedmiller†
Ilary Riedmiller† Hamza Ur Rehman†
Hamza Ur Rehman† Annalisa Santini†Valerio Succetti†
Annalisa Santini†Valerio Succetti† Lorenzo Volpini†
Lorenzo Volpini†The ability of a virus to spread between individuals, its replication capacity and the clinical course of the infection are macroscopic consequences of a multifaceted molecular interaction of viral components with the host cell. The heavy impact of COVID-19 on the world population, economics and sanitary systems calls for therapeutic and prophylactic solutions that require a deep characterization of the interactions occurring between virus and host cells. Unveiling how SARS-CoV-2 engages with host factors throughout its life cycle is therefore fundamental to understand the pathogenic mechanisms underlying the viral infection and to design antiviral therapies and prophylactic strategies. Two years into the SARS-CoV-2 pandemic, this review provides an overview of the interplay between SARS-CoV-2 and the host cell, with focus on the machinery and compartments pivotal for virus replication and the antiviral cellular response. Starting with the interaction with the cell surface, following the virus replicative cycle through the characterization of the entry pathways, the survival and replication in the cytoplasm, to the mechanisms of egress from the infected cell, this review unravels the complex network of interactions between SARS-CoV-2 and the host cell, highlighting the knowledge that has the potential to set the basis for the development of innovative antiviral strategies.
In December 2019 several cases of an atypical viral respiratory infection, later named COVID-19, emerged in Wuhan, China (1). A month later, in January 2020, a novel coronavirus was isolated from clinical specimens, phylogenetically related to betacoronaviruses that recently caused acute respiratory syndromes i.e., SARS-CoV and MERS-CoV (2). The novel coronavirus was named SARS-CoV-2 and, in March 2020, COVID-19 was declared pandemic by the World Health Organization (WHO). The origin of SARS-CoV-2 remains unclear, as reiterated by the WHO (3). Initially, a zoonotic origin was proposed, according to which “wet” markets in Wuhan would have been the first source for animal-human transmission (4), but the market samples that were collected did not allow to establish the exact zoonotic predecessor strain (5). Therefore, the exact place of origin and the possible intermediate hosts need to be further investigated. One of the viral strains closest to SARS-CoV-2, RaTG13, has been found in horseshoe bats (Rhinolophus affinis from Yunnan Province in China) and has a genome sequence identity of 96.2% (2). Interestingly, the variable loop region of the spike protein has a unique evolutionary history compared to the rest of the SARS-CoV-2 genome overall, since it is similar to the same region observed in the coronavirus strain derived from the Malayan pangolin (pangolin-CoV-2020). It was therefore proposed that the Malayan pangolin may have been the intermediate host of SARS-CoV-2 (6). However, a direct origin from pangolin was challenged by the discovery of other viruses similarly close to SARS-CoV-2, found in bats from different locations of Southeast Asia (7).
An ancestral recombination event between the lineages leading to SARS-CoV-2 could therefore also have taken place in bats or in another intermediate host. Although pangolin-CoV-2020, RaTG13, and other bat CoVs are phylogenetically close to SARS-CoV-2, the viral genome underwent complex recombination events between divergent strains residing in different host species during its evolution, explaining the evolutionary histories of different genomic segments (8). Therefore, while bats are probably the reservoir hosts for this virus, it is likely that other mammalian species acted as intermediate hosts, as documented with civets and camels for SARS and MERS, respectively. Within these unknown intermediate hosts, SARS-CoV-2 acquired some or all the mutations needed for efficient transmission and replication into humans (9). In conclusion, although the molecular and phylogenetic analyses indicate the zoonotic hypothesis as the most likely, the origin and intermediate host species of SARS-CoV-2 remain uncertain and an unnatural origin of this coronavirus cannot be formally excluded [for a detailed review, see (10)].
According to the current best estimate of the Center for Disease Control and Prevention (CDC), the infection fatality ratio (estimated number of deaths per 1,000,000 infections) ranges from 20 in children and adolescents to 90,000 in adults older than 65, demonstrating high morbidity for the elderly population. Accordingly, it has been shown that the aging process predisposes older people to greater morbidity and mortality rates (11). It is estimated that 30% of the infections are asymptomatic (12) with more than 50% of transmission occurring in the pre-symptomatic phase (13), making the infection difficult to identify and contain. Similarly to SARS-CoV and MERS-CoV, SARS-CoV-2 may cause a severe respiratory syndrome, frequently associated with comorbidities (14, 15). The pathogenesis is characterized by diffuse alveolar damage occasionally accompanied by microthrombi and vascular damage with immune depletion [for a review see (16)]. Different transmission modes have been described for SARS-CoV-2, including aerosol, surface contamination, and the fecal–oral route (17–19) leading to severe flu-like symptoms that include fever, cough, and dyspnea. The incubation period ranges between 1 and 14 days and can progress to acute respiratory distress, pneumonia, renal failure and death [see (20)]. Respiratory failure in severe SARS-CoV-2 illness has been found to be associated with a hyper inflammation, which may be caused by a cytokine storm syndrome. Interleukin 6, interleukin 8, E-cadherin, MCP-1, VEGF, among other molecules, are involved in the cytokine release syndrome aggravated by trans signaling [reviewed in (21)], similarly to what has been observed in the case of SARS-CoV (22). These proinflammatory mediators can, in turn, perpetuate lung disease by elevating C-reactive protein from the liver through STAT3-IL-6 signaling (23), contributing to lung tissue damage.
SARS-CoV-2 is an enveloped, single-stranded positive-sense RNA virus with a diameter of 60–140 nm and spikes of 9–12 nm in length (Figure 1). It is part of the betacoronavirus genus, which includes MERS-CoV and SARS-CoV (24). The virus particle is made of structural viral proteins including spike (S), envelope (E), membrane (M), and nucleocapsid (N) protein (Figure 1 and Table 1). The 419 amino acid-long N protein is the only structural protein inside the virion, associated with the viral genomic RNA via electrostatic interactions driven by positively charged amino acid residues and modulates RNA unwinding after entry into the cell (33). Other structural proteins are inserted into the lipidic viral envelope. The E protein forms an ion channel and participates in viral assembly, while the M protein is critical for incorporating essential viral components into new virions during morphogenesis. The S protein binds the receptor expressed by host cells and promotes fusion of the viral and cellular membrane [see (34) for a review]. The SARS-CoV-2 genome is ~30 kb and encodes 14 ORFs (Figure 2). The genome is flanked by 5′ and 3′ untranslated regions (UTRs) that contain cis-acting secondary RNA structures essential for RNA synthesis. At the 5′ end, the genomic RNA features two large open reading frames (ORF1a and ORF1b) that occupy two-thirds of the capped and polyadenylated genome and encode 16 non-structural proteins (Nsps 1–16) that make up the replicase complex (Table 2). Nine accessory proteins—termed ORF3a, 3b, 6, 7a, 7b, 8, 9a, 9b, and 10—are encoded by homonymous orfs and, although deemed as non-essential for the virus replication in vitro (Table 3), are thought to exert important functions in modulating the host cell metabolism and antiviral immunity [see (78) for a review].
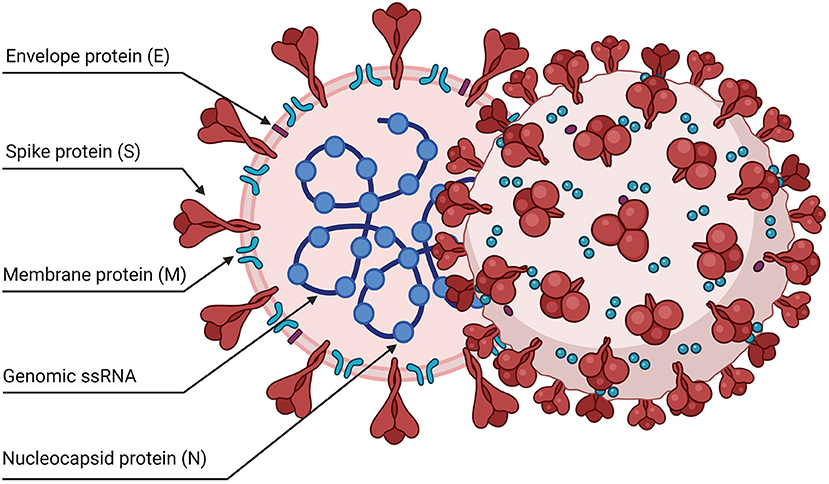
Figure 1. Schematic representation of the SARS-CoV-2 viral particle. The virion contains a positive-sense, single-stranded RNA genome (+ssRNA) enclosed by a lipidic envelope and by structural viral proteins. The nucleocapsid protein (N) is associated with the RNA genome inside the virus particles. Other proteins are inserted in the lipid envelope: the spike trimers (S), the envelope (E), and membrane (M) proteins.
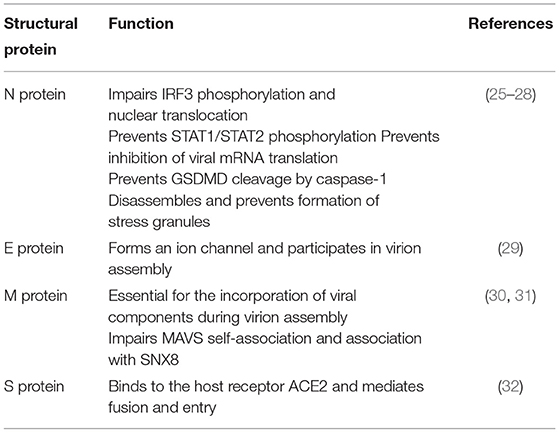
Table 1. Structural proteins encoded by SARS-CoV-2 genome and their functions.
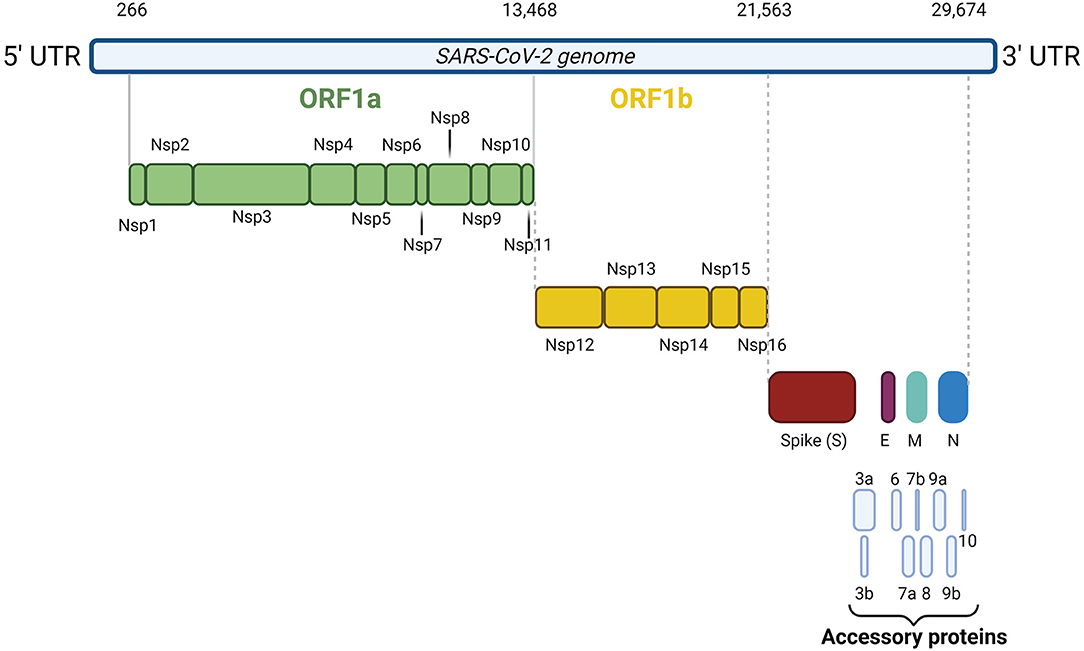
Figure 2. SARS-CoV-2 genome organization. The 30 kb SARS-CoV-2 genome is flanked by 5′ and 3′ untranslated regions. At the 5′ end, the genomic RNA contains two large open reading frames (ORF1a and ORF1b) encoding 16 non-structural proteins (Nsps 1–16). At the 3' end, the genome encodes the four structural proteins S, N, M, and E and nine accessory proteins, namely ORF3a, 3b, 6, 7a, 7b, 8, 9a, 9b, and 10.

Table 2. Non-structural proteins encoded by SARS-CoV-2 genome and their functions.
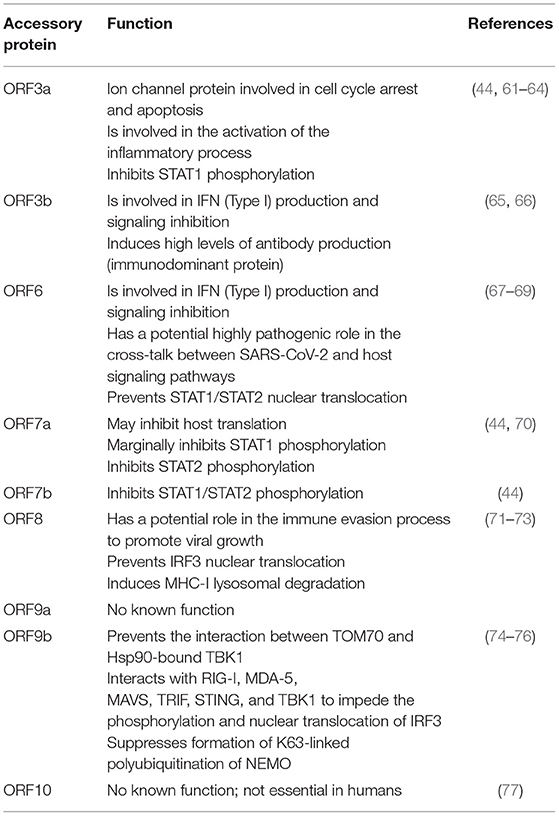
Table 3. Accessory proteins encoded by SARS-CoV-2 genome and their functions.
In the respiratory tract, SARS-CoV-2 invades preferentially mucus-producing goblet cells and the ciliated cells, as indicated indirectly by the topology of expression of host entry factors, by in vitro and ex vivo tropism studies and by post-mortem examinations (79–82). In addition, endothelial cells in the lung were also shown to be susceptible to infection in vivo (81, 83). The spike protein interacts with the host cell receptor hACE2 with the receptor-binding domain (RBD) (84, 85) followed by important conformational changes required to achieve infection (Figures 3, 4). Accordingly, SARS-CoV-2 relies on cellular proteases for priming the spike protein (14, 84, 86). Conformational changes follow receptor binding and trigger the events leading to fusion with the cellular membrane followed by the penetration of the viral ribonucleoprotein complex into the cytoplasm [for a review on SARS-CoV-2 replication see (87)]. Once into the cytoplasm, the virus releases its RNA genome which is first translated to produce the viral replicase polyproteins pp1a and 1ab and subsequently cleaved into smaller products by virus-encoded proteases. The viral polymerase transcribes a series of subgenomic mRNAs by discontinuous transcription, which are then translated into the viral structural proteins. The N protein forms a complex with the genomic RNA while the S, E and M proteins are inserted into the viral envelope at the ER and Golgi intermediate compartments. The newly assembled viral particles are then released from the infected cells by exocytosis (Figure 3).
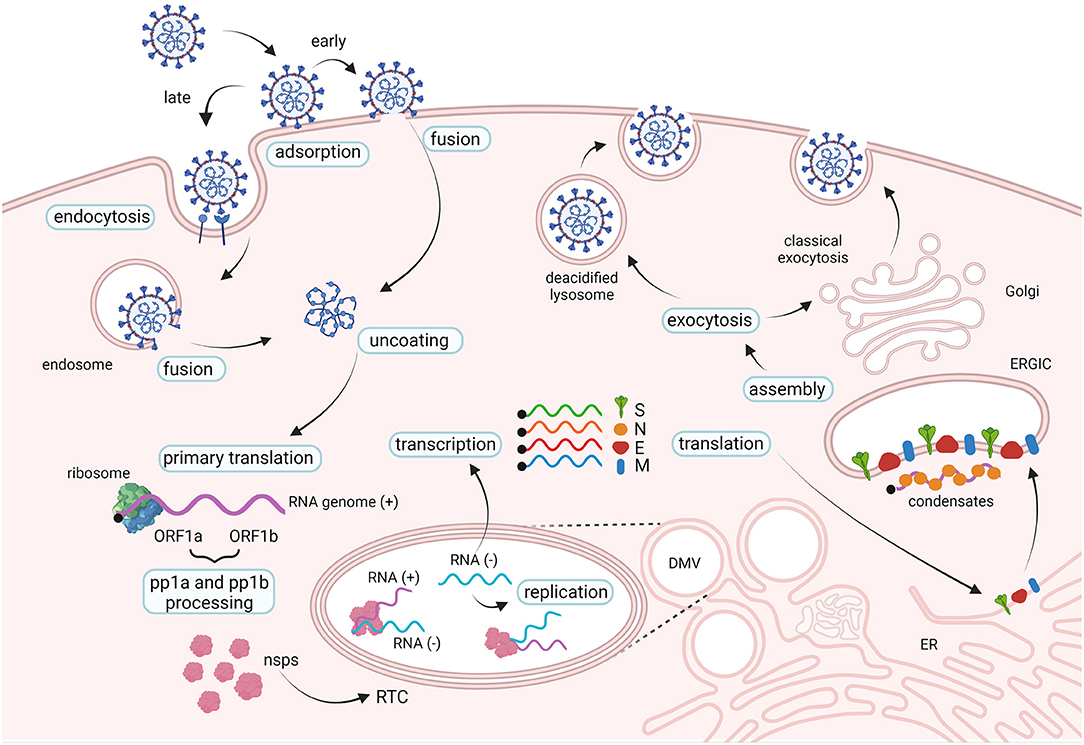
Figure 3. The life cycle of SARS-CoV-2. SARS-CoV-2 interacts with cellular receptors such as human angiotensin-converting enzyme 2 (hACE2) and with host proteases which activate the spike proteins (not shown). Entry in the cell cytoplasm may occur in two ways: the viral particle is endocytosed before fusing with the endosomal membrane (late pathway), or the viral membrane fuses with the cell membrane at the cell surface (early pathway). Two large open reading frames, ORF1a and ORF1b, are immediately translated in polyproteins pp1a and pp1ab, that are processed into the individual non-structural proteins (nsps) which form the viral replication and transcription complex (RTC). Viral genomic RNA replication occurs in protective double-membrane vesicles (DMVs). Transcription and translation of the negative template result in the formation of structural proteins that are inserted into the endoplasmic reticulum (ER) membrane and transit through the ER-to-Golgi intermediate compartment (ERGIC). Here, condensates of newly produced genomic RNA and N proteins interact with E and M proteins resulting in assembly of viral particles, which bud into the lumen of secretory vesicular compartments. Virions are secreted from the infected cell by exocytosis in two ways: through the classical exocytosis pathway via the Golgi compartment or through the incorporation in deacidified lysosomes that fuse with the cellular surface membrane.
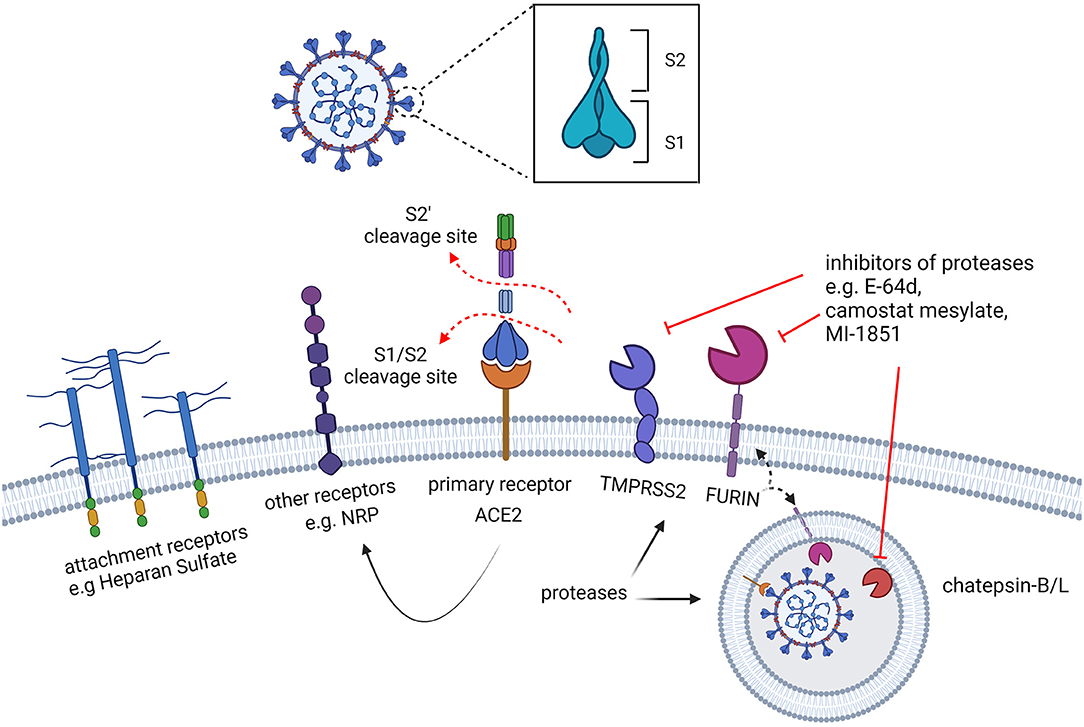
Figure 4. Early interactions of SARS-CoV-2 with the target cells. The spike protein of SARS-CoV-2 recognizes surface receptors to bind and fuse with the cell. A preliminary interaction of the virus particle with the cell is mediated by attachment to cell surface glycans, such as heparan sulphate, followed by the interaction with specific receptors. While the human angiotensin-converting enzyme 2 (hACE2) is the main receptor recognized by the spike, other cell surface proteins, such as NRP, are also known to serve as receptors capable of triggering virus entry. The activation of the fusogenic activity of the spike protein also requires proteolytic processing by cellular proteases which are located on the cell surface or in the endosomal vesicles. Furin is required for the cleavage which separates the S1 and S2 subunits of the spike, predisposing the protein to the interaction with ACE2 and to the subsequent cleavage (S2') by the activating proteases: TMPRSS2, exclusively located at the cell surface, and cathepsins, in the endosomes.
As for many pathogens, glycoconjugates surrounding mammalian cells are also exploited by SARS-CoV-2 as attachment factors driven by non-specific electrostatic interactions which promote the primary virion-cell surface binding (88). Accordingly, different coronaviruses have been documented to bind host glycans, such as Heparan sulfate (HS) (89–91). The SARS-CoV-2 RBD contains a strongly electro-positive surface, which can accommodate 20 monosaccharides from heparin via hydrogen bonds and hydrophobic interactions. Notably, the glycan-binding surface on the RBD is adjacent to, but separate from, the ACE2-binding site, suggesting that ACE2 and HS interactions with the spike glycoprotein are not mutually exclusive (92–94). Despite 73% identity between SARS-CoV and SARS-CoV-2 RBDs, the electrostatic potential of SARS-CoV-2 is much higher, probably mediated by two amino acid substitutions (Thr to Lys 444, Glu to Asn 354) that enhance the predicted coordination with the 20 monosaccharide residues from heparin. HS is thought to enhance binding to ACE2 by promoting an RBD open conformation state (see below), therefore acting as a priming co-receptor that favors the interaction with ACE2 (92).
The important role of such primary interactions suggests pathogenic mechanisms as well as antiviral strategies. On one side it has been hypothesized that secretion of polycations from neutrophils may worsen SARS-CoV-2 infection by facilitating receptor-mediated entry following neutralization of electrostatic repulsive forces between the cell and the viral membranes (95). On the other side such preliminary interactions inspire strategies that could be exploited in therapies aimed at blocking the electrostatic binding (96): negatively charged polysulfates, such as heparin or hyperbranched polyglycerol sulfate (HPGS), can bind to the spike protein, providing valid models to design polyanionic inhibitors of viral infection. Accordingly, heparin-derivatives such as unfractionated heparin, non-anticoagulant heparin, heparin lyases and low molecular weight heparin have been proposed for COVID-19 treatment, not only to treat thrombotic complications ranging from vascular micro-thromboses, venous thromboembolic disease, and stroke, but also to interfere with viral infection (97, 98). Preliminary attachment to cell surface glycoconjugates was shown for different viruses for which the inhibitory activity of heparan sulfate and similar polyanionic compounds has been observed in vitro. For SARS-CoV-2, as well as for other different viruses, the real therapeutic activity of these strategies awaits clinical demonstration (99).
While the cell surface molecule ACE2 was soon identified to be the prominent receptor bound by the spike protein, other cell surface molecules have been later proposed to function as alternative receptors or co-receptors (Figure 4), including KIM1, AXL, L-SIGN, and DC-SIGN, and SR-B1 (100–104). For most of these molecules it remains unclear whether these are alternative receptors to ACE2, or whether their activity is limited to facilitating viral entry. In addition to describing the interaction with the main receptor ACE2, here we discuss the potential role of Neuropilin-1, independently reported by two research groups (105, 106).
The human angiotensin-converting enzyme 2 (hACE2) was found to be the main host cellular receptor recognized by the S protein (84–86). ACE2 is a blood pressure and kidney function regulator of the renin-angiotensin-aldosterone system, expressed in most tissues of the body. It is essential for processing angiotensin 2 and therefore it is involved in vasoconstriction as well as in pro-fibrotic and pro-inflammatory processes (107). This receptor is organized as a homodimer, stabilized by B0AT1 (SLC6A19), a transporter which mediates uptake of neutral amino acids into intestinal cells (108). The spike glycoprotein recognizes the N-terminal peptidase domain of ACE2 (PD, 19–615 residues), which is also the catalytic domain of the protein. Hence, there is the possibility that viral infection could interfere with the angiotensin 2 pathway by contributing to pathogenesis. Given the widespread use of ACE2 inhibitors to treat hypertension and diabetes, a contribution of such medications to COVID-19 pathogenesis has also been investigated, but so far without finding clinical support (109).
The virion spike glycoprotein (S) is a class I viral fusion protein which forms a trimer and is processed by host proteases in two domains (S1 and S2) folding into a metastable pre-fusion conformation. The S1 subunit binds ACE2 while the S2 subunit catalysez fusion with the target cell (110). The S1 C-terminal domain (CTD) contains the receptor binding domain (RBD, Figure 5), which is sufficient for binding to the ACE2-PD domain and is the main determinant of viral host range and tropism. The glycoprotein can be found in two distinct conformations, which make the RBD differently accessible to the receptor. While an “up” state readily exposes the RBD to receptor binding, a “down” state, which protects the RBD from crucial neutralizing antibodies, makes it less available for the interaction with ACE2 (Figure 5B). The SARS-CoV-2 spike predominantly acquires the latter conformation (32, 111) which makes receptor recognition less favorable while also contributing to decreasing the virus vulnerability to neutralization.
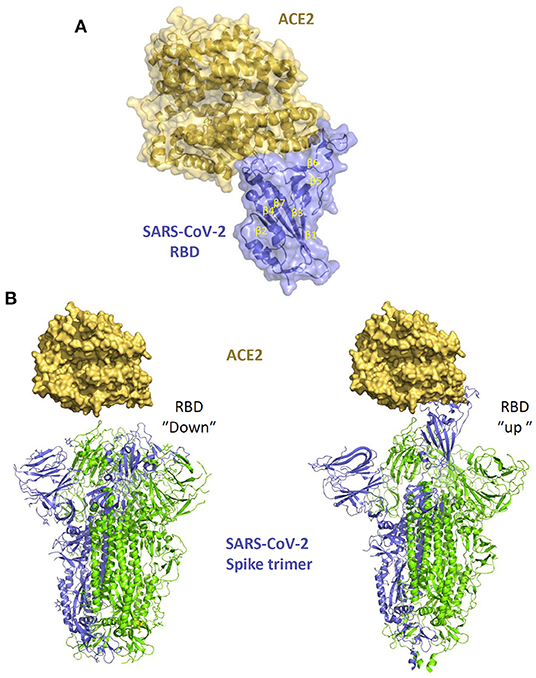
Figure 5. The interaction of SARS-CoV-2 spike with ACE2. (A) structure of SARS-CoV-2 spike RBD in complex with ACE2 (PDB 6M0J). (B) different conformations of the spike trimer with RBD “down” (PDB 6ZB5) and RBD “up” (PDB 6zGG) which shows the exposure of the RBM within the RBB which facilitates the interaction with ACE2.
Our molecular understanding of the interaction between the spike protein and ACE2 builds from the knowledge gained from SARS-CoV studies (112), later confirmed by structural studies with SARS-CoV-2 (Figure 5A) (84, 85, 113). The RBD contains 2 subdomains: the “core loop” made of five-stranded anti-parallel β sheets (β1 to β4 and β7), with three short-connecting α helices (αA to αC) and the “extended loop” subdomain, which is a concave surface formed by two-stranded β sheets (β5 and β6) forming the interface with ACE2. Variations of ACE2 sequences in different animal species impact on the affinity of the binding with the S protein determining different degree of susceptibility to infection and virus transmission. Of note, within β6, residues Leu472, Asn479, Thr487 are critical for cross-species and human-to-human transmission of SARS-CoV. Met82 of human ACE2 interacts with Leu472, while the Asn82 in rat ACE2 introduces a glycan that produces steric hindrance, disrupting the binding. Indeed, some amino acidic variations in the recognition interfaces of SARS-CoV-2-RBD/ACE2 are likely to increase the binding affinity compared with SARS-CoV-RBD/ACE2 (85), possibly explaining the higher infection efficiency of SARS-CoV-2.
Neuropilin-1 (NRP1), is a multifunctional surface protein which binds secreted peptides such as VEGF and semaphoring (114). NRP1 plays an important role for the development of neurons and the cardiovascular system as well as for tumor growth and tumor vascularization (115). The protein recognizes furin-cleaved 192 substrates which, upon cleavage, expose a polybasic conserved C-end rule peptide (TQTNSPRRAROH). Accordingly, furin cleavage of the spike glycoprotein generates a C-end rule peptide, which was found to bind NRP1 and mediate cell entry through endocytosis (116). NRP1-driven virus entry is thought to facilitate infection of cells with low ACE2 expression level, such as olfactory endothelial cells (105), possibly by facilitating the separation of S1 and S2 (117). Indeed, entry promoted by NRP1 could have important neurological implications, especially in the olfactory-related region of the CNS, supporting the role of NRP1 in the neurological symptoms of SARS-CoV-2 (118). In vitro studies have shown that ACE2 expression in pulmonary and olfactory cells is low (119), while NRP1 appears to be abundantly expressed, consistent with an important role of the latter as a coreceptor to enhance cell tropism for the upper respiratory tract. Understanding the molecular details of the interaction between NRP1 and the furin-cleaved S1 domain may therefore help future studies for developing SARS-CoV-2 inhibitors (106). For over 20 years there has been strong interest in interfering with NRP1 binding to its ligands. For example, different strategies have been pursued to interfere with NRP1 binding to VEGF to block endothelial cell migration, including NRP1-based antibody therapy (120) and small molecules-based therapy to target the NRP1-b1 domain (121), the same found to interact with the SARS-CoV-2 spike. Monoclonal antibodies were therefore tested to establish the functional relevance of the NRP1-b1-S1 molecular interaction. Therapies which block NRP1, such as antibodies-based therapy, small peptides-based therapy as well as small molecules-based therapy (122), may potentially be exploited to target also SARS-CoV-2. Accordingly, a selective NRP1 210 antagonist that interacts with the NRP1-b1 binding pocket was shown to reduce NRP1-b1/S1 binding and to inhibit viral entry (105).
In addition to receptor binding, efficient entry of coronaviruses also requires processing of the spike protein by cellular proteases. In the case of SARS-CoV-2, the proteolytic processing includes both a “priming” and an “activation” cleavage (86, 116). The first “primimg” cleavage reshapes the spike by cleaving S1 and S2, which remain non-covalently attached. The second cleavage (S2') occurs within S2, exposing the fusion peptide and activating its fusogenic potential (Figure 6). The priming processing predisposes the spike protein to the interaction with ACE2 and to the cleavage by the activating protease (86, 116). Proteolytic processing is therefore crucial for SARS-CoV-2 infectivity and has been intensively investigated to reveal which cellular factors are involved and in which cellular compartment they act. The spike of SARS-CoV-2 can be targeted by proteases during biogenesis in the Golgi of virus-producing cells and in target cells, either on the cell surface or inside an intracellular vesicle, depending on the entry pathway followed by the virus. After emerging from the producer cell, coronaviruses are known to infect target cells either by fusing with the host cell at the plasma membrane (“early pathway”), or within an intracellular vesicle following endosomal uptake (“late pathway”). SARS-CoV-2 is documented to exploit both pathways [Figure 3, see (123) for a review] and takes advantage of different host proteases encountered on its way including furin, transmembrane protease serine S1 member 2 (TMPRSS2) and endosomal cysteine protease Cathepsin B and L (CatB/L) (124). Such proteases play a crucial role during infection since they modulate viral infectivity, tropism, transmission and pathogenesis (125).
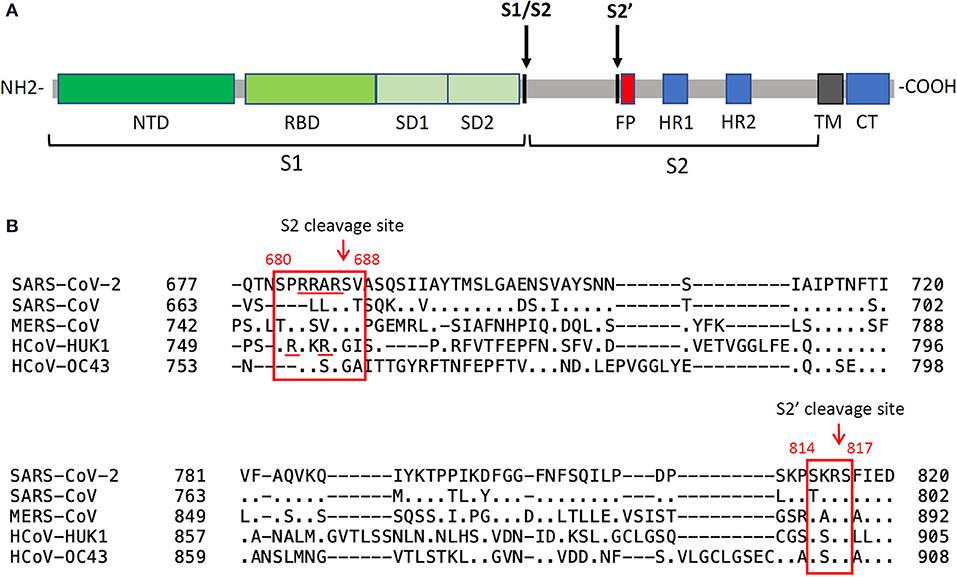
Figure 6. Spike cleavage in betacoronaviruses. (A) schematic organization of the SARS-CoV-2 spike protein domains and cleavages by host proteases. The priming cleavage at the S2 site separates the subunit S1 from S2, while the activating cleavage occurs at the S2' site in close proximity to the fusion peptide. NTD, N-terminal domain; RBD, receptor-binding domain; SD1, subdomain 1; SD2, subdomain 2; FP, fusion peptide; CH, central helix; SD3, subdomain 3; TM, transmembrane domain; CT, cytoplasmic tail. (B) sequence alignments of the regions containing the protease cleavage sites in human betacoronaviruses. The priming cleavage of SARS-CoV-2 involves a polybasic S2 furin cleavage sequence (RXXR) which is absent in other sarbecoviruses, such as in SARS-CoV, but is found in other human betacoronaviruses. Processing of S1/S2 exposes the S2' site which can be cleaved by type II transmembrane serine protease and cathepsins.
The first protease cleavage (“priming”) occurs at the boundary between the two functional subunits of the spike extracellular domain (S1 and S2, Figure 6) by recognition of a unique multibasic furin cleavage site (FCS) rich in Arginine (680SPRRAR↓SV688) which has been structurally well-characterized in complex with the protease (126). The presence of this sequence at the S1/S2 boundary is a crucial virulence determinant peculiar to SARS-CoV-2 since it is absent in most other coronaviruses, including SARS-CoV (127, 128). Only MERS-CoV contains a furin pseudo-binding site, though not as efficiently processed by the protease (128). In SARS-CoV-2, the FCS is a well-exposed loop within the spike structure, fully accessible to protease recognition. Indeed, from biophysical analysis (126) it was demonstrated that the cleavage loop (N657 to Q690) perfectly fits in the canyon-like crevice which identifies the substrate-binding pocket of furin. The higher proteolytic processing susceptibility of the spike from SARS-CoV-2 compared to that from others HCoV S, together with the ubiquitous expression of furin, may therefore facilitate viral entry into cells, leading to increased infectivity and transmissibility of the virus compared to SARS-CoV and MERS-CoV.
Furin is a membrane-bound protease and as such it is produced in the ER and transported through the secretory pathway. Like glycoproteins of other viruses (e.g., retroviruses), the spike glycoprotein can be processed by furin during biogenesis in producer cells. However, membrane-bound furin can also process the spike glycoprotein in target cells during the early steps of infection and, being also shed from the membranes, viruses can be primed by the protease in the extracellular space before attachment to target cells. Of note, the S1/S2 sequence boundary can be quickly lost because of lab adaptation to cell culture (129–131). Accordingly, virus replication in vitro results in deletion of the—RRAR—motif and increases the dependence of the virus on the endosomal pathway. This indicates that the emergence of the FCS could have been driven by in vivo selective pressure to fuse at the cell surface rather than in the endosomes, a pathway that allows the virus to avoid inhibition by IFITM proteins (132). Irrespective of the cellular site of cleavage, furin is expressed in different tissues and contributes to expanding the virus tropism to sites and organs usually refractory to infection by other coronaviruses (132). The increased tropism mediated by furin could also favor viral dissemination among individuals, since the virus can be shed in different body fluids and secretions (125).
In addition to increasing virus pathogenicity by altering virus particles infection, the ability of the spike protein to engage furin was also suggested to affect the normal physiology of the lungs because of molecular mimicry. An amino acid sequence similar to the FCS found within the SARS-CoV-2 spike is present in the human epithelial sodium channel α-subunit (ENaC-α), a protein involved in the homeostasis of airway surface liquid (133). This structural mimicry could therefore lead to a competition for furin in infected lung cells, with the virus interfering with fluid clearance from alveoli and contributing to edema and pulmonary pathology (134).
The priming step gives rise to a spike structure made of S1–S2 non-covalently bound in a “pre-fusion” conformation which is also processed by other proteases, either on the target cell surface or within an endosome (127). This processing activates the fusogenicity of the spike protein, leading also to the formation of syncytia, which, as explain later, may contribute to pathogenesis and to spreading of the virus to neighboring cells.
Following protease-mediated “priming,” the S1 subunit is released, exposing a S2' cleavage site (814KR↓SF817) located at the N terminal of the fusion peptide. Such “activation” cleavage can be performed by diverse proteases which are found in different cellular compartments. As already well-characterized for SARS-CoV and MERS-CoV, TMPRSS2 plays a major role in activating the spike glycoprotein at the cell surface of target cells [see (135) for a review]. In addition, Cathepsin L, a member of the lysosomal cysteine protease, activated at low pH, was also found to target the spike protein of incoming virions taken up into the endosomal pathway (136–138). Of note, while activation by TMPRSS2 requires the prior furin priming cleavage, the spike can be activated independently on priming by cathepsins within an endosomal vesicle (139), giving SARS-CoV-2 the possibility to exploit different entry pathways. Accordingly, it has been observed that in Calu-3 cells the virus exploits a pH-independent early pathway by exploiting proteases such as TMPRSS2, enriched in airway epithelial cells, while in simian epithelial Vero cells SARS-CoV-2 is suggested to follow a low pH endosomal entry pathway supported by proteases such as Cathepsin L (132, 140). The observation that TMPRSS2 is enriched in nasal and bronchial tissues may therefore explain the high efficiency of SARS-CoV-2 infection of these tissues via respiratory droplets (80).
Given the importance of proteases during the early phases of the infection process, inhibition of furin, TMPRSS2 or cathepsins has been proposed in order to interfere with “priming” and “activation” cleavages of the trimeric S glycoprotein (141–143). Proving this concept, Hoffmann and coworkers (127) were the first to report the ability of a TMPRSS2 inhibitor, camostat mesylate, in preventing viral entry in lung Calu-3 cells, possibly by interfering with the activating cleavage.
Furin-specific inhibitors, such as decanoyl-RVKR-chloromethylketone (CKM) and naphthofluorescein, were also identified as potential antiviral leads able to prevent both priming of S glycoprotein and syncytia formation (144). In contrast, other studies highlighted how inhibitors of furin may be not sufficient to block virus infection, given the redundancy with other proteases (145). Along the same line, E-64d (a Cathepsin B and L inhibitor) was found to act efficiently against the virus only when used simultaneously with a TMPRSS2 inhibitor, further corroborating the concept that SARS-CoV-2 can exploit more than one protease to proteolytically activate the S glycoprotein. This is consistent with the evidence that simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors in vitro prevents SARS-CoV entry into cells expressing ACE2 and TMPRSS2 (146). The cooperation of different protease inhibitors could therefore be a promising strategy to treat COVID-19 (144, 145, 147), as shown with a combination of camostat mesylate and MI-1851 (a synthetic furin inhibitor) which promoted a strong and synergic reduction of viral replication in human airway epithelial cells. Of note, the experimental system used could lead to discordant results given that SARS-CoV-2 can follow different entry routes in different cells and therefore be processed by different proteases. However, even though TMPRSS2 and lysosomal cathepsins have been both demonstrated to have cumulative effects with furin on activating SARS-CoV-2 entry, inhibition of TMPRSS2 was found to be sufficient to prevent SARS-CoV-2 entry in lung cell lines and primary lung cells (148), raising hopes for their future use as antivirals in vivo. Accordingly, promising preclinical results obtained with Nafamostat, another TMPRSS2 inhibitor (149, 150), are currently awaiting clinical confirmation (151).
As observed with many enveloped viruses, such as HIV-1, RSV, and herpesviruses, cell surface expression of viral glycoproteins may promote fusion between infected cells and non-infected cells that express a functional virus receptor [see (152) for a review]. This interaction causes the formation of multinucleated syncytia which may contribute to virus propagation and to tissue damage. While syncytia were never observed with SARS-CoV-1, infection with SARS-CoV-2 is reported to induce the formation of multinucleated cells in vitro (153–156) as well as in vivo (154, 157–159), leading to the fusion of pneumocytes (157) with the contribution of lymphocytes (159) in the lungs. The propensity of the SARS-CoV-2 spike to induce the formation of multinucleated cells could be related to the processing by cellular proteases, such as furin and TMPRSS2 (156, 159), which activates and unleash the fusogenic potential of the viral glycoprotein not only on the viral particle but also on the cell surface. Of note, as well as requiring recognition of the cognate receptor and the activation by proteolytic processing, fusion depends also on the engagement of the calcium-activated ion channel TMEM16 in cells expressing the spike protein (158). On this basis, approved drugs inhibiting TMEM16 were found not only to block syncytia formation but also to prevent cytopathic effects and inhibit virus replication in vitro, suggesting a novel antiviral target.
The success of the fusion process also depends upon the composition of the viral and host membranes, given that the biophysical properties of different lipid species determines membrane fluidity and curvature [see (160) for a review]. If on one side phosphatidylethanolamine and cholesterol are found to enhance membrane fluidity and promote a negative curvature critical for viral fusion, lysophospholipids (LPLs) promote positive curvature and inhibit fusion (161). Accordingly, compounds interfering with lipid metabolism are proposed as drugs for the treatment of COVID-19, capable of inhibiting virus replication and syncytia formation. In particular, 25-hydrocholesterol (25HC) was shown to block membrane fusion by depleting cholesterol from the plasma membrane following activation of acyl-CoA:cholesterol acyltransferase (154, 162). The importance of the cholesterol metabolism for SARS-CoV-2 infection was also demonstrated by the inhibition of virus replication in vitro upon treatment of 27-hydroxycholesterol, an endogenous oxysterol metabolite of cholesterol targeting lipid rafts and the late endosomal compartment (163) with no cytotoxic effects in vitro. The potential of targeting viral fusion by altering the lipid composition of biological membranes is therefore an antiviral strategy that should be further explored to test its applicability in vivo.
Soon after SARS-CoV-2 was isolated, serious concerns were raised about the possibility that the virus could take advantage of antibody dependent enhancement of infection (ADE, Figure 7), posing a problem for vaccine development and therapeutic approaches in patients infected with different SARS-CoV-2 variants (164). The molecular bases of the ADE rely on poorly neutralizing or non-neutralizing antibodies binding to surface viral antigens and promoting viral particle uptake into cells that express an Fc receptor, thus enhancing the infection of the host (165). Of note, ADE could be potentially caused by the presence of suboptimal concentrations of neutralizing antibodies or antibodies with decreased affinity produced in the host either following an earlier infection, vaccination, or passive transfer (166). The ADE phenomenon has been reported for some human viruses, such as dengue, Zika and respiratory syncytial virus (RSV), and also in some veterinary pathogens such as foot-and-mouth disease virus (FMDV), porcine reproductive and respiratory syndrome virus (PRRSV) [for a review see (167)].
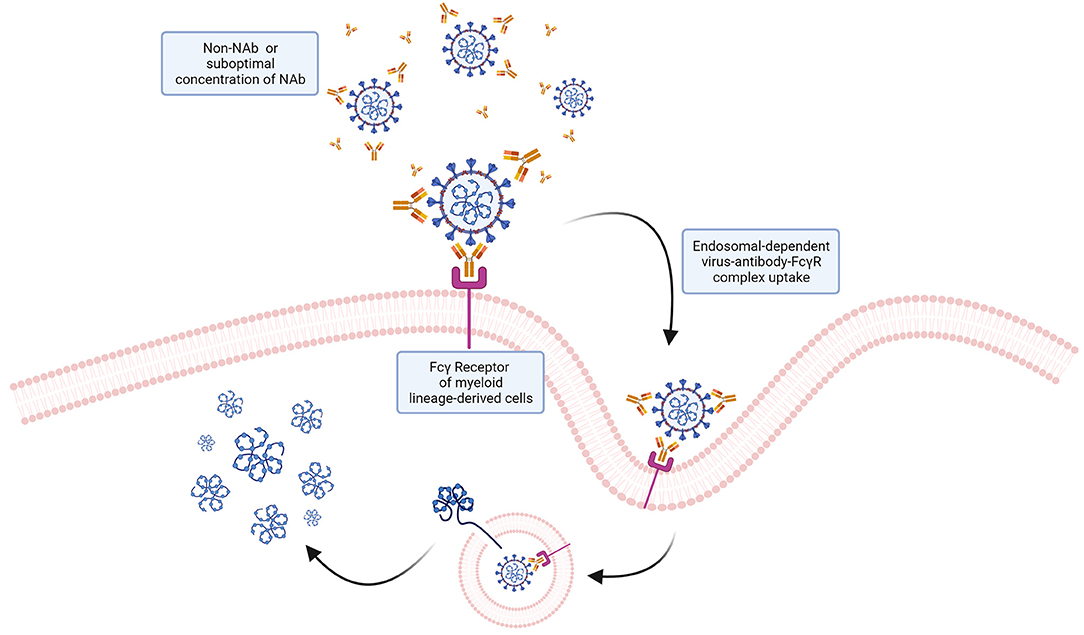
Figure 7. ADE: a potential mechanism of ADE for SARS-CoV-2? Antibodies interacting with virions without neutralizing the ability of the virus to infect target cells could potentially mediate viral particles uptake by cells expressing Fcγ receptors via endocytosis. Internalized virions could then fuse with the endosomal membrane and release the genome into the cytoplasm. While this mechanism, known to favor infection of other viruses, has been hypothesized, no evidence so far has demonstrated that ADE occurs with SARS-CoV-2.
Myeloid cells, including monocytes, macrophages and dendritic cells (DCs), are considered likely targets of ADE infection, since they express the FcγR which could confer the virus the ability to infect cells even in the absence of the spike cognate receptor (167). For non-macrophage-tropic respiratory viruses (such as RSV), the formation of immune complexes (i.e., virus-antibody complexes) in lung and airway tissues induce a local activation of the complement system and release of cytokines resulting in heavy inflammation and airway obstruction that eventually can lead to acute respiratory distress syndrome (168) closely similar to COVID-19 clinical manifestations, which could involve the complement system activation (169).
Concerns about the possibility of ADE for SARS-CoV-2 were raised looking at its close relative SARS-CoV. The two viruses have a genome sequence similarity of 79.5% and they also share the canonical receptor for viral entry (ACE2), which is bound by SARS-CoV-2 with 10–20 times increased affinity compared to SARS-CoV. ADE could result from poorly neutralizing antibodies raised by previous infections with other coronaviruses, since the S protein sequence similarity between different coronaviruses suggests the presence of cross-reactive epitopes (169). Indeed, in the case of SARS-CoV, anti-spike antibodies were reported to bind Fcγ receptors on the surface of M2 macrophages normally devoted to anti-inflammatory functions (170). Accordingly, anti-spike antibodies were shown to increase the infection of monocytes and lymphocytes by SARS-CoV in vitro in the absence of the canonical viral receptor, indicating an ACE2 independent entry mechanism (171). Understanding whether ADE can occur during SARS-CoV-2 infection has been fundamental for the evaluation of vaccine safety and the development of immunomodulatory therapies. Fortunately, up to date, there is no proof that ADE occurs in SARS-CoV-2 infections and there is no evidence that the available vaccines for COVID-19 are eliciting ADE in subjects that are reinfected. Indeed, it appears that vaccinated subjects are less severely affected by the disease. However, it will be important to keep monitoring for signs of ADE to optimize and update current prophylactic and therapeutic interventions.
Following receptor binding and the possible uptake into a vesicle, the fusion mechanism is triggered by S2 [see (172) for a review]. The S2 sequence contains regions conserved among CoVs required for the fusion machinery, notably a fusion peptide and two conserved heptad repeats (HR) instrumental for bringing viral and cellular membranes in close proximity for fusion. After activation of the spike glycoprotein, a conformational change occurs leading to the release of the fusion peptide and the interaction with the cellular membrane (172). The collapse of S2, which bridges the virus and cellular membranes, pulls the two membranes together with HR1 and HR2 forming the canonical 6-helix bundle first reported for CoVs in mouse hepatitis virus (MHV) (173) and then for other coronaviruses (141). As mentioned earlier, SARS-CoV-2 can fuse either at the cell membrane or after having been taken up by endocytosis. Understanding which entry pathway plays a crucial role in vivo remains an important issue which also indicates whether endo-lysosomotropic compounds can be used as possible therapeutic agents. In the meantime, the possibility of inhibiting virus infection by interfering with the maturation of endosomes has been investigated intensely. As mentioned earlier, CatB and CatL are the main proteases required for SARS-CoV-2 activation in the endosomes and both are active only at an acidic pH. Accordingly, as the endosomal vesicle is formed and clathrin breaks off, the pH of the vesicle decreases and the endosome matures to late endosome (pH of 5.5–6), which eventually fuses with the lysosome reaching a pH of ~4.6 (174). Understanding which cellular factors govern endosome acidification has become crucial to identify cellular functions required for the virus to infect cells using this pathway. Viral envelope fusion with the endolysosomal membrane cells depends, among other factors, on the lysosomal two-pore channel 2 (TPC2) (175). TPCs are dimeric ion channels composed of a duplicated domain architecture and are considered an evolutionary bridge to four-domain voltage-gated Ca2+ and Na+ channels [for a review see (176)]. The opening of TPCs is known to induce a strong sodium-driven depolarization in the endo-lysosomal membrane, which has been demonstrated to facilitate membrane fusion (177). Even though the specific role of TPC2 for virus escape into the cytoplasm is not completely clear, inhibition of TPCs should both impair the fusogenic potential of the endo-lysosomal system and disrupt the correct intracellular vesicle trafficking, resulting in inhibition of viral replication (178, 179). Accordingly, it has been demonstrated that knockdown and pharmacological inhibition of both TPC2, mainly expressed in late endosomes/lysosomes, and TPC1, which mainly localizes to early endosomes, attenuate intracellular trafficking of MERS-CoV through the endolysosomal system (180). Accordingly, Naringenin (Nar), which is the predominant flavanone in grapefruit and was found to impair TPC2 (181), has been investigated for its effects against SARS-CoV-2, after its activity had previously been observed against viruses that enter via a pH-dependent pathway such as Hepatitis C virus (182), influenza A virus (183), dengue virus (184), and Zika virus (185). Interestingly, Nar suppresses also inflammatory cytokine production through both transcriptional and post-transcriptional mechanisms, by regulating lysosome function, resulting in the inhibition of TNF-α and IL-6 secretion by macrophages and T cells (186, 187), which could contribute to prevent the excess of inflammation in COVID-19 patients. Like Nar, the TPC-specific blocker tetrandrine was also found to prevent cellular entry, viral-endosome membrane fusion and capsid disassembly required for successful virus entry (176, 188, 189). In addition, the endo-lysosomal lipid phosphatidylinositol-3,5-bisphosphate [PI(3, 5)P2] has also emerged as a direct channel activator that binds to TPCs (190) suggesting that inhibiting PI(3, 5)P2 could have antiviral effects. Accordingly, inhibition of the phosphoinositide kinase PIKfyve has been demonstrated to prevent infection by Zaire ebolavirus and SARS-CoV-2 (191). While these results obtained in vitro suggest the strong antiviral power of inhibiting the correct endolysosomal functionality, the toxicity and efficacy of these treatments in vivo remain to be tested, especially considering the ability of SARS-CoV-2 to exploit both pH-dependent and -independent pathways. Such adaptability of SARS-CoV-2 could be at the basis of the controversy surrounding the use of hydroxychloroquine for treating COVD-19. Quinine-derivative drugs, and hydroxychloroquine in particular, are well-known compounds with lysosomotropic activity which inhibit the replication of coronaviruses in vitro (192, 193) and were soon proposed as possible treatments for SARS-CoV-2 (194). However, the antiviral efficacy of the drug in cell culture (195–197) is in stark contrast with its inefficacy as a prophylactic and therapeutic drug in clinical settings (198–200). Accordingly, the ability of SARS-CoV-2 to exploit TMPRSS2, avoid the endosomal compartment and fuse at the cell surface could explain such discrepancy and limit the efficacy of drugs targeting the pH acidification (201).
After the virus accesses the cytoplasm, the replicative cycle begins with the translation of single-stranded positive-sense genomic RNA (ssRNA+). The translation of ORF1a and ORF1b from the genomic RNA produces two polyproteins, pp1a and pp1ab. Like all viruses, SARS-CoV-2 depends on the host cell translation machinery (Figure 8). Accordingly, RNA-interactome studies revealed the association of the viral RNA with components of the eukaryotic translation machinery, such as initiation factors, the cap- and poly(A)-binding proteins and ribosomal proteins (202–205). Furthermore, coronaviruses have evolved specialized mechanisms to hijack the host gene expression machinery and employ cellular resources to regulate viral protein production. Such mechanisms include inhibition of host protein synthesis and endonucleolytic cleavage of host mRNAs (206). A pivotal role in regulating the translation machinery of the host cell is played by the viral protein Nsp1, which is among the first proteins to be expressed after cell entry and causes repression of host translation (207). For SARS-CoV, Nsp1 has also been demonstrated to induce an endonucleolytic cleavage of the host RNA (208), while sparing from degradation viral mRNA by a yet unknown mechanism, likely mediated by the 5' UTR of the virus transcript (209). By combining cryo-electron microscopy and biochemistry, SARS-CoV-2 Nsp1, the host shutoff factor, has been recently demonstrated to bind the ribosomal mRNA channel (210). The protein was shown to interact with a range of different ribosomal conformations, as it co-migrates with both the 40S and the 80S complexes, competing with mRNA for ribosome binding. Nsp1 associates with the 40S ribosomal subunit with its C terminus interfering with the level of mRNA entry into the ribosome channel, thereby inhibiting host translation (210). As observed in the high-resolution structure of the 40S–Nsp1 complex, the C-terminal part of Nsp1 in the mRNA entrance channel folds into two helices. The first helix, composed of C-terminal residues 153–160, interacts with uS5 and uS3 [as reviewed (211)] through multiple hydrophobic side chains. The two helices are connected by a short loop containing the KH motif that establishes stacking interactions with helix h18 of the 18S rRNA through U607 and U630, as well as backbone binding. The second helix (residues 166–178), interacts with the phosphate backbone of h18 via two conserved arginines, R171 and R175 (210). Overall, these interactions tightly bind Nsp1 to the 40S subunit to cause translation inhibition by sterically occluding the entrance region of the mRNA channel. Interestingly, this inhibition mechanism may be unique to SARS-CoV-2 and other closely related beta-coronaviruses because the C-terminal region of Nsp1 is shorter in alpha-coronaviruses and is not highly conserved among other beta-coronaviruses, including MERS-CoV [as reviewed (212)]. Nevertheless, the viral mRNA has been demonstrated to be more efficiently translated than host mRNAs in the presence of this mechanism. Further details were emerged by studying the eukaryotic ribosome during initiation with the cricket paralysis virus (CrPV) internal ribosome entry site (IRES), which can directly recruit and assemble with the 40S or 80S ribosome without requiring any eIFs (213). Cryo-EM analyses with CrPV IRES have suggested that Nsp1 may act by changing the conformation of ribosomal subunit resulting in incorrect positioning of the mRNA 3' region (211). The conformation of the 40S ribosomal subunit in the Nsp1-40S complex appears to be in a “closed state,” suggesting that Nsp1 not only plugs the mRNA channel but also changes the 40S subunit conformation making it incompatible with loading of mRNA. Nsp1 is proposed to dislodge the canonical tRNA-mRNA interaction from the 40S subunit, interfering with the joining of the 60S subunit to form the 80S initiation complex (211). Overall, these results indicate that the C-terminal domain of Nsp1 is necessary and sufficient for inhibition of translation. Instead, its N terminus may play a role in suppressing host gene expression of Type I interferon in infected cells, as described later (214), and regulating cellular mRNA stability (215). Accordingly, an attenuating mutation of Nsp1 was found to decrease the ability of SARS-CoV to replicate in cells with an intact interferon response (216). However, how SARS-CoV-2 RNA escapes inhibition of translation remains unclear, even though it has been postulated that interactions involving the viral 5' UTR may result in the “unplugging” of Nsp1 from the 40S ribosome during initiation of viral translation (211). The SL1 hairpin structure found at the 5' leader sequence is required for the interaction of the RNA molecule with the Nsp1 C-terminal domain, allowing viral RNA to be translated despite Nsp1 binding to the ribosome (37).
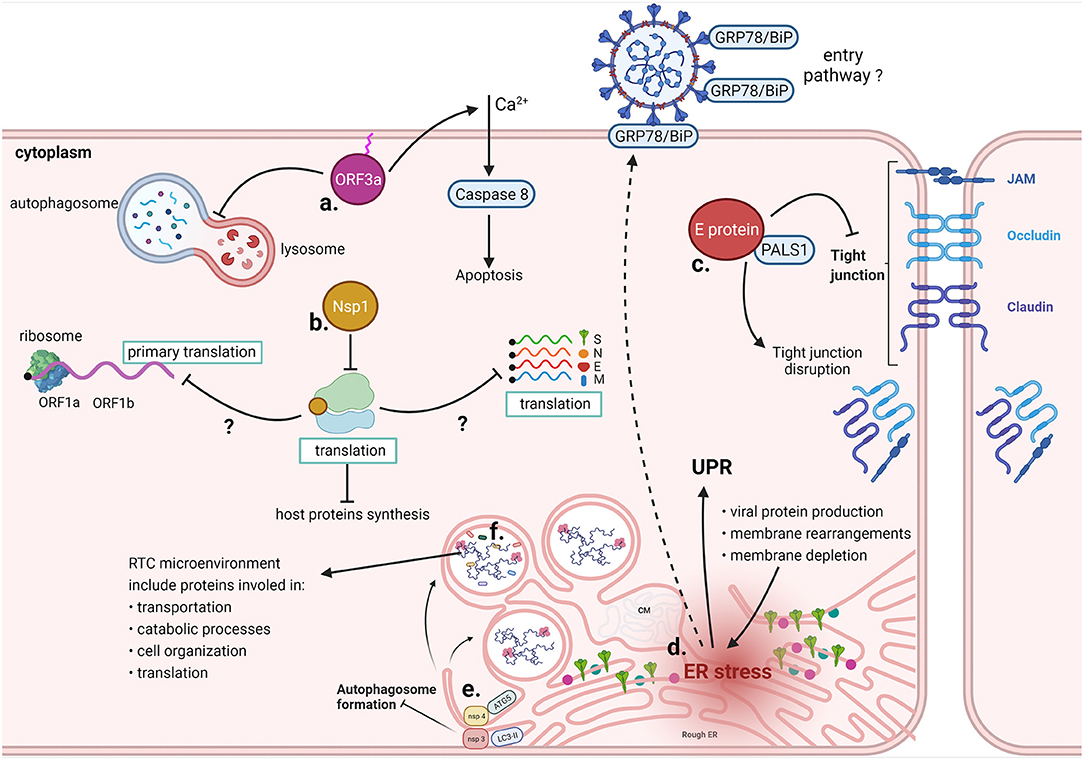
Figure 8. Cytoplasmic interactions of SARS-CoV-2. a. ORF3a requires binding to the plasma membrane to promote Ca2+-dependent apoptosis and prevents autophagosome-lysosome fusion. b. Nsp1 sterically occludes the mRNA entrance channel of the ribosome, prevents the physiological conformation of 48S PIC, and reduces mRNA transport through the NPC. Molecular mechanisms whereby Nsp1 specifically blocks only host translation have yet to be clarified. c. Computational predictions indicate that SARS-CoV-2 E protein binds to PALS1, normally involved in tight junctions maintenance, leading to their disruption. d. Excessive viral protein production, membrane rearrangements leading to the formation of DMVs and membrane depletion due to virus budding from ERGIC contribute to ER stress, which results in the UPR. Prolonged ER stress induces missorting of GRP78/BiP to the cell membrane, likely increasing infectivity upon binding of the chaperone with the SARS-CoV-2 spike protein. e. Nsp3 and Nsp4 prevent the formation of autophagosomes by inducing the formation of DMVs; ATG5, LC3 and other cellar factors involved in the autophagosome formation colocalize with the non-structural proteins. f. Different host proteins have been identified in the RTC microenvironment, including proteins involved in transportation, catabolic processes, cell organization and translation.
In addition to altering the initiation of translation, the observation that the overall transcriptome profile is perturbed by Nsp1 in infected cells has led to the hypothesis that this viral protein functions autonomously as an mRNA export inhibitor (217). Nsp1 directly binds the mRNA export factor NXF1 and reduces its interaction with the nuclear pore complex (NPC), thereby reducing expression of host mRNAs, including those which encode antiviral factors. Nsp1 disrupts NXF1 interaction with adaptor proteins which regulate mRNA interaction, and with the NPC (217). Accordingly, Nsp1 has been observed in the vicinity of the NPCs, raising the possibility that Nsp1 may shuttle between the nucleus and the cytoplasm, with its steady-state distribution being predominantly cytoplasmic. The effect on mRNA nuclear export results in an increased availability of the host translational machinery for viral mRNAs, thus favoring virus replication. SARS-CoV-2 Nsp1 therefore suppresses host gene expression by both inhibiting translation and blocking mRNA nuclear export, two functions performed by distinct domains within the C- and N-terminal regions of the protein (217).
Following translation, a Replication and Transcription Complex (RTC) is assembled, involving several non-structural proteins including Nsp12, the RNA-dependent RNA polymerase (RdRp), and its two accessory factors Nsp7 and Nsp8, that all together form the RdRp core complex. It is thought that additional subunits bind this complex, including Nsp9, Nsp10, Nsp13, Nsp14, and Nsp16 (218). The RTC is involved in two processes of the coronavirus replicative cycle: genome replication and transcription of subgenomic mRNAs, which are then translated into structural and accessory proteins (219). In analogy with our knowledge from other coronaviruses, we can deduce that several host factors associate with the RTC of SARS-CoV-2 to modulate transcription. The involvement of host factors in the functionality of coronaviruses RTC was investigated recently using proximity labeling with the mouse hepatitis virus (MHV) model (220). More than 500 host proteins were found associated with the RTC microenvironment, including factors involved in transportation, catabolic processes, cell organization and translation. The latter category was enriched by factors involved in translation initiation, such as the ribosomal proteins Rpl13a and Rls24d1 and several subunits of the eIF3 complex, which were confirmed to be crucial for MHV replication. The association of factors orchestrating translation initiation was confirmed for SARS-CoV-2 by a recent study which found Nsp9 interacting with eIF4H, a partner of the initiation factor eIF4A (221). Accordingly, the eiF4 inhibitor zotatifin was observed to exert a strong antiviral activity and on these bases was approved for a phase-I clinical trial for the treatment of COVID-19 (222), leading the way to exploit the interface between the viral replication machinery and the host factors as target for therapeutic intervention.
The replication of coronaviruses and many other positive-sense RNA viruses occurs in the cytoplasm of infected cells, in association with modified membranes that are transformed into distinctive structures called viral replication organelles (ROs). ROs provide platforms that facilitate viral RNA synthesis by concentrating relevant factors, while preventing the exposure of viral replication intermediates to cytosolic innate immune sensors (223). The formation of invaginations can occur at different membranes of various organelles, including the endoplasmic reticulum (ER), the endolysosomes, and mitochondria (224, 225). Such membranous structures may take the shape of convoluted membranes (CVs) and double-membrane vesicles (DMVs), originating from the endoplasmic reticulum, and the recently discovered double-membrane spherules (226). Viral RNA synthesis was shown to predominantly occur within such DMVs, with a pore connecting the interior of the vesicles to the cytosol for transport of viral RNAs and metabolites required for RNA synthesis.
The membrane-spanning Nsp3, Nsp4, and Nsp6 have been proven to be implicated in diverting cellular endomembranes into replication organelles, with other non-structural proteins and unknown cellular factors possibly involved in the process (224, 227). Such proliferation and reorganization of intracellular membranes by viruses is reminiscent of the formation of autophagosomes, which nucleate in the cytoplasm to originate a double membrane organelle. It is therefore plausible that SARS-CoV-2 exploits cellular factors required for autophagy alongside viral proteins for the biogenesis of DMV. Accordingly, coronaviral Nsp3 and Nsp4, which are co-translationally inserted in the ER, are sufficient to induce the formation of convoluted membranous structures in human cells and by inducing the formation of DMVs they inhibit the formation of canonical autophagosomes (45, 228). Cellular factors involved in autophagosome formation, such as ATG5 (229), LC3 (230), and ptdIns3P (231), were observed to colocalize with non-structural viral proteins on DMVs, and to be required for coronavirus replication, though perhaps in a cell-type specific fashion.
The ability of coronaviruses to induce formation of DMVs associates also with the modulation of lipid biosynthesis, resulting in a selective lipid composition with alteration of glycerophospholipids and cholesterol levels. To this end, coronaviruses hijack host factors to manipulate lipid mobilization and synthesis. Accordingly, sterol regulatory element-binding proteins (SREBPs), which regulate the biosynthesis of cellular cholesterol and fatty acids, were found to play an essential role for DMV formation by MERS-CoV (232). In addition, TMEM41B, an ER resident protein involved in induction of autophagy (233) and thought to be required for coronavirus replication, was identified as a crucial factor for the replication of diverse coronaviruses possibly by mobilizing cholesterol and other lipids for DMV formation (234). The antiviral effect of drugs targeting lipid biosynthesis, such as AM580 (232), demonstrate that interfering with lipid homeostasis could become an effective antiviral strategy. Further elucidation of the mechanisms and factors required for DMV formation will however be crucial to understand to what extent this process can be targeted to interfere with virus replication.
As recently proposed in a cryo-ET study (235), nucleocapsid assembly of SARS-CoV-2 is driven by the interaction between the N protein and the viral gRNA, giving rise to viral ribonucleoparticles (vRNPs) in a beads-on-a-string like conformation, resembling the helical filaments already observed for other coronaviruses (236). This conformation allows maintenance of high steric flexibility between the vRNPs, ideal for the incorporation of the SARS-CoV-2 large genome. It has been estimated that vRNPs are made of ~12 N monomers and 800 nt RNA and that each virion contains ~35–40 vRNPs (237). The pivotal role of the N protein in nucleocapsid assembly is further supported by its ability to undergo phase separation with RNA, as reported by several independent studies (238–243). Specific gRNA elements were proposed to differentially interact with the N protein, either promoting liquid-liquid phase separation (LLPS) or impeding it. A mixture of LLPS promoting elements, such as the 5'end and the nucleocapsid encoding region found at the 3'end, characterize the full lenght gRNA and favor a selective LLPS resulting in the exclusion of host mRNA from packaging (239). An L/Q rich region found in the central intrinsically disordered region (IDR) of N is supposed to be responsible for these interactions (243). As suggested by others and despite previous indications of phase separation being implicated in other viral contexts (242), it is currently impossible to exclude that phase separation is just an epiphenomenon due to the high concentration and multivalency of N proteins. For this reason, further studies are needed to elucidate the implication of LLPS in the viral replication cycle, eventually addressing its physiological relevance in SARS-CoV-2 assembly (239), as already proposed (242). Interestingly, recent analyses using fluorescent microscopy and FRAP indicate accumulation of RdRp at the level of N/RNA condensates (241). Even though this seems in contrast with the concept that replication occurs in ER DMVs, a dual involvement of N protein in both assembly and replication was recently demonstrated. It appears that phosphorylation of the N proteins in a particular S/R rich region, found in proximity of the L/Q-rich region of the central IDR, allows it to recruit host factors (e.g., RNA helicases), to eventually promote viral RNA template switching and subgenomic mRNA transcription. On the other hand, hypophosphorylation of the N protein in the same domain is responsible for its incorporation into virions (238, 243).
Altogether, these findings are pointing at the central role of the condensates involving the N protein, indicating this as a potential therapeutic target for future antiviral therapies against SARS-CoV-2. Possible compounds could either induce condensate dissolution (e.g., 1,6-hexanediol) and size modifications (e.g., kanamycin, lipoic acid) and/or alter protein/RNA ratio (e.g., kanamycin) (239). It is reasonable to assume that condensate formation could be advantageous to concentrate viral components in an isolated compartment in the cytosol, possibly improving nucleocapsid assembly. However, the mechanism underlying the recruitment of these condensates to the membranes, where subsequent steps in virion assembly take place, remains unclear (242).
The final assembly of SARS-CoV-2 virions requires M, E, S and N proteins and takes place at the level of intracellular membranes belonging to the ER-Golgi intermediate compartment (ERGIC), as previously shown for SARS-CoV-1 and other coronaviruses (236, 244). Virus budding occurs mainly at the level of the ERGIC membrane arrangements where vRNPs were observed to accumulate on the cytosolic side (235), while S trimers were found to be facing the luminal side. S trimers and M protein alone were shown to be unable to induce membrane curvature, consistent with previous evidence from SARS-CoV (244). Nonetheless, it appears that the M protein interacts with vRNPs (243) possibly driving their accumulation at the ERGIC membrane and together causing its curvature, which might also be dependent on the E protein, as suggested by studies with other coronaviruses (236). The evidence that some individual vRNPs are oriented on the viral envelope in an “eggs-in-a-nest” shaped assembly supports the role of specific M-N interactions driving packaging of new virions (237). Moreover, additional evidence shows that the N protein undergoes phase separation with soluble fragments of the membrane associated M protein, highlighting an important interaction that reinforces the pivotal role of M protein in recruiting the assembled nucleocapsid to the ERGIC compartment (243). Furthermore, the simultaneous expression of M, N, S, and E was shown to be pivotal for efficient viral assembly and it was also demonstrated that both E and M play a central role in inducing the retention of S in the ERGIC compartment. A recent study shows that M is able to directly interact with S through a C terminal retention signal, while E indirectly induces S retention by altering the cell secretory pathway, possibly by acting as a viroporin (245). The accumulation of M and E at the level of ERGIC/Golgi membranes could alter the activity of glycosylases, hence disrupting the N glycosylation profile of the S protein. The advantage given by the modulation of S maturation might be related to the ability of the virion to attach to lectins found at the surface of permissive cells, as proposed for SARS-CoV (246). Interestingly, a recent publication (247) suggests that the glycosylation profile of the Spike protein can be affected by inhibitors of protein glycosylation such as Miglustat and Celgosivir, resulting in misfolded S protein accumulation and reduction of infectious viruses release, highlighting the antiviral potential of these drugs.
The completion of the first step along the egress pathway of SARS-CoV-2 is established by the virus budding in the lumen of the ERGIC compartment. From this point on, it was originally believed that SARS-CoV-2 would follow the biosynthetic secretory pathway engaged by other enveloped viruses. However, recent findings suggest that SARS-CoV-2 and other betacoronaviruses exploit a lysosomal exocytic pathway that results in non-lytic release of the virions (248) confirmed also by recent cryo-imaging studies which indicate the formation of tunnels connecting multi virus-containing vesicles to the cell membrane, possibly supporting the lysosomal mediated exocytosis (249). These tunnels were not observed in another independent study, which instead indicates that virus egress follows a secretory pathway involving fusion of small vesicles generally containing only one viral particle (250). Nevertheless, by following such unconventional non-lytic egress pathway, viruses are co-trafficked together with the ER chaperone GRP78/BIP which could therefore assist the correct folding of newly synthesized viral protein. Interestingly, given the low pH, the lysosomal egress pathway could cause a premature activation of the viral particles. However, late endosomes/lysosomes of infected cells were shown to be deacidified and carry lysosomal enzymes with reduced activity. The exact deacidification mechanism has not yet been elucidated, with one possibility being that this is actively promoted by the infected cells to relieve viral-induced stress (248, 251). However, one deacidification pathway involves ORF3a which may act as a viroporin, consistent with previous knowledge involving SARS-CoV (63, 252, 253).
Interestingly, perturbation of lysosomal pH could also disrupt the antigen cross-presentation pathway and the endolysosomal Toll-like receptor signaling (248), benefiting virus survival. Since the lysosomal exocytic pathway depends on small GTPases, such as Arl8b and Rab7, which regulate intracellular trafficking, these have also become possible targets to inhibit the latest stage of the virus life cycle (248, 254).
It has been well-established that the SARS-CoV accessory protein ORF3a induces apoptosis, Golgi fragmentation, and accumulation of intracellular vesicles (255). Even though data with SARS-CoV-2 remain scarce, the pro-apoptotic activity of SARS-CoV-2 ORF3a has been confirmed in different cell lines, where the viral protein was shown to activate mainly the extrinsic apoptotic pathway by promoting cleavage of caspase-8 (64). Features that regulate SARS-CoV ORF3a intracellular transport include a tyrosine-based sorting motif (YXXΦ) and a diacidic EXD domain (256), a cysteine-rich sequence and a region with potassium channel activity (257). The protein ORF3a from SARS-CoV-2 shares 73% amino acid homology with its counterpart in SARS-CoV, maintaining the cysteine-rich sequence, the YXXΦ motif and the potassium channel region (64) and exerting a relatively weaker apoptotic activity than that associated with SARS-CoV ORF3a, correlating with the lower virulence of the pandemic virus. Interestingly, some of the mutations naturally occurring in the SARS-CoV-2 ORF3a coding sequence have been recently demonstrated to significantly affect protein stability and secondary structure (258), possibly affecting the virus pro-apoptotic activity. In-silico studies suggest that these mutations may help the virus to evade the host immune system because of the loss of B-cell putative epitopes (258), indicating a possible trade-off between the loss of apoptotic activity in favor of immune evasion. Nevertheless, residues of SARS-CoV-2 ORF3a predicted to favor stability of the protein central pore remain unaltered conserved (259) indicating that, though lower than in SARS-CoV, the ability to induce apoptosis remain important. Since the ion channel activity of ORF3a is required for its pro-apoptotic activity, inhibiting its function may still provide a direction toward interfering with coronaviruses cytopathology (257).
As a catabolic pathway of mammalian cells, autophagy controls viral infections at multiple levels by causing destruction of viruses, regulating inflammatory responses, and promoting antigen presentation. Moreover, viruses manipulate autophagy for their immune evasion, replication, and release from infected cells [for a review see (260)]. SARS-CoV-2 ORF3a has been shown to inhibit autophagosome-lysosome fusion by disrupting the assembly of the RAB7-HOPS fusion machinery, evidence obtained with ectopic expression of ORF3a (261). The HOPS tethering complex regulates the activity of SNARE proteins which govern autophagosome fusion with lysosomes (262). The C-terminal region of SARS-CoV-2 ORF3a was reported to interact with VPS39, a component of the HOPS complex, and to disrupt the assembly with the SNARE proteins (263). Interestingly, given the conformation similarity between DMVs and autophagosomes, ORF3a disruption of RAB7-HOPS interaction may also be beneficial for the virus to prevent DMVs from fusing with lysosomes, thereby promoting the conditions for optimal viral replication (261).
Infections caused by coronaviruses, such as SARS-CoV, MHV, IBV, and HCoV-HKU1, are known to induce ER stress, resulting in the activation of the unfolded protein response (UPR). This is regulated by ER transmembrane sensors PKR-like ER protein kinase (PERK), inositol-requiring protein 1 (IRE1) and activating transcriptional factor 6 (ATF6), leading to increased expression of ER chaperons, to global repression of protein synthesis or even to apoptosis when the ER stress becomes persistent [reviewed in (264, 265)]. ER stress is demonstrated by the induction of Immunoglobulin heavy chain-binding protein (BiP, also known as glucose-regulated protein 78, or GRP78) or glucose-regulated protein 94 (GRP94) in cells infected with SARS-CoV (265). From our knowledge based on SARS-CoV, ER stress is induced by different mechanisms which are consequences of the viral replicative cycle in the host cell. First, the UPR can be triggered by ER saturation following the extensive production of viral proteins, particularly the spike and the accessory proteins ORF3a, ORF6, ORF7a, and ORF8ab (266–268). In addition, contributions to ER stress originate from the formation of DMVs and other membrane rearrangements as well as from the ER membrane depletion following virus budding from the ERGIC and autophagy (265). More directly, the central region of the S1 subunit (amino acids 201–400) of SARS-CoV was found responsible for activating the transcriptional activity of GRP78 and GRP94 promoters, eventually leading to UPR (269). ORF8b was instead shown to contribute to ER stress by forming aggregates dependent on a C-terminal VLVVL motif (253). The contribution of ORF8 to ER stress was additionally confirmed by studying two genotypes of SARS-CoV-2, namely ORF8L and ORF8S, which carry a Leucine or a Serine at position 84 and were identified during the early stages of the pandemic in China. Despite lacking the aggregation motif found in SARS-CoV (270), both ORF8L and ORF8S are able to trigger the activation of ATF6 and IRE1 (71) and PERK (271). SARS-CoV-2 ORF8 is encoded by a hypervariable gene and several different polymorphisms have already been identified, as reported by a recent review (73). A large deletion (Δ382) which abolishes ORF8 expression associates with a favorable clinical outcome despite no appreciable effects on viral replication (272) demonstrating the contribution of ORF8 to SARS-CoV-2 virulence. Given all these indications that point toward a crucial role in pathogenesis, inhibiting ORF8 effector functions could be used to improve the clinical course of the infection (273).
Another contribution to the induction of UPR comes from the region IV of the SARS-CoV-2 spike protein which was observed to interact with the substrate-binding domain (SBD) of BiP (254). Conditions of prolonged ER stress result in an altered trafficking of BiP to the cell surface, causing its accumulation on the cell membrane or in extracellular secretions. Indeed, increased levels of BiP in lung pneumocytes and macrophages was observed in patients with COVID-19 (274). Interestingly, cell surface accumulated BiP has been shown to function as attachment factors for MERS-CoV, promoting viral infection (275). SARS-CoV-2 spike protein might also bind BiP, presumably facilitating viral entry and boosting infection (254). In addition to characterizing several pathological conditions, such as diabetes, obesity, and neurodegeneration, persistence of ER stress resulting from the accumulation of misfolded proteins can be elicited during aging by a decline of ER chaperone activity. Age and comorbidities therefore may predispose to ER stress caused by coronavirus infection, as well as lead to an increased susceptibility to infection due to accumulation of BiP on the cell surface (276, 277), therefore contributing to the severity of the coronavirus diseases in elderly or chronically diseased individuals.
If from one side it appears that the interaction between SARS-CoV-2 proteins and UPR effectors leads to the activation of this stress response, another recent article suggests that, even though CoVs infection initiate ER stress signaling and induces UPR components at the mRNA level, these are inhibited at the protein level (278). This could indicate that coronaviruses, including SARS-CoV-2, might have evolved strategies acting at posttranscriptional or translational level to escape antiviral response placed by BiP, IRE1α, and HERPUD. The evolution of a viral mechanism to counteract the effects of ER-stress is consistent with the emergent notion that UPR provides a crucial contribution to the activation of innate antiviral signaling, as seen with flaviviruses (279). Strikingly, treatment of cells with thapsigargin, a guaianolide which induces ER-stress by inhibiting the ER Calcium ATPase, represses replication of SARS-CoV-2 and other CoVs, counteracts virus mediated BiP downregulation, activates IRE1α and outweighs coronavirus mediated inhibition of global protein synthesis, thus becoming an attractive antiviral drug candidate (278, 280).
The enigmatic envelope (E) protein is the smallest among SARS-CoV-2 structural components acting as a viroporin and a virulence factor proposed to affect assembly, budding and envelope formation (29). Other than assisting virus replication, the E protein was also observed to interfere with the control of cell polarity and cell-cell junction integrity in epithelial cells. For both SARS-CoV and SARS-CoV-2, the E protein was shown to bind PALS1 PDZ domain, via the C-terminal DLLV motif (281–283). PALS1 is part of the CRUMBS3-PALS1-PATJ polarity complex, which is crucial for the establishment and maintenance of epithelial polarity. Accordingly, in cells infected with SARS-CoV, PALS1 was shown to relocalize to virus assembly sites resulting in the disruption of tight junctions. Disruption of tight junctions could also be the result of an interaction between the C-terminal domain of SARS-CoV-2 E protein and the PDZ domain of Zona Occludens-1 (ZO1), known for its pivotal role in tight junction formation (283). At the same time, the E protein was shown to also cause disruption of adherens junctions by impairing E-cadherin cell surface delivery (281, 282, 284). The impairment of both types of junctions could therefore explain the alveolar desquamation observed in SARS-CoV infection and the disruption of the epithelial barrier eventually favoring viral dissemination through systemic circulation and the amplification of the inflammatory response (281). Based on the genomic differences with SARS-CoV, a stronger interaction with PALS1 was predicted in silico based on two polymorphic residues at the C-terminus domain, postulating a crucial pathogenic role of the E protein of SARS-CoV-2 (282). Accordingly, the complex between SARS-CoV-2 E protein and PALS1 was characterized by cryo-EM indicating a possible target for novel peptide and small molecule inhibitors (285).
Upon infection, SARS-CoV-2 pathogen associated molecular patterns (PAMPs) can be recognized by several host pathogen recognition receptors (PRR), predominantly including Toll-like receptors (TLRs), retinoic-acid inducible gene I (RIG-I) and melanoma differentiation-associated 5 (MDA-5). Receptors intercepting SARS-CoV-2 can be found either in the endosomal compartment, where TLR-7 and TLR-8 can recognize the ssRNA viral genome, or in the cytoplasm, where RIG-I and MDA-5 initiate a signaling cascade in response to the ssRNA genome and the replication-intermediate dsRNA, respectively (Figure 9). RIG-I and MDA-5 signal through the same cascade: upon target RNA recognition, the CARD domain of these sensors recruit E3 ligases (TRIM25 and Riplet) that form a K63-linked polyubiquitin scaffold, allowing RIG-I and MDA-5 to interact with mitochondrial antiviral signaling protein (MAVS) on the outer mitochondrial membrane and form MAVS aggregates. These aggregates will in turn recruit TRAF family E3 ubiquitin ligases, which form new K63-linked polyubiquitin that activate TANK binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3), eventually leading to the production of type 1 interferons (IFNα/β) and activation of NF-κB [for a review see (286)].
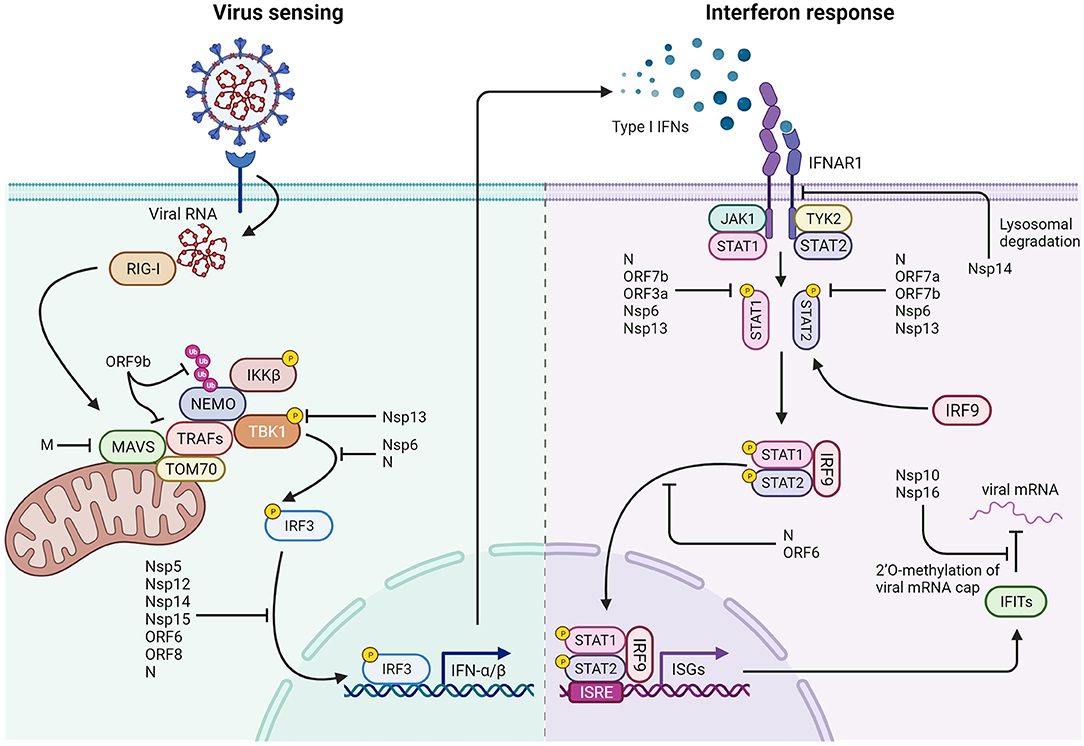
Figure 9. Interactions of SARS-CoV-2 with cellular components of the immune system. Left: SARS-CoV-2 triggers several mechanisms that hinder type 1 interferon production by preventing both viral RNA detection and activity of the IRF3 transcription factor. ORF9b interacts with TOM70 on the mitochondrial surface and prevents K63-polyubiquitination of NEMO. The M protein prevents MAVS aggregation and interaction with downstream proteins. Nsp13 blocks TBK1 phosphorylation by inhibiting its interaction with MAVS, while Nsp6 binds TBK1, and N protein prevents IRF3 phosphorylation. Nsp5, Nsp12, Nsp14, Nsp15, ORF6, ORF8, and N are reported to impair nuclear localization of phosphorylated IRF3. Right: Mechanisms that impair the response to type I interferons: Nsp14 induces lysosomal degradation of type 1 IFN receptor. Nsp6, Nsp13, ORF3a, ORF7a, ORF7b, and N prevent STAT1 and STAT2 phosphorylation. ORF6 and N protein hamper phosphorylated STATs nuclear translocation. In addition, Nsp10 and Nsp16 are responsible for 2'O-methylation of viral mRNA cap, limiting IFITs activation and translation.
Interferon production is pivotal for the induction of an antiviral state and the activation of adaptive immunity in both the infected cell and the surrounding cells, as it has both autocrine and paracrine effects. Interferon molecules, upon binding to their corresponding receptors IFNAR1/IFNAR2 on cells surfaces, activate a signaling cascade mediated by the Janus kinase 1 (JAK1), Tyrosine kinase 2 (TYK2) and signal transducer and activator of transcription (STAT1 and STAT2) (213). Upon phosphorylation, these mediators undergo dimerization and nuclear translocation, ultimately leading to the activation of several IFN-stimulated genes (ISGs) and contributing to the establishment of the antiviral state (287).
Several SARS-CoV-2 proteins have been shown to inhibit IFN-β promoter activation (54). Screening evidence reports that 11 viral proteins, both structural and non-structural, can antagonize the RNA-dependent IFN-β induction by targeting different components of the intracellular antiviral response triggered by RIG-I.
SARS-CoV-2 ORF9b was shown to localize on mitochondrial membrane and elicit the suppression of type I IFN response through association with TOM70, one of RIG-I mitochondrial interacting proteins besides MAVS, TRAF3 and TRAF6 (288) (Figure 9). In particular, upon viral RNA recognition, TOM70 mediates the interaction between MAVS and Hsp90-bound TBK1 and IRF3 (289). ORF9b can prevent the interaction of TOM70 with Hsp90, thus impairing this signaling pathway. The residues found to be crucial for the interaction between TOM70 and ORF9b are TOM70 E477 and ORF9b S53, and the interaction is facilitated by the binding between TOM70 R192 and the ORF9b EEVD motif (74). Since SARS-CoV-2 is detected mainly by RIG-I (76), the inactivation of this pathway strongly affects the host immune response. Consequently, the development of molecules inhibiting the interaction between ORF9b and TOM70 represents a possible therapeutic approach. Besides interacting with TOM70, ORF9b interrupts the K63-linked polyubiquitination of NEMO, hence preventing IKK activation and IKKβ phosphorylation, ultimately blocking NF-κB activation (76). In addition, ORF9b has been suggested to act at the convergence point among the RIG-I/MDA-5-MAVS, TLR3-TRIF, cGAS-STING pathways, that is TBK1, and inhibit them all. This protein also interacts with STING preventing TBK1 phosphorylation and TRIF interaction with TBK1. The presence of anti-cGAS-STING pathway proteins in SARS-CoV-2, despite it being an RNA virus, suggests that this pathway is involved in SARS-CoV-2 clearance (75).
Recent results also suggested that the M protein inhibits the RIG-I mediated antiviral response by interacting with the MAVS modulator. Mechanistically, the M protein impairs the MAVS aggregation in particular by disrupting MAVS self-association and its association with SNX8, thus altering the recruitment of downstream TRAF3, TBK1, and IRF3 which are required for an efficient innate antiviral response (31).
Additional proteins interfere with IFN-β induction by interacting with TBK1, which is essential for the phosphorylation and subsequent activation of IRF3. Indeed, Nsp6 has been shown to reduce the activity of genes driven by the IFN-β promoter in vitro (44) by binding TBK1 and reducing phosphorylation of IRF3. Interestingly, the naturally occurring L37F mutation of this protein is associated with a milder outcome of the infection (290), although the impact of this mutation on RIG-I signaling has not yet been directly documented.
Nsp13, the SARS-CoV-2 helicase, is another inhibitor of the type I IFN response (54). Nsp13 binds and blocks TBK1 phosphorylation, thus resulting in decreased IRF3 activation and IFN-β production. Recent results suggest that Nsp13 suppresses type I IFN production by disrupting the association of TBK1 with MAVS (52). Furthermore, Nsp13 hijacks the deubiquitinase USP13, which deubiquitinates and therefore stabilizes Nsp13. Interestingly, knockout of USP13 or treatment with USP13 inhibitor mitigates the Nsp13 inhibition of type I IFN response and suppresses viral replication. Therefore, USP13 inhibitors could be used to suppress virus replication by targeting Nsp13 for degradation (52).
Several proteins have been shown to impair either phosphorylation or nuclear translocation of IRF3. Nsp5 counteracts RNA-dependent IFN-β induction by acting downstream of IRF3 cytoplasmic phosphorylation mediated by TBK1 and IKKε. Indeed, Nsp5 was shown to impair the nuclear localization of phosphorylated IRF3 rather than its stabilization or phosphorylation (42). Nsp12, an RNA dependent RNA polymerase, was also reported to inhibit the IFN signaling (54), by suppressing the nuclear translocation of IRF3 which is essential for the promotion of production of IFN-α/β. The mechanism was shown to be independent from its polymerase activity (50), though results from a recent report confuted this activity (291). Nsp14 and Nsp15 have also been reported to block IRF3 nuclear translocation (57), SARS-CoV-2 Nsp15 being less potent in counteracting both IFN-β induction and signaling than the SARS-CoV analog, perhaps contributing to the more favorable clinical outcome of SARS-CoV-2 (292).
ORF6 and ORF8 are also reported to inhibit the nuclear translocation of IRF3. Two of the genotypes of ORF8, ORF8L, and ORF8S, were shown to be capable of down-regulating IFN-β, interferon-stimulated genes (ISGs) ISG15 and ISG56 production, and impaired IRF3 nuclear translocation in response to poly (I:C). This down-regulating effect was found at the level of both protein abundance and mRNA levels, but it remains unclear whether SARS-CoV-2 ORF8 physically interacts with transcription factors involved in IFN-β regulation (71). The ability of ORF6 to inhibit IRF3 nuclear translocation is conserved between SARS-CoV-1 and SARS-CoV-2, depending on the sequence DEEQPMEID found at its C-terminus, which could indicate another possible therapeutic target for anti-COVID-19 therapy (54).
Finally, the N protein co-localizes with RIG-I by interacting with its DExD/H domain and, with still undefined mechanisms, impairs both IRF3 phosphorylation and nuclear translocation rather than IRF3 production, thus impacting IFN-β production (293).
Interferon signaling is inhibited in host cells by a number of SARS-CoV-2 proteins that generally modulate STAT1 or STAT2 phosphorylation, thus preventing their nuclear translocation (Figure 9). So far, the viral proteins that have been reported to have such activity include N, ORF3a, ORF6, ORF7 (including both ORF7a and ORF7b), Nsp6, Nsp13, and Nsp14. The nucleocapsid (N) protein antagonizes IFN-I signaling by preventing STAT1 and STAT2 phosphorylation and interfering with the interaction of STAT1 with JAK1 and of STAT2 with TYK2, suggesting a competitive binding to STAT molecules with the respective downstream kinases. In addition, the interaction between SARS-CoV-2 N and STAT1/STAT2 prevents their IFN-induced nuclear translocation. The N-terminal 361 amino acids of N were found to be sufficient for its activity, including cytoplasmic localization, suppression of phosphorylation and interaction with STAT1/STAT2 (25).
ORF6 exerts its immune escape activity by inhibiting both IRF3 activation and STAT1 nuclear translocation. Nuclear translocation of STAT proteins is known to occur upon their phosphorylation and dimerization and to depend on the activity of the KPNA1-KPNB1 complex, which docks the import complex to the nuclear pore complex (NPC) (294). The formation of an ORF6-Nup98-Rae1 complex leads to the inability of the cargo-KPNA1-KPNB1 complex to dock at the NPC, and thus promotes cytoplasmic accumulation of phosphorylated STATs (68). ORF7a marginally inhibits STAT1 phosphorylation, while the effect of ORF7a and ORF7b on STAT2 is more pronounced and ORF7b is active also against STAT1 (44). Further analysis found the phosphorylation antagonizing effect of ORF7a to be mediated and enhanced by K63-linked polyubiquitin chains at lysine 119 (52). ORF3a contributes to the inhibition of STAT1 phosphorylation and marginally suppresses STAT2 phosphorylation (44). Nsp6 and Nsp13 act similarly on interferon signaling by suppressing STAT1 and STAT2 phosphorylation. Instead, Nsp14 acts upstream of STAT1 and STAT2 phosphorylation by inducing the lysosomal degradation of the type I IFN receptor IFNAR1, thus preventing the activation of STAT1 and STAT2 transcription factors (295).
Pyroptosis has been determined to play a critical role in establishing an early antiviral response. The process is dependent on the activation of the inflammasome, a multiprotein complex consisting of pro-caspase-1, ASC and inflammasome nucleators such as NOD-like receptors (NLRs), AIM2 and pyrin. Of the many NLRs described so far, NLRP3 is strongly induced by viral infection, thus leading to inflammasome formation (296). NLRP3 inflammasome activation leads to pro-caspase-1 auto-cleavage and generation of active caspase-1 that, in turn, will process pro-IL-1β and pro-IL-18 into IL-1β, and IL-18, respectively. The secretion of these cytokines is pivotal for the recruitment of effector immune cells to the site of infection and for the establishment of an adaptive immune response. In addition to the processing of pro-IL-1β and pro-IL-18, active caspase-1 also cleaves Gasdermin D (GSDMD) (296), releasing a N-terminus fragment which interacts with membrane lipids for the formation of transmembrane pores, that cause the release of cytokines and danger signals and ultimately lead to cell death (297). SARS-CoV-2 nucleocapsid protein can interact with GSDMD and prevent caspase-1-mediated cleavage (Figure 10). Because NLRP3 inflammasome formation is unaffected by SARS-CoV-2 nucleocapsid protein, the natural consequence of GSDMD protection is the intracellular accumulation of IL-1β and IL-18 that cannot be released. This mechanism could therefore delay the initial antiviral response and contribute to the asymptomatic phase of COVID-19 infection (298). Opposing this evidence, multiple studies have in contrast shown that the inflammasome is activated in COVID-19 patients, linking this activation to a more severe disease (299–301). It was proposed that this can trigger a feedback loop of IL-1β signaling that also induces interleukin-6 (IL-6) production [reviewed in (302)]. Since high levels of IL-6 are found in severe COVID-19 cases, this step may provide a crucial contribution to the cytokine storm observed in patients (303).
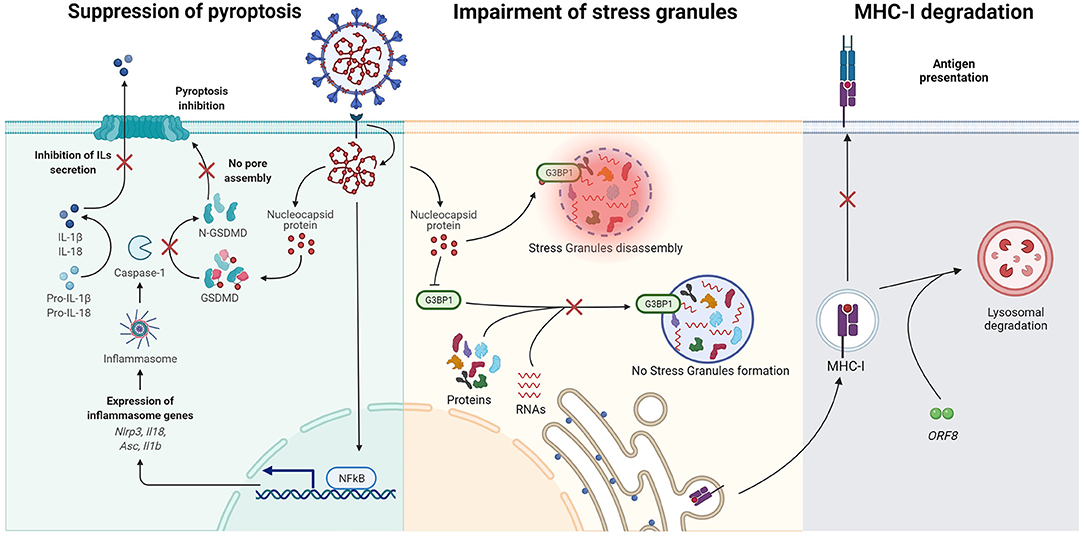
Figure 10. Additional mechanisms for immune evasion. Left: impairment of pyroptosis. The interaction of the N protein with the linker peptide of GSDMD prevents cleavage of GSDMD by inflammasome-activated caspase-1, thus preventing N-GSDMD from assembling pores in the cell membrane and triggering pyroptosis. Center: impairment of Stress Granules. Binding of N protein to G3BP1 prevents G3BP1 interaction with other stress granules proteins, hampering formation of new stress granules and disassembling those already formed. Right: MHC-I degradation. SARS-CoV-2 ORF8 induces MHC-I lysosomal degradation, thus reducing antigen presentation on host cells.
Besides the mechanisms employed by SARS-CoV-2 to evade the action of the innate immune system, SARS-CoV-2 adopts additional strategies to escape the multifaceted immunity of infected cells (Figure 10). As we have described earlier, SARS-CoV-2 non-structural protein 1 (Nsp1) binds to the 40S subunit of the ribosome and shuts off host RNA translation (304), a mechanism that was also observed in other coronaviruses including SARS-CoV [reviewed in (214, 305)], and which limits the ability of the host cells to express antiviral activities.
An additional mechanism is provided by Nsp16, which has been identified to be responsible for viral mRNA cap 2'O-methylation in other coronaviruses (306). 2'O-methylation of mRNA cap allows viruses to avoid restriction by IFN-induced proteins with tetratricopeptide repeats (IFIT) by mimicking the structure and post-transcriptional modifications of endogenous, eukaryotic mRNA (307). Nsp10 has been observed to associate with Nsp16 and to be required to achieve mRNA cap methylation in SARS-CoV (308). The same complex with the same methyltransferase activity is found in SARS-CoV-2 (309).
Nsp15 was shown to inhibit autophagy induction (295), while the N protein prevents the inhibition of viral mRNA translation. Accordingly, one of the innate mechanisms employed by eukaryotic cells to hamper viral infection is based on the formation of stress granules (SGs), cytoplasmic mRNA-protein aggregates. These ribonucleoprotein complexes are capable of trapping viral mRNA [reviewed in (310)], thus limiting their translation. Most viruses have evolved mechanisms to escape this inhibition by prevention of SG formation [reviewed in (305)], and SARS-CoV-2 makes no exception. SARS-COV-2 N protein was found to bind the core protein of SGs Ras-GTPase activating SH3-domain-binding-protein 1/2 (G3BP1/2) (262, 306). By specifically binding G3BP1/2, the N protein prevents the formation of stress granules and disassembles already formed granules by blocking the interaction between G3BP1/2 and other SG-related proteins, thus facilitating viral assembly. The N protein exerts this activity via formation of liquid-liquid phase separation (LLPS) condensates. In particular, N protein intrinsically disordered region 1 (IDR1) is responsible for phase separation with G3BP1 (307). G3BP1/2 additionally functions as a positive regulator of RIG-I and significantly improves RIG-I-mediated IFN-β synthesis (308, 309) and of cGAS (308). This may be a potential mechanism by which the SARS-CoV-2 N protein affects IFN-β via the RIG-I signaling pathway (50).
MHC-I expression seems also to be dysregulated by SARS-CoV-2, a unique feature not shared with other SARS-CoV strains (72). HEK cells infected with SARS-CoV-2 were observed to decrease their MHC-I expression after infection. A similar effect was observed in mice, where MHC-I expression in lung epithelial cells was lower in animals with ongoing virus replication compared to animals that had fully recovered. ORF8 was found the most likely culprit for this process by selectively targeting MHC-I for lysosomal degradation via autophagy, protecting cells from cytotoxic T lymphocytes-mediated killing. Some in vitro evidence suggests that ORF8 may have more roles in immune evasion as previously described. Its ectopic expression has been correlated with a reduced expression of several IFNγ-induced antiviral molecules with different biochemical activities in lung epithelial cells (311). Interestingly, the same group observed that ectopic expression of ORF8 in HEK293 cells reduced the expression of different antiviral proteins, suggesting that the effects of ORF8 on ISGs may be cell type specific.
After SARS-CoV-2 emerged in 2019, the scientific community has engaged in an unprecedented effort to elucidate the virus replication mechanism, decipher the host immune response and understand the immunopathogenesis caused by the infection. Accordingly, a clearer picture of the complex pattern of molecular interactions of SARS-CoV-2 with host cells is now helping to prepare novel strategies for prevention and treatment. Here we have shown how in <2 years researchers have produced 3D structures revealing the fine details of the interaction between viral proteins and host factors, obtained exhaustive proteomics, transcriptomics, and interactomics profiles which have mapped the intricate interplay of the virus with the cells, and identified novel host factors crucial for the outcome of the infection. All this would not have been possible in such a short period of time without the prior knowledge built after years of coronavirus research triggered by previous outbreaks and epidemics, especially those involving SARS-CoV and MERS-CoV. However, some aspects of the interaction between the virus and the host cell require an improved understanding to unveil the mechanisms supporting virus replication and identify novel viral inhibitors.
The precise understanding of SARS-CoV-2 entry pathways remain essential to identify new antiviral targets, which together with neutralizing antibodies could prevent infection of the host cell. Moreover, a key early event that deserves further investigation is the specific host translation inhibition by Nsp1, an activity which could reveal a crucial Achille's heel of the virus. In general, much mystery still surrounds some ORFs and Nsps of SARS-CoV-2, which are likely to play important roles in regulating cellular pathways and in tuning the cellular environment for optimal virus replication while avoiding immunity. This is for example the case of ORF3a and its effect on the apoptotic machinery. Indeed, blocking this viral protein could re-establish the defective autophagosome-lysosome fusion of infected cells, thereby preventing formation of DMVs and favoring recognition by the immune system. At the same time, the mechanism of viral exit from the infected cell remains uncertain and insights on this step of the infectious cycle are pivotal to understand how spreading of the virus in the host can be inhibited, possibly leading to the development of new drugs.
In conclusion, while the pandemic is slowing down thanks to the global administration of vaccines, the research should continue in order to fully understand the mechanisms of SARS-CoV-2 interaction with the cell, providing results that can be translated into treatments and preparing the world for future viral outbreaks.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We wish to thank Graziano Lolli for his help with processing the structure images in Figure 5.
1. Worobey M. Dissecting the early COVID-19 cases in Wuhan. Science. (2021) 374:eabm4454. doi: 10.1126/science.abm4454
2. Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
3. WHO Calls for Further Studies, Data on Origin of SARS-CoV-2 Virus, Reiterates That All Hypotheses Remain Open. Available online at: https://www.who.int/news/item/30-03-2021-who-calls-for-further-studies-data-on-origin-of-sars-cov-2-virus-reiterates-that-all-hypotheses-remain-open (accessed December 13, 2021).
4. Report of the WHO-China Joint Mission on Coronavirus Disease 2019 (COVID-19). Available online at: https://www.who.int/publications-detail-redirect/report-of-the-who-china-joint-mission-on-coronavirus-disease-2019-(covid-19) (accessed December 13, 2021).
5. Malaiyan J, Arumugam S, Mohan K, Radhakrishnan GG. An update on the origin of SARS-CoV-2: despite closest identity, bat (RaTG13) and pangolin derived coronaviruses varied in the critical binding site and O-linked glycan residues. J Med Virol. (2021) 93:499–505. doi: 10.1002/jmv.26261
6. Lam TT-Y, Jia N, Zhang Y-W, Shum MH-H, Jiang J-F, Zhu H-C, et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature. (2020) 583:282–5. doi: 10.1038/s41586-020-2169-0
7. Zhou H, Ji J, Chen X, Bi Y, Li J, Wang Q, et al. Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. Cell. (2021) 184:4380–91.e14. doi: 10.1016/j.cell.2021.06.008
8. Singh D, Yi SV. On the origin and evolution of SARS-CoV-2. Exp Mol Med. (2021) 53:537–47. doi: 10.1038/s12276-021-00604-z
9. Zhang Y-Z, Holmes EC. A genomic perspective on the origin and emergence of SARS-CoV-2. Cell. (2020) 181:223–7. doi: 10.1016/j.cell.2020.03.035
10. Domingo JL. What we know and what we need to know about the origin of SARS-CoV-2. Environ Res. (2021) 200:111785. doi: 10.1016/j.envres.2021.111785
11. Tiruneh SA, Tesema ZT, Azanaw MM, Angaw DA. The effect of age on the incidence of COVID-19 complications: a systematic review and meta-analysis. Syst Rev. (2021) 10:80. doi: 10.1186/s13643-021-01636-2
12. Oran DP, Topol EJ. The proportion of SARS-CoV-2 infections that are asymptomatic. Ann Intern Med. (2021) 174:655–62. doi: 10.7326/M20-6976
13. Johansson MA, Quandelacy TM, Kada S, Prasad PV, Steele M, Brooks JT, et al. SARS-CoV-2 transmission from people without COVID-19 symptoms. JAMA Netw Open. (2021) 4:e2035057. doi: 10.1001/jamanetworkopen.2020.35057
14. Hussain M, Jabeen N, Amanullah A, Baig AA, Aziz B, Shabbir S, et al. Molecular docking between human TMPRSS2 and SARS-CoV-2 spike protein: conformation and intermolecular interactions. AIMS Microbiol. (2020) 6:350–60. doi: 10.3934/microbiol.2020021
15. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. (2020) 180:934–43. doi: 10.1001/jamainternmed.2020.0994
16. Batah SS, Fabro AT. Pulmonary pathology of ARDS in COVID-19: a pathological review for clinicians. Respir Med. (2021) 176:106239. doi: 10.1016/j.rmed.2020.106239
17. Harrison AG, Lin T, Wang P. Mechanisms of SARS-CoV-2 transmission and pathogenesis. Trends Immunol. (2020) 41:1100–15. doi: 10.1016/j.it.2020.10.004
18. Karia R, Gupta I, Khandait H, Yadav A, Yadav A. COVID-19 and its modes of transmission. SN Comprehens Clin Med. (2020) 2:1798–801. doi: 10.1007/s42399-020-00498-4
19. Zhou L, Ayeh SK, Chidambaram V, Karakousis PC. Modes of transmission of SARS-CoV-2 and evidence for preventive behavioral interventions. BMC Infect Dis. (2021) 21:1–9. doi: 10.1186/s12879-021-06222-4
20. Mehta OP, Bhandari P, Raut A, Kacimi SEO, Huy NT. Coronavirus Disease (COVID-19): comprehensive review of clinical presentation. Front Public Health. (2021) 8:1034. doi: 10.3389/fpubh.2020.582932
21. Yang L, Xie X, Tu Z, Fu J, Xu D, Zhou Y. The signal pathways and treatment of cytokine storm in COVID-19. Sig Transduct Target Ther. (2021) 6:1–20. doi: 10.1038/s41392-021-00679-0
22. Wong CK, Lam CWK, Wu AKL, Ip WK, Lee NLS, Chan IHS, et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol. (2004) 136:95–103. doi: 10.1111/j.1365-2249.2004.02415.x
23. Mosquera-Sulbaran JA, Pedreañez A, Carrero Y, Callejas D. C-reactive protein as an effector molecule in Covid-19 pathogenesis. Rev Med Virol. (2021) 31:e2221. doi: 10.1002/rmv.2221
24. Coronaviridae - Positive Sense RNA Viruses - Positive Sense RNA Viruses. ICTV (2011). Available online at: https://talk.ictvonline.org/ictv-reports/ictv_9th_report/positive-sense-rna-viruses-2011/w/posrna_viruses/222/coronaviridae (accessed December 13, 2021).
25. Mu J, Fang Y, Yang Q, Shu T, Wang A, Huang M, et al. SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discov. (2020) 6:1–4. doi: 10.1038/s41421-020-00208-3
26. Zhao Y, Sui L, Wu P, Wang W, Wang Z, Yu Y, et al. A dual-role of SARS-CoV-2 nucleocapsid protein in regulating innate immune response. Sig Transduct Target Ther. (2021) 6:1–14. doi: 10.1038/s41392-021-00742-w
27. Zheng Z-Q, Wang S-Y, Xu Z-S, Fu Y-Z, Wang Y-Y. SARS-CoV-2 nucleocapsid protein impairs stress granule formation to promote viral replication. Cell Discov. (2021) 7:1–11. doi: 10.1038/s41421-021-00275-0
28. Cui L, Wang H, Ji Y, Yang J, Xu S, Huang X, et al. The nucleocapsid protein of coronaviruses acts as a viral suppressor of RNA silencing in mammalian cells. J Virol. (2015) 89:9029–43. doi: 10.1128/JVI.01331-15
29. Pervushin K, Tan E, Parthasarathy K, Lin X, Jiang FL, Yu D, et al. Structure and inhibition of the SARS coronavirus envelope protein ion channel. PLoS Pathog. (2009) 5:e1000511. doi: 10.1371/journal.ppat.1000511
30. Neuman BW, Kiss G, Kunding AH, Bhella D, Baksh MF, Connelly S, et al. A structural analysis of M protein in coronavirus assembly and morphology. J Struct Biol. (2011) 174:11–22. doi: 10.1016/j.jsb.2010.11.021
31. Fu Y-Z, Wang S-Y, Zheng Z-Q, Huang Y, Li W-W, Xu Z-S, et al. SARS-CoV-2 membrane glycoprotein M antagonizes the MAVS-mediated innate antiviral response. Cell Mol Immunol. (2020) 18:613–20. doi: 10.1038/s41423-020-00571-x
32. Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, et al. Structural basis of receptor recognition by SARS-CoV-2. Nature. (2020) 581:221–4. doi: 10.1038/s41586-020-2179-y
33. Rahman MS, Islam MR, Alam ASMRU, Islam I, Hoque MN, Akter S, et al. Evolutionary dynamics of SARS-CoV-2 nucleocapsid protein and its consequences. J Med Virol. (2021) 93:2177–95. doi: 10.1002/jmv.26626
34. Hartenian E, Nandakumar D, Lari A, Ly M, Tucker JM, Glaunsinger BA. The molecular virology of coronaviruses. J Biol Chem. (2020) 295:12910–34. doi: 10.1074/jbc.REV120.013930
35. Huang C, Lokugamage KG, Rozovics JM, Narayanan K, Semler BL, Makino S. SARS coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mRNAs: viral mRNAs are resistant to nsp1-Induced RNA Cleavage. PLoS Pathog. (2011) 7:e1002433. doi: 10.1371/journal.ppat.1002433
36. Tanaka T, Kamitani W, DeDiego ML, Enjuanes L, Matsuura Y. Severe acute respiratory syndrome coronavirus nsp1 facilitates efficient propagation in cells through a specific translational shutoff of host mRNA. J Virol. (2012) 86:11128–37. doi: 10.1128/JVI.01700-12
37. Tidu A, Janvier A, Schaeffer L, Sosnowski P, Kuhn L, Hammann P, et al. The viral protein NSP1 acts as a ribosome gatekeeper for shutting down host translation and fostering SARS-CoV-2 translation. RNA. (2021) 27:253–64. doi: 10.1261/rna.078121.120
38. Slavin M, Zamel J, Zohar K, Eliyahu T, Braitbard M, Brielle E, et al. Targeted in situ cross-linking mass spectrometry and integrative modeling reveal the architectures of three proteins from SARS-CoV-2. PNAS. (2021) 118:e2103554118. doi: 10.1073/pnas.2103554118
39. Osipiuk J, Azizi S-A, Dvorkin S, Endres M, Jedrzejczak R, Jones KA, et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat Commun. (2021) 12:743. doi: 10.1038/s41467-021-21060-3
40. Gadlage MJ, Sparks JS, Beachboard DC, Cox RG, Doyle JD, Stobart CC, et al. Murine hepatitis virus nonstructural protein 4 regulates virus-induced membrane modifications and replication complex function. J Virol. (2010) 84:280–90. doi: 10.1128/JVI.01772-09
41. Lee J, Worrall LJ, Vuckovic M, Rosell FI, Gentile F, Ton A-T, et al. Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. Nat Commun. (2020) 11:5877. doi: 10.1038/s41467-020-19662-4
42. Fung SY, Siu KL, Lin H, Yeung ML, Jin DY. SARS-CoV-2 main protease suppresses type I interferon production by preventing nuclear translocation of phosphorylated IRF3. Int J Biol Sci. (2021) 17:1547–54. doi: 10.7150/ijbs.59943
43. Cottam EM, Whelband MC, Wileman T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy. (2014) 10:1426–41. doi: 10.4161/auto.29309
44. Xia H, Cao Z, Xie X, Zhang X, Chen JY-C, Wang H, et al. Evasion of type I interferon by SARS-CoV-2. Cell Rep. (2020) 33:108234. doi: 10.1016/j.celrep.2020.108234
45. Angelini MM, Akhlaghpour M, Neuman BW, Buchmeier MJ. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio. (2013) 4:e00524-13. doi: 10.1128/mBio.00524-13
46. te Velthuis AJW, van den Worm SHE, Snijder EJ. The SARS-coronavirus nsp7+nsp8 complex is a unique multimeric RNA polymerase capable of both de novo initiation and primer extension. Nucleic Acids Res. (2012) 40:1737–47. doi: 10.1093/nar/gkr893
47. Yan L, Ge J, Zheng L, Zhang Y, Gao Y, Wang T, et al. Cryo-EM structure of an extended SARS-CoV-2 replication and transcription complex reveals an intermediate state in cap synthesis. Cell. (2021) 184:184–93.e10. doi: 10.1016/j.cell.2020.11.016
48. Decroly E, Debarnot C, Ferron F, Bouvet M, Coutard B, Imbert I, et al. Crystal structure and functional analysis of the SARS-coronavirus RNA Cap 2′-O-methyltransferase nsp10/nsp16 Complex. PLoS Pathog. (2011) 7:e1002059. doi: 10.1371/journal.ppat.1002059
49. te Velthuis AJW, Arnold JJ, Cameron CE, van den Worm SHE, Snijder EJ. The RNA polymerase activity of SARS-coronavirus nsp12 is primer dependent. Nucleic Acids Res. (2010) 38:203–214. doi: 10.1093/nar/gkp904
50. Wang W, Zhou Z, Xiao X, Tian Z, Dong X, Wang C, et al. SARS-CoV-2 nsp12 attenuates type I interferon production by inhibiting IRF3 nuclear translocation. Cell Mol Immunol. (2021) 18:945–53. doi: 10.1038/s41423-020-00619-y
51. Jia Z, Yan L, Ren Z, Wu L, Wang J, Guo J, et al. Delicate structural coordination of the Severe Acute Respiratory Syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. (2019) 47:6538–50. doi: 10.1093/nar/gkz409
52. Guo G, Gao M, Gao X, Zhu B, Huang J, Luo K, et al. SARS-CoV-2 non-structural protein 13 (nsp13) hijacks host deubiquitinase USP13 and counteracts host antiviral immune response. Signal Transduct Targeted Ther. (2021) 6:1–3. doi: 10.1038/s41392-021-00509-3
53. Minskaia E, Hertzig T, Gorbalenya AE, Campanacci V, Cambillau C, Canard B, et al. Discovery of an RNA virus 3′ → 5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. PNAS. (2006) 103:5108–13. doi: 10.1073/pnas.0508200103
54. Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun. (2020) 11:1–2. doi: 10.1038/s41467-020-17665-9
55. Palermo E, Di Carlo D, Sgarbanti M, Hiscott J. Type I interferons in COVID-19 pathogenesis. Biology. (2021) 10:829. doi: 10.3390/biology10090829
56. Bhardwaj K, Sun J, Holzenburg A, Guarino LA, Kao CC. RNA recognition and cleavage by the SARS coronavirus endoribonuclease. J Mol Biol. (2006) 361:243–56. doi: 10.1016/j.jmb.2006.06.021
57. Yuen C-K, Lam J-Y, Wong W-M, Mak L-F, Wang X, Chu H, et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg Microbes Infect. (2020) 9:1418–28. doi: 10.1080/22221751.2020.1780953
58. Koepke L, Hirschenberger M, Hayn M, Kirchhoff F, Sparrer KM. Manipulation of autophagy by SARS-CoV-2 proteins. Autophagy. (2021) 17:2659–61. doi: 10.1080/15548627.2021.1953847
59. Shi P, Su Y, Li R, Liang Z, Dong S, Huang J. PEDV nsp16 negatively regulates innate immunity to promote viral proliferation. Virus Res. (2019) 265:57–66. doi: 10.1016/j.virusres.2019.03.005
60. Vithani N, Ward MD, Zimmerman MI, Novak B, Borowsky JH, Singh S, et al. SARS-CoV-2 Nsp16 activation mechanism and a cryptic pocket with pan-coronavirus antiviral potential. Biophys J. (2021) 120:2880–9. doi: 10.1016/j.bpj.2021.03.024
61. Yuan X, Li J, Shan Y, Yang Z, Zhao Z, Chen B, et al. Subcellular localization and membrane association of SARS-CoV 3a protein. Virus Res. (2005) 109:191–202. doi: 10.1016/j.virusres.2005.01.001
62. Yuan X, Yao Z, Wu J, Zhou Y, Shan Y, Dong B, et al. G1 phase cell cycle arrest induced by SARS-CoV 3a protein via the cyclin D3/pRb pathway. Am J Respir Cell Mol Biol. (2007) 37:9–19. doi: 10.1165/rcmb.2005-0345RC
63. Castaño-Rodriguez C, Honrubia JM, Gutiérrez-Álvarez J, DeDiego ML, Nieto-Torres JL, Jimenez-Guardeño JM, et al. Role of severe acute respiratory syndrome coronavirus viroporins E, 3a, and 8a in replication and pathogenesis. mBio. (2018) 9. doi: 10.1128/mBio.02325-17
64. Ren Y, Shu T, Wu D, Mu J, Wang C, Huang M, et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol Immunol. (2020) 17:881–3. doi: 10.1038/s41423-020-0485-9
65. Hachim A, Kavian N, Cohen CA, Chin AWH, Chu DKW, Mok CKP, et al. ORF8 and ORF3b antibodies are accurate serological markers of early and late SARS-CoV-2 infection. Nat Immunol. (2020) 21:1293–301. doi: 10.1038/s41590-020-0773-7
66. Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y, et al. SARS-CoV-2 ORF3b Is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. (2020) 32:108185. doi: 10.1016/j.celrep.2020.108185
67. Li J-Y, Liao C-H, Wang Q, Tan Y-J, Luo R, Qiu Y, et al. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. (2020) 286:198074. doi: 10.1016/j.virusres.2020.198074
68. Miorin L, Kehrer T, Sanchez-Aparicio MT, Zhang K, Cohen P, Patel RS, et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc Natl Acad Sci. (2020) 117:28344–54. doi: 10.1073/pnas.2016650117
69. Lee J-G, Huang W, Lee H, van de Leemput J, Kane MA, Han Z. Characterization of SARS-CoV-2 proteins reveals Orf6 pathogenicity, subcellular localization, host interactions and attenuation by Selinexor. Cell Biosci. (2021) 11:58. doi: 10.1186/s13578-021-00568-7
70. Kopecky-Bromberg SA, Martinez-Sobrido L, Palese P. 7a Protein of severe acute respiratory syndrome coronavirus inhibits cellular protein synthesis and activates p38 mitogen-activated protein kinase. J Virol. (2006) 80:785–93. doi: 10.1128/JVI.80.2.785-793.2006
71. Rashid F, Dzakah EE, Wang H, Tang S. The ORF8 protein of SARS-CoV-2 induced endoplasmic reticulum stress and mediated immune evasion by antagonizing production of interferon beta. Virus Res. (2021) 296:198350. doi: 10.1016/j.virusres.2021.198350
72. Zhang Y, Chen Y, Li Y, Huang F, Luo B, Yuan Y, et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-?. Proc Natl Acad Sci. (2021) 118:e2024202118. doi: 10.1073/pnas.2024202118
73. Zinzula L. Lost in deletion: the enigmatic ORF8 protein of SARS-CoV-2. Biochem Biophys Res Commun. (2021) 538:116–24. doi: 10.1016/j.bbrc.2020.10.045
74. Brandherm L, Kobaš AM, Klöhn M, Brüggemann Y, Pfaender S, Rassow J, et al. Phosphorylation of SARS-CoV-2 Orf9b regulates its targeting to two binding sites in TOM70 and recruitment of Hsp90. Int J Mol Sci. (2021) 22:9233. doi: 10.3390/ijms22179233
75. Han L, Zhuang MW, Deng J, Zheng Y, Zhang J, Nan ML, et al. SARS-CoV-2 ORF9b antagonizes type I and III interferons by targeting multiple components of the RIG-I/MDA-5–MAVS, TLR3–TRIF, and cGAS–STING signaling pathways. J Med Virol. (2021) 93:5376–89. doi: 10.1002/jmv.27050
76. Wu J, Shi Y, Pan X, Wu S, Hou R, Zhang Y, et al. SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep. (2021) 34:108761. doi: 10.1016/j.celrep.2021.108761
77. Pancer K, Milewska A, Owczarek K, Dabrowska A, Kowalski M, Łabaj PP, Branicki W, et al. The SARS-CoV-2 ORF10 is not essential in vitro or in vivo in humans. PLoS Pathog. (2020) 16:e1008959. doi: 10.1371/journal.ppat.1008959
78. Redondo N, Zaldívar-López S, Garrido JJ, Montoya M. SARS-CoV-2 accessory proteins in viral pathogenesis: knowns and unknowns. Front Immunol. (2021) 12:2698. doi: 10.3389/fimmu.2021.708264
79. Hui KPY, Cheung M-C, Perera RAPM, Ng K-C, Bui CHT, Ho JCW, et al. Tropism, replication competence, and innate immune responses of the coronavirus SARS-CoV-2 in human respiratory tract and conjunctiva: an analysis in ex-vivo and in-vitro cultures. Lancet Respir Med. (2020) 8:687–95. doi: 10.1016/S2213-2600(20)30193-4
80. Sungnak W, Huang N, Bécavin C, Berg M, Queen R, Litvinukova M, et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. (2020) 26:681–7. doi: 10.1038/s41591-020-0868-6
81. Liu J, Li Y, Liu Q, Yao Q, Wang X, Zhang H, et al. SARS-CoV-2 cell tropism and multiorgan infection. Cell Discov. (2021) 7:1–4. doi: 10.1038/s41421-021-00249-2
82. Muus C, Luecken MD, Eraslan G, Sikkema L, Waghray A, Heimberg G, et al. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat Med. (2021) 27:546–59. doi: 10.1038/s41591-020-01227-z
83. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. (2020) 395:1417–8. doi: 10.1016/S0140-6736(20)30937-5
84. Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z, et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. (2020) 181:894–904.e9. doi: 10.1016/j.cell.2020.03.045
85. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. (2020) 367:1444–8. doi: 10.1126/science.abb2762
86. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–80.e8. doi: 10.1016/j.cell.2020.02.052
87. V'kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. (2020) 19:155–70. doi: 10.1038/s41579-020-00468-6
88. Koehler M, Delguste M, Sieben C, Gillet L, Alsteens D. Initial step of virus entry: virion binding to cell-surface glycans. Annu Rev Virol. (2020) 7:143–65. doi: 10.1146/annurev-virology-122019-070025
89. Madu IG, Chu VC, Lee H, Regan AD, Bauman BE, Whittaker GR. Heparan sulfate is a selective attachment factor for the avian coronavirus infectious bronchitis virus Beaudette. Avian Dis. (2007) 51:45–51. doi: 10.1637/0005-2086(2007)051[0045:HSIASA]2.0.CO;2
90. Watanabe R, Sawicki SG, Taguchi F. Heparan sulfate is a binding molecule but not a receptor for CEACAM1-independent infection of murine coronavirus. Virology. (2007) 366:16–22. doi: 10.1016/j.virol.2007.06.034
91. Milewska A, Zarebski M, Nowak P, Stozek K, Potempa J, Pyrc K. Human coronavirus NL63 utilizes heparan sulfate proteoglycans for attachment to target cells. J Virol. (2014) 88:13221–30. doi: 10.1128/JVI.02078-14
92. Clausen TM. SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell. (2020) 183:1043–57.e15. doi: 10.1016/j.cell.2020.09.033
93. Kalra RS, Kandimalla R. Engaging the spikes: heparan sulfate facilitates SARS-CoV-2 spike protein binding to ACE2 and potentiates viral infection. Signal Transduct Targeted Ther. (2021) 6:1–2. doi: 10.1038/s41392-021-00470-1
94. Mycroft-West CJ, Su D, Pagani I, Rudd TR, Elli S, Gandhi NS, et al. Heparin inhibits cellular invasion by SARS-CoV-2: structural dependence of the interaction of the spike S1 receptor-binding domain with heparin. Thromb Haemost. (2020) 120:1700–15. doi: 10.1055/s-0040-1721319
95. Ginsburg I, Fibach E. Polycations and polyanions in SARS-CoV-2 infection. Med Hypotheses. (2021) 146:110470. doi: 10.1016/j.mehy.2020.110470
96. Vert M. The non-specific antiviral activity of polysulfates to fight SARS-CoV-2, its mutants and viruses with cationic spikes. J Biomater Sci Polym Ed. (2021) 32:1466–71. doi: 10.1080/09205063.2021.1925391
97. Thachil J. The versatile heparin in COVID-19. J Thromb Haemost. (2020) 18:1020–2. doi: 10.1111/jth.14821
98. Wu D, Koganti R, Lambe UP, Yadavalli T, Nandi SS, Shukla D. Vaccines and therapies in development for SARS-CoV-2 infections. J Clin Med. (2020) 9:1885. doi: 10.3390/jcm9061885
99. Bianculli RH, Mase JD, Schulz MD. Antiviral polymers: past approaches and future possibilities. Macromolecules. (2020) 53:9158–86. doi: 10.1021/acs.macromol.0c01273
100. Wei C, Wan L, Yan Q, Wang X, Zhang J, Yang X, et al. HDL-scavenger receptor B type 1 facilitates SARS-CoV-2 entry. Nat Metab. (2020) 2:1391–400. doi: 10.1038/s42255-020-00324-0
101. Wang K, Chen W, Zhang Z, Deng Y, Lian J-Q, Du P, et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct Target Ther. (2020) 5:1–10. doi: 10.1038/s41392-020-00426-x
102. Wang S, Qiu Z, Hou Y, Deng X, Xu W, Zheng T, et al. AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res. (2021) 31:126–40. doi: 10.1038/s41422-020-00460-y
103. Amraei R, Yin W, Napoleon MA, Suder EL, Berrigan J, Zhao Q, et al. CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2. ACS Central Sci. (2021) 7:1156–65. doi: 10.1021/acscentsci.0c01537
104. Yang C, Zhang Y, Zeng X, Chen H, Chen Y, Yang D, et al. Kidney injury molecule-1 is a potential receptor for SARS-CoV-2. J Mol Cell Biol. (2021) 13:185–96. doi: 10.1093/jmcb/mjab003
105. Cantuti-Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S, et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. (2020) 370:856–60. doi: 10.1126/science.abd2985
106. Daly JL, Simonetti B, Klein K, Chen K-E, Williamson MK, Antón-Plágaro C, et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science. (2020) 370:861–5. doi: 10.1126/science.abd3072
107. Hamming I, Cooper ME, Haagmans BL, Hooper NM, Korstanje R, Osterhaus A, et al. The emerging role of ACE2 in physiology and disease. J Pathol. (2007) 212:1–11. doi: 10.1002/path.2162
108. Kowalczuk S, Bröer A, Tietze N, Vanslambrouck JM, Rasko JEJ, Bröer S. A protein complex in the brush-border membrane explains a Hartnup disorder allele. FASEB J. (2008) 22:2880–7. doi: 10.1096/fj.08-107300
109. Pang J, Liu M, Ling W, Jin T. Friend or foe? ACE2 inhibitors and GLP-1R agonists in COVID-19 treatment. Obes Med. (2021) 22:100312. doi: 10.1016/j.obmed.2020.100312
110. Duan L, Zheng Q, Zhang H, Niu Y, Lou Y, Wang H. The SARS-CoV-2 spike glycoprotein biosynthesis, structure, function, and antigenicity: implications for the design of spike-based vaccine immunogens. Front Immunol. (2020) 11:2593. doi: 10.3389/fimmu.2020.576622
111. Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. (2020) 181:281–292.e6. doi: 10.1016/j.cell.2020.02.058
112. Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. (2005) 309:1864–8. doi: 10.1126/science.1116480
113. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. (2020) 367:1260–3. doi: 10.1126/science.abb2507
114. Pellet-Many C, Frankel P, Jia H, Zachary I. Neuropilins: structure, function and role in disease. Biochem J. (2008) 411:211–26. doi: 10.1042/BJ20071639
115. Zachary I. Neuropilins: role in signalling, angiogenesis and disease. Chem Immunol Allergy. (2014) 99:37–70. doi: 10.1159/000354169
116. Coutard B, Valle C, Lamballerie X de, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. (2020) 176:104742. doi: 10.1016/j.antiviral.2020.104742
117. Li Z-L, Buck M. Neuropilin-1 assists SARS-CoV-2 infection by stimulating the separation of Spike protein S1 and S2. Biophys J. (2021) 120:2828–37. doi: 10.1016/j.bpj.2021.05.026
118. Davies J, Randeva HS, Chatha K, Hall M, Spandidos DA, Karteris E, et al. Neuropilin-1 as a new potential SARS-CoV-2 infection mediator implicated in the neurologic features and central nervous system involvement of COVID-19. Mol Med Rep. (2020) 22:4221–6. doi: 10.3892/mmr.2020.11510
119. Hikmet F, Méar L, Edvinsson Å, Micke P, Uhlén M, Lindskog C. The protein expression profile of ACE2 in human tissues. Mol Syst Biol. (2020) 16:e9610. doi: 10.15252/msb.20209610
120. Ko J-H, Kwon H-S, Kim B, Min G, Shin C, Yang S-W, et al. Preclinical efficacy and safety of an anti-human VEGFA and anti-human NRP1 dual-targeting bispecific antibody (IDB0076). Biomolecules. (2020) 10:919. doi: 10.3390/biom10060919
121. Jarvis A, Allerston CK, Jia H, Herzog B, Garza-Garcia A, Winfield N, et al. Small molecule inhibitors of the neuropilin-1 Vascular Endothelial Growth Factor A (VEGF-A) interaction. J Med Chem. (2010) 53:2215–26. doi: 10.1021/jm901755g
122. Parker MW, Xu P, Li X, Kooi CWV. Structural basis for selective Vascular Endothelial Growth Factor-A (VEGF-A) binding to neuropilin-1 *. J Biol Chem. (2012) 287:11082–9. doi: 10.1074/jbc.M111.331140
123. Jackson CB, Farzan M, Chen B, Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol. (2022) 23:3–20. doi: 10.1038/s41580-021-00418-x
124. Zhu Y, Feng F, Hu G, Wang Y, Yu Y, Zhu Y, et al. A genome-wide CRISPR screen identifies host factors that regulate SARS-CoV-2 entry. Nat Commun. (2021) 12:1–11. doi: 10.1038/s41467-021-21213-4
125. Wang Q, Qiu Y, Li J-Y, Zhou Z-J, Liao C-H, Ge X-Y. A unique protease cleavage site predicted in the spike protein of the novel pneumonia coronavirus (2019-nCoV) potentially related to viral transmissibility. Virol Sin. (2020) 35:337–9. doi: 10.1007/s12250-020-00212-7
126. Vankadari N. Structure of furin protease binding to SARS-CoV-2 spike glycoprotein and implications for potential targets and virulence. J Phys Chem Lett. (2020) 11:6655–63. doi: 10.1021/acs.jpclett.0c01698
127. Hoffmann M, Kleine-Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell. (2020) 78:779–84.e5. doi: 10.1016/j.molcel.2020.04.022
128. Örd M, Faustova I, Loog M. The sequence at Spike S1/S2 site enables cleavage by furin and phospho-regulation in SARS-CoV2 but not in SARS-CoV1 or MERS-CoV. Sci Rep. (2020) 10:1–10. doi: 10.1038/s41598-020-74101-0
129. Davidson AD, Williamson MK, Lewis S, Shoemark D, Carroll MW, Heesom KJ, et al. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. (2020) 12:1–15. doi: 10.1186/s13073-020-00763-0
130. Lau S-Y, Wang P, Mok BW-Y, Zhang AJ, Chu H, Lee AC-Y, et al. Attenuated SARS-CoV-2 variants with deletions at the S1/S2 junction. Emerg Microbes Infect. (2020) 9:837–42. doi: 10.1080/22221751.2020.1756700
131. Ogando NS, Dalebout TJ, Zevenhoven-Dobbe JC, Limpens RWAL, Meer Y van der, Caly L, et al. SARS-coronavirus-2 replication in Vero E6 cells: replication kinetics, rapid adaptation and cytopathology. J Gen Virol. (2020) 101:925–40. doi: 10.1099/jgv.0.001453
132. Peacock TP, Goldhill DH, Zhou J, Baillon L, Frise R, Swann OC, et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat Microbiol. (2021) 6:899–909. doi: 10.1038/s41564-021-00908-w
133. Anand P, Puranik A, Aravamudan M, Venkatakrishnan AJ, Soundararajan V. SARS-CoV-2 strategically mimics proteolytic activation of human ENaC. eLife. (2020) 9:e58603. doi: 10.7554/eLife.58603
134. Wynne BM, Zou L, Linck V, Hoover RS, Ma H-P, Eaton DC. Regulation of lung epithelial sodium channels by cytokines and chemokines. Front Immunol. (2017) 8:766. doi: 10.3389/fimmu.2017.00766
135. Takeda M. Proteolytic activation of SARS-CoV-2 spike protein. Microbiol Immunol. (2021). doi: 10.1111/1348-0421.12945. [Epub ahead of print].
136. Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci. (2005) 102:11876–81. doi: 10.1073/pnas.0505577102
137. Huang IC, Bosch BJ, Li F, Li W, Kyoung HL, Ghiran S, et al. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J Biol Chem. (2006) 281:3198–203. doi: 10.1074/jbc.M508381200
138. Liu T, Luo S, Libby P, Shi GP. Cathepsin L-selective inhibitors: a potentially promising treatment for COVID-19 patients. Pharmacol Ther. (2020) 213:107587. doi: 10.1016/j.pharmthera.2020.107587
139. Tang T, Jaimes JA, Bidon MK, Straus MR, Daniel S, Whittaker GR. Proteolytic activation of SARS-CoV-2 spike at the S1/S2 boundary: potential role of proteases beyond furin. ACS Infect Dis. (2021) 7:264–72. doi: 10.1021/acsinfecdis.0c00701
140. Koch J, Uckeley ZM, Doldan P, Stanifer M, Boulant S, Lozach P-Y. TMPRSS2 expression dictates the entry route used by SARS-CoV-2 to infect host cells. EMBO J. (2021) 40:e107821. doi: 10.15252/embj.2021107821
141. Wang L, Xiang Y. Spike glycoprotein-mediated entry of SARS coronaviruses. Viruses. (2020) 12:1289. doi: 10.3390/v12111289
142. Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. (2020) 10:766–88. doi: 10.1016/j.apsb.2020.02.008
143. Xia S, Lan Q, Su S, Wang X, Xu W, Liu Z, et al. The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin. Signal Transduct Target Ther. (2020) 5:1–3. doi: 10.1038/s41392-020-0184-0
144. Cheng YW, Chao TL, Li CL, Chiu MF, Kao HC, Wang SH, et al. Furin inhibitors block SARS-CoV-2 spike protein cleavage to suppress virus production and cytopathic effects. Cell Rep. (2020) 33:108254. doi: 10.1016/j.celrep.2020.108254
145. Papa G, Mallery DL, Albecka A, Welch LG, Cattin-Ortolá J, Luptak J, et al. Furin cleavage of SARS-CoV-2 spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog. (2021) 17:e1009246. doi: 10.1371/journal.ppat.1009246
146. Kawase M, Shirato K, Hoek L van der, Taguchi F, Matsuyama S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J Virol. (2012) 86:6537–45. doi: 10.1128/JVI.00094-12
147. Bestle D, Heindl MR, Limburg H, van TVL, Pilgram O, Moulton H, et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci Alliance. (2020) 3. doi: 10.1101/2020.04.15.042085
148. Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci. (2020) 117:11727–34. doi: 10.1073/pnas.2003138117
149. Yamamoto M, Kiso M, Sakai-Tagawa Y, Iwatsuki-Horimoto K, Imai M, Takeda M, et al. The anticoagulant nafamostat potently inhibits SARS-CoV-2 S protein-mediated fusion in a cell fusion assay system and viral infection in vitro in a cell-type-dependent manner. Viruses. (2020) 12:629. doi: 10.3390/v12060629
150. Li K, Meyerholz DK, Bartlett JA, McCray PB. The TMPRSS2 inhibitor nafamostat reduces SARS-CoV-2 pulmonary infection in mouse models of COVID-19. mBio. (2021) 12:e0097021. doi: 10.1128/mBio.00970-21
151. Zhuravel SV, Khmelnitskiy OK, Burlaka OO, Gritsan AI, Goloshchekin BM, Kim S, et al. Nafamostat in hospitalized patients with moderate to severe COVID-19 pneumonia: a randomised Phase II clinical trial. EClinicalMedicine. (2021) 41:101169. doi: 10.1016/j.eclinm.2021.101169
152. Leroy H, Han M, Woottum M, Bracq L, Bouchet J, Xie M, et al. Virus-mediated cell-cell fusion. Int J Mol Sci. (2020) 21:9644. doi: 10.3390/ijms21249644
153. Buchrieser J, Dufloo J, Hubert M, Monel B, Planas D, Rajah MM, et al. Syncytia formation by SARS-CoV-2-infected cells. EMBO J. (2020) 39:e106267. doi: 10.15252/embj.2020106267
154. Sanders DW, Jumper CC, Ackerman PJ, Bracha D, Donlic A, Kim H, et al. SARS-CoV-2 requires cholesterol for viral entry and pathological syncytia formation. eLife. (2021) 10:e65962. doi: 10.7554/eLife.65962
155. Li D, Liu Y, Lu Y, Gao S, Zhang L. Palmitoylation of SARS-CoV-2 S protein is critical for S-mediated syncytia formation and virus entry. J Med Virol. (2022) 94:342–8. doi: 10.1002/jmv.27339
156. Cheng Y-W, Chao T-L, Li C-L, Wang S-H, Kao H-C, Tsai Y-M, et al. D614G Substitution of SARS-CoV-2 spike protein increases syncytium formation and virus titer via enhanced furin-mediated spike cleavage. mBio. (2021) 12:e00587-21. doi: 10.1128/mBio.00587-21
157. Bussani R, Schneider E, Zentilin L, Collesi C, Ali H, Braga L, et al. Persistence of viral RNA, pneumocyte syncytia and thrombosis are hallmarks of advanced COVID-19 pathology. EBioMedicine. (2020) 61:103104. doi: 10.1016/j.ebiom.2020.103104
158. Braga L, Ali H, Secco I, Chiavacci E, Neves G, Goldhill D, et al. Drugs that inhibit TMEM16 proteins block SARS-CoV-2 spike-induced syncytia. Nature. (2021) 594:88–93. doi: 10.1038/s41586-021-03491-6
159. Zhang Z, Zheng Y, Niu Z, Zhang B, Wang C, Yao X, et al. SARS-CoV-2 spike protein dictates syncytium-mediated lymphocyte elimination. Cell Death Differ. (2021) 28:2765–77. doi: 10.1038/s41418-021-00782-3
160. Theken KN, Tang SY, Sengupta S, FitzGerald GA. The roles of lipids in SARS-CoV-2 viral replication and the host immune response. J Lipid Res. (2021) 62:100129. doi: 10.1016/j.jlr.2021.100129
161. Teissier E, Pécheur E-I. Lipids as modulators of membrane fusion mediated by viral fusion proteins. Eur Biophys J. (2007) 36:887–99. doi: 10.1007/s00249-007-0201-z
162. Zang R, Case JB, Yutuc E, Ma X, Shen S, Castro MFG, et al. Cholesterol 25-hydroxylase suppresses SARS-CoV-2 replication by blocking membrane fusion. PNAS. (2020) 117:32105–13. doi: 10.1073/pnas.2012197117
163. Marcello A, Civra A, Milan Bonotto R, Nascimento Alves L, Rajasekharan S, Giacobone C, et al. The cholesterol metabolite 27-hydroxycholesterol inhibits SARS-CoV-2 and is markedly decreased in COVID-19 patients. Redox Biol. (2020) 36:101682. doi: 10.1016/j.redox.2020.101682
164. Lee WS, Wheatley AK, Kent SJ, DeKosky BJ. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat Microbiol. (2020) 5:1185–191. doi: 10.1038/s41564-020-00789-5
165. Taylor A, Foo S-S, Bruzzone R, Dinh LV, King NJC, Mahalingam S. Fc receptors in antibody-dependent enhancement of viral infections. Immunol Rev. (2015) 268:340–64. doi: 10.1111/imr.12367
166. Arvin AM, Fink K, Schmid MA, Cathcart A, Spreafico R, Havenar-Daughton C, et al. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature. (2020) 584:353–63. doi: 10.1038/s41586-020-2538-8
167. Bournazos S, Gupta A, Ravetch JV. The role of IgG Fc receptors in antibody-dependent enhancement. Nat Rev Immunol. (2020) 20:633–43. doi: 10.1038/s41577-020-00410-0
168. Polack FP, Teng MN L., Collins P, Prince GA, Exner M, Regele H, et al. A role for immune complexes in enhanced respiratory syncytial virus disease. J Exp Med. (2002) 196:859–65. doi: 10.1084/jem.20020781
169. Karthik K, Senthilkumar TMA, Udhayavel S, Raj GD. Role of antibody-dependent enhancement (ADE) in the virulence of SARS-CoV-2 and its mitigation strategies for the development of vaccines and immunotherapies to counter COVID-19. Hum Vaccin Immunother. (2020) 16:3055–60. doi: 10.1080/21645515.2020.1796425
170. Liu L, Wei Q, Lin Q, Fang J, Wang H, Kwok H, et al. Anti–spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight. (2019) 4:e123158. doi: 10.1172/jci.insight.123158
171. Yip MS, Leung NHL, Cheung CY, Li PH, Lee HHY, Daëron M, et al. Antibody-dependent infection of human macrophages by severe acute respiratory syndrome coronavirus. Virol J. (2014) 11:82. doi: 10.1186/1743-422X-11-82
172. Alsaadi EAJ, Jones IM. Membrane binding proteins of coronaviruses. Fut Virol. (2019) 14:275–86. doi: 10.2217/fvl-2018-0144
173. Bosch BJ, Zee R van der, Haan CAM de, Rottier PJM. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol. (2003) 77:8801–811. doi: 10.1128/JVI.77.16.8801-8811.2003
174. Huotari J, Helenius A. Endosome maturation. EMBO J. (2011) 30:3481–500. doi: 10.1038/emboj.2011.286
175. Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. (2020) 11:1–12. doi: 10.1038/s41467-020-15562-9
176. Zhao Z, Qin P, Huang YW. Lysosomal ion channels involved in cellular entry and uncoating of enveloped viruses: implications for therapeutic strategies against SARS-CoV-2. Cell Calcium. (2021) 94:102360. doi: 10.1016/j.ceca.2021.102360
177. Wang X, Zhang X, Dong XP, Samie M, Li X, Cheng X, et al. TPC proteins are phosphoinositide- activated sodium-selective ion channels in endosomes and lysosomes. Cell. (2012) 151:372–83. doi: 10.1016/j.cell.2012.08.036
178. Filippini A, D'Amore A, Palombi F, Carpaneto A. Could the inhibition of endo-lysosomal Two-Pore Channels (TPCs) by the natural flavonoid naringenin represent an option to fight SARS-CoV-2 infection? Front Microbiol. (2020) 11:970. doi: 10.3389/fmicb.2020.00970
179. Moccia F, Negri S, Faris P, Perna A, Luca AD, Soda T, et al. Targeting endolysosomal two-pore channels to treat cardiovascular disorders in the novel COronaVIrus Disease 2019. Front Physiol. (2021) 12:690189. doi: 10.3389/fphys.2021.690189
180. Gunaratne G. Characterization Modulation of NAADP-Dependent Calcium Signaling Events Supporting Middle East Respiratory Syndrome Coronavirus Infectivity. (2018). Available online at: http://conservancy.umn.edu/handle/11299/201699 (accessed December 12, 2021).
181. Pafumi I, Festa M, Papacci F, Lagostena L, Giunta C, Gutla V, et al. Naringenin impairs two-pore channel 2 activity and inhibits VEGF-induced angiogenesis. Sci Rep. (2017) 7:1–11. doi: 10.1038/s41598-017-04974-1
182. Nahmias Y, Goldwasser J, Casali M, Poll D van, Wakita T, Chung RT, et al. Apolipoprotein B–dependent hepatitis C virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatology. (2008) 47:1437–45. doi: 10.1002/hep.22197
183. Dong W, Wei X, Zhang F, Hao J, Huang F, Zhang C, et al. A dual character of flavonoids in influenza A virus replication and spread through modulating cell-autonomous immunity by MAPK signaling pathways. Sci Rep. (2014) 4:1–12. doi: 10.1038/srep07237
184. Frabasile S, Koishi AC, Kuczera D, Silveira GF, Verri WA, Santos CND dos, et al. The citrus flavanone naringenin impairs dengue virus replication in human cells. Sci Rep. (2017) 7:1–11. doi: 10.1038/srep41864
185. Cataneo AHD, Kuczera D, Koishi AC, Zanluca C, Silveira GF, Arruda TB de, et al. The citrus flavonoid naringenin impairs the in vitro infection of human cells by Zika virus. Sci Rep. (2019) 9:1–15. doi: 10.1038/s41598-019-52626-3
186. Jin L, Zeng W, Zhang F, Zhang C, Liang W. Naringenin ameliorates acute inflammation by regulating intracellular cytokine degradation. J Immunol. (2017) 199:3466–77. doi: 10.4049/jimmunol.1602016
187. Zeng W, Jin L, Zhang F, Zhang C, Liang W. Naringenin as a potential immunomodulator in therapeutics. Pharmacol Res. (2018) 135:122–6. doi: 10.1016/j.phrs.2018.08.002
188. Sakurai Y, Kolokoltsov AA, Chen C-C, Tidwell MW, Bauta WE, Klugbauer N, et al. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science. (2015) 347:995–8. doi: 10.1126/science.1258758
189. Simmons JA, D'Souza RS, Ruas M, Galione A, Casanova JE, White JM. Ebolavirus glycoprotein directs fusion through NPC1 + endolysosomes. J Virol. (2016) 90:605–10. doi: 10.1128/JVI.01828-15
190. She J, Guo J, Chen Q, Zeng W, Jiang Y, Bai X. Structural insights into the voltage and phospholipid activation of the mammalian TPC1 channel. Nature. (2018) 556:130–4. doi: 10.1038/nature26139
191. Kang Y-L, Chou Y, Rothlauf PW, Liu Z, Soh TK, Cureton D, et al. Inhibition of PIKfyve kinase prevents infection by Zaire ebolavirus and SARS-CoV-2. Proc Natl Acad Sci. (2020) 117:20803–13. doi: 10.1073/pnas.2007837117
192. Keyaerts E, Vijgen L, Maes P, Neyts J, Ranst MV. In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine. Biochem Biophys Res Commun. (2004) 323:264–8. doi: 10.1016/j.bbrc.2004.08.085
193. Vincent MJ, Bergeron E, Benjannet S, Erickson BR, Rollin PE, Ksiazek TG, et al. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J. (2005) 2:1–10. doi: 10.1186/1743-422X-2-69
194. Hu TY, Frieman M, Wolfram J. Insights from nanomedicine into chloroquine efficacy against COVID-19. Nat Nanotechnol. (2020) 15:247–9. doi: 10.1038/s41565-020-0674-9
195. Liu J, Cao R, Xu M, Wang X, Zhang H, Hu H, et al. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. (2020) 6:1–4. doi: 10.1038/s41421-020-0156-0
196. Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. (2020) 30:269–71. doi: 10.1038/s41422-020-0282-0
197. Yao X, Ye F, Zhang M, Cui C, Huang B, Niu P, et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Clin Infect Dis. (2020) 71:732–9. doi: 10.1093/cid/ciaa237
198. Boulware DR, Pullen MF, Bangdiwala AS, Pastick KA, Lofgren SM, Okafor EC, et al. A randomized trial of hydroxychloroquine as postexposure prophylaxis for Covid-19. N Engl J Med. (2020) 383:517–25. doi: 10.1056/NEJMoa2016638
199. Gautret P, Lagier JC, Parola P, Hoang VT, Meddeb L, Mailhe M, et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int J Antimicrob Agents. (2020) 56:105949. doi: 10.1016/j.ijantimicag.2020.105949
200. Self WH, Semler MW, Leither LM, Casey JD, Angus DC, Brower RG, et al. Effect of hydroxychloroquine on clinical status at 14 days in hospitalized patients with COVID-19: a randomized clinical trial. JAMA. (2020) 324:2165–76. doi: 10.1001/jama.2020.22240
201. Ou T, Mou H, Zhang L, Ojha A, Choe H, Farzan M. Hydroxychloroquine-mediated inhibition of SARS-CoV-2 entry is attenuated by TMPRSS2. PLoS Pathog. (2021) 17:e1009212. doi: 10.1371/journal.ppat.1009212
202. Schmidt N, Lareau CA, Keshishian H, Ganskih S, Schneider C, Hennig T, et al. The SARS-CoV-2 RNA–protein interactome in infected human cells. Nat Microbiol. (2020) 6:339–53. doi: 10.1038/s41564-020-00846-z
203. Flynn RA, Belk JA, Qi Y, Yasumoto Y, Wei J, Alfajaro MM, et al. Discovery and functional interrogation of SARS-CoV-2 RNA-host protein interactions. Cell. (2021) 184:2394–411.e16. doi: 10.1016/j.cell.2021.03.012
204. Kamel W, Noerenberg M, Cerikan B, Chen H, Järvelin AI, Kammoun M, et al. Global analysis of protein-RNA interactions in SARS-CoV-2-infected cells reveals key regulators of infection. Mol Cell. (2021) 81:2851–67.e7. doi: 10.1016/j.molcel.2021.05.023
205. Lee S, Lee Y, Choi Y, Son A, Park Y, Lee K-M, et al. The SARS-CoV-2 RNA interactome. Mol Cell. (2021) 81:2838–50.e6. doi: 10.1016/j.molcel.2021.04.022
206. Thiel V, Ivanov KA, Putics Á, Hertzig T, Schelle B, Bayer S, et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J Gen Virol. (2003) 84:2305–15. doi: 10.1099/vir.0.19424-0
207. Lokugamage KG, Narayanan K, Huang C, Makino S. Severe acute respiratory syndrome coronavirus protein nsp1 is a novel eukaryotic translation inhibitor that represses multiple steps of translation initiation. J Virol. (2012) 86:13598–608. doi: 10.1128/JVI.01958-12
208. Kamitani W, Huang C, Narayanan K, Lokugamage KG, Makino S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat Struct Mol Biol. (2009) 16:1134–40. doi: 10.1038/nsmb.1680
209. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. (2008) 4:44–57. doi: 10.1038/nprot.2008.211
210. Schubert K, Karousis ED, Jomaa A, Scaiola A, Echeverria B, Gurzeler L-A, et al. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat Struct Mol Biol. (2020) 27:959–66. doi: 10.1038/s41594-020-0511-8
211. Yuan S, Peng L, Park JJ, Hu Y, Devarkar SC, Dong MB, et al. Nonstructural protein 1 of SARS-CoV-2 is a potent pathogenicity factor redirecting host protein synthesis machinery toward viral RNA. Mol Cell. (2020) 80:1055–66.e6. doi: 10.1016/j.molcel.2020.10.034
212. Yuan S, Balaji S, Lomakin IB, Xiong Y. Coronavirus Nsp1: immune response suppression and protein expression inhibition. Front Microbiol. (2021) 12:2683. doi: 10.3389/fmicb.2021.752214
213. Martinez-Salas E, Francisco-Velilla R, Fernandez-Chamorro J, Embarek AM. Insights into structural and mechanistic features of viral IRES elements. Front Microbiol. (2018) 8:2629. doi: 10.3389/fmicb.2017.02629
214. Narayanan K, Huang C, Lokugamage K, Kamitani W, Ikegami T, Tseng C-TK, et al. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type i interferon, in infected cells. J Virol. (2008) 82:4471–9. doi: 10.1128/JVI.02472-07
215. Almeida MS, Johnson MA, Herrmann T, Geralt M, Wüthrich K. Novel β-barrel fold in the nuclear magnetic resonance structure of the replicase nonstructural protein 1 from the severe acute respiratory syndrome coronavirus. J Virol. (2007) 81:3151–61. doi: 10.1128/JVI.01939-06
216. Wathelet MG, Orr M, Frieman MB, Baric RS. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J Virol. (2007) 81:11620–33. doi: 10.1128/JVI.00702-07
217. Zhang K, Miorin L, Makio T, Dehghan I, Gao S, Xie Y, et al. Nsp1 protein of SARS-CoV-2 disrupts the mRNA export machinery to inhibit host gene expression. Sci Adv. (2021) 7:eabe7386. doi: 10.1126/sciadv.abe7386
218. Hillen HS. Structure and function of SARS-CoV-2 polymerase. Curr Opin Virol. (2021) 48:82–90. doi: 10.1016/j.coviro.2021.03.010
219. Sola I, Almazán F, Zúñiga S, Enjuanes L. Continuous and discontinuous RNA synthesis in coronaviruses. Annu Rev Virol. (2015) 2:265–88. doi: 10.1146/annurev-virology-100114-055218
220. V'kovski P, Gerber M, Kelly J, Pfaender S, Ebert N, Lagache SB, Simillion C, et al. Determination of host proteins composing the microenvironment of coronavirus replicase complexes by proximity-labeling. eLife. (2019) 8:e42037. doi: 10.7554/eLife.42037
221. Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. (2020) 583:459–68. doi: 10.1038/s41586-020-2286-9
222. Effector Therapeutics. A Phase 1b, Randomized, Double-Blind, Placebo-Controlled, Dose Escalation Trial of Intravenous Zotatifin in Adults With Mild or Moderate Coronavirus Disease 2019 (COVID-19). https://Clinicaltrials.gov (2021). Available online at: https://clinicaltrials.gov/ct2/show/NCT04632381 (accessed December 12, 2021).
223. Stertz S, Reichelt M, Spiegel M, Kuri T, Martínez-Sobrido L, García-Sastre A, et al. The intracellular sites of early replication and budding of SARS-coronavirus. Virology. (2007) 361:304–15. doi: 10.1016/j.virol.2006.11.027
224. Knoops K, Kikkert M, Worm SHE van den, Zevenhoven-Dobbe JC, Meer Y van der, Koster AJ, et al. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. (2008) 6:e226. doi: 10.1371/journal.pbio.0060226
225. Castro IF de, Fernández JJ, Barajas D, Nagy PD, Risco C, Ewald A. Three-dimensional imaging of the intracellular assembly of a functional viral RNA replicase complex. J Cell Sci. (2017) 130:260–68. doi: 10.1242/jcs.181586
226. Snijder EJ, Limpens RWAL, Wilde AH de, Jong AWM de, Zevenhoven-Dobbe JC, Maier HJ, et al. A unifying structural and functional model of the coronavirus replication organelle: tracking down RNA synthesis. PLoS Biol. (2020) 18:e3000715. doi: 10.1371/journal.pbio.3000715
227. Wolff G, Melia CE, Snijder EJ, Bárcena M. Double-membrane vesicles as platforms for viral replication. Trends Microbiol. (2020) 28:1022–33. doi: 10.1016/j.tim.2020.05.009
228. Oudshoorn D, Rijs K, Limpens RWAL, Groen K, Koster AJ, Snijder EJ, et al. Expression and cleavage of middle east respiratory syndrome coronavirus nsp3-4 polyprotein induce the formation of double-membrane vesicles that mimic those associated with coronaviral RNA replication. mBio. (2017) 8. doi: 10.1128/mBio.01658-17
229. Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem. (2004) 279:10136–41. doi: 10.1074/jbc.M306124200
230. Reggiori F, Monastyrska I, Verheije MH, Calì T, Ulasli M, Bianchi S, et al. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe. (2010) 7:500–8. doi: 10.1016/j.chom.2010.05.013
231. Cottam EM, Maier HJ, Manifava M, Vaux LC, Chandra-Schoenfelder P, Gerner W, et al. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy. (2011) 7:1335–47. doi: 10.4161/auto.7.11.16642
232. Yuan S, Chu H, Chan JF-W, Ye Z-W, Wen L, Yan B, et al. SREBP-dependent lipidomic reprogramming as a broad-spectrum antiviral target. Nat Commun. (2019) 10:1–15. doi: 10.1038/s41467-018-08015-x
233. Moretti F, Bergman P, Dodgson S, Marcellin D, Claerr I, Goodwin JM, et al. TMEM41B is a novel regulator of autophagy and lipid mobilization. EMBO Rep. (2018) 19:e45889. doi: 10.15252/embr.201845889
234. Trimarco JD, Heaton BE, Chaparian RR, Burke KN, Binder RA, Gray GC, et al. TMEM41B is a host factor required for the replication of diverse coronaviruses including SARS-CoV-2. PLoS Pathog. (2021) 17:e1009599. doi: 10.1371/journal.ppat.1009599
235. Klein S, Cortese M, Winter SL, Wachsmuth-Melm M, Neufeldt CJ, Cerikan B, et al. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat Commun. (2020) 11:1–10. doi: 10.1038/s41467-020-19619-7
236. Haan CAM de, Rottier PJM. Molecular interactions in the assembly of coronaviruses. Adv Virus Res. (2005) 64:165–230. doi: 10.1016/S0065-3527(05)64006-7
237. Yao H, Song Y, Chen Y, Wu N, Xu J, Sun C, et al. Molecular architecture of the SARS-CoV-2 virus. Cell. (2020) 183:730–8.e13. doi: 10.1016/j.cell.2020.09.018
238. Carlson CR, Asfaha JB, Ghent CM, Howard CJ, Hartooni N, Safari M, et al. Phosphoregulation of phase separation by the SARS-CoV-2 N protein suggests a biophysical basis for its dual functions. Mol Cell. (2020) 80:1092–103.e4. doi: 10.1016/j.molcel.2020.11.025
239. Iserman C, Roden CA, Boerneke MA, Sealfon RSG, McLaughlin GA, Jungreis I, et al. Genomic RNA elements drive phase separation of the SARS-CoV-2 nucleocapsid. Mol Cell. (2020) 80:1078–91.e6. doi: 10.1016/j.molcel.2020.11.041
240. Perdikari TM, Murthy AC, Ryan VH, Watters S, Naik MT, Fawzi NL. SARS-CoV-2 nucleocapsid protein phase-separates with RNA and with human hnRNPs. EMBO J. (2020) 39:e106478. doi: 10.15252/embj.2020106478
241. Savastano A, Opakua AI de, Rankovic M, Zweckstetter M. Nucleocapsid protein of SARS-CoV-2 phase separates into RNA-rich polymerase-containing condensates. Nat Commun. (2020) 11:1–10. doi: 10.1038/s41467-020-19843-1
242. Cubuk J, Alston JJ, Incicco JJ, Singh S, Stuchell-Brereton MD, Ward MD, et al. The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. Nat Commun. (2021) 12:1–17. doi: 10.1038/s41467-021-21953-3
243. Lu S, Ye Q, Singh D, Cao Y, Diedrich JK, Yates JR, et al. The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein. Nat Commun. (2021) 12:1–15. doi: 10.1038/s41467-020-20768-y
244. Siu YL, Teoh KT, Lo J, Chan CM, Kien F, Escriou N, et al. The M, E, and N structural proteins of the severe acute respiratory syndrome coronavirus are required for efficient assembly, trafficking, and release of virus-like particles. J Virol. (2008) 82:11318–330. doi: 10.1128/JVI.01052-08
245. Boson B, Legros V, Zhou B, Siret E, Mathieu C, Cosset FL, et al. The SARS-CoV-2 envelope and membrane proteins modulate maturation and retention of the spike protein, allowing assembly of virus-like particles. J Biol Chem. (2021) 296:100111. doi: 10.1074/jbc.RA120.016175
246. Gramberg T, Hofmann H, Möller P, Lalor PF, Marzi A, Geier M, et al. LSECtin interacts with filovirus glycoproteins and the spike protein of SARS coronavirus. Virology. (2005) 340:224–36. doi: 10.1016/j.virol.2005.06.026
247. Rajasekharan S, Milan Bonotto R, Nascimento Alves L, Kazungu Y, Poggianella M, Martinez-Orellana P, et al. Inhibitors of protein glycosylation are active against the coronavirus severe acute respiratory syndrome coronavirus SARS-CoV-2. Viruses. (2021) 13:808. doi: 10.3390/v13050808
248. Ghosh S, Dellibovi-Ragheb TA, Kerviel A, Pak E, Qiu Q, Fisher M, et al. β-coronaviruses use lysosomes for egress instead of the biosynthetic secretory pathway. Cell. (2020) 183:1520–35.e14. doi: 10.1016/j.cell.2020.10.039
249. Mendonça L, Howe A, Gilchrist JB, Sheng Y, Sun D, Knight ML, et al. Correlative multi-scale cryo-imaging unveils SARS-CoV-2 assembly and egress. Nat Commun. (2021) 12:1–10. doi: 10.1038/s41467-021-24887-y
250. Eymieux S, Uzbekov R, Rouillé Y, Blanchard E, Hourioux C, Dubuisson J, et al. Secretory vesicles are the principal means of SARS-CoV-2 egress. Cells. (2021) 10:2047. doi: 10.3390/cells10082047
251. Wang X, Melino G, Shi Y. Actively or passively deacidified lysosomes push β-coronavirus egress. Cell Death Dis. (2021) 12:1–3. doi: 10.1038/s41419-021-03501-5
252. Lu W, Zheng B-J, Xu K, Schwarz W, Du L, Wong CKL, et al. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc Natl Acad Sci. (2006) 103:12540–5. doi: 10.1073/pnas.0605402103
253. Yue Y, Nabar NR, Shi C-S, Kamenyeva O, Xiao X, Hwang I-Y, et al. SARS-coronavirus open reading frame-3a drives multimodal necrotic cell death. Cell Death Dis. (2018) 9:1–15. doi: 10.1038/s41419-018-0917-y
254. Ibrahim IM, Abdelmalek DH, Elshahat ME, Elfiky AA. COVID-19 spike-host cell receptor GRP78 binding site prediction. J Infect. (2020) 80:554–62. doi: 10.1016/j.jinf.2020.02.026
255. Freundt EC, Yu L, Goldsmith CS, Welsh S, Cheng A, Yount B, et al. The open reading frame 3a protein of severe acute respiratory syndrome-associated coronavirus promotes membrane rearrangement and cell death. J Virol. (2010) 84:1097–109. doi: 10.1128/JVI.01662-09
256. Minakshi R, Padhan K. The YXXΦ motif within the severe acute respiratory syndrome coronavirus (SARS-CoV) 3a protein is crucial for its intracellular transport. Virol J. (2014) 11:1–10. doi: 10.1186/1743-422X-11-75
257. Chan CM, Tsoi H, Chan WM, Zhai S, Wong CO, Yao X, et al. The ion channel activity of the SARS-coronavirus 3a protein is linked to its pro-apoptotic function. Int J Biochem Cell Biol. (2009) 41:2232–9. doi: 10.1016/j.biocel.2009.04.019
258. Azad GK, Khan PK. Variations in Orf3a protein of SARS-CoV-2 alter its structure and function. Biochem Biophys Rep. (2021) 26:100933. doi: 10.1016/j.bbrep.2021.100933
259. Bianchi M, Borsetti A, Ciccozzi M, Pascarella S. SARS-Cov-2 ORF3a: mutability and function. Int J Biol Macromolecules. (2021) 170:820–6. doi: 10.1016/j.ijbiomac.2020.12.142
260. Choi Y, Bowman JW, Jung JU. Autophagy during viral infection — a double-edged sword. Nat Rev Microbiol. (2018) 16:341–54. doi: 10.1038/s41579-018-0003-6
261. Zhang Y, Sun H, Pei R, Mao B, Zhao Z, Li H, et al. The SARS-CoV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell Discov. (2021) 7:1–12. doi: 10.1038/s41421-021-00268-z
262. Yim WW-Y, Mizushima N. Lysosome biology in autophagy. Cell Discov. (2020) 6:1–12. doi: 10.1038/s41421-020-0141-7
263. Miao G, Zhao H, Li Y, Ji M, Chen Y, Shi Y, et al. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev Cell. (2021) 56:427–42.e5. doi: 10.1016/j.devcel.2020.12.010
264. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. (2007) 8:519–29. doi: 10.1038/nrm2199
265. Fung TS, Huang M, Liu DX. Coronavirus-induced ER stress response and its involvement in regulation of coronavirus–host interactions. Virus Res. (2014) 194:110–23. doi: 10.1016/j.virusres.2014.09.016
266. Chan C-P, Siu K-L, Chin K-T, Yuen K-Y, Zheng B, Jin D-Y. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J Virol. (2006) 80:9279–87. doi: 10.1128/JVI.00659-06
267. Ye Z, Wong CK, Li P, Xie Y. A SARS-CoV protein, ORF-6, induces caspase-3 mediated, ER stress and JNK-dependent apoptosis. Biochim Biophys Acta Gen Subj. (2008) 1780:1383–7. doi: 10.1016/j.bbagen.2008.07.009
268. Sung SC, Chao CY, Jeng KS, Yang JY, Lai MMC. The 8ab protein of SARS-CoV is a luminal ER membrane-associated protein and induces the activation of ATF6. Virology. (2009) 387:402–13. doi: 10.1016/j.virol.2009.02.021
269. Siu K-L, Chan C-P, Kok K-H, Woo PC-Y, Jin D-Y. Comparative analysis of the activation of unfolded protein response by spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus HKU1. Cell Biosci. (2014) 4:1–9. doi: 10.1186/2045-3701-4-3
270. Chan JF-W, Kok K-H, Zhu Z, Chu H, To KK-W, Yuan S, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. (2020) 9:221–36. doi: 10.1080/22221751.2020.1719902
271. Echavarría-Consuegra L, Cook GM, Busnadiego I, Lefèvre C, Keep S, Brown K, et al. Manipulation of the unfolded protein response: a pharmacological strategy against coronavirus infection. PLoS Pathog. (2021) 17:e1009644. doi: 10.1371/journal.ppat.1009644
272. Young BE, Fong SW, Chan YH, Mak TM, Ang LW, Anderson DE, et al. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: an observational cohort study. Lancet. (2020) 396:603–11. doi: 10.1016/S0140-6736(20)31757-8
273. Valcarcel A, Bensussen A, Álvarez-Buylla ER, Díaz J. Structural analysis of SARS-CoV-2 ORF8 protein: pathogenic and therapeutic implications. Front Genet. (2021) 12:693227. doi: 10.3389/fgene.2021.693227
274. Puzyrenko A, Jacobs ER, Sun Y, Felix JC, Sheinin Y, Ge L, et al. Pneumocytes are distinguished by highly elevated expression of the ER stress biomarker GRP78, a co-receptor for SARS-CoV-2, in COVID-19 autopsies. Cell Stress Chaperones. (2021) 26:859–68. doi: 10.1007/s12192-021-01230-4
275. Chu H, Chan CM, Zhang X, Wang Y, Yuan S, Zhou J, et al. Middle East respiratory syndrome coronavirus and bat coronavirus HKU9 both can utilize GRP78 for attachment onto host cells. J Biol Chem. (2018) 293:11709–26. doi: 10.1074/jbc.RA118.001897
276. Naidoo N, Brown M. The endoplasmic reticulum stress response in aging and age-related diseases. Front Physiol. (2012) 3:263. doi: 10.3389/fphys.2012.00263
277. Aoe T. Pathological aspects of COVID-19 as a conformational disease and the use of pharmacological chaperones as a potential therapeutic strategy. Front Pharmacol. (2020) 11:1095. doi: 10.3389/fphar.2020.01095
278. Shaban MS, Müller C, Mayr-Buro C, Weiser H, Meier-Soelch J, Albert BV, et al. Multi-level inhibition of coronavirus replication by chemical ER stress. Nat Commun. (2021) 12:5536. doi: 10.1038/s41467-021-25551-1
279. Carletti T, Zakaria MK, Faoro V, Reale L, Kazungu Y, Licastro D, et al. Viral priming of cell intrinsic innate antiviral signaling by the unfolded protein response. Nat Commun. (2019) 10:3889. doi: 10.1038/s41467-019-11663-2
280. Al-Beltagi S, Preda CA, Goulding LV, James J, Pu J, Skinner P, et al. Thapsigargin is a broad-spectrum inhibitor of major human respiratory viruses: coronavirus, respiratory syncytial virus and influenza A virus. Viruses. (2021) 13:234. doi: 10.3390/v13020234
281. Teoh K-T, Siu Y-L, Chan W-L, Schlüter MA, Liu C-J, Peiris JSM, et al. The SARS coronavirus E protein interacts with PALS1 and alters tight junction formation and epithelial morphogenesis. Mol Biol Cell. (2010) 21:3838–52. doi: 10.1091/mbc.E10-04-0338
282. Maio FD, Cascio EL, Babini G, Sali M, Longa SD, Tilocca B, et al. Improved binding of SARS-CoV-2 Envelope protein to tight junction-associated PALS1 could play a key role in COVID-19 pathogenesis. Microbes Infect. (2020) 22:592–7. doi: 10.1016/j.micinf.2020.08.006
283. Shepley-McTaggart A, Sagum CA, Oliva I, Rybakovsky E, DiGuilio K, Liang J, et al. SARS-CoV-2 Envelope (E) protein interacts with PDZ-domain-2 of host tight junction protein ZO1. PLoS ONE. (2021) 16:e0251955. doi: 10.1371/journal.pone.0251955
284. Wang Q, Chen X-W, Margolis B. PALS1 regulates E-cadherin trafficking in mammalian epithelial cells. Mol Biol Cell. (2007) 18:874–85. doi: 10.1091/mbc.e06-07-0651
285. Chai J, Cai Y, Pang C, Wang L, McSweeney S, Shanklin J, et al. Structural basis for SARS-CoV-2 envelope protein recognition of human cell junction protein PALS1. Nat Commun. (2021) 12:1–6. doi: 10.1038/s41467-021-23533-x
286. Mak TW, Saunders ME, Jett BD editors. Chapter 3 - innate immunity. In: Primer to the Immune Response. 2nd ed. Boston, MA: Academic Cell (2013). p. 55–83.
287. Stanifer ML, Pervolaraki K, Boulant S. Differential regulation of type I and type III interferon signaling. Int J Mol Sci. (2019) 20:1445. doi: 10.3390/ijms20061445
288. Jiang H, Zhang H, Meng Q, Xie J, Li Y, Chen H, et al. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell Mol Immunol. (2020) 17:998–1000. doi: 10.1038/s41423-020-0514-8
289. Liu XY, Wei B, Shi HX, Shan YF, Wang C. Tom70 mediates activation of interferon regulatory factor 3 on mitochondria. Cell Res. (2010) 20:994–1011. doi: 10.1038/cr.2010.103
290. Nagy Á, Pongor S, Gyorffy B. Different mutations in SARS-CoV-2 associate with severe and mild outcome. Int J Antimicrob Agents. (2021) 57:106272. doi: 10.1016/j.ijantimicag.2020.106272
291. Li A, Zhao K, Zhang B, Hua R, Fang Y, Jiang W, et al. SARS-CoV-2 NSP12 protein is not an interferon-β antagonist. J Virol. (2021) 95:e0074721. doi: 10.1128/JVI.00747-21
292. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
293. Chen K, Xiao F, Hu D, Ge W, Tian M, Wang W, et al. SARS-CoV-2 nucleocapsid protein interacts with RIG-I and represses RIG-mediated IFN-β production. Viruses. (2020) 13:47. doi: 10.3390/v13010047
294. Moroianu J, Hijikata M, Blobel G, Radu A. Mammalian karyopherin alpha 1 beta and alpha 2 beta heterodimers: alpha 1 or alpha 2 subunit binds nuclear localization signal and beta subunit interacts with peptide repeat-containing nucleoporins. Proc Natl Acad Sci. (1995) 92:6532–6. doi: 10.1073/pnas.92.14.6532
295. Hayn M, Hirschenberger M, Koepke L, Nchioua R, Straub JH, Klute S, et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. (2021) 35:109126. doi: 10.1016/j.celrep.2021.109126
296. Zhao C, Zhao W. NLRP3 inflammasome—a key player in antiviral responses. Front Immunol. (2020) 11:211. doi: 10.3389/fimmu.2020.00211
297. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. (2016) 535:153–58. doi: 10.1038/nature18629
298. Ma J, Zhu F, Zhao M, Shao F, Yu D, Ma J, et al. SARS-CoV-2 nucleocapsid suppresses host pyroptosis by blocking Gasdermin D cleavage. EMBO J. (2021) 40:e108249. doi: 10.15252/embj.2021108249
299. Rodrigues TS, de Sá KSG, Ishimoto AY, Becerra A, Oliveira S, Almeida L, et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J Exp Med. (2020) 218:e20201707. doi: 10.1084/jem.20201707
300. Ferreira AC, Soares VC, de Azevedo-Quintanilha IG, Dias S da SG, Fintelman-Rodrigues N, Sacramento CQ, et al. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discov. (2021) 7:1–12. doi: 10.1038/s41420-021-00477-1
301. Zheng J, Wang Y, Li K, Meyerholz DK, Allamargot C, Perlman S. Severe acute respiratory syndrome coronavirus 2–induced immune activation and death of monocyte-derived human macrophages and dendritic cells. J Infect Dis. (2021) 223:785–95. doi: 10.1093/infdis/jiaa753
302. van de Veerdonk FL, Netea MG. Blocking IL-1 to prevent respiratory failure in COVID-19. Crit Care. (2020) 24:445. doi: 10.1186/s13054-020-03166-0
303. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
304. Thoms M, Buschauer R, Ameismeier M, Koepke L, Denk T, Hirschenberger M, et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science. (2020) 369:1249–56. doi: 10.1126/science.abc8665
305. Narayanan K, Ramirez SI, Lokugamage KG, Makino S. Coronavirus nonstructural protein 1: common and distinct functions in the regulation of host and viral gene expression. Virus Res. (2015) 202:89–100. doi: 10.1016/j.virusres.2014.11.019
306. Decroly E, Imbert I, Coutard B, Bouvet M, Selisko B, Alvarez K, et al. Coronavirus nonstructural protein 16 is a cap-0 binding enzyme possessing (Nucleoside-2′ O)-methyltransferase activity. J Virol. (2008) 82:8071–84. doi: 10.1128/JVI.00407-08
307. Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. (2010) 468:452–6. doi: 10.1038/nature09489
308. Bouvet M, Debarnot C, Imbert I, Selisko B, Snijder EJ, Canard B, et al. In vitro Reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog. (2010) 6:e1000863. doi: 10.1371/journal.ppat.1000863
309. Viswanathan T, Arya S, Chan S-H, Qi S, Dai N, Misra A, et al. Structural basis of RNA cap modification by SARS-CoV-2. Nat Commun. (2020) 11:1–7. doi: 10.1038/s41467-020-17496-8
310. Lloyd RE. Regulation of stress granules and P-bodies during RNA virus infection. Wiley Interdiscipl Rev RNA. (2013) 4:317–31. doi: 10.1002/wrna.1162
Keywords: SARS-CoV-2, virus-cell interactions, innate immunity, receptors, virus replication
Citation: Pizzato M, Baraldi C, Boscato Sopetto G, Finozzi D, Gentile C, Gentile MD, Marconi R, Paladino D, Raoss A, Riedmiller I, Ur Rehman H, Santini A, Succetti V and Volpini L (2022) SARS-CoV-2 and the Host Cell: A Tale of Interactions. Front. Virol. 1:815388. doi: 10.3389/fviro.2021.815388
Received: 15 November 2021; Accepted: 17 December 2021;
Published: 12 January 2022.
Edited by:
Alessandro Marcello, International Centre for Genetic Engineering and Biotechnology, ItalyReviewed by:
Elisa Vicenzi, San Raffaele Hospital (IRCCS), ItalyCopyright © 2022 Pizzato, Baraldi, Boscato Sopetto, Finozzi, Gentile, Gentile, Marconi, Paladino, Raoss, Riedmiller, Ur Rehman, Santini, Succetti and Volpini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Massimo Pizzato, bWFzc2ltby5waXp6YXRvQHVuaXRuLml0
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.