 Eden Ozer
Eden Ozer Lital Alfonta
Lital Alfonta
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Bioeng. Biotechnol. , 26 February 2021
Sec. Synthetic Biology
Volume 9 - 2021 | https://doi.org/10.3389/fbioe.2021.594429
Escherichia coli has been considered as the most used model bacteria in the majority of studies for several decades. However, a new, faster chassis for synthetic biology is emerging in the form of the fast-growing gram-negative bacterium Vibrio natriegens. Different methodologies, well established in E. coli, are currently being adapted for V. natriegens in the hope to enable a much faster platform for general molecular biology studies. Amongst the vast technologies available for E. coli, genetic code expansion, the incorporation of unnatural amino acids into proteins, serves as a robust tool for protein engineering and biorthogonal modifications. Here we designed and adapted the genetic code expansion methodology for V. natriegens and demonstrate an unnatural amino acid incorporation into a protein for the first time in this organism.
Vibrio natriegens (V. natriegens), is a fast-growing gram-negative bacterium identified in the 1960s of the 20th century with a remarkable division time of less than 10 min (Eagon, 1962). Despite the fact that Escherichia coli (E. coli) is considered the most convenient organism for lab work, it has a division time that is roughly double that of V. natriegens (Clark and Maaløe, 1967). Considering the need to optimize the efficiency of molecular and synthetic biology studies in the lab, V. natriegens presents an unprecedented opportunity to speed-up lab work and emerges as a novel synthetic biology and biotechnological chassis. Apart from its rapid growth, V. natriegens can also reach higher cell densities than E. coli, it can tolerate a broad range of pH conditions, possess natural competence for environmental DNA receival, utilize a wide range of substrate and grow on various carbon sources, has a higher substrate uptake, can serve as a safer expression host for pathogen related proteins and express proteins in high yields. Recently, multiple studies have emerged, demonstrating the adaptation of methodologies used in E. coli, for their utilization in V. natriegens (Hoff et al., 2020; Peng et al., 2020). As such, protein expression (Weinstock et al., 2016; Schleicher et al., 2018), molecular biology (Weinstock et al., 2016; Dalia et al., 2017; Tschirhart et al., 2019), and cell-free protein expression platforms (Des Soye et al., 2018; Failmezger et al., 2018; Wiegand et al., 2018) were successfully developed in V. natriegens.
The incorporation of unnatural amino acids (Uaas) into proteins is part of the genetic code expansion experimental efforts and is considered a powerful tool in protein engineering (Xiao and Schultz, 2016). The insertion of a Uaa in a site-specific manner allows the introduction of new functions in a protein in a controlled manner and their use in research that involves fluorescence resonance energy transfer (FRET) (Ozer et al., 2017), transcription/translation kinetic analysis (Schlesinger et al., 2017), engineering of oxygen tolerant enzymes (Bhagat et al., 2020), and cellular localization (Cohen et al., 2020). To date, over 200 Uaas were incorporated into different proteins (Xiao and Schultz, 2016) in different organisms including the primary E. coli system, amongst them are Salmonella (Gan et al., 2016), cyanobacteria (Chemla et al., 2017), Yeast (Hancock et al., 2010), Caenorhabditis elegans (Greiss and Chin, 2011), mouse (Ernst et al., 2016), mammalian cells (Italia et al., 2017), and cell-free systems (Ozer et al., 2017). While evolving into a common biotechnological platform, it is yet to be adapted to V. natriegens. Genetic code expansion in V. natriegens can serve as an improved and a faster platform for future protein engineering in this new chassis, possibly for much-needed research in the organism itself but mainly as a powerhouse for production of proteins with expanded amino acid repertoire.
Propargyl-L-lysine (PrK) was purchased from Syn- Chem (Elk Grove Village, IL, United States). Tris(3-hydroxypropyltriazolylmethyl) amine (THPTA), Tetramethyl- rhodamine-azide (TAMRA-Az), and TAMRA-alkyne were purchased from Sigma-Aldrich (Rehovot, Israel). All restriction enzymes were purchased from Thermo Fisher Scientific (Waltham, MA, United States), while all DNA oligonucleotides were obtained from Syntezza Bioscience (Jerusalem, Israel).
All E. coli molecular biology experiments in this study were performed in DH5a strain while all V. natriegens molecular biology was performed in ATCC strain # 14048.
pBEST-P70b-UTR1-deGFP-6xHis-T500 was used as the basis for all plasmid designs (Schlesinger et al., 2017). All PCR generated amplicons were separated on an agarose gel and purified using nucleospin gel and PCR clean-up (Macherey-Nagel, Germany).
The final plasmid contains the Methanosarcina mazei pyrrolysine system (MmPyl), composed of MmPyl-tRNA and MmPyl tRNA-synthetase (MmPylRS), and was constructed in a two-step procedure. The first plasmid that was generated was with the addition of MmPyl-tRNA to the pBEST vector, and was named pRaGE-Pyl tRNA plasmid. tRNA native promoter and terminator regions were chosen from the V. natriegens genome and were amplified using primers 1 + 2 and 3 + 4 (Supplementary Table 2). MmPyl-tRNA was generated through overlapping segments between primers 2 and 3 using a Gibson assembly. pBEST vector amplification was performed using primers 5 + 6 (Supplementary Table 2) and DpnI restriction enzyme. Following a Gibson assembly, the assembly mix was transformed into DH5a E. coli cells using a standard heat-shock protocol and plated on selective media (100 μg/mL carbenicillin).
The next plasmid generation was pRaGE harboring the full MmPyl system named pRaGE-Pyl: MmPyl-tRNA and MmPylRS together. The design was as an extension to the previously generated pRaGE-Pyl tRNA variant. Vector amplicon was generated using primers 6 + 7 (Supplementary Table 2) and underwent DpnI treatment. Yeast origin of replication and selection marker (designed for yeast assembly) were amplified using primers 8 + 9 (Supplementary Table 2). V. natriegens native regions upstream and downstream of the glutamic acid tRNA synthetase (GluRS) were chosen to surround the MmPylRS gene. GluRS promoter and terminator were amplified from V. natriegens genome using primer 10 + 11 and 12 + 13, respectively (Supplementary Table 2). Finally, the MmPylRS fragment was amplified using primers 14 + 15 (Supplementary Table 2). Standard yeast assembly protocol (Chemla et al., 2017) was performed for all amplicons. The assembled plasmid was extracted from yeast (ZYMO research, Irvine, CA, United States) and was transformed into DH5a E. coli using a standard heat-shock protocol and plated on selective media (100 μg/mL carbenicillin).
The final plasmid assembly was performed in order to generate a 2nd pRaGE plasmid harboring the partial OTS, this time with MmPylRS only, named pRaGE-PylRS. Using the previously generated pRaGE-Pyl plasmid, containing the full OTS, the fragment was amplified using primers 5 + 16 (Supplementary Table 2), assembled by a yeast assembly. All plasmids were sequenced for proper assembly validation.
Electroporation protocol was based on a published protocol by Lee et al. (2019). Liquid culture was grown in LB3 (LB-Miller with extra salt for final NaCl concentration of 3%) at 30°C for overnight incubation. For each transformation, 100 μL of overnight culture were pelleted down and resuspended with 100 μL of fresh LB3 and added to 10 mL of LB3. Subculture was grown at 37°C for 1:10 h (approximately at a 0.4 OD600) and harvested at 4°C, 3,500 × g for 5 min. From this point on, all steps were performed on ice. Cells were resuspended with 1 M cold sorbitol and transferred into a chilled 1.5 mL Eppendorf tube where they were pelleted down at 4°C, 20,000 × g for 1 min. Cells were then washed twice more with 1 M cold sorbitol and 4°C, 20,000 × g for 1 min centrifugation. Finally, electrocompetent cells were resuspended with 50 μL 1 M cold sorbitol and used fresh for electroporation.
Approximately 100 ng of the desired plasmid were added to the electrocompetent cells and stirred gently. Following 2 min incubation on ice, cells were then transferred into a chilled 1 mm cuvette (Cell Projects Ltd., Kent, United Kingdom) and left on ice for five additional minutes. Cells were electroporated using Gene Pulser® II (Bio-Rad, Hercules, CA, United States) using the following parameters: 0.4 kV, 1 kΩ, 25 μF and were immediately recovered with 1 mL LB3 media for 1–2 h at 37°C. Cells were plated on selective LB3 agar plates carrying 100 μg/mL carbenicillin. Plates were grown at 30°C overnight.
A swab from a glycerol stock was grown in LB3 at 30°C overnight (when bacteria were harboring a plasmid, carbenicillin was added to a final concentration of 100 μL/mL). Fresh culture of 3 mL LB3 was made from previously grown stationery culture using 1:100 dilution and incubated at 30°C overnight. Cultures were supplemented with a final concentration of 1 mM PrK. Following liquid culture growth, 1 mL of the culture was lysed. The culture was then washed once with 1 mL of phosphate buffer 100 mM pH = 7. The cells were pelleted down and resuspended with 100 μL lysis solution composed of 90% phosphate buffer, 10% BugBuster® 10X protein extraction reagent (Merck, Billerica, MA, United States), Turbonuclease (Sigma, St. Louis, MO, United States), Lysozyme (Sigma-Aldrich, Rehovot, Israel) and protease inhibitor (Merck, Darmstadt, Germany). Samples were incubated at 25°C for 30 min with mild shaking, followed by 4°C centrifugation at 10,000 × g for 10 min. Supernatant was collected for further analysis.
Lysed samples were separated using SDS-PAGE 4–20% Expressplus protein gel (GeneScript, Nanjing, China). Transferring gel stamp to an Immun-Blot® PVDF membrane (Bio-Rad, Hercules, CA, United States) using eBlot® protein transfer system (GenScript, Nanjing, China), samples underwent a standard Western-blot protocol with GFP (B-2) mouse monoclonal antibody (sc-9996, Santa Cruz, CA, United States) as primary antibody and Goat anti-mouse IgG H&L (HRP) (ab97023, abcam, Cambridge, United Kingdom) as a secondary antibody. Membrane imaging was done using ImageQuant LAS 4000 imager with a maximum exposure time of 10 s (Fujifilm, Tokyo, Japan).
Three separate sets of liquid cultures were grown at 30°C for 16 h. Each set was composed of three V. natriegens strains: native strain, wild type (WT) GFP expression strain and TAG mutant GFP expression strain grown in the presence of 1 mM PrK. All samples were tested for OD600 and fluorescence (488 nm excitation and 507 nm emission) in three technical repeats, using Synergy HT plate reader (Biotek, Winooski, VT, United States). Each sample fluorescence value was divided by its OD600 value. Values of WT and mutant expression were normalized to the native strain’s value of fluorescence/OD600. Finally, the suppression efficiency of each set out of the three was determined through the value of normalized fluorescence/OD600 of mutant divided by the value of normalized fluorescence/OD600 of WT. Error bar in Figure 1D could only be calculated for mutant expression (since WT value was always set to 100% by definition), representing a standard deviation between three different suppression efficiencies values calculated to be ± 0.037%.
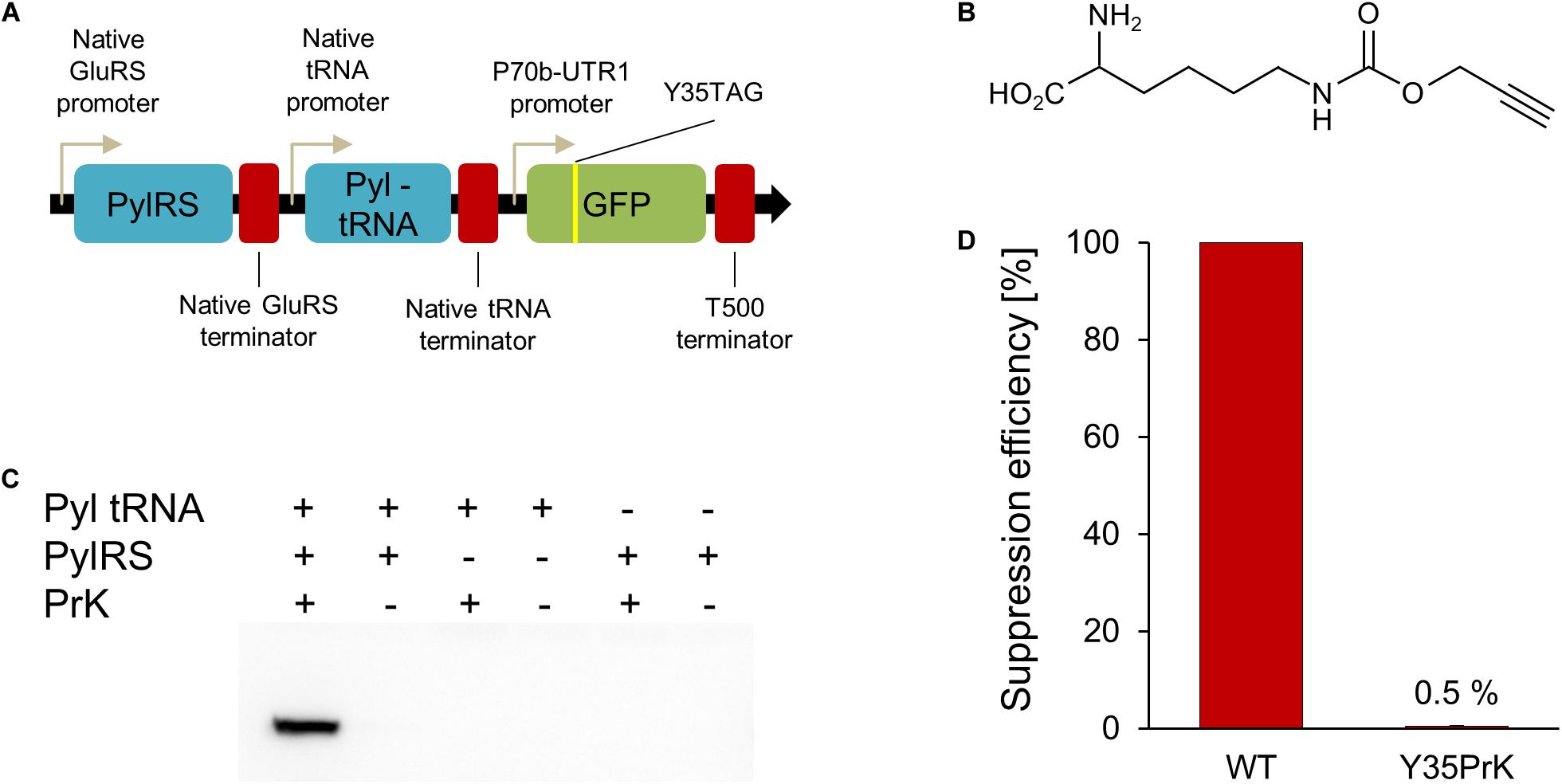
Figure 1. V. natriegens genetic code expansion system. (A) Genetic map of constructed pRaGE-Pyl. (B) Unnatural amino acid (Uaa) used in this study, propargyl-L-lysine (PrK). (C) Anti-GFP Western-blot analysis to test orthogonality and Uaa incorporation into GFP. (D) Calculated suppression efficiency for genetic code expansion in V. natriegens based on GFP Y35PrK mutant compared to WT GFP.
A 100 mL of LB3 culture of V. natriegens harboring the pRaGE-Pyl TAG-deGFP-Y35TAG-NHis plasmid, was grown at 30°C overnight. The culture was lysed using standard needle sonication, followed by IMAC purification (Novagene, Madison, WI, United States) according to manufacturer guidelines. The elution fraction was concentrated using Vivaspin 6, 10000 MWCO PES (Sartorius, Goettingen, Germany). Purification products were run on SDS-PAGE 4–20% Expressplus protein gel (GeneScript, Nanjing, China) and a standard Coomassie staining protocol.
A band of ∼27 kDa corresponding to GFP size was incised and digested in-gel by trypsin according to manufacturer’s protocol (Promega). Peptides were then extracted from the gel and loaded onto a liquid chromatography mass spectrometer (LC-MS). LC-MS analysis was performed using an Eksigent nano−HPLC (model nanoLC-2D, Netherlands) connected to an LTQ Orbitrap XL ETD (Thermo Fisher Scientific, Germany and United States). Reverse−phase chromatography of peptides was performed using a C−18 column [IntegraFritTM, 360 μm OD × 75 ID (μm); New Objective United States]. Peptides were separated by a 70 min linear gradient, starting with 100% buffer A (5% acetonitrile, 0.1% formic acid) and ending with 80% buffer B (80% acetonitrile, 0.1% formic acid), at a flow rate of 300 mL/min. A full scan, acquired at 60,000 resolution, was followed by CID MS/MS analysis performed for the five most abundant peaks, in a data−dependent mode. Fragmentation (with minimum signal trigger threshold set at 500) and detection of fragments were carried out in a linear ion trap. Maximum ion fill time settings were 500 ms for a high−resolution full scan in an Orbitrap analyzer and 200 ms for MS/MS analysis in the ion trap. The AGC settings were 5 × 105 and 1 × 104 (MS/MS) for Orbitrap and linear ion trap analyzers, respectively. Proteins were identified and validated using SEQUEST software and data sets operated under the Proteome Discoverer 2.3 software (Thermo Fisher Scientific). Mass tolerance for precursors and fragmentations was set to 10 ppm and 0.8 Da, respectively.
Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAC) click reaction (Kolb et al., 2001) was performed on purified GFP Y35PrK protein for PrK incorporation validation. Fluorescent dye was added to a concentration of 50 μM, while THPTA, sodium ascorbate, and CuCl2 were added to a final concentration of 1.2 mM, 2.5 mM, and 200 μM, respectively. A volume of 38 μL concentrated purified protein was added to the reaction, followed by 1 h incubation at room temperature with mild shaking. After the reaction, samples were examined through SDS-PAGE 4–20% Expressplus protein gel (GeneScript, Nanjing, China). Fluorescent SDS-PAGE images were obtained using Typhoon FLA 9500 imager (GE Healthcare Life Sciences, Uppsala, Sweden) 532 nm laser and LPG green filter on TAMRA setting. Following fluorescence imaging, the experiment has proceeded to anti-GFP Western-blot analysis as was described earlier.
In order to establish Uaa incorporation in proteins in an organismfor the first time, an orthogonal translation system (OTS) composed of a tRNA and tRNA-synthetase pair is needed (Xiao and Schultz, 2016). In order to confirm the orthogonality of the OTS in V. natriegens, the OTS needs to interact with a specific Uaa and to have no cross-reactivity with other components: native amino acids, endogenous tRNAs or tRNA-synthetases. The Methanosarcina mazei pyrrolysyl (MmPyl) OTS was employed and proven orthogonal in several gram-negative bacteria (Greiss and Chin, 2011; Chemla et al., 2017; Ozer et al., 2017). In addition, the MmPyl OTS was demonstrated to incorporate a large variety of Uaas and was therefore chosen to be tested in V. natriegens as well (Dumas et al., 2015). In order for the OTS to be expressed under the regulation of native promoters and terminators, the upstream and downstream sequences of genomic regions of V. natriegens were used. According to the genomic analysis on codon usage in V. natriegens (Lee et al., 2019), we have found that the GAA codon, encoding for glutamic acid, is the most abundant codon in the genome of this organism. Since promoters and terminators are not yet fully annotated and characterized in V. natriegens, we have used an educated guess as of their identity. Hence, glutamic acid tRNA-synthetase (GluRS) upstream and downstream regions were cloned before and after MmPyl tRNA-synthetase (MmPylRS) gene, respectively. In addition, the upstream and downstream regions of a sequence of three native V. natriegens’s tRNAs (glutamyl-tRNA, lysyl-tRNA, and valyl-tRNA) were chosen to control the expression of MmPyl-tRNA.
The MmPyl-tRNA anti-codon loop was to correspond to a TAG stop-codon mutation encoded in a gene of interest, specifically, a TAG mutation was introduced to a deGFP reporter gene. The TAG mutation site was introduced instead of a tyrosine at position 35 of GFP, a known permissive site (Chemla et al., 2018) for proteins in E. coli. Based on reports that V. natriegens can harbor plasmids with the pBR322 origin of replication (Tschirhart et al., 2019), the pBEST plasmid (Schlesinger et al., 2017) was used as a vector encoding for the MmPyl OTS and mutant GFP gene, which were combined into a new plasmid which name reflects V. natriegens fast-growing nature: rapid genetic code expansion plasmid (pRaGE-Pyl, Figure 1A) (the full plasmid sequence is available in the Supplementary Material section).
The Uaa that we have chosen to incorporate into GFP was propargyl-L-lysine (PrK, Figure 1B). We have first tested the effect of PrK on V. natriegens growth. PrK supplement as well as pRaGE variants transformation into V. natriegens did not impair bacterial growth (Supplementary Figures 1, 2, respectively), hence orthogonality and incorporation validation were followed. The best way to test orthogonality is by testing each OTS component separately. For that purpose, two additional variants of the pRaGE-Pyl plasmid were generated and named pRaGE-Pyl tRNA and pRaGE-PylRS. Both variants encode for the mutant GFP reporter protein, as in the original pRaGE-Pyl construct, however, each variant carried only a single component out of the orthogonal translational pair, either orthogonal MmPyl-tRNA or orthogonal MmPylRS. Protein expression was compared for all plasmid variants, either harboring the full system or a partial system, in the presence or absence of a Uaa.
Through its alkyne side group, PrK can be conjugated to an azide using a click reaction (Kolb et al., 2001). Ideally, we would have liked to perform click chemistry on V. natriegens whole cell lysate and determine site-specific PrK incorporation, along with orthogonality validation. This means that PrK should only be incorporated into GFP and not anywhere else in the proteome of the host organism. Unfortunately, click chemistry did not work on V. natriegens whole cell lysate, even when a purified protein, containing PrK, was added exogenously as a control. We speculate that an unknown component in the V. natriegens lysate is inhibiting the click reaction, and further research is needed to better understand the mechanism involved. Despite the fact that we could not confirm whether PrK is incorporated elsewhere in the proteome, it was still possible to determine the fidelity of PrK incorporation into GFP or whether it is one of the natural amino acids that was being incorporated instead. Upon GFP expression attempts, we have conducted a Western-blot analysis using an anti-GFP antibody. This analysis has established that only in the presence of the full MmPyl OTS and PrK, was a full-length GFP expressed (Figure 1C) (A complete Western-blot is available as Supplementary Figure 3). This result indicate that MmPyl OTS is orthogonal to the translational machinery of the host organism, as there was no full-length GFP expression when partial OTS was used or in the absence of a Uaa. Moreover, fully-elongated GFP expression was detected only in the presence of the entire OTS and PrK, suggesting positive Uaa incorporation as well and no misincorporation events.
In genetic code expansion, mutant protein yield is usually lower when compared to the wild type form (WT) and is commonly referred to as suppression efficiency. It is measured as the relative fraction of mutant protein levels, when incorporating Uaas, from WT protein production levels. Taking into consideration the context effects (Chemla et al., 2018), it is important to note that Uaa incorporation “events” are different from one another by multiple variables. For example, the context of the site of Uaa incorporation, target protein, codon usage, expression promoters and host organism may result in significant differences in protein yields. While protein quantification calculations are not very accurate and are affected by many different factors, for the time being V. natriegens genetic code expansion of GFP at the 35th site, suppression efficiency was calculated to be ∼0.5% using GFP fluorescence (Figure 1D). Densitometry analysis of a Western blot with anti-GFP antibody of WT GFP and Y35TAG GFP mutant in the presence of PrK, although less accurate, resulted in a similar suppression efficiency as that determined by fluorescence measurements (Supplementary Figure 4). It is known that Uaa incorporation is a slower process than native amino acids incorporation, therefore mutant proteins are usually produced in lower quantities than WT proteins (Schlesinger et al., 2017). Also taking into consideration the use of endogenous promoters and terminators in this study, along with the fact that the MmPylRS and GFP genes are not codon-optimized to the V. natriegens codon usage, could explain the observed low suppression efficiency. Presented here is the first attempt at genetic code expansion in V. natriegens, hence we believe the system can be further modified and optimized for future research. In the meantime, V. natriegens WT protein production is vast (Weinstock et al., 2016; Tschirhart et al., 2019), therefore the very low 0.5% suppression efficiency still resulted in the production of 104 μg of mutant protein from 100 mL of culture [calculations of protein yields were based on Ozer et al. (2017)]. Produced mutant protein was sufficient for downstream characterization and analyses.
Mutant GFP protein, expressed in the presence of the complete OTS set and PrK, was purified and was further examined for validation of PrK incorporation. As mentioned, if incorporated, the alkyne moiety of PrK could undergo a click reaction. Using the click reaction to a TAMRA-azide fluorescent marker, a fluorescent SDS-PAGE band corresponded to PrK presence in the purified protein, a clear indication for Uaa incorporation (Figure 2A). Furthermore, the fluorescent band was ruled out as a possible artifact from the protein itself as there was no signal without a click reaction in the TAMRA-Az wavelengths, for the same protein concentrations in both samples identified by Western-blot (Figure 2B). Final validation for PrK incorporation into the purified mutant GFP was through peptide mass fingerprinting. The relevant peptide that included the 35th position of the protein with an incorporated Uaa, was identified with the correct modification of PrK instead of tyrosine in the WT proteins using MS/MS analysis (Figure 2C). These results, the azide conjugation and the peptide mass fingerprinting validated the desired incorporation of PrK in a site-specific manner and were therefore a direct proof for the desired genetic code expansion.
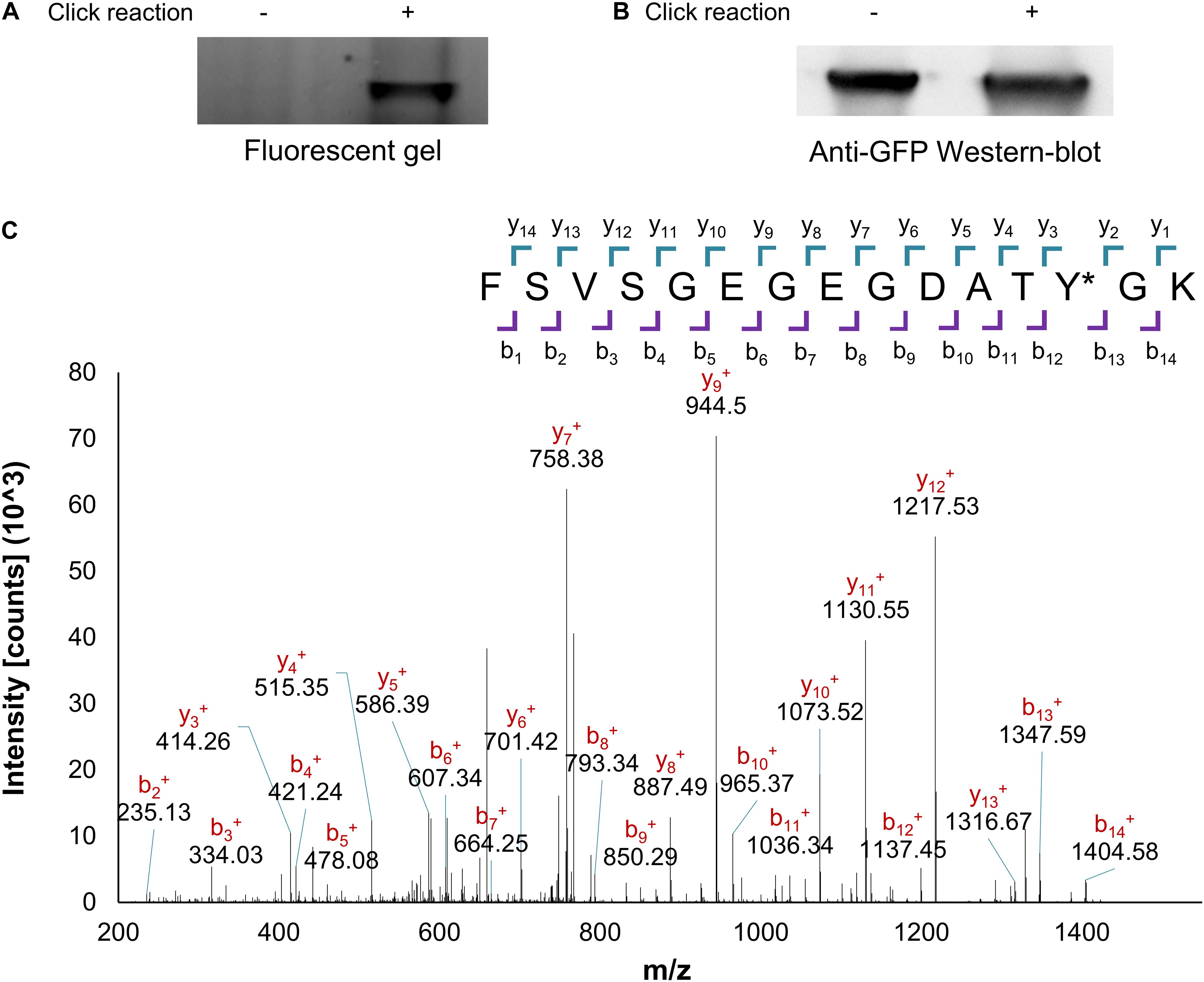
Figure 2. PrK incorporation validation. Purified protein with an incorporated alkyne bearing Uaa (GFP Y35PrK) with or without click reaction to an azide-bearing fluorophore analyzed through Fluorescent SDS-PAGE (A) or anti-GFP Western-blot analysis (B). (C) Peptide mass fingerprinting fragmentation analysis of the peptide with incorporated PrK in the GFP protein, confirms the presence of PrK at the 35th position instead of tyrosine (labeled as Y∗, raw data available in Supplementary Material).
The results presented herein of PrK incorporation validation proved our ability to successfully expand the genetic code of V. natriegens for the first time. The system’s establishment was performed using a GFP reporter protein, yet now that the OTS proved functional it would be possible to employ it for other incorporation sites and target proteins in future research utilizing V. natriegens. As the employed system is the first one for genetic code expansion in V. natriegens it is expected to serve as a stepping stone for further developments and system optimization. However, in the meantime, despite a low calculated suppression efficiency, the amount of produced protein containing Uaa is satisfactory for protein purification and downstream analyses. Proteins with Uaas expressed in V. natriegens may replace production from E. coli and serve as a future tool for various research such as the production of electrochemically active enzymes, oxygen-tolerant enzymes, etc. As V. natriegens is being referred to as the most promising future chassis in synthetic biology, it holds great potential for promoting research in microbiology as well as convenient molecular biology, protein expression and biotechnological platform. The system presented herein has the potential to serve as a powerful tool and may be used for future, much-needed Uaa containing protein expression. We expect that genetic code expansion in V. natriegens will join previously developed methodologies in the growing tool-box relevant to this organism for the benefit of future research.
All additional data including primers and gene sequences, plasmid maps and additional validation results could be found in the supplementary material section. pRAGE plasmid is available through addgene with the following accession number and link: (https://www.addgene.org/160041/).
EO conceived, performed, analyzed experiments, and authored the manuscript. LA conceived experiments, supervised the research, provided facilities, written, and edited the manuscript. Both authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ashok Kumar Bhagat and Michael Shaferman are acknowledged for technical assistance. We would like to thank the Ilse Katz Institute for Nanoscale Science and Technology Shared Resource Facility for their technical contribution in peptide mass finger printing (Mark M. Karpasas). Dr. Anna Bakhrat’s contribution to genetic engineering is thankfully acknowledged. We would also like to thank the Kreitmann School for graduate students for a Ph.D. fellowship (EO) and the Ben-Gurion University of the Negev for a continued support of our research (LA).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2021.594429/full#supplementary-material
Bhagat, A. K., Buium, H., Shmul, G., and Alfonta, L. (2020). Genetically expanded reactive-oxygen-tolerant alcohol dehydrogenase II. ACS Catal. 10, 3094–3102. doi: 10.1021/acscatal.9b03739
Chemla, Y., Friedman, M., Heltberg, M., Bakhrat, A., Nagar, E., Schwarz, R., et al. (2017). Expanding the genetic code of a photoautotrophic organism. Biochemistry 56, 2161–2165. doi: 10.1021/acs.biochem.7b00131
Chemla, Y., Ozer, E., Algov, I., and Alfonta, L. (2018). Context effects of genetic code expansion by stop codon suppression. Curr. Opin. Chem. Biol. 46, 146–155. doi: 10.1016/j.cbpa.2018.07.012
Clark, D. J., and Maaløe, O. (1967). DNA replication and the division cycle in Escherichia coli. J. Mol. Biol. 23, 99–112. doi: 10.1016/S0022-2836(67)80070-6
Cohen, M., Ozer, E., Kushmaro, A., and Alfonta, L. (2020). Cellular localization of cytochrome bd in cyanobacteria using genetic code expansion. Biotechnol. Bioeng. 117, 523–530. doi: 10.1002/bit.27194
Dalia, T. N., Hayes, C. A., Stolyar, S., Marx, C. J., McKinlay, J. B., and Dalia, A. B. (2017). Multiplex genome editing by natural transformation (MuGENT) for Synthetic Biology in Vibrio natriegens. ACS Synth. Biol. 6, 1650–1655. doi: 10.1021/acssynbio.7b00116
Des Soye, B. J., Davidson, S. R., Weinstock, M. T., Gibson, D. G., and Jewett, M. C. (2018). Establishing a high-yielding cell-free protein synthesis platform derived from Vibrio natriegens. ACS Synth. Biol. 7, 2245–2255. doi: 10.1021/acssynbio.8b00252
Dumas, A., Lercher, L., Spicer, C. D., and Davis, B. G. (2015). Designing logical codon reassignment – Expanding the chemistry in biology. Chem. Sci. 6, 50–69. doi: 10.1039/C4SC01534G
Eagon, R. G. (1962). Pseudomonas natriegens, a marine bacterium with a generation time of less than 10 minutes. J. Bacteriol. 83, 736–737. doi: 10.1128/JB.83.4.736-737.1962
Ernst, R. J., Krogager, T. P., Maywood, E. S., Zanchi, R., Beránek, V., Elliott, T. S., et al. (2016). Genetic code expansion in the mouse brain. Nat. Chem. Biol. 12, 776–778. doi: 10.1038/nchembio.2160
Failmezger, J., Scholz, S., Blombach, B., and Siemann-Herzberg, M. (2018). Cell-free protein synthesis from fast-growing Vibrio natriegens. Front. Microbiol 9:1146. doi: 10.3389/fmicb.2018.01146
Gan, Q., Lehman, B. P., Bobik, T. A., and Fan, C. (2016). Expanding the genetic code of Salmonella with non-canonical amino acids. Sci. Rep. 6, 39920–39926. doi: 10.1038/srep39920
Greiss, S., and Chin, J. W. (2011). Expanding the genetic code of an animal. J. Am. Chem. Soc. 133, 14196–14199. doi: 10.1021/ja2054034
Hancock, S. M., Uprety, R., Deiters, A., and Chin, J. W. (2010). Expanding the genetic code of yeast for incorporation of diverse unnatural amino acids via a pyrrolysyl-tRNA synthetase/tRNA pair. J. Am. Chem. Soc. 132, 14819–14824. doi: 10.1021/ja104609m
Hoff, J., Daniel, B., Stukenberg, D., Thuronyi, B. W., Waldminghaus, T., and Fritz, G. (2020). Vibrio natriegens: an ultrafast−growing marine bacterium as emerging synthetic biology chassis. Environ. Microbiol. 22, 4394–4408. doi: 10.1111/1462-2920.15128
Italia, J. S., Zheng, Y., Kelemen, R. E., Erickson, S. B., Addy, P. S., and Chatterjee, A. (2017). Expanding the genetic code of mammalian cells. Biochem. Soc. Trans. 45, 555–562. doi: 10.1042/BST20160336
Kolb, H. C., Finn, M. G., and Sharpless, K. B. (2001). Click Chemistry: diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 40, 2004–2021.
Lee, H. H., Ostrov, N., Wong, B. G., Gold, M. A., Khalil, A. S., and Church, G. M. (2019). Functional genomics of the rapidly replicating bacterium Vibrio natriegens by CRISPRi. Nat. Microbiol. 4, 1105–1113. doi: 10.1038/s41564-019-0423-8
Ozer, E., Chemla, Y., Schlesinger, O., Aviram, H. Y., Riven, I., Haran, G., et al. (2017). In vitro suppression of two different stop codons. Biotechnol. Bioeng. 114, 1065–1073. doi: 10.1002/bit.26226
Peng, Y., Han, X., Xu, P., and Tao, F. (2020). Next−generation microbial workhorses: comparative genomic analysis of fast−growing vibrio strains reveals their biotechnological potential. Biotechnol. J. 15:1900499. doi: 10.1002/biot.201900499
Schleicher, L., Muras, V., Claussen, B., Pfannstiel, J., Blombach, B., Dibrov, P., et al. (2018). Vibrio natriegens as host for expression of multisubunit membrane protein complexes. Front. Microbiol. 9:2537. doi: 10.3389/fmicb.2018.02537
Schlesinger, O., Chemla, Y., Heltberg, M., Ozer, E., Marshall, R., Noireaux, V., et al. (2017). Tuning of recombinant protein expression in Escherichia coli by manipulating transcription, translation initiation rates, and incorporation of noncanonical amino acids. ACS Synth. Biol. 6, 1076–1085. doi: 10.1021/acssynbio.7b00019
Tschirhart, T., Shukla, V., Kelly, E. E., Schultzhaus, Z., Newringeisen, E., Erickson, J. S., et al. (2019). Synthetic biology tools for the fast-growing marine bacterium Vibrio natriegens. ACS Synth. Biol. 8, 2069–2079. doi: 10.1021/acssynbio.9b00176
Weinstock, M. T., Hesek, E. D., Wilson, C. M., and Gibson, D. G. (2016). Vibrio natriegens as a fast-growing host for molecular biology. Nat. Methods 13, 849–851. doi: 10.1038/nmeth.3970
Wiegand, D. J., Lee, H. H., Ostrov, N., and Church, G. M. (2018). Establishing a cell-free Vibrio natriegens expression system. ACS Synth. Biol. 7, 2475–2479. doi: 10.1021/acssynbio.8b00222
Keywords: Vibrio natriegens, genetic code expansion, unnatural amino acids, propargyl-L-lysine, synthetic biology
Citation: Ozer E and Alfonta L (2021) Genetic Code Expansion of Vibrio natriegens. Front. Bioeng. Biotechnol. 9:594429. doi: 10.3389/fbioe.2021.594429
Received: 13 August 2020; Accepted: 26 January 2021;
Published: 26 February 2021.
Edited by:
Hao Song, Tianjin University, ChinaReviewed by:
Dae-Hee Lee, Korea Research Institute of Bioscience and Biotechnology (KRIBB), South KoreaCopyright © 2021 Ozer and Alfonta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lital Alfonta, YWxmb250YWxAYmd1LmFjLmls; orcid.org/0000-0002-3805-8625
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.