 Victor Margelidon-Cozzolino
Victor Margelidon-Cozzolino Anne Tsicopoulos
Anne Tsicopoulos Cécile Chenivesse
Cécile Chenivesse Patricia de Nadai1
Patricia de Nadai1- 1Univ. Lille, CNRS, INSERM, CHU de Lille, Institut Pasteur de Lille, Unité INSERM U1019-UMR9017-CIIL-Centre d'Infection et d'Immunité de Lille, Lille, France
- 2CRISALIS (Clinical Research Initiative in Severe Asthma: a Lever for Innovation & Science), F-CRIN Network, INSERM US015, Toulouse, France
Airway remodeling is a frequent pathological feature of severe asthma leading to permanent airway obstruction in up to 50% of cases and to respiratory disability. Although structural changes related to airway remodeling are well-characterized, immunological processes triggering and maintaining this phenomenon are still poorly understood. As a consequence, no biotherapy targeting cytokines are currently efficient to treat airway remodeling and only bronchial thermoplasty may have an effect on bronchial nerves and smooth muscles with uncertain clinical relevance. Th17 cytokines, including interleukin (IL)-17 and IL-22, play a role in neutrophilic inflammation in severe asthma and may be involved in airway remodeling. Indeed, IL-17 is increased in sputum from severe asthmatic patients, induces the expression of “profibrotic” cytokines by epithelial, endothelial cells and fibroblasts, and provokes human airway smooth muscle cell migration in in vitro studies. IL-22 is also increased in asthmatic samples, promotes myofibroblast differentiation, epithelial-mesenchymal transition and proliferation and migration of smooth muscle cells in vitro. Accordingly, we also found high levels of IL-17 and IL-22 in a mouse model of dog-allergen induced asthma characterized by a strong airway remodeling. Clinical trials found no effect of therapy targeting IL-17 in an unselected population of asthmatic patients but showed a potential benefit in a sub-population of patients exhibiting a high level of airway reversibility, suggesting a potential role on airway remodeling. Anti-IL-22 therapies have not been evaluated in asthma yet but were demonstrated efficient in severe atopic dermatitis including an effect on skin remodeling. In this review, we will address the role of Th17 cytokines in airway remodeling through data from in vitro, in vivo and translational studies, and examine the potential place of Th17-targeting therapies in the treatment of asthma with airway remodeling.
Introduction
Asthma is a chronic respiratory disease characterized by lower airway inflammation, leading to airway hyperresponsiveness (AHR) and airway obstruction, resulting in recurrent symptoms (chest wheezing, cough, dyspnea), exacerbations (flare-up) and altered quality of life. As one of the most frequent chronic respiratory disease, asthma is estimated to affect around 5–10% of adults (up to 20% in high-income countries) and from 5 to 20% of children. As a consequence, asthma is a major public health issue (1–4). The cornerstone of the treatment of asthma is inhaled steroids (ICS). Severe asthma is defined as asthma requiring at least high doses of ICS in association with another controller (generally long-acting beta-agonist bronchodilators) and/or oral steroids more than half a year to be controlled, or that remains uncontrolled despite these therapies (5–7). The burden of asthma is particularly heavy in patients with severe asthma who experience a severely impaired quality of life, respiratory disability, frequent and severe exacerbations, poorer control of symptoms, and hospitalisations. Severe asthma is also associated with lower respiratory function with persistent airway obstruction, representing about 50% of severe asthmatic patients, and sometimes loss of reversibility of airway obstruction (6–8). These patients account for the major part of asthma-related healthcare cost (6, 7).
From a pathophysiological point of view, persistent airway obstruction and loss of reversibility are the clinical hallmark of airway remodeling. Airway remodeling is a complex biological process, found in most chronic inflammatory airway diseases, which contributes to airway obstruction (9). In asthma, airway remodeling is characterized by several elementary structural changes in bronchial mucosa: loss of epithelial integrity, epithelial-mesenchymal transition (EMT), goblet cell hyperplasia, mucus hypersecretion, subepithelial fibrosis with thickening of epithelial basement membrane, smooth muscle cell proliferation, and hypertrophy. Inflammatory cell infiltration in bronchial mucosa can also be associated with airway remodeling and contribute to this process (10–12). These changes lead to increased lower airway resistances and reduced bronchial lumen, thus contributing to persistent airway obstruction. Although airway remodeling is present in asthma in all degrees of severity, airway remodeling intensity correlates with asthma severity (11, 13–17).
Asthma has long been considered as an allergic Th2-driven inflammatory disease, involving the “canonical” Th2 cytokines [interleukines (IL)-4, IL-5, IL-13], eosinophilic inflammation, and mediators of allergic inflammation (mainly immunoglobulins E and mast cells) (18). Over the last 20 years, major advances have been made in the understanding of asthma immunobiology and heterogeneity. Cluster analyses of large cohorts of patients allowed to identify several homogeneous subpopulations of asthmatic patients according to clinical characteristics (phenotypes), and their correlates in inflammatory and immunobiological processes (endotypes) (18–20). Hence, asthmatic patients are currently categorized according to the dominant inflammatory pathway driving bronchial inflammation, and particularly the degree of involvement of T2 inflammation: T2high asthma (typically allergic and non-allergic eosinophilic asthma) vs. T2low asthma (21). T2low asthma refers to a heterogeneous group of patients with distinct endotypes, currently poorly characterized, like neutrophilic or paucigranulocytic inflammation.
Both patients with T2high or T2low asthma can present severe asthma. However, the neutrophilic endotype, characterized by a predominance of neutrophils in airway inflammation, has been particularly associated with severe asthma and resistance to ICS and oral steroids (22–25). In contrast, T2high asthma is more commonly associated with response to steroids. Additionally, patients with T2high severe asthma have now access to effective biotherapies that have emerged over the last decade, based on monoclonal antibodies targeting Th2 cytokines and immunoglobulins E (26). Although involvement of non-T2 pathways in asthma remains not fully understood, recent works have highlighted the role of Th17-driven inflammation in T2low asthma (27–29). Th17 CD4+ cells produce IL-17 and IL-22, two key cytokines involved in neutrophilic inflammation in the context of antimicrobial host defense, and in the regulation of epithelial function and repairing. These cytokines are both also implicated in inflammatory disease pathogeny such as rheumatoid arthritis or psoriasis (30). As neutrophilic inflammation has been associated with T2low asthma and severe asthma (22, 23), a role for Th17-type cytokines in asthma and airway remodeling has been rapidly postulated and gradually investigated over the past few years.
In this review, after going over the molecular and cellular mechanisms of the Th17 pathway, we will further scrutinize the existing evidences of the involvement of the Th17-type cytokines in asthma and airway remodeling. We will also comprehensively focus on the biological clues involving IL-17 and IL-22 in each of the elementary pathological component of airway remodeling. Eventually, an updated overview of the perspectives of Th-17 targeted therapies in asthma will be provided.
TH17-Cytokine Producing Cells and TH17 Differentiation
In the 2000's, a subpopulation of CD4+ T cells, distinct from Th1 et Th2 cells, and characterized by the production of IL-17A (also named IL-17), IL-17F and IL-22 was identified and named accordingly to these cytokines “Th17 cells” (31–35). Their role in general inflammatory pathophysiology and more specifically in respiratory diseases and asthma has rapidly been investigated and led to the concept of a “Th17 pathway” and Th17-type cytokines (IL-17, IL-17F, and IL-22). Alongside Th17 cells, other cell types were subsequently found to be sources of Th17-type cytokines. Moreover, a subtype of CD4+ T cells producing only IL-22 but not IL-17 (Th22 cells), was later discovered (36, 37). More recently, a specific subset of innate lymphoid cells (ILC), named ILC3, has emerged as another important local cellular source of IL-17 and IL-22, especially in asthma (38, 39). Eosinophils were also found to produce IL-17 in asthma (40). Other cell types have been shown to produce Th17-type cytokines but their contribution to asthma pathophysiology is not fully elucidated: γΔ T cells, CD8+ T cells, B cells, Natural Killer T cells (41–46). Of note, mixed cytokine phenotypes have been identified in asthmatic patients such as Th2/Th17 (co-expressing IL-4 and IL-17) and Th1/Th22 [co-expressing Interferon (IFN) and IL-22] profiles (47, 48). This might be of interest as in vitro data seem to indicate that IL-4 and IFN-g play a role in Th1/Th2 polarization of human bronchial epithelial cells and may contribute to airway remodeling particularly through IL-4-induced expression of RUNX2 in epithelial cells which stimulates TGFβ production (49).
Th17 differentiation is dependent on the high expression of the master transcription factor Retinoic acid receptor- related Orphan Receptor-γt (RORγt) (and to a lesser extent RORα), leading to Th17-type cytokine production (50, 51). Th17 differentiation is initially promoted either synergistically by IL-6 and TGF-β, or by IL-1ß, respectively through STAT3, SMADs, and AKT/mTOR and p38 pathway activations (52–61) (Figure 1). Aryl hydrocarbon receptor transcription factor ligands can also promote Th17 differentiation by binding their cytosolic receptor (62, 63). Amplifying auto-feedback loops involving IL-21 and IL-23 ensure stabilization of Th17 differentiation and proliferation of Th17 cells (44, 52). Of note, type 1 and 2 interferons, IL-27, and IL-4 cytokines have the ability to inhibit Th17 differentiation (64–67). Triggers of Th17 differentiation are multiple, including microbial environment (of which microbiome composition), sodium homeostasis, allergy, complement activation and inorganic particle exposure (61). Interestingly, in mice, the exposure to microbiota promotes the expansion of Th17 cells, ILC3, and regulatory T cells exhibiting characteristics of Th17 differentiation (expression of RORγt), which negatively regulates Th2 response, in intestinal mucosa (68). Th17 regular function is currently thought to promote tissue inflammation in order to ensure early clearance of extracellular pathogens for which Th1 and Th2 responses are insufficient (61, 69, 70). Th22 differentiation requires IL-6 and TNFa co-stimulation (Figure 1). IL-22 can also be co-secreted by Th1 and Th2 cells. Aryl hydrocarbon receptor has been suggested as the master transcriptional factor of Th22 cells but is not the only Th22 differentiation determinant (71).
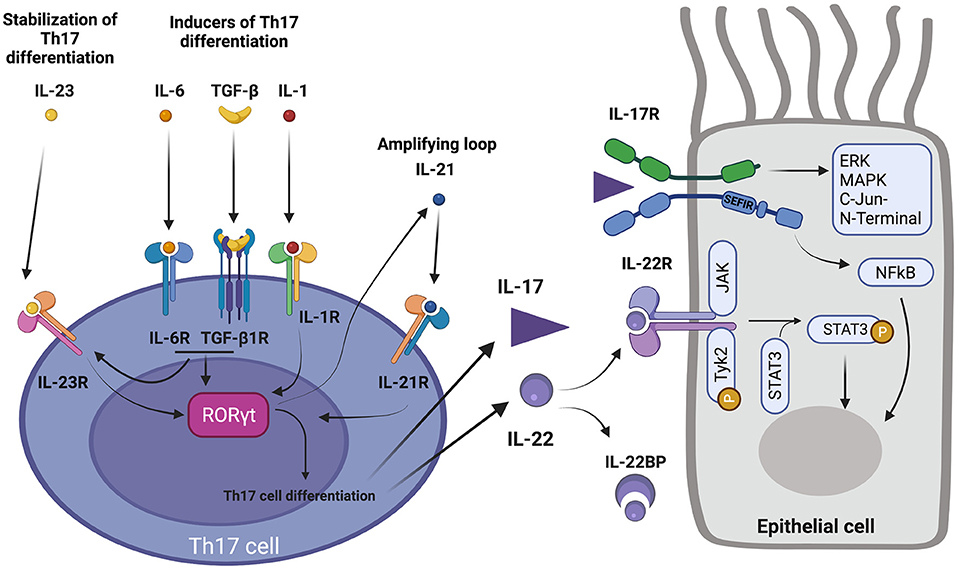
Figure 1. Th17 pathway. (Left) acquisition of Th17 differentiation. 1-IL-6 and TGFβ, and IL-1β promote RORγt expression and Th17 differentiation. 2-IL-6 and TGFβ induce IL-23R expression which allows IL-23 to stabilize Th17 differentiation. 3-Th17 differentiation also includes secretion of IL-21 which in turn enhances Th17 differentiation, acting as a positive feedback loop. 4- Secretion of IL-17 and IL-22 by Th17 cell. (Right) action of IL-17 and IL-22 on their respective receptors and activation of corresponding downstream pathways in an epithelial cell. 5- IL-22 binding protein (IL-22BP) regulates action of IL-22. 6-IL-22R activation induce Janus Kinase 1 (JAK) and Tyk2 associated kinases which lead to activation of transcription factor STAT3 by phosphorylation. 7-IL-17R binding by IL-17 activates SEFIR intracellular domain which activates ERK, MAPK, C-Jun N-terminal pathways, and transcription factor NF-κB.
TH17 Cytokines and Pathways
IL-17A, initially known as CTLA-8, belongs to a family of cytokines including IL-17A, IL-17B, IL-17C, IL-17D, IL-17E (IL-25), and IL-17F. Among them, IL-17A (also known as IL-17) and IL-17F have been the most comprehensively studied members, and have been shown to play a physiological and pathophysiological role in humans (61). IL-17A mainly exists under a homodimeric form (72). IL-17A promotes inflammation through stimulation of granulopoiesis, induction of neutrophil-attractant and neutrophil-activating cytokines and chemokines (CXCL1, CXCL2, CXCL5, CXCL8, CXCL9, CXCL10, IL-6, G-CSF, and GM-CSF) (73–77). IL-17 effects are driven through its dimeric receptor IL-17R. In humans, IL-17R exists under several forms, of which IL-17RA and IL-17RC are respectively homodimeric cognate receptors for IL-17A and IL-17F. IL-17RA expression is ubiquitary (hematopoietic cells and structural non-hematopoietic cells like epithelial cells, endothelial cells, fibroblasts) conversely to IL-17RC, which is exclusively expressed in structural non-hematopoietic cells. Signaling via heterodimeric cytokine IL-17A/F and heterodimeric receptor IL-17RAC is also possible (78–80). IL-17 effects are transduced by the SEFIR intracellular domain of IL-17R. Downstream IL-17R signaling noticeably activates NFκB, ERK, and MAPK and c-Jun N-terminal kinase pathways (61) (Figure 1).
IL-22 belongs to the IL-10 cytokine family. It plays an important role in host defense in barrier tissues, and in epithelial protection and regeneration after injury (42, 46, 81–85). It enhances proliferation and migration of cells expressing IL-22R, while it inhibits their apoptosis and differentiation (86–89). The antimicrobial activity of IL-22 includes the stimulation of S100 protein and defensin productions (90). Interestingly, IL-22 production is dependent of the Aryl hydrocarbon receptor transcription factor (62, 91). IL-22 binds a heterodimeric receptor made up of the IL-22R1 and IL-10R2 subunits. Interestingly, IL-22R is only expressed on structural cells (epithelial and endothelial cells, fibroblasts, and smooth muscle cells) and not in hematopoietic cells. This repartition of IL-22R is highly relevant regarding the potential role of IL-22 in airway remodeling as these cell types are involved in airway remodeling processes. IL-22R mainly activates downstream STAT3 through transduction signals involving the receptor associated Janus kinases Jak1 and Tyk2 (92–94) (Figure 1). A soluble receptor for IL-22 [IL-22 binding protein (IL-22BP)], sharing high homology with IL-22R, contributes to IL-22 pathway regulation (95). IL-22 also plays a role in the tissue recruitment of neutrophils by inducing the production of neutrophil attracting chemokines, notably that of CXCL1 and CXCL5 by bronchial epithelial cells (96).
Interactions between IL-17 and IL-22 remain mostly not well-understood. Initially thought to be exclusively part of the same immunological response because of their common cellular source (Th17 cells, ILC3), each of these cytokines is also produced independently by many other cells (Th1 cells, Th22 cells, mixed Th1/Th22 cells for IL-22; Th2/Th17 for IL-17) (42, 48). Several studies suggest a mutual ability of IL-17 and IL-22 to inhibit each other production and respective Th17 and Th22 differentiation (97–99).
Beyond the homeostatic functions of IL-17 and IL-22, Th17 response is known to be involved in several chronic inflammatory diseases such as rheumatoid arthritis, psoriasis, psoriatic arthritis, ankylosing spondylitis, Crohn's disease, and atopic dermatitis (100–103). Among these diseases, some are particularly characterized by neutrophilic inflammation and tissue remodeling, such as psoriasis and atopic dermatitis. As a consequence, the role of IL-17 and IL-22 in asthma, has been questioned over the last two decades.
Involvement of IL-17 and IL-22 in Asthma
The contribution of IL-17 in asthma was first suspected from the identification of asthmatic patients with neutrophilic inflammation in sputum as a distinct cluster of non-eosinophilic severe asthma (104, 105). Knowing the central role of IL-17 in neutrophil migration, recruitment and activation, multiple studies investigated the association between IL-17 levels in serum, sputum, bronchial biopsies and asthma. Most studies found not only increased IL-17 levels in asthmatic patients (28, 40, 106–108), but also positive correlation between IL-17 levels and asthma severity and association with airway neutrophilia (106, 109, 110). The presence of cells expressing IL-17 (ILC3 cells, Th17 cells) was also positively correlated with asthma severity (22, 39). Interestingly, specific Th2/Th17 CD4+ T cells, producing both Th2 and Th17 cytokines were identified in bronchoalveolar lavage (BAL) of asthmatic patients. Severe asthma was associated with increased Th2/Th17 cells predominantly polarized toward IL-17 production (Th2/Th17high) in BAL (111). The involvement of IL-17 in asthma pathogenesis was further confirmed in murine models, which globally found that ovalbumin sensitization (inhaled, intraperitoneal, or epicutaneous route) followed by inhaled challenge was associated with Th17 response, up-regulation of IL-17 in airways, and that up-regulation of IL-17 enhanced airway neutrophilia and steroid-resistant AHR (112–115). IL-17 production is also increased in obesity-associated murine models of asthma and asthmatic pediatric patients (38, 39, 116). Data from murine models are summarized in Table 1.
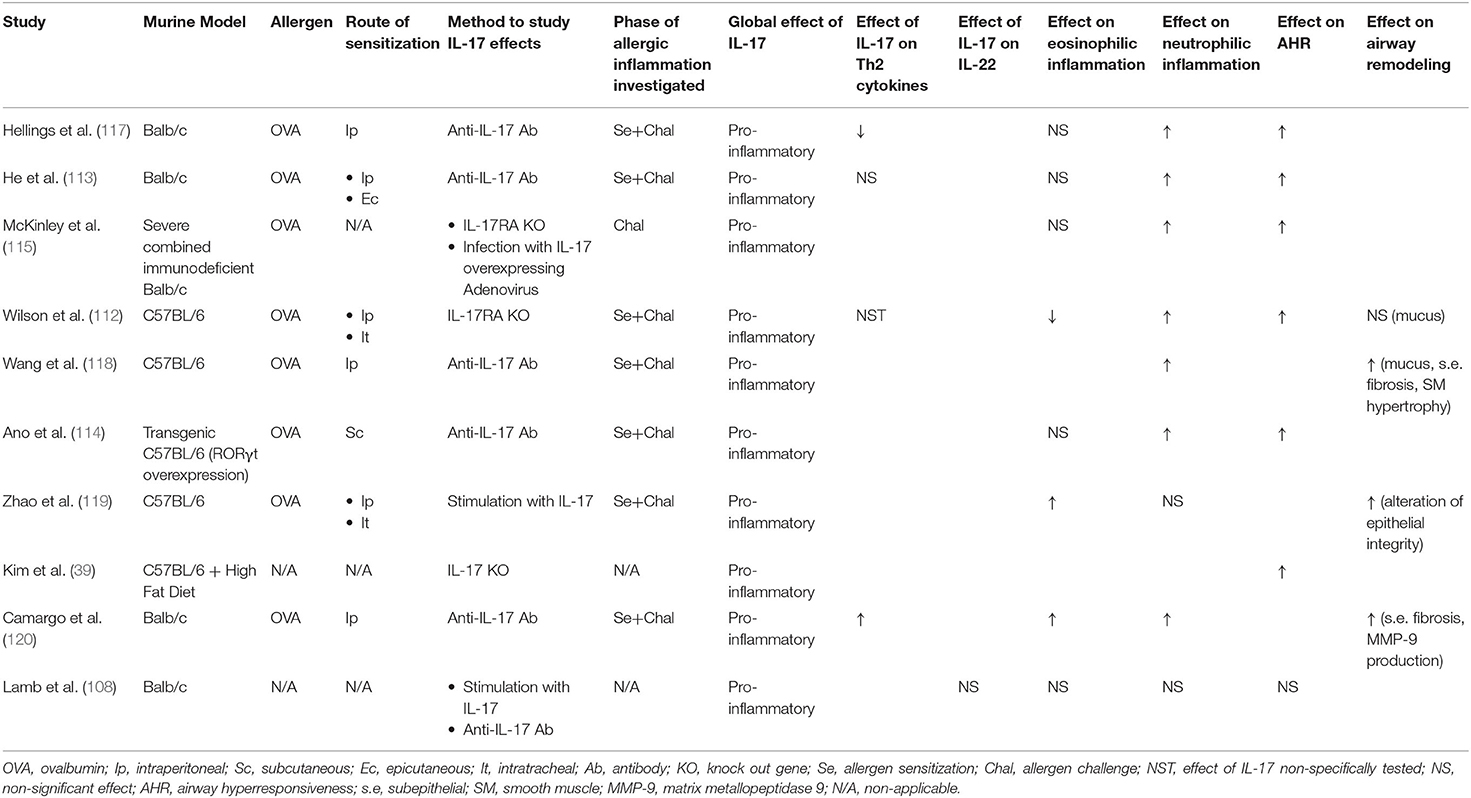
Table 1. Summary of data about the role of IL-17 in asthma from murine models.
Similarly, IL-22 and IL-22R upregulation was suggested in several studies in serum and in bronchial biopsies from asthmatic adults and children (predominantly in severe asthmatic patients) and in murine models of asthma, as well as in patients with other allergic respiratory diseases such as atopic rhinitis (99, 121–124). IL-22 producing cells, particularly lymphocytes, are also increased in bronchial biopsies from asthmatic children (123).
However, the role of IL-22 as a protective or a pathogenic actor in asthma remains less clear than that of IL-17, as pathogenic effect failed to be constantly reproduced in murine models. Indeed, conflicting evidences have shown that IL-22 could have deleterious but also beneficial effects on airway epithelium in asthma (42, 95, 125, 126) (Table 2). Hence, a protective action of IL-22 on airway allergic inflammation, in particular on airway eosinophilic inflammation and Th2 cytokine production, on AHR and even on some pathological feature of airway remodeling (goblet cell hyperplasia) has been described in several studies (127–129, 131). On the other hand, as underlined by Hirose et al. in an extensive review about dual effects of IL-22 on airway epithelium, atopic dermatitis models and some murine models of asthma found pro-inflammatory effects of IL-22, with enhanced AHR and airway remodeling (mucus hyperproduction and goblet cell hyperplasia, and smooth muscle cell hyperplasia) (95, 99, 133, 134). These contrasting results have been discussed in a previous review (125) (Table 2). One of the main differences between these studies is the route of sensitization, which may account for a differential effect of IL-22 with a protective effect associated with the intraperitoneal route, and pro-inflammatory effects associated with the subcutaneous route. Additionally, the protective or pro-inflammatory effects of IL-22 were differently exerted according to the phase of the allergic reaction (sensitization or challenge). In this regard, the work of Leyva-Castillo et al., which tried to mimick the atopic march, is interesting. They found that epicutaneous sensitization with ovalbumin in mice further challenged with intranasal ovalbumin led to increased expression of IL-22 in serum and lungs, which was associated with enhanced AHR and mixed neutrophilic and eosinophilic airway inflammation. This was mainly observed with epicutaneous sensitization and not intraperitoneal sensitization (132). Overall, the precise role of IL-22 in asthma is still unclear, probably dependent on many parameters, such as the timing of action during the allergic process, the route of sensitization, as well as the immunological microenvironment regulating the Th22 response (Table 2). One can hypothesize that when IL-22 response is imbalanced, its physiological role may turn into a harmful action.
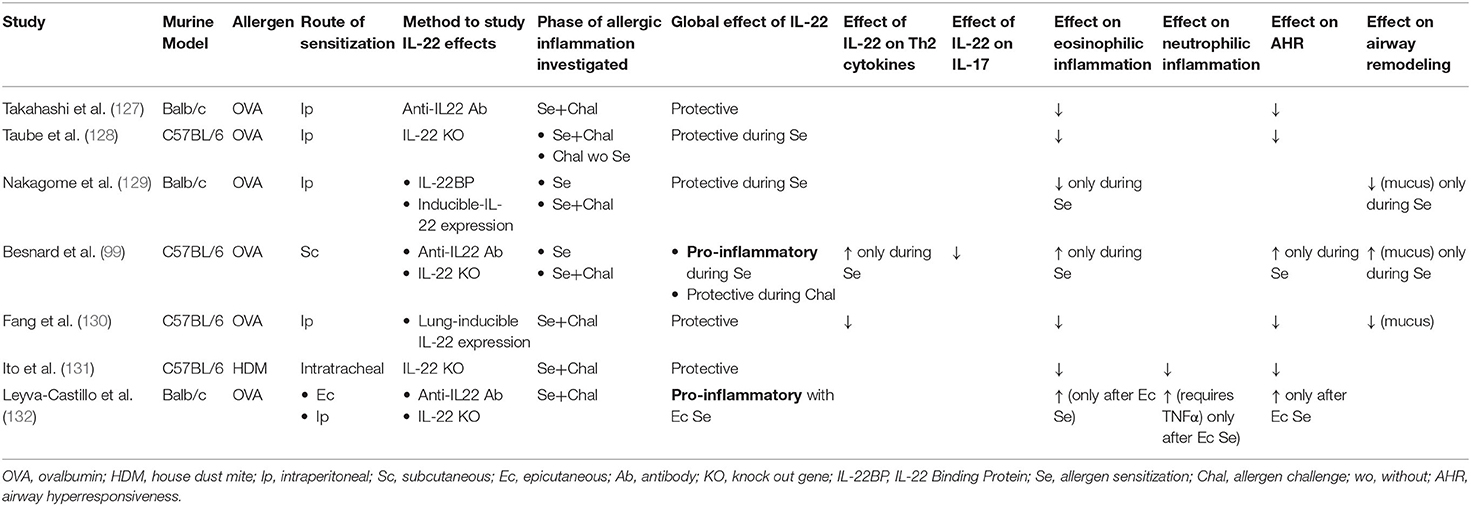
Table 2. Summary of studies showing conflicting evidence about the role of IL-22 in asthma murine models.
Of interest, several studies have shown a global stimulation of the Th17 response in asthma, with simultaneously increased expression of IL-17 and IL-22 in peripheral mononuclear blood cells (PMBC) and bronchial biopsies from asthmatic patients, this increase being even higher in severe asthmatic patients. Furthermore, increased levels of IL-17 and IL-22 were insensitive to steroids, a characteristic frequently found in patients with neutrophilic airway inflammation and with airway remodeling and specific to severe asthma (25, 116, 135, 136). In a recent work, our group also reported airway remodeling and neutrophilia in a murine model of allergic asthma induced by dog allergen. In this model, IL-17 and IL-22 expression were both upregulated (137).
In the era of “Omics,” two studies looked at transcriptomics in blood, sputum and bronchial biopsies of asthmatic patients, mainly in the U-BIOPRED cohort. Thereby, Badi et al. identified in asthmatic patients a transcriptomic signature previously identified in atopic dermatitis, a well-known Th22-driven inflammatory disease. This signature included upregulation of Th17 and Th22 pathway-related genes. Enrichment of this signature in biological samples from asthmatic patients was positively correlated with asthma severity (138). Similarly, Lamb et al. found an upregulated Th17 transcriptomic signature in bronchial biopsies from asthmatic patients (108).
Finally, both IL-17 and IL-22 levels have been found to be negatively correlated with FEV1 (25, 136, 139), a hallmark of severe asthma with airway remodeling. Overall, the involvement of IL-17 in asthma is well-established. Although the involvement of IL-22 in asthma seems very likely, its exact role is still to be clarified.
IL-17, IL-22, and Neutrophilic Inflammation in Asthma
Airway inflammation contributes to the airway remodeling process (10). In asthma, airway inflammation is usually characterized by T2 inflammation with eosinophilic cell infiltration of airways. Nevertheless, airway neutrophilia is classically associated with severe asthma (23). Neutrophilic asthma is also associated with steroid resistance, poorer lung function (FEV1) and lower reversibility of airway obstruction, which can be accounted by airway remodeling (22, 25, 140–142).
IL-17 and IL-22 are involved in neutrophilic inflammation in asthma. As demonstrated on bronchial biopsies from a large cohort of asthmatic patients, airway neutrophilia is associated with both increased IL-17 and IL-22-producing cells (22, 108). More recently, Badi et al. showed in a cohort of asthmatic patients that the degree of enrichment in atopic dermatitis-similar transcriptomic signature, including overexpression of Th17 and Th22-related genes, was strongly associated with severe neutrophilic asthma (138).
Indeed, IL-17 is a key cytokine in the recruitment and activation of neutrophils (69, 76). Neutrophilia is correlated with IL-17 expression in airway epithelium (108). In murine models of asthma, IL-17 stimulates and orchestrates airway neutrophilic infiltration by inducing the expression of neutrophil-attractant chemokines like CXCL1, CXCL2, IL-6, and IL-8 in humans (75, 108, 112–115, 117, 143).
Though its place still needs to be clarified in airway neutrophilia, IL-22 seems also to be an important factor in neutrophil recruitment (144, 145). It is able to also enhance production of neutrophil recruitment associated chemokines like CXCL8 (in humans), CXCL1, CXCL2, CXCL3, CXCL5, and IL-1b, particularly in mold allergen-induced asthma murine models (132, 137, 143, 144, 146). Interestingly, several data suggest a pro-inflammatory effect of IL-22 synergistically with IL-17 on airway neutrophilia by inducing the expression of chemokines by epithelial cells but also by airway smooth muscle cells (98, 108, 143, 147).
Overall, the association between both of these cytokines and neutrophilic inflammation is highly relevant, knowing that neutrophils are thought to be an effector cell of airway remodeling, particularly through the release of proteolytic enzymes like elastases and metalloproteases (notably MMP-9), thus participating in structural change of the extracellular matrix of bronchial epithelium (148, 149).
Involvement of IL-17 and IL-22 in Airway Remodeling
Experimental data provides substantial arguments for an involvement of IL-17 and IL-22 in airway remodeling through their contribution to the diverse pathobiological processes that lead to airway remodeling: subepithelial fibrosis, mucus hyperproduction, airway inflammation and smooth muscle cell hyperplasia and hypertrophia.
Out of the field of asthma, IL-17 and IL-22 enhance airway remodeling in murine models of lung injury by pollutants (airway neutrophilic infiltration, epithelial desquamation, subepithelial fibrosis) (150). They are also involved in tissue remodeling in other inflammatory diseases (bone remodeling in psoriatic arthritis, skin remodeling in atopic dermatitis) as well as in other chronic respiratory diseases, such as chronic obstructive pulmonary disease (COPD) (76, 100, 151–158).
The enhancement of airway remodeling by IL-17 has been confirmed in murine models of asthma in which its blockade by anti-IL-17 antibodies or genetic deficiency reduced mucus hypersecretion, goblet cell hyperplasia, subepithelial collagen deposition and airway smooth muscle layer thickening together with airway neutrophilia (118, 119). Interestingly, IL-17 seemed to increase epithelium permeability to allergen, indicating a loss of epithelial integrity, also observed in airway remodeling (119). IL-17-induced airway remodeling seemed to be dependent on IL-6 (119), and to be partially mediated through heparin-binding epidermal growth factor (HB-EGF) (118).
Regarding the specific topic of airway remodeling, the understanding of the role of IL-22 suffers from conflicting evidences. Fang et al. found a modulatory effect of IL-22 with decreased eosinophilic inflammation and Th2 cytokine secretion, decreased AHR but also reduced mucus production and goblet cell hyperplasia in lung-specific IL-22 transgenic model of ovalbumin-induced asthma (130). Paradoxically, in another murine model of dog allergen-induced asthma, which reproduces the characteristics of airway remodeling, Aryl hydrocarbon receptor antagonism decreased IL-22 levels (but not IL-17) in lungs together with decreased airway remodeling. The reduction of subepithelial fibrosis, mucus hyperproduction and smooth muscle thickness after treatment with Aryl hydrocarbon receptor antagonist suggests an association between airway remodeling and IL-22 (137). Interestingly, as previously underlined, IL-22R is not expressed on hematopoietic cells but only on structural cells (epithelial, endothelial cells, fibroblasts, smooth muscle cells) which are major contributors of airway remodeling (42).
Interestingly, B cells, particularly regulatory B cells, seem to have a beneficial regulatory effect on AHR and airway remodeling in a murine model of asthma (159). Although there is no specific data on the role of Th17 cytokines in this regulatory process, several works have showed that Th17 cells, IL-17 and IL-22 (though this is an indirect effect as B cells do not express IL-22R) may exert an important role of recruitment, stimulation and maturation of B cells and a regulatory role of B cell response in mucosal barriers (160–166). Subsequently, Th17 cytokines might also contribute to airway remodeling by an indirect effect of regulation of B cell response.
Respiratory tract infections, and in particular viral infections, may also contribute to the pathophysiology of asthma and the development of airway remodeling as a bronchial chronic inflammation trigger, especially in T2low asthma (167–169). In this setting, knowing the role of IL-17 and IL-22 in antimicrobial host defenses in mucosal barriers (170–172), infections may play an important role of trigger of Th17-induced airway remodeling in patients with recurrent infection phenotype, by inducing chronically high levels of Th17 cytokines in lungs.
Overall, IL-17 involvement in airway remodeling seems well-established while it is less clear for IL-22. However, in vitro data provide numerous clues in favor of the involvement of both of these cytokines in several biological processes contributing to airway remodeling (Figure 2).
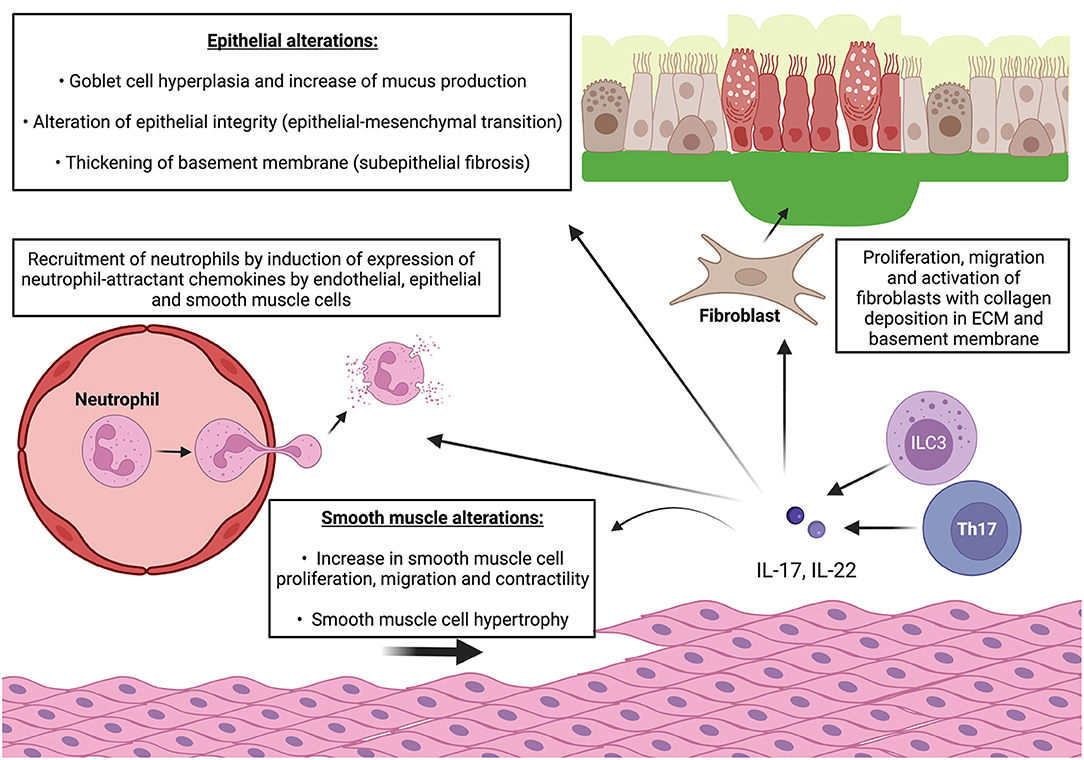
Figure 2. IL-17 and IL-22 cooperate to induce features of airway remodeling in asthma. ECM: extracellular matrix. Pro-inflammatory effect: release of IL-17 and IL-22 by Th17 cells and ILC-3 induce expression of neutrophil-attractant chemokines by endothelial cells and enhance neutrophil recruitment and diapedesis. IL-17 and IL-22 also induce expression of neutrophil-attractant and neutrophil-activating chemokines by epithelial cells and smooth muscle cells which lead to local neutrophilic inflammation with release of proteolytic enzymes (elastase, metalloproteases). Epithelial alterations: IL-17 and IL-22 also increase production of mucus by goblet cells and promotes goblet cell hyperplasia. They also alter epithelial integrity by enhancing E/N-cadherin switch which leads to epithelial-mesenchymal transition. Smooth muscle alterations: IL-17 and IL-22 promote smooth muscle cell proliferation, migration, and contractility (only IL-17). Subepithelial fibrosis: IL-17 and IL-22 stimulate fibroblast proliferation and switch to pro-fibrotic phenotype and promote collagen production and deposition in ECM relsulting in thickening of basement membrane. IL-22 also promotes migration of myofibroblast in smooth muscle cell bundles which contributes to local fibrosis and smooth muscle contractility.
Subepithelial Fibrosis
Subepithelial fibrosis is a major feature of airway remodeling in asthma. It is mainly characterized by increased collagen deposition in the extracellular matrix (ECM), resulting in thickening of basement epithelial membrane of bronchial mucosa and reduced airway compliance. This phenomenon contributes to airway obstruction. Mainly fibroblasts, but also smooth muscle cells are the cellular effectors of subepithelial fibrosis in asthma. TGF-β and IL-11 are the main profibrotic cytokines which stimulate collagen secretion by fibroblasts (9–12).
In chronic inflammatory bowel diseases, the effect of Th17-type cytokines on subepithelial fibrosis through their action on myofibroblasts is well-described (173). If Th2-type cytokines IL-4 and IL-13 involvement in subepithelial fibrosis in asthma is acknowledged (174, 175), pro-fibrotic effects of IL-17 are also well-documented. Indeed, IL-17 induces the production of pro-fibrotic cytokines IL-6 and IL-11 by fibroblasts obtained from bronchial biopsies of asthmatic patients (40). Additionally, Bellini et al. showed that circulating fibrocytes isolated from exacerbating asthmatic patients had increased expression of IL-17RA as compared to fibrocytes from control healthy subjects (175). Both IL-17A and IL-17F (but not Th2 cytokines) have demonstrated the ability to induce release of pro-neutrophilic chemokines (CXCL1, CXCL8, and TNFa) by fibrocytes, and to enhance fibrocytes switching toward a profibrotic phenotype (increased production of α-smooth muscle actin (SMA) mediated by CXCL8, of collagen, and of proangiogenic factors like VEGF and angiogenin) and fibrocyte proliferation (175, 176). This proliferative effect could be mediated by IL-6 and the Leukemia inhibitory factor (LIF), which were both found increased after IL-17 stimulation in this model (175). Al-Muhsen et al. suggested that eosinophils could also contribute to the IL-17-driven subepithelial fibrosis process. In vitro, the stimulation of eosinophils from asthmatic patients by IL-17 (but not by Th1 nor Th2 cytokines) induces the expression of the pro-fibrotic cytokines IL-11 and TGF-β1. However, this effect requires a co-stimulation with both IL-17A and IL-17F and can be synergistically enhanced by co-stimulation with IL-23, a known inducer and upregulator of IL-17R. This effect is driven by the p38 MAPKinase pathway (177). More recently, an original work highlighted that IL-17 induces autophagy and mitochondrial dysfunction in human fibroblasts from asthmatic patients, associated with impaired apoptosis and increased pro-fibrotic response (178).
Similar data suggests an involvement of IL-22 in subepithelial fibrosis in asthma. Liu et al. showed that blood from asthmatic children containing high levels of IL-22 and IL-22-producing lymphocytes had the ability to stimulate embryonic fibroblast proliferation and production by fibroblasts of collagen (α-1 and α-2 chains of collagen 1), thus suggesting a contribution of IL-22 to subepithelial fibrosis in asthma through its action on fibroblasts. This effect is linked to the activation of JAK/STAT3 signaling pathway by IL-22R1 (123).
Moreover, fibroblasts from bronchial biopsies of asthmatic patients have also been shown to promote Th17 differentiation of naïve CD4+ lymphocytes and secretion of IL-17 and IL-22, which in turn stimulates the production of pro-fibrotic cytokines (IL-6, TGFb) by fibroblasts but also cytokines favoring Th17 differentiation (IL-23, IL-6, and IL-1b). This suggests that the microenvironment can also promote a Th17 response in asthmatic patients, through the existence of amplifying loops in the fibrosing processes (179).
Subepithelial fibrosis also requires ECM remodeling involving proteolytic activity. In this setting, several studies interestingly showed, in murine models of asthma exacerbation, that IL-17 enhanced the production of metalloproteases MMP-9 and MMP-12, neutrophil elastase, and the myeloperoxidase activity, as well as ECM collagen deposition (120, 180).
Finally, biopsies from moderate to severe asthmatic patients showed increased expression of IL-17 in epithelial and specifically in subepithelial compartments, the latter being the location of subepithelial fibrosis, thus arguing in favor of a likely involvement of IL-17 in subepithelial fibrosis in humans. However, downregulation of IL-17 by a 2 week-course of oral steroids did not affect collagen deposition (181).
Taken together, these data suggest that IL-17 and IL-22 can promote subepithelial fibrosis in asthma by a direct action on fibrocytes and fibroblasts, activating collagen production and deposition in ECM, promoting metalloprotease activity, angiogenesis and cell proliferation, enhancing Th17 response and in situ neutrophil recruitment, and inhibiting fibroblasts apoptosis.
Mucus Hyperproduction and Goblet Cell Hyperplasia
Another critical aspect of airway remodeling is represented by changes in mucus secretion function. From a pathological point of view, in asthma, airway remodeling is associated with goblet cell hyperplasia/metaplasia and mucus hypersecretion. From a biochemical point of view, changes in mucus rheology and composition are also observed (9–12).
Normal mucus composition is dominated by mucin glycoproteins, of which MUC5AC and MUC5B are the most abundant. It has been demonstrated in vitro that IL-17 (but not Th2 cytokines) was able to increase MUC5AC and MUC5B expression by bronchial epithelial cells (182–184). Stimulation of MUC5AC production is dependent on NfκB transcription factor (183). IL-17 also induces IL-6 production through activation of the JAK2 pathway, which in turn activates the ERK pathway, responsible for enhancement of MUC5B production (182).
At the tissue-scale, IL-17 stimulation increases the number of goblet cells which are the cellular source of mucus in airways (185, 186). Moreover, a recent work seems to implicate IL-17 in goblet cell metaplasia (187).
In addition, a chronic exposition to Aspergillus-induced model of allergic asthma in mice showed features of airway remodeling, noticeably airway inflammation and goblet cell hyperplasia with mucus hyperproduction, associated with lung function alteration. Dectin-1 is a C-type lectin receptor (CLR) that is primarily responsible for β-glucan recognition and control of fungal infection. Interestingly, the features of asthma in this model of chronic exposure to aspergillus were reduced by Dectin-1 deficiency which also downregulated IL-17 and IL-22 production. Of note, neutralization of IL-22 improved lung function in this model. These data suggests that IL-22 could be also involved in alteration of mucus secretion in asthma (146).
Epithelial-Mesenchymal Transition (EMT) and Epithelial Alterations
Epithelial structure alteration is a key component of airway remodeling. In this process, most structural changes lead to altered epithelium integrity, epithelial cell damage and apoptosis, as well as increased epithelial permeability. Among these changes, a phenotypic switch to mesenchymal cell characteristics is observed in epithelial cells, thus achieving the epithelial-mensenchymal transition (EMT). One of the most critical changes concerns junction proteins, with loss of intercellular tight junction, especially downregulation of the expression of E-cadherin, leading to increase in epithelial permeability (9–12). Additionally, acquisition of a mesenchymal phenotype is likely to contribute to airway fibrosis (188).
In this setting, a potential role for IL-22 is very likely regarding its role in protection, restauration and maintenance of epithelial barrier homeostasis (125, 156). In vitro, TGF-β1-induced EMT in primary epithelial bronchial cells of severe asthmatic patients is enhanced by addition of IL-22. IL-22-enhanced EMT is characterized by acquisition of morphologic characteristics of mesenchymal cells (“spindle cells”), further decrease in E-cadherin expression and further increase in N-cadherin expression. This effect seems to be driven by Zeb1 transcription factor (121).
IL-17 has been involved in EMT in various respiratory diseases, like idiopathic pulmonary fibrosis, lung cancer, or COPD (189–192). In a translational work on obliterans bronchiolitis, Vittal et al. found that IL-17 could induce EMT, partially through TGF-β production, resulting in decreased expression of E-cadherin (193). In in vitro models, IL-17 induces, synergistically with IL-4 and TGF-β, reentering in cell cycle (proliferation) of epithelial cells, fibroblast morphotype switch, downregulation of E-cadherin, and upregulation of α-SMA (157, 194). A synergistic effect of cigarette smoke together with IL-17 on EMT was also reported by Ma et al. (191).
Interestingly, neutrophils from asthmatic patients were recently shown to have the ability to induce EMT (195). Given the preponderant role of IL-17 and IL-22 in neutrophil recruitment, this supports the involvement of both of these cytokines in EMT in asthma.
Increase of Airway Smooth Muscle Mass
Increase of airway smooth muscle mass participates to airway obstruction in asthma and is one of the hallmark of airway remodeling (9–12). It results from several mechanisms including increased smooth muscle cell proliferation (hyperplasia), smooth muscle cell hypertrophy, abnormal migration properties leading to increased number of cells in smooth muscle bundles, especially myofibroblasts which also contributes to local fibrosis (12).
In in vitro models, IL-17A and IL-17F, as well as IL-22 promote airway smooth muscle cell (ASMC) proliferation, induce ASMC migration through activation of their respective cognate receptors and downstream activation of MAPKinases and NFκB pathways, and inhibit ASMC apoptosis (196, 197). IL-17-induced ASMC migration is also mediated by Growth-Related Oncogens (GRO), GROα being CXCL1, also involved in neutrophil migration (198). Neutrophils contribute to smooth muscle cell hyperplasia, which is critical given that IL-17 and IL-22 play an important role in neutrophil recruitment, particularly in airway smooth muscle bundles (199).
Additionally, translational data from two studies indicate that IL-17 but not IL-22 can enhance smooth muscle cell contraction via NF-κB/RhoA/ROCK2 signaling cascades, which are involved in regulation of phosphorylation of myosin light chain and thus ASMC contractility (200–202).
Overall, IL-17 and IL-22 stimulates ASMC proliferation and contractility, smooth muscle cell infiltration by neutrophils, migration of myofibroblasts into the smooth muscle layer, inhibit ASMC apoptosis, leading to thickening of airway smooth muscle mass.
Therapeutic Directions
As they are tightly linked to asthma immunobiology, and especially to severe asthma, IL-17 and IL-22 appear as promising targets in the treatment of severe asthma. In regard to the overview of the very likely role of IL-17 and IL-22 in airway remodeling in asthma that we provide in this review, this therapeutic perspective is particularly interesting as there is currently no specific pharmacological treatment targeting airway remodeling in asthma, whereas several biotherapies are available for patients with severe T2high asthma. To date, mainly bronchial thermoplasty, an interventional endoscopic procedure, offers a treatment option for these patients (203). Recently, tezepelumab, a human monoclonal antibody blocking thymic stromal lymphopoietin (TSLP), an epithelial-cell–derived cytokine which, as an alarmin, plays an early role in the inflammatory cascade in asthma, showed significant reduction of exacerbation rate in non-selected patients with severe asthma, including patients with non-T2 asthma. The effect was consistent in the subgroup of patients with low T2 biomarkers (low blood eosinophil count and low FeNO), suggesting that the effectiveness of this biotherapy is maintained in patients with non-T2 asthma (204). Of note a recent phase 2 clinical trial did not show any improvement on airway remodeling (epithelial integrity and basement membrane thickness) (205). Nevertheless, tezepelumab may offer a therapeutic option for patients with severe non-T2 asthma and could prevent the development of non-already established airway remodeling by early reduction of bronchial inflammation.
In the setting of Th17-targeting treatment of severe asthma, IL-17 blockade strategy has been the most studied so far (Table 3). According to the results obtained in the previously mentioned murine models of asthma, the effect of brodalumab, a humanized anti-IL17 monoclonal antibody already approved in the treatment of psoriasis, was investigated in a randomized controlled trial (RCT) in non-selected patients with moderate-to-severe asthma (206). No significant difference was found on the primary outcome criteria, which was the control of asthma based on the Asthma Control Questionnaire (ACQ). Nevertheless, the results of this study have been extensively discussed on two points. First, there was no selection of patients according to phenotype or endotype, and, as a consequence, many patients with T2high asthma may have been included, a category of patients for whom a beneficial effect of anti-IL-17 treatment would be less expected than for patients with neutrophilic asthma. Second, the primary outcome criteria was improvement of asthma control, a criteria which has not been found improved by any other already-registered biotherapies in severe asthma, and which also may not reflect improvement in airway remodeling. Interestingly, a subgroup of patients with high FEV1 reversibility at inclusion demonstrated a significant difference in asthma control, suggesting that patients with not-yet-established permanent obstruction and airway remodeling might benefit from IL-17 blockade strategy in the early phase of airway remodeling (206, 209, 210). Recently, a phase I trial studying the effect of a bispecific anti-IL-13 and anti-IL-17A antibody in asthma was early terminated due to high frequency of treatment immunization (207). Additionally, experimental data suggest that blockade of IL-17 may restore sensitivity to steroids, including airway remodeling features (211). Lastly, a phase 2a RCT found that risankizumab, an anti-IL-23 monoclonal antibody, induced downregulation of Th17 associated transcription factors, and showed deleterious effects of outcomes (time to first worsening, annual rate of asthma worsening) in an unselected population of severe asthmatic patients (208). This result may put in perspective the protective aspects of Th17 response in asthma (anti-microbial defense in particular, although the proportion of infectious exacerbations was not specifically reported in this study) (209). Therefore, more data are needed on IL-17 blockade strategy. Trials selecting patients based on an “IL-17 phenotype” using biomarkers like sputum neutrophilia, IL-17 levels in blood or sputum, or airway remodeling biomarkers are expected to make progresses in this field.

Table 3. Summary of results from pre-clinical and clinical studies on Th17-related therapies.
Additionally, beyond the non-T2 asthma field, specific allergen immunotherapy in allergic rhinitis is associated with decreased serum levels of IL-17 mRNA and IL-17 as compared to non-treated controls (212, 213). Furthermore, the ratio between B regulatory cells (Breg) and Th17 cells (Breg/Th17 ratio) obtained from blood samples taken in the early phase of immunotherapy, positively correlates with a symptom score of response to this treatment at 3 years (214). This effect of immunotherapy on Th17 response might be driven through transdifferentiation of Th17 cells toward Treg cells, contributing to immunotolerance to allergens (215, 216). Interestingly, a recent work suggested that specific allergen immunotherapy-induced immunotolerance was associated with a decrease in “exhausted-like” Th2 cells in a murine model of chronic exposure to ovalbumin allergen and in patients with allergic rhinitis, a specific phenotype of Th2 cells characterized by the expression of CTLA-4 and PD-1 (217), which has paradoxically been proposed to contribute to the enhancement of Th2 response (218, 219). Of note, in their murine model, immunotherapy was associated with decreased neutrophil count and levels of IL-17 in BAL but increased expression of IL-17 and IL-22 in lung homogenates (217). Similarly, modulation of ILC2 exhausted phenotype might be a relevant target, particularly knowing their involvement in regulation of chronic allergic inflammation and that a subset of these cells can produce IL-17 (220, 221). Improvement in the understanding of the regulatory role of exhausted phenotype of T2 inflammation-associated cells in allergic inflammation and of their interaction with Th17 pathway may contribute to the development of therapies indirectly targeting Th17 pathway which may also impact Th17-associated airway remodeling.
Although there are no currently clinical studies of IL-22 targeting therapies in asthma, some data suggest a potential role for such therapies. First, the efficacy of the anti-IL-22 antibody fezakinumab was demonstrated in a randomized controlled trial in atopic dermatitis, a disease which shares many pathophysiological features with asthma and especially neutrophilic asthma (tissue remodeling, neutrophilic inflammation) (222). As previously mentioned, data on IL-22 blockade strategy from murine models provide conflicting evidence of the benefit of this strategy. Interestingly, Badi et al. found a predictive transcriptomic signature of response to fezakinumab in atopic dermatitis which was enriched in blood of patients with severe asthma from U-BIOPRED cohort. This enrichment was especially associated with neutrophilic asthma. The response to fezakinumab signature was correlated with Th22 related gene expression in blood and sputum (especially IL-22, CCR10, Aryl hydrocarbon receptor). IL-22 expression in sputum correlated with the enrichment of the response signature (138). Given the established role of IL-22 in epithelial physiology and in tissue remodeling (157), it might be an interesting target for the treatment of airway remodeling in asthma.
Finally, a new and potent RORγt inhibitor, administered in a mouse model of house dust mite-induced asthma which exhibited a transcriptomic signature overlapping with that of a cluster of patients with severe neutrophilic asthma from the U-BIOPRED cohort, showed a decrease in IL-22, IL-17, and CXCL1 levels in BAL fluid, in BAL neutrophilia and more interestingly an improvement in airway resistances in a dose dependent manner (108). This effect was not achieved by an anti-IL-17 antibody alone. This result may confirm the synergistic action of IL-17 and IL-22 that has been suggested in several studies (98, 108, 143, 147). An anti-inflammatory effect targeting the Th17 response might require an abolition of both IL-17 and IL-22 actions. Considering this result and this hypothesis, the inhibition of RORγt, the master driver of Th17 differentiation may be a promising track in asthma.
Overall, substantial progresses are still to be done in the field of treatment targeting the Th17 response in asthma and airway remodeling. Clinical trials relying on relevant outcome criteria and with endotype-based selection of patients are needed. Moreover, timing of administration of biotherapies targeting airway remodeling might be a critical parameter affecting results of clinical trials. Have such therapies to be administered preventively before establishment of severe and fixed (maybe irreversible) airway remodeling and obstruction? Or should we expect to reverse already-established airway remodeling? Further responses are awaited on this burning topic.
Conclusion
Many evidences are accumulating to indicate a leading role of Th17 cytokines in patients with severe T2low asthma, particularly in patients with neutrophilic asthma and airway remodeling. However, the exact role of Th17 cytokines, especially IL-22, still needs to be clarified in vivo, though an important number of experimental in vitro data show their involvement in elementary biological processes that contribute to airway remodeling (airway inflammation, subepithelial fibrosis, mucus hyperproduction, thickening of airway smooth muscle, epithelial-mesenchymal transition and alteration of epithelial integrity). Therefore, IL-17 and IL-22 are attractive therapeutic targets for new biotherapies in severe asthma while there is currently no pharmacological treatment targeting airway remodeling. Future clinical trials involving molecules targeting Th17 cytokines using appropriate outcomes and selected populations will be required to progress in this direction.
Author Contributions
VM-C wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
All figures in this manuscript were created with BioRender.com.
References
1. Pearce N, Aït-Khaled N, Beasley R, Mallol J, Keil U, Mitchell EA, et al. Worldwide trends in the prevalence of asthma symptoms: phase III of the International Study of Asthma and Allergies in Childhood (ISAAC). Thorax. (2007) 62:757–65. doi: 10.1136/thx.2006.070169
2. To T, Stanojevic S, Moores G, Gershon AS, Bateman ED, Cruz AA, et al. Global asthma prevalence in adults: findings from the cross-sectional world health survey. BMC Public Health. (2012) 12:204. doi: 10.1186/1471-2458-12-204
3. Uphoff EP, Bird PK, Antó JM, Basterrechea M, von Berg A, Bergström A, et al. Variations in the prevalence of childhood asthma and wheeze in MeDALL cohorts in Europe. ERJ Open Res. (2017) 3:00150–2016. doi: 10.1183/23120541.00150-2016
4. Reddel HK, Bateman ED, Becker A, Boulet LP, Cruz AA, Drazen JM, et al. A summary of the new GINA strategy: a roadmap to asthma control. Eur Respir J. (2015) 46:622–39. doi: 10.1183/13993003.00853-2015
5. Nordon C, Grimaldi-Bensouda L, Pribil C, Nachbaur G, Amzal B, Thabut G, et al. Clinical and economic burden of severe asthma: a French cohort study. Respir Med. (2018) 144:42–9. doi: 10.1016/j.rmed.2018.10.002
6. Bourdin A, Fabry-Vendrand C, Ostinelli J, Ait-Yahia M, Darnal E, Bouee S, et al. The burden of severe asthma in france: a case-control study using a medical claims database. J Allergy Clin Immunol Pract. (2019) 7:1477–87. doi: 10.1016/j.jaip.2018.12.029
7. Chung KF, Wenzel SE, Brozek JL, Bush A, Castro M, Sterk PJ, et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J. (2014) 43:343–73. doi: 10.1183/09031936.00202013
8. Moore WC, Meyers DA, Wenzel SE, Teague WG Li H, Li X, D'Agostino R, et al. Identification of asthma phenotypes using cluster analysis in the severe asthma research program. Am J Respir Crit Care Med. (2010) 181:315–23. doi: 10.1164/rccm.200906-0896OC
9. Hough KP, Curtiss ML, Blain TJ, Liu RM, Trevor J, Deshane JS, et al. Airway remodeling in asthma. Front Med. (2020) 7:191. doi: 10.3389/fmed.2020.00191
10. Bergeron C, Tulic MK, Hamid Q. Airway remodelling in asthma: from benchside to clinical practice. Can Respir J. (2010) 17:318029. doi: 10.1155/2010/318029
11. Bergeron C, Al-Ramli W, Hamid Q. Remodeling in asthma. Proc Am Thorac Soc. (2009) 6:301–5. doi: 10.1513/pats.200808-089RM
12. Girodet PO, Ozier A, Bara I, Tunon De Lara JM, Marthan R, Berger P. Airway remodeling in asthma: new mechanisms and potential for pharmacological intervention. Pharmacol Ther. (2011) 130:325–37. doi: 10.1016/j.pharmthera.2011.02.001
13. Boulet LP, Laviolette M, Turcotte H, Cartier A, Dugas M, Malo JL, et al. Bronchial subepithelial fibrosis correlates with airway responsiveness to methacholine. Chest. (1997) 112:45–52. doi: 10.1378/chest.112.1.45
14. Tepper JS, Costa DL, Lehmann JR, Weber MF, Hatch GE. Unattenuated structural and biochemical alterations in the rat lung during functional adaptation to ozone. Am Rev Respir Dis. (1989) 140:493–501. doi: 10.1164/ajrccm/140.2.493
15. Minshall E, Chakir J, Laviolette M, Molet S, Zhu Z, Olivenstein R, et al. IL-11 expression is increased in severe asthma: association with epithelial cells and eosinophils. J Allergy Clin Immunol. (2000) 105:232–8. doi: 10.1016/S0091-6749(00)90070-8
16. Benayoun L, Druilhe A, Dombret MC, Aubier M, Pretolani M. Airway structural alterations selectively associated with severe asthma. Am J Respir Crit Care Med. (2003) 167:1360–8. doi: 10.1164/rccm.200209-1030OC
17. Little SA, Sproule MW, Cowan MD, Macleod KJ, Robertson M, Love JG, et al. High resolution computed tomographic assessment of airway wall thickness in chronic asthma: reproducibility and relationship with lung function and severity. Thorax. (2002) 57:247–53. doi: 10.1136/thorax.57.3.247
18. Takahashi K, Pavlidis S, Ng Kee Kwong F, Hoda U, Rossios C, Sun K, et al. Sputum proteomics and airway cell transcripts of current and ex-smokers with severe asthma in U-BIOPRED: an exploratory analysis. Eur Respir J. (2018) 51:2017. doi: 10.1183/13993003.02173-2017
19. Agache I, Akdis C, Jutel M, Virchow JC. Untangling asthma phenotypes and endotypes. Allergy Eur J Allergy Clin Immunol. (2012) 67:835–46. doi: 10.1111/j.1398-9995.2012.02832.x
20. Shaw DE, Sousa AR, Fowler SJ, Fleming LJ, Roberts G, Corfield J, et al. Clinical and inflammatory characteristics of the European U-BIOPRED adult severe asthma cohort. Eur Respir J. 46:1308–21. doi: 10.1183/13993003.00779-2015
21. Kuruvilla ME, Lee FE-H, Lee GB. Understanding asthma phenotypes, endotypes, and mechanisms of disease. Clin Rev Allergy Immunol. (2019) 56:219. doi: 10.1007/s12016-018-8712-1
22. Bullone M, Carriero V, Bertolini F, Folino A, Mannelli A, Stefano AD, et al. Elevated serum IgE, oral corticosteroid dependence and IL-17/22 expression in highly neutrophilic asthma. Eur Respir J. (2019) 54:2019. doi: 10.1183/13993003.00068-2019
23. Ray A, Kolls JK. Neutrophilic inflammation in asthma and association with disease severity. Trends Immunol. (2017) 38:942–54. doi: 10.1016/j.it.2017.07.003
24. Fajt ML, Wenzel SE. Asthma phenotypes and the use of biologic medications in asthma and allergic disease: the next steps toward personalized care. J Allergy Clin Immunol. (2015) 135:299–310. doi: 10.1016/j.jaci.2014.12.1871
25. Ricciardolo FLM, Sorbello V, Folino A, Gallo F, Massaglia GM, Favatà G, et al. Identification of IL-17F/frequent exacerbator endotype in asthma. J Allergy Clin Immunol. (2017) 140:395–406. doi: 10.1016/j.jaci.2016.10.034
26. Rupani H, Fong WCG, Kyyaly MA, Kurukulaaratchy RJ. Recent insights into the management of inflammation in asthma. J Inflamm Res. (2021) 14:4371–97. doi: 10.2147/JIR.S295038
27. Lindén A, Dahlén B. Interleukin-17 cytokine signalling in patients with asthma. Eur Respir J. (2014) 44:1319–31. doi: 10.1183/09031936.00002314
28. Sorbello V, Ciprandi G, Di Stefano A, Massaglia GM, Favatà G, Conticello S, et al. Nasal IL-17F is related to bronchial IL-17F/neutrophilia and exacerbations in stable atopic severe asthma. Allergy Eur J Allergy Clin Immunol. (2015) 70:236–40. doi: 10.1111/all.12547
29. Sze E, Bhalla A, Nair P. Mechanisms and therapeutic strategies for non-T2 asthma. Allergy Eur J Allergy Clin Immunol. (2020) 75:311–25. doi: 10.1111/all.13985
30. Akdis M, Palomares O, Van De Veen W, Van Splunter M, Akdis CA. TH17 and TH22 cells: a confusion of antimicrobial response with tissue inflammation versus protection. J Allergy Clin Immunol. (2012) 129:1438–49. doi: 10.1016/j.jaci.2012.05.003
31. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. (2003) 421:744–8. doi: 10.1038/nature01355
32. Harrington LE, Mangan PR, Weaver CT. Expanding the effector CD4 T-cell repertoire: the Th17 lineage. Curr Opin Immunol. (2006) 18:349–56. doi: 10.1016/j.coi.2006.03.017
33. Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. (2003) 198:1951–7. doi: 10.1084/jem.20030896
34. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. (2005) 6:1133–41. doi: 10.1038/ni1261
35. Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of TH17 cells. Nature. (2008) 453:1051–7. doi: 10.1038/nature07036
36. Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol. (2009) 10:857–63. doi: 10.1038/ni.1767
37. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells-a proposal for uniform nomenclature. Nat Rev Immunol. (2013) 13:145–9. doi: 10.1038/nri3365
38. Everaere L, Ait-Yahia S, Molendi-Coste O, Vorng H, Quemener S, LeVu P, et al. Innate lymphoid cells contribute to allergic airway disease exacerbation by obesity. J Allergy Clin Immunol. (2016) 138:1309–18.e11. doi: 10.1016/j.jaci.2016.03.019
39. Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med. (2014) 20:54–61. doi: 10.1038/nm.3423
40. Molet S, Hamid Q, Davoine F, Nutku E, Taha R, Pagé N, et al. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol. (2001) 108:430–8. doi: 10.1067/mai.2001.117929
41. Hamada H, Garcia-Hernandez ML, Reome JB, Misra SK, Strutt TM, McKinstry KK, et al. Tc17, a unique subset of CD8 T cells that can protect against lethal influenza challenge. J Immunol. (2009) 182:3469–81. doi: 10.4049/jimmunol.0801814
42. Dudakov JA, Hanash AM, Van Den Brink MRM. Interleukin-22: immunobiology and pathology. Annu Rev Immunol. (2015) 33:747–85. doi: 10.1146/annurev-immunol-032414-112123
43. Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing γδ T cells selectively expand in response to pathogen products and environmental signals. Immunity. (2009) 31:321–30. doi: 10.1016/j.immuni.2009.06.020
44. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KHG. Interleukin-1 and IL-23 induce innate IL-17 production from γδ T cells, amplifying Th17 responses and autoimmunity. Immunity. (2009) 31:331–41. doi: 10.1016/j.immuni.2009.08.001
45. Simonian PL, Wehrmann F, Roark CL, Born WK, O'Brien RL, Fontenot AP. γδ T cells protect against lung fibrosis via IL-22. J Exp Med. (2010) 207:2239–53. doi: 10.1084/jem.20100061
46. Paget C, Ivanov S, Fontaine J, Renneson J, Blanc F, Pichavant M, et al. Interleukin-22 is produced by invariant natural killer T lymphocytes during influenza A virus infection: potential role in protection against lung epithelial damages. J Biol Chem. (2012) 287:8816–29. doi: 10.1074/jbc.M111.304758
47. Cosmi L, Maggi L, Santarlasci V, Capone M, Cardilicchia E, Frosali F, et al. Identification of a novel subset of human circulating memory CD4+ T cells that produce both IL-17A and IL-4. J Allergy Clin Immunol. (2010) 125:12. doi: 10.1016/j.jaci.2009.10.012
48. Pennino D, Bhavsar PK, Effner R, Avitabile S, Venn P, Quaranta M, et al. IL-22 suppresses IFN-γ-mediated lung inflammation in asthmatic patients. J Allergy Clin Immunol. (2013) 131:562–70. doi: 10.1016/j.jaci.2012.09.036
49. Zissler UM, Chaker AM, Effner R, Ulrich M, Guerth F, Piontek G, et al. Interleukin-4 and interferon-γ orchestrate an epithelial polarization in the airways. Mucosal Immunol. (2016) 9:917–26. doi: 10.1038/mi.2015.110
50. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORγt directs the differentiation program of Proinflammatory IL-17+ T helper cells. Cell. (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
51. Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors RORα and RORγ. Immunity. (2008) 28:29–39. doi: 10.1016/j.immuni.2007.11.016
52. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. (2007) 8:967–74. doi: 10.1038/ni1488
53. Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, Mckenzie B, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. (2006) 116:1310–6. doi: 10.1172/JCI21404
54. Robinson KM, Manni ML, Biswas PS, Alcorn JF. Clinical consequences of targeting IL-17 and TH17 in autoimmune and allergic disorders. Curr Allergy Asthma Rep. (2013) 13:587–95. doi: 10.1007/s11882-013-0361-0
55. Yang XO, Panopoulos AD, Nurieva R, Seon HC, Wang D, Watowich SS, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. (2007) 282:9358–63. doi: 10.1074/jbc.C600321200
56. Korn T, Bettelli E, Gao W, Awasthi A, Jäger A, Strom TB, et al. IL-21 initiates an alternative pathway to induce proinflammatory T H17 cells. Nature. (2007) 448:484–7. doi: 10.1038/nature05970
57. Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. (2008) 8:337–48. doi: 10.1038/nri2295
58. Gulen MF, Kang Z, Bulek K, Youzhong W, Kim TW, Chen Y, et al. The receptor SIGIRR suppresses Th17 cell proliferation via inhibition of the interleukin-1 receptor pathway and mTOR kinase activation. Immunity. (2010) 32:54–66. doi: 10.1016/j.immuni.2009.12.003
59. Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. (2009) 30:576–87. doi: 10.1016/j.immuni.2009.02.007
60. Chen Q, Yang W, Gupta S, Biswas P, Smith P, Bhagat G, et al. IRF-4-binding protein inhibits interleukin-17 and interleukin-21 production by controlling the activity of IRF-4 transcription factor. Immunity. (2008) 29:899–911. doi: 10.1016/j.immuni.2008.10.011
61. Manni ML, Robinson KM, Alcorn JF. A tale of two cytokines: IL-17 and IL-22 in asthma and infection. Expert Rev Respir Med. (2014) 8:25–42. doi: 10.1586/17476348.2014.854167
62. Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. (2008) 453:106–9. doi: 10.1038/nature06881
63. Stockinger B, Hirota K, Duarte J, Veldhoen M. External influences on the immune system via activation of the aryl hydrocarbon receptor. Semin Immunol. (2011) 23:99–105. doi: 10.1016/j.smim.2011.01.008
64. Diveu C, McGeachy MJ, Boniface K, Stumhofer JS, Sathe M, Joyce-Shaikh B, et al. IL-27 blocks RORc expression to inhibit lineage commitment of Th17 cells. J Immunol. (2009) 182:5748–56. doi: 10.4049/jimmunol.0801162
65. Ramgolam VS, Sha Y, Jin J, Zhang X, Markovic-Plese S. IFN-β inhibits human Th17 cell differentiation. J Immunol. (2009) 183:5418–27. doi: 10.4049/jimmunol.0803227
66. Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. (2011) 12:247–54. doi: 10.1038/ni.1995
67. Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. (2007) 26:371–81. doi: 10.1016/j.immuni.2007.02.009
68. Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y, et al. The microbiota regulates type 2 immunity through RORγt+ T cells. Science. (2015) 349:989–93. doi: 10.1126/science.aac4263
69. Wang YH, Wills-Karp M. The potential role of interleukin-17 in severe asthma. Curr Allergy Asthma Rep. (2011) 11:388–94. doi: 10.1007/s11882-011-0210-y
70. Ramakrishnan RK, Al Heialy S, Hamid Q. Role of IL-17 in asthma pathogenesis and its implications for the clinic. Expert Rev Respir Med. (2019) 13:1057–68. doi: 10.1080/17476348.2019.1666002
71. Eyerich K, Eyerich S. Th22 cells in allergic disease. Allergo J Int. (2015) 24:3. doi: 10.1007/s40629-015-0039-3
72. Kolls JK, Lindén A. Interleukin-17 family members and inflammation. Immunity. (2004) 21:467–76. doi: 10.1016/j.immuni.2004.08.018
73. Laan M, Kisand K, Kont V, Möll K, Tserel L, Scott HS, et al. Autoimmune regulator deficiency results in decreased expression of CCR4 and CCR7 ligands and in delayed migration of CD4 + thymocytes. J Immunol. (2009) 183:7682–91. doi: 10.4049/jimmunol.0804133
74. Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. (1996) 183:2593–603. doi: 10.1084/jem.183.6.2593
75. Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, et al. Requirement of endogenous stem cell factor and granulocyte-colony-stimulating factor for IL-17-mediated granulopoiesis. J Immunol. (2000) 194:519–27. doi: 10.4049/jimmunol.164.9.4783
76. Nembrini C, Marsland BJ, Kopf M. IL-17-producing T cells in lung immunity and inflammation. J Allergy Clin Immunol. (2009) 123:986–94. doi: 10.1016/j.jaci.2009.03.033
77. Pelletier M, Maggi L, Micheletti A, Lazzeri E, Tamassia N, Costantini C, et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood. (2010) 115:335–43. doi: 10.1182/blood-2009-04-216085
78. Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. (2009) 9:556–67. doi: 10.1038/nri2586
79. Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, et al. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. (1995) 3:811–21. doi: 10.1016/1074-7613(95)90070-5
80. Ho AW, Gaffen SL. IL-17RC: a partner in IL-17 signaling and beyond. Semin Immunopathol. (2010) 32:33–42. doi: 10.1007/s00281-009-0185-0
81. Dumoutier L, Van Roost E, Ameye G, Michaux L, Renauld J-C. IL-TIF/IL-22: genomic organization and mapping of the human and mouse genes. Genes Immun. (2000) 1:488–94. doi: 10.1038/sj.gene.6363716
82. Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from TH-17, TH1 and TH2 cells. Nat Immunol. (2009) 10:864–71. doi: 10.1038/ni.1770
83. Lindemans CA, Calafiore M, Mertelsmann AM, O'Connor MH, Dudakov JA, Jenq RR, et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature. (2015) 528:560–4. doi: 10.1038/nature16460
84. Hanash AM, Dudakov JA, Hua G, O'Connor MH, Young LF, Singer N V, et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity. (2012) 37:339–50. doi: 10.1016/j.immuni.2012.05.028
85. Alcorn JF. IL-22 plays a critical role in maintaining epithelial integrity during pulmonary infection. Front Immunol. (2020) 11:1160. doi: 10.3389/fimmu.2020.01160
86. Boniface K, Bernard F-X, Garcia M, Gurney AL, Lecron J-C, Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. (2005) 174:3695–702. doi: 10.4049/jimmunol.174.6.3695
87. Wolk K, Sabat R. Interleukin-22: a novel T- and NK-cell derived cytokine that regulates the biology of tissue cells. Cytokine Growth Factor Rev. (2006) 17:367–80. doi: 10.1016/j.cytogfr.2006.09.001
88. Wolk K, Witte E, Wallace E, Döcke WD, Kunz S, Asadullah K, et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. (2006) 36:1309–23. doi: 10.1002/eji.200535503
89. Nograles KE, Zaba LC, Guttman-Yassky E, Fuentes-Duculan J, Suárez-Fariñas M, Cardinale I, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol. (2008) 159:1092–102. doi: 10.1111/j.1365-2133.2008.08769.x
90. Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. (2004) 21:241–54. doi: 10.1016/j.immuni.2004.07.007
91. Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol. (2012) 13:144–52. doi: 10.1038/ni.2187
92. Xie MH, Aggarwal S, Ho WH, Foster J, Zhang Z, Stinson J, et al. Interleukin (IL)-22, a novel human cytokine that signals through the interferon receptor-related proteins CRF2-4 and IL-22R. J Biol Chem. (2000) 275:31335–9. doi: 10.1074/jbc.M005304200
93. Kotenko SV, Izotova LS, Mirochnitchenko OV, Esterova E, Dickensheets H, Donnelly RP, et al. Identification of the functional interleukin-22 (IL-22) receptor complex. The IL-10R2 chain (IL-10Rβ) is a common chain of both the IL-10 and IL-22 (IL-10-related T cell-derived inducible factor, IL-TIF) receptor complexes. J Biol Chem. (2001) 276:2725–32. doi: 10.1074/jbc.M007837200
94. Lejeune D, Dumoutier L, Constantinescu S, Kruijer W, Schuringa JJ, Renauld JC. Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line: pathways that are shared with and distinct from IL-10. J Biol Chem. (2002) 277:33676–82. doi: 10.1074/jbc.M204204200
95. Hirose K, Ito T, Nakajima H. Roles of IL-22 in allergic airway inflammation in mice and humans. Int Immunol. (2018) 30:413–8. doi: 10.1093/intimm/dxy010
96. Aujla SJ, Kolls JK. IL-22: a critical mediator in mucosal host defense. J Mol Med. (2009) 87:451–4. doi: 10.1007/s00109-009-0448-1
97. Plé C, Fan Y, Yahia SA, Vorng H, Everaere L, Chenivesse C, et al. Polycyclic aromatic hydrocarbons reciprocally regulate IL-22 and IL-17 cytokines in peripheral blood mononuclear cells from both healthy and asthmatic subjects. PLoS ONE. (2015) 10:e0122372. doi: 10.1371/journal.pone.0122372
98. Sonnenberg GF, Nair MG, Kirn TJ, Zaph C, Fouser LA, Artis D. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J Exp Med. (2010) 207:1293–305. doi: 10.1084/jem.20092054
99. Besnard AG, Sabat R, Dumoutier L, Renauld JC, Willart M, Lambrecht B, et al. Dual role of IL-22 in allergic airway inflammation and its cross-talk with IL-17A. Am J Respir Crit Care Med. (2011) 183:1153–63. doi: 10.1164/rccm.201008-1383OC
100. Krueger JG, Fretzin S, Suárez-Fariñas M, Haslett PA, Phipps KM, Cameron GS, et al. IL-17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J Allergy Clin Immunol. (2012) 130:24. doi: 10.1016/j.jaci.2012.04.024
101. Baeten D, Baraliakos X, Braun J, Sieper J, Emery P, Van Der Heijde D, et al. Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: a randomised, double-blind, placebo-controlled trial. Lancet. (2013) 382:1705–13. doi: 10.1016/S0140-6736(13)61134-4
102. Akiyama S, Sakuraba A. Distinct roles of interleukin-17 and T helper 17 cells among autoimmune diseases. J Transl Autoimmun. (2021) 4:100104–100104. doi: 10.1016/j.jtauto.2021.100104
103. Eyerich K, Dimartino V, Cavani A. IL-17 and IL-22 in immunity: driving protection and pathology. Eur J Immunol. (2017) 47:607–14. doi: 10.1002/eji.201646723
104. Wenzel SE, Schwartz LB, Langmack EL, Halliday JL, Trudeau JB, Gibbs RL, et al. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med. (1999) 160:1001–8. doi: 10.1164/ajrccm.160.3.9812110
105. Moore WC, Hastie AT Li X, Li H, Busse WW, Jarjour NN, Wenzel SE, et al. Sputum neutrophil counts are associated with more severe asthma phenotypes using cluster analysis. J Allergy Clin Immunol. (2014) 133:1557. doi: 10.1016/j.jaci.2013.10.011
106. Al-Ramli W, Préfontaine D, Chouiali F, Martin JG, Olivenstein R, Lemière C, et al. TH17-associated cytokines (IL-17A and IL-17F) in severe asthma. J Allergy Clin Immunol. (2009) 123:1185–7. doi: 10.1016/j.jaci.2009.02.024
107. Doe C, Bafadhel M, Siddiqui S, Desai D, Mistry V, Rugman P, et al. Expression of the T helper 17-associated cytokines IL-17A and IL-17F in asthma and COPD. Chest. (2010) 138:1140–7. doi: 10.1378/chest.09-3058
108. Lamb D, De Sousa D, Quast K, Fundel-Clemens K, Erjefält JS, Sandén C, et al. RORγt inhibitors block both IL-17 and IL-22 conferring a potential advantage over anti-IL-17 alone to treat severe asthma. Respir Res. (2021) 22:7. doi: 10.1186/s12931-021-01743-7
109. Agache I, Ciobanu C, Agache C, Anghel M. Increased serum IL-17 is an independent risk factor for severe asthma. Respir Med. (2010) 104:1131–7. doi: 10.1016/j.rmed.2010.02.018
110. Bullens DMA, Truyen E, Coteur L, Dilissen E, Hellings PW, Dupont LJ, et al. IL-17 mRNA in sputum of asthmatic patients: linking T cell driven inflammation and granulocytic influx? Respir Res. (2006) 7:135. doi: 10.1186/1465-9921-7-135
111. Irvin C, Zafar I, Good J, Rollins D, Christianson C, Gorska MM, et al. Increased frequency of dual-positive TH2/TH17 cells in bronchoalveolar lavage fluid characterizes a population of patients with severe asthma. J Allergy Clin Immunol. (2014) 134:1175–86.e7. doi: 10.1016/j.jaci.2014.05.038
112. Wilson RH, Whitehead GS, Nakano H, Free ME, Kolls JK, Cook DN. Allergic sensitization through the airway primes Th17-dependent neutrophilia and airway hyperresponsiveness. Am J Respir Crit Care Med. (2009) 180:720. doi: 10.1164/rccm.200904-0573OC
113. He R, Oyoshi MK, Jin H, Geha RS. Epicutaneous antigen exposure induces a Th17 response that drives airway inflammation after inhalation challenge. Proc Natl Acad Sci USA. (2007) 104:15817–22. doi: 10.1073/pnas.0706942104
114. Ano S, Morishima Y, Ishii Y, Yoh K, Yageta Y, Ohtsuka S, et al. Transcription factors GATA-3 and RORγt are important for determining the phenotype of allergic airway inflammation in a murine model of asthma. J Immunol. (2013) 190:1056–65. doi: 10.4049/jimmunol.1202386
115. McKinley L, Alcorn JF, Peterson A, DuPont RB, Kapadia S, Logar A, et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol. (2008) 181:4089–97. doi: 10.4049/jimmunol.181.6.4089
116. Wu Y, Yue J, Wu J, Zhou W, Li D, Ding K, et al. Obesity may provide Pro-ILC3 development inflammatory environment in asthmatic children. J Immunol Res. (2018) 2018:1628620. doi: 10.1155/2018/1628620
117. Hellings PW, Kasran A, Liu Z, Vandekerckhove P, Wuyts A, Overbergh L, et al. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am J Respir Cell Mol Biol. (2003) 28:42–50. doi: 10.1165/rcmb.4832
118. Wang Q, Li H, Yao Y, Xia D, Zhou J. The overexpression of heparin-binding epidermal growth factor is responsible for Th17-induced airway remodeling in an experimental asthma model. J Immunol. (2010) 185:834–41. doi: 10.4049/jimmunol.0901490
119. Zhao J, Lloyd CM, Noble A. Th17 responses in chronic allergic airway inflammation abrogate regulatory T-cell-mediated tolerance and contribute to airway remodeling. Mucosal Immunol. (2013) 6:335–46. doi: 10.1038/mi.2012.76
120. Camargo LN, Righetti RF, Aristóteles LR, dos Santos TM, de Souza FCR, Fukuzaki S, et al. Effects of anti-IL-17 on inflammation, remodeling, and oxidative stress in an experimental model of asthma exacerbated by LPS. Front Immunol. (2018) 8:1835. doi: 10.3389/fimmu.2017.01835
121. Johnson JR, Nishioka M, Chakir J, Risse PA, Almaghlouth I, Bazarbashi AN, et al. IL-22 contributes to TGF-β1-mediated epithelial-mesenchymal transition in asthmatic bronchial epithelial cells. Respir Res. (2013) 14:1–12. doi: 10.1186/1465-9921-14-118
122. Tamasauskiene L, Gintauskiene VM, Bastyte D, Sitkauskiene B. Role of IL-22 in persistent allergic airway diseases caused by house dust mite: a pilot study. BMC Pulm Med. (2021) 21:1–8. doi: 10.1186/s12890-021-01410-z
123. Liu J, Shang B, Bai J. IL-22/IL-22R1 promotes proliferation and collagen synthesis of MRC-5 cells via the JAK/STAT3 signaling pathway and regulates airway subepithelial fibrosis. Exp Ther Med. (2020) 20:2148–56. doi: 10.3892/etm.2020.8931
124. Farfariello V, Amantini C, Nabissi M, Morelli MB, Aperio C, Caprodossi S, et al. IL-22 mRNA in peripheral blood mononuclear cells from allergic rhinitic and asthmatic pediatric patients. Pediatr Allergy Immunol. (2011) 22:419–23. doi: 10.1111/j.1399-3038.2010.01116.x
125. Ito T, Hirose K, Nakajima H. Bidirectional roles of IL-22 in the pathogenesis of allergic airway inflammation. Allergol Int. (2019) 68:4–8. doi: 10.1016/j.alit.2018.10.002
126. Arshad T, Mansur F, Palek R, Manzoor S, Liska V. A double edged sword role of interleukin-22 in wound healing and tissue regeneration. Front Immunol. (2020) 11:2148. doi: 10.3389/fimmu.2020.02148
127. Takahashi K, Hirose K, Kawashima S, Niwa Y, Wakashin H, Iwata A, et al. IL-22 attenuates IL-25 production by lung epithelial cells and inhibits antigen-induced eosinophilic airway inflammation. J Allergy Clin Immunol. (2011) 128:18. doi: 10.1016/j.jaci.2011.06.018
128. Taube C, Tertilt C, Gyülveszi G, Dehzad N, Kreymborg K, Schneeweiss K, et al. IL-22 is produced by innate lymphoid cells and limits inflammation in allergic airway disease. PLoS ONE. (2011) 6:e21799. doi: 10.1371/journal.pone.0021799
129. Nakagome K, Imamura M, Kawahata K, Harada H, Okunishi K, Matsumoto T, et al. High expression of IL-22 suppresses antigen-induced immune responses and eosinophilic airway inflammation via an IL-10–associated mechanism. J Immunol. (2011) 187:5077–89. doi: 10.4049/jimmunol.1001560
130. Fang P, Zhou L, Zhou Y, Kolls JK, Zheng T, Zhu Z. Immune modulatory effects of IL-22 on allergen-induced pulmonary inflammation. PLoS ONE. (2014) 9:e107454. doi: 10.1371/journal.pone.0107454
131. Ito T, Hirose K, Saku A, Kono K, Takatori H, Tamachi T, et al. IL-22 induces Reg3γ and inhibits allergic inflammation in house dust mite-induced asthma models. J Exp Med. (2017) 214:3037–50. doi: 10.1084/jem.20162108
132. Leyva-Castillo JM, Yoon J, Geha RS. IL-22 promotes allergic airway inflammation in epicutaneously sensitized mice. J Allergy Clin Immunol. (2019) 143:619–30.e7. doi: 10.1016/j.jaci.2018.05.032
133. Lou H, Lu J, Choi EB, Oh MH, Jeong M, Barmettler S, et al. Expression of IL-22 in the skin causes Th2-biased immunity, epidermal barrier dysfunction, and pruritus via stimulating epithelial Th2 cytokines and the GRP pathway. J Immunol. (2017) 198:2543–55. doi: 10.4049/jimmunol.1600126
134. Glocova I, Brück J, Geisel J, Müller-Hermelink E, Widmaier K, Yazdi AS, et al. Induction of skin-pathogenic Th22 cells by epicutaneous allergen exposure. J Dermatol Sci. (2017) 87:268–77. doi: 10.1016/j.jdermsci.2017.06.006
135. Nanzer AM, Chambers ES, Ryanna K, Richards DF, Black C, Timms PM, et al. Enhanced production of IL-17A in patients with severe asthma is inhibited by 1α,25-dihydroxyvitamin D3 in a glucocorticoid-independent fashion. J Allergy Clin Immunol. (2013) 132:37. doi: 10.1016/j.jaci.2013.03.037
136. Zhu J, Cao Y, Li K, Wang Z, Zuo P, Xiong W, et al. Increased expression of aryl hydrocarbon receptor and interleukin 22 in patients with allergic asthma. Asian Pacific J Allergy Immunol. (2011) 29:266–72.
137. Bouté M, Ait Yahia S, Nanou J, Alvarez-Simon D, Audousset C, Vorng H, et al. Direct activation of the aryl hydrocarbon receptor by dog allergen participates in airway neutrophilic inflammation. Allergy Eur J Allergy Clin Immunol. (2021) 76:2245–9. doi: 10.1111/all.14740
138. Badi YE, Pavel AB, Pavlidis S, Riley JH, Bates S, Kermani NZ, et al. Mapping atopic dermatitis and anti–IL-22 response signatures to type 2–low severe neutrophilic asthma. J Allergy Clin Immunol. (2021) 149:89–101. doi: 10.1016/j.jaci.2021.04.010
139. Ciprandi G, Cuppari C, Salpietro AM, Tosca MA, Rigoli L, Grasso L, et al. Serum IL-23 strongly and inversely correlates with FEV1 in asthmatic children. Int Arch Allergy Immunol. (2012) 159:183–6. doi: 10.1159/000336418
140. Louis R, Lau LCK, Bron AO, Roldaan AC, Radermecker M, Djukanović R. The relationship between airways inflammation and asthma severity. Am J Respir Crit Care Med. (2000) 161:9–16. doi: 10.1164/ajrccm.161.1.9802048
141. Cundall M, Sun Y, Miranda C, Trudeau JB, Barnes S, Wenzel SE. Neutrophil-derived matrix metalloproteinase-9 is increased in severe asthma and poorly inhibited by glucocorticoids. J Allergy Clin Immunol. (2003) 112:1064–71. doi: 10.1016/j.jaci.2003.08.013
142. Jatakanon A, Uasuf C, Maziak W, Lim S, Chung KF, Barnes PJ. Neutrophilic inflammation in severe persistent asthma. Am J Respir Crit Care Med. (1999) 160:1532–9. doi: 10.1164/ajrccm.160.5.9806170
143. Dragon S, Rahman MS, Yang J, Unruh H, Halayko AJ, Gounni AS. IL-17 enhances IL-1β-mediated CXCL-8 release from human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. (2007) 292:2006. doi: 10.1152/ajplung.00306.2006
144. Wolk K, Haugen HS, Xu W, Witte E, Waggie K, Anderson M, et al. IL-22 and IL-20 are key mediators of the epidermal alterations in psoriasis while IL-17 and IFN-γ are not. J Mol Med. (2009) 87:523–36. doi: 10.1007/s00109-009-0457-0
145. Stacey MA, Marsden M, Pham N TA, Clare S, Dolton G, Stack G, et al. Neutrophils recruited by IL-22 in peripheral tissues function as TRAIL-dependent antiviral effectors against MCMV. Cell Host Microbe. (2014) 15:471–83. doi: 10.1016/j.chom.2014.03.003
146. Lilly LM, Gessner MA, Dunaway CW, Metz AE, Schwiebert L, Weaver CT, et al. The β-glucan receptor dectin-1 promotes lung immunopathology during fungal allergy via IL-22. J Immunol. (2012) 189:3653–60. doi: 10.4049/jimmunol.1201797
147. Yang L, Zheng Y, Miao Y, Yan W, Geng Y, Dai Y, et al. Bergenin, a PPARγ agonist, inhibits Th17 differentiation and subsequent neutrophilic asthma by preventing GLS1-dependent glutaminolysis. Acta Pharmacol Sin. (2021) 21:717. doi: 10.1038/s41401-021-00717-1
148. Ventura I, Vega A, Chacõn P, Chamorro C, Aroca R, Gõmez E, et al. Neutrophils from allergic asthmatic patients produce and release metalloproteinase-9 upon direct exposure to allergens. Allergy Eur J Allergy Clin Immunol. (2014) 69:898–905. doi: 10.1111/all.12414
149. Voynow JA, Shinbashi M. Neutrophil elastase and chronic lung disease. Biomolecules. (2021) 11:65. doi: 10.3390/biom11081065
150. Michaudel C, Bataille F, Maillet I, Fauconnier L, Colas C, Sokol H, Straube M, et al. Ozone-induced aryl hydrocarbon receptor activation controls lung inflammation via interleukin-22 modulation. Front Immunol. (2020) 11:144. doi: 10.3389/fimmu.2020.00144
151. Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. (2007) 445:648–51. doi: 10.1038/nature05505
152. Starkey MR, Plank MW, Casolari P, Papi A, Pavlidis S, Guo Y, et al. IL-22 and its receptors are increased in human and experimental COPD and contribute to pathogenesis. Eur Respir J. (2019) 54:2018. doi: 10.1183/13993003.00174-2018
153. Yu Y, Zhao L, Xie Y, Xu Y, Jiao W, Wu J, et al. Th1/th17 cytokine profiles are associated with disease severity and exacerbation frequency in copd patients. Int J COPD. (2020) 15:1287–99. doi: 10.2147/COPD.S252097
154. Papp KA, Leonardi C, Menter A, Ortonne J-P, Krueger JG, Kricorian G, et al. Brodalumab, an anti–interleukin-17–receptor antibody for psoriasis. N Engl J Med. (2012) 366:1181–9. doi: 10.1056/NEJMoa1109017
155. Simon D, Aeberhard C, Erdemoglu Y, Simon HU. Th17 cells and tissue remodeling in atopic and contact dermatitis. Allergy Eur J Allergy Clin Immunol. (2014) 69:125–31. doi: 10.1111/all.12351
156. Eyerich S, Eyerich K, Pennino D, Carbone T, Nasorri F, Pallotta S, et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest. (2009) 119:3573–85. doi: 10.1172/JCI40202
157. Ji X, Li J, Xu L, Wang W, Luo M, Luo S, et al. IL4 and IL-17A provide a Th2/Th17-polarized inflammatory milieu in favor of TGF-β1 to induce bronchial epithelial-mesenchymal transition (EMT). Int J Clin Exp Pathol. (2013) 6:1481–92. Available online at: /pmc/articles/PMC3726963/ (accessed January 5, 2022).
158. Brunner PM, Pavel AB, Khattri S, Leonard A, Malik K, Rose S, et al. Baseline IL-22 expression in patients with atopic dermatitis stratifies tissue responses to fezakinumab. J Allergy Clin Immunol. (2019) 143:142–54. doi: 10.1016/j.jaci.2018.07.028
159. Habener A, Happle C, Grychtol R, Skuljec J, Busse M, Dalüge K, et al. Regulatory B cells control airway hyperreactivity and lung remodeling in a murine asthma model. J Allergy Clin Immunol. (2021) 147:2281–94.e7. doi: 10.1016/j.jaci.2020.09.041
160. Patakas A, Benson RA, Withers DR, Conigliaro P, McInnes IB, Brewer JM, et al. Th17 effector cells support B cell responses outside of germinal centres. PLoS ONE. (2012) 7:49715. doi: 10.1371/journal.pone.0049715
161. Mitsdoerffer M, Lee Y, Jäger A, Kim HJ, Korn T, Kolls JK, et al. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci USA. (2010) 107:14292–7. doi: 10.1073/pnas.1009234107
162. Halwani R, Al-Kufaidy R, Vazquez-Tello A, Pureza MA, BaHammam AS, Al-Jahdali H, et al. IL-17 enhances chemotaxis of primary human B cells during asthma. PLoS ONE. (2014) 9:114604. doi: 10.1371/journal.pone.0114604
163. Scanlon KM, Hawksworth RJ, Lane SJ, Mahon BP. IL-17A induces CCL28, supporting the chemotaxis of IgE-secreting B cells. Int Arch Allergy Immunol. (2011) 156:51–61. doi: 10.1159/000322178
164. Al-Kufaidy R, Vazquez-Tello A, BaHammam AS, Al-Muhsen S, Hamid Q, Halwani R. IL-17 enhances the migration of B cells during asthma by inducing CXCL13 chemokine production in structural lung cells. J Allergy Clin Immunol. (2017) 139:696–9.e5. doi: 10.1016/j.jaci.2016.07.037
165. Barone F, Nayara S, Camposa J, Cloakea T, Withers DR, Toellner KM, et al. IL-22 regulates lymphoid chemokine production and assembly of tertiary lymphoid organs. Proc Natl Acad Sci USA. (2015) 112:11024–9. doi: 10.1073/pnas.1503315112
166. Rheum A, Corneth O, Mus A-M, Asmawidjaja P, Ouyang W, Kil L, et al. Impaired B cell immunity in IL-22 knock-out mice in collagen induced arthritis. Ann Rheum Dis. (2011) 70:A58–9. doi: 10.1136/ard.2010.149005.3
167. Kuo C, Lim S, King NJC, Bartlett NW, Walton RP, Zhu J, et al. Rhinovirus infection induces expression of airway remodelling factors in vitro and in vivo. Respirology. (2011) 16:367–77. doi: 10.1111/j.1440-1843.2010.01918.x
168. Becnel D, You D, Erskin J, Dimina DM, Cormier SA. A role for airway remodeling during respiratory syncytial virus infection. Respir Res. (2005) 6:1–11. doi: 10.1186/1465-9921-6-122
169. Lloyd CM, Robinson DS. Allergen-induced airway remodelling. Eur Respir J. (2007) 29:1020–32. doi: 10.1183/09031936.00150305
170. Rahmawati SF, te Velde M, Kerstjens HAM, Dömling ASS, Groves MR, Gosens R. Pharmacological rationale for targeting IL-17 in asthma. Front Allergy. (2021) 2:40. doi: 10.3389/falgy.2021.694514
171. Nightingale S. Role of IL-17 and IL-23 in the pathogenesis of neutrophilic asthma. Int J Immunol Immunother. (2020) 7:1410049. doi: 10.23937/2378-3672/1410049
172. Hirose K, Takahashi K, Nakajima H. Roles of IL-22 in allergic airway inflammation. J Allergy. (2013) 2013:1–5. doi: 10.1155/2013/260518
173. Filidou E, Valatas V, Drygiannakis I, Arvanitidis K, Vradelis S, Kouklakis G, et al. Cytokine receptor profiling in human colonic subepithelial myofibroblasts: a differential effect of Th polarization-associated cytokines in intestinal fibrosis. Inflamm Bowel Dis. (2018) 24:2224–41. doi: 10.1093/ibd/izy204
174. Komai M, Tanaka H, Masuda T, Nagao K, Ishizaki M, Sawada M, et al. Role of Th2 responses in the development of allergen-induced airway remodelling in a murine model of allergic asthma. Br J Pharmacol. (2003) 138:912–20. doi: 10.1038/sj.bjp.0705105
175. Bellini A, Marini MA, Bianchetti L, Barczyk M, Schmidt M, Mattoli S. Interleukin (IL)-4, IL-13, and IL-17A differentially affect the profibrotic and proinflammatory functions of fibrocytes from asthmatic patients. Mucosal Immunol. (2012) 5:140–9. doi: 10.1038/mi.2011.60
176. Hayashi H, Kawakita A, Okazaki S, Yasutomi M, Murai H, Ohshima Y. IL-17A/F modulates fibrocyte functions in cooperation with CD40-mediated signaling. Inflammation. (2013) 36:830–8. doi: 10.1007/s10753-013-9609-z
177. Al-Muhsen S, Letuve S, Vazquez-Tello A, Pureza MA, Al-Jahdali H, Bahammam AS, Hamid Q, et al. Th17 cytokines induce pro-fibrotic cytokines release from human eosinophils. Respir Res. (2013) 14:34. doi: 10.1186/1465-9921-14-34
178. Ramakrishnan RK, Bajbouj K, Al Heialy S, Mahboub B, Ansari AW, Hachim IY, et al. IL-17 induced autophagy regulates mitochondrial dysfunction and fibrosis in severe asthmatic bronchial fibroblasts. Front Immunol. (2020) 11:1002. doi: 10.3389/fimmu.2020.01002
179. Loubaki L, Hadj-Salem I, Fakhfakh R, Jacques E, Plante S, Boisvert M, et al. Co-culture of human bronchial fibroblasts and CD4+ T cells increases Th17 cytokine signature. PLoS ONE. (2013) 8:e81983. doi: 10.1371/journal.pone.0081983
180. Hoshino H, Laan M, Sjöstrand M, Lötvall J, Skoogh BE, Lindén A. Increased elastase and myeloperoxidase activity associated with neutrophil recruitment by IL-17 in airways in vivo. J Allergy Clin Immunol. (2000) 105:143–9. doi: 10.1016/S0091-6749(00)90189-1
181. Chakir J, Shannon J, Molet S, Fukakusa M, Elias J, Laviolette M, et al. Airway remodeling-associated mediators in moderate to severe asthma: effect of steroids on TGF-β, IL-11, IL-17, and type I and type III collagen expression. J Allergy Clin Immunol. (2003) 111:1293–8. doi: 10.1067/mai.2003.1557
182. Chen Y, Thai P, Zhao YH, Ho YS, DeSouza MM, Wu R. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J Biol Chem. (2003) 278:17036–43. doi: 10.1074/jbc.M210429200
183. Fujisawa T, Velichko S, Thai P, Hung L-Y, Huang F, Wu R. Regulation of airway MUC5AC expression by IL-1β and IL-17A; the NF-κB paradigm. J Immunol. (2009) 183:6236–43. doi: 10.4049/jimmunol.0900614
184. Mammen MJ, Ali J, Aurora A, Sharma UC, Aalinkeel R, Mahajan SD, et L. IL-17 is a key regulator of mucin-galectin-3 interactions in asthma. Int J Cell Biol. (2021) 2021:9997625. doi: 10.1155/2021/9997625
185. Herbert C, Shadie AM, Kumar RK. Interleukin-17 signalling in a murine model of mild chronic asthma. Int Arch Allergy Immunol. (2013) 162:253–62. doi: 10.1159/000353247
186. Newcomb DC, Boswell MG, Sherrill TP, Polosukhin VV, Boyd KL, Goleniewska K, et al. IL-17A induces signal transducers and activators of transcription-6- independent airway mucous cell metaplasia. Am J Respir Cell Mol Biol. (2013) 48:711–6. doi: 10.1165/rcmb.2013-0017OC
187. Pezzulo AA, Tudas RA, Stewart CG, Vargas Buonfiglio LG, Lindsay BD, Taft PJ, et al. HSP90 inhibitor geldanamycin reverts IL-13–and IL-17–induced airway goblet cell metaplasia. J Clin Invest. (2019) 129:744–58. doi: 10.1172/JCI123524
188. Sohal SS, Ward C, Walters EH. Importance of epithelial mesenchymal transition (EMT) in COPD and asthma. Thorax. (2014) 69:768. doi: 10.1136/thoraxjnl-2014-205582
189. Wang T, Liu Y, Zou JF, Cheng ZS. Interleukin-17 induces human alveolar epithelial to mesenchymal cell transition via the TGF-β1 mediated Smad2/3 and ERK1/2 activation. PLoS ONE. (2017) 12:183972. doi: 10.1371/journal.pone.0183972
190. Gu K, Li M-M, Shen J, Liu F, Cao J-Y, Jin S, et al. Interleukin-17-induced EMT promotes lung cancer cell migration and invasion via NF-κB/ZEB1 signal pathway. Am J Cancer Res. (2015) 5:1169–79.
191. Ma L, Jiang M, Zhao X, Sun J, Pan Q, Chu S. Cigarette and IL-17A synergistically induce bronchial epithelial-mesenchymal transition via activating IL-17R/NF-κB signaling. BMC Pulm Med. (2020) 20:1–7. doi: 10.1186/s12890-020-1057-6
192. Jiang G, Liu CT, Zhang WD. IL-17A and GDF15 are able to induce epithelial-mesenchymal transition of lung epithelial cells in response to cigarette smoke. Exp Ther Med. (2018) 16:12–20. doi: 10.3892/etm.2018.6145
193. Vittal R, Fan L, Greenspan DS, Mickler EA, Gopalakrishnan B, Gu H, et al. IL-17 induces type V collagen overexpression and EMT via TGF-β-dependent pathways in obliterative bronchiolitis. Am J Physiol Lung Cell Mol Physiol. (2013) 304:L401. doi: 10.1152/ajplung.00080.2012
194. Sun J, Chu S, Lu M, Pan Q, Li D, Zheng S, et al. The roles of dipeptidyl peptidase-4 and its inhibitors in the regulation of airway epithelial-mesenchymal transition. Exp Lung Res. (2020) 46:163–73. doi: 10.1080/01902148.2020.1753853
195. Haddad A, Gaudet M, Plesa M, Allakhverdi Z, Mogas AK, Audusseau S, et al. Neutrophils from severe asthmatic patients induce epithelial to mesenchymal transition in healthy bronchial epithelial cells. Respir Res. (2019) 20:1–14. doi: 10.1186/s12931-019-1186-8
196. Chang Y, Al-Alwan L, Risse PA, Halayko AJ, Martin JG, Baglole CJ, et al. Th17-associated cytokines promote human airway smooth muscle cell proliferation. FASEB J. (2012) 26:5152–60. doi: 10.1096/fj.12-208033
197. Chang Y, Al-Alwan L, Risse PA, Roussel L, Rousseau S, Halayko AJ, et al. TH17 cytokines induce human airway smooth muscle cell migration. J Allergy Clin Immunol. (2011) 127:1046–53.e2. doi: 10.1016/j.jaci.2010.12.1117
198. Al-Alwan LA, Chang Y, Baglole CJ, Risse PA, Halayko AJ, Martin JG, et al. Autocrine-regulated airway smooth muscle cell migration is dependent on IL-17-induced growth-related oncogenes. J Allergy Clin Immunol. (2012) 130:977–85.e6. doi: 10.1016/j.jaci.2012.04.042
199. Ogawa H, Azuma M, Tsunematsu T, Morimoto Y, Kondo M, Tezuka T, et al. Neutrophils induce smooth muscle hyperplasia via neutrophil elastase-induced FGF-2 in a mouse model of asthma with mixed inflammation. Clin Exp Allergy. (2018) 48:1715–25. doi: 10.1111/cea.13263
200. Kudo M, Melton AC, Chen C, Engler MB, Huang KE, Ren X, et al. IL-17A produced by αβ T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat Med. (2012) 18:547–54. doi: 10.1038/nm.2684
201. Chiba Y, Tanoue G, Suto R, Suto W, Hanazaki M, Katayama H, et al. Interleukin-17A directly acts on bronchial smooth muscle cells and augments the contractility. Pharmacol Rep. (2017) 69:377–85. doi: 10.1016/j.pharep.2016.12.007
202. Fong V, Hsu A, Wu E, Looney AP, Ganesan P, Ren X, et al. Arhgef12 drives IL17A-induced airway contractility and airway hyperresponsiveness in mice. JCI Insight. (2018) 3:123578. doi: 10.1172/jci.insight.123578
203. Jendzjowsky N, Laing A, Malig M, Matyas J, Heuvel E, Dumonceaux C, et al. Long-term modulation of airway remodeling in severe asthma following bronchial thermoplasty. Eur Respir J. (2021) 2021:2100622. doi: 10.1183/13993003.00622-2021
204. Menzies-Gow A, Corren J, Bourdin A, Chupp G, Israel E, Wechsler ME, et al. Tezepelumab in adults and adolescents with severe, uncontrolled asthma. N Engl J Med. (2021) 384:1800–9. doi: 10.1056/NEJMoa2034975
205. Diver S, Khalfaoui L, Emson C, Wenzel SE, Menzies-Gow A, Wechsler ME, et al. Effect of tezepelumab on airway inflammatory cells, remodelling, and hyperresponsiveness in patients with moderate-to-severe uncontrolled asthma (CASCADE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med. (2021) 9:1299–312. doi: 10.1016/S2213-2600(21)00226-5
206. Busse WW, Holgate S, Kerwin E, Chon Y, Feng JY, Lin J, et al. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med. (2013) 188:1294–302. doi: 10.1164/rccm.201212-2318OC
207. Staton TL, Peng K, Owen R, Choy DF, Cabanski CR, Fong A, et al. A phase I, randomized, observer-blinded, single and multiple ascending-dose study to investigate the safety, pharmacokinetics, and immunogenicity of BITS7201A, a bispecific antibody targeting IL-13 and IL-17, in healthy volunteers. BMC Pulm Med. (2019) 19:9. doi: 10.1186/s12890-018-0763-9
208. Brightling CE, Nair P, Cousins DJ, Louis R, Singh D. Risankizumab in severe asthma — a phase 2a, placebo-controlled trial. N Engl J Med. (2021) 385:1669–79. doi: 10.1056/NEJMoa2030880
209. Hynes GM, Hinks TSC. The role of interleukin-17 in asthma: a protective response? ERJ Open Res. (2020) 6:00364–2019. doi: 10.1183/23120541.00364-2019
210. Chesné J, Braza F, Mahay G, Brouard S, Aronica M, Magnan A. IL-17 in severe asthma: where do we stand? Am J Respir Crit Care Med. (2014) 190:1094–101. doi: 10.1164/rccm.201405-0859PP
211. Ouyang S, Liu C, Xiao J, Chen X, Lui AC, Li X. Targeting IL-17A/glucocorticoid synergy to CSF3 expression in neutrophilic airway diseases. JCI Insight. (2020) 5:132836. doi: 10.1172/jci.insight.132836
212. Ciprandi G, Fenoglio D, De Amici M, Marseglia G, Murdaca G, Di Gioacchino M. Serum IL-17 after one course of sublingual immunotherapy in allergic rhinitis to birch. Eur J Inflamm. (2009) 7:49–51. doi: 10.1177/1721727X0900700107
213. Li CW, Lu HG, Chen DH, Lin Z, Bin A, Wang DY, et al. In vivo and in vitro studies of Th17 response to specific immunotherapy in house dust mite-induced allergic rhinitis patients. PLoS ONE. (2014) 9:e91950. doi: 10.1371/journal.pone.0091950
214. Zissler UM, Jakwerth CA, Guerth FM, Pechtold L, Aguilar-Pimentel JA, Dietz K, et al. Early IL-10 producing B-cells and coinciding Th/Tr17 shifts during three year grass-pollen AIT. EBioMedicine. (2018) 36:475–88. doi: 10.1016/j.ebiom.2018.09.016
215. Zissler UM, Schmidt-Weber CB. Predicting success of allergen-specific immunotherapy. Front Immunol. (2020) 11:1826. doi: 10.3389/fimmu.2020.01826
216. Wang J, Qiu L, Chen Y, Chen M. Sublingual immunotherapy increases Treg/Th17 ratio in allergic rhinitis. Open Med. (2021) 16:826–32. doi: 10.1515/med-2021-0285
217. Wang SH, Zissler UM, Buettner M, Heine S, Heldner A, Kotz S, et al. An exhausted phenotype of TH2 cells is primed by allergen exposure, but not reinforced by allergen-specific immunotherapy. Allergy Eur J Allergy Clin Immunol. (2021) 76:2827–39. doi: 10.1111/all.14896
218. Mcalees JW, Lajoie S, Dienger K, Sproles AA, Richgels PK, Yang Y, et al. Differential control of CD4+ T-cell subsets by the PD-1/PD-L1 axis in a mouse model of allergic asthma. Eur J Immunol. (2015) 45:1019–29. doi: 10.1002/eji.201444778
219. Akbari O, Stock P, Singh AK, Lombardi V, Lee WL, Freeman GJ, et al. PD-L1 and PD-L2 modulate airway inflammation and iNKT-cell-dependent airway hyperreactivity in opposing directions. Mucosal Immunol. (2010) 3:81–91. doi: 10.1038/mi.2009.112
220. Miyamoto C, Kojo S, Yamashita M, Moro K, Lacaud G, Shiroguchi K, et al. Runx/Cbfβ complexes protect group 2 innate lymphoid cells from exhausted-like hyporesponsiveness during allergic airway inflammation. Nat Commun. (2019) 10:1–13. doi: 10.1038/s41467-019-08365-0
221. Ebihara T, Taniuchi I. Exhausted-like group 2 innate lymphoid cells in chronic allergic inflammation. Trends Immunol. (2019) 40:1095–104. doi: 10.1016/j.it.2019.10.007
222. Guttman-Yassky E, Brunner PM, Neumann AU, Khattri S, Pavel AB, Malik K, et al. Efficacy and safety of fezakinumab (an IL-22 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by conventional treatments: a randomized, double-blind, phase 2a trial. J Am Acad Dermatol. (2018) 78:872–81.e6. doi: 10.1016/j.jaad.2018.01.016
Keywords: asthma, airway remodeling, Th17 inflammation, interleukine-17, interleukine-22
Citation: Margelidon-Cozzolino V, Tsicopoulos A, Chenivesse C and de Nadai P (2022) Role of Th17 Cytokines in Airway Remodeling in Asthma and Therapy Perspectives. Front. Allergy 3:806391. doi: 10.3389/falgy.2022.806391
Received: 31 October 2021; Accepted: 10 January 2022;
Published: 02 February 2022.
Edited by:
Qutayba Hamid, McGill University, CanadaReviewed by:
Ulrich Matthias Zissler, Technical University of Munich, GermanyManali Mukherjee, McMaster University, Canada
Delphine Gras, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2022 Margelidon-Cozzolino, Tsicopoulos, Chenivesse and de Nadai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Victor Margelidon-Cozzolino, dm1hcmdlbGlkb25AZ21haWwuY29t