 Rafał A. Bachorz
Rafał A. Bachorz Damian Nowak
Damian Nowak Marcin Ratajewski
Marcin Ratajewski- 1Institute of Medical Biology, Polish Academy of Sciences, Łódź, Poland
- 2Department of Quantum Chemistry, Faculty of Chemistry, Adam Mickiewicz University, Poznań, Poland
The drug design process can be successfully supported using a variety of in silico methods. Some of these are oriented toward molecular property prediction, which is a key step in the early drug discovery stage. Before experimental validation, drug candidates are usually compared with known experimental data. Technically, this can be achieved using machine learning approaches, in which selected experimental data are used to train the predictive models. The proposed Python software is designed for this purpose. It supports the entire workflow of molecular data processing, starting from raw data preparation followed by molecular descriptor creation and machine learning model training. The predictive capabilities of the resulting models were carefully validated internally and externally. These models can be easily applied to new compounds, including within more complex workflows involving generative approaches.
1 Introduction
Drug discovery is a process oriented towards the identification and development of biologically active compounds. These molecules are expected to act selectively on certain biological targets, such as enzymes or protein receptors, to influence their biological behavior. A critical element of the drug design process is experimental verification of the ability of a molecule to achieve the desired biological effect. In the early drug discovery stage, this can lead to significant costs because of the large number of drug candidates considered. To avoid this, one can attempt to predict the properties based on existing experimental data. This allows the removal of many compounds and ultimately leaves only the most promising candidates. To achieve this goal, it is necessary to develop a predictive model that properly captures the relationship between the structure of a molecule and its properties. The acronym QSAR represents quantitative structure–activity relationship and relates to a set of techniques capable of predicting the biological activities of compounds based on their structural features. A similar term QSPR, which represents quantitative structure–property relationship, is somewhat more generic and covers any molecular property that can be inferred from the underlying molecular features. The first attempts to QSAR modeling were carried out more than 60 years ago Hansch et al. (1962); Free and Wilson (1964); Hansch and Fujita (1964), and till now are still one of the most important computational tools in the hands of medicinal chemists Tropsha et al. (2003); Murphy (2011). The applications of QSAR/QSPR models are broad and include toxicity Ariëns (1984); Hansch et al. (1995, 1989); Votano (2004); Patlewicz et al. (2008) and metabolism predictions Chohan et al. (2006); Sridhar et al. (2012). QSAR studies are often oriented toward model development that supports virtual screening for promising drug candidates for certain diseases, such as malaria Zhang et al. (2013), schistosomiasis Neves et al. (2016), and influenza Lian et al. (2016). QSAR/QSPR approaches are specifically used in machine learning applications. There are several commercially available programs that cover either partially or completely the QSAR/QSPR workflow. Examples include ADMET Predictor™ from Simulations-Plus (2023), Deep AutoQSAR from Schrödinger Suite Dixon et al. (2016), Biovia Discovery Studio from Dassault Systémes BIOVIA Dassault Systémes (2023), MOE from the Chemical Computing Group (CCG) Chemical Computing Group (2023), VLifeQSAR from VLife Sciences VLife (2023), and Flare™ from Cresset Cresset (2023). All these tools are capable of creating QSPAR/QSAR predictive models based on the entire portfolio of Machine Learning methodologies and various flavors of molecular descriptors/fingerprints. For instance, DeepAutoQSAR provides Deep Neural Network methodologies based on custom implementation of molecular descriptors, allowing for the training and application of state-of-the-art quantitative structure-activity relationship (QSAR) models. The Flare ™ module from Cresset provides a Multilayer Perceptron method and a set of other Machine Learning methodologies supporting the development of consensus regression and classification models. Other vendors provide tools that differ slightly in various aspects; however, their common denominator is the commercial nature of their programs. The main goal of the proposed software is to provide an open-source alternative in the form of Python scripts based exclusively on available open-source cheminformatics and machine learning libraries, which implement a complete QSAR/QSPR workflow. In addition, the functional scope of the proposed QSPRmodeler software is beyond the scope of standard machine learning or cheminformatics libraries that are considered separately. The main novelty of the proposed solution is that it combines these two worlds into a single entity, allowing for the straightforward management of chemical information and efficient extraction of predictive signals. Moreover, the proposed functional design enables the incorporation of new machine learning methodologies, which makes the open-source society a toolset capable of exploring novel predictive approaches in a chemical context.
2 Software description
The proposed software combines existing Python libraries to cover all the key steps of the QSAR/QSPR modeling process. The workflow is shown in Figure 1. The entire calculation depends on three files: the data file with experimental values in csv form (denoted as Experimental_data.csv), the training configuration (denoted as Training_configuration.json), and the data processing pipeline file (denoted as Pipeline_configuration.json). The incoming data must be prepared in a simple form of a csv file, with the SMILES Weininger (1988) code accompanied by the experimental data. The experimental data are usually IC50 or EC50 values expressed in molar units. Multiple experimental values often exist for the same compound. These values may have been derived from an entirely independent experimental investigation involving qualitatively different biological assays. Thus, an unwanted effect is the potential inconsistency in the experimental endpoints for the same compound. The raw data prepossessing phase (denoted as “1” in Figure 1) measures the level of inconsistency as a standard deviation and removes cases in which it exceeds a certain threshold value, as defined in the Training_configuration.json file (set at the level of 100 nM). For the remaining consistent cases, the chosen aggregation strategy is applied, for example, the arithmetic mean, median, maximum, or minimum function. The resulting dataset is then a simple table associating a certain molecule in the SMILES form Weininger (1988) with single, potentially aggregated, experimental values. In the next step, denoted as “2” in Figure 1, the molecular features are calculated. In particular, various types of molecular fingerprints, such as daylight fingerprints Daylight Chemical Information Systems (2019), atom-pair fingerprints Carhart et al. (1985), topological torsion fingerprints Nilakantan et al. (1987), Morgan fingerprints Rogers and Hahn (2010) and MACCS keys Durant et al. (2002) can be obtained. All these are calculated using the open-source RDKit chemical informatics library Landrum (2022). Selected molecular descriptors can also be incorporated to augment the molecular feature space further. To achieve this, we integrated the Mordreds library Moriwaki et al. (2018) offering an implementation of 1825 molecular descriptors. The next step of the workflow, denoted as “3” in Figure 1 shows the standard data processing steps for molecular descriptors, such as scaling and Principal Component Analysis (PCA) transformation. The latter can be applied to both the molecular fingerprints and descriptors. This step is accomplished using the Scikit-learn library Pedregosa et al. (2011). Step “4” of the workflow takes care of the processing of the target value, in particular logarithm transformation of the numerical value in the case of regression models or target binarization in the case of the binary classifiers. Upon completion of this step, all data are prepared for predictive analytics. The next step, denoted as “5,” applies the chosen machine learning methodology to the prepared, earlier data. Currently, six common predictive model types are available: extreme gradient boosting (XGBoost) Chen and Guestrin (2016), artificial neural networks in the form of multilayer perceptrons Bishop (1995), support vector machines Cortes and Vapnik (1995); Vapnik (1998), random forests Ho (1995), ridges Hoerl and Kennard (1970), and bagging models. With each of these methodologies, a definition of the hyperparameter space is provided. The predictive model creation step involves hyperparameter optimization within the Hyperopt framework Bergstra et al. (2013), which implements the heuristics of the Tree of Parzen Estimators Bergstra et al. (2011). The last step of the workflow, denoted as ”6,” involves final quality measures calculation and model serialization. The final predictive model is stored in a dedicated file together with all the auxiliary information required for the subsequent standalone application in the inference mode. In particular, the entire data-processing pipeline is serialized such that the only information required to perform the prediction is the molecule provided in the form of a SMILES code. The program automatically turns the SMILES representation into a feature space compliant with the model interface and ultimately provides the prediction. A serialized model is an autonomous artifact that is easily integrated into various workflows and molecular predictive use cases. The associated Github repository contains a set of Jupyter notebooks that illustrate how QSPR models can be used in the inference mode, for example, to predict the molecular/biological properties of new compounds. One can imagine multiple, more complex use cases, such as virtual screening of molecular databases, application of unsupervised learning to molecular data, or integration with generative chemistry workflows where models are created that are responsible for criticizing new species against optimized properties. The module is also prone to potential extensions, and an intermediate Python programmer can easily add a new predictive methodology or adopt the provided scripts for particular needs.
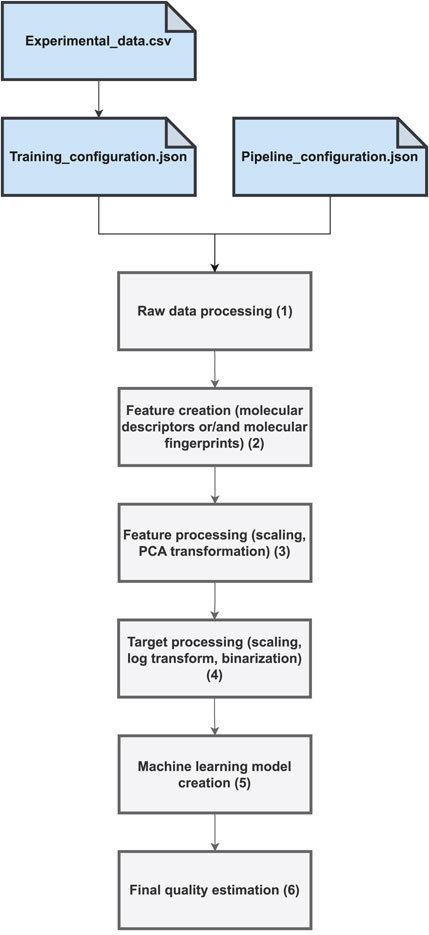
Figure 1. General workflow of the QSPRmodeler program.
3 Illustrative examples
3.1 Introduction
As illustrative examples, we applied the presented methodology to QSAR modeling of the inhibitory effects of the human androgen receptor (AR) and the activation effects of the pregnane X receptor (PXR) receptor. The former (AR, NR3C4) belongs to the nuclear receptor subfamily 3, a group C nuclear receptor superfamily of proteins Burris et al. (2013). AR acts as a transcription factor that regulates genes important for the development and maintenance of primary and secondary male characteristics Heemers and Tindall (2007). Similar to other nuclear receptors, AR activity is regulated by low-molecular-weight ligands. In the absence of a ligand, the AR resides in the cytoplasm bind to heat shock proteins (HSPs). Upon binding to the ligand, the receptor changes its conformation, homodimerizes, and translocates into the nucleus to regulate AR-dependent genes Prescott and Coetzee (2006). Testosterone and dihydrotestosterone are the endogenous ligands of AR. Under physiological conditions, AR is involved in the development of prostate; however, the disturbed function of these receptor leads to uncontrolled proliferation of prostate cells and the appearance of cancer Lonergan and Tindall (2011), Jernberg et al. (2017). Prostate cancer cells require androgens for survival and proliferation, which is why therapies that use anti-androgens targeting the function of AR are generally effective Kokal et al. (2020).
The second receptor, PXR (NR1I2), regulates xenobiotic metabolism and is involved in the maintenance of liver physiology Cai et al. (2021). This receptor recognizes a wide range of structurally diverse compounds, including endogenous metabolites such as bile acids Staudinger et al. (2001), phthalates Hurst and Waxman (2004), and mycotoxins Ratajewski et al. (2011), and responds to various pharmacologic compounds, including but not limited to rifampicin, dexamethasone, clotrimazole, etoposide, trifluridine, and mycophenolic acid Moore et al. (2000); Ratajewski et al. (2015); Yim et al. (2023). Interactions between pharmacological compounds and PXR are crucial, as PXR recognition can markedly enhance the liver transformation rate of various xenobiotics, leading to potential drug-to-drug interactions Lehmann et al. (1998); Fuhr (2000); Ratajewski et al. (2015).
This encouraged us to use the presented computer-based QSPRmodeler environment by applying machine learning to identify novel chemical structures targeting the ligand-binding domains (LBD) of both receptors. All available experimental values, the IC50 values for AR, and the EC50 values for PXR (half-maximal inhibitory and effective concentrations, respectively, for IC50 and EC50), were retrieved from the ChEMBL 3.3 database Gaulton et al. (2012). The experimental IC50 data for the AR reflected the IC50 measurements of 1575 different chemical species. For the other receptor, we found 1187 entries of EC50 values. Multiple experimental values are often obtained for the same molecule using different biochemical assays. The experimental values obtained for the same molecule cannot always be combined; therefore, we carefully analyzed the available data and excluded doubtful experimental endpoints. The resulting datasets were used to create regression and binary classification models using the QSPRmodeler toolset presented herein. The target value of the regression model was chosen as the negative logarithm of IC50 or EC50, denoted as pIC50 or pEC50, respectively. The classification model was trained on the binarized data, that is, each molecule was assigned to the ACTIVE or INACTIVE class with the class-determining threshold value assumed to be 1000 and 12,000 nM, respectively, for the IC50 values of the AR and EC50 values of the PXR. According to the configured workflows, the models were trained using the XGBoost method within a 5-fold crossvalidation scheme. The space of molecular features was limited to 8 descriptors: SLogP (octanol-water partition coefficient), SMR (molar refractivity), naRing (number of aromatic rings), nHBAcc (number of hydrogen bond acceptors), nHBDon (number of hydrogen bond donors), nRot (number of rotatable bonds), MW (molecular weight), and TopoPSA (topological polar surface area). In addition, the feature space was augmented with two drug-like filters, the Lipinsky rule-of-five and Ghose filters, and 50 most important principal components were calculated based on a 1024-bit long Morgan fingerprint. Thus, each molecule was characterized by a vector of 60 numbers, reflecting its topological and physiochemical properties. The goal functions of the hyperparameter optimization were chosen as the mean square error (MSE) and accuracy for the regressor and classifier, respectively. The Hyperopt module was used with default settings, with the maximum number of evaluations set at 100. Training was performed using 90% of the available data, whereas the remaining 10% of data were used as a test set for final quality estimation.
3.2 Androgen receptor
Figure 2 compares the experimental and predicted values calculated for the molecules from the test set containing 158 compounds. Approximately 80% of these molecules were predicted within a range of 1.0 log unit, and the average MSE for the entire test set was 0.61 log unit, which reflects the common predictive strength of the QSAR models. The predictive capabilities of the classification models, calculated using the hold-out test set, are listed in Table 1. The classifier reached a satisfactory level of accuracy of 82% with reasonable levels of both sensitivity and specificity. The ROC curves presented in Figure 3, and the relatively high values of the area under this curve, i.e. 0.81, clearly demonstrate the presence of a predictive signal in the data as well as the ability of the tool to extract this signal. It is worth mentioning that although the discussed models were prepared mainly for presentation purposes, they compared well relative to, or even outperformed, the available models. For instance, the accuracy, sensitivity, and specificity of the AR model were 82%, 85.4%, and 76.4%, respectively, which can be compared to the Random Forest model with 73%, 72%, and 72% as described in Piir et al. (2021). As an additional feature supporting the Machine Learning model interpretability, we delivered the feature importance capability. It is available for decision-tree-based models and provides quantitative insights into the strength of the features involved in model creation. As an example, Figure 4 shows the most influential features with the strongest contributions to the predictions for the XGBoost classifier developed for AR. The most important feature is “nRot,” which represents the number of rotatable bonds. This molecular descriptor reflects the compound flexibility, which is important from the perspective of ligand binding to the binding pocket of the receptor. Molecular descriptors and the PCA features derived from molecular fingerprints were among the 40 most important features, reflecting the topological aspects of the molecule. All configuration files and data are available in the associated GitHub repository.
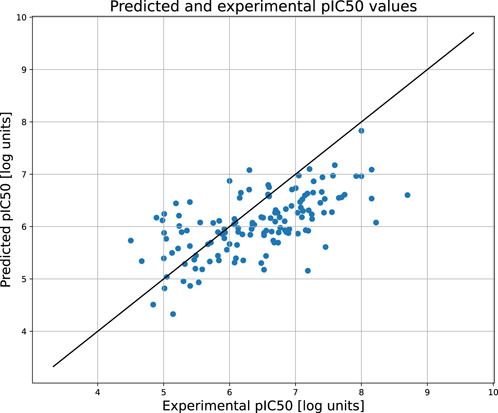
Figure 2. The scatter plot showing the relation between the predicted and experimental pIC50 values of androgen receptor. The predictions were obtained for the held out test set.

Table 1. Predictive capabilities of the androgen and pregnane X receptors classification model. The results were obtained with the held out test set representing 10% of entire available data.
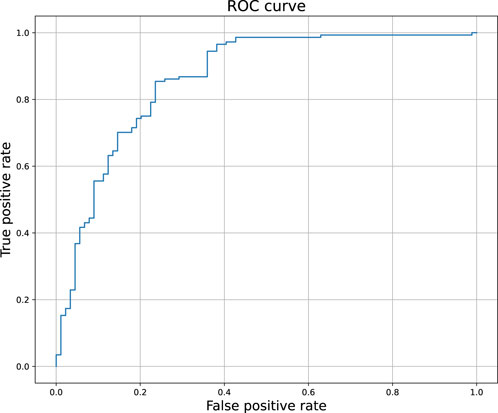
Figure 3. The ROC curve of the binary classifier calculated for androgen receptor. The predictions were obtained for the held out test set.
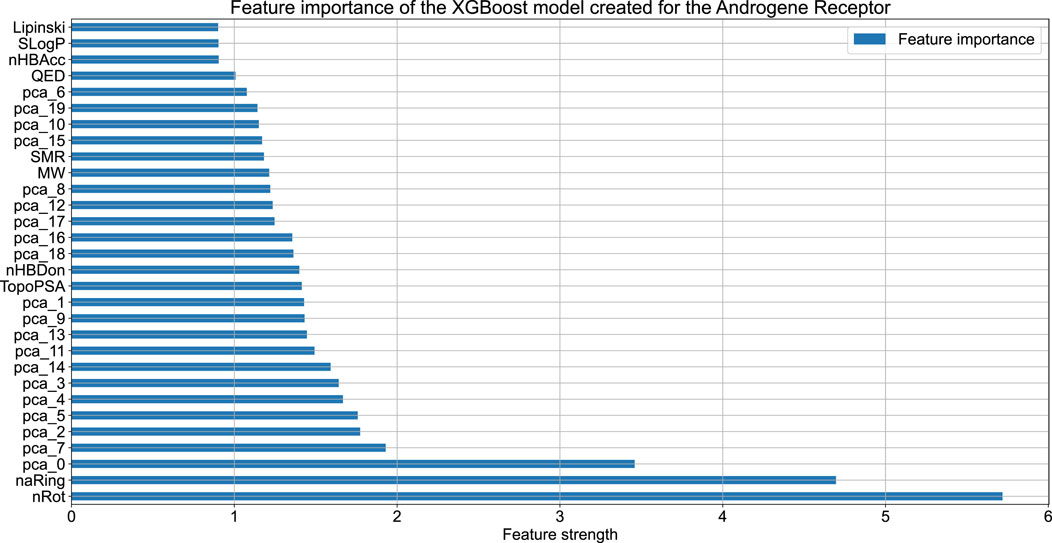
Figure 4. The feature importance plot obtained for the XGBoost model trained on the AR data. The plot contains the information about the 40 features having the highest influence on model predictions.
3.3 Pregnane X receptor
Similarly to the previous case, Figure 5 provides a detailed comparison between the experimental and predicted values calculated for the test set molecules associated with the PXR receptor containing 112 compounds. This figure highlights that the vast majority of these molecules - over 83% - were predicted with relatively high degree of accuracy, falling within a range of 1.0 log unit of the experimental values. This level of precision reflects the expected and consistent performance of QSAR models, reinforcing their reliability in this context. The predictive capabilities of the classification models, which were rigorously evaluated using the hold-out test set, are summarized in Table 1. The classifiers demonstrated a commendable level of efficiency, achieving sensitivity and specificity rates of 83%, indicating that the models are equally proficient at identifying both true positives and true negatives. Additionally, the model achieved an overall accuracy of 82%, further underscoring the robustness in predicting the biological activity of molecules within the PXR receptor dataset. Moreover, the ROC curve presented in Figure 6, along with the relatively high area under the curve (AUC) value of 0.82, provides evidence of a predictive signal within the data. This high AUC value not only confirms the presence of a meaningful relationship between the molecular descriptors and biological activity but also attests to the tool’s effectiveness in capturing and utilizing this signal to make accurate predictions. Overall, these results affirm the model’s practical utility and its potential for application in drug discovery and other related fields. All configuration files and data are available in the associated GitHub repository.

Figure 5. The scatter plot showing the relation between the predicted and experimental pEC50 values of the pregnane X receptror. The predictions were obtained for the held out test set.
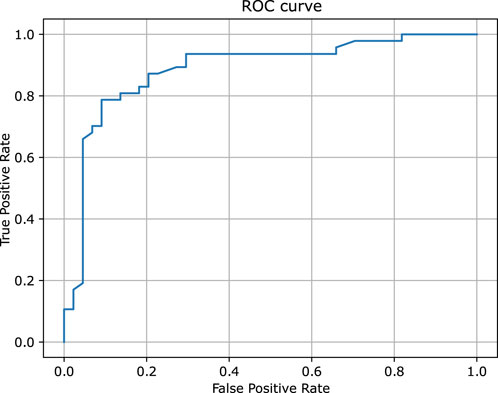
Figure 6. The ROC curve of the binary classifier calculated for pregnane X receptor. The predictions were obtained for the held out test set.
4 Summary
Here, we present a Python module called QSPRmodeler, a tool dedicated to the creation of binary classifiers and regression predictive models oriented toward predicting the biological and molecular properties of molecules. The tool utilizes the Python libraries RDKit and Mordred available from the cheminformatics site and a set of popular machine learning libraries to solve predictive analytics problems (extreme gradient boosting (XGBoost) Chen and Guestrin (2016), artificial neural networks in the form of multilayer perceptrons Bishop (1995), support vector machines Cortes and Vapnik (1995); Vapnik (1998), random forests Ho (1995), ridges Hoerl and Kennard (1970), and bagging models). The user input is limited to providing the data in an expected manner, creating the configuration files, and managing the data transformation and hyperparameter optimization. The proposed solution implements well-established machine learning practices in the context of molecules. The capabilities of the QSPRmodeler were illustrated using an exemplary application to human androgen and pregnane X receptors based on publicly available data. The resulting regression and classification models exhibited predictive capabilities and could be easily applied to various custom workflows. The implementation was provided within a permissive open-source licensing model and is available in the public GitHub repository. It is worth mentioning that the proposed tool was recently applied to the virtual screening of a large database of compounds, resulting in the discovery and experimental verification of new biologically active ligands for the ROR
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://github.com/rafalbachorz/qsprmodeler.
Author contributions
RB: Conceptualization, Data curation, Methodology, Software, Validation, Writing - original draft. DN: Data curation, Writing - original draft, Software. MR: Conceptualization, Software, Writing - original draft, Funding acquisition.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Science Center, Project number 2019/33/B/NZ7/00795.
Conflict of interest
The authors declare that this study was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ariëns, E. J. (1984). Domestication of chemistry by design of safer chemicals: structure-activity relationships. Drug Metab. Rev. 15, 425–504. doi:10.3109/03602538409029970
Bachorz, R. A., Pastwinska, J., Nowak, D., Karas, K., Karwaciak, I., and Ratajewski, M. (2023). The application of machine learning methods to the prediction of novel ligands for ROR γ/ROR γ T receptors. Comput. Struct. Biotechnol. J. 21, 5491–5505. doi:10.1016/j.csbj.2023.10.021
Baniecki, H., Kretowicz, W., Piatyszek, P., Wisniewski, J., and Biecek, P. (2021). dalex: responsible machine learning with interactive explainability and fairness in python. J. Mach. Learn. Res. 22, 1–7.
Bergstra, J., Bardenet, R., Bengio, Y., and Kégl, B. (2011). “Algorithms for hyper-parameter optimization,”. Advances in neural information processing Systems. Editors J. Shawe-Taylor, R. Zemel, P. Bartlett, F. Pereira, and K. Weinberger (USA: Curran Associates, Inc.), 24, 1–9.
Bergstra, J., Yamins, D., and Cox, D. (2013). “Making a science of model search: hyperparameter optimization in hundreds of dimensions for vision architectures,” in Proceedings of the 30th international conference on machine learning. 28 of Proceedings of machine learning research. Editors S. Dasgupta, and D. McAllester (Atlanta, Georgia, USA: PMLR), 115–123.
Burris, T. P., Solt, L. A., Wang, Y., Crumbley, C., Banerjee, S., Griffett, K., et al. (2013). Nuclear receptors and their selective pharmacologic modulators. Pharmacol. Rev. 65, 710–778. doi:10.1124/pr.112.006833
Cai, X., Young, G. M., and Xie, W. (2021). The xenobiotic receptors pxr and car in liver physiology, an update. Biochimica Biophysica Acta. Mol. Basis Dis. 1867, 166101. doi:10.1016/j.bbadis.2021.166101
Carhart, R. E., Smith, D. H., and Venkataraghavan, R. (1985). Atom pairs as molecular features in structure-activity studies: definition and applications. J. Chem. Inf. Comput. Sci. 25, 64–73. doi:10.1021/ci00046a002
Chemical Computing Group (2023). MOE: molecular operating environment. Chem. Comput. Group. Available at: https://www.chemcomp.com/.
Chen, T., and Guestrin, C. (2016). “Xgboost: a scalable tree boosting system,” in Proceedings of the 22nd ACM SIGKDD international Conference on knowledge Discovery and data mining (san francisco California USA: acm), 785–794. doi:10.1145/2939672.2939785
Chohan, K., Paine, S., and Waters, N. (2006). Quantitative structure activity relationships in drug metabolism. Curr. Top. Med. Chem. 6, 1569–1578. doi:10.2174/156802606778108960
Cortes, C., and Vapnik, V. (1995). Support-vector networks. Mach. Learn. 20, 273–297. doi:10.1007/bf00994018
Cresset (2023). Flare. United Kingdom: Cresset Ltd. Available at: https://www.cresset-group.com/software/flare/.
Dixon, S. L., Duan, J., Smith, E., Von Bargen, C. D., Sherman, W., and Repasky, M. P. (2016). Autoqsar: an automated machine learning tool for best-practice quantitative structure–activity relationship modeling. Future Med. Chem. 8, 1825–1839. doi:10.4155/fmc-2016-0093
Durant, J. L., Leland, B. A., Henry, D. R., and Nourse, J. G. (2002). Reoptimization of mdl keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 42, 1273–1280. doi:10.1021/ci010132r
Free, S. M., and Wilson, J. W. (1964). A mathematical contribution to structure-activity studies. J. Med. Chem. 7, 395–399. doi:10.1021/jm00334a001
Fuhr, U. (2000). Induction of drug metabolising enzymes: pharmacokinetic and toxicological consequences in humans. Clin. Pharmacokinet. 38, 493–504. doi:10.2165/00003088-200038060-00003
Gaulton, A., Bellis, L. J., Bento, A. P., Chambers, J., Davies, M., Hersey, A., et al. (2012). Chembl: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40, D1100–D1107. doi:10.1093/nar/gkr777
Hansch, C., and Fujita, T. (1964). p-σ-π Analysis. A method for the correlation of biological activity and chemical structure. J. Am. Chem. Soc. 86, 1616–1626. doi:10.1021/ja01062a035
Hansch, C., Hoekman, D., Leo, A., Zhang, L., and Li, P. (1995). The expanding role of quantitative structure-activity relationships (qsar) in toxicology. Toxicol. Lett. 79, 45–53. doi:10.1016/0378-4274(95)03356-P
Hansch, C., Kim, D., Leo, A. J., Novellino, E., Silipo, C., Vittoria, A., et al. (1989). Toward a quantitative comparative toxicology of organic compounds. CRC Crit. Rev. Toxicol. 19, 185–226. doi:10.3109/10408448909037471
Hansch, C., Maloney, P. P., Fujita, T., and Muir, R. M. (1962). Correlation of biological activity of phenoxyacetic acids with hammett substituent constants and partition coefficients. Nature 194, 178–180. doi:10.1038/194178b0
Heemers, H. V., and Tindall, D. J. (2007). Androgen receptor (ar) coregulators: a diversity of functions converging on and regulating the ar transcriptional complex. Endocr. Rev. 28, 778–808. doi:10.1210/er.2007-0019
Ho, T. K. (1995). Random decision forests. Proc. 3rd Int. Conf. document analysis Recognit. (IEEE) 1, 278–282. doi:10.1109/ICDAR.1995.598994
Hoerl, A. E., and Kennard, R. W. (1970). Ridge regression: biased estimation for nonorthogonal problems. Technometrics 12, 55–67. doi:10.1080/00401706.1970.10488634
Hu, L., Pan, X., Tang, Z., and Luo, X. (2022). A fast fuzzy clustering algorithm for complex networks via a generalized momentum method. IEEE Trans. Fuzzy Syst. 30, 3473–3485. doi:10.1109/TFUZZ.2021.3117442
Hurst, C. H., and Waxman, D. J. (2004). Environmental phthalate monoesters activate pregnane x receptor-mediated transcription. Toxicol. Appl. Pharmacol. 199, 266–274. doi:10.1016/j.taap.2003.11.028
Jernberg, E., Bergh, A., and Wikström, P. (2017). Clinical relevance of androgen receptor alterations in prostate cancer. Endocr. Connect. 6, R146–R161. doi:10.1530/EC-17-0118
Kokal, M., Mirzakhani, K., Pungsrinont, T., and Baniahmad, A. (2020). Mechanisms of androgen receptor agonist- and antagonist-mediated cellular senescence in prostate cancer. Cancers 12, 1833. doi:10.3390/cancers12071833
Lehmann, J. M., McKee, D. D., Watson, M. A., Willson, T. M., Moore, J. T., and Kliewer, S. A. (1998). The human orphan nuclear receptor pxr is activated by compounds that regulate cyp3a4 gene expression and cause drug interactions. J. Clin. Investigation 102, 1016–1023. doi:10.1172/JCI3703
Li, G., Zhao, B., Su, X., Yang, Y., Hu, P., Zhou, X., et al. (2024). Discovering consensus regions for interpretable identification of rna n6-methyladenosine modification sites via graph contrastive clustering. IEEE J. Biomed. Health Inf. 28, 2362–2372. doi:10.1109/JBHI.2024.3357979
Lian, W., Fang, J., Li, C., Pang, X., Liu, A.-L., and Du, G.-H. (2016). Discovery of influenza a virus neuraminidase inhibitors using support vector machine and naïve bayesian models. Mol. Divers. 20, 439–451. doi:10.1007/s11030-015-9641-z
Lonergan, P. E., and Tindall, D. J. (2011). Androgen receptor signaling in prostate cancer development and progression. J. Carcinog. 10, 20. doi:10.4103/1477-3163.83937
Moore, L. B., Parks, D. J., Jones, S. A., Bledsoe, R. K., Consler, T. G., Stimmel, J. B., et al. (2000). Orphan nuclear receptors constitutive androstane receptor and pregnane x receptor share xenobiotic and steroid ligands. J. Biol. Chem. 275, 15122–15127. doi:10.1074/jbc.M001215200
Moriwaki, H., Tian, Y.-S., Kawashita, N., and Takagi, T. (2018). Mordred: a molecular descriptor calculator. J. Cheminformatics 10, 4. doi:10.1186/s13321-018-0258-y
Murphy, R. F. (2011). An active role for machine learning in drug development. Nat. Chem. Biol. 7, 327–330. doi:10.1038/nchembio.576
Neves, B. J., Dantas, R. F., Senger, M. R., Melo-Filho, C. C., Valente, W. C. G., de Almeida, A. C. M., et al. (2016). Discovery of new anti-schistosomal hits by integration of qsar-based virtual screening and high content screening. J. Med. Chem. 59, 7075–7088. doi:10.1021/acs.jmedchem.5b02038
Nilakantan, R., Bauman, N., Dixon, J. S., and Venkataraghavan, R. (1987). Topological torsion: a new molecular descriptor for sar applications. comparison with other descriptors. J. Chem. Inf. Comput. Sci. 27, 82–85. doi:10.1021/ci00054a008
Patlewicz, G., Roberts, D. W., and Uriarte, E. (2008). A comparison of reactivity schemes for the prediction skin sensitization potential. Chem. Res. Toxicol. 21, 521–541. doi:10.1021/tx700338q
Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., Grisel, O., et al. (2011). Scikit-learn: machine learning in python. J. Mach. Learn. Res. 12, 2825–2830.
Piir, G., Sild, S., and Maran, U. (2021). Binary and multi-class classification for androgen receptor agonists, antagonists and binders. Chemosphere 262, 128313. doi:10.1016/j.chemosphere.2020.128313
Prescott, J., and Coetzee, G. A. (2006). Molecular chaperones throughout the life cycle of the androgen receptor. Cancer Lett. 231, 12–19. doi:10.1016/j.canlet.2004.12.037
Ratajewski, M., Grzelak, I., Wiśniewska, K., Ryba, K., Gorzkiewicz, M., Walczak-Drzewiecka, A., et al. (2015). Screening of a chemical library reveals novel pxr-activating pharmacologic compounds. Toxicol. Lett. 232, 193–202. doi:10.1016/j.toxlet.2014.10.009
Ratajewski, M., Walczak-Drzewiecka, A., Sałkowska, A., and Dastych, J. (2011). Aflatoxins upregulate cyp3a4 mrna expression in a process that involves the pxr transcription factor. Toxicol. Lett. 205, 146–153. doi:10.1016/j.toxlet.2011.05.1034
Rogers, D., and Hahn, M. (2010). Extended-connectivity fingerprints. J. Chem. Inf. Model. 50, 742–754. doi:10.1021/ci100050t
Simulations-Plus (2023). ADMET predictor. Simulations Plus. Available at: https://www.simulations-plus.com/software/admetpredictor/.
Sridhar, J., Liu, J., Foroozesh, M., and Stevens, C. L. K. (2012). Insights on cytochrome p450 enzymes and inhibitors obtained through qsar studies. Molecules 17, 9283–9305. doi:10.3390/molecules17089283
Staudinger, J. L., Goodwin, B., Jones, S. A., Hawkins-Brown, D., MacKenzie, K. I., LaTour, A., et al. (2001). The nuclear receptor pxr is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. U. S. A. 98, 3369–3374. doi:10.1073/pnas.051551698
Tropsha, A., Gramatica, P., and Gombar, V. (2003). The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. QSAR & Comb. Sci. 22, 69–77. doi:10.1002/qsar.200390007
Vapnik, V. (1998). The support vector method of function estimation. Boston, MA: Springer US, 55–85. doi:10.1007/978-1-4615-5703-6_3
VLife (2023). VLife technologie. QSARpro, Accurate activity prediction; new molecule design. Available at: https://www.vlifesciences.com/products/QSARPro/Product_QSARpro.php.
Votano, J. R. (2004). Three new consensus qsar models for the prediction of ames genotoxicity. Mutagenesis 19, 365–377. doi:10.1093/mutage/geh043
Weininger, D. (1988). Smiles, a chemical language and information system. 1. introduction to methodology and encoding rules. J. Chem. Inf. Model. 28, 31–36. doi:10.1021/ci00057a005
Yim, R. M., Sahni, V. N., and Mathis, J. G. (2023). Mycophenolate mofetil-induced hyperlipidemia with cutaneous manifestations. Clin. Case Rep. 11, e7056. doi:10.1002/ccr3.7056
Keywords: QSPR, machine learning, drug design, biological activity, ADMET
Citation: Bachorz RA, Nowak D and Ratajewski M (2024) QSPRmodeler - An open source application for molecular predictive analytics. Front. Bioinform. 4:1441024. doi: 10.3389/fbinf.2024.1441024
Received: 30 May 2024; Accepted: 27 August 2024;
Published: 23 September 2024.
Edited by:
Mubashir Hassan, The Research Institute at the Nationwide Children’s Hospital, United StatesReviewed by:
Jiashun Mao, Yonsei University, Republic of KoreaSaeed Ahmad, Nationwide Children’s Hospital, United States
Copyright © 2024 Bachorz, Nowak and Ratajewski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rafał A. Bachorz, rbachorz@cbm.pan.pl