Corrigendum: Negatively regulated by miR-29c-3p, MTFR1 promotes the progression and glycolysis in lung adenocarcinoma via the AMPK/mTOR signalling pathway
 Yongmeng Li
Yongmeng Li Yanfei Liu
Yanfei Liu Kai Jin
Kai Jin Rui Dong
Rui Dong Cun Gao
Cun Gao Libo Si
Libo Si Zitong Feng
Zitong Feng Huiying Zhang
Huiying Zhang Hui Tian
Hui Tian- 1Department of Thoracic Surgery, Qilu Hospital, Cheeloo College of Medicine, Shandong University, Jinan, China
- 2Department of Anesthesiology, Qilu Children’s Hospital of Shandong University, Jinan, China
Background: Lung adenocarcinoma (LUAD) is the major form of lung cancer that presents a major peril to public health. Owing to the high rates of morbidity, mortality and chemoresistance, it is necessary to develop more effective therapeutic targets of LUAD. Mitochondrial fission regulator 1 (MTFR1) affects the occurrence and development of some diseases by regulating mitochondrial dynamics and is dysregulated in LUAD. However, the functions and molecular mechanisms of MTFR1 in LUAD have not been investigated.
Methods: Immunohistochemical (IHC) analysis, real-time quantitative polymerase chain reaction (RT-qPCR), bioinformatic analysis and western blot (WB) were performed to assess the expression of MTFR1 at both protein and mRNA levels. The biological functions of MTFR1 in LUAD cells were assessed based on various in vivo and in vitro experiments. The dual-luciferase reporter assay and some rescue experiments were performed to evaluate the underlying mechanism of MTFR1 in LUAD.
Results: MTFR1 was upregulated in LUAD cells and tissues and correlated with dismal clinicopathologic features and a worse prognosis of patients with LUAD. Functionally, MTFR1 overexpression stimulated the proliferation, invasion, migration and glycolytic capacity and impeded the apoptosis of LUAD cells; however, opposite results were obtained when MTFR1 expression was knocked down. MTFR1, which was directly targeted by miR-29c-3p, may exert its biological functions through the AMPK/mTOR signalling pathway.
Conclusion: MTFR1 promotes the progression of LUAD. Therefore, targeting MTFR1 can offer an effective therapeutic strategy for LUAD treatment.
Introduction
Lung cancer is a major contributor to cancer-associated mortality worldwide, and non-small cell lung cancer (NSCLC) is the major subtype constituting approximately 85% of all lung cancer cases (Chen et al., 2016; Sung et al., 2021). LUAD has been recognised as the most common NSCLC subtype which has high morbidity and fatality rates (Herbst et al., 2018). Although patients with LUAD have benefited significantly from the development of targeted therapy and immunotherapy in recent years, their clinical prognosis remains unsatisfactory. Therefore, it is necessary to discover novel therapeutic targets and biomarkers for LUAD.
Studies have revealed that abnormal metabolism is a typical characteristic of cancer cells and a key contributor to cancer progression (Feng and Levine, 2010; Hanahan and Weinberg, 2011). Mitochondria are the main energy-generating organelles, which are responsible for cellular metabolism in eukaryotic cells (McBride et al., 2006). Mitochondria are in constant dynamic equilibrium between fission and fusion to adapt to functional changes (Chan, 2006; Knott et al., 2008; Chan, 2012). The disruption of this balance may contribute to the occurrence of different diseases, including different types of cancers (Lin and Beal, 2006; Guo et al., 2020). Recent studies have indicated that dysfunctional mitochondrial fission is involved in the progression of some malignancies, including glioblastoma, liver cancer and gastric cancer (Hernandez-Alvarez and Zorzano, 2021; Lenzi et al., 2021; Zhu et al., 2021). Furthermore, some studies have indicated that mitochondrial dynamics have an imperative function in the chemoresistance of cancer cells (Kong et al., 2015; Cai et al., 2016; Huang et al., 2018). Mitochondrial fission regulator 1 (MTFR1), previously called CHPPR or FAM54A2, is a mitochondrial protein with a polyproline-rich region (Tonachini et al., 2004). Some studies have indicated that MTFR1 can promote mitochondrial fission and regulate cellular respiration (Tonachini et al., 2004; Monticone et al., 2010). In addition, MTFR1 has a vital function in oral squamous cell carcinoma, acute myocardial infarction (AMI) and head and neck squamous cell carcinoma (HNSCC) (Wang et al., 2015; Reddy et al., 2019; Huang et al., 2020). Moreover, bioinformatic analyses in a study have indicated that MTFR1 is dysregulated in LUAD (Sanada et al., 2019). However, the function and mechanism of action of MTFR1 in LUAD remain investigated.
MicroRNAs (miRNAs), which are small non-coding RNAs spanning approximately 21 nucleotides, usually impede gene expression by binding to the 3′ untranslated region (3′-UTR) of their target mRNAs (Bartel, 2004). A study has indicated that miRNAs are involved in the regulation of most mammalian mRNAs (Friedman et al., 2009). Therefore, it does not come as a surprise that the dysregulation of miRNA expression may cause a multitude of diseases including cancers (Jiang et al., 2009; Li et al., 2021). miR-29c-3p suppresses the progression of various cancer types, including intrahepatic cholangiocarcinoma, cervical cancer, ovarian cancer and oesophageal cancer (Wang H. et al., 2020; Feng et al., 2020; Hozaka et al., 2021; Zou et al., 2021). However, to date, the expression level and function of miR-29c-3p in LUAD have yet to be investigated.
In this study, we aimed to assess the expression level, biological role and underlying molecular mechanism of MTFR1 in LUAD. We reported for the first time that MTFR1 is overexpressed in LUAD cells and tissues. Furthermore, MTFR1 stimulated the progression of LUAD both in vivo and in vitro. We propose that MTFR1 is a direct target of miR-29c-3p and may exert its biological effects via the AMPK/mTOR signalling pathway. Therefore, this study offers novel insights into the role of MTFR1 as an effective therapeutic target for LUAD treatment.
Methods
Data Processing
Gene expression data were downloaded from The Cancer Genome Atlas (TCGA, https://tcga-data.nci.nih.gov/tcga/), Genome–Tissue Expression (GTEx, https://www.ncbi.nlm.nih.gov/geo/) and Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) databases. The target miRNAs were predicted using the following bioinformatics software: miRbase (www.mirbase.org), starBase (http://starbase.sysu.edu.cn), miRWalk (www.umm.heidelberg.de/apps/zmf/mirwalk) and miRDB (http://mirdb.org/miRDB).
Collection of Clinical Tissue Specimens
A total of 85 LUAD tissues and matched paracancerous normal tissues of the lung were obtained from our tissue bank to establish the tissue microarray. All tissue samples were extracted through surgical resection at the Department of Thoracic Surgery, Qilu Hospital of Shandong University from 2004 to 2014. In addition, 20 paired fresh LUAD tissues and corresponding paracancerous tissues were obtained immediately after surgical resection in patients with LUAD admitted to our department. This study was reviewed and approved by the Medical Ethics Committee of Qilu Hospital of Shandong University (Approval No. KYLL-2016-097). The patients included in this study or their family members were requested to sign an informed consent document.
Cell Culture and Treatment
A total of five LUAD cell lines (H1299, PC9, A549, H1975, and H157), human embryonic kidney 293 (HEK293) cells and human bronchial epithelial (HBE) cells were provided by the Shanghai Academy of Science (Shanghai, China). The cell lines were cultured in RPMI-1640 medium supplemented with 10% foetal bovine serum (FBS; Gibco, NY, United States). The cells were incubated in a moistened environment with 5% CO2 at 37°C. The sh-NC, sh-MTFR1, oe-NC and oe-MTFR1 lentiviruses (Jikai Co., Shanghai, China) were transduced into the corresponding cells. The sh-RNA sequences are listed in Supplementary Table S1. The cells that were stably transduced were selected using puromycin (2 μg/ml) for 7 days after transduction. Mimics-miR-29c-3p, inhibitor-miR-29c-3p and their corresponding negative controls (NCs) were obtained from GenePharma Co., Ltd. (Shanghai, China). The oligonucleotide sequences were as follows: mimics-miR-29c-3p (5′-UAGCACCAUUUGAAAUCGGUUA-3′), mimics-NC (5′-UUCUCCGAACGUGUCACGUTT-3′), inhibitor-miR-29c-3p (5′-UAACCGAUUUCAAAUGGUGCUA-3′) and inhibitor-NC (5′-CAGUACUUUUGUGUAGUACAA-3′). The cells were transfected with miRNAs using the jetPRIME transfection reagent (Polyplus-transfection, Illkirch, France) according to the manufacturer’s protocol.
Western Blot
Proteins extracted using the radioimmunoprecipitation assay (RIPA) reagent were quantified using a BCA kit (Biyuntian, China). Subsequently, the protein lysates were separated using 6% or 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred onto polyvinylidene difluoride (PVDF) membranes. Furthermore, 5% non-fat milk was used to block the membranes at ambient temperature for 1 h. The membranes were subsequently washed with TBST (thrice for 10 min each) and were incubated overnight with primary antibodies at 4°C, followed by incubation with a secondary antibody at ambient temperature for 1 h. Eventually, bands were visualised using an enhanced chemiluminescence (ECL) system. β-tubulin was used as the internal control. The following antibodies were used in this study: anti-N-cadherin (ab18203, Abcam), anti-MTFR1 (ab198192, Abcam), anti-Vimentin (ab92547, Abcam), anti-p-mTOR (ab109268, Abcam), anti-E-cadherin (ab40772, Abcam), anti-mTOR (ab32028, Abcam), anti-Snail (ab216347, Abcam), anti-p-AMPK (ab133448, Abcam), anti-Slug (ab27568, Abcam), anti-Ki-67 (ab16667, Abcam), anti-AMPK (ab32047, Abcam), anti-PARP (ab32064, Abcam), anti-BAX (32503, Abcam), anti-BCL-2 (ab32124, Abcam) and anti-β-Tubulin (ab18207, Abcam).
RNA Isolation and Real-Time Quantitative Polymerase Chain Reaction
Extraction of Total RNA was extracted using the RNAfast200 kit (Fastagen, Shanghai, China). Complementary DNA (cDNA) was synthesised using a reverse transcription kit (Toyobo, Osaka, Japan) according to the manufacturer’s instructions. RT-qPCR was then executed in Bio-Rad IQ 5 system (Bio-Rad) using the SYBR Green Supermix (Toyobo, Osaka, Japan). The primers used include the following: MTFR1 (F, 5′-TGCAACAGAATGGAGTCCCA-3′ and R, 5′-AAGGGGTGGCCTTGATCTGA-3′); GAPDH (F, 5′-GCACCGTCAAGGCTGAGAAC-3′ and R, 5′-TGGTGAAGACGCCAGTGGA-3′); miR-29c-3p (F, 5′-CTCCTCCTTTTAGCACCATTTG-3′ and R, 5′-TATGCTTGTTCTCGTCTCTGTGTC-3′) and U6 (F, 5′-CAGCACATATACTAAAATTGGAACG-3′ and R, 5′-ACGAATTTGCGTGTCATCC-3′). U6 and GAPDH were used as internal references, and all the aforementioned primers were obtained from GenePharma Co., Ltd. (Shanghai, China). The experiment was repeated thrice, and data were analysed using the 2−△△Ct method.
Haematoxylin–Eosin Staining and Immunohistochemical Analysis
First, the formalin-fixed and paraffin-embedded tissues samples were split into smaller sections of 4 µm. For HE staining, each section was dewaxed, rehydrated and stained with HE, and images were captured using an inverted microscope. With regards to IHC, each section was dewaxed, rehydrated and incubated with primary antibodies overnight at 4°C, followed by incubation with a secondary antibody at ambient temperature for 1 h. Subsequently, the sections were viewed, and an inverted microscope was used to capture images. Lastly, histochemical scoring (H-score) was used to assess the results of IHC analysis as previously described (Xie et al., 2014). The following antibodies were used: anti-MTFR1 (ab198192, Abcam) and anti-Ki-67 (ab16667, Abcam).
Cell Counting Kit-8 Cell Proliferation Assay
The transduced cells were seeded into 96-well plates (2,000 cells/well) and cultured for 24, 48, 72, and 96 h, sequentially. Subsequently, 10 µL of CCK-8 solution (5 mg/ml; Solarbio, China) was added to each well. After incubating the cells for 2 h in dark, absorbance was recorded at 450 nm using a microplate reader. The experiment was performed in triplicates.
EdU Staining
The transduced cells were seeded into 96-well plates (5 × 103/well) and incubated with complete RPMI-1640 medium containing 5-ethynyl-2-′deoxyuridine (EdU; 1000:1) for 2 h. EdU staining was performed using an EdU kit (RiboBio, Guangzhou, China) according to the manufacturer’s protocol. Lastly, the stained cells were photographed using a fluorescence microscope. The experiment was performed in triplicates.
Colony Formation Assay
The transduced cells were seeded in 6-well plates (1,000 cells/well) and incubated for 15 days. Then the cells were fixed with 4% paraformaldehyde for 30 min and stained with 0.1% crystal violet for 15 min. Subsequently, the cells were washed thrice with phosphate-buffered saline (PBS). Lastly, a digital camera was used to capture images. The experiment was performed in triplicates.
Analysis of Cell Cycle and Apoptosis
The cell cycle and apoptosis were assessed using a flow cytometer (BD Biosciences, United States). A propidium iodide (PI) staining kit (Yeasen, Shanghai, China) was used to analyse the cell cycle, whereas the Annexin V-APC Apoptosis Detection Kit (eBioscience, Thermo Fisher) was used to assess cell apoptosis according to the manufacturer’s protocol. The experiments were performed in triplicates.
Wound-Healing Assay
The transduced cells were seeded in 6-well plates until they attained confluency. Subsequently, a graze in the monolayer was created using the tip of a 200-μL pipette. After three PBS washes, the medium was replaced with serum-free media (SFM). Subsequently, the width of the wound was visualised and assessed under a microscope at 0 and 24 h. The experiment was performed in triplicates.
Transwell Assay
Cell suspensions (5 × 104 cells/200 uL of SFM) were added to the upper Transwell chamber with or without Matrigel, whereas 600 uL of a medium supplemented with 20% FBS was added to the lower Transwell chamber. After 24 h, the cells in the upper chamber were removed, and those that entered or moved to the lower chamber were fixed using 75% ice-cold alcohol for 30 min. Subsequently, the cells were stained with 0.1% crystal violet for 20 min, and an inverted microscope was used to capture images. The experiment was performed in triplicates.
Measurement of Glucose, Lactate and Extracellular Acidification Rate
The transduced cells were seeded in 6-well plates (1 × 105 cells/well) and incubated for 48 h. Subsequently, the culture medium was harvested, and the levels of glucose and lactate were measured using the CheKine™ Glucose Assay Kit (Abbkine, Wuhan, Hubei, China) and CheKine™ Lactate Assay Kit (Abbkine, Wuhan, Hubei, China), respectively, according to the manufacturer’s protocol. For ECAR measurement, cells were seeded into a Seahorse XF 96 cell culture micro-plate and incubated overnight (1 × 104/well; Agilent Technologies). The next day, a Seahorse XFe96 Analyzer (Agilent Technologies) was used to measure the ECAR with a Seahorse XF Glycolysis Stress Test Kit (Agilent Technologies) following the manufacturer’s protocols. Data were analysed by the Seahorse XF 96 Wave software. The above experiments were performed in triplicates.
Dual-Luciferase Reporter Assay
HEK-293T cells were seeded into 24-well plates (5 × 104 cells/well). After incubating the cells for 24 h, they were transiently co-transfected with mimics-NC or mimics-miR-29c-3p together with the mutated (MUT) 3′-UTR or wild-type (WT) MTFR1 mRNA. After 48 h, the luciferase activity was assessed using a dual-luciferase assay kit (Promega). The experiment was replicated in triplicates.
In Vivo Experiments
We procured 4-week-old BALB/c nude mice from GemPharmatech Co., Ltd. (Nanjing, China). To establish a xenograft tumour model, 20 nude mice were categorised randomly into four cohorts. Subsequently, 5 × 106 transduced A549 cells (LV-sh-MTFR1 and LV-sh-NC) or PC9 cells (LV-oe-MTFR1 and LV-oe-NC) were hypodermically implanted into the right armpit area of each mouse. The tumour size was measured every 4 days. The tumour volumes were computed using the following formula: V = (length × width2)/2. The nude mice were sacrificed on the 28th day, and all tumours were collected and weighed. To construct an experimental model for lung metastasis, 20 nude mice were randomly divided into four cohorts, and 2 × 106 transduced cells were introduced into the mice via an injection in the tail vein. The mice were euthanised after 2 months, and their lungs were obtained. All specimens were further stained with IHC or HE. The animal experiments were approved by the Shandong University Animal Research Ethics Committee under a project license (SCXK Lu 20090001).
Statistical Analysis
Data were expressed as the mean ± standard deviation (SD). Data were analysed using the GraphPad Prism (GraphPad version 8) and R (version 4.0.3) software. The Student’s t-test (two-sided), chi-square test, Pearson’s correlation analysis and Kaplan–Meier (KM) survival analysis were performed as indicated. Multiple groups were compared for identifying statistically significant differences using one-way analysis of variance (ANOVA). The observed differences were considered statistically significant at p < 0.05.
Results
MTFR1 was Overexpressed in LUAD Cells and Tissues and Associated With a Poor Prognosis
First, we compared MTFR1 gene expression between normal lung tissues and LUAD tissues using high-throughput data from the GEO, TCGA and GTEx databases. MTFR1 was considerably overexpressed in LUAD tissues as compared with normal lung tissues (p < 0.0001, Figures 1A,B). The KM analysis revealed that elevated MTFR1 expression was associated with a poor prognosis in patients with LUAD in the TCGA cohort (p = 0.0088, Figure 1C). Furthermore, the mRNA and protein expression of MTFR1 was evaluated in 20 pairs of clinical samples. As demonstrated in Figures 1D,E, compared with those in the adjacent normal lung tissues, the protein and mRNA levels of MTFR1 were considerably elevated in LUAD tumour tissues. A total of 85 pairs of LUAD tissues and their corresponding adjoining normal lung tissues were subjected to IHC analysis with MTFR1 antibody, and the representative pictures are shown in Figure 1F. Furthermore, the expression of MTFR1 was quantified in each tissue sample. The result of IHC scoring revealed that the expression of MTFR1 was considerably elevated in LUAD tissues as compared with that in adjoining normal lung tissues. (p < 0.001, Figure 1G). The correlation between MTFR1 expression and clinical features of patients with LUAD was further examined. High MTFR1 expression was significantly related to the clinical stage (p = 0.007), lymph node metastasis (p = 0.016) and tumour size (p = 0.023) (Table 1). In addition, survival analysis revealed that patients with an elevated MTFR1 expression level exhibited worse clinical outcomes (p = 0.002, Figure 1H). Lastly, MTFR1 expression was examined in various cell lines. At both protein and mRNA levels, the expression of MTFR1 in LUAD cell lines was considerably higher than that in HBE cells (Figures 1I,IJ). Altogether, the results indicated that MTFR1 was upregulated in LUAD cells and tissues and associated with a poor prognosis in patients with LUAD.
FIGURE 1
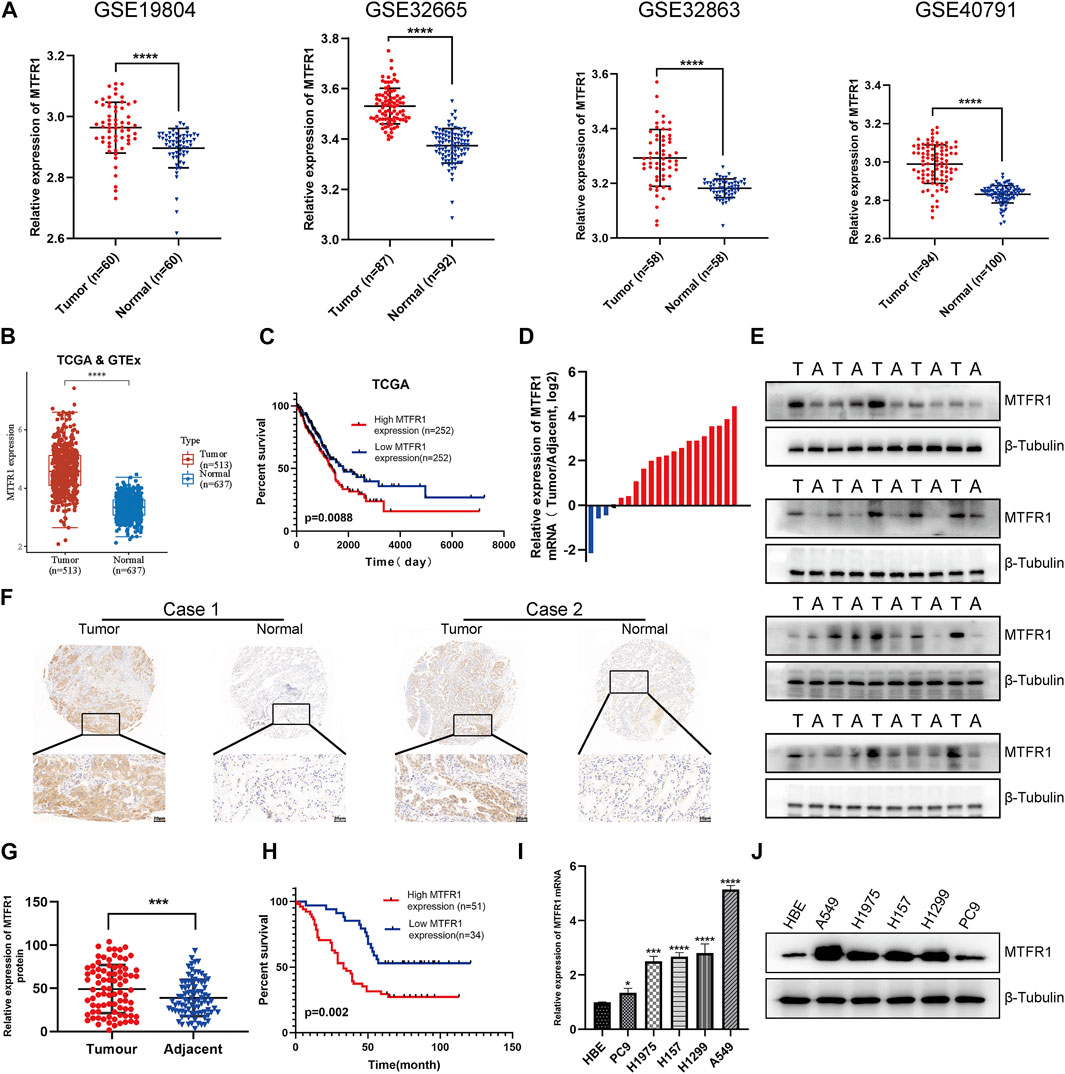
FIGURE 1. Mitochondrial fission regulator 1 (MTFR1) was overexpressed in lung adenocarcinoma (LUAD) cell lines and tissues. (A) The expression levels of MTFR1 in four Gene Expression Omnibus (GEO) datasets. (B) MTFR1 expression levels in The Cancer Genome Atlas (TCGA) and Genome–Tissue Expression (GTEx) databases. (C) Kaplan–Meier (KM) analysis for TCGA-LUAD samples. (D, E) The relative mRNA and protein expression of MTFR1 in 20 pairs of LUAD tumour (T) tissues and their matching adjoining (A) normal tissues. (F) Representative immunohistochemical images of LUAD and adjacent nonmalignant specimens stained for analysing MTFR1 expression. (G) The immunohistochemical score of 85 pairs of LUAD tissues and adjoining paracancerous tissues (p < 0.001). (H) Kaplan–Meier survival analyses of 85 patients with LUAD. (I, J) The relative protein and mRNA expression of MTFR1 in different cell lines. Data were articulated as the mean ± standard deviation (SD; *p < 0.05, ***p < 0.001, ****p < 0.0001).
TABLE 1

TABLE 1. Correlation between MTFR1 expression and clinicopathological features in LUAD.
MTFR1 Promoted the Proliferation and Inhibited the Apoptosis of LUAD Cells
sh-RNA 2 had the best knockdown efficiency among these three sh-RNAs (Supplementary Table S1; Supplementary Figure S1A), so that it was selected for the subsequent experiments. Because MTFR1 was expressed at high levels in A549 and H1299 cells but at low levels in PC9 cells (Figures 1I,J). Therefore, we downregulated the expression of MTFR1 in A549 and H1299 cells and upregulated its expression in PC9 cells using lentiviral transduction. After transduction, MTFR1 expression was considerably downregulated in A549 and H1299 cells and upregulated in PC9 cells (Figures 2A,B). The proliferative ability of the stably transduced A549, H1299, and PC9 cells were then detected using EdU, colony formation and CCK-8 assays. The results of these assays revealed that the proliferative ability of sh-MTFR1 A549 and H1299 cells was significantly reduced as compared with that of the cells in the sh-NC groups. However, the proliferative ability of oe-MTFR1 PC9 cells was higher than that of PC9 cells in the oe-NC group (Figures 2C, 3A,B). Furthermore, flow cytometry was employed to identify the effects of MTFR1 on the cell cycle progression and apoptosis of LUAD cells. The results revealed that MTFR1 knockdown could promote cell apoptosis in A549 and H1299 cells, whereas MTFR1 upregulation could reduce apoptosis in PC9 cells (Figure 3C). In addition, we observed that MTFR1 had no apparent effect on cell cycle progression (figure not shown). Lastly, cell proliferation and apoptosis-related proteins were identified in transduced A549, H1299 and PC9 cells. The expression of Ki-67 and Bcl-2 proteins was decreased, whereas that of PARP and BAX proteins was increased after the knockdown of MTFR1 in H1299 and A549 cells. However, we observed opposite results after upregulating MTFR1 expression in PC9 cells (Figure 3D). These results indicated that MTFR1 promoted the proliferation and inhibited the apoptosis of LUAD cells.
FIGURE 2
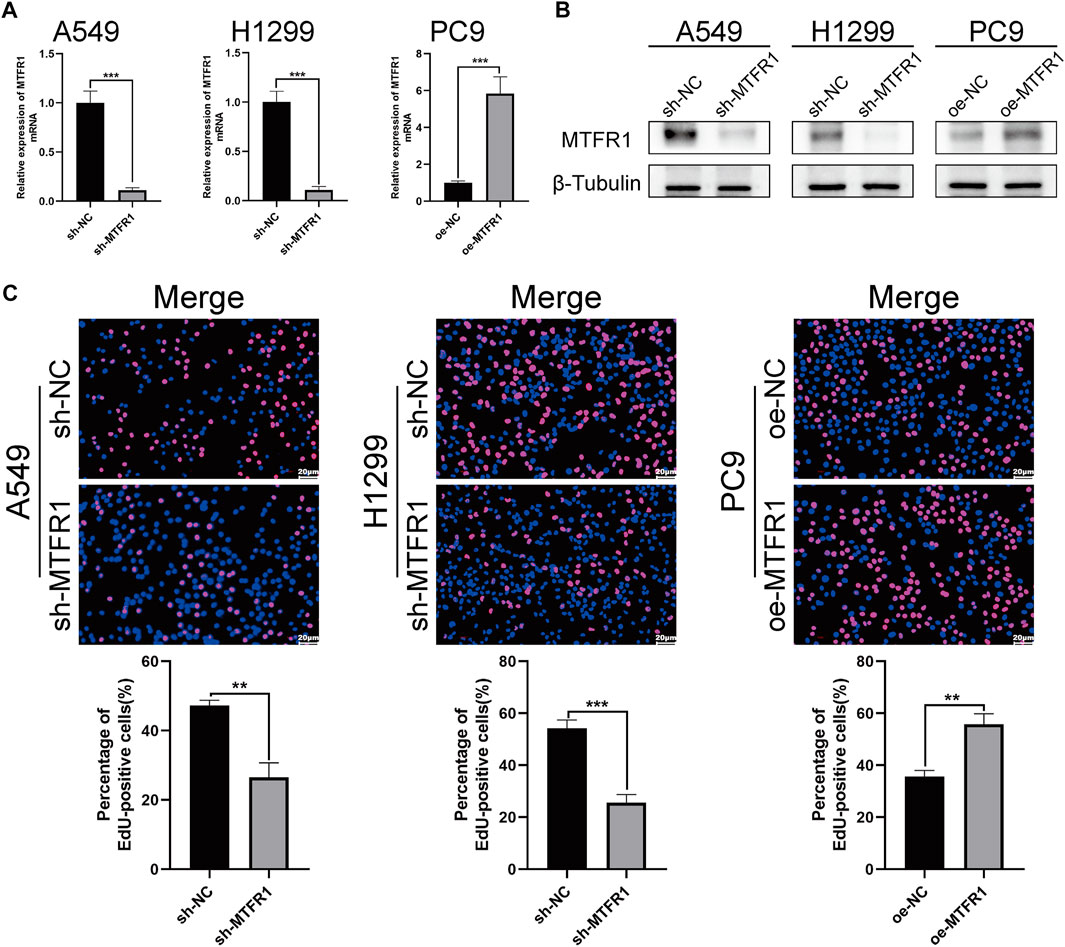
FIGURE 2. MTFR1 stimulated the proliferation of LUAD cells. (A, B) Evaluation of the effectiveness of lentiviral transduction by real-time quantitative polymerase chain reaction (RT-qPCR) and western blotting (WB). (C) EdU staining of the transduced cells. Data were articulated as the mean ± standard deviation (SD; **p < 0.01, ***p < 0.001).
FIGURE 3
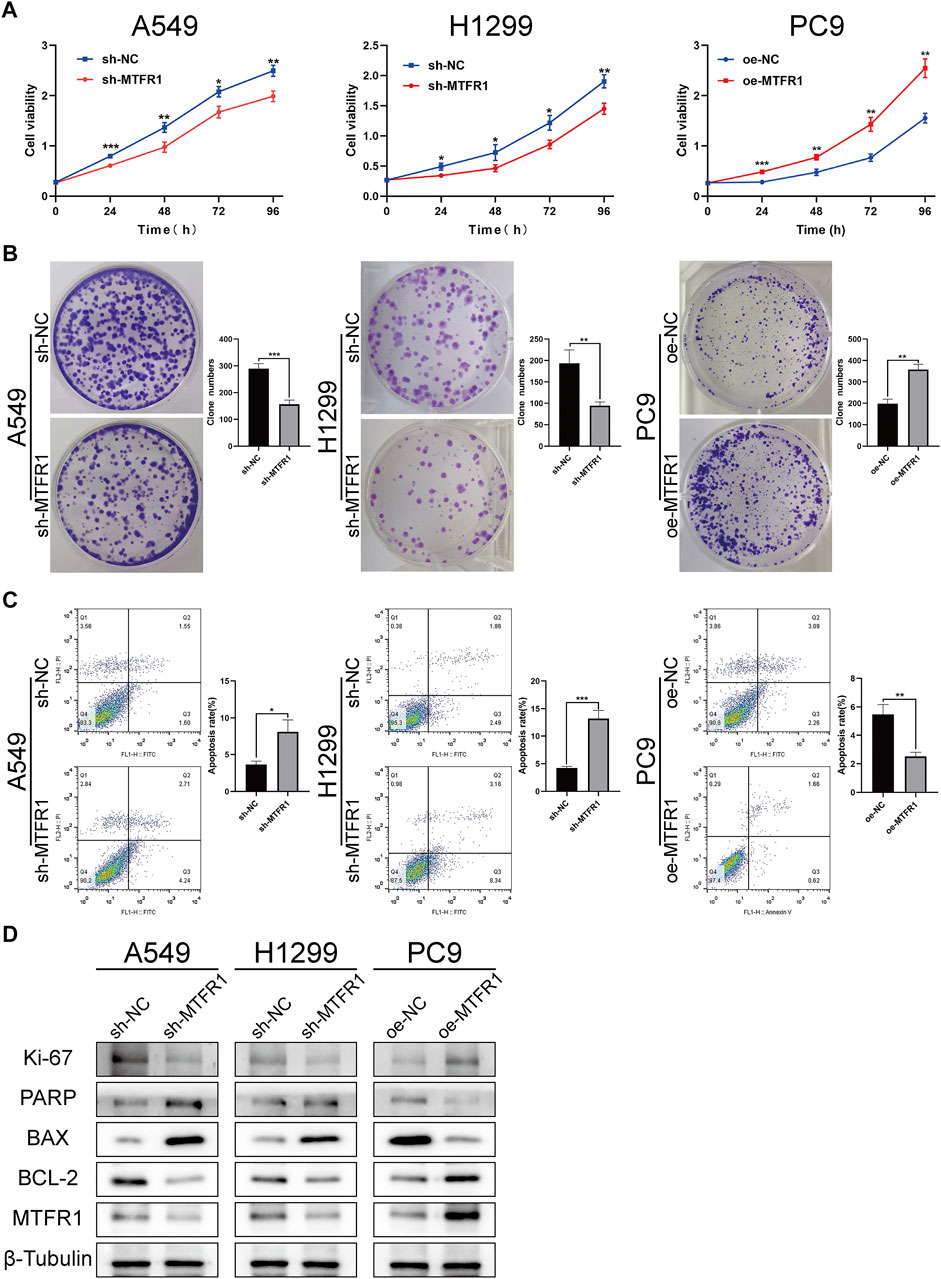
FIGURE 3. MTFR1 stimulated proliferation and suppressed apoptosis of LUAD cells. (A) Cell Counting Kit-8 (CCK-8) assay of the transduced cells. (B) Colony formation assay of the transduced cells. (C) Evaluation of apoptosis by flow cytometry. (D) Evaluation of the expression of cell proliferation and apoptosis-related protein by WB. Data were articulated as the mean ± standard deviation (SD; *p < 0.05, **p < 0.01 ***p < 0.001).
MTFR1 Promoted the Migration and Invasion of LUAD Cells
To assess the impact of MTFR1 on the migratory and invasive abilities of LUAD cancer cells, we performed Transwell and wound-healing assays. The results of the Transwell assay revealed that the numbers of invaded and migrated cells were considerably decreased after MTFR1 knockdown in H1299 and A549 cells (Figures 4A,B). Consistently, the numbers of invaded and migrated cells were considerably higher in the oe-MTFR1 cohort than in the oe-NC cohort (Figure 4C; p < 0.01). The results of the wound-healing assay revealed that MTFR1 downregulation in H1299 and A549 cells was associated with a considerably smaller wound closure area in the sh-MTFR1 cohort than in the NC cohort (Figures 4D,E; p < 0.001). However, when MTFR1 was upregulated in PC9 cells, the wound closure area was considerably larger in the oe-MTFR1 cohort than in the oe-NC cohort (Figure 4F; p < 0.01). Because epithelial–mesenchymal transition (EMT) is vital for tumour invasion and metastasis, we assessed the expression of EMT-related proteins in the transduced cells. The results revealed that MTFR1 knockdown decreased the expression of vimentin, N-cadherin, snail, and slug proteins but increased the expression of E-cadherin in H1299 and A549 cells. However, contradictory results were obtained when MTFR1 was upregulated in PC9 cells (Figure 4G). Altogether, these results suggested that MTFR1 stimulated the migration and invasion of LUAD cells.
FIGURE 4
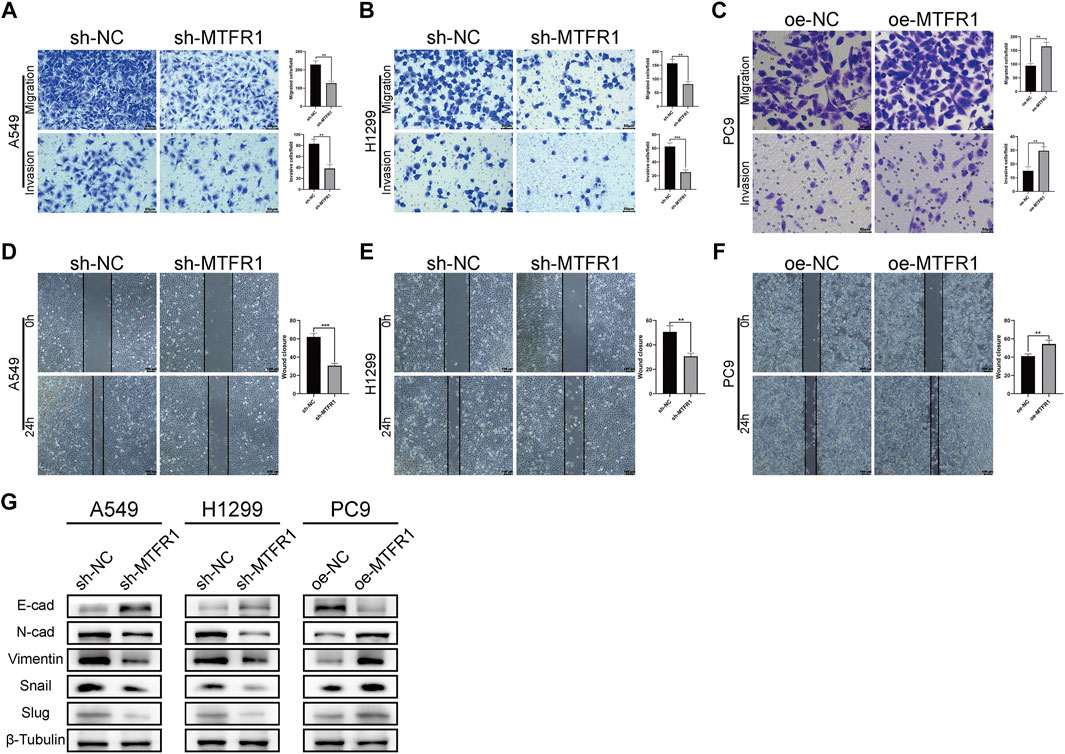
FIGURE 4. (A–F) Evaluation of the invasion and migration abilities of transduced cells by Transwell and wound-healing assays. (G) Evaluation of the levels of epithelial–mesenchymal transition (EMT)-related proteins by WB. Data were articulated as the mean ± standard deviation (SD; **p < 0.01 ***p < 0.001).
MTFR1 Regulated Glycolysis and the AMPK/mTOR Signalling Pathway in LUAD Cells
To investigate whether MTFR1 affected glycolysis in LUAD cells, we measured the ECAR, the glucose consumption and lactate production rates in the transduced LUAD cells. The rates were increased in PC9 cells after MTFR1 overexpression whereas decreased in H1299 and A549 cells after MTFR1 downregulation (Figures 5A–C). These findings indicated that MTFR1 promoted aerobic glycolysis in LUAD cells.
FIGURE 5
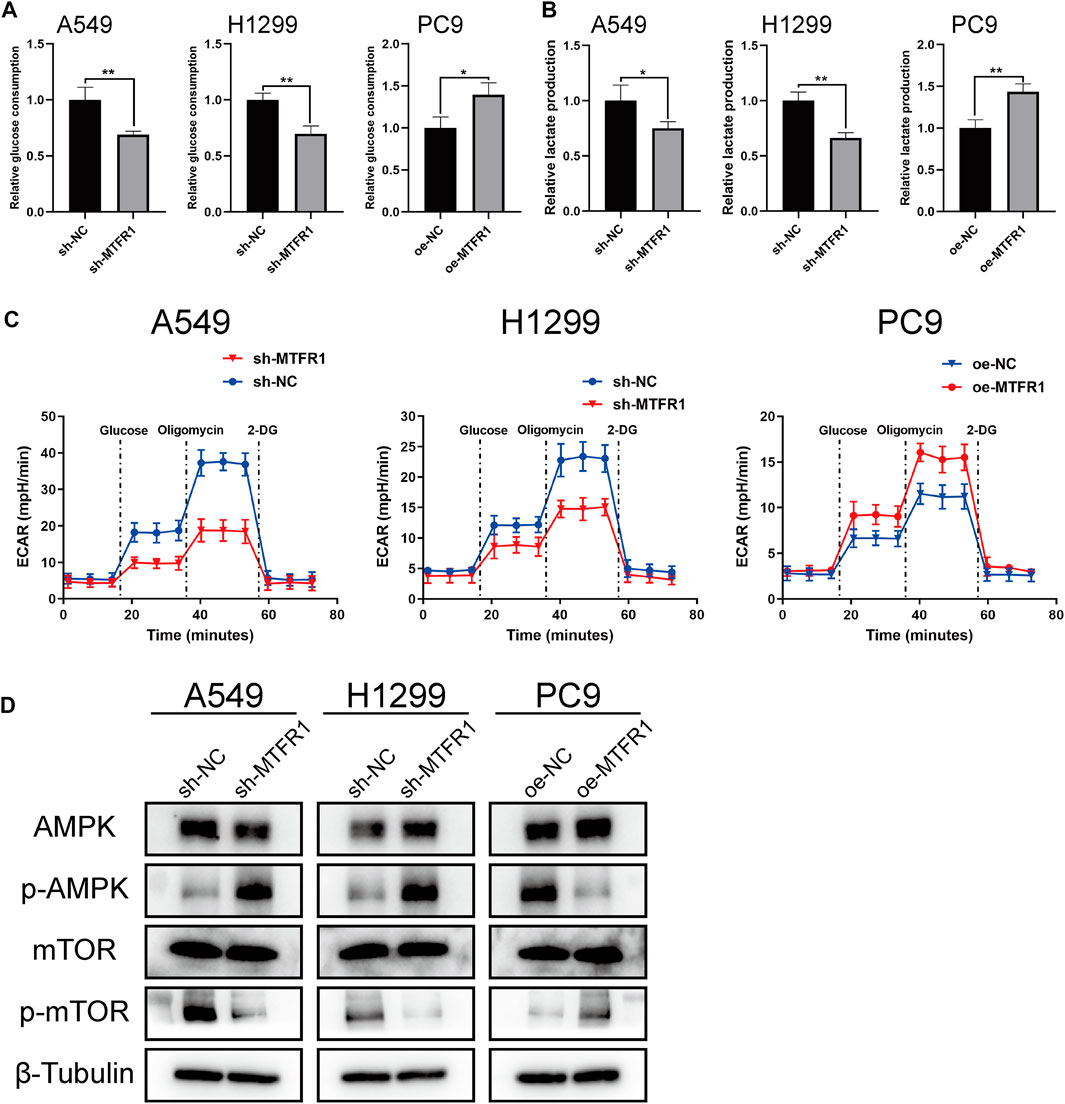
FIGURE 5. MTFR1 regulated glycolysis and AMPK/mTOR signalling pathway in LUAD cells. (A) Relative glucose consumption of the transduced cells. (B) Relative lactate production of the transduced cells. (C) Relative extracellular acidification rate (ECAR) of the transduced cells. (D) Evaluation of the expression of AMPK, p-AMPK, mTOR, and p-mTOR in the transduced cells by WB. Data were articulated as the mean ± standard deviation (SD; *p < 0.05, **p < 0.01).
Many studies have reported that the AMPK/mTOR signalling pathway is involved in the regulation of tumour metabolism. Therefore, we detected the expression of the major molecules that participate in the AMPK/mTOR pathway (mTOR, p-mTOR, AMPK, and p-AMPK). The results indicated that the expression of p-MTOR was reduced whereas that of p-AMPK was elevated when MTFR1 expression was downregulated in H1299 and A549 cells. However, contrasting effects were obtained when MTFR1 expression was upregulated in PC9 cells (Figure 5D). In conclusion, MTFR1 may exert its pro-oncogenic functions by targeting the AMPK/mTOR signalling pathway in LUAD cells.
MTFR1 was Directly Targeted by miR-29c-3p
First, we predicted the upstream miRNAs of MTFR1 based on bioinformatic analysis and chose three of them for primary study (Supplementary Tables S2, S3). After transfecting the inhibitors and mimics of these three miRNAs into the corresponding cells, we observed that miR-29c-3p had the most obvious effects on MTFR1 expression (Supplementary Figure S1B–D). Thus, we eventually selected miR-29c-3p for further study. At the cellular level, miR-29c-3p expression was noticeably higher in HBE cells than in the four LUAD cell lines (Figure 6A). At the tissue level, miR-29c-3p expression was higher in paracancerous tissues than that in the LUAD tissues (Figure 6B). Moreover, KM survival analysis demonstrated that elevated expression of miR-29c-3p was associated with a good prognosis in patients with LUAD in the TCGA cohort (p = 0.018; Figure 6C). Furthermore, we searched the starBase database to assess the relationship between MTFR1 and miR-29c-3p expression in patients with LUAD. We found miR-29c-3p expression exhibited an inverse relationship with MTFR1 expression (Figure 6D; r = −0.283, p < 0.0001). Subsequently, we performed the dual-luciferase reporter assay to further assess whether a direct regulatory correlation existed between MTFR1 and miR-29c-3p. Fluorescence intensity in the mimics-miR-29c-3p + MTFR1 WT cohort was lower than that in the other cohorts (p < 0.0001) as demonstrated in Figure 6E. These results indicated that miR-29c-3p could directly bind to the 3′-UTR of MTFR1 mRNA. Figure 6F demonstrates the predicted binding sites between MTFR1 mRNA and miR-29c-3p. Lastly, we transfected A549 and H1299 cells with inhibitor-miR-29c-3p and PC9 cells with mimics-miR-29c-3p to evaluate the expression of miR-29c-3p and MTFR1. We observed that when miR-29c-3p expression was upregulated in H1299 and A549 cells, MTFR1 was downregulated at both protein and mRNA levels; however, when miR-29c-3p expression was downregulated in PC9 cells, MTFRI was upregulated at both protein and mRNA levels (Figures 6G–I). In conclusion, these results revealed that MTFR1 was directly targeted by miR-29c-3p.
FIGURE 6
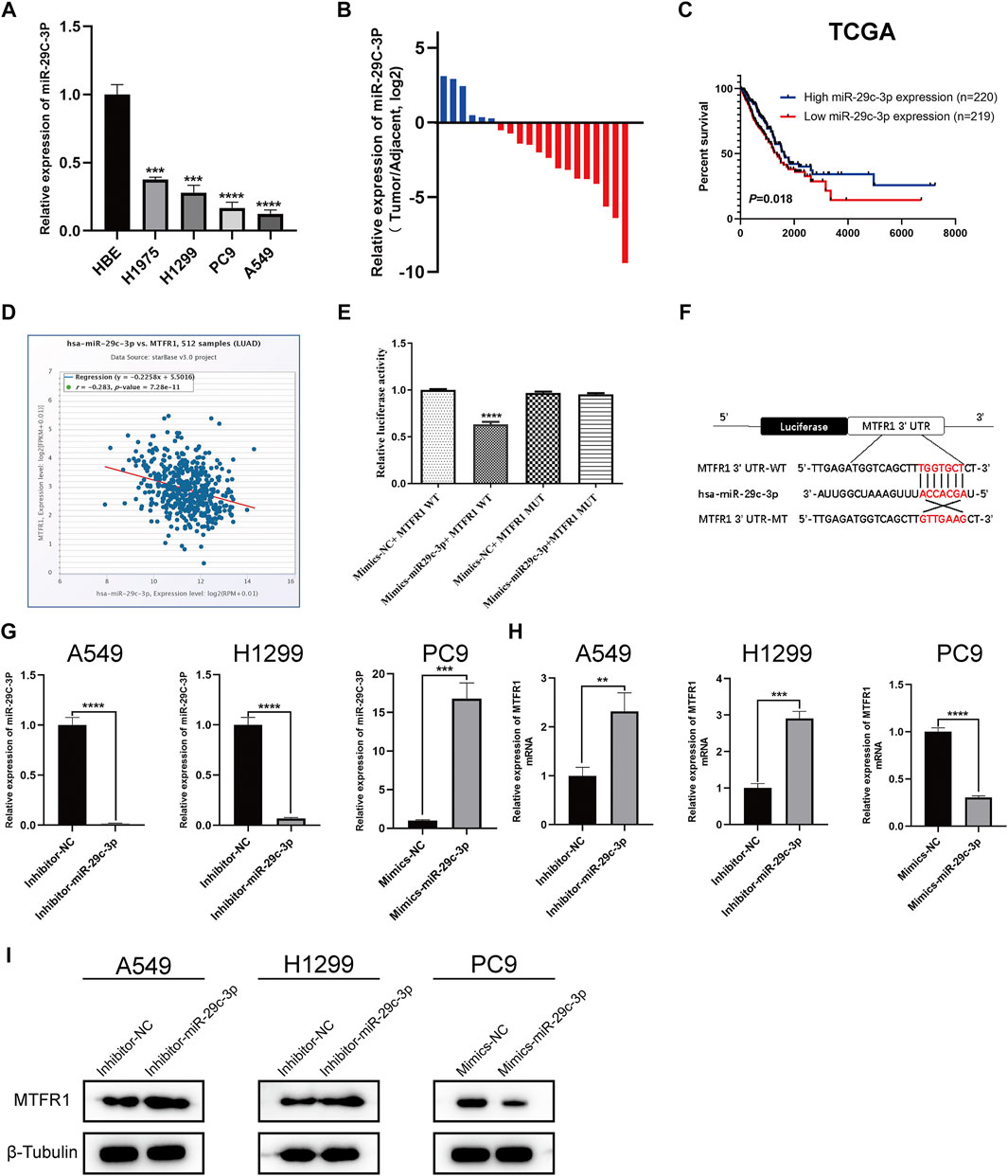
FIGURE 6. MTFR1 is directly targeted by miR-29c-3p. (A) miR-29c-3p expression in different cell lines. (B) The relative expression of miR-29c-3p in 20 pairs of LUAD tissues and their corresponding adjoining paracancerous tissues. (C) Kaplan–Meier analysis of TCGA LUAD samples based on miR-29c-3p expression. (D) Pearson’s correlation analysis for assessing the relationship between miR-29c-3p and MTFR1 expression. (E, F) Results of the dual-luciferase reporter assay and the predicted binding site between miR-29c-3p and MTFR1 3′-UTR. (G–I) The expression of miR-29c-3p and MTFR1 in cells transfected with inhibitor-miR-29c-3p or mimic-miR-29c-3p. Data were articulated as the mean ± standard deviation (SD; **p < 0.01, ***p < 0.001, ****p < 0.0001).
miR-29c-3p Could Partly Rescue the Phenotypic Changes Caused by MTFR1
We performed rescue experiments to assess whether MTFR1 was functionally regulated by miR-29c-3p. First, we transfected sh-MTFR1 A549 and H1299 cells with inhibitor-miR-29c-3p and oe-MTFR1 PC9 cells with mimics-miR-29c-3p and evaluated the transfection efficiency at both protein and mRNA levels (Figures 7A,B). Furthermore, the results of CCK-8 and Transwell assays revealed that miR-29c-3p could reverse the phenotypic changes on the proliferation, migration and invasion of LUAD cells caused by MTFR1 (Figures 7C,D). Moreover, the evaluation of ECAR, glucose and lactate levels indicated that miR-29c-3p could rescue the changes in glycolytic capacity induced by MTFR1 (Figures 8A–C). Moreover, the expression of proteins associated with cell proliferation, apoptosis, EMT and AMPK/mTOR pathway changed accordingly (Figures 8D–F). Altogether, the results indicated that phenotype changes induced by MTFR1 could be reversed by miR-29c-3p.
FIGURE 7
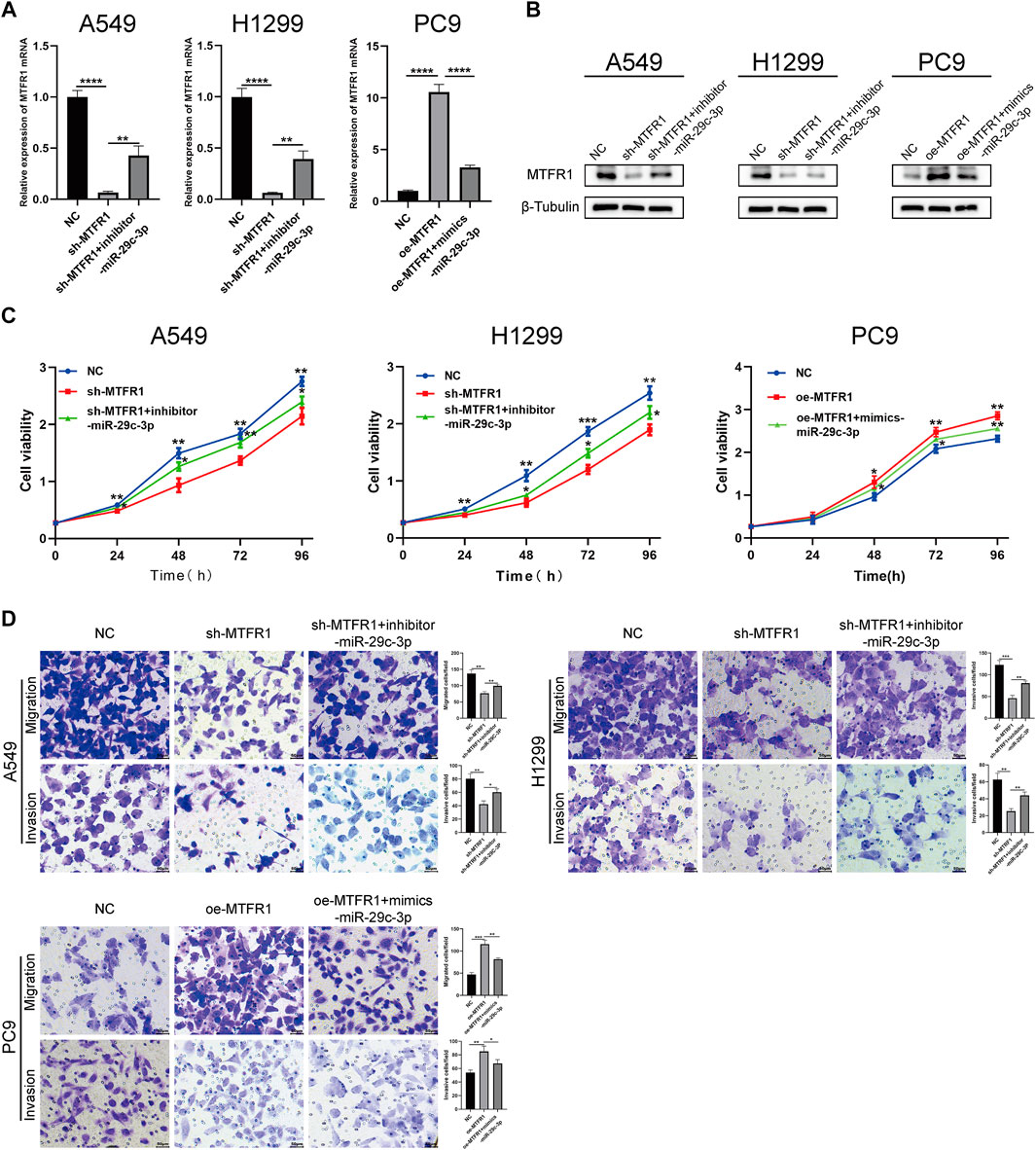
FIGURE 7. miR-29c-3p could partly rescue the phenotypic changes caused by MTFR1. (A, B) Transfection efficacy validated using real-time quantitative polymerase chain reaction (RT-qPCR) and WB. (C) miR-29c-3p could rescue the effect of MTFR1 on the proliferation of LUAD cells. (D) miR-29c-3p could rescue the effect of MTFR1 on the invasion and migration of LUAD cells. Data were articulated as the mean ± standard deviation (SD; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
FIGURE 8
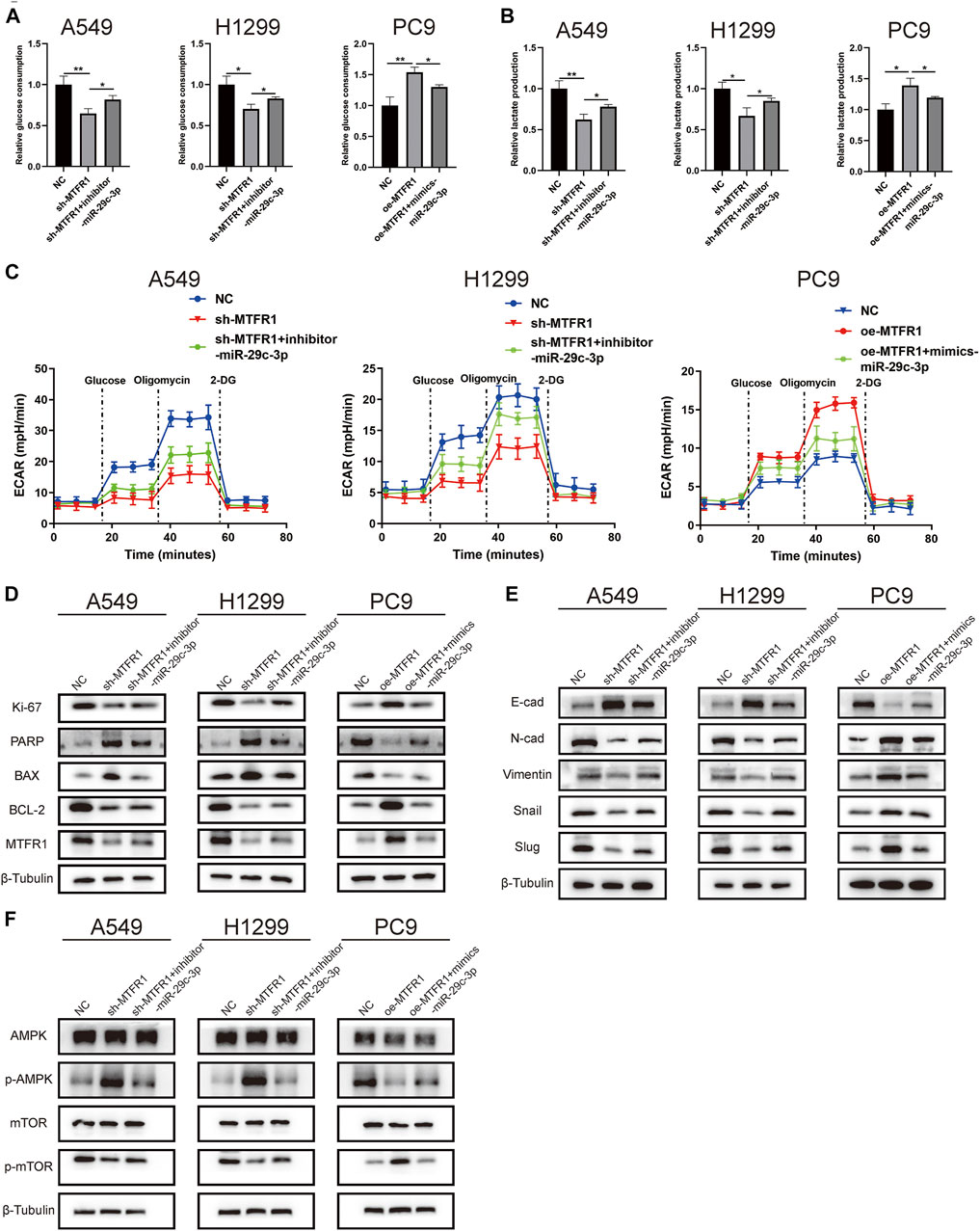
FIGURE 8. miR-29c-3p could partly rescue the changes of glycolytic capacity and the expression of the phenotype-associated proteins caused by MTFR1 in LUAD cells. (A–C) miR-29c-3p could rescue the effect of MTFR1 on the glycolytic capacity of LUAD cells. (D–F) miR-29c-3p could reverse MTFR1-induced alterations in the expression of proteins associated with cell proliferation, apoptosis, EMT and the AMPK/mTOR signalling pathway in LUAD cells. Data were articulated as the mean ± standard deviation (SD; *p < 0.05, **p < 0.01).
MTFR1 Promoted Tumour Growth and Metastasis in Vivo
A tumour xenograft-harbouring nude mouse model and a tail-vein injection-induced metastasis model were constructed to assess the in vivo impacts of MTFR1 on tumour growth and metastasis. Tumours in the sh-NC cohort exhibited a faster growth rate than that of the sh-MTFR1 cohort. Moreover, the average tumour weight and volume in the sh-NC cohort were significantly higher than those in the sh-MTFR1 cohort (Figure 9A). Simultaneously, tumours in the oe-MTFR1 cohort exhibited a faster growth rate and a significantly higher average weight and volume than those in the oe-NC group (Figure 9B). Furthermore, HE staining and IHC analysis were performed on tumour xenografts to analyse Ki-67 and MTFR1 expressions. Groups with high MTFR1 expression exhibited higher Ki-67 expression than that exhibited by groups with low MTFR1 expression (Figure 9C). The results of the lung metastasis model assay revealed that MTFR1 overexpression increased and MTFR1 knockdown decreased the number of lung metastatic nodules (Figure 9D). Altogether, the results indicated that MTFR1 promoted LUAD tumour growth and metastasis in vivo.
FIGURE 9
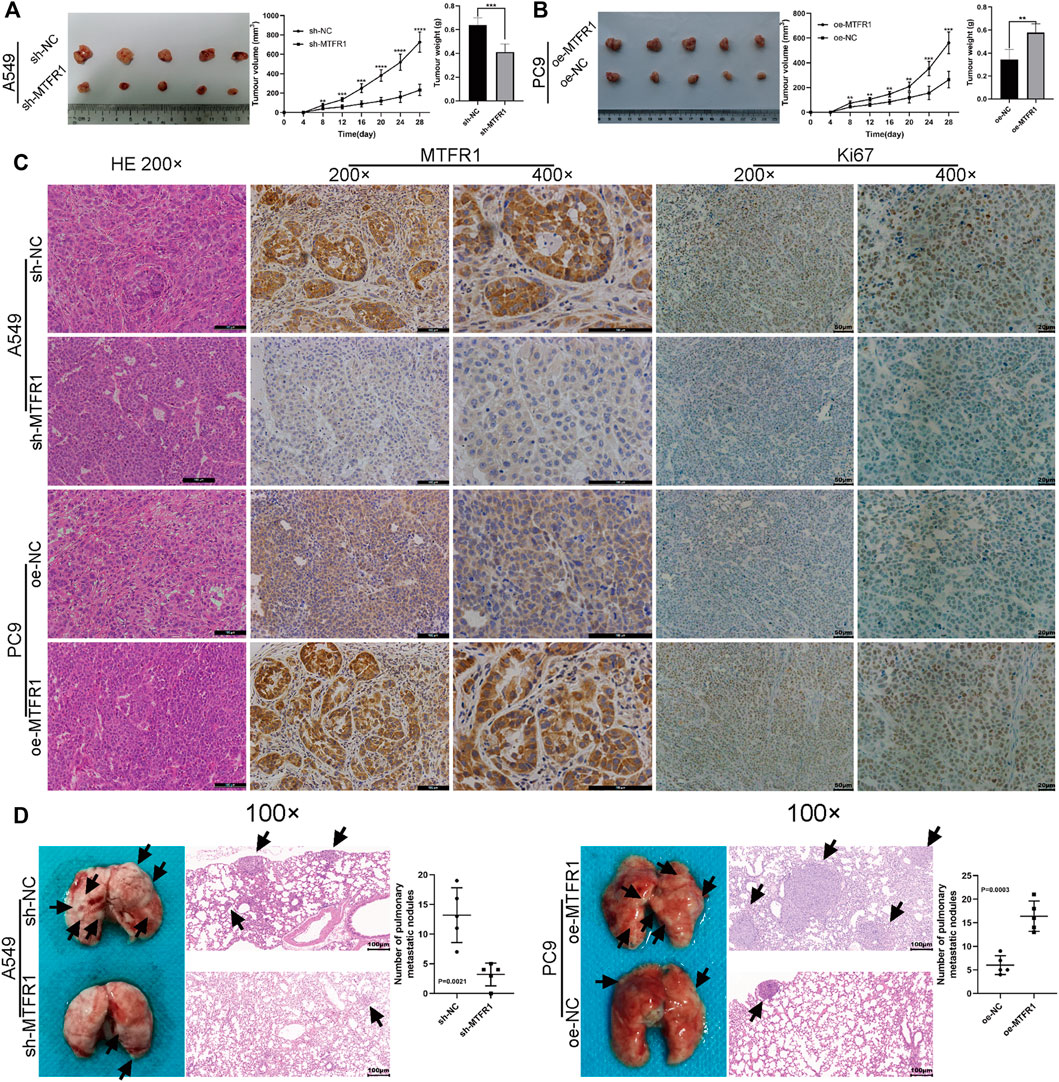
FIGURE 9. MTFR1 promoted the progression of LUAD cells in vivo. (A,B) Tumour images, tumour growth curves and tumour weights of each group. (C) Haematoxylin–eosin (HE) staining and immunohistochemical analysis of the Ki-67 and MTFR1 proteins of tumours in each group. (D) Photographs (left) and HE-stained images (right) of the lungs in each group; the black arrowheads denote lung metastasis nodules (**p < 0.01, ***p < 0.001, ****p < 0.0001).
Discussion
This study reported for the first time that MTFR1 was markedly overexpressed in LUAD cell lines and tissue samples. Moreover, high expression of MTFR1 was associated with adverse clinicopathological features and poor overall survival of LUAD patients. Findings from both in vivo and in vitro experiments demonstrated that overexpressed MTFR1 stimulated the proliferation, invasion, migration and glycolytic capacity and repressed the apoptosis of LUAD cells, whereas MTFR1 knockdown exerted contradictory effects. In addition, we discovered that miR-29c-3p was a direct upstream target of MTFR1 and could inhibit the cancer-promoting functions of MTFR1. Furthermore, we also discovered that MTFR1 exerts its biological functions by regulating the AMPK/mTOR signalling pathway. All these results indicate that MTFR1 suppression may serve as a new therapeutic strategy for treating patients with LUAD.
In recent years, the essential role of mitochondrial dynamics (fusion/fission) in the development of cancers has gained increasing recognition. Rehman et al. reported that mitochondrial fission promotes the progression of lung cancer (Rehman et al., 2012). In addition, Zhao et al. suggested that mitochondrial fission also promotes the progression of breast cancer. MTFR1, a mitochondrial protein, facilitates mitochondrial fission. In addition, MTFR2, which belongs to the same family as MTFR1, promotes the progression of breast cancer and oral squamous carcinoma when its expression is dysregulated (Lu et al., 2019; Wang W. et al., 2020). However, to date, only a few studies have used bioinformatic analyses to assess whether MTFR1 is involved in the development of cancers (Wang et al., 2015; Reddy et al., 2019; Sanada et al., 2019). In our study, we demonstrated that MTFR1 was abnormally upregulated in patients with LUAD and might act as a prognostic marker. Moreover, MTFR1 promoted the progression of LUAD. Therefore, our study provided an ideal biomarker for the diagnosis, therapeutic intervention and prognosis of LUAD.
Metabolic reprogramming has been recognised as one of the most remarkable hallmarks of cancer cells. Metabolic reprogramming in cancer cells can be best explained by the Warburg effect, in which cancer cells preferentially use glycolysis for energy generation regardless of the presence of oxygen (Vander Heiden et al., 2009). As mentioned previously, cancer cells can regulate mitochondrial dynamics to meet their metabolic requirements. Therefore, we hypothesised that MTFR1 could regulate the Warburg effect by altering mitochondrial dynamics in LUAD cells. To verify our hypothesis, we analysed the ECAR, the glucose consumption and lactate production rates of the transduced cells. We found that when MTFR1 expression was upregulated, the ECAR and the rates of glucose consumption and lactate production in LUAD cells were concomitantly elevated. However, the opposite result was observed when MTFR1 expression was downregulated. These findings indicate that MTFR1 stimulates the Warburg effect of LUAD cells. Our findings offer a new understanding of the treatment of LUAD by targeting cancer metabolism.
The occurrence and development of LUAD are accompanied by the alteration of multiple signalling pathways. Among these pathways, the AMPK/mTOR pathway is closely associated with tumour metabolism (Han et al., 2013). AMPK is considered an energy status sensor that contributes to maintaining cellular energy homeostasis. In addition, AMPK performs other functions such as regulation of cell growth and proliferation, autophagy, mitochondrial biogenesis and recycling and maintenance of cell polarity (Hardie, 2011). mTOR, which is negatively regulated by AMPK, is a central integrator of both growth factors and nutrient signals. It regulates numerous cellular processes including cell growth, cell cycle and angiogenesis (Chapuis et al., 2010). Previous studies have indicated that targeting AMPK/mTOR may be an efficient strategy for treating NSCLC (Han et al., 2013). Based on these studies and our finding that MTFR1 could promote aerobic glycolysis in LUAD, we hypothesised that MTFR1 could perform its biological function by regulating the AMPK/mTOR signalling pathway. Therefore, we identified the expression levels of several critical proteins of the AMPK/mTOR signalling pathway. As expected, MTFR1 overexpression elevated p-AMPK expression and reduced p-mTOR expression. Opposing findings were observed when MTFR1 expression was downregulated. These results confirmed our hypothesis that MTFR1 expression changes could change the expression of AMPK/mTOR signalling pathway and it may perform its biological through this pathway.
miRNAs are a class of non-coding RNAs that have been extensively studied, and aberrant miRNA expression is associated with many human diseases, including various cancers (Jiang et al., 2009). In our study, we predicted and validated miR-29c-3p as a direct upstream miRNA of MTFR1. miR-29c-3p expression was not only downregulated in LUAD cells but also inversely correlated with MTFR1. Furthermore, we performed rescue experiments to confirm that miR-29c-3p might partly rescue the malignant phenotypes induced by MTFR1. These findings indicate that the miR-29c-3p/MTFR1/AMPK/mTOR axis plays an important role in LUAD. However, because MTFR1 exerts robust cancer-promoting effects on LUAD, further investigation is required to identify proteins that interact with MTFR1 to exercise such effects.
In conclusion, MTFR1 serves as a strong tumour-promoting factor in LUAD, and targeting MTFR1 may provide a better disease outcome for patients with LUAD.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Medical Ethics Committee of Qilu Hospital of Shandong University. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Shandong University Animal Research Ethics Committee.
Author Contributions
YL, LS, and HT designed this study. KJ, RD, and ZF collected samples and information of LUAD patients. YL performed the experiments and wrote the manuscript. YLI analyzed the data. CG and HZ revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the Youth Fund of Qilu Hospital of Shandong University (No. 2019QLQN50), the Natural Science Foundation of Shandong Province (No. ZR2020QH213), and the Taishan Scholar Program of Shandong Province (No. ts201712087).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Professor Qingjie Wang for the guidance of the written manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.771824/full#supplementary-material
References
Bartel, D. (2004). MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 116, 281–297. doi:10.1016/s0092-8674(04)00045-5
Cai, J., Wang, J., Huang, Y., Wu, H., Xia, T., Xiao, J., et al. (2016). ERK/Drp1-dependent Mitochondrial Fission Is Involved in the MSC-Induced Drug Resistance of T-Cell Acute Lymphoblastic Leukemia Cells. Cell Death Dis 7 (11), e2459. doi:10.1038/cddis.2016.370
Chan, D. C. (2012). Fusion and Fission: Interlinked Processes Critical for Mitochondrial Health. Annu. Rev. Genet. 46, 265–287. doi:10.1146/annurev-genet-110410-132529
Chan, D. C. (2006). Mitochondria: Dynamic Organelles in Disease, Aging, and Development. Cell 125 (7), 1241–1252. doi:10.1016/j.cell.2006.06.010
Chapuis, N., Tamburini, J., Green, A. S., Willems, L., Bardet, V., Park, S., et al. (2010). Perspectives on Inhibiting mTOR as a Future Treatment Strategy for Hematological Malignancies. Leukemia 24 (10), 1686–1699. doi:10.1038/leu.2010.170
Chen, W., Zheng, R., Baade, P. D., Zhang, S., Zeng, H., Bray, F., et al. (2016). Cancer Statistics in China, 2015. CA: A Cancer J. Clinicians 66 (2), 115–132. doi:10.3322/caac.21338
Feng, S., Luo, S., Ji, C., and Shi, J. (2020). miR-29c-3p Regulates Proliferation and Migration in Ovarian Cancer by Targeting KIF4A. World J. Surg. Onc 18 (1), 315. doi:10.1186/s12957-020-02088-z
Feng, Z., and Levine, A. J. (2010). The Regulation of Energy Metabolism and the IGF-1/mTOR Pathways by the P53 Protein. Trends Cel Biol. 20 (7), 427–434. doi:10.1016/j.tcb.2010.03.004
Friedman, R. C., Farh, K. K.-H., Burge, C. B., and Bartel, D. P. (2009). Most Mammalian mRNAs Are Conserved Targets of microRNAs. Genome Res. 19 (1), 92–105. doi:10.1101/gr.082701.108
Guo, L., Cui, C., Wang, J., Yuan, J., Yang, Q., Zhang, P., et al. (2020). PINCH-1 Regulates Mitochondrial Dynamics to Promote Proline Synthesis and Tumor Growth. Nat. Commun. 11 (1), 4913. doi:10.1038/s41467-020-18753-6
Han, D., Li, S.-J., Zhu, Y.-T., Liu, L., and Li, M.-X. (2013). LKB1/AMPK/mTOR Signaling Pathway in Non-small-cell Lung Cancer. Asian Pac. J. Cancer Prev. 14 (7), 4033–4039. doi:10.7314/apjcp.2013.14.7.4033
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of Cancer: the Next Generation. Cell 144 (5), 646–674. doi:10.1016/j.cell.2011.02.013
Hardie, D. G. (2011). AMP-activated Protein Kinase-An Energy Sensor that Regulates All Aspects of Cell Function. Genes Dev. 25 (18), 1895–1908. doi:10.1101/gad.17420111
Herbst, R. S., Morgensztern, D., and Boshoff, C. (2018). The Biology and Management of Non-small Cell Lung Cancer. Nature 553 (7689), 446–454. doi:10.1038/nature25183
Hernández-Alvarez, M. I., and Zorzano, A. (2021). Mitochondrial Dynamics and Liver Cancer. Cancers 13 (11), 2571. doi:10.3390/cancers13112571
Hozaka, Y., Seki, N., Tanaka, T., Asai, S., Moriya, S., Idichi, T., et al. (2021). Molecular Pathogenesis and Regulation of the miR-29-3p-Family: Involvement of ITGA6 and ITGB1 in Intra-hepatic Cholangiocarcinoma. Cancers 13 (11), 2804. doi:10.3390/cancers13112804
Huang, L., Guo, B., Liu, S., Miao, C., and Li, Y. (2020). Inhibition of the LncRNA Gpr19 Attenuates Ischemia‐reperfusion Injury after Acute Myocardial Infarction by Inhibiting Apoptosis and Oxidative Stress via the miR‐324‐5p/Mtfr1 axis. IUBMB Life 72 (3), 373–383. doi:10.1002/iub.2187
Huang, L., Luan, T., Chen, Y., Bao, X., Huang, Y., Fu, S., et al. (2018). LASS2 Regulates Invasion and Chemoresistance via ERK/Drp1 Modulated Mitochondrial Dynamics in Bladder Cancer Cells. J. Cancer 9 (6), 1017–1024. doi:10.7150/jca.23087
Jiang, Q., Wang, Y., Hao, Y., Juan, L., Teng, M., Zhang, X., et al. (2009). miR2Disease: a Manually Curated Database for microRNA Deregulation in Human Disease. Nucleic Acids Res. 37 (Database issue), D98–D104. doi:10.1093/nar/gkn714
Knott, A. B., Perkins, G., Schwarzenbacher, R., and Bossy-Wetzel, E. (2008). Mitochondrial Fragmentation in Neurodegeneration. Nat. Rev. Neurosci. 9 (7), 505–518. doi:10.1038/nrn2417
Kong, B., Tsuyoshi, H., Orisaka, M., Shieh, D.-B., Yoshida, Y., and Tsang, B. K. (2015). Mitochondrial Dynamics Regulating Chemoresistance in Gynecological Cancers. Ann. N.Y. Acad. Sci. 1350, 1–16. doi:10.1111/nyas.12883
Lenzi, P., Ferese, R., Biagioni, F., Fulceri, F., Busceti, C. L., Falleni, A., et al. (2021). Rapamycin Ameliorates Defects in Mitochondrial Fission and Mitophagy in Glioblastoma Cells. Int. J. Mol. Sci. 22 (10), 5379. doi:10.3390/ijms22105379
Li, Y., Dong, R., Lu, M., Cheng, C., Feng, Z., Zhao, R., et al. (2021). Let-7b-3p Inhibits Tumor Growth and Metastasis by Targeting the BRF2-Mediated MAPK/ERK Pathway in Human Lung Adenocarcinoma. Transl Lung Cancer Res. 10 (4), 1841–1856. doi:10.21037/tlcr-21-299
Lin, M. T., and Beal, M. F. (2006). Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases. Nature 443 (7113), 787–795. doi:10.1038/nature05292
Lu, G., Lai, Y., Wang, T., Lin, W., Lu, J., Ma, Y., et al. (2019). Mitochondrial Fission Regulator 2 (MTFR2) Promotes Growth, Migration, Invasion and Tumour Progression in Breast Cancer Cells. Aging (Albany NY) 11 (22), 10203–10219. doi:10.18632/aging.102442
McBride, H. M., Neuspiel, M., and Wasiak, S. (2006). Mitochondria: More Than Just a Powerhouse. Curr. Biol. 16 (14), R551–R560. doi:10.1016/j.cub.2006.06.054
Monticone, M., Panfoli, I., Ravera, S., Puglisi, R., Jiang, M.-M., Morello, R., et al. (2010). The Nuclear Genes Mtfr1 and Dufd1 Regulate Mitochondrial Dynamic and Cellular Respiration. J. Cel. Physiol. 225 (3), 767–776. doi:10.1002/jcp.22279
Reddy, R. B., Khora, S. S., and Suresh, A. (2019). Molecular Prognosticators in Clinically and Pathologically Distinct Cohorts of Head and Neck Squamous Cell Carcinoma-A Meta-Analysis Approach. PLoS One 14 (7), e0218989. doi:10.1371/journal.pone.0218989
Rehman, J., Zhang, H. J., Toth, P. T., Zhang, Y., Marsboom, G., Hong, Z., et al. (2012). Inhibition of Mitochondrial Fission Prevents Cell Cycle Progression in Lung Cancer. FASEB j. 26 (5), 2175–2186. doi:10.1096/fj.11-196543
Sanada, H., Seki, N., Mizuno, K., Misono, S., Uchida, A., Yamada, Y., et al. (2019). Involvement of Dual Strands of miR-143 (miR-143-5p and miR-143-3p) and Their Target Oncogenes in the Molecular Pathogenesis of Lung Adenocarcinoma. Int. J. Mol. Sci. 20 (18), 4482. doi:10.3390/ijms20184482
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A. Cancer J. Clin. 71 (3), 209–249. doi:10.3322/caac.21660
Tonachini, L., Monticone, M., Puri, C., Tacchetti, C., Pinton, P., Rizzuto, R., et al. (2004). Chondrocyte Protein with a Poly-Proline Region (CHPPR) Is a Novel Mitochondrial Protein and Promotes Mitochondrial Fission. J. Cel. Physiol. 201 (3), 470–482. doi:10.1002/jcp.20126
Vander Heiden, M. G., Cantley, L. C., and Thompson, C. B. (2009). Understanding the Warburg Effect: the Metabolic Requirements of Cell Proliferation. Science 324 (5930), 1029–1033. doi:10.1126/science.1160809
Wang, H., Fu, L., Wei, D., Wang, B., Zhang, C., Zhu, T., et al. (2020a). MiR-29c-3p Suppresses the Migration, Invasion and Cell Cycle in Esophageal Carcinoma via CCNA2/p53 Axis. Front. Bioeng. Biotechnol. 8, 75. doi:10.3389/fbioe.2020.00075
Wang, W., Xiong, M., Jiang, L., Chen, Z., and Shao, Y. (2020b). MTFR2 Promotes the Proliferation, Migration, and Invasion of Oral Squamous Carcinoma by Switching OXPHOS to Glycolysis. Front. Oncol. 10, 858. doi:10.3389/fonc.2020.00858
Wang, W., Lim, W. K., Leong, H. S., Chong, F. T., Lim, T. K. H., Tan, D. S. W., et al. (2015). An Eleven Gene Molecular Signature for Extra-capsular Spread in Oral Squamous Cell Carcinoma Serves as a Prognosticator of Outcome in Patients without Nodal Metastases. Oral Oncol. 51 (4), 355–362. doi:10.1016/j.oraloncology.2014.12.012
Xie, P., Zhang, M., He, S., Lu, K., Chen, Y., Xing, G., et al. (2014). The Covalent Modifier Nedd8 Is Critical for the Activation of Smurf1 Ubiquitin Ligase in Tumorigenesis. Nat. Commun. 5, 3733. doi:10.1038/ncomms4733
Zhu, H., Wang, G., Zhu, H., and Xu, A. (2021). MTFR2, A Potential Biomarker for Prognosis and Immune Infiltrates, Promotes Progression of Gastric Cancer Based on Bioinformatics Analysis and Experiments. J. Cancer 12 (12), 3611–3625. doi:10.7150/jca.58158
Keywords: lung cancer, microRNA, mitochondrial fission regulator 1, Warburg effect, proliferation, invasion, migration
Citation: Li Y, Liu Y, Jin K, Dong R, Gao C, Si L, Feng Z, Zhang H and Tian H (2021) Negatively Regulated by miR-29c-3p, MTFR1 Promotes the Progression and Glycolysis in Lung Adenocarcinoma via the AMPK/mTOR Signalling Pathway. Front. Cell Dev. Biol. 9:771824. doi: 10.3389/fcell.2021.771824
Received: 07 September 2021; Accepted: 18 November 2021;
Published: 01 December 2021.
Edited by:
Jiayi Wang, Shanghai Jiaotong University, ChinaReviewed by:
Li Zhang, Huazhong University of Science and Technology, ChinaLifang Ma, Shanghai Jiaotong University, China
Behzad Mansoori, Wistar Institute, United States
Copyright © 2021 Li, Liu, Jin, Dong, Gao, Si, Feng, Zhang and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Tian, tianhuiql@126.com