 David A. Lewis1*
David A. Lewis1* Tony Ly2,3*
Tony Ly2,3*- 1Ashworth Laboratories, Institute of Immunology and Infectious Research, University of Edinburgh, Edinburgh, United Kingdom
- 2Wellcome Centre for Cell Biology, University of Edinburgh, Edinburgh, United Kingdom
- 3Centre for Gene Regulation and Expression, University of Dundee, Dundee, United Kingdom
CD8+ T cells play important roles in immunity and immuno-oncology. Upon antigen recognition and co-stimulation, naïve CD8+ T cells escape from dormancy to engage in a complex programme of cellular growth, cell cycle entry and differentiation, resulting in rapid proliferation cycles that has the net effect of producing clonally expanded, antigen-specific cytotoxic T lymphocytes (CTLs). A fraction of activated T cells will re-enter dormancy by differentiating into memory T cells, which have essential roles in adaptive immunity. In this review, we discuss the current understanding of cell cycle entry control in CD8+ T cells and crosstalk between these mechanisms and pathways regulating immunological phenotypes.
Introduction
CD8+ T cells are an important component of the acquired immune response. They have a role in limiting viral infection and cancer progression through recognising key viral/tumour antigens on the infected cell surface and targeting them for destruction (Kim and Ahmed, 2010). T cells are the product of central selection, a process in which newly developed progenitor T cells go through differentiation and numerous divisions, becoming lineage restricted toward the naïve CD8+ T cell phenotype. These naive T cells then lie dormant predominately within the lymph nodes (Carpenter and Bosselut, 2010). Naïve CD8+ T cells are activated by antigen presenting cells. The activation phase is characterised by major rewiring of their cellular proteomes and metabolism. Activated T cells then rapidly proliferate in what is known as the expansion phase (Figure 1A). During this phase, the doubling rate averages at 6–8 h with some studies demonstrating a peak doubling time of 4.5 h, or even 2 h (Kurts et al., 1997; Yoon et al., 2010). Also during this phase, T cells will begin to differentiate toward one of two T effector (Teff) phenotypes, the short lived effector cell (SLEC) which die during the subsequent contraction phase, and the memory progenitor cells (MPEC) which survive beyond the contraction phase. MPECs then differentiate toward one of the memory T cell phenotypes, e.g., the central memory T cell (Tcm), and effector memory T cell (Tem). The role of SLECs is to clear the infection, which is done by targeted release of cytolytic granzymes and perforins to infected cells. Following the clearance of infection, memory T cells remain in a state of dormancy, with Tcm localising to the lymph nodes, and Tem remaining in the periphery. Like naïve T cells, they await antigen stimulation in order to activate and proliferate, but have a higher antigenic threshold and more quickly initiate proliferation (Mehlhop-Williams and Bevan, 2014). This enables memory T cells to act as early responders to repeat infections.
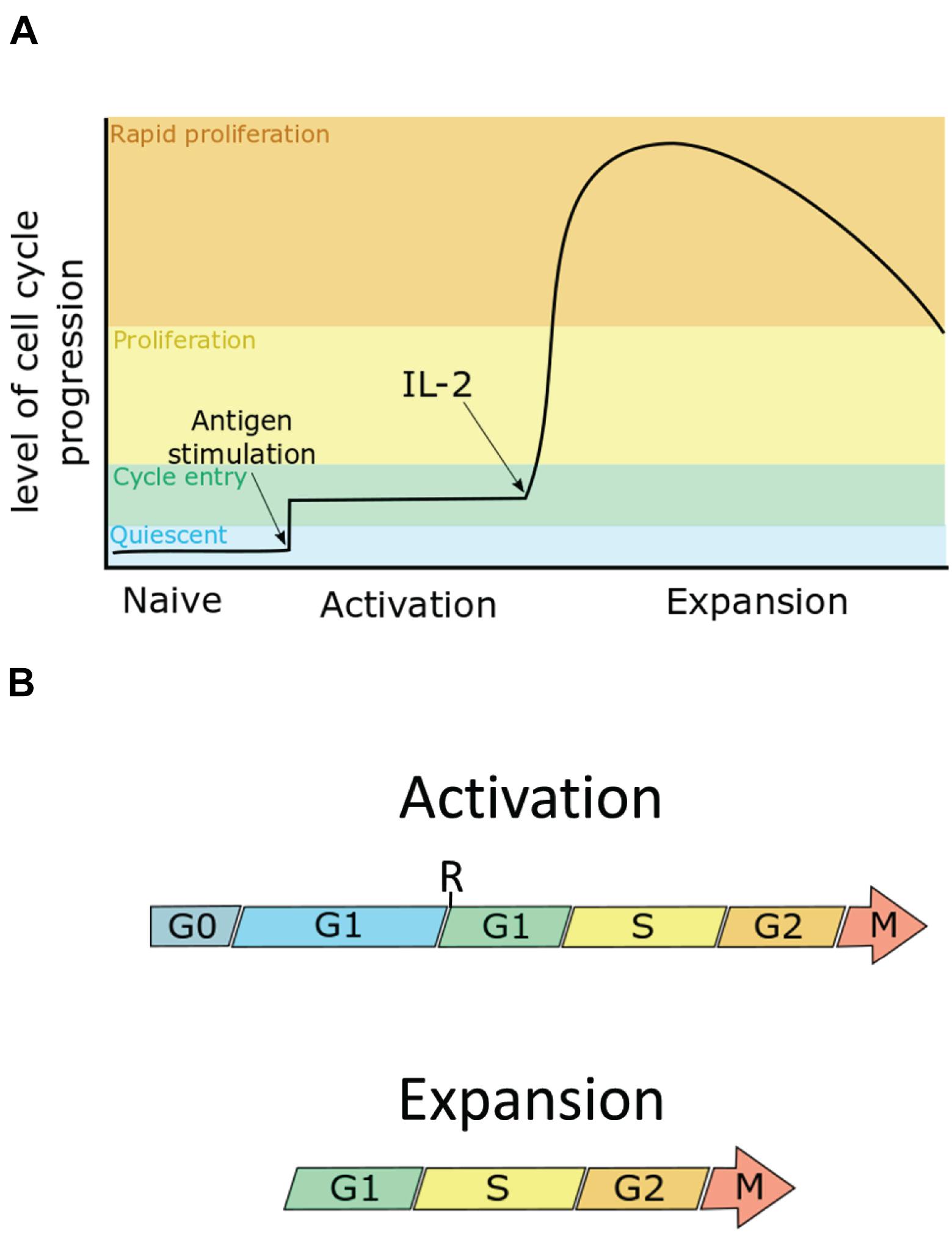
Figure 1. The cell cycle can be split into four phases: the first gap phase (G1), DNA synthesis phase (S), the second gap phase (G2), and mitosis (M). G1 can be further subdivided into pre- and post- restriction point G1 (pre-R G1, post-R G1). Quiescent cells are in a G0 state. (A) Naïve T cells are cells in G0. Upon maximum stimulation by T-cell receptor-antigen and co-stimulation, murine CD8+ T cells enter the cell cycle, going through only a few divisions until they receive the IL-2 signal, at which point they proliferate rapidly as they enter the expansion phase. (B) During activation, T cells enter an extended G1 phase, in which cells increase in size by ∼3–4-fold, entering their first S-phase within 30 h. During the expansion phase, cells then undergo rapid proliferation, dividing as quickly as once every 4 h, to clonally expand the antigen-specific T cell pool. Arrows indicate continuation beyond mitosis into the next G1.
What factors determine whether daughter cells from an activated naïve T cell move toward the MPEC or SLEC phenotype are a topic of active debate. It is generally agreed, however, that SLECs represent a fully differentiated form of T cell, which develop full effector function in exchange of proliferative potential and survival that typify MPECS. Complementing this model are studies that demonstrate differences in cell cycle progression between the phenotypes, with SLECs progressing at a much faster rate than MPEC (Kinjyo et al., 2015; Kretschmer et al., 2020) demonstrating a link between control of proliferation and differentiation.
In this review, we will first present an overview on cell cycle regulation based on various mammalian model systems. We then examine the pathways controlling CD8+ T cell differentiation and proliferation with reference to the cell cycle regulation pathways outlined prior and explore potential crosstalk between these two key processes.
Cell Cycle Regulation
The cell cycle is divided into four phases (Figure 1B). In the first gap phase (G1), the cell prepares for DNA replication (S). This is followed by a second gap phase (G2) when the cell prepares for mitosis (M). Quiescent, or G0 cells enter the cell cycle by first transiting through G1. During G1, cells pass through a commitment step called the restriction point (R). Before R, cells are susceptible to cell cycle arrest by mitogen starvation. However, beyond R, progression through the cell cycle is mitogen-independent in human fibroblasts. The time cells spend in pre-R G1 is heterogeneous compared with the time spent in the remaining phases of the cell cycle (Zetterberg and Larsson, 1985; Zetterberg et al., 1995). Additionally mitogen starved cells which exit to G0 and are reintroduced to mitogen have been observed to take up to an additional 8 h to reach and complete mitosis compared to cells cultured in mitogen-replete conditions (Zetterberg and Larsson, 1991). Thus, G0, pre-R and post-R G1 can be considered distinct phases that are distinguished in their mitogen responsiveness and in the length of time needed to progress to S-phase. It should be noted, however, that it is as yet unclear if the restriction point as described in fibroblasts exists within T cells. This is due to the differences in mitogenic signalling within T cells which make the model established in fibroblasts more challenging to test in T cells.
Key proteins that control cell cycle progression include (1) the cyclin proteins, which rise and fall in abundance at key stages of cell cycle, (2) the cyclin-dependent kinases (CDKs) which become active when bound to their cognate cyclins, and (3) the CDK inhibitor proteins (CDKi) which control cell cycle progression at key check points in response to mitogen starvation or cellular stress (e.g., DNA damage).
CDK-Rb-E2F
Cyclins, CDKs, and CDKis have key roles in regulating the activity of E2 factor F (E2F), a transcription factor that promotes the expression of cell cycle genes, including the cyclins and CDKs themselves.
In G0 and pre-R G1, E2F mediated transcription will be inhibited by pocket proteins Retinoblastoma protein (Rb), p107, and p130, preventing the cell from progressing to S phase (Stengel et al., 2009). An active kinase complex is formed by interaction of cyclin D with either CDK4 or CDK6, and this kinase activity mediates hyperphosphorylation of Rb, causing Rb to decouple from E2F, and thereby enable E2F to promote upregulation of its targets. Such targets include a large number of S phase-promoting proteins, including cyclin E and cyclin A. Cyclin E will form a complex with CDK2 which amongst other processes will contribute to Rb hyperphosphorylation, resulting in a positive feedback loop that activates E2F (Dyson, 1998). Recent work suggests that CDK4/6 regulates cellular processes other than those downstream of E2F. Cyclin D3-CDK6 phosphorylates key enzymes in the glycolysis pathway, diverting metabolites from glycolysis into the pentose phosphate pathway to promote redox balance (via production of NADPH) and cellular anabolism (e.g., by generating nucleotide precursors) (Wang et al., 2017).
Cyclin-dependent kinases activities can be inhibited by stoichiometric binding to CDKis, of which there are two main families: the INK4 family consisting of p16, p15, p18, and p19 which inhibit Cyclin D-CDK4/6 complex; and the Cip/Kip family which consists of p27, p21, and p57 that inhibit cyclin-CDK complexes, and are able to induce cell cycle arrest at G1 phase (Sherr and Roberts, 1999). The regulation of CDK4/6 complexes by the Cip/Kip family members are more complex. Cip/Kip proteins can promote the formation of cyclin-CDK complexes and in the absence of p21 or p27, cyclin D-CDK complexes do not form (Labaer et al., 1997; Cheng et al., 1999). Trimeric species containing p27 phosphorylated on tyrosine 74, cyclin D, and CDK4 retain kinase activity (Guiley et al., 2019). On the other hand, high levels of p21 inhibit CDK4 (Labaer et al., 1997). Cip/Kip proteins are inactivated by post-translational modification and degradation. For example, tyrosine phosphorylation on p27 by mitogen-activated tyrosine kinases disrupts the inhibitory interaction of p27 with CDK2 and promotes p27 degradation (Grimmler et al., 2007),
Anaphase Promoting Complex/Cyclosome
Stability of cyclin, CDK and CDKi proteins are controlled post-translationally in a cell cycle regulated manner by E3 ubiquitin ligases. The anaphase promoting complex/cyclosome (APC/C) is a large, multi-subunit E3 ligase that targets many proteins, including cyclins and other E2F targets, for destruction in mitosis and G1. The APC/C has two co-activators, Cell Division Cycle (Cdc) 20 and Fizzy-related protein homolog-1 (Fzr1) (also known as Cdh1). The substrate adaptor functions of Cdc20 are primarily in mitosis, whereas Cdh1 is important in mitotic exit and G1. Indeed, inactivation of the APC/C-Cdh1 has been shown to be a second crucial step in promoting the transition from G1 to S (Cappell et al., 2016). Thus, proteins required for G1/S are upregulated by increased synthesis via transcription by E2F and increased stability via APC/C inactivation. APC/C inactivation in G1 is mediated by multiple mechanisms, including phosphorylation of Cdh1 (Kramer et al., 2000) by CDK2 (Lukas et al., 1999) and by the accumulation the pseudosubstrate inhibitor protein, F-box only protein 5 (Fbxo5) [also known as the early mitotic inhibitor 1 (Emi1)] (Cappell et al., 2018). Additionally, there are deubiquitinases (DUBs), including ubiquitin specific peptidase 37 (Usp37), which stabilise APC/C substrates by removing ubiquitin chains that would otherwise target these proteins for proteasomal destruction (Huang et al., 2011).
Activation Phase (TCR Induced Proliferation)
T cell activation and proliferation requires three stimulation signals, antigenic stimulation via the T cell receptor (TCR), co-receptor signalling from professional antigen presenting cells via CD28, and cytokine stimulation primarily driven by interleukin (IL)-2. Stimulation of the TCR alone results in anergy. Anergic cells do not produce IL-2, fail to proliferate, and become unresponsive to further stimulation attempts. A dual signal of TCR and CD28 induces production of IL-2 and proliferation within the first 24 h. IL-2 stimulation through the IL-2 receptor (IL-2R) enables rapid proliferation as the cells enter the expansion phase (Mondino et al., 2006).
As depicted in Figure 2A, TCR stimulation leads to phosphorylation of lymphocyte-specific protein tyrosine kinase (Lck), which in return phosphorylates the ζ chains of the TCR along the immunoreceptor tyrosine-based activation motifs (ITAMS). This enables recruitment of TCR-associated protein 70 (Zap70), which is subsequently phosphorylated by Lck. Zap70 then phosphorylates four key sites on the linker for activation of T cells (LAT) which allows for recruitment of proteins into the LAT signalosome. The downstream effect is activation of the Rat sarcoma (Ras)/extracellular signal-related kinase (Erk)/Activator protein 1 (AP-1) pathway, Protein kinase C–θ (PKCθ)/κB kinase (IKK)/nuclear factor-κB (Nf-κB) pathway, and the calcium-dependent Calcineurin/nuclear factor of activated T cells (NFAT) pathway (Brownlie and Zamoyska, 2013; Hwang et al., 2020). The transcription factors downstream of these pathways, NFAT, Nf-κB, and AP-1 all contribute to the transcription of IL2 and also contribute to the transcription of IL2RA, which encodes the α-chain of the IL-2 receptor (CD25) (Liao et al., 2013).
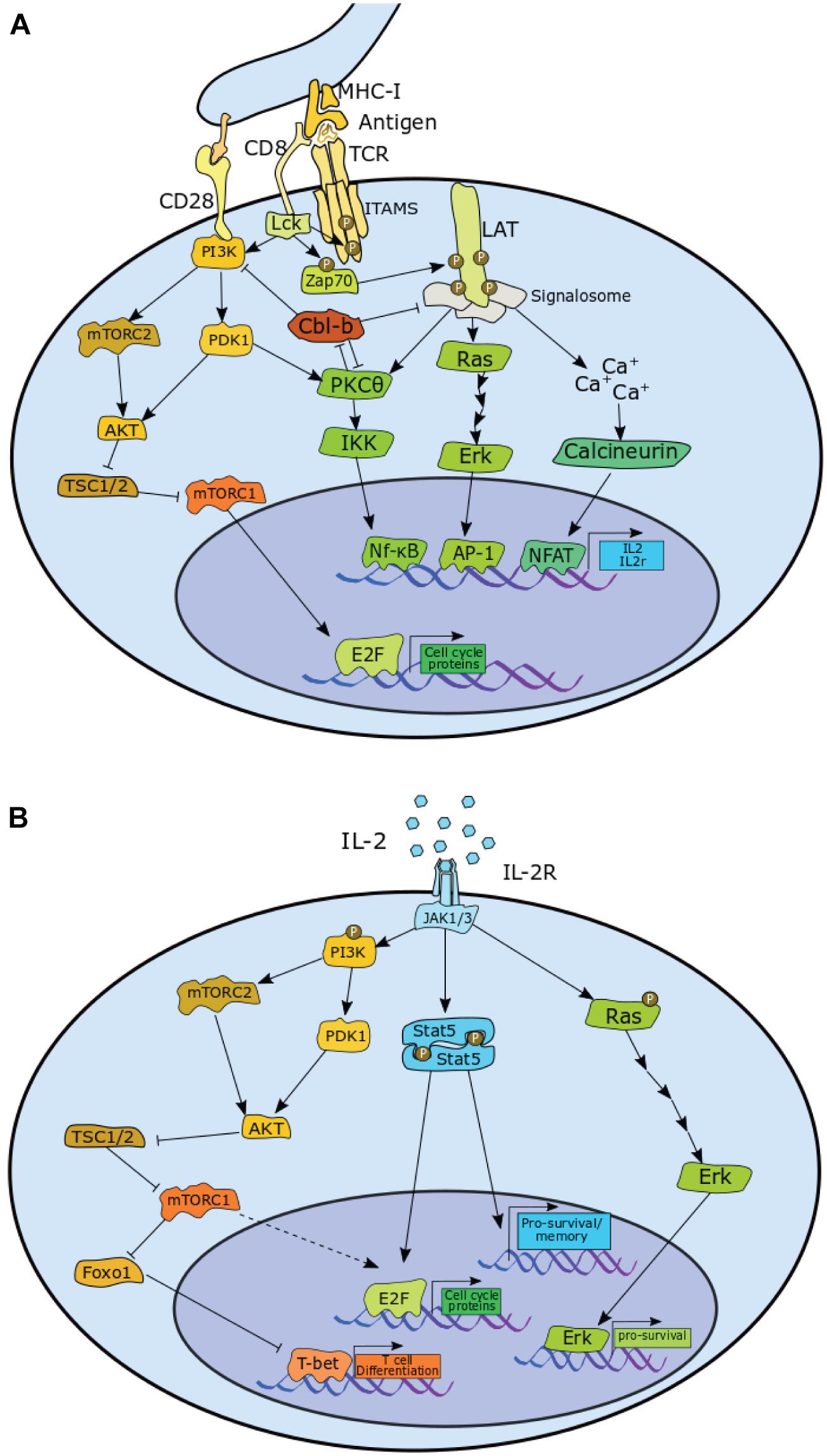
Figure 2. Pathways in CD8+ T cells that contribute to the proliferation via upregulation of E2F and pro-survival signals, or contribute toward differentiation in both (A) antigen induced activation phase and (B) the IL-2 mediated expansion phase CD8+ T cells. P, phosphorylation. A detailed description is provided in the main text.
Protein tyrosine kinase positively regulates phosphoinositide-dependent kinase 1 (PDK1) activity, which stimulates two major pathways: PKCθ/IKK/Nf-κB pathway and the Ak strain transforming (AKT)/ mammalian target of rapamycin complex 1 (mTORC1) pathway. Lck mediates this via activation of phosphoinositide 3-Kinase (PI3K), which is enhanced by co-receptor CD28 stimulation. PI3K is also responsible for enabling the assembly of mTOR complex 2 (mTORC2), which activates AKT, enabling it to target its substrates, such as the mTORC1 inhibitor, Hamartin (TSC1)/Tuberin (TSC2), thereby upregulating mTORC1 activity (Brownlie and Zamoyska, 2013; Jutz et al., 2016; Spolski et al., 2018; Hwang et al., 2020).
With TCR stimulation alone, however, the components of the LAT signalosome, as well as PKCθ are targeted for degradation by the E3-Ubiquitin ligase casitas B-lineage lymphoma proto-oncogene (Cbl)-b. Cbl-b also has the effect of reducing PI3K activity by preventing formation of phosphatidylinositol-3-phosphate (PIP3). The net effect of this is a sharp reduction in NFAT, NFκB, AP-1, and mTOR activity, resulting in the cell becoming anergic (Liu et al., 2014).
CD28 stimulation counteracts this effect by enhancing PKCθ activity, which promotes degradation of Cbl-b. Indeed, loss of PKCθ activity further decreases transcription factor activity downstream of LAT (Gruber et al., 2009), while knockout of CD28 leads to a reduction in proliferation (Li et al., 2004). Conversely, both Cbl-b deletion and Cbl-b inactive mutation enables T cell proliferation and production of IL-2 in the absence of CD28 signal. Loss of Cbl-b activity greatly enhances CD28-mediated proliferation and IL-2 production (Paolino et al., 2011). Consistent with a role in negatively regulating activation, Cbl-b also emerged as a top hit in a CRIPSR genetic screen for regulators of secondary activation in human CD8+ T cells (Shifrut et al., 2018).
Interestingly, there is no requirement for Lck in the activation of memory T cells upon secondary infection by LCMV in vivo. The frequency of antigen-specific T cells after secondary challenge to antigen was unaffected in memory cells depleted of Lck compared to Lck-replete cells, suggesting that proliferation is unaffected by Lck depletion. Re-challenged Lck-depleted cells also produced comparable levels of interferon (IFN)-γ to that of Lck-replete cells. This is in direct contrast to naïve cells depleted of Lck, which do not proliferate in response to antigen (Tewari et al., 2006). In addition, ex vivo memory T cells seem more reliant on cytokine signalling to induce proliferation, with antigen and CD28 stimulation alone proving insufficient to trigger proliferation in memory T cells (Cho et al., 1999).
Mammalian Target of Rapamycin Complex, Cell Growth, and E2 Factor F
T cell receptor/CD28 stimulation of naïve cells is sufficient to initiate proliferation. TCR/CD28 stimulation leads to phosphorylation of Rb, expression of cyclin E, cyclin A, and CDK2, and the degradation of p27 (Appleman et al., 2000). Rapamycin, an inhibitor of mTOR, induces a severe delay in proliferation (D’Souza and Lefrançois, 2003), resulting in decreased levels of cyclin D3 and cyclin E. And while cyclin D3 levels recover 3–5 days following rapamycin treatment, cyclin E remains low, suggesting a dependence on mTOR for antigen-induced E2F activation. Although the exact mechanism is unknown, expression of a rapamycin resistant mutant of p70S6k (also known as S6K) rescues E2F activity in rapamycin-treated cells; thereby demonstrating that mTOR activity promotes cell cycle progression by activating E2F through S6K (Brennan et al., 1999). In contrast to wild-type cells, proliferation of IL-2–/– cells is arrested completely by rapamycin. In addition, stimulator of interferon genes (STING) activity reduces levels of cyclins A and E, and Cdk1, leading to a reduction in activation induced proliferation. STING inhibits mTOR, reduces S6K activity, reduces phosphorylation of eukaryotic translation initiation factor 4E (eIF4E)-binding protein (4E-BP1), and reduces STAT5 phosphorylation, leading to decreased levels of S-phase promoting proteins (Imanishi et al., 2019). Together these results suggest that IL-2 signalling contributes to proliferation in an mTOR-independent manner as outlined in Figure 2B. Interestingly, mTOR inhibition does not delay cells in the expansion phase (Colombetti et al., 2006).
Canonically there are three E2F isoforms which promote proliferation, E2F1-3, and that activation of these transcription factors are essential to cell cycle progression (Wu et al., 2001). However, it has been shown that in CD8+ T cells, E2F1-3 can have both positive and negative effects on cell cycle progression. Single knockout of either E2F1 or E2F2 results in reduction in proliferation, while a double knockout yields a hyperproliferative phenotype and reduced antigenic threshold (Murga et al., 2001; Zhu et al., 2001). In addition to this, CD8+ T cells deficient in E2F1 show much reduced activation induced cell death (Gao et al., 2004). CD8+ T cells are not the only cell line in which E2F behaves in a non-canonical fashion. In retinal cells Myc mediated proliferation has been shown to persist even in the absence of E2F1–3 proving these E2Fs to be largely non-essential for these cells (Chen et al., 2009), while in progenitor cell lines approaching terminal differentiation, E2F1-3 seem to have a cell cycle repressive role upon forming a complex with Rb (Chong et al., 2009). It is, however, difficult to say whether similar processes are occurring within CD8+ T cells post activation as the precise molecular regulators of E2F transcription targets have yet to be fully explored.
mTOR also upregulates the c-Myc transcription factor, which has a crucial role in T cell metabolism (Vartanian et al., 2011; Wang et al., 2011; Waickman and Powell, 2012) and expression of several cell cycle regulators, including p27, cyclins, and CDKs (Dang et al., 2006). T cells lacking c-Myc have defects in glycolysis, glutaminolysis, cell growth, and proliferation. Indeed, compared to wild type, c-Myc knockout cells show decreased levels of CDK4, CDK2, and Cdc25A (Wang et al., 2011). Furthermore, Raptor deficient cells that have impaired mTORC1 activity show reduced levels of key transcription factors including Myc, but also GA-binding protein alpha chain (Gabpa), yin yang (YY)1, and Sterol Regulatory Element Binding Transcription Factor (Srebf)1, all of which are associated with mitochondrial function (Tan et al., 2017).
Cyclin-Dependent Kinase Regulation of T Cell Activation
CDKi proteins interfere with the pathways downstream of TCR stimulation. p27 is a key negative regulator of IL-2 production in anergic CD4+ T cells (Boussiotis et al., 2000). Ectopic expression of p27, but not p21, suppresses IL-2 production in CD4+ T cells. Depletion of p27 in stimulated Jurkat T cells enhances transcription of an IL-2 luciferase reporter construct and enhanced cellular AP-1 activity. The proposed mechanism is that p27 directly binds JAB1/COPS5, a subunit of the COP9 signalosome and positive regulates AP-1, inducing translocation of AP-1 from the nucleus to the cytoplasm (Tomoda et al., 1999), thereby suppressing IL-2 transcription (Boussiotis et al., 2000). Whether this effect extends to CD8+ T cells is yet to be determined.
There is significant interest in the roles of CDK4/6 in immunomodulation, and how small molecule inhibitors of CDK4/6 affect immune cell phenotypes. Recent data using CDK4/6 inhibitors (CDK4/6i) suggest a direct role of Cyclin-CDKs in controlling T cell differentiation. Three CDK4/6i are FDA-approved for HR+ breast cancer: palbociclib (PD-0332991), abemaciclib (LY2835219), and ribociclib (LEE011). Numerous clinical trials are ongoing for the treatment of other solid tumours (Álvarez-Fernández and Malumbres, 2020). Extensive reviews on this topic already exist and lie outside the scope of this review (Chaikovsky and Sage, 2018; Ameratunga et al., 2019). Interestingly, recent evidence suggests these inhibitors function similarly to the INK4 family of proteins by binding to monomeric CDK4/6 (Guiley et al., 2019). Proliferation of CD4+ Treg cells is inhibited by CDK4/6 inhibition whereas proliferation of CD8+ cells is relatively unaffected (Goel et al., 2017). Indeed, CD8+ T cell function can be augmented by CDK4/6 inhibition. Treatment of CD8+ T cells with CDK4/6 inhibitors enhances NFAT activity, increasing expression of CD25 and production of IL-2 and granzyme B, indicating that CDK6 may have a role in limiting T cell effector differentiation (Deng et al., 2018). Indeed, loss of CDK6 in T cells led to an increase in IL-2 production, and was also seen to impair type I interferon signalling events and increased metabolic processes resulting in enhanced ATP production and maximal respiration in addition to affecting proliferation (Klein et al., 2021).
In summary, T cell activation produces a signalling cascade that triggers a change in gene expression that promotes cell growth, increased anabolic metabolism, and stimulation of nascent CDK/E2F activities. The significant rewiring of the proteome and metabolism is important for supporting the next phase of CD8+ T cell differentiation, in which activated cells rapidly proliferate to clonally expand antigen-specific CTLs.
Expansion Phase (Il-2 Driven Proliferation)
A key regulator of CD8+ T cell proliferation in the expansion phase is IL-2, a cytokine and potent T cell mitogen (Smith and Ruscetti, 1981) that is produced primarily by activated CD4+ and CD8+ T cells (Nelson, 2004). Upon activation, CD8+ T cells secrete IL-2 and express a high affinity IL-2 receptor subunit, CD25. Blockade of IL-2 leads to reduced proliferation (Mishima et al., 2017). While TCR stimulation alone has been shown to be sufficient to induce proliferation of CD8+ T cells, the absence of IL-2 signal results in suboptimal expansion and cell cycle slower progression (D’Souza and Lefrançois, 2003). In addition, the presence of IL-2 in culture reduces the minimum threshold of TCR signalling required to enter cell cycle (Au-Yeung et al., 2017).
Continuous exposure to IL-2 is essential for prolonged expansion. IL-2 starvation leads to reduced cell viability, loss of CD25 expression, and decreased cellular protein content, including many major cell cycle regulators: cyclin D2, cyclin D3, cyclin A, cyclin B1, cyclin B2, CDK4, CDK6, CDK1, p15, and p21. Notably, p27 is one of the few proteins to increase as a result of IL-2 starvation (Rollings et al., 2018). p27 is implicated in controlling cell cycle exit in the contraction phase. In vivo experiments utilising p27 KO CD8+ T cells continued to expand up to day 11 post immunisation, while WT began contraction by day 8. The effect of p27 is more pronounced for MPECs than SLECs. MPEC cells maintained their higher populations as late as 30 days post immunisation, while SLEC p27 knockouts generally returned to similar cell numbers as WT cells by day 15. This difference in response is likely due to MPECs expressing higher pro-survival factors and having a greater capacity for IL-2 production than SLECs (Singh et al., 2010).
Whereas studies consistently show that p27–/– mice exhibit splenic and thymic hyperplasia (Fero et al., 1996; Kiyokawa et al., 1996; Nakayama et al., 1996), the data for p21–/– mice are conflicting (Balomenos et al., 2000). Activated p21–/–, p27–/– double knockout (DKO) splenocytes are hyperproliferative in vitro. Freshly isolated splenocytes from p21–/– but not p27–/– mice, had a higher frequency of CD25 expressing naïve CD4+ and CD8+ T cells. The percentage of CD25+ cells in activated DKO or p27–/– splenocyte cultures were elevated by ∼10% relative to wild type, or to p21–/– (Wolfraim et al., 2004), which may enhance T cell sensitivity to IL-2, and therefore mitogen-induced proliferation.
Downstream of IL-2 receptor activation is the STAT5 pathway (Moriggl et al., 1999a), which is known to positively regulate proliferation. STAT5 activation induces homeostatic proliferation in naïve CD8+ T cells, even in the absence of cytokine stimulation (Burchill et al., 2003), and in activated T cells, like mTOR, STAT5 enhances E2F activity by activating S6K (Lockyer et al., 2007). STAT5 KO T cells have reduced cell cycle regulators, including cyclin D2, cyclin D3, cyclin E, cyclin A, and CDK6 (Moriggl et al., 1999b) while inhibition of STAT5 results in a dose dependent loss of Cyclin E (Lai et al., 2009). All of these proteins play important roles in G1-S phase progression. STAT5 signalling upregulates levels of pro-survival proteins Bcl-2 and Bcl-XL. Additionally, STAT5 upregulates c-Myc, further implicating it as an important mediator of CD8+ T cell proliferation (Lord et al., 2000). Constitutively active STAT5 promotes homeostatic proliferation and survival of CD8+ T cells. Interestingly, STAT5 also enhances presence of T memory cell phenotype, increasing the frequency of cells expressing key memory markers including CD127, CD122, CD62L, and Bcl-2 (Hand et al., 2010).
As previously noted, CDK4/6 inhibitors are shown to enhance CD25 expression during activation. However, post expansion phase, cells that have been previously exposed to the inhibitors produce a greater pool of memory T cells by upregulating Max Dimerization Protein (Mxd)4, a negative regulator of Myc/Max formation (Heckler et al., 2021). Interestingly, this event seems to be independent of cell cycle arrest, suggesting a role for CDK4/6 in affecting T cell development (Heckler et al., 2021; Lelliott et al., 2021).
mTOR is also upregulated by IL-2 via the PI3K/AKT pathway. However, other pathways contribute more strongly to T cell proliferation in the expansion phase, as inhibiting mTOR in this phase does not arrest proliferation (Howden et al., 2019). Similarly, naïve CD8+ treated with AKT inhibitors responded less to stimulation, with proliferation severely reduced by day 2 of the culture in a dose dependent manner (Cho et al., 2013). If treated 3 days post activation, however, AKT inhibition resulted in little impact on proliferation (Macintyre et al., 2011).
Conversely, T cells expressing constitutively activated AKT were defective in memory T cell development in vivo. These cells have reduced CD127, CD122, CD62L, and Bcl-2 (Hand et al., 2010). CD127 reduction is predominately controlled by PI3K/AKT activity in a STAT5 independent way, as STAT5 knockout cells had little impact on CD127 (Xue et al., 2002). Constitutively active AKT cells also showed poor STAT5 phosphorylation in response to any cytokine stimulation, and as a result had a defect in homeostatic proliferation.
Taken together, these data suggest that a principal role of the IL2R/PI3K/AKT/mTOR pathway is the differentiation of T cells toward the SLEC phenotype, which occurs in parallel to IL2R/STAT5. The latter is the dominant pathway that promotes upregulation of cell cycle regulatory proteins and therefore proliferation of CD8+ T cells.
Cyclin E/A-CDK2 complexes phosphorylate numerous substrates, including targets outside of canonical cell cycle pathways that directly regulate DNA replication and mitosis. For example, cyclin E/A-CDK2 phosphorylates and inhibits Foxo1 within a number of cancer cell lines (Huang et al., 2006). In T cells, mTOR drives differentiation toward SLEC phenotype by phosphorylation of Foxo1, an inhibitor of SLEC phenotype transcription factor T-box expressed in T cells (T-bet) (Rao et al., 2010; Michelini et al., 2013; Pollizzi et al., 2015). Thus, CDK2-mediated inhibition of Foxo1 raises the intriguing possibility that CDK2 may also have a role in enhancing T-bet activity, and thus provide a mechanism by which high CDK2 expression can enhance differentiation. However, this has yet to be demonstrated in T cells.
The Cell Cycle Control Protein Network in T Cells
T cell activation and their differentiation into effector and memory cells provides exemplar systems to study the G0 to G1 transition in non-immortalised cells in the physiological context of the adaptive immune response. The proteomes of these cells have been characterised to high depth using mass spectrometry-based proteomics, enabling detailed measurements of protein copies per cell in naïve and activated murine CD8+ T cells (Howden et al., 2019; Marchingo et al., 2020). The Immunological Proteomic Resource (ImmPRes) is an open access public resource consolidating proteomics experiments carried out on murine immune cell populations, including effector and memory CD8+ T cell states.1 We therefore used these publicly available data to evaluate the copies of key cell cycle proteins in naïve, activated (24 h), CTL (IL-2), and memory (IL-15) cell populations (Figure 3).
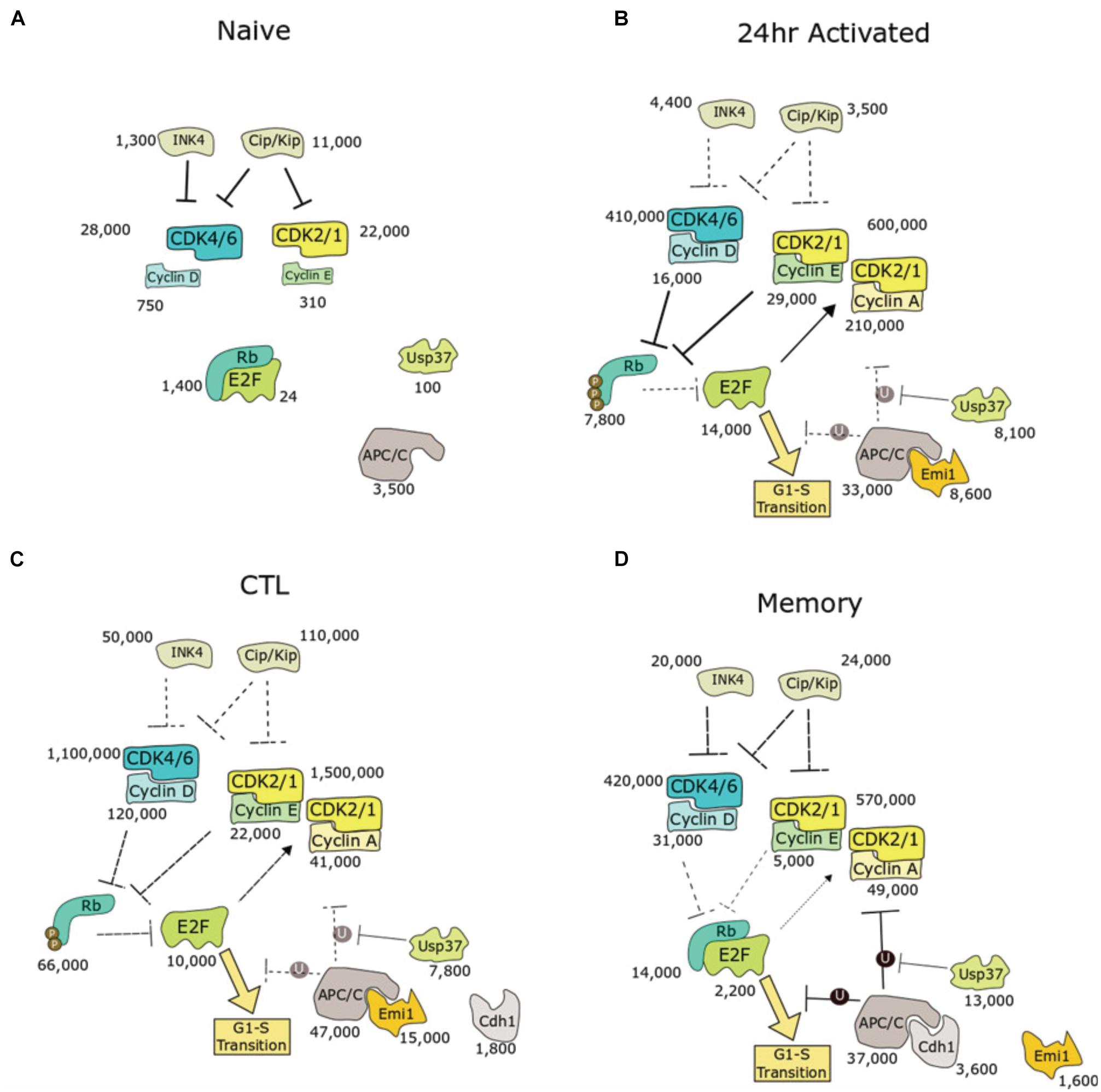
Figure 3. Biochemical pathway analysis of cell cycle entry in murine CD8+ T cells with protein copies measured by quantitative proteomics. Four populations were presented: (A) quiescent naive, (B) activation phase, (C) expansion phase toward SLEC phenotype, or (D) expansion phase toward MPEC phenotype. Numbers indicate copy number of each protein as determined by quantitative proteomics, acquired from Immpress (“ImmPRes.co.uk,” University of Dundee). For protein families (e.g., INK4, CIP/KIP, cyclins, and CDKs), summed copies are shown. E2F copies represent the sum of E2Fs 1–3. Median subunit copy number is shown for the APC/C. Mechanisms predicted to be dominant are highlighted in bold lines, whereas suppressed pathways are indicated by dotted lines. P, phosphorylation; U, ubiquitination.
These data show that naïve cells have proportionally higher numbers of CDKis for each CDK (CIP/KIP and INK4) and very low abundance of cyclins. For example, naïve cells express 3,800 copies of p27 and less than 1,000 copies each of cyclin D2 and cyclin D3. In addition, levels of activating E2F transcription factors (20 copies) are two orders of magnitude lower than their stoichiometric inhibitor, Rb (1,400 copies). E2F activity is therefore strongly inhibited, which maintains naïve cells in quiescence. Interestingly, of the CIP/KIP family of proteins, p27 is higher expressed than p21 except during IL-2 mediated expansion, supporting a role of p27 over p21 as a regulator of quiescence and homeostatic proliferation in T cells.
Upon activation, E2F levels increase 500-fold to 14,000 copies, contributing to a gene expression programme that promotes G1/S progression. For example, levels of CDK1/2/4/6, cyclin D, cyclin E, and cyclin A are all upregulated. Intriguingly, while Rb levels increase in activated T cells to 7,800 copies. There is a 3-fold increase in cyclin D2, and 8-fold increase in cyclin D3, and around a 30-fold increase in CDK4/6 copies, while p27 decreases 19-fold. Interestingly, CD8+ T cells can bypass CDK4/6 inhibition (Goel et al., 2017), leading to slowed proliferation (Heckler et al., 2021; Lelliott et al., 2021), but not arrest. The major changes observed copies in the Rb-E2F pathway may contribute to this resistance to G1 arrest by CDK4/6 inhibitors.
Inactivation of the APC/C is another mechanism to promote G1/S transition (Cappell et al., 2018). Median copies of the core APC/C subunits increase from 3,500 copies in naïve to 33,000 copies in activated T cells. Interestingly, Cdh1, the co-activator and substrate adaptor subunit of APC/C during G1, is undetectable in naïve and activated T cells. Likewise, Emi1 is undetectable in naïve. However, Emi1, an E2F target, increases to 8,600 copies in activated T cells.
Interleukin-2-mediated differentiation into CTLs is accompanied by a nearly 10-fold increase in Rb levels relative to 24 h activated T cells. Unlike newly activated T cells, which have nearly 2-fold excess E2F to Rb, CTLs contain far higher copies of Rb (66,000) to E2F (10,000). Thus, in CTLs there are sufficient levels of Rb, when not phosphorylated by CDK, to suppress all copies of E2F. However, copies of cyclins and CDKs are much higher in CTLs, leading to high activity of CDKs, which phosphorylate and inactivate Rb. These two mechanisms of E2F activation, Rb phosphorylation and excess E2F, suggest that newly activated T cells and CTLs may have differential sensitivity to pharmacological inhibition of CDK2/4/6.
Memory T cells produced by culture with IL-15 proliferate slower. In contrast to CTLs, memory T cells have reduced levels of cyclins and CDK2/4/6. However, like CTLs, copies of Rb (14,000) exceed copies of E2F (2,200). This may make memory T cells more sensitive toward Rb-mediated inhibition of E2F activity compared to naïve and 24-activated T cells. Memory T cells have increased copies of APC-C/Cdh1 (3,600) compared to CTLs, which also may contribute to extending G1 phase by keeping cyclin levels low.
Concluding Remarks
Immune cells play critical roles in normal physiology, pathology, and the development of new therapies to treat disease. Understanding fundamental mechanisms for how proliferation is controlled in T cells will be important in making advances in these areas of immunological research. This is highlighted by recent studies showing that CDK4/6 inhibitors affect proliferation of CD4+ Treg cells, but not CTLs (Goel et al., 2017), and enhance functions of checkpoint-activated CD8+ T cells (Deng et al., 2018). These results suggest that there are major differences between CD4+ and CD8+ T cells in G1 control, which will be interesting to explore in future studies. Deeper insights into how the cell cycle control network is structured in different immune subsets may provide important clues into the mode of action of these inhibitors on immune cells and an opportunity to identify new targets for modulating immune cell proliferation and function.
Author Contributions
DL prepared the figures. DL and TL wrote the manuscript. Both authors contributed to the article and approved the submitted version.
Funding
TL was supported by a Sir Henry Dale Fellowship funded by the Wellcome Trust and the Royal Society (206211/A/17/Z). DL was supported by a BBSRC EASTBIO Ph.D. Studentship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the Ly, Cantrell, and Zamoyska groups for helpful discussions.
Footnotes
References
Álvarez-Fernández, M., and Malumbres, M. (2020). Mechanisms of sensitivity and resistance to CDK4/6 inhibition. Cancer Cell 37, 514–529.
Ameratunga, M., Kipps, E., Okines, A. F. C., and Lopez, J. S. (2019). To cycle or fight—CDK4/6 inhibitors at the crossroads of anticancer immunity. Clin. Cancer Res. 25, 21–28.
Appleman, L. J., Berezovskaya, A., Grass, I., and Boussiotis, V. A. (2000). CD28 costimulation mediates T cell expansion via IL-2-independent and IL-2-dependent regulation of cell cycle progression. J. Immunol. 164, 144–151. doi: 10.4049/jimmunol.164.1.144
Au-Yeung, B. B., Smith, G. A., Mueller, J. L., Heyn, C. S., Jaszczak, R. G., Weiss, A., et al. (2017). IL-2 modulates the TCR signaling threshold for CD8 but Not CD4 T cell proliferation on a single-cell level. J. Immunol. 198, 2445–2456. doi: 10.4049/jimmunol.1601453
Balomenos, D., Martín-Caballero, J., García, M. I., Prieto, I., Flores, J. M., Serrano, M., et al. (2000). The cell cycle inhibitor P21 controls T-cell proliferation and sex- linked lupus development. Nat. Med. 6, 171–176. doi: 10.1038/72272
Boussiotis, V. A., Freeman, G. J., Taylor, P. A., Berezovskaya, A., Grass, I., Blazar, B. R., et al. (2000). P27(Kip1) functions as an anergy factor inhibiting interleukin 2 transcription and clonal expansion of alloreactive human and mouse helper T lymphocytes. Nat. Med. 6, 290–297. doi: 10.1038/73144
Brennan, P., Babbage, J. W., Thomas, G., and Cantrell, D. (1999). P70s6k integrates phosphatidylinositol 3-kinase and rapamycin-regulated signals for E2F regulation in T lymphocytes. Mol. Cell. Biol. 19, 4729–4738. doi: 10.1128/mcb.19.7.4729
Brownlie, R. J., and Zamoyska, R. (2013). T cell receptor signalling networks: branched, diversified and bounded. Nat. Rev. Immunol. 13, 257–269.
Burchill, M. A., Goetz, C. A., Prlic, M., O’Neil, J. J., Harmon, I. R., Bensinger, S. J., et al. (2003). Distinct effects of STAT5 activation on CD4 + and CD8 + T Cell homeostasis: development of CD4 + CD25 + regulatory T cells versus CD8 + memory T cells. J. Immunol. 171, 5853–5864. doi: 10.4049/jimmunol.171.11.5853
Cappell, S. D., Chung, M., Jaimovich, A., Spencer, S. L., and Meyer, T. (2016). Irreversible APCCdh1 inactivation underlies the point of no return for cell-cycle entry. Cell 166, 167–180. doi: 10.1016/j.cell.2016.05.077
Cappell, S. D., Mark, K. G., Garbett, D., Pack, L. R., Rape, M., and Meyer, T. (2018). EMI1 switches from being a substrate to an inhibitor of APC/CCDH1 to start the cell cycle. Nature 558, 313–317. doi: 10.1038/s41586-018-0199-7
Carpenter, A. C., and Bosselut, R. (2010). Decision checkpoints in the thymus. Nat. Immunol. 11, 666–673.
Chaikovsky, A. C., and Sage, J. (2018). Beyond the cell cycle: enhancing the immune surveillance of tumors via CDK4/6 inhibition. Mol. Cancer Res. 16, 1454–1457.
Chen, D., Pacal, M., Wenzel, P., Knoepfler, P. S., Leone, G., and Bremner, R. (2009). Division and apoptosis in the E2f-Deficient retina. Nature 462:925. doi: 10.1038/NATURE08544
Cheng, M., Olivier, P., Diehl, J. A., Fero, M., Roussel, M. F., Roberts, J. M., et al. (1999). “The P21(Cip1) and P27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 18, 1571–1583. doi: 10.1093/emboj/18.6.1571
Cho, B. K., Wang, C., Sugawa, S., Eisen, H. N., and Chen, J. (1999). Functional differences between memory and naive CD8 T Cells. Proc. Natl. Acad. Sci. U.S.A. 96, 2976–2981. doi: 10.1073/pnas.96.6.2976
Cho, J.-H., Kim, H.-O., Kim, K.-S., Yang, D.-H., Surh, C. D., and Sprent, J. (2013). Unique features of naive CD8 + T Cell activation by IL-2. J. Immunol. 191, 5559–5573. doi: 10.4049/jimmunol.1302293
Chong, J.-L., Wenzel, P. L., Sáenz-Robles, M. T., Nair, V., Ferrey, A., Hagan, J. P., et al. (2009). E2F1-3 switch from activators in progenitor cells to repressors in differentiating cells. Nature 462:930. doi: 10.1038/NATURE08677
Colombetti, S., Basso, V., Mueller, D. L., and Mondino, A. (2006). Prolonged TCR/CD28 engagement drives IL-2-independent t cell clonal expansion through signaling mediated by the mammalian target of rapamycin. J. Immunol. 176, 2730–2738. doi: 10.4049/jimmunol.176.5.2730
Dang, C. V., O’Donnell, K. A., Zeller, K. I., Nguyen, T., Osthus, R. C., and Li, F. (2006). The C-Myc target gene network. Semin. Cancer Biol. 16, 253–264.
Deng, J., Wang, E. S., Jenkins, R. W., Li, S., Dries, R., Yates, K., et al. (2018). CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 8, 216–233. doi: 10.1158/2159-8290.CD-17-0915
D’Souza, W. N., and Lefrançois, L. (2003). IL-2 Is not required for the initiation of CD8 T cell cycling but sustains expansion. J. Immunol. 171, 5727–5735. doi: 10.4049/jimmunol.171.11.5727
Fero, M. L., Rivkin, M., Tasch, M., Porter, P., Carow, C. E., Firpo, E., et al. (1996). A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in P27Kip1-deficient mice. Cell 85, 733–744. doi: 10.1016/S0092-8674(00)81239-8
Gao, X., Tewari, K., Svaren, J., and Suresh, M. (2004). Role of cell cycle regulator E2F1 in regulating CD8 T Cell responses during acute and chronic viral infection. Virology 324, 567–576. doi: 10.1016/J.VIROL.2004.04.012
Goel, S., DeCristo, M. J., Watt, A. C., BrinJones, H., Sceneay, J., Li, B. B., et al. (2017). CDK4/6 inhibition triggers anti-tumor immunity. Nature 548:471. doi: 10.1038/NATURE23465
Grimmler, M., Wang, Y., Mund, T., Cilenšek, Z., Keidel, E. M., Waddell, M. B., et al. (2007). Cdk-inhibitory activity and stability of P27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell 128, 269–280. doi: 10.1016/j.cell.2006.11.047
Gruber, T., Hermann-Kleiter, N., Hinterleitner, R., Fresser, F., Schneider, R., Gastl, G., et al. (2009). PKC-θ modulates the strength of t cell responses by targeting Cbl-b for ubiquitination and degradation. Sci. Signal. 2, ra30–ra30. doi: 10.1126/scisignal.2000046
Guiley, K. Z., Stevenson, J. W., Lou, K., Barkovich, K. J., Kumarasamy, V., Wijeratne, T. U., et al. (2019). P27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 366:6471. doi: 10.1126/science.aaw2106
Hand, T. W., Cui, W., Woo Jung, Y., Sefik, E., Joshi, N. S., Chandele, A., et al. (2010). Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc. Natl. Acad. Sci. U. S. A. 107, 16601–16606. doi: 10.1073/pnas
Heckler, M., Ali, L. R., Clancy-Thompson, E., Qiang, L., Ventre, K. S., Lenehan, P., et al. (2021). Inhibition of CDK4/6 promotes CD8 T cell memory formation 1 2. Cancer Discov. [Epub ahead of print]. doi: 10.1158/2159-8290.CD-20-1540
Howden, A. J. M., Hukelmann, J. L., Brenes, A., Spinelli, L., Sinclair, L. V., Lamond, A. I., et al. (2019). Quantitative analysis of T cell proteomes and environmental sensors during T cell differentiation. Nat. Immunol. 20, 1542–1554. doi: 10.1038/s41590-019-0495-x
Huang, H., Regan, K. M., Lou, Z., Chen, J., and Tindall, D. J. (2006). CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science (80-.). 314, 294–297. doi: 10.1126/science.1130512
Huang, X. D., Summers, M. K., Pham, V., Lill, J. R., Liu, J., Lee, G., et al. (2011). Deubiquitinase USP37 Is activated by CDK2 to antagonize APCCDH1 and promote S phase entry. Mol. Cell 42, 511–523. doi: 10.1016/j.molcel.2011.03.027
Hwang, J. R., Byeon, Y., Kim, D., and Park, S. G. (2020). Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp. Mol. Med. 52, 750–761.
Imanishi, T., Unno, M., Kobayashi, W., Yoneda, N., Matsuda, S., Ikeda, K., et al. (2019). Reciprocal regulation of STING and TCR signaling by MTORC1 for T-cell activation and function. Life Sci Alliance 2:e201800282. doi: 10.26508/lsa.201800282
Jutz, S., Leitner, J., Schmetterer, K., Doel-Perez, I., Majdic, O., Grabmeier-Pfistershammer, K., et al. (2016). Assessment of costimulation and coinhibition in a triple parameter T cell reporter line: simultaneous measurement of NF-K B, NFAT and AP-1. J. Immunol. Methods 430, 10–20. doi: 10.1016/j.jim.2016.01.007
Kim, P. S., and Ahmed, R. (2010). Features of responding T cells in cancer and chronic infection. Curr. Opin. Immunol. 22, 223–230. doi: 10.1016/j.coi.2010.02.005
Kinjyo, I., Qin, J., Tan, S.-Y., Wellard, C. J., Mrass, P., Ritchie, W., et al. (2015). Real-time tracking of cell cycle progression during CD8+ effector and memory T-cell differentiation. Nat. Commun. 6:6301. doi: 10.1038/ncomms7301
Kiyokawa, H., Kineman, R. D., Manova-Todorova, K. O., Soares, V. C., Huffman, E. S., Ono, M., et al. (1996). Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of P27Kip1. Cell 85, 721–732. doi: 10.1016/S0092-8674(00)81238-6
Klein, K., Witalisz-Siepracka, A., Gotthardt, D., Agerer, B., Locker, F., Grausenburger, R., et al. (2021). T cell-intrinsic CDK6 Is dispensable for anti-viral and anti-tumor responses in vivo. Front. Immunol. 12:650977. doi: 10.3389/FIMMU.2021.650977
Kramer, E. R., Scheuringer, N., Podtelejnikov, A. V., Mann, M., and Peters, J. M. (2000). Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol. Biol. Cell 11, 1555–1569. doi: 10.1091/mbc.11.5.1555
Kretschmer, L., Flossdorf, M., Mir, J., Cho, Y. L., Plambeck, M., Treise, I., et al. (2020). Differential expansion of t central memory precursor and effector subsets is regulated by division speed. Nat. Commun. 11, 1–12. doi: 10.1038/s41467-019-13788-w
Kurts, C., Kosaka, H., Carbone, F. R., Miller, J. F. A. P., and Heath, W. R. (1997). Class I-restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8+ T cells. J. Exp. Med. 186, 239–245. doi: 10.1084/jem.186.2.239
Labaer, J., Garrett, M. D., Stevenson, L. F., Slingerland, J. M., Sandhu, C., Chou, H. S., et al. (1997). New functional activities for the P21 family of CDK inhibitors. Genes Dev. 11, 847–862. doi: 10.1101/gad.11.7.847
Lai, Y.-P., Lin, C.-C., Liao, W.-J., Tang, C.-Y., and Chen, S.-C. (2009). CD4 + T Cell-derived IL-2 signals during early priming advances primary CD8 + T cell responses. PLoS One 4:7766. doi: 10.1371/journal.pone.0007766
Lelliott, E. J., Kong, I. Y., Zethoven, M., Ramsbottom, K. M., Martelotto, L. G., Meyran, D., et al. (2021). CDK4/6 inhibition promotes anti-tumor immunity through the induction of T Cell memory. Cancer Discov. [Epub ahead of print]. doi: 10.1158/2159-8290.CD-20-1554
Li, D., Gál, I., Vermes, C., Alegre, M.-L., Chong, A. S. F., Chen, L., et al. (2004). Cutting edge: Cbl-b: one of the key molecules tuning CD28- and CTLA-4-mediated T cell costimulation. J. Immunol. 173, 7135–7139. doi: 10.4049/jimmunol.173.12.7135
Liao, W., Lin, J. X., and Leonard, W. J. (2013). Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity 38, 13–25.
Liu, Q., Langdon, W. Y., and Zhang, J. (2014). E3 ubiquitin ligase Cbl-b in innate and adaptive immunity. Cell Cycle 13, 1875–1884.
Lockyer, H. M., Tran, E., and Nelson, B. H. (2007). STAT5 is essential for Akt/P70S6 kinase activity during IL-2-induced lymphocyte proliferation. J. Immunol. 179, 5301–5308. doi: 10.4049/jimmunol.179.8.5301
Lord, J. D., McIntosh, B. C., Greenberg, P. D., and Nelson, B. H. (2000). The IL-2 receptor promotes lymphocyte proliferation and induction of the c- Myc, Bcl-2, and Bcl-x genes through the trans- activation domain of stat5. J. Immunol. 164, 2533–2541. doi: 10.4049/jimmunol.164.5.2533
Lukas, C., Sørensen, C. S., Kramer, E., Santoni-Ruglu, E., Lindeneg, C., Peters, J. M., et al. (1999). Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase-promoting complex. Nature 401, 815–818. doi: 10.1038/44611
Macintyre, A. N., Finlay, D., Preston, G., Sinclair, L. V., Waugh, C. M., Tamas, P., et al. (2011). Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity 34, 224–236. doi: 10.1016/j.immuni.2011.01.012
Marchingo, J. M., Sinclair, L. V., Howden, A. J., and Cantrell, D. A. (2020). Quantitative analysis of how Myc controls T cell proteomes and metabolic pathways during T cell activation. Elife 9:e53725. doi: 10.7554/ELIFE.53725
Mehlhop-Williams, E. R., and Bevan, M. J. (2014). Memory CD8+ T cells exhibit increased antigen threshold requirements for recall proliferation. J. Exp. Med. 211, 345–356. doi: 10.1084/jem.20131271
Michelini, R. H., Doedens, A. L., Goldrath, A. W., and Hedrick, S. M. (2013). Differentiation of CD8 memory T cells depends on foxo1. J. Exp. Med. 210, 1189–1200. doi: 10.1084/jem.20130392
Mishima, T., Fukaya, S., Toda, S., Ando, Y., Matsunaga, T., and Inobe, M. (2017). Rapid G0/1 transition and cell cycle progression in CD8+ T cells compared to CD4+ T cells following in vitro stimulation. Microbiol. Immunol. 61, 168–175. doi: 10.1111/1348-0421.12479
Mondino, A., Colombetti, S., Basso, V., and Mueller, D. L. (2006). Mammalian target of rapamycin through signaling mediated by the IL-2-independent T cell clonal expansion prolonged TCR/CD28 engagement drives. J. Immunol. 176, 2730–2738.
Moriggl, R., Sexl, V., Piekorz, R., Topham, D., and Ihle, J. N. (1999a). Stat5 activation is uniquely associated with cytokine signaling in peripheral T cells. Immunity 11, 225–230. doi: 10.1016/S1074-7613(00)80097-7
Moriggl, R., Topham, D. J., Teglund, S., Sexl, V., McKay, C., Wang, D., et al. (1999b). Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity 10, 249–259. doi: 10.1016/S1074-7613(00)80025-4
Murga, M., Fernández-Capetillo, O., Field, S., Moreno, B., Borlado, L., Fujiwara, Y., et al. (2001). Mutation of E2F2 in mice causes enhanced T lymphocyte proliferation, leading to the development of autoimmunity. Immunity 15, 959–970. doi: 10.1016/S1074-7613(01)00254-0
Nakayama, K., Ishida, N., Shirane, M., Inomata, A., Inoue, T., Shishido, N., et al. (1996). Mice lacking P27Kip1 display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 85, 707–720. doi: 10.1016/S0092-8674(00)81237-4
Nelson, B. H. (2004). IL-2, regulatory T cells, and tolerance. J. Immunol. 172, 3983–3988. doi: 10.4049/jimmunol.172.7.3983
Paolino, M., Thien, C. B. F., Gruber, T., Hinterleitner, R., Baier, G., Langdon, W. Y., et al. (2011). Essential role of E3 ubiquitin ligase activity in Cbl-b– regulated T cell functions. J. Immunol. 186, 2138–2147. doi: 10.4049/jimmunol.1003390
Pollizzi, K. N., Patel, C. H., Sun, I. H., Oh, M. H., Waickman, A. T., Wen, J., et al. (2015). MTORC1 and MTORC2 selectively regulate CD8+ T cell differentiation. J. Clin. Invest. 125, 2090–2108. doi: 10.1172/JCI77746
Rao, R. R., Li, Q., Odunsi, K., and Shrikant, P. A. (2010). The mtor kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and eomesodermin. Immunity 32, 67–78. doi: 10.1016/j.immuni.2009.10.010
Rollings, C. M., Sinclair, L. V., Brady, H. J. M., Cantrell, D. A., and Ross, S. H. (2018). Interleukin-2 shapes the cytotoxic T cell proteome and immune environment–sensing programs. Sci. Signal 11:eaa8112.
Sherr, C. J., and Roberts, J. M. (1999). CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 13, 1501–1512. doi: 10.1101/gad.13.12.1501
Shifrut, E., Carnevale, J., Tobin, V., Roth, T. L., Woo, J. M., Bui, C. T., et al. (2018). Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell 175, 1958–1971.e15. doi: 10.1016/j.cell.2018.10.024
Singh, A., Jatzek, A., Plisch, E. H., Srinivasan, R., Svaren, J., and Suresh, M. (2010). Regulation of memory CD8 T-cell differentiation by cyclin-dependent kinase inhibitor P27Kip1. Mol. Cell. Biol. 30, 5145–5159. doi: 10.1128/mcb.01045-09
Smith, K. A., and Ruscetti, F. W. (1981). T-cell growth factor and the culture of cloned functional T cells. Adv. Immunol 31, 137–175. doi: 10.1016/S0065-2776(08)60920-7
Spolski, R., Li, P., and Leonard, W. J. (2018). Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat. Rev. Immunol. 18, 648–659.
Stengel, K. R., Thangavel, C., Solomon, D. A., Angus, S. P., Zheng, Y., and Knudsen, E. S. (2009). Retinoblastoma/P107/P130 pocket proteins. proteins dynamics and interactions with target gene promoters. J. Biol. Chem. 284, 19265–19271. doi: 10.1074/jbc.M808740200
Tan, H., Yang, K., Li, Y., Shaw, T. I., Wang, Y., Blanco, D. B., et al. (2017). Integrative proteomics and phosphoproteomics profiling reveals dynamic signaling networks and bioenergetics pathways underlying T cell activation. Immunity 46, 488–503. doi: 10.1016/J.IMMUNI.2017.02.010
Tewari, K., Walent, J., Svaren, J., Zamoyska, R., and Suresh, M. (2006). Differential requirement for Lck during primary and memory CD8+ T cell responses. Proc. Natl. Acad. Sci. U.S.A. 103, 16388–16393. doi: 10.1073/pnas.0602565103
Tomoda, K., Kubota, Y., and Kato, J. Y. (1999). Degradation of the cyclin-dependent-kinase inhibitor P27(Kip1) is instigated by Jab1. Nature 398, 160–165. doi: 10.1038/18230
Vartanian, R., Masri, J., Martin, J., Cloninger, C., Holmes, B., Artinian, N., et al. (2011). AP-1 regulates cyclin D1 and c-MYC transcription in an AKT-dependent manner in response to MTOR inhibition: role of AIP4/Itch-mediated JUNB degradation. Mol. Cancer Res. 9, 115–130. doi: 10.1158/1541-7786.MCR-10-0105
Waickman, A. T., and Powell, J. D. (2012). MTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol. Rev. 249, 43–58. doi: 10.1111/j.1600-065X.2012.01152.x
Wang, H., Nicolay, B. N., Chick, J. M., Gao, X., Geng, Y., Ren, H., et al. (2017). The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature 546, 426–430. doi: 10.1038/nature22797
Wang, R., Dillon, C. P., Shi, L. Z., Milasta, S., Carter, R., Finkelstein, D., et al. (2011). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882. doi: 10.1016/j.immuni.2011.09.021
Wolfraim, L. A., Walz, T. M., James, Z., Fernandez, T., and Letterio, J. J. (2004). P21 Cip1 and P27 Kip1 act in synergy to alter the sensitivity of naive T cells to TGF-β-mediated G 1 arrest through modulation of IL-2 responsiveness. J. Immunol. 173, 3093–3102. doi: 10.4049/jimmunol.173.5.3093
Wu, L., Timmers, C., Maiti, B., Saavedra, H. I., Sang, L., Chong, G. T., et al. (2001). The E2F1–3 transcription factors are essential for cellular proliferation. Nature 414, 457–462. doi: 10.1038/35106593
Xue, H. H., Kovanen, P. E., Pise-Masison, C. A., Berg, M., Radovich, M. F., Brady, J. N., et al. (2002). Il-2 negatively regulates IL-7 receptor α chain expression in activated T lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 99, 13759–13764. doi: 10.1073/pnas.212214999
Yoon, H., Kim, T. S., and Braciale, T. J. (2010). The cell cycle time of CD8+ T cells responding in vivo is controlled by the type of antigenic stimulus. PLoS One 5:e15423. doi: 10.1371/journal.pone.0015423
Zetterberg, A., and Larsson, O. (1985). Kinetic analysis of regulatory events in G1 leading to proliferation or quiescence of swiss 3T3 cells. Proc. Natl. Acad. Sci. U.S.A. 82, 5365–5369. doi: 10.1073/pnas.82.16.5365
Zetterberg, A., and Larsson, O. (1991). Coordination between cell growth and cell cycle transit in animal cells. Cold Spring Harb. Symp. Quant. Biol. 56, 137–147. doi: 10.1101/SQB.1991.056.01.018
Zetterberg, A., Larsson, O., and Wiman, K. G. (1995). What is the restriction point? Curr. Opin. Cell Biol. 7, 835–842. doi: 10.1016/0955-0674(95)80067-0
Keywords: quiescence, T cell, proliferation, cell cycle, T cell activation
Citation: Lewis DA and Ly T (2021) Cell Cycle Entry Control in Naïve and Memory CD8+ T Cells. Front. Cell Dev. Biol. 9:727441. doi: 10.3389/fcell.2021.727441
Received: 18 June 2021; Accepted: 07 September 2021;
Published: 06 October 2021.
Edited by:
Guang Yao, University of Arizona, United StatesReviewed by:
Sander Van Den Heuvel, Utrecht University, NetherlandsJing Qu, Institute of Zoology, (CAS), China
Copyright © 2021 Lewis and Ly. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David A. Lewis, RC5BLkxld2lzLTFAc21zLmVkLmFjLnVr; Tony Ly, dGx5QGR1bmRlZS5hYy51aw==