 Beatriz León
Beatriz León- Department of Microbiology, University of Alabama at Birmingham, Birmingham, AL, United States
Allergic diseases, including atopic dermatitis, allergic rhinitis, asthma, and food allergy, are caused by abnormal responses to relatively harmless foreign proteins called allergens found in pollen, fungal spores, house dust mites (HDM), animal dander, or certain foods. In particular, the activation of allergen-specific helper T cells towards a type 2 (Th2) phenotype during the first encounters with the allergen, also known as the sensitization phase, is the leading cause of the subsequent development of allergic disease. Infants and children are especially prone to developing Th2 cell responses after initial contact with allergens. But in addition, the rates of allergic sensitization and the development of allergic diseases among children are increasing in the industrialized world and have been associated with living in urban settings. Particularly for respiratory allergies, greater susceptibility to developing allergic Th2 cell responses has been shown in children living in urban environments containing low levels of microbial contaminants, principally bacterial endotoxins [lipopolysaccharide (LPS)], in the causative aeroallergens. This review highlights the current understanding of the factors that balance Th2 cell immunity to environmental allergens, with a particular focus on the determinants that program conventional dendritic cells (cDCs) toward or away from a Th2 stimulatory function. In this context, it discusses transcription factor-guided functional specialization of type-2 cDCs (cDC2s) and how the integration of signals derived from the environment drives this process. In addition, it analyzes observational and mechanistic studies supporting an essential role for innate sensing of microbial-derived products contained in aeroallergens in modulating allergic Th2 cell immune responses. Finally, this review examines whether hyporesponsiveness to microbial stimulation, particularly to LPS, is a risk factor for the induction of Th2 cell responses and allergic sensitization during infancy and early childhood and the potential factors that may affect early-age response to LPS and other environmental microbial components.
Introduction
Allergic diseases are chronic inflammatory disorders caused by aberrant immune reactions to harmless foreign proteins called allergens. Allergic diseases can affect the respiratory tract, skin, and digestive system, causing airway allergy, atopic dermatitis, and food allergy. This review focuses on respiratory allergies, which are induced by exposure to airborne allergens. The most common aeroallergens are pollen, fungal spores, house dust mites (HDMs), and animal allergens. Respiratory allergies include allergic rhinitis and asthma, which affect the upper and lower airways, respectively. Inflammation triggered by exposure to allergens in the upper respiratory tract causes sneezing, congestion, and itchy nose, mouth, and eyes. Inflammation in the lower respiratory tract in patients with asthma can cause breathing problems and obstruct airflow when the airways swell and narrow and produce excess mucus.
Allergic respiratory disorders are increasingly prevalent in the developed world and are the most common chronic immunological diseases affecting children (1, 2). Approximately 7% of the population of the United States suffers from respiratory allergies. Among children, the frequency is higher, with around 10% of children affected by allergic disorders of the respiratory tract, of which approximately 6% have asthma (3–6). In particular, allergic airway disorders often have an early onset and usually develop before the age of 5 years and then persist throughout life (7–9). Current trends predict that pediatric respiratory allergy incidence rates will continue to rise, particularly in developed countries (4, 5, 10), providing further evidence that lifestyle change associated with urbanization and industrialization is contributing to this increase in allergic airway disease in children.
The characteristic pattern of inflammation in the airways of children with allergy is mediated by type 2 immune responses mainly regulated by T helper 2 (Th2) and type 2 T follicular helper (Tfh2) cells. Tfh2 cells secrete the cytokines interleukin (IL)-4, IL-21, and IL-13 in B cell follicles, which promotes the production of immunoglobulin E (IgE) and IgG1 by B cells (11, 12), leading to the activation of mast cells and basophils (13). Th2 effector cells secrete IL-4, IL-13, IL-5, and IL-9 upon allergen stimulation (14, 15), promoting inflammation and tissue remodeling by inducing the infiltration and activation of eosinophils (16) and excessive secretion of mucus (17). A fundamental characteristic of the development of airway inflammation upon allergen contact is that it requires an initial exposure to allergen or “sensitization” that does not necessarily cause symptoms or pathology. However, once a person becomes “sensitized” to an allergen, they will develop pathology following secondary or subsequent allergen exposures or “challenges.” During this initial sensitization phase, allergens trigger an immune reaction that ultimately activates conventional dendritic cells (cDCs) to prime allergen-specific CD4+ T cells with a Th2-like cytokine profile. The presence and persistence of these allergen-specific T cells after initial priming marks the predisposition to develop allergic responses, as these T cells can be subsequently reactivated upon re-exposure to the same inhaled allergen, causing their migration to the airways, where they locally produce Th2 cytokines (15, 18). Accumulation of Th2 effector cells in the lungs ultimately stimulates the hallmarks features of allergic inflammation, such as tissue swelling, mucus hypersecretion, and bronchial hyperresponsiveness (14).
An accumulating piece of evidence has shown that during early childhood, there is a predisposition to prime Th2-biased immune responses compared to later in life (19–21). These data suggest that environmental factors specifically impacting during a time window in early life would instigate the initial priming of aeroallergen-specific T cells toward a Th2-like cytokine profile, thus promoting allergic sensitization. Furthermore, lifestyle changes in developed countries are contributing to this trend (5, 10). Living in developed countries is associated with industrialization, urbanization, and changes in hygiene habits. The most hygienic environment and practices apply particularly to infants and young children. These changes lead to a restriction in exposure to inhalant dust-related microbial products during the first years of life. In particular, a number of epidemiologic studies have drawn attention to decreased exposure to inhalant house-dust bacterial endotoxins (lipopolysaccharides, LPS) and endotoxin-contaminated aeroallergens in childhood and the risk of developing Th2-driven, allergen-induced airway inflammation (22–30). This review discusses the mechanisms that promote and prevent Th2 cell immunity—especially highlighting the diverse signals elicited by the environmental allergens that lead to the activation of different cells in the exposed tissue, including epithelial/stromal cells, monocyte subsets, and innate lymphoid cell group 2 (ILC2). It further analyzes how signals derived from these cells are ultimately integrated by cDCs, particularly type 2 cDCs (cDC2s), to program their function to induce or prevent Th2 cell responses. Moreover, this review examines the fundamental role of endotoxin/LPS contained in airborne allergens in preventing allergen-specific T cell priming toward a Th2 cell phenotype and, thus, in preventing new allergen sensitizations. Finally, it analyzes the mechanisms that promote a state of hyporesponsiveness to endotoxins/LPS during early childhood, favoring Th2 cell sensitization to aeroallergens with low endotoxin contamination and ultimately contributing to increased susceptibility to developing respiratory allergic diseases in early life.
Mechanisms that promote Th2 cell priming by dendritic cells
cDCs are the principal antigen-presenting cells (APCs) to initiate CD4+ T cell activation and proliferation by presenting protein-derived peptides on major histocompatibility complex (MHC) molecules (also known as “signal 1”) and by providing potent co-stimulatory signals through the engagement of CD28 expressed by the T cells (also known as “signal 2”). In addition, cDCs provide polarizing cytokines that determine the differentiation of naïve CD4+ T cells into the various effector T helper (Th) subsets (also known as “signal 3”). For example, cDC-derived IL-12 induces Th1 cell differentiation by promoting interferon-γ (IFNγ) production and expression of the Th1 cell-associated transcription factor T-bet (also known as TBX21) (31–34). Transforming grow factor-β (TGFβ) induces the transcription factor forkhead box P3 (FOXP3) and thus drives T regulatory T (Treg) cell differentiation (35). But, the presence of TGFβ along with IL-6 during T cell priming promotes the expression of the retinoic acid receptor-related orphan receptor-γt (RORγt; also known as RORC) that drives Th17 cell differentiation, thus subverting differentiation of Treg cells (36, 37). IL-23 then maintains the Th17 phenotype by stabilizing RORγt, thereby allowing Th17 cells to release their effector cytokines (38). Since IL-12, TGFβ, IL-6, and IL-23 are cytokines produced by cDCs in response to pathogen or danger recognition (39–42), it is believed that cDCs provide all three signals for the differentiation of Th1, Th17, and induced Treg responses.
Although many studies have made clear that cDCs are required for the induction of Th2 cell responses (15, 18, 43–53), an equivalent cDC-derived cytokine or “signal 3” that induces Th2 cell differentiation has not been found. Instead, early studies proposed that Th2 cell differentiation may be controlled by the strength of the “signal 1” delivered through the T cell receptor (TCR). This includes the affinity of the interactions between MHC and TCR molecules and the amount of antigen. This hypothesis was initially based on in vitro studies showing that weak TCR signaling as a result of stimulation with low doses of antigen or low-affinity peptides favored IL-4 over IFNγ production (54–58). And although in vivo studies using adoptive transfer of DCs loaded with a range of peptide concentrations have likewise suggested that low TCR signal strength favors Th2 cell induction (59), in vivo models of infection or immunization have not reproduced these findings, as many studies have shown that high doses of antigen enhance the generation of Th2 cells (60–68) and that TCR affinity does not affect IL-4 production by activated T cells (69). Therefore, current evidence does not clearly indicate whether TCR signal intensity controls the induction of Th2 cell responses upon in vivo exposure to Th2-inducing stimuli. Other studies have tested whether specific costimulatory signals influence Th2 cell differentiation. Particularly, some studies have provided evidence that in vitro generated DCs expressing membrane-bound Notch ligands, particularly Jagged 1 and Jagged 2, gain the capacity to instruct Th2 cell polarization (70, 71). However, other studies have shown that Jagged expression by DCs is insufficient or not required for Th2 cell differentiation (72, 73). Furthermore, in vivo studies have shown that expression of Jagged Notch ligands on cDCs is dispensable for induction of Th2 cell responses to natural allergens and subsequent allergic airway inflammation (74). Thus, current evidence does not support a role for Jagged expression on cDCs in directing Th2 cell differentiation to allergens. Instead, Notch has been found to orchestrate multiple Th cell programs, including Th1, Th2, Th17, Treg, and Tfh, and has been suggested to be a general and unbiased amplifier of Th cell responses (75).
Importantly, however, initial Th2 cell lineage commitment has been shown to require IL-2 signaling and IL-2-induced activation of signal transducer and activation of transcription 5 (STAT5) (76, 77). Specifically, IL-2-STAT5 signaling is needed to promote transcription of the Il4ra gene, leading to increased cell surface expression of IL-4Rα (also known as CD124) and subsequently increased responsiveness to IL-4 (77). In addition, IL-2 stabilizes the accessibility of the Il4 locus, allowing for early IL-4 production (76–78). Thus, IL-2-induced signaling during early Th2 cell differentiation is required to support increased IL-4 production and increased IL-4 responsiveness, allowing for an IL-4-positive feedback amplification loop that preserves the Th2 cell phenotype. cDCs are not a critical source of IL-2. And although group 3 ILCs are a dominant source of IL-2 in peripheral tissues (79, 80), in the secondary lymphoid organs, where T cell priming occurs, IL-2 is thought to be produced primarily by CD4+ T cells shortly after their activation by cDCs. IL-2 signaling is first required for the initial clonal T-cell expansion (81, 82). However, IL-2-deficient T cells also show defective Th2 cell differentiation in vitro (76, 77), supporting that IL-2 produced by activated T cells is necessary and sufficient to initiate the Th2 cell differentiation program. Remarkably, in vitro IL-2 production and responsiveness are favored by weaker TCR signaling (55, 83), which may be one mechanism to explain how TCR signal intensity can control in vitro Th2 cell differentiation. However, IL-2 is not exclusively produced during Th2 cell differentiation. Therefore, Th2 cell commitment must be regulated in vivo by additional mechanisms. One possibility is that CD4+ T cells default into the Th2 cell pathway in the presence of strong IL-2 signaling but the absence of positive signals (or cytokines) that drive differentiation into other Th lineages, particularly into Th1 cells (31–33, 84, 85). The regulatory roles of Th1-inducing signals in inhibiting Th2 cell differentiation will be discussed in detail below.
Accumulating evidence has shown that interactions between naïve CD4 T cells and antigen-presenting cDCs can occur within distinct sub-anatomic regions of secondary lymphoid tissues, leading to the differentiation of specific Th subsets (86, 87). These data suggest that T-cell lineage commitment and the acquisition of district cytokine profile may be controlled by particular localization within secondary lymphoid organs. In particular, Th1-directing stimulations induce cDCs to localize into the deep T cell area of the lymph node via a CC-chemokine receptor 7 (CCR7)-dependent mechanism (88–90). This positioning of cDC allows for efficient initial priming of Th1 cell responses (88). On the contrary, during Th2-directing stimulations, antigen-bearing cDCs are preferentially attracted to the border of B cell follicles and T cell zone (T-B border) and to the area between B cell follicles (perifollicular region) rather than to the T cell zone (18, 86, 89, 91, 92). The encounter of cDCs and responding CD4+ T cells, specifically within the T-B/perifollicular zone of the lymph node, was found necessary for the establishment of Th2 cell responses, as repositioning of cDCs or T cells to the T cell area compromised Th2 cell priming (86, 89). Thus, this evidence suggests that the T cell area of the lymph node is a specialized microenvironment that contributes to the effective priming of Th1 cells but also is an unfavorable environment for the priming of Th2 cell responses. In contrast, Th2 cell responses are efficiently primed in the T-B/perifollicular region of the lymph nodes. Since the signals that induce Th1 and Th2 cells have antagonistic effects on each other, it is understandable that the differentiation of these two subsets occurs in anatomically separate settings. However, it is more than possible that these particular lymph node microenvironments optimize the differentiation of individual T cell responses. As an example, chemokine signals expressed in the T area that induce the positioning of cDCs within this location also enhance the ability of the cDCs to produce IL-12 and differentiate Th1 cells (93). Besides, T cell priming in the T cell area allows for interaction with blood-recruited monocytes and monocyte-derived DCs (moDCs) (94–96), providing an additional source of IL-12 for optimal Th1 cell differentiation (88, 97–99). On the other hand, Th2 cell differentiation requires strong and sustained IL-2 signaling, which is perhaps best achieved in the T-B/perifollicular region. For example, IL-2 could be more available in the T-B/perifollicular area due to the low presence of Treg cells that consume IL-2 (100). However, questions remain as to how the cDCs integrate signals provided in the environment after Th2-directing stimulations to migrate and be specifically retained in the T-B/perifollicular areas of secondary lymphoid tissues, to provide signals that efficiently support autocrine IL-2 production and signaling in T cells, without providing cytokines that inhibit the Th2 cell differentiation program.
Innate sensing of microbial pathogens or derived products is principally mediated by pattern recognition receptors (PRRs), which recognize molecules frequently found in pathogens (the so-called pathogen associated molecular patterns or PAMPs). PRRs include Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), NOD-like receptors (NLRs), C-type lectin receptors (CLRs), and cytosolic DNA sensors (101). Upon ligand engagement, PRRs trigger intracellular signal transduction pathways, which ultimately result in the expression of a variety of pro-inflammatory and polarizing cytokines and up-regulation of co-stimulatory molecules. These gene products orchestrate the early host response to infection and also are a prerequisite for the subsequent activation and shaping of adaptive immunity. Particularly, pathogen recognition by PRRs and subsequent expression of polarizing cytokines is essential for initiating Th1 and Th17 responses. In contrast, Th2 cell responses can develop or even get enhanced in the absence of PRR-mediated signaling (102–108). Indeed, as it will be discussed in below sections, the trigger of PRR-mediated signaling and subsequent induction of pro-inflammatory cytokine response usually suppresses Th2 cell priming. This is in line with the idea that no positive signal (or cytokine) controls Th2 polarization; instead, it is the absence of one that causes Th2 cell responses to develop. However, the activation of certain PRR in specific cell types or by certain PAMPs can elicit attenuated rather than promote inflammatory cytokine responses and consequently have been shown to act as adjuvants that favor or enhance biased type 2-immunity. For example, TLR2 stimulation promotes weak IL-12 production (109–113), and thus TLR2-mediated signals preferentially stimulate Th2 cell polarization (111, 113, 114). Likewise, polysaccharides commonly found in allergens can bind CLRs such as Dectin-1/CLEC7A and Dectin-2 (115–117). Dectin-1 and Dectin-2 ligands usually prompt attenuated IL-12 responses (118–120), but still can act as adjuvants that promote cDC activation and migration for the subsequent priming of allergen-specific Th2 cells (121–125). But among all PRRs, TLR4 is probably the most ambiguous regulator in the context of type 2-immunity, as its activation can lead to suppression of Th2 cell differentiation or, conversely, be a driving force of Th2 bias. How TLR4 activation can cause these two opposite outcomes in the context of type 2-immunity is still a topic of research. In general, strong stimulation of the TLR4-driven pro-inflammatory response is associated with IL-12 release and suppression of Th2 cell priming (103, 126–128). However, several factors can influence TLR4-mediated signaling and cause an attenuated pro-inflammatory cytokine response that promotes type 2-inflammation. LPS is the best-known and most potent TLR4 agonist and is, therefore, a powerful inducer of pro-inflammatory cytokines (129). But it should be noted that in this context, host sensitivity to LPS is critical when it comes to eliciting a robust pro-inflammatory cytokine response leading to IL-12-producing cDCs and suppression of Th2 cell responses, particularly in response to low amounts of LPS. And therefore, suboptimal responses to low amounts of LPS may promote rather than prevent Th2 responses, thereby favoring allergic airway inflammation (103, 130–132). As will be discussed below, moDCs are the primary sensor cells for low-dose LPS and a key source of pro-inflammatory cytokines. Thus, factors influencing moDC differentiation and activation can switch LPS activity from protective to pro-pathogenic in the context of Th2 cell allergic responses. In this regard, activation of TLR4 in the absence of moDCs initiates, rather than prevents, Th2 allergic inflammation in the lungs (103). When moDCs are absent, LPS primarily drives TLR4 activation in the stroma/epithelium (103, 130, 131, 133). And this context favor Th2 cell development since the TLR4-driven response in structural cells has a low pro-inflammatory component but induces cytokines or “alarmins” that condition cDCs to initiate Th2 cell responses (134). On the other hand, although TLR4 primarily recognizes and is activated by LPS, alternative agonists for TLR4 have also been described (135). Importantly, TLR4 signaling induced by some of these alternative agonists has been shown to promote, rather than suppress, type 2-driven inflammation (136–139). A common feature of these TLR4 agonist ligands is that they induce alternative activation of TLR4, resulting in weakened inflammatory response. Canonical activation of TLR4 by LPS is absolutely dependent on the presence of myeloid differentiation factor-2 (MD-2), which binds LPS and initiates TLR4 pro-inflammatory cytokine signaling (140–142). However, alternative TLR4 ligands that elicit type 2-immunity activate TLR4 in the absence of MD-2 (137–139). The proposed mechanism for this alternative/non-canonical TLR4 activation is that these ligands are proteins with structural and functional homology to MD-2 that can reconstruct TLR4 signaling in the absence of MD-2 (137). Future studies will be required to understand precisely how canonical and non-canonical TLR4 activation pathways can elicit different cytokine profiles and thus enable distinct TLR4 functions.
In addition to their ability to activate PRRs, allergens with strong ability to elicit Th2 cell responses, such as HDM, pollen, fungi, or cockroaches, share the common feature of containing proteases (143). Moreover, proteolytic enzymes extracted from these allergens have the ability to trigger potent Th2 cell responses (116, 144–146). Thus, the presence of protease activity in allergens is thought to play an important role in initiating Th2 cell responses. Additionally, several cell populations and mediators have been shown to support cDC activation and migration and, thus, have been found to be central regulators of Th2 cell immunity. Principally, activation of epithelial cells by microbial products, cytokines, or allergens with proteolytic activity and the consequent release of the “alarmin” cytokines thymic stromal lymphopoietin (TSLP), IL-33, and IL-25 is thought to be the initial step for the subsequent activation of cDCs and priming of allergen-specific Th2 cells (134, 147, 148). In support, alarmin cytokines and their receptors are major susceptibility loci for human asthma (149–151). Importantly, TSLP, IL-33 and IL-25 are potent activators of ILC2, which produce IL-13 in response to these stimulations (53, 152–158). IL-13 production by ILC2 has been shown to be important for the initiation of Th2 cell responses after allergic sensitization or immunization through the skin or the respiratory tract (53, 152). Mechanistically, IL-13 produced by ILC2 has been shown to promote the migration of cDCs into the draining lymph node and their functional specialization to stimulate naïve T cells to differentiate into Th2 cells (53, 152). cDCs can be divided into two ontogenically distinct subsets, the type-1 cDCs (cDC1s) and type-2 cDCs (cDC2s) (159). In the context of induction of allergen-specific CD4+ T-cell responses, cDC2s are especially adept at capturing and transporting allergen-derived proteins from the tissue into the draining lymph node and presenting them to CD4+ T-cell to induce their activation and polarization (18, 46, 103, 128). The integration of the signals in the environment defines the functional specialization of cDC2s and ultimately determines their ability to either promote (18, 46–51, 53) or prevent (103, 128) Th2 cell responses to allergens. The specific signals driving cDC2 diversification are gradually being characterized. The expression of the transcription factors Interferon regulatory factor 4 (IRF4) (48, 50), Krüppel-like factor 4 (KLF4) (51), and STAT6 (53) promote the functional specialization of cDC2s to favor Th2 cell differentiation. In contrast, and as will be discussed below, the induced expression of the transcription factor T-bet is intrinsically necessary for the ability of cDC2s to produce sustained IL-12 and suppress Th2 cell priming (103, 128). Thus, these data suggest transcription factor-guided functional specialization of cDC2s and the influence of environment-derived signals in driving this process. Indeed, IRF4 controls unresponsiveness to TLR4 stimulation in cDC2s and, therefore, imprints a low capacity to produce IL-12 in response to LPS (48, 160–162). Additionally, KLF4 and STAT6 enable cDC2s to respond to IL-13, thus guiding the function of cDC2s to promote Th2 cell differentiation, most likely by similarly reducing the cDC2 ability to produce IL-12 in response to PAMPs, while preserving their CD4+ T cell stimulatory capacity (53, 163, 164). Overall, the data suggest that the ability of cDC2 to suppress IL-12 production and the prevention of naïve CD4+ T cells from receiving IL-12 signaling are decisive for inducing Th2 cell responses. Furthermore, the data suggest that this can be controlled by the transcriptional programming of cDC2s and by particular location of DC-T cell interactions away from the T cell areas of secondary lymphoid tissues (Figure 1).
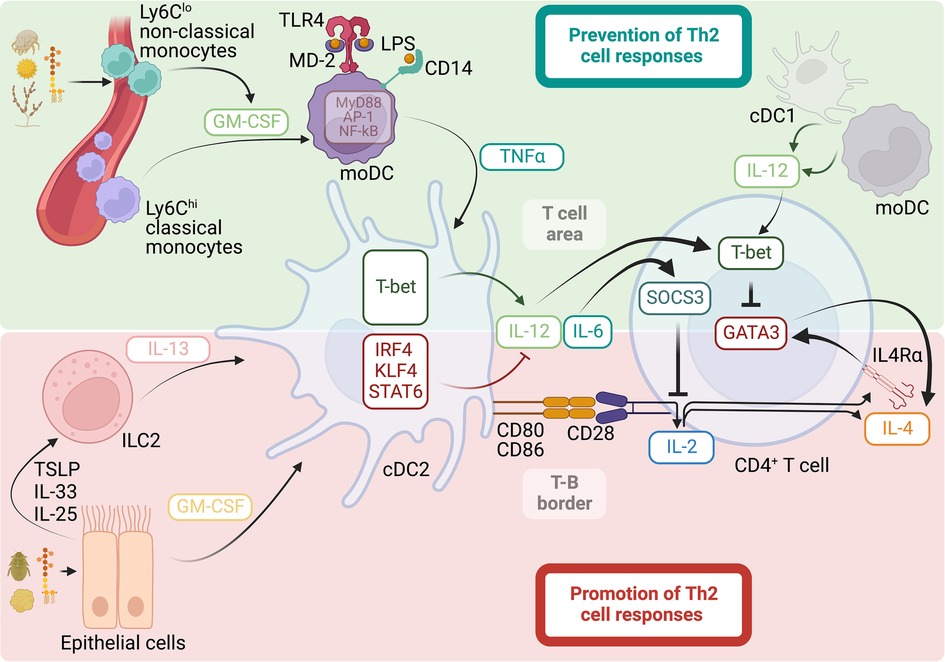
Figure 1. Mechanisms that promote and prevent Th2 cell responses to allergens. Promotion of Th2 cell responses: allergens with proteolytic activity, cytokines, and microbial products, such as LPS, can activate epithelial cells for subsequent release of the cytokines GM-CSF, TSLP, IL-33, and IL-25. This leads to the activation of ILC2s and the release of IL-13. GM-CSF and IL-13 stimulate the migration and expression of the transcription factors IRF4, KLF4, and STAT6 in cDC2s, ultimately promoting the functional specialization of the cDC2s to support Th2 cell differentiation by reducing the ability of the cDC2s to produce IL-12, while retaining their co-stimulatory ability to foster strong IL-2 responses in CD4+ T cells. Strong and sustained IL-2 signaling in the absence of IL-12 promotes Th2 cell lineage commitment by promoting the expression of IL-4Rα and IL-4, allowing for an IL-4-positive feedback loop that initiates and preserves the Th2 cell phenotype. Prevention of Th2 cell responses: allergens with cysteine protease activity stimulate GM-CSF release from perivascular Ly6Clo non-classical monocytes, guiding the differentiation of Ly6Chi classical monocytes into moDCs. GM-CSF licenses an inflammatory signature in moDCs by increasing the expression of TLR4, CD14, and intracellular signaling members involved in the MyD88/NF-kB/AP-1-dependent pathway. Functional programming of moDCs by GM-CSF allows these cells to increase their sensitivity to LPS and stimulate the production of the pro-inflammatory cytokine TNFα, which guides cDC2 activation for Th2 cell suppression rather than promotion. In particular, TNFα induces the expression of the transcription factor T-bet in cDC2s, which is intrinsically necessary for the ability of cDC2s to produce sustained IL-12 and suppress Th2 cell priming by inducing T-bet and inhibiting GATA3 in CD4+ T cells. cDC2s can also produce IL-6, upregulating SOCS3 in CD4+ T cells and ultimately suppressing IL-2 signaling and early Th2 cell commitment. cDC1s and moDCs are additional sources of IL-12 that contribute to the suppression of Th2 cell responses.
Generation of effector Th2 cell responses in the lung
Experimental mouse models of allergic response to inhaled allergens have been instrumental in investigating the mechanisms underlying the initiation and maintenance of allergen-specific Th2 cell responses. Using these models, it has been shown that the development of allergic Th2 cell responses through the intranasal (i.n.) route usually occurs in two steps. The first step is called “sensitization” and is initiated after the primary i.n. exposure to an allergen. During the initial sensitization lung-migratory cDC2s traffic into the lung-draining lymph nodes to prime allergen-specific CD4+ T cells with a type 2/Th2-biased cytokine profile (15, 18, 46, 47, 50, 51). Importantly, however, this initial exposure does not typically result in the accumulation of effector allergen-specific Th2 cells in the airways (15, 18) and therefore does not necessarily cause clinical manifestations. Instead, allergen sensitization triggers a strongly biased Tfh2 cell response that is restricted to the lung-draining lymph nodes (15, 18, 165). The second step is characterized by the development of pathology and clinical features following secondary or subsequent allergen exposures or “challenges.” This phase is characterized by the accumulation of effector allergen-specific Th2 cells in the airways and Th2 cytokine production (15, 18).
Tfh2 cells can produce large amounts of Th2 cytokines, including IL-4 and IL-13, in response to allergens and helminths (12, 18, 89, 166–170) and, in fact, are the primary sources of IL-4 and IL-13 during the sensitization phase (12, 18). IL-4/IL-13-producing Tfh2 cells are critical for the sustained production of IgG1 and IgE class switching (166, 167, 169) and high-affinity IgE (12). A division of labor between IL-4-producing Tfh2 and IL-13-producing Tfh2 [also known as Tfh13 (12)] has been proposed, where IL-4-producing Tfh2 instruct the production of IgG1 and low-affinity IgE, but IL-13 production drives high-affinity IgE secretion and anaphylaxis (12). In animal models, IL-13-producing Tfh2 are not induced after infection by helminths (12, 170), but they can be generated to aeroallergens, including HDM and fungal allergens in some studies (12), but not in others (171). But, besides controlling B cell isotype switching, Tfh2 cells generated during the sensitization phase can survive in the lymph nodes as memory cells and have the unique ability to give rise to effector Th2 cells upon allergen rechallenge (18). These data suggest lineage flexibility of Tfh2 cells in allergic disease and identify these cells as a crucial reservoir of Th2 cell progenitors. Tfh cell development depends on the expression of the transcription factor B cell lymphoma 6 (Bcl6), which functions as a transcriptional repressor that prevents the acquisition of T effector programs (172). In contrast, the generation of effector Th2 cells in the lung requires expression of the transcription factor B-lymphocyte-induced maturation protein 1 (Blimp1), encoded by the Prdm1 gene (173). Bcl6 and Blimp1 are reciprocally antagonistic transcription factors (174, 175). Thus, the balance between Blimp1 and Bcl6 expression likely controls the relative commitment of CD4 T cells to the effector Th2 or Tfh2 cell pathways. Taken together, the two-step model for the development of allergic Th2 cell responses predicts that induced expression of Bcl6 in T cells (favored during sensitization to inhalant allergens) drives Tfh2 cell differentiation and central memory. In contrast, induced expression of Blimp1 during allergen re-exposure promotes effector Th2 cells that migrate to the lungs (Figure 2). Consistent with this model, upregulation of Bcl6 expression upon allergen exposure inhibits effector Th2 cell fate choice (171), whereas the up-regulation of Blimp1 favors the differentiation of effector Th2 cells at the expense of Tfh cells (173). Understanding what signals control Bcl6 and Blimp expression in allergen-specific T cells will be essential for understanding Tfh vs. effector fate choice. Like in other immune responses (172), the acquisition of Bcl6 by allergen-specific T cells is initiated by cDC priming but then maintained by cognate interactions with B cells (15, 18). Cognate B cells preserve the pool of allergen-specific Tfh2 and provide a niche for memory development and maintenance (18). In contrast, the loss of B cell interactions favors the effector Th2 cell pathway (171). Thus, the crosstalk between T cells and B cells likely regulates the balance between Tfh and effector Th2 cell differentiation. On the other hand, Blimp1 expression increases in response to IL-2 acting through STAT5 (176, 177), which has been shown to be a potent inhibitory mechanism of Bcl6 and Tfh cell responses (178, 179); but it enhances the generation of effector Th2 cells migrating to the lung (171). Similarly, TSLP activates STAT5, which represses Bcl6 and stimulates effector pathogenic Th2 cell responses (180). IL-10 can also upregulate Blimp1 via STAT3 (181) and has been shown to promote effector Th2 responses and the development of allergic lung disease by repressing Bcl6 expression (173). Finally, IL-33 can likewise promote Blimp1 expression in T cells and exacerbate allergic airway inflammation (182) (Figure 2). Future studies will need to determine the factors that control the production of these cytokines, their source, and their relative contribution to the effector Th2 and Tfh2 cell pathways. Notably, the antagonism between Bcl6 and Blimp1 has been shown to control the balance of Tfh2 vs. effector Th2 pathways during respiratory but not cutaneous allergen sensitization (173, 183). Moreover, the nature of the allergen can also affect the differential development of Tfh2 and effector Th2 cells (166). Thus, it will also be necessary to address how different mechanisms govern during sensitization with diverse natural allergens and different routes.
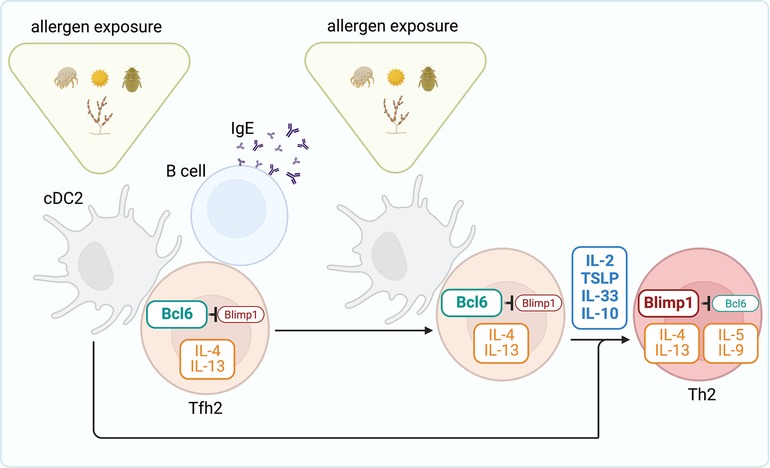
Figure 2. Steps toward generating effector Th2 cell responses to allergens. During the sensitization phase, cDC2s prime allergen-specific Tfh2 cell responses in the draining lymph node. Through interactions with B cells, Tfh2 cells promote IgE secretion. Following allergen re-exposure, Tfh2 cells differentiate into effector Th2 cells that subsequently migrate to the lung and promote allergic airway inflammation. Tfh2 cell development depends on the expression of the transcription factor Bcl6, which functions as a transcriptional repressor that prevents Blimp1 expression and the acquisition of the Th2 effector program. The cytokines IL-2, TSLP, IL-33, and IL-10 promote Blimp1 expression and thus drive differentiation of Th2 effectors from tfh2 cells.
Mechanisms that suppress Th2 cell priming by dendritic cells
T-bet is a critical transcription factor for Th1 cell differentiation. T cells lacking T-bet fail to develop into Th1 cells (85). But, loss of T-bet also results in the activation of the Th2 cell differentiation program (33, 85, 184, 185). Moreover, overexpression of T-bet in Th2 cells results in the loss of the Th2 cell phenotype (85, 185). Thus, T-bet plays a crucial role in suppressing the Th2 cell-associated program. GATA Binding Protein 3 (GATA3) is the critical transcription factor for Th2 cell differentiation (186–190). IL-4-mediated STAT6 activation is the main inducing signal for GATA3 upregulation and maintenance (188, 191). Mechanistically, T-bet suppresses the Th2 cell differentiation program by directly inhibiting GATA3 expression through epigenetic repression at the Gata3 locus (33, 185) and inhibiting GATA3 function through protein-protein interactions (192).
In vitro and in vivo animal models have essentially contributed to the understanding of the role of T-bet in preventing Th2 cell development. However, an important question is to demonstrate whether T-bet plays a significant role in actively preventing allergen-specific Th2 cell development in healthy non-atopic individuals. Recently, the first patient with autosomal recessive complete T-bet deficiency has been reported (193–195). This human T-bet deficiency causes disrupting development of IFNγ-producing cells (193). But importantly, the patient also developed upper airway inflammation, peripheral eosinophilia, and increased production of the Th2 cytokines IL-4, IL-5, and IL-13 due to Th2 skewing of T-bet-deficient CD4+ T cells (194). Additionally, a B cell class switching to IgG1, IgG4, and IgE has been observed in this patient (195), possibly due to the favored induction of Tfh2 and Th2 cell cytokine production (194). Thus, T-bet contributes to preventing Th2-biased differentiation in humans. Interestingly, T-bet protein is downregulated in T cells from the airways of human patients with Th2-driven asthma (184). More recent human studies have further found that activated allergen-specific T cells exist in both healthy and allergic/asthmatic subjects (196). However, the polarization profile of the reactive T cells was different. Allergic/asthmatic had allergen-specific T cells with a Th2 cell profile, whereas allergen-responsive T cells in healthy/non-allergic subjects were characterized by a Th1/IFN signature (196). Overall, these data suggest that T-bet plays an essential role in preventing Th2 cell priming and Th2 cell-driven allergic airway disease in humans.
Allergens contain endogenous PRR agonists or can be contaminated with exogenous environmental PRR ligands. Several studies have indicated that exposure to allergens with high content of microbial products, particularly bacterial endotoxins or LPS, inversely correlates with the development of allergen-induced, Th2-driven diseases such as allergic asthma and atopy (22–30). This data suggests that LPS detection is linked to the suppression of Th2 cell immune responses to allergens. As a note, among all the pro-inflammatory PRR ligands that can be found in allergens, it is not surprising that LPS shows the most robust correlations due to its extreme potency. But, since TLRs have redundant functions, various TLR ligands could likely suppress th2 responses through similar mechanisms. Using animal models, we have demonstrated that sensitization with HDM allergens containing low amounts of LPS can very efficiently prevent Th2 cell-driven allergic inflammation, particularly in adult mice (128). LPS prevents Th2-dependent allergic responses by enabling IL-12 production by lung-migratory cDC2s. Consequently, allergen-specific T cells interacting with IL-12-expressing cDC2s can upregulate T-bet (128), which precludes the Th2 cell differentiation program and subsequent pathogenic allergic response to HDM allergens. T-bet can be redundantly upregulated by IL-12 and IFNγ (33, 197–199). IFNγ produced by T cells induces T-bet expression, which serves as a positive feedback loop that allows complete Th1 cell differentiation (198, 200). But during initial priming, it is widely accepted that IL-12, produced primarily by cDCs, initiates Th1 cell differentiation by promoting the early upregulation of T-bet. T-bet then stimulates IFNγ production, allowing for the positive feedback loop that maximizes Th1 immunity when IL-12 becomes limited (31, 33, 201, 202). Besides controlling Th1 cell differentiation, T-bet suppresses the Th2 cell differentiation program to aeroallergens, where IL-12 produced by cDC2s plays a crucial role in the process (128). Importantly, sensitization with allergens containing low amounts of LPS induces moderate levels of T-bet in allergen-responsive T cells. Although these moderate levels of T-bet expression are sufficient to inhibit Th2 cell differentiation, they do not cause substantial IFNγ production or Th1 cell differentiation (128). Thus, different levels of T-bet expression may differentially control Th2 cell suppression, Th1 cell generation, and the acquisition of effector functions (203).
Migratory cDC2s can either promote (18, 46–51, 53) or suppress (103, 128) Th2 cell responses to allergens, and in this regard, the activation state of cDC2s is crucial to define their role. As previously discussed, the transcription factors IRF4, KLF4, and STAT6 program cDC2s to support Th2 cell differentiation, most likely by reducing the ability of cDC2s to produce IL-12 (48, 50, 51, 53, 160–162). We have found, otherwise, that the transcription factor T-bet promotes the functional specialization of cDC2s to suppress Th2 cell differentiation by promoting the ability of cDC2s to produce sustained IL-12 after sensitization with allergens containing low amounts of LPS (103, 128). Therefore, the ability of cDC2s to promote or prevent Th2 cell responses is highly dependent on their acquired ability to express T-bet and produce IL-12 (Figure 1). Although the ability of cDC2s to produce IL-12 appears to be tightly regulated and correlates with its functional ability, cDC1s constitutively express high levels of IL-12 (128, 204, 205). As such, cDC1s are commonly associated with the Th1-inducing cDC subset (159). Additionally, cDC1s have been shown to contribute to ameliorating Th2 cell responses to allergens (206, 207). However, cDC2s can also produce large amounts of IL-12 when properly stimulated (97, 208–210). Furthermore, IL-12-producing DC2s are the main contributors to the prevention of Th2 cell responses upon host detection of low levels of LPS in aeroallergens (103, 128). As such, cDC2s can normally prevent Th2 cell differentiation in mice deficient in Basic leucine zipper ATF-like transcription factor 3 (BATF3) despite the absence of cDC1s; but they fail to suppress allergic Th2 cell responses when they cannot express T-bet and produce IL-12 (128). In conclusion, cDC2s can activate either a pro- or anti-Th2 cell phenotype (Figure 1). Future studies should determine the full picture by integrating the heterogeneity of transcriptional programs that underlie functional specification in cDC2s.
As mentioned, host recognition of LPS and robust induction of pro-inflammatory cytokine responses are linked to the suppression of Th2 cell immune responses to allergens. Thus, in this context, factors that influence or modify LPS responsiveness have the potential to alter Th2-driven allergen sensitization. LPS can be detected by multiple cells expressing TLR4. Predominantly, myeloid cells such as neutrophils, macrophages, cDCs, and inflammatory monocytes can very efficiently respond to LPS since they express optimal levels of TLR4 and the co-receptors CD14 and MD-2. Among myeloid cells, classical monocytes express the highest levels of these proteins (211). CD14 binds LPS and transfers the bound LPS to the TLR4-MD-2 complex (212). This transfer cascade is essential for the efficient triggering of the TLR4-induced inflammatory cytokine response involving activation of the TIRAP-MyD88 and TRAM-TRIF signaling pathways (141, 213, 214). Non-hematopoietic cells such as vascular (215), epithelial (216), and adipose cells (217) can also express TLR4. However, unlike myeloid innate immune cells, these structural cells express low levels of CD14 and MD-2 and have been shown to be poorly responsive to LPS under normal conditions (218–221). Given the diversity of cells that can respond to LPS, this can lead to highly variable sensitivities to LPS and qualitatively and quantitatively very different cytokine responses depending on the cell type(s) involved. In this regard, as discussed previously, it has been shown that LPS-driven TLR4 signaling in stromal cells, most likely in airway epithelial cells, may favor the initiation of airway Th2 cell responses to proteins that would otherwise behave as inert antigens (103, 130–132, 222). Stromal or epithelial cell-guided LPS responses have a low inflammatory component; however, they could still stimulate the production of proallergic cytokines or “alarmins” and the consequent activation and migration of antigen-containing cDCs (15). Conversely, LPS-induced TLR4 activation in hematopoietic cells is needed to suppress Th2 cell sensitization to natural allergens (103). However, the inhibitory functions of cDC2s on Th2 cell allergic responses do not require direct recognition of LPS (103). Instead, this function in cDC2s is controlled by inflammatory mediators produced by classical Ly6Chi monocytes that activate the differentiation program into moDCs (103). Granulocyte-macrophage colony-stimulating factor (GM-CSF) is critical for moDC differentiation (103, 223, 224), and GM-CSF-driven functional programming of moDCs allows these cells to increase their sensitivity to LPS and their ability to elicit a pro-inflammatory cytokine response (103, 225–228). In particular, GM-CSF licenses the inflammatory signature in moDC by increasing the expression of TLR4, CD14, and intracellular signaling members involved in the TIRAP-MyD88-dependent pathway (103, 229). In contrast, the TRIF-TRAM-dependent pathway appears to be downregulated by GM-CSF signaling in classical monocytes (103). The TIRAP-MyD88-dependent pathway induces rapid activation of nuclear factor κB (NF-kB) and activator protein 1 (AP-1), ultimately leading to the production of inflammatory cytokines such as tumor necrosis factor-α (TNFα) (230, 231). The TRIF-TRAM-dependent pathway is otherwise responsible for mediating type-1 IFN production and signaling (232, 233). Thus, GM-CSF preferentially licenses for the production of TNFα and other pro-inflammatory cytokines in classical Ly6Chi monocytes while inhibiting their type 1 IFN response. Overall, the functional programming of moDCs by GM-CSF allows these cells to increase their sensitivity to LPS and stimulate the production of pro-inflammatory cytokines that allow indirect activation of lung-migratory cDC2s for Th2 cell suppression rather than promotion (Figure 1). Therefore, GM-CSF-driven moDCs have an essential role as amplifiers and modifiers of LPS functions in allergic inflammation.
GM-CSF can be produced by multiple cells types, including hematopoietic (e.g., T cells, macrophages, monocytes, mast cells) and non-hematopoietic (e.g., vascular endothelial cells, epithelial cells, and fibroblasts) cells (234). GM-CSF is produced locally, where it regulates cell function in close proximity. Homeostatic GM-CSF production is required for the differentiation of alveolar macrophages (235, 236). But, in general, GM-CSF production is usually linked to infection, inflammation, or pathological conditions. As such, a number of pro-inflammatory mediators can induce the expression of GM-CSF (237, 238). Allergens can also induce GM-CSF production by different cell types (103, 133, 239). Allergens with cysteine protease activity have a strong capacity to trigger robust GM-CSF response and promote moDC differentiation. In particular, cysteine protease activity on allergens prompts GM-CSF production by perivascular non-classical Ly6Clo monocytes, thereby guiding the differentiation of classical Ly6Chi monocyte into moDCs (103). Conversely, trypsin-like serine protease activity or the presence of endogenous TLR4 ligands on allergens activate lung epithelial cells to induce GM-CSF production (133, 240). Importantly, the production of GM-CSF by these two different cellular compartments, which release GM-CSF at different locations and in different amounts, appears to have completely opposite results in allergen sensitization. Epithelial-derived GM-CSF has been shown to play a critical role in allergic sensitization, as this source of GM-CSF promotes the activation and migration of cDCs from the lung to the lymph node, where they prime Th2 cell responses (103, 133, 240–243). In contrast, production of GM-CSF in perivascular areas by non-classical monocytes regulates the de novo generation of moDCs from newly-recruited classical monocytes, which ultimately instigates cDC2s for Th2 cell suppression to LPS-contaminated allergens (103) (Figure 1). Although classical Ly6Chi monocytes are critical mediators of inflammatory responses, non-classical Ly6Clo monocytes have been widely viewed as anti-inflammatory, as they maintain vascular homeostasis and promote reparative processes (244–248). However, non-classical monocytes are also a first line in the recognition of immunological insults and can contribute to inflammation (249–254). Future research should determine whether nonclassical monocytes could control the inflammatory functions of classical monocytes in settings other than allergen exposure and cysteine protease-mediated activation. Overall, the published mechanistic data suggest the interplay between the GM-CSF response elicited after allergen exposure by different cellular sources and the amount of endotoxin/microbial products contained in those inhaled allergens can oppositely balance the activation of mDC2s and, in the end, establish whether the stimulated allergen-specific T cell response will be protective or pathogenic. In view of this, an altered response to TLR ligands or GM-CSF conditioned by polymorphisms that affect the function of these signaling pathways might be associated with altered susceptibility to allergen sensitization. Many studies have tested this hypothesis and have found an association between TLR or GM-CSF polymorphisms and sensitization to airborne environmental allergens and allergy development. For example, single nucleotide polymorphisms (SNPs) in CD14, TLR4, and TLR2 genes have been correlated with the development of allergy sensitization and symptoms of allergy in children (255–264). Likewise, variants in the gene for GM-CSF have been associated with the development of atopic diseases in children (265–267). In contrast, other studies have shown no association between CD14, TLR2, TLR4, or GM-CSF polymorphisms and allergy (268–272). Thus, although polymorphism in genes encoding GM-CSF and TLR signaling members is associated in many cases with allergic phenotypes, results are variable. In general, stronger associations have been found in children living in environments that expose them to high levels or a different diversity of microbial agents, suggesting that genetic variants on allergy susceptibility appear to be modified by quantitative factors (levels of exposure) and qualitative components (the type of microbial products/TLR ligands present in the environment) (255, 258, 260, 261, 263). Therefore, gene-by-environment interactions between functional gene polymorphisms and environmental exposures should be considered together to better understand susceptibility to allergic sensitization [reviewed in (273)].
Although, as discussed in this section, IL-12 and T-bet play essential roles in the suppression of Th2 cell differentiation, we have recently discovered that IL-6 also plays a crucial and T-bet-independent role in inhibiting the Th2 cell differentiation program [preprint (274)]. Loss-of-function mutations that affect IL-6 signaling, including IL6 receptor (IL-6R) (275), Glycoprotein 130 (GP130) (276, 277), and STAT3 (278–281), lead to increased Th2 bias and manifestations of allergy (282). Moreover, the IL6R locus has been associated with allergy (283, 284). Although these studies with patients suggest a significant role for IL-6 in controlling Th2 bias, the specific contribution of IL-6 and the underlying mechanism remain largely undefined. As discussed, Th2 cell lineage commitment requires strong and sustained IL-2 signaling (76, 77). Our data found that IL-6 suppresses IL-2 signaling during early T cell activation, thereby inhibiting Th2 priming [preprint (274)]. IL-6-driven inhibition of IL-2 signaling in allergen-responding T cells is mediated by the upregulation of the suppressor of cytokine signaling 3 (SOCS3). Thus, the IL-12-T-bet and IL-6-SOCS3 axes cooperate to inhibit the Th2 cell differentiation program by inhibiting GATA-3 and IL-2 signaling in allergen-responsive T cells, respectively (Figure 1).
Mechanisms that favor Th2 biased immune responses at an early age
It is well known that infants and children are at a higher risk of developing Th2 cell allergic responses than adults. Therefore, they suffer from a high prevalence of respiratory allergic diseases (19, 285). Indeed, observational studies have shown that primary sensitizations to airborne allergens develop gradually and peak in children one to three years of age (7), and are associated with the later development of allergic diseases and asthma, usually before the age of five (5, 6, 8). Importantly, the incidence of childhood allergic rhinitis and asthma has dramatically increased over the past decades in the U.S. and other industrialized nations (6, 10, 286). As such, pediatric asthma is considered the leading chronic disease of childhood. However, the immunological mechanisms underlying the high susceptibility to allergic airway disease in infants and children and the sharply increased burden of allergies and asthma in children in the developed world remain poorly understood. Urbanization is thought to play a major role, as allergic lung diseases are becoming more common in children in urban areas, although the prevalence remains low in rural areas (287–289). Rural/traditional farming settings are associated with close contact with livestock and increased exposure to endotoxin-contaminated aeroallergens in dust (290). Furthermore, epidemiological studies indicate that early exposure to endotoxin-contaminated aeroallergens in those traditional rural/farming settings protects against asthma/allergic reactions in school-age children, whereas infants sensitized to common circulating allergens in low-LPS environments are at increased risk of becoming atopic and developing symptoms of allergic disease later in life (22–30, 291). As discussed previously, laboratory mechanistic studies have established that exposure to airborne allergens containing endotoxin protects against allergic inflammation by preventing the Th2 cell differentiation program in allergen-specific T cells (103, 107, 128). These mechanistic studies have further shown that while low-LPS exposure during allergen sensitization is sufficient to protect adult mice from developing Th2-driven allergic inflammation in the lungs, infant mice require higher doses of LPS (128). These data indicate that LPS prevents Th2-dependent allergic responses with different thresholds in adults and infants, thus providing a plausible mechanism underlying the higher susceptibility to allergic disease and asthma, particularly observed in children living in urban areas and exposed to low-LPS contaminated aeroallergens. As mentioned, mouse studies have further demonstrated that suppression of Th2-driven immune responses to allergens depends on the production of pro-inflammatory cytokines by classical monocytes, particularly TNFα, upon TLR4 engagement (103, 128). However, during infancy, LPS hyporesponsiveness has been observed. In particular, human studies have shown that monocytes from newborns and infants produce significantly fewer pro-inflammatory cytokines, with markedly impaired TNFα release, after stimulation with low concentrations of LPS (292–295), which has been associated with decreased expression of TLR4, CD14, and MyD88 in neonatal monocytes in some studies (293), but not all (296). Corresponding with this, we have also observed that classical monocytes from infant mice produce less TNFα in the lungs after low-LPS sensitization. Furthermore, we have observed that impaired production of TNFα at low doses of LPS is particularly evident in mice during a time window from day 7 to day 20 after birth, after which LPS-driven TNFα production rapidly reaches adult levels (128). Since TNFα released by monocyte-derived cells tunes cDC2 activation to produce IL-12 for subsequent inhibition of Th2 cell sensitization to inhaled allergens (103, 128), it is easy to infer that the inability of infant monocytes to respond to LPS and produce optimal amounts of TNFα is a major cause underlying the increased risk of developing Th2 responses to allergens during infancy (Figure 3). However, how age modulates the ability of classical monocytes to respond to LPS is a topic of further investigation. GM-CSF is a critical factor controlling the expression of CD14, TLR4, and TLR signal transduction proteins in monocytes and, thus, their LPS sensitivity and responsiveness (103). Reduced GM-CSF production and GM-CSF mRNA expression have been observed in blood mononuclear cells from human newborns compared to adults (297–300). Furthermore, studies in mice have shown that innate myeloid cells in the lung respond poorly to GM-CSF (301). These studies suggest that the GM-CSF axis as an amplifier of LPS responses may be functioning poorly at early ages. The cause of these changes during early life is unknown. The microbiome plays an essential role in the maturation of the infant immune response, and indeed, the establishment and development of the gut, lung, and skin microbiota in early life occurs in parallel with the acquisition of immune functions (302–312). Therefore, it is possible that early infant intestinal, lung, and skin exposure to commensal bacteria and fungi may modulate the GM-CSF-TLR4 axis and, thus, LPS responsiveness. Some evidence detailed below supports this hypothesis. Farming-related exposures, which, as discussed above, decrease the risk for allergic outcomes, have been associated with early upregulation of CD14 and TLR expression in the first year of life (313–316). These data suggest that although newborns naturally develop dampened TLR responses, specific environmental exposures during the first year of life contribute to the upregulation of innate immune receptors. Early upregulation of TLR and CD14 expression likely leads to an increased ability to generate inhibitory signals for Th2 cell development and ultimately to reduced risk of allergic diseases. Many recent studies have attempted to identify early life exposures associated with protection against allergies. Children living on farms are exposed to a wider range of environmental bacteria and fungi. The greater diversity of environmental microbial exposure is correlated with protection from allergic airway disease (317). In addition, consumption of raw farm milk (313, 316, 318, 319) and dairy products (320–322) have been identified as exposures that contribute to the protective effect of farm life on childhood allergies. Furthermore, the diversity of the foods introduced in the first two years of life has also been associated with protection from allergic diseases (320, 321, 323). All of these exposures are suggested to play a pivotal role in the evolution and establishment of the microbiome from infancy toward the acquisition of adult-like communities (324–326). Mouse studies have evaluated how specific changes in the composition of the gut and lung microbiota with age, diet, or exposure to environmental microorganisms are associated with protection against allergic diseases (310, 312, 325). In particular, the increase in bacteria from the phylum Bacteroidetes and their ability to ferment fiber in short-chain fatty acids (SCFA) have been shown to modulate the cDC2 and moDC compartments, leading them to a maturation profile that is ineffective in driving Th2 cell responses (310, 325). Thus, metabolites produced by the microbiota can influence the functional specialization of cDC2s and moDCs to prevent Th2 cell responses to allergens. Interestingly, early-life dietary practices associated with protection from allergic disease have also been correlated with the maturation of the gut microbiome and elevated SCFA levels (325, 327, 328). Overall, dietary and environmental microbial exposures shape the maturation of the infant microbiome toward an adult-like structure. The data indicate that early maturation of the infant microbiome helps prevent the development of Th2 cell responses, thus protecting against allergic airway diseases. One of the plausible mechanisms by which the maturation of the infant gut microbiota would protect against allergic diseases could be by promoting the upregulation of innate immune receptors, including TLR4 and CD14, which would help to acquire greater sensitivity and response to microbial products, particularly to LPS.
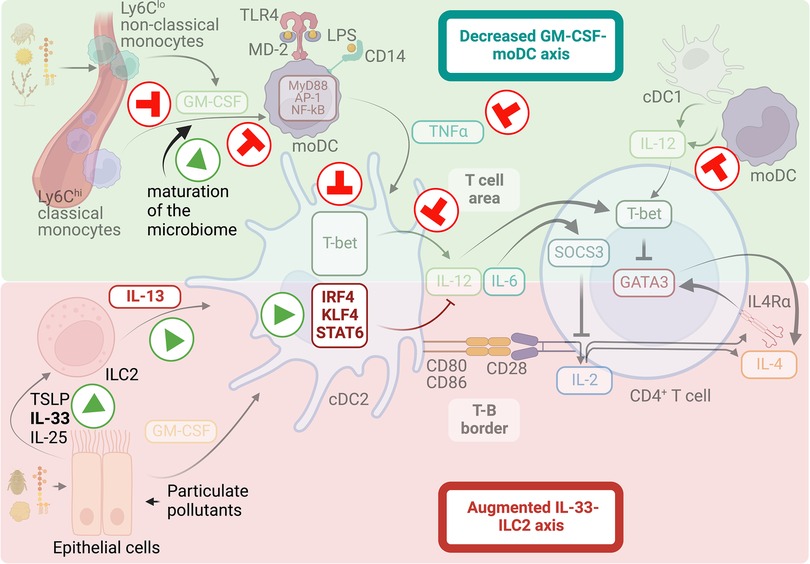
Figure 3. Mechanisms that favor Th2-biased immune responses at early ages. Infants and young children are at higher risk of developing Th2 cell allergic responses than adults. The lack of inhibitory signals and the intensification of triggering factors contribute to the increase in Th2-biased immunity and sensitization to allergens during early childhood. A malfunctioning GM-CSF-moDC axis conditions for LPS hyporesponsiveness during infancy, hindering the induction of T-bet expression on cDC2s and thus driving deficient IL-12 responses. Early dietary practices and exposure to environmental microorganism may shape the maturation of the infant microbiome into compositions that enhance the GM-CSF-moDC axis and protect against sensitization to airborne allergens. On the other hand, a hyperactive IL-33 axis operating in the lung during the early postnatal period increases IL-13 production by ILC2, which activates the migration and pro-Th2 function of cDC2s. Additionally, exposure to inhalable particulate pollutants can intensify this tendency.
In addition to the low ability to counteract Th2 cell responses, infant environments have been shown to produce pro-Th2 factors that contribute to the propensity to develop allergic sensitization during infancy. A hyperactive IL-33 axis has been shown to operate in the lung during the early postnatal period, increasing cytokine production by ILC2s, which in turn activates the migration and pro-Th2 function of cDC2 (152, 329, 330). This augmented IL-33 activity has been associated with the postnatal phase of lung alveolarization, suggesting that normal postnatal lung development can predispose to Th2 cell immunity. On the other hand, urbanization is linked to increased exposure to airborne particle pollution (also called particulate matter or PM). Recent studies have shown that early-life exposure to PM is associated, in many cases, with increased susceptibility to allergic sensitization, airway allergy, and asthma [reviewed in (331, 332)]. Although the exact mechanism by which PM may influence the onset of allergic disease is not yet fully understood, the airway epithelium has been shown to be the primary site for PM deposition, and it is believed that PM-driven activation of airway epithelium is central in the effects derived from PM exposure (333). The key mechanism by which PM is thought to exert its effects is the generation of reactive oxygen species (ROS), which induce antioxidant and inflammatory responses in exposed epithelial cells (334, 335). Moreover, PM can affect the integrity of the airway epithelial barrier (334). Therefore, the increased susceptibility to develop allergic disease in the presence of PM may arise from impaired barrier function of the epithelium, leading to increased permeability of the airway mucosa to allergens, and increased PM-stimulated release of alarmin cytokines that promote type-2 immunity to allergens (333). Still, more studies are needed to identify the exact mechanisms by which PM may be a risk factor for allergic diseases, particularly during childhood. Furthermore, the critical age window for exposure has yet to be established. In conclusion, both the lack of inhibitory signals and the increase in triggering factors contribute to augmented type 2 immunity and allergen sensitization during infancy (Figure 3). The onset of allergic sensitization coincides with the early establishment and evolution of the gut, lung, and skin microbiome and the structural development of the lungs in early childhood. Additionally, it is influenced by dietary behaviors and environmental exposures, including biological and chemical contaminants. How these factors may contribute to the risk of developing Th2 responses early in life and the underlying mechanisms remain to be fully explored. Obtaining this knowledge would help establish interventions and recommendations that seek to prevent sensitization to airborne allergens in the first years of life.
Finally, it is important to note that although exposure to highly LPS-contaminated inhalant allergens protects from the development of allergic Th2 cell responses during infancy and childhood, high endotoxin exposure can be a respiratory hazard in adulthood (336). The airway response in adults exposed to dust containing high levels of LPS is generally characterized by increased production of the pro-inflammatory cytokines IL-1, IL-6, and TFNα, the development of Th1 and Th17 responses, and infiltration of neutrophils (337–343). Overall, although hyporesponsiveness to LPS during infancy makes high LPS exposure necessary to prevent Th2 allergic responses, increased sensitivity to LPS with age implies that same high exposure to LPS may be detrimental by inducing an excessive pro-inflammatory response. Thus, hygienic perspectives should be different in childhood and adulthood and according to the degree of sensitivity to endotoxin.
Concluding remarks
The early window of life is particularly predisposed to developing Th2 cell responses after initial contact with aeroallergens. This appears to be due to an imbalance between signals that counteract and promote Th2 cell development. cDCs elicit Th2 cell responses after a complex interaction with epithelial cells, monocytes, ILC2, and probably other immune cells that occur in the context of exposure to allergens harboring protease activity and PRR ligands. Additionally, it is now recognized that the succession of microbiota in early life and the exposure to chemical and biological contaminants make essential contributions to the infant response to allergens by altering intercellular communications. In the future, it will be crucial to understand how the infant microenvironment controls the specific signals delivered upon encountering an allergen and how these are influenced by diet and different environmental exposures, ultimately allowing us to understand better how all these signals shape the functionality of the cDCs and the specific T cell responses elicited.
Author contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was supported the University of Alabama at Birmingham (UAB) and the National Institutes of Health grant 2R01AI116584 to B. León.
Acknowledgments
Figures were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Umetsu DT, McIntire JJ, Akbari O, Macaubas C, DeKruyff RH. Asthma: an epidemic of dysregulated immunity. Nat Immunol. (2002) 3:715–20. doi: 10.1038/ni0802-715
2. Eder W, Ege MJ, von Mutius E. The asthma epidemic. N Engl J Med. (2006) 355:2226–35. doi: 10.1056/NEJMra054308
3. Pols DH, Wartna JB, van Alphen EI, Moed H, Rasenberg N, Bindels PJ, et al. Interrelationships between atopic disorders in children: a meta-analysis based on ISAAC questionnaires. PLoS One. (2015) 10:e0131869. doi: 10.1371/journal.pone.0131869
4. Pawankar R. Allergic diseases and asthma: a global public health concern and a call to action. World Allergy Organ J. (2014) 7:12. doi: 10.1186/1939-4551-7-12
5. Masoli M, Fabian D, Holt S, Beasley R, P. Global Initiative for Asthma. The global burden of asthma: executive summary of the GINA dissemination committee report. Allergy. (2004) 59:469–78. doi: 10.1111/j.1398-9995.2004.00526.x.
6. Akinbami LJ, Moorman JE, Bailey C, Zahran HS, King M, Johnson CA, Liu X. Trends in asthma prevalence, health care use, and mortality in the United States, 2001-2010. NCHS Data Brief (2012) 1–8.22617340
7. Holt PG. Primary allergic sensitization to environmental antigens: perinatal T cell priming as a determinant of responder phenotype in adulthood. J Exp Med. (1996) 183:1297–301. doi: 10.1084/jem.183.4.1297
8. Lodge CJ, Lowe AJ, Gurrin LC, Hill DJ, Hosking CS, Khalafzai RU, et al. House dust mite sensitization in toddlers predicts current wheeze at age 12 years. J Allergy Clin Immunol. (2011) 128:782–788.e9. doi: 10.1016/j.jaci.2011.06.038
9. Arshad SH, Tariq SM, Matthews S, Hakim E. Sensitization to common allergens and its association with allergic disorders at age 4 years: a whole population birth cohort study. Pediatrics. (2001) 108:E33. doi: 10.1542/peds.108.2.e33
10. Pawankar R, Canonica GW, Holgate ST, Lockey RF. Allergic diseases and asthma: a major global health concern. Curr Opin Allergy Clin Immunol. (2012) 12:39–41. doi: 10.1097/ACI.0b013e32834ec13b
11. Yang Z, Wu CM, Targ S, Allen CDC. IL-21 is a broad negative regulator of IgE class switch recombination in mouse and human B cells. J Exp Med. (2020) 217.
12. Gowthaman U, Chen JS, Zhang B, Flynn WF, Lu Y, Song W, et al. Identification of a T follicular helper cell subset that drives anaphylactic IgE. Science. (2019) 365. doi: 10.1126/science.aaw6433
13. Krystel-Whittemore M, Dileepan KN, Wood JG. Mast cell: a multi-functional master cell. Front Immunol. (2015) 6:620.26779180
14. Leon B, Ballesteros-Tato A. Modulating Th2 cell immunity for the treatment of asthma. Front Immunol. (2021) 12:637948. doi: 10.3389/fimmu.2021.637948
15. Leomicronn B. T cells in allergic asthma: key players beyond the Th2 pathway. Curr Allergy Asthma Rep. (2017) 17:43. doi: 10.1007/s11882-017-0714-1
16. Coffman RL, Seymour BW, Hudak S, Jackson J, Rennick D. Antibody to interleukin-5 inhibits helminth-induced eosinophilia in mice. Science. (1989) 245:308–10. doi: 10.1126/science.2787531
17. Kuperman DA, Huang X, Koth LL, Chang GH, Dolganov GM, Zhu Z, et al. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med. (2002) 8:885–9. doi: 10.1038/nm734
18. Ballesteros-Tato A, Randall TD, Lund FE, Spolski R, Leonard WJ, Leon B. T follicular helper cell plasticity shapes pathogenic T helper 2 cell-mediated immunity to inhaled house dust Mite. Immunity. (2016) 44:259–73. doi: 10.1016/j.immuni.2015.11.017
19. Debock I, Flamand V. Unbalanced neonatal CD4(+) T-cell immunity. Front Immunol. (2014) 5:393. doi: 10.3389/fimmu.2014.00393
20. Zaghouani H, Hoeman CM, Adkins B. Neonatal immunity: faulty T-helpers and the shortcomings of dendritic cells. Trends Immunol. (2009) 30:585–91. doi: 10.1016/j.it.2009.09.002
21. Adkins B, Leclerc C, Marshall-Clarke S. Neonatal adaptive immunity comes of age. Nat Rev Immunol. (2004) 4:553–64. doi: 10.1038/nri1394
22. Abraham JH, Finn PW, Milton DK, Ryan LM, Perkins DL, Gold DR. Infant home endotoxin is associated with reduced allergen-stimulated lymphocyte proliferation and IL-13 production in childhood. J Allergy Clin Immunol. (2005) 116:431–7. doi: 10.1016/j.jaci.2005.05.015
23. Braun-Fahrlander C, Gassner M, Grize L, Neu U, Sennhauser FH, Varonier HS, et al. Prevalence of hay fever and allergic sensitization in farmer's children and their peers living in the same rural community. SCARPOL team. Swiss study on childhood allergy and respiratory symptoms with respect to air pollution. Clin Exp Allergy. (1999) 29:28–34. doi: 10.1046/j.1365-2222.1999.00479.x
24. Braun-Fahrlander C, Riedler J, Herz U, Eder W, Waser M, Grize L, et al. Environmental exposure to endotoxin and its relation to asthma in school-age children. N Engl J Med. (2002) 347:869–77. doi: 10.1056/NEJMoa020057
25. Gehring U, Bischof W, Fahlbusch B, Wichmann HE, Heinrich J. House dust endotoxin and allergic sensitization in children. Am J Respir Crit Care Med. (2002) 166:939–44. doi: 10.1164/rccm.200203-256OC
26. Gereda JE, Leung DY, Thatayatikom A, Streib JE, Price MR, Klinnert MD, et al. Relation between house-dust endotoxin exposure, type 1 T-cell development, and allergen sensitisation in infants at high risk of asthma. Lancet. (2000) 355:1680–3. doi: 10.1016/S0140-6736(00)02239-X
27. Stein MM, Hrusch CL, Gozdz J, Igartua C, Pivniouk V, Murray SE, et al. Innate immunity and asthma risk in amish and hutterite farm children. N Engl J Med. (2016) 375:411–21. doi: 10.1056/NEJMoa1508749
28. von Mutius E, Braun-Fahrlander C, Schierl R, Riedler J, Ehlermann S, Maisch S, et al. Exposure to endotoxin or other bacterial components might protect against the development of atopy. Clin Exp Allergy. (2000) 30:1230–4. doi: 10.1046/j.1365-2222.2000.00959.x
29. Loss GJ, Depner M, Hose AJ, Genuneit J, Karvonen AM, Hyvarinen A, et al. The early development of wheeze. Environmental determinants and genetic susceptibility at 17q21. Am J Respir Crit Care Med. (2016) 193:889–97. doi: 10.1164/rccm.201507-1493OC
30. House JS, Wyss AB, Hoppin JA, Richards M, Long S, Umbach DM, et al. Early-life farm exposures and adult asthma and atopy in the agricultural lung health study. J Allergy Clin Immunol. (2017) 140:249–256.e14. doi: 10.1016/j.jaci.2016.09.036
31. Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. (1996) 382:174–7. doi: 10.1038/382174a0
32. Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, et al. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature. (1996) 382:171–4. doi: 10.1038/382171a0
33. Zhu J, Jankovic D, Oler AJ, Wei G, Sharma S, Hu G, et al. The transcription factor T-bet is induced by multiple pathways and prevents an endogenous Th2 cell program during Th1 cell responses. Immunity. (2012) 37:660–73. doi: 10.1016/j.immuni.2012.09.007
34. Macatonia SE, Hosken NA, Litton M, Vieira P, Hsieh CS, Culpepper JA, et al. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J Immunol. (1995) 154:5071–9.7730613
35. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4 + CD25- naive T cells to CD4 + CD25 + regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. (2003) 198:1875–86. doi: 10.1084/jem.20030152
36. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
37. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
38. Stritesky GL, Yeh N, Kaplan MH. IL-23 promotes maintenance but not commitment to the Th17 lineage. J Immunol. (2008) 181:5948–55. doi: 10.4049/jimmunol.181.9.5948
39. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. (2011) 34:637–50. doi: 10.1016/j.immuni.2011.05.006
40. Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. (2000) 407:784–8. doi: 10.1038/35037722
41. Kinnebrew MA, Buffie CG, Diehl GE, Zenewicz LA, Leiner I, Hohl TM, et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity. (2012) 36:276–87. doi: 10.1016/j.immuni.2011.12.011
42. Lacy-Hulbert A, Smith AM, Tissire H, Barry M, Crowley D, Bronson RT, et al. Ulcerative colitis and autoimmunity induced by loss of myeloid alphav integrins. Proc Natl Acad Sci U S A. (2007) 104:15823–8. doi: 10.1073/pnas.0707421104
43. van Rijt LS, Jung S, Kleinjan A, Vos N, Willart M, Duez C, et al. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J Exp Med. (2005) 201:981–91. doi: 10.1084/jem.20042311
44. Lim H, Kim YU, Yun K, Drouin SM, Chung Y. Distinct regulation of Th2 and Th17 responses to allergens by pulmonary antigen presenting cells in vivo. Immunol Lett. (2013) 156:140–8. doi: 10.1016/j.imlet.2013.10.003
45. Phythian-Adams AT, Cook PC, Lundie RJ, Jones LH, Smith KA, Barr TA, et al. CD11c Depletion severely disrupts Th2 induction and development in vivo. J Exp Med. (2010) 207:2089–96. doi: 10.1084/jem.20100734
46. Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco-Madeira F, Toussaint W, et al. Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity. (2013) 38:322–35. doi: 10.1016/j.immuni.2012.10.016
47. Mesnil C, Sabatel CM, Marichal T, Toussaint M, Cataldo D, Drion PV, et al. Resident CD11b(+)Ly6C(-) lung dendritic cells are responsible for allergic airway sensitization to house dust mite in mice. PLoS One. (2012) 7:e53242. doi: 10.1371/journal.pone.0053242
48. Gao Y, Nish SA, Jiang R, Hou L, Licona-Limon P, Weinstein JS, et al. Control of T helper 2 responses by transcription factor IRF4-dependent dendritic cells. Immunity. (2013) 39:722–32. doi: 10.1016/j.immuni.2013.08.028
49. Kumamoto Y, Linehan M, Weinstein JS, Laidlaw BJ, Craft JE, Iwasaki A. CD301b(+) Dermal dendritic cells drive T helper 2 cell-mediated immunity. Immunity. (2013) 39:733–43. doi: 10.1016/j.immuni.2013.08.029
50. Williams JW, Tjota MY, Clay BS, Vander Lugt B, Bandukwala HS, Hrusch CL, et al. Transcription factor IRF4 drives dendritic cells to promote Th2 differentiation. Nat Commun. (2013) 4:2990. doi: 10.1038/ncomms3990
51. Tussiwand R, Everts B, Grajales-Reyes GE, Kretzer NM, Iwata A, Bagaitkar J, et al. Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity. (2015) 42:916–28. doi: 10.1016/j.immuni.2015.04.017
52. Deckers J, Sichien D, Plantinga M, Van Moorleghem J, Vanheerswynghels M, Hoste E, et al. Epicutaneous sensitization to house dust mite allergen requires interferon regulatory factor 4-dependent dermal dendritic cells. J Allergy Clin Immunol. (2017) 140:1364–1377.e2. doi: 10.1016/j.jaci.2016.12.970
53. Mayer JU, Hilligan KL, Chandler JS, Eccles DA, Old SI, Domingues RG, et al. Homeostatic IL-13 in healthy skin directs dendritic cell differentiation to promote TH2 and inhibit TH17 cell polarization. Nat Immunol. (2021) 22:1538–50. doi: 10.1038/s41590-021-01067-0
54. Constant S, Pfeiffer C, Woodard A, Pasqualini T, Bottomly K. Extent of T cell receptor ligation can determine the functional differentiation of naive CD4+ T cells. J Exp Med. (1995) 182:1591–6. doi: 10.1084/jem.182.5.1591
55. Rogers PR, Croft M. Peptide dose, affinity, and time of differentiation can contribute to the Th1/Th2 cytokine balance. J Immunol. (1999) 163:1205–13.10415015
56. Tao X, Grant C, Constant S, Bottomly K. Induction of IL-4-producing CD4+ T cells by antigenic peptides altered for TCR binding. J Immunol. (1997) 158:4237–44.9126985
57. Milner JD, Fazilleau N, McHeyzer-Williams M, Paul W. Cutting edge: lack of high affinity competition for peptide in polyclonal CD4 + responses unmasks IL-4 production. J Immunol. (2010) 184:6569–73. doi: 10.4049/jimmunol.1000674
58. Hosken NA, Shibuya K, Heath AW, Murphy KM, O'Garra A. The effect of antigen dose on CD4+ T helper cell phenotype development in a T cell receptor-alpha beta-transgenic model. J Exp Med. (1995) 182:1579–84. doi: 10.1084/jem.182.5.1579
59. van Panhuys N, Klauschen F, Germain RN. T-cell-receptor-dependent signal intensity dominantly controls CD4(+) T cell polarization in vivo. Immunity. (2014) 41:63–74. doi: 10.1016/j.immuni.2014.06.003
60. Ismail N, Bretscher PA. The Th1/Th2 nature of concurrent immune responses to unrelated antigens can be independent. J Immunol. (1999) 163:4842–50.10528185
61. Menon JN, Bretscher PA. Parasite dose determines the Th1/Th2 nature of the response to leishmania major independently of infection route and strain of host or parasite. Eur J Immunol. (1998) 28:4020–8. doi: 10.1002/(SICI)1521-4141(199812)28:12%3C4020::AID-IMMU4020%3E3.0.CO;2-3
62. Parish CR, Liew FY. Immune response to chemically modified flagellin. 3. Enhanced cell-mediated immunity during high and low zone antibody tolerance to flagellin. J Exp Med. (1972) 135:298–311. doi: 10.1084/jem.135.2.298
63. Bretscher PA, Wei G, Menon JN, Bielefeldt-Ohmann H. Establishment of stable, cell-mediated immunity that makes “susceptible” mice resistant to leishmania major. Science. (1992) 257:539–42. doi: 10.1126/science.1636090
64. Bancroft AJ, Else KJ, Grencis RK. Low-level infection with trichuris muris significantly affects the polarization of the CD4 response. Eur J Immunol. (1994) 24:3113–8. doi: 10.1002/eji.1830241230
65. Sarzotti M, Robbins DS, Hoffman PM. Induction of protective CTL responses in newborn mice by a murine retrovirus. Science. (1996) 271:1726–8. doi: 10.1126/science.271.5256.1726
66. Wang LF, Lin JY, Hsieh KH, Lin RH. Epicutaneous exposure of protein antigen induces a predominant Th2-like response with high IgE production in mice. J Immunol. (1996) 156:4077–82.8666772
67. Power CA, Wei G, Bretscher PA. Mycobacterial dose defines the Th1/Th2 nature of the immune response independently of whether immunization is administered by the intravenous, subcutaneous, or intradermal route. Infect Immun. (1998) 66:5743–50. doi: 10.1128/IAI.66.12.5743-5750.1998
68. Ismail N, Bretscher PA. More antigen-dependent CD4(+) T cell/CD4(+) T cell interactions are required for the primary generation of Th2 than of Th1 cells. Eur J Immunol. (2001) 31:1765–71. doi: 10.1002/1521-4141(200106)31:6%3C1765::AID-IMMU1765%3E3.0.CO;2-T
69. Keck S, Schmaler M, Ganter S, Wyss L, Oberle S, Huseby ES, et al. Antigen affinity and antigen dose exert distinct influences on CD4 T-cell differentiation. Proc Natl Acad Sci U S A. (2014) 111:14852–7. doi: 10.1073/pnas.1403271111
70. Okamoto M, Matsuda H, Joetham A, Lucas JJ, Domenico J, Yasutomo K, et al. Jagged1 on dendritic cells and notch on CD4+ T cells initiate lung allergic responsiveness by inducing IL-4 production. J Immunol. (2009) 183:2995–3003. doi: 10.4049/jimmunol.0900692
71. Amsen D, Blander JM, Lee GR, Tanigaki K, Honjo T, Flavell RA. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell. (2004) 117:515–26. doi: 10.1016/S0092-8674(04)00451-9
72. Krawczyk CM, Sun J, Pearce EJ. Th2 differentiation is unaffected by Jagged2 expression on dendritic cells. J Immunol. (2008) 180:7931–7. doi: 10.4049/jimmunol.180.12.7931
73. Worsley AG, LeibundGut-Landmann S, Slack E, Phng LK, Gerhardt H, Reis e Sousa C, et al. Dendritic cell expression of the notch ligand jagged2 is not essential for Th2 response induction in vivo. Eur J Immunol. (2008) 38:1043–9. doi: 10.1002/eji.200737335
74. Tindemans I, Lukkes M, de Bruijn MJW, Li BWS, van Nimwegen M, Amsen D, et al. Notch signaling in T cells is essential for allergic airway inflammation, but expression of the notch ligands jagged 1 and jagged 2 on dendritic cells is dispensable. J Allergy Clin Immunol. (2017) 140:1079–89. doi: 10.1016/j.jaci.2016.11.046
75. Tindemans I, Peeters MJW, Hendriks RW. Notch signaling in T helper cell subsets: instructor or unbiased amplifier? Front Immunol. (2017) 8:419. doi: 10.3389/fimmu.2017.00419
76. Cote-Sierra J, Foucras G, Guo L, Chiodetti L, Young HA, Hu-Li J, et al. Interleukin 2 plays a central role in Th2 differentiation. Proc Natl Acad Sci U S A. (2004) 101:3880–5. doi: 10.1073/pnas.0400339101
77. Liao W, Schones DE, Oh J, Cui Y, Cui K, Roh TY, et al. Priming for T helper type 2 differentiation by interleukin 2-mediated induction of interleukin 4 receptor alpha-chain expression. Nat Immunol. (2008) 9:1288–96. doi: 10.1038/ni.1656
78. Zhu J, Cote-Sierra J, Guo L, Paul WE. Stat5 activation plays a critical role in Th2 differentiation. Immunity. (2003) 19:739–48. doi: 10.1016/S1074-7613(03)00292-9
79. Zhou L, Chu C, Teng F, Bessman NJ, Goc J, Santosa EK, et al. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature. (2019) 568:405–9. doi: 10.1038/s41586-019-1082-x
80. Roediger B, Kyle R, Tay SS, Mitchell AJ, Bolton HA, Guy TV, et al. IL-2 is a critical regulator of group 2 innate lymphoid cell function during pulmonary inflammation. J Allergy Clin Immunol. (2015) 136:1653–1663.e7. doi: 10.1016/j.jaci.2015.03.043
81. Fujii H, Ogasawara K, Otsuka H, Suzuki M, Yamamura K, Yokochi T, et al. Functional dissection of the cytoplasmic subregions of the IL-2 receptor betac chain in primary lymphocyte populations. EMBO J. (1998) 17:6551–7. doi: 10.1093/emboj/17.22.6551
82. Moriggl R, Topham DJ, Teglund S, Sexl V, McKay C, Wang D, et al. Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity. (1999) 10:249–59. doi: 10.1016/S1074-7613(00)80025-4
83. Yamane H, Zhu J, Paul WE. Independent roles for IL-2 and GATA-3 in stimulating naive CD4+ T cells to generate a Th2-inducing cytokine environment. J Exp Med. (2005) 202:793–804.16172258
84. Wang ZE, Reiner SL, Zheng S, Dalton DK, Locksley RM. CD4 + effector Cells default to the Th2 pathway in interferon gamma-deficient mice infected with leishmania major. J Exp Med. (1994) 179:1367–71. doi: 10.1084/jem.179.4.1367
85. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. (2000) 100:655–69. doi: 10.1016/S0092-8674(00)80702-3
86. Leon B, Lund FE. Compartmentalization of dendritic cell and T-cell interactions in the lymph node: anatomy of T-cell fate decisions. Immunol Rev. (2019) 289:84–100. doi: 10.1111/imr.12758
87. Huang JY, Lyons-Cohen MR, Gerner MY. Information flow in the spatiotemporal organization of immune responses. Immunol Rev. (2022) 306:93–107. doi: 10.1111/imr.13046
88. Leal JM, Huang JY, Kohli K, Stoltzfus C, Lyons-Cohen MR, Olin BE, et al. Innate cell microenvironments in lymph nodes shape the generation of T cell responses during type I inflammation. Sci Immunol. (2021) 6.33579750
89. Leon B, Ballesteros-Tato A, Browning JL, Dunn R, Randall TD, Lund FE. Regulation of T(H)2 development by CXCR5 + dendritic cells and lymphotoxin-expressing B cells. Nat Immunol. (2012) 13:681–90. doi: 10.1038/ni.2309
90. Gunn MD, Kyuwa S, Tam C, Kakiuchi T, Matsuzawa A, Williams LT, et al. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J Exp Med. (1999) 189:451–60. doi: 10.1084/jem.189.3.451
91. Saeki H, Wu MT, Olasz E, Hwang ST. A migratory population of skin-derived dendritic cells expresses CXCR5, responds to B lymphocyte chemoattractant in vitro, and co-localizes to B cell zones in lymph nodes in vivo. Eur J Immunol. (2000) 30:2808–14. doi: 10.1002/1521-4141(200010)30:10%3C2808::AID-IMMU2808%3E3.0.CO;2-K
92. Sokol CL, Camire RB, Jones MC, Luster AD. The chemokine receptor CCR8 promotes the migration of dendritic cells into the lymph node parenchyma to initiate the allergic immune response. Immunity. (2018) 49:449–463.e6. doi: 10.1016/j.immuni.2018.07.012
93. Marsland BJ, Battig P, Bauer M, Ruedl C, Lassing U, Beerli RR, et al. CCL19 And CCL21 induce a potent proinflammatory differentiation program in licensed dendritic cells. Immunity. (2005) 22:493–505. doi: 10.1016/j.immuni.2005.02.010
94. Leon B, Ardavin C. Monocyte migration to inflamed skin and lymph nodes is differentially controlled by L-selectin and PSGL-1. Blood. (2008) 111:3126–30. doi: 10.1182/blood-2007-07-100610
95. Leon B, Ardavin C. Monocyte-derived dendritic cells in innate and adaptive immunity. Immunol Cell Biol. (2008) 86:320–4. doi: 10.1038/icb.2008.14
96. Leon B, Lopez-Bravo M, Ardavin C. Monocyte-derived dendritic cells. Semin Immunol. (2005) 17:313–8. doi: 10.1016/j.smim.2005.05.013
97. Leon B, Lopez-Bravo M, Ardavin C. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against leishmania. Immunity. (2007) 26:519–31. doi: 10.1016/j.immuni.2007.01.017
98. Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. (2003) 19:59–70. doi: 10.1016/S1074-7613(03)00171-7
99. Hilligan KL, Tang SC, Hyde EJ, Roussel E, Mayer JU, Yang J, et al. Dermal IRF4 + dendritic cells and monocytes license CD4+ T helper cells to distinct cytokine profiles. Nat Commun. (2020) 11:5637. doi: 10.1038/s41467-020-19463-9
100. Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, et al. CCR7 Provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med. (2014) 211:121–36. doi: 10.1084/jem.20131142
101. Pandey S, Kawai T, Akira S. Microbial sensing by toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb Perspect Biol. (2014) 7:a016246.25301932
102. Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. (2001) 2:947–50. doi: 10.1038/ni712
103. Kaur K, Bachus H, Lewis C, Papillion AM, Rosenberg AF, Ballesteros-Tato A, et al. GM-CSF production by non-classical monocytes controls antagonistic LPS-driven functions in allergic inflammation. Cell Rep. (2021) 37:110178. doi: 10.1016/j.celrep.2021.110178
104. Kaisho T, Hoshino K, Iwabe T, Takeuchi O, Yasui T, Akira S. Endotoxin can induce MyD88-deficient dendritic cells to support T(h)2 cell differentiation. Int Immunol. (2002) 14:695–700. doi: 10.1093/intimm/dxf039
105. Ochi A, Nguyen AH, Bedrosian AS, Mushlin HM, Zarbakhsh S, Barilla R, et al. Myd88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J Exp Med. (2012) 209:1671–87. doi: 10.1084/jem.20111706
106. Gaddis DE, Michalek SM, Katz J. TLR4 Signaling via MyD88 and TRIF differentially shape the CD4+ T cell response to Porphyromonas gingivalis hemagglutinin B. J Immunol. (2011) 186:5772–83. doi: 10.4049/jimmunol.1003192
107. Bortolatto J, Borducchi E, Rodriguez D, Keller AC, Faquim-Mauro E, Bortoluci KR, et al. Toll-like receptor 4 agonists adsorbed to aluminium hydroxide adjuvant attenuate ovalbumin-specific allergic airway disease: role of MyD88 adaptor molecule and interleukin-12/interferon-gamma axis. Clin Exp Allergy. (2008) 38:1668–79. doi: 10.1111/j.1365-2222.2008.03036.x
108. Shalaby KH, Jo T, Nakada E, Allard-Coutu A, Tsuchiya K, Hirota N, et al. ICOS-expressing CD4 T cells induced via TLR4 in the nasal mucosa are capable of inhibiting experimental allergic asthma. J Immunol. (2012) 189:2793–804. doi: 10.4049/jimmunol.1201194
109. Oliveira-Nascimento L, Massari P, Wetzler LM. The role of TLR2 in infection and immunity. Front Immunol. (2012) 3:79. doi: 10.3389/fimmu.2012.00079
110. Hirschfeld M, Weis JJ, Toshchakov V, Salkowski CA, Cody MJ, Ward DC, et al. Signaling by toll-like receptor 2 and 4 agonists results in differential gene expression in murine macrophages. Infect Immun. (2001) 69:1477–82. doi: 10.1128/IAI.69.3.1477-1482.2001
111. Dillon S, Agrawal A, Van Dyke T, Landreth G, McCauley L, Koh A, et al. A toll-like receptor 2 ligand stimulates Th2 responses in vivo, via induction of extracellular signal-regulated kinase mitogen-activated protein kinase and c-fos in dendritic cells. J Immunol. (2004) 172:4733–43. doi: 10.4049/jimmunol.172.8.4733
112. Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, et al. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science. (1999) 285:736–9. doi: 10.1126/science.285.5428.736
113. Re F, Strominger JL. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human dendritic cells. J Biol Chem. (2001) 276:37692–9. doi: 10.1074/jbc.M105927200
114. Pulendran B, Kumar P, Cutler CW, Mohamadzadeh M, Van Dyke T, Banchereau J. Lipopolysaccharides from distinct pathogens induce different classes of immune responses in vivo. J Immunol. (2001) 167:5067–76. doi: 10.4049/jimmunol.167.9.5067
115. Barrett NA, Maekawa A, Rahman OM, Austen KF, Kanaoka Y. Dectin-2 recognition of house dust mite triggers cysteinyl leukotriene generation by dendritic cells. J Immunol. (2009) 182:1119–28. doi: 10.4049/jimmunol.182.2.1119
116. Kheradmand F, Kiss A, Xu J, Lee SH, Kolattukudy PE, Corry DB. A protease-activated pathway underlying th cell type 2 activation and allergic lung disease. J Immunol. (2002) 169:5904–11. doi: 10.4049/jimmunol.169.10.5904
117. Van Dyken SJ, Garcia D, Porter P, Huang X, Quinlan PJ, Blanc PD, et al. Fungal chitin from asthma-associated home environments induces eosinophilic lung infiltration. J Immunol. (2011) 187:2261–7. doi: 10.4049/jimmunol.1100972
118. Gerosa F, Baldani-Guerra B, Lyakh LA, Batoni G, Esin S, Winkler-Pickett RT, et al. Differential regulation of interleukin 12 and interleukin 23 production in human dendritic cells. J Exp Med. (2008) 205:1447–61. doi: 10.1084/jem.20071450
119. Yonekawa A, Saijo S, Hoshino Y, Miyake Y, Ishikawa E, Suzukawa M, et al. Dectin-2 is a direct receptor for mannose-capped lipoarabinomannan of mycobacteria. Immunity. (2014) 41:402–13. doi: 10.1016/j.immuni.2014.08.005
120. Robinson MJ, Osorio F, Rosas M, Freitas RP, Schweighoffer E, Gross O, et al. Dectin-2 is a syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J Exp Med. (2009) 206:2037–51. doi: 10.1084/jem.20082818
121. Hadebe S, Kirstein F, Fierens K, Redelinghuys P, Murray GI, Williams DL, et al. beta-Glucan exacerbates allergic airway responses to house dust mite allergen. Respir Res. (2016) 17:35. doi: 10.1186/s12931-016-0352-5
122. Ito T, Hirose K, Norimoto A, Tamachi T, Yokota M, Saku A, et al. Dectin-1 plays an important role in house dust Mite-induced allergic airway inflammation through the activation of CD11b+ dendritic cells. J Immunol. (2017) 198:61–70. doi: 10.4049/jimmunol.1502393
123. Norimoto A, Hirose K, Iwata A, Tamachi T, Yokota M, Takahashi K, et al. Dectin-2 promotes house dust mite-induced T helper type 2 and type 17 cell differentiation and allergic airway inflammation in mice. Am J Respir Cell Mol Biol. (2014) 51:201–9. doi: 10.1165/rcmb.2013-0522OC
124. Clarke DL, Davis NH, Campion CL, Foster ML, Heasman SC, Lewis AR, et al. Dectin-2 sensing of house dust mite is critical for the initiation of airway inflammation. Mucosal Immunol. (2014) 7:558–67. doi: 10.1038/mi.2013.74
125. Barrett NA, Rahman OM, Fernandez JM, Parsons MW, Xing W, Austen KF, et al. Dectin-2 mediates Th2 immunity through the generation of cysteinyl leukotrienes. J Exp Med. (2011) 208:593–604. doi: 10.1084/jem.20100793
126. Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. (2001) 1:135–45. doi: 10.1038/35100529
127. Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. (2001) 2:675–80. doi: 10.1038/90609
128. Bachus H, Kaur K, Papillion AM, Marquez-Lago TT, Yu Z, Ballesteros-Tato A, et al. Impaired tumor-necrosis-factor-alpha-driven dendritic cell activation limits lipopolysaccharide-induced protection from allergic inflammation in infants. Immunity. (2019) 50:225–240.e4. doi: 10.1016/j.immuni.2018.11.012
129. Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. (2008) 42:145–51. doi: 10.1016/j.cyto.2008.01.006
130. Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via toll-like receptor 4 triggering of airway structural cells. Nat Med. (2009) 15:410–6. doi: 10.1038/nm.1946
131. Tan AM, Chen HC, Pochard P, Eisenbarth SC, Herrick CA, Bottomly HK. TLR4 Signaling in stromal cells is critical for the initiation of allergic Th2 responses to inhaled antigen. J Immunol. (2010) 184:3535–44. doi: 10.4049/jimmunol.0900340
132. Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med. (2002) 196:1645–51. doi: 10.1084/jem.20021340
133. Sheih A, Parks WC, Ziegler SF. GM-CSF produced by the airway epithelium is required for sensitization to cockroach allergen. Mucosal Immunol. (2017) 10:705–15. doi: 10.1038/mi.2016.90
134. Roan F, Obata-Ninomiya K, Ziegler SF. Epithelial cell-derived cytokines: more than just signaling the alarm. J Clin Invest. (2019) 129:1441–51. doi: 10.1172/JCI124606
135. Erridge C. Endogenous ligands of TLR2 and TLR4: agonists or assistants? J Leukoc Biol. (2010) 87:989–99. doi: 10.1189/jlb.1209775
136. Millien VO, Lu W, Shaw J, Yuan X, Mak G, Roberts L, et al. Cleavage of fibrinogen by proteinases elicits allergic responses through toll-like receptor 4. Science. (2013) 341:792–6. doi: 10.1126/science.1240342
137. Trompette A, Divanovic S, Visintin A, Blanchard C, Hegde RS, Madan R, et al. Allergenicity resulting from functional mimicry of a toll-like receptor complex protein. Nature. (2009) 457:585–8. doi: 10.1038/nature07548
138. Dang EV, Lei S, Radkov A, Volk RF, Zaro BW, Madhani HD. Secreted fungal virulence effector triggers allergic inflammation via TLR4. Nature. (2022) 608:161–7. doi: 10.1038/s41586-022-05005-4
139. Leon B. Fooling TLR4 to promote fungal virulence. Immunity. (2022) 55:1591–3. doi: 10.1016/j.immuni.2022.08.015
140. Bryant CE, Spring DR, Gangloff M, Gay NJ. The molecular basis of the host response to lipopolysaccharide. Nat Rev Microbiol. (2010) 8:8–14. doi: 10.1038/nrmicro2266
141. Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. (2009) 458:1191–5. doi: 10.1038/nature07830
142. Ryu JK, Kim SJ, Rah SH, Kang JI, Jung HE, Lee D, et al. Reconstruction of LPS transfer cascade reveals structural determinants within LBP, CD14, and TLR4-MD2 for efficient LPS recognition and transfer. Immunity. (2017) 46:38–50. doi: 10.1016/j.immuni.2016.11.007
143. Stewart GA, Thompson PJ. The biochemistry of common aeroallergens. Clin Exp Allergy. (1996) 26:1020–44. doi: 10.1111/j.1365-2222.1996.tb00641.x
144. Cayrol C, Duval A, Schmitt P, Roga S, Camus M, Stella A, et al. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat Immunol. (2018) 19:375–85. doi: 10.1038/s41590-018-0067-5
145. Kamijo S, Takeda H, Tokura T, Suzuki M, Inui K, Hara M, et al. IL-33-mediated innate response and adaptive immune cells contribute to maximum responses of protease allergen-induced allergic airway inflammation. J Immunol. (2013) 190:4489–99. doi: 10.4049/jimmunol.1201212
146. Snelgrove RJ, Gregory LG, Peiro T, Akthar S, Campbell GA, Walker SA, et al. Alternaria-derived serine protease activity drives IL-33-mediated asthma exacerbations. J Allergy Clin Immunol. (2014) 134:583–592.e6. doi: 10.1016/j.jaci.2014.02.002
147. Cayrol C, Girard JP. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol. (2014) 31:31–7. doi: 10.1016/j.coi.2014.09.004
148. Cayrol C, Girard JP. Interleukin-33 (IL-33): a nuclear cytokine from the IL-1 family. Immunol Rev. (2018) 281:154–68. doi: 10.1111/imr.12619
149. Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. (2010) 363:1211–21. doi: 10.1056/NEJMoa0906312
150. Waage J, Standl M, Curtin JA, Jessen LE, Thorsen J, Tian C, et al. Genome-wide association and HLA fine-mapping studies identify risk loci and genetic pathways underlying allergic rhinitis. Nat Genet. (2018) 50:1072–80. doi: 10.1038/s41588-018-0157-1
151. Portelli MA, Hodge E, Sayers I. Genetic risk factors for the development of allergic disease identified by genome-wide association. Clin Exp Allergy. (2015) 45:21–31. doi: 10.1111/cea.12327
152. Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity. (2014) 40:425–35. doi: 10.1016/j.immuni.2014.01.011
153. Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med. (2013) 210:2939–50. doi: 10.1084/jem.20130351
154. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. (2010) 464:1367–70. doi: 10.1038/nature08900
155. Leyva-Castillo JM, Galand C, Mashiko S, Bissonnette R, McGurk A, Ziegler SF, et al. ILC2 Activation by keratinocyte-derived IL-25 drives IL-13 production at sites of allergic skin inflammation. J Allergy Clin Immunol. (2020) 145:1606–1614.e4. doi: 10.1016/j.jaci.2020.02.026
156. Kabata H, Flamar AL, Mahlakoiv T, Moriyama S, Rodewald HR, Ziegler SF, et al. Targeted deletion of the TSLP receptor reveals cellular mechanisms that promote type 2 airway inflammation. Mucosal Immunol. (2020) 13:626–36. doi: 10.1038/s41385-020-0266-x
157. Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity. (2012) 36:451–63. doi: 10.1016/j.immuni.2011.12.020
158. Mohapatra A, Van Dyken SJ, Schneider C, Nussbaum JC, Liang HE, Locksley RM. Group 2 innate lymphoid cells utilize the IRF4-IL-9 module to coordinate epithelial cell maintenance of lung homeostasis. Mucosal Immunol. (2016) 9:275–86. doi: 10.1038/mi.2015.59
159. Cabeza-Cabrerizo M, Cardoso A, Minutti CM, Pereira da Costa M, Reis e Sousa C. Dendritic cells revisited. Annu Rev Immunol. (2021) 39:131–66. doi: 10.1146/annurev-immunol-061020-053707
160. Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K, et al. Negative regulation of toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci U S A. (2005) 102:15989–94. doi: 10.1073/pnas.0508327102
161. Akbari M, Honma K, Kimura D, Miyakoda M, Kimura K, Matsuyama T, et al. IRF4 In dendritic cells inhibits IL-12 production and controls Th1 immune responses against leishmania major. J Immunol. (2014) 192:2271–9. doi: 10.4049/jimmunol.1301914
162. Honma K, Udono H, Kohno T, Yamamoto K, Ogawa A, Takemori T, et al. Interferon regulatory factor 4 negatively regulates the production of proinflammatory cytokines by macrophages in response to LPS. Proc Natl Acad Sci U S A. (2005) 102:16001–6. doi: 10.1073/pnas.0504226102
163. Leyva-Castillo JM, Das M, Artru E, Yoon J, Galand C, Geha RS. Mast cell-derived IL-13 downregulates IL-12 production by skin dendritic cells to inhibit the TH1 cell response to cutaneous antigen exposure. J Allergy Clin Immunol. (2021) 147:2305–2315.e3. doi: 10.1016/j.jaci.2020.11.036
164. Dhakal M, Miller MM, Zaghouani AA, Sherman MP, Zaghouani H. Neonatal basophils stifle the function of early-life dendritic cells to curtail Th1 immunity in newborn mice. J Immunol. (2015) 195:507–18. doi: 10.4049/jimmunol.1500027
165. Coquet JM, Schuijs MJ, Smyth MJ, Deswarte K, Beyaert R, Braun H, et al. Interleukin-21-Producing CD4(+) T cells promote type 2 immunity to house dust mites. Immunity. (2015) 43:318–30. doi: 10.1016/j.immuni.2015.07.015
166. Kobayashi T, Iijima K, Dent AL, Kita H. Follicular helper T cells mediate IgE antibody response to airborne allergens. J Allergy Clin Immunol. (2017) 139:300–313.e7. doi: 10.1016/j.jaci.2016.04.021
167. Meli AP, Fontes G, Leung Soo C, King IL. T follicular helper cell-derived IL-4 is required for IgE production during intestinal helminth infection. J Immunol. (2017) 199:244–52. doi: 10.4049/jimmunol.1700141
168. Weinstein JS, Herman EI, Lainez B, Licona-Limon P, Esplugues E, Flavell R, et al. TFH Cells progressively differentiate to regulate the germinal center response. Nat Immunol. (2016) 17:1197–205. doi: 10.1038/ni.3554
169. Reinhardt RL, Liang HE, Locksley RM. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol. (2009) 10:385–93. doi: 10.1038/ni.1715
170. Liang HE, Reinhardt RL, Bando JK, Sullivan BM, Ho IC, Locksley RM. Divergent expression patterns of IL-4 and IL-13 define unique functions in allergic immunity. Nat Immunol. (2011) 13:58–66. doi: 10.1038/ni.2182
171. Hondowicz BD, An D, Schenkel JM, Kim KS, Steach HR, Krishnamurty AT, et al. Interleukin-2-Dependent allergen-specific tissue-resident memory cells drive asthma. Immunity. (2016) 44:155–66. doi: 10.1016/j.immuni.2015.11.004
172. Choi J, Crotty S. Bcl6-Mediated transcriptional regulation of follicular helper T cells (TFH). Trends Immunol. (2021) 42:336–49. doi: 10.1016/j.it.2021.02.002
173. He K, Hettinga A, Kale SL, Hu S, Xie MM, Dent AL, et al. Blimp-1 is essential for allergen-induced asthma and Th2 cell development in the lung. J Exp Med. (2020) 217.
174. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. (2009) 325:1006–10. doi: 10.1126/science.1175870
175. Crotty S, Johnston RJ, Schoenberger SP. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol. (2010) 11:114–20. doi: 10.1038/ni.1837
176. Martins GA, Cimmino L, Liao J, Magnusdottir E, Calame K. Blimp-1 directly represses Il2 and the Il2 activator fos, attenuating T cell proliferation and survival. J Exp Med. (2008) 205:1959–65. doi: 10.1084/jem.20080526
177. Gong D, Malek TR. Cytokine-dependent blimp-1 expression in activated T cells inhibits IL-2 production. J Immunol. (2007) 178:242–52. doi: 10.4049/jimmunol.178.1.242
178. Ballesteros-Tato A, Leon B, Graf BA, Moquin A, Adams PS, Lund FE, et al. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity. (2012) 36:847–56. doi: 10.1016/j.immuni.2012.02.012
179. Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 Is a potent negative regulator of TFH cell differentiation. J Exp Med. (2012) 209:243–50. doi: 10.1084/jem.20111174
180. Rochman Y, Dienger-Stambaugh K, Richgels PK, Lewkowich IP, Kartashov AV, Barski A, et al. TSLP Signaling in CD4(+) T cells programs a pathogenic T helper 2 cell state. Sci Signal. (2018) 11. doi: 10.1126/scisignal.aam8858
181. Poholek AC, Jankovic D, Villarino AV, Petermann F, Hettinga A, Shouval DS, et al. IL-10 induces a STAT3-dependent autoregulatory loop in TH2 cells that promotes blimp-1 restriction of cell expansion via antagonism of STAT5 target genes. Sci Immunol. (2016) 1. doi: 10.1126/sciimmunol.aaf8612
182. Koh B, Ulrich BJ, Nelson AS, Panangipalli G, Kharwadkar R, Wu W, et al. Bcl6 and Blimp1 reciprocally regulate ST2(+) treg-cell development in the context of allergic airway inflammation. J Allergy Clin Immunol. (2020) 146:1121–1136.e9. doi: 10.1016/j.jaci.2020.03.002
183. Chandler J, Prout M, Old S, Morgan C, Ronchese F, Benoist C, et al. BCL6 Deletion in CD4 T cells does not affect Th2 effector mediated immunity in the skin. Immunol Cell Biol. (2022).36177669
184. Finotto S, Neurath MF, Glickman JN, Qin S, Lehr HA, Green FH, et al. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science. (2002) 295:336–8. doi: 10.1126/science.1065544
185. Usui T, Preiss JC, Kanno Y, Yao ZJ, Bream JH, O'Shea JJ, et al. T-bet regulates Th1 responses through essential effects on GATA-3 function rather than on IFNG gene acetylation and transcription. J Exp Med. (2006) 203:755–66. doi: 10.1084/jem.20052165
186. Pai SY, Truitt ML, Ho IC. GATA-3 deficiency abrogates the development and maintenance of T helper type 2 cells. Proc Natl Acad Sci U S A. (2004) 101:1993–8. doi: 10.1073/pnas.0308697100
187. Zhang DH, Cohn L, Ray P, Bottomly K, Ray A. Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J Biol Chem. (1997) 272:21597–603. doi: 10.1074/jbc.272.34.21597
188. Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. (1997) 89:587–96. doi: 10.1016/S0092-8674(00)80240-8
189. Zhu J, Min B, Hu-Li J, Watson CJ, Grinberg A, Wang Q, et al. Conditional deletion of Gata3 shows its essential function in T(H)1-T(H)2 responses. Nat Immunol. (2004) 5:1157–65. doi: 10.1038/ni1128
190. Zhang DH, Yang L, Cohn L, Parkyn L, Homer R, Ray P, et al. Inhibition of allergic inflammation in a murine model of asthma by expression of a dominant-negative mutant of GATA-3. Immunity. (1999) 11:473–82. doi: 10.1016/S1074-7613(00)80122-3
191. Ho IC, Tai TS, Pai SY. GATA3 And the T-cell lineage: essential functions before and after T-helper-2-cell differentiation. Nat Rev Immunol. (2009) 9:125–35. doi: 10.1038/nri2476
192. Hwang ES, Szabo SJ, Schwartzberg PL, Glimcher LH. T helper cell fate specified by kinase-mediated interaction of T-bet with GATA-3. Science. (2005) 307:430–3. doi: 10.1126/science.1103336
193. Yang R, Mele F, Worley L, Langlais D, Rosain J, Benhsaien I, et al. Human T-bet governs innate and innate-like adaptive IFN-gamma immunity against mycobacteria. Cell. (2020) 183:1826–1847.e31. doi: 10.1016/j.cell.2020.10.046
194. Yang R, Weisshaar M, Mele F, Benhsaien I, Dorgham K, Han J, et al. High Th2 cytokine levels and upper airway inflammation in human inherited T-bet deficiency. J Exp Med. (2021) 218.
195. Yang R, Avery DT, Jackson KJL, Ogishi M, Benhsaien I, Du L, et al. Human T-bet governs the generation of a distinct subset of CD11c(high)CD21(low) B cells. Sci Immunol. (2022) 7:eabq3277. doi: 10.1126/sciimmunol.abq3277
196. Seumois G, Ramirez-Suastegui C, Schmiedel BJ, Liang S, Peters B, Sette A, et al. Single-cell transcriptomic analysis of allergen-specific T cells in allergy and asthma. Sci Immunol. (2020) 5. doi: 10.1126/sciimmunol.aba6087
197. Ouyang W, Ranganath SH, Weindel K, Bhattacharya D, Murphy TL, Sha WC, et al. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity. (1998) 9:745–55. doi: 10.1016/S1074-7613(00)80671-8
198. Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, et al. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol. (2002) 3:549–57. doi: 10.1038/ni794
199. Lighvani AA, Frucht DM, Jankovic D, Yamane H, Aliberti J, Hissong BD, et al. T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proc Natl Acad Sci U S A. (2001) 98:15137–42. doi: 10.1073/pnas.261570598
200. Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. (2002) 295:338–42. doi: 10.1126/science.1065543
201. Trinchieri G. Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu Rev Immunol. (1995) 13:251–76. doi: 10.1146/annurev.iy.13.040195.001343
202. Butcher MJ, Zhu J. Recent advances in understanding the Th1/Th2 effector choice. Fac Rev. (2021) 10:30. doi: 10.12703/r/10-30
203. Kallies A, Good-Jacobson KL. Transcription factor T-bet orchestrates lineage development and function in the immune system. Trends Immunol. (2017) 38:287–97. doi: 10.1016/j.it.2017.02.003
204. Mashayekhi M, Sandau MM, Dunay IR, Frickel EM, Khan A, Goldszmid RS, et al. CD8alpha(+) Dendritic cells are the critical source of interleukin-12 that controls acute infection by toxoplasma gondii tachyzoites. Immunity. (2011) 35:249–59. doi: 10.1016/j.immuni.2011.08.008
205. Everts B, Tussiwand R, Dreesen L, Fairfax KC, Huang SC, Smith AM, et al. Migratory CD103 + dendritic cells suppress helminth-driven type 2 immunity through constitutive expression of IL-12. J Exp Med. (2016) 213:35–51. doi: 10.1084/jem.20150235
206. Han M, Ma J, Ouyang S, Wang Y, Zheng T, Lu P, et al. The kinase p38alpha functions in dendritic cells to regulate Th2-cell differentiation and allergic inflammation. Cell Mol Immunol. (2022) 19:805–19. doi: 10.1038/s41423-022-00873-2
207. Conejero L, Khouili SC, Martinez-Cano S, Izquierdo HM, Brandi P, Sancho D. Lung CD103 + dendritic cells restrain allergic airway inflammation through IL-12 production. JCI Insight. (2017) 2. doi: 10.1172/jci.insight.90420
208. Igyarto BZ, Haley K, Ortner D, Bobr A, Gerami-Nejad M, Edelson BT, et al. Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity. (2011) 35:260–72. doi: 10.1016/j.immuni.2011.06.005
209. Bosteels C, Neyt K, Vanheerswynghels M, van Helden MJ, Sichien D, Debeuf N, et al. Inflammatory type 2 cDCs acquire features of cDC1s and macrophages to orchestrate immunity to respiratory virus infection. Immunity. (2020) 52:1039–1056.e9. doi: 10.1016/j.immuni.2020.04.005
210. Edwards AD, Manickasingham SP, Sporri R, Diebold SS, Schulz O, Sher A, et al. Microbial recognition via toll-like receptor-dependent and -independent pathways determines the cytokine response of murine dendritic cell subsets to CD40 triggering. J Immunol. (2002) 169:3652–60. doi: 10.4049/jimmunol.169.7.3652
211. Sabroe I, Jones EC, Usher LR, Whyte MK, Dower SK. Toll-like receptor (TLR)2 and TLR4 in human peripheral blood granulocytes: a critical role for monocytes in leukocyte lipopolysaccharide responses. J Immunol. (2002) 168:4701–10. doi: 10.4049/jimmunol.168.9.4701
212. Park BS, Lee JO. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med. (2013) 45:e66. doi: 10.1038/emm.2013.97
213. Meng J, Lien E, Golenbock DT. MD-2-mediated ionic interactions between lipid A and TLR4 are essential for receptor activation. J Biol Chem. (2010) 285:8695–702. doi: 10.1074/jbc.M109.075127
214. Meng J, Gong M, Bjorkbacka H, Golenbock DT. Genome-wide expression profiling and mutagenesis studies reveal that lipopolysaccharide responsiveness appears to be absolutely dependent on TLR4 and MD-2 expression and is dependent upon intermolecular ionic interactions. J Immunol. (2011) 187:3683–93. doi: 10.4049/jimmunol.1101397
215. Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Lab Invest. (2006) 86:9–22. doi: 10.1038/labinvest.3700366
216. McClure R, Massari P. TLR-Dependent Human mucosal epithelial cell responses to microbial pathogens. Front Immunol. (2014) 5:386. doi: 10.3389/fimmu.2014.00386
217. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 Links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. (2006) 116:3015–25. doi: 10.1172/JCI28898
218. Jersmann HP, Hii CS, Hodge GL, Ferrante A. Synthesis and surface expression of CD14 by human endothelial cells. Infect Immun. (2001) 69:479–85. doi: 10.1128/IAI.69.1.479-485.2001
219. Funda DP, Tuckova L, Farre MA, Iwase T, Moro I, Tlaskalova-Hogenova H. CD14 Is expressed and released as soluble CD14 by human intestinal epithelial cells in vitro: lipopolysaccharide activation of epithelial cells revisited. Infect Immun. (2001) 69:3772–81. doi: 10.1128/IAI.69.6.3772-3781.2001
220. Fernandez-Real JM, Perez del Pulgar S, Luche E, Moreno-Navarrete JM, Waget A, Serino M, et al. CD14 Modulates inflammation-driven insulin resistance. Diabetes. (2011) 60:2179–86. doi: 10.2337/db10-1210
221. Jia HP, Kline JN, Penisten A, Apicella MA, Gioannini TL, Weiss J, et al. Endotoxin responsiveness of human airway epithelia is limited by low expression of MD-2. Am J Physiol Lung Cell Mol Physiol. (2004) 287:L428–37. doi: 10.1152/ajplung.00377.2003
222. McAlees JW, Whitehead GS, Harley IT, Cappelletti M, Rewerts CL, Holdcroft AM, et al. Distinct Tlr4-expressing cell compartments control neutrophilic and eosinophilic airway inflammation. Mucosal Immunol. (2015) 8:863–73. doi: 10.1038/mi.2014.117
223. Menezes S, Melandri D, Anselmi G, Perchet T, Loschko J, Dubrot J, et al. The heterogeneity of Ly6C(hi) monocytes controls their differentiation into iNOS(+) macrophages or monocyte-derived dendritic cells. Immunity. (2016) 45:1205–18. doi: 10.1016/j.immuni.2016.12.001
224. Louis C, Cook AD, Lacey D, Fleetwood AJ, Vlahos R, Anderson GP, et al. Specific contributions of CSF-1 and GM-CSF to the dynamics of the mononuclear phagocyte system. J Immunol. (2015) 195:134–44. doi: 10.4049/jimmunol.1500369
225. Amorim A, De Feo D, Friebel E, Ingelfinger F, Anderfuhren CD, Krishnarajah S, et al. IFNgamma and GM-CSF control complementary differentiation programs in the monocyte-to-phagocyte transition during neuroinflammation. Nat Immunol. (2022) 23:217–28. doi: 10.1038/s41590-021-01117-7
226. Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, et al. The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity. (2015) 43:502–14. doi: 10.1016/j.immuni.2015.08.010
227. Spath S, Komuczki J, Hermann M, Pelczar P, Mair F, Schreiner B, et al. Dysregulation of the cytokine GM-CSF induces spontaneous phagocyte invasion and immunopathology in the central nervous system. Immunity. (2017) 46:245–60. doi: 10.1016/j.immuni.2017.01.007
228. Lotfi N, Zhang GX, Esmaeil N, Rostami A. Evaluation of the effect of GM-CSF blocking on the phenotype and function of human monocytes. Sci Rep. (2020) 10:1567. doi: 10.1038/s41598-020-58131-2
229. Parajuli B, Sonobe Y, Kawanokuchi J, Doi Y, Noda M, Takeuchi H, et al. GM-CSF increases LPS-induced production of proinflammatory mediators via upregulation of TLR4 and CD14 in murine microglia. J Neuroinflammation. (2012) 9:268. doi: 10.1186/1742-2094-9-268
230. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. (2014) 5:461. doi: 10.3389/fimmu.2014.00461
231. Takeda K, Akira S. TLR Signaling pathways. Semin Immunol. (2004) 16:3–9. doi: 10.1016/j.smim.2003.10.003
232. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. (2003) 301:640–3. doi: 10.1126/science.1087262
233. Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. (2003) 424:743–8. doi: 10.1038/nature01889
235. Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. (2001) 15:557–67. doi: 10.1016/S1074-7613(01)00218-7
236. Gschwend J, Sherman SPM, Ridder F, Feng X, Liang HE, Locksley RM, et al. Alveolar macrophages rely on GM-CSF from alveolar epithelial type 2 cells before and after birth. J Exp Med. (2021) 218. doi: 10.1084/jem.20210745
237. Ushach I, Zlotnik A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J Leukoc Biol. (2016) 100:481–9. doi: 10.1189/jlb.3RU0316-144R
238. Wicks IP, Roberts AW. Targeting GM-CSF in inflammatory diseases. Nat Rev Rheumatol. (2016) 12:37–48. doi: 10.1038/nrrheum.2015.161
239. Llop-Guevara A, Chu DK, Walker TD, Goncharova S, Fattouh R, Silver JS, et al. A GM-CSF/IL-33 pathway facilitates allergic airway responses to sub-threshold house dust mite exposure. PLoS One. (2014) 9:e88714. doi: 10.1371/journal.pone.0088714
240. Day SB, Ledford JR, Zhou P, Lewkowich IP, Page K. German Cockroach proteases and protease-activated receptor-2 regulate chemokine production and dendritic cell recruitment. J Innate Immun. (2012) 4:100–10. doi: 10.1159/000329132
241. Nobs SP, Pohlmeier L, Li F, Kayhan M, Becher B, Kopf M. GM-CSF instigates a dendritic cell-T-cell inflammatory circuit that drives chronic asthma development. J Allergy Clin Immunol. (2021) 147:2118–2133.e3. doi: 10.1016/j.jaci.2020.12.638
242. Zhou Q, Ho AW, Schlitzer A, Tang Y, Wong KH, Wong FH, et al. GM-CSF-licensed CD11b+ lung dendritic cells orchestrate Th2 immunity to Blomia tropicalis. J Immunol. (2014) 193:496–509. doi: 10.4049/jimmunol.1303138
243. Cates EC, Fattouh R, Wattie J, Inman MD, Goncharova S, Coyle AJ, et al. Intranasal exposure of mice to house dust mite elicits allergic airway inflammation via a GM-CSF-mediated mechanism. J Immunol. (2004) 173:6384–92. doi: 10.4049/jimmunol.173.10.6384
244. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. (2003) 19:71–82. doi: 10.1016/S1074-7613(03)00174-2
245. Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, et al. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell. (2013) 153:362–75. doi: 10.1016/j.cell.2013.03.010
246. Quintar A, McArdle S, Wolf D, Marki A, Ehinger E, Vassallo M, et al. Endothelial protective monocyte patrolling in large arteries intensified by western diet and atherosclerosis. Circ Res. (2017) 120:1789–99. doi: 10.1161/CIRCRESAHA.117.310739
247. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. (2007) 204:3037–47. doi: 10.1084/jem.20070885
248. Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. (2014) 114:1611–22. doi: 10.1161/CIRCRESAHA.114.303204
249. Cros J, Cagnard N, Woollard K, Patey N, Zhang SY, Senechal B, et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. (2010) 33:375–86. doi: 10.1016/j.immuni.2010.08.012
250. Narasimhan PB, Marcovecchio P, Hamers AAJ, Hedrick CC. Nonclassical monocytes in health and disease. Annu Rev Immunol. (2019) 37:439–56. doi: 10.1146/annurev-immunol-042617-053119
251. Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK 3rd, et al. Nonclassical Ly6C(-) monocytes drive the development of inflammatory arthritis in mice. Cell Rep. (2014) 9:591–604. doi: 10.1016/j.celrep.2014.09.032
252. Waschbisch A, Schroder S, Schraudner D, Sammet L, Weksler B, Melms A, et al. Pivotal role for CD16+ monocytes in immune surveillance of the central nervous system. J Immunol. (2016) 196:1558–67. doi: 10.4049/jimmunol.1501960
253. Finsterbusch M, Hall P, Li A, Devi S, Westhorpe CL, Kitching AR, et al. Patrolling monocytes promote intravascular neutrophil activation and glomerular injury in the acutely inflamed glomerulus. Proc Natl Acad Sci U S A. (2016) 113:E5172–81. doi: 10.1073/pnas.1606253113
254. Zhu H, Hu F, Sun X, Zhang X, Zhu L, Liu X, et al. CD16(+) Monocyte subset was enriched and functionally exacerbated in driving T-cell activation and B-cell response in systemic lupus erythematosus. Front Immunol. (2016) 7:512.27917174
255. Kurowski M, Majkowska-Wojciechowska B, Wardzynska A, Kowalski ML. Associations of allergic sensitization and clinical phenotypes with innate immune response genes polymorphisms are modified by house dust mite allergen exposure. Arch Med Sci. (2011) 7:1029–36. doi: 10.5114/aoms.2011.26616
256. Sackesen C, Karaaslan C, Keskin O, Tokol N, Tahan F, Civelek E, et al. The effect of polymorphisms at the CD14 promoter and the TLR4 gene on asthma phenotypes in turkish children with asthma. Allergy. (2005) 60:1485–92. doi: 10.1111/j.1398-9995.2005.00874.x
257. Fageras Bottcher M, Hmani-Aifa M, Lindstrom A, Jenmalm MC, Mai XM, Nilsson L, et al. A TLR4 polymorphism is associated with asthma and reduced lipopolysaccharide-induced interleukin-12(p70) responses in Swedish children. J Allergy Clin Immunol. (2004) 114:561–7. doi: 10.1016/j.jaci.2004.04.050
258. Smit LA, Bongers SI, Ruven HJ, Rijkers GT, Wouters IM, Heederik D, et al. Atopy and new-onset asthma in young danish farmers and CD14, TLR2, and TLR4 genetic polymorphisms: a nested case-control study. Clin Exp Allergy. (2007) 37:1602–8. doi: 10.1111/j.1365-2222.2007.02831.x
259. Lachheb J, Dhifallah IB, Chelbi H, Hamzaoui K, Hamzaoui A. Toll-like receptors and CD14 genes polymorphisms and susceptibility to asthma in Tunisian children. Tissue Antigens. (2008) 71:417–25. doi: 10.1111/j.1399-0039.2008.01011.x
260. Smit LA, Siroux V, Bouzigon E, Oryszczyn MP, Lathrop M, Demenais F, et al. CD14 And toll-like receptor gene polymorphisms, country living, and asthma in adults. Am J Respir Crit Care Med. (2009) 179:363–8. doi: 10.1164/rccm.200810-1533OC
261. Eder W, Klimecki W, Yu L, von Mutius E, Riedler J, Braun-Fahrlander C, et al. Toll-like receptor 2 as a major gene for asthma in children of European farmers. J Allergy Clin Immunol. (2004) 113:482–8. doi: 10.1016/j.jaci.2003.12.374
262. Baldini M, Lohman IC, Halonen M, Erickson RP, Holt PG, Martinez FD. A polymorphism* in the 5’ flanking region of the CD14 gene is associated with circulating soluble CD14 levels and with total serum immunoglobulin E. Am J Respir Cell Mol Biol. (1999) 20:976–83. doi: 10.1165/ajrcmb.20.5.3494
263. Eder W, Klimecki W, Yu L, von Mutius E, Riedler J, Braun-Fahrlander C, et al. Opposite effects of CD 14/-260 on serum IgE levels in children raised in different environments. J Allergy Clin Immunol. (2005) 116:601–7. doi: 10.1016/j.jaci.2005.05.003
264. Leynaert B, Guilloud-Bataille M, Soussan D, Benessiano J, Guenegou A, Pin I, et al. Association between farm exposure and atopy, according to the CD14 C-159 T polymorphism. J Allergy Clin Immunol. (2006) 118:658–65. doi: 10.1016/j.jaci.2006.06.015
265. Rohrbach M, Frey U, Kraemer R, Liechti-Gallati S. A variant in the gene for GM-CSF, I117T, is associated with atopic asthma in a Swiss population of asthmatic children. J Allergy Clin Immunol. (1999) 104:247–8. doi: 10.1016/S0091-6749(99)70147-8
266. He JQ, Ruan J, Chan-Yeung M, Becker AB, Dimich-Ward H, Pare PD, et al. Polymorphisms of the GM-CSF genes and the development of atopic diseases in at-risk children. Chest. (2003) 123:438S. doi: 10.1378/chest.123.3_suppl.438S
267. Rafatpanah H, Bennett E, Pravica V, McCoy MJ, David TJ, Hutchinson IV, et al. Association between novel GM-CSF gene polymorphisms and the frequency and severity of atopic dermatitis. J Allergy Clin Immunol. (2003) 112:593–8. doi: 10.1016/S0091-6749(03)01797-4
268. Kabesch M, Hasemann K, Schickinger V, Tzotcheva I, Bohnert A, Carr D, et al. A promoter polymorphism in the CD14 gene is associated with elevated levels of soluble CD14 but not with IgE or atopic diseases. Allergy. (2004) 59:520–5. doi: 10.1111/j.1398-9995.2004.00439.x
269. Hussein YM, Awad HA, Shalaby SM, Ali AS, Alzahrani SS. Toll-like receptor 2 and toll-like receptor 4 polymorphisms and susceptibility to asthma and allergic rhinitis: a case-control analysis. Cell Immunol. (2012) 274:34–8. doi: 10.1016/j.cellimm.2012.02.006
270. Galli E, Ciucci A, Cersosimo S, Pagnini C, Avitabile S, Mancino G, et al. Eczema and food allergy in an Italian pediatric cohort: no association with TLR-2 and TLR-4 polymorphisms. Int J Immunopathol Pharmacol. (2010) 23:671–5. doi: 10.1177/039463201002300233
271. Noguchi E, Nishimura F, Fukai H, Kim J, Ichikawa K, Shibasaki M, et al. An association study of asthma and total serum immunoglobin E levels for toll-like receptor polymorphisms in a Japanese population. Clin Exp Allergy. (2004) 34:177–83. doi: 10.1111/j.1365-2222.2004.01839.x
272. Saeki H, Tsunemi Y, Asano N, Nakamura K, Sekiya T, Hirai K, et al. Analysis of GM-CSF gene polymorphisms (3606 T/C and 3928C/T) in Japanese patients with atopic dermatitis. Clin Exp Dermatol. (2006) 31:278–80. doi: 10.1111/j.1365-2230.2005.02052.x
273. von Mutius E. Influences in allergy: epidemiology and the environment. J Allergy Clin Immunol. (2004) 113:373–9. quiz 380. doi: 10.1016/j.jaci.2003.12.040
274. Bachus H, McLaughlin E, Lewis C, Papillion AM, Benveniste EN, Hill DD, et al. IL-6 prevents Th2 cell polarization by promoting SOCS3-dependent suppression of IL-2 signaling. BioRxiv. (2022.
275. Spencer S, Kostel Bal S, Egner W, Lango Allen H, Raza SI, Ma CA, et al. Loss of the interleukin-6 receptor causes immunodeficiency, atopy, and abnormal inflammatory responses. J Exp Med. (2019) 216:1986–98. doi: 10.1084/jem.20190344
276. Schwerd T, Twigg SRF, Aschenbrenner D, Manrique S, Miller KA, Taylor IB, et al. A biallelic mutation in IL6ST encoding the GP130 co-receptor causes immunodeficiency and craniosynostosis. J Exp Med. (2017) 214:2547–62. doi: 10.1084/jem.20161810
277. Beziat V, Tavernier SJ, Chen YH, Ma CS, Materna M, Laurence A, et al. Dominant-negative mutations in human IL6ST underlie hyper-IgE syndrome. J Exp Med. (2020) 217.
278. Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 Mutations in the hyper-IgE syndrome. N Engl J Med. (2007) 357:1608–19. doi: 10.1056/NEJMoa073687
279. Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. (2007) 448:1058–62. doi: 10.1038/nature06096
280. Chandesris MO, Melki I, Natividad A, Puel A, Fieschi C, Yun L, et al. Autosomal dominant STAT3 deficiency and hyper-IgE syndrome: molecular, cellular, and clinical features from a French national survey. Medicine (Baltimore). (2012) 91:e1–e19. doi: 10.1097/MD.0b013e31825f95b9
281. Beziat V, Li J, Lin JX, Ma CS, Li P, Bousfiha A, et al. A recessive form of hyper-IgE syndrome by disruption of ZNF341-dependent STAT3 transcription and activity. Sci Immunol. (2018) 3. doi: 10.1126/sciimmunol.aat4956
282. Chen YH, Spencer S, Laurence A, Thaventhiran JE, Uhlig HH. Inborn errors of IL-6 family cytokine responses. Curr Opin Immunol. (2021) 72:135–45. doi: 10.1016/j.coi.2021.04.007
283. Ferreira MA, Matheson MC, Duffy DL, Marks GB, Hui J, Le Souef P, et al. Identification of IL6R and chromosome 11q13.5 as risk loci for asthma. Lancet. (2011) 378:1006–14. doi: 10.1016/S0140-6736(11)60874-X
284. Hinds DA, McMahon G, Kiefer AK, Do CB, Eriksson N, Evans DM, et al. A genome-wide association meta-analysis of self-reported allergy identifies shared and allergy-specific susceptibility loci. Nat Genet. (2013) 45:907–11. doi: 10.1038/ng.2686
285. Prescott SL, Macaubas C, Smallacombe T, Holt BJ, Sly PD, Holt PG. Development of allergen-specific T-cell memory in atopic and normal children. Lancet. (1999) 353:196–200. doi: 10.1016/S0140-6736(98)05104-6
286. Pate CA, Zahran HS, Qin X, Johnson C, Hummelman E, Malilay J. Asthma surveillance - United States, 2006-2018. MMWR Surveill Summ. (2021) 70:1–32. doi: 10.15585/mmwr.ss7005a1
287. Nicolaou N, Siddique N, Custovic A. Allergic disease in urban and rural populations: increasing prevalence with increasing urbanization. Allergy. (2005) 60:1357–60. doi: 10.1111/j.1398-9995.2005.00961.x
288. Majkowska-Wojciechowska B, Pelka J, Korzon L, Kozlowska A, Kaczala M, Jarzebska M, et al. Prevalence of allergy, patterns of allergic sensitization and allergy risk factors in rural and urban children. Allergy. (2007) 62:1044–50. doi: 10.1111/j.1398-9995.2007.01457.x
289. Beasley R, Crane J, Lai CK, Pearce N. Prevalence and etiology of asthma. J Allergy Clin Immunol. (2000) 105:S466–72. doi: 10.1016/S0091-6749(00)90044-7
290. Radon K, Danuser B, Iversen M, Monso E, Weber C, Hartung J, et al. Air contaminants in different European farming environments. Ann Agric Environ Med. (2002) 9:41–8.12088396
291. Strieker S, Weinmann T, Gerlich J, von Mutius E, Nowak D, Radon K, et al. Farm living and allergic rhinitis from childhood to young adulthood: prospective results of the GABRIEL study. J Allergy Clin Immunol. (2022.35779667
292. Yerkovich ST, Wikstrom ME, Suriyaarachchi D, Prescott SL, Upham JW, Holt PG. Postnatal development of monocyte cytokine responses to bacterial lipopolysaccharide. Pediatr Res. (2007) 62:547–52. doi: 10.1203/PDR.0b013e3181568105
293. Yan SR, Qing G, Byers DM, Stadnyk AW, Al-Hertani W, Bortolussi R. Role of MyD88 in diminished tumor necrosis factor alpha production by newborn mononuclear cells in response to lipopolysaccharide. Infect Immun. (2004) 72:1223–9. doi: 10.1128/IAI.72.3.1223-1229.2004
294. Levy O, Zarember KA, Roy RM, Cywes C, Godowski PJ, Wessels MR. Selective impairment of TLR-mediated innate immunity in human newborns: neonatal blood plasma reduces monocyte TNF-alpha induction by bacterial lipopeptides, lipopolysaccharide, and imiquimod, but preserves the response to R-848. J Immunol. (2004) 173:4627–34. doi: 10.4049/jimmunol.173.7.4627
295. Sadeghi K, Berger A, Langgartner M, Prusa AR, Hayde M, Herkner K, et al. Immaturity of infection control in preterm and term newborns is associated with impaired toll-like receptor signaling. J Infect Dis. (2007) 195:296–302. doi: 10.1086/509892
296. Levy O. Innate immunity of the human newborn: distinct cytokine responses to LPS and other toll-like receptor agonists. J Endotoxin Res. (2005) 11:113–6. doi: 10.1177/09680519050110020701
297. Cairo MS, Suen Y, Knoppel E, van de Ven C, Nguyen A, Sender L. Decreased stimulated GM-CSF production and GM-CSF gene expression but normal numbers of GM-CSF receptors in human term newborns compared with adults. Pediatr Res. (1991) 30:362–7. doi: 10.1203/00006450-199110000-00013
298. Cairo MS, Suen Y, Knoppel E, Dana R, Park L, Clark S, et al. Decreased G-CSF and IL-3 production and gene expression from mononuclear cells of newborn infants. Pediatr Res. (1992) 31:574–8. doi: 10.1203/00006450-199206000-00007
299. Appelt AW, Welty JD, Peterson MB. Changes in sarcolemmal and sarcoplasmic reticulum ATPase activities with age in the cardiomyopathic Syrian hamster. J Mol Cell Cardiol. (1976) 8:901–7. doi: 10.1016/0022-2828(76)90072-9
300. Lee SM, Knoppel E, van de Ven C, Cairo MS. Transcriptional rates of granulocyte-macrophage colony-stimulating factor, granulocyte colony-stimulating factor, interleukin-3, and macrophage colony-stimulating factor genes in activated cord versus adult mononuclear cells: alteration in cytokine expression may be secondary to posttranscriptional instability. Pediatr Res. (1993) 34:560–4. doi: 10.1203/00006450-199311000-00002
301. Nelson DJ, Holt PG. Defective regional immunity in the respiratory tract of neonates is attributable to hyporesponsiveness of local dendritic cells to activation signals. J Immunol. (1995) 155:3517–24.7561047
302. Dzidic M, Boix-Amoros A, Selma-Royo M, Mira A, Collado MC. Gut Microbiota and mucosal immunity in the neonate. Med Sci (Basel). (2018) 6.30018263
303. Gensollen T, Blumberg RS. Correlation between early-life regulation of the immune system by microbiota and allergy development. J Allergy Clin Immunol. (2017) 139:1084–91. doi: 10.1016/j.jaci.2017.02.011
304. Gensollen T, Iyer SS, Kasper DL, Blumberg RS. How colonization by microbiota in early life shapes the immune system. Science. (2016) 352:539–44. doi: 10.1126/science.aad9378
305. Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature. (2016) 535:65–74. doi: 10.1038/nature18847
306. Hanski I, von Hertzen L, Fyhrquist N, Koskinen K, Torppa K, Laatikainen T, et al. Environmental biodiversity, human microbiota, and allergy are interrelated. Proc Natl Acad Sci U S A. (2012) 109:8334–9. doi: 10.1073/pnas.1205624109
307. Arrieta MC, Stiemsma LT, Dimitriu PA, Thorson L, Russell S, Yurist-Doutsch S, et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci Transl Med. (2015) 7:307ra152. doi: 10.1126/scitranslmed.aab2271
308. Depner M, Ege MJ, Cox MJ, Dwyer S, Walker AW, Birzele LT, et al. Bacterial microbiota of the upper respiratory tract and childhood asthma. J Allergy Clin Immunol. (2017) 139:826–834.e13. doi: 10.1016/j.jaci.2016.05.050
309. Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y, et al. The microbiota regulates type 2 immunity through RORgammat(+) T cells. Science. (2015) 349:989–93. doi: 10.1126/science.aac4263
310. Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med. (2014) 20:159–66. doi: 10.1038/nm.3444
311. Derrien M, Alvarez AS, de Vos WM. The gut Microbiota in the first decade of life. Trends Microbiol. (2019) 27:997–1010. doi: 10.1016/j.tim.2019.08.001
312. Gollwitzer ES, Saglani S, Trompette A, Yadava K, Sherburn R, McCoy KD, et al. Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat Med. (2014) 20:642–7. doi: 10.1038/nm.3568
313. Loss G, Bitter S, Wohlgensinger J, Frei R, Roduit C, Genuneit J, et al. Prenatal and early-life exposures alter expression of innate immunity genes: the PASTURE cohort study. J Allergy Clin Immunol. (2012) 130:523–30.e9. doi: 10.1016/j.jaci.2012.05.049
314. Lauener RP, Birchler T, Adamski J, Braun-Fahrlander C, Bufe A, Herz U, et al. Expression of CD14 and toll-like receptor 2 in farmers’ and non-farmers’ children. Lancet. (2002) 360:465–6. doi: 10.1016/S0140-6736(02)09641-1
315. Ege MJ, Frei R, Bieli C, Schram-Bijkerk D, Waser M, Benz MR, et al. Not all farming environments protect against the development of asthma and wheeze in children. J Allergy Clin Immunol. (2007) 119:1140–7. doi: 10.1016/j.jaci.2007.01.037
316. Bieli C, Eder W, Frei R, Braun-Fahrlander C, Klimecki W, Waser M, et al. A polymorphism in CD14 modifies the effect of farm milk consumption on allergic diseases and CD14 gene expression. J Allergy Clin Immunol. (2007) 120:1308–15. doi: 10.1016/j.jaci.2007.07.034
317. Ege MJ, Mayer M, Normand AC, Genuneit J, Cookson WO, Braun-Fahrlander C, et al. Exposure to environmental microorganisms and childhood asthma. N Engl J Med. (2011) 364:701–9. doi: 10.1056/NEJMoa1007302
318. Loss G, Apprich S, Waser M, Kneifel W, Genuneit J, Buchele G, et al. The protective effect of farm milk consumption on childhood asthma and atopy: the GABRIELA study. J Allergy Clin Immunol. (2011) 128:766–773.e4. doi: 10.1016/j.jaci.2011.07.048
319. Waser M, Michels KB, Bieli C, Floistrup H, Pershagen G, von Mutius E, et al. Inverse association of farm milk consumption with asthma and allergy in rural and suburban populations across Europe. Clin Exp Allergy. (2007) 37:661–70. doi: 10.1111/j.1365-2222.2006.02640.x
320. Stampfli M, Frei R, Divaret-Chauveau A, Schmausser-Hechfellner E, Karvonen AM, Pekkanen J, et al. Inverse associations between food diversity in the second year of life and allergic diseases. Ann Allergy Asthma Immunol. (2022) 128:39–45. doi: 10.1016/j.anai.2021.10.005
321. Roduit C, Frei R, Loss G, Buchele G, Weber J, Depner M, et al. Development of atopic dermatitis according to age of onset and association with early-life exposures. J Allergy Clin Immunol. (2012) 130:130–6.e5. doi: 10.1016/j.jaci.2012.02.043
322. Wijga AH, Smit HA, Kerkhof M, de Jongste JC, Gerritsen J, Neijens HJ, et al. Association of consumption of products containing milk fat with reduced asthma risk in pre-school children: the PIAMA birth cohort study. Thorax. (2003) 58:567–72. doi: 10.1136/thorax.58.7.567
323. Roduit C, Frei R, Depner M, Schaub B, Loss G, Genuneit J, et al. Increased food diversity in the first year of life is inversely associated with allergic diseases. J Allergy Clin Immunol. (2014) 133:1056–64. doi: 10.1016/j.jaci.2013.12.1044
324. von Mutius E. The microbial environment and its influence on asthma prevention in early life. J Allergy Clin Immunol. (2016) 137:680–9. doi: 10.1016/j.jaci.2015.12.1301
325. Depner M, Taft DH, Kirjavainen PV, Kalanetra KM, Karvonen AM, Peschel S, et al. Maturation of the gut microbiome during the first year of life contributes to the protective farm effect on childhood asthma. Nat Med. (2020) 26:1766–75. doi: 10.1038/s41591-020-1095-x
326. Illi S, Depner M, Pfefferle PI, Renz H, Roduit C, Taft DH, et al. Immune responsiveness to LPS determines risk of childhood wheeze and asthma in 17q21 risk allele carriers. Am J Respir Crit Care Med. (2022) 205:641–50. doi: 10.1164/rccm.202106-1458OC
327. Roduit C, Frei R, Ferstl R, Loeliger S, Westermann P, Rhyner C, et al. High levels of butyrate and propionate in early life are associated with protection against atopy. Allergy. (2019) 74:799–809. doi: 10.1111/all.13660
328. De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A. (2010) 107:14691–6. doi: 10.1073/pnas.1005963107
329. de Kleer IM, Kool M, de Bruijn MJ, Willart M, van Moorleghem J, Schuijs MJ, et al. Perinatal activation of the interleukin-33 pathway promotes type 2 immunity in the developing lung. Immunity. (2016) 45:1285–98. doi: 10.1016/j.immuni.2016.10.031
330. Steer CA, Martinez-Gonzalez I, Ghaedi M, Allinger P, Matha L, Takei F. Group 2 innate lymphoid cell activation in the neonatal lung drives type 2 immunity and allergen sensitization. J Allergy Clin Immunol. (2017) 140:593–595.e3. doi: 10.1016/j.jaci.2016.12.984
331. Yang SI. Particulate matter and childhood allergic diseases. Korean J Pediatr. (2019) 62:22–9. doi: 10.3345/kjp.2018.07045
332. Guarnieri M, Balmes JR. Outdoor air pollution and asthma. Lancet. (2014) 383:1581–92. doi: 10.1016/S0140-6736(14)60617-6
333. De Grove KC, Provoost S, Brusselle GG, Joos GF, Maes T. Insights in particulate matter-induced allergic airway inflammation: focus on the epithelium. Clin Exp Allergy. (2018) 48:773–86. doi: 10.1111/cea.13178
334. Cooper DM, Loxham M. Particulate matter and the airway epithelium: the special case of the underground? Eur Respir Rev. (2019) 28. doi: 10.1183/16000617.0066-2019
335. Li N, Xia T, Nel AE. The role of oxidative stress in ambient particulate matter-induced lung diseases and its implications in the toxicity of engineered nanoparticles. Free Radic Biol Med. (2008) 44:1689–99. doi: 10.1016/j.freeradbiomed.2008.01.028
336. Basinas I, Sigsgaard T, Kromhout H, Heederik D, Wouters IM, Schlunssen V. A comprehensive review of levels and determinants of personal exposure to dust and endotoxin in livestock farming. J Expo Sci Environ Epidemiol. (2015) 25:123–37. doi: 10.1038/jes.2013.83
337. Wang Z, Larsson K, Palmberg L, Malmberg P, Larsson P, Larsson L. Inhalation of swine dust induces cytokine release in the upper and lower airways. Eur Respir J. (1997) 10:381–7. doi: 10.1183/09031936.97.10020381
338. Wang Z, Malmberg P, Ek A, Larsson K, Palmberg L. Swine dust induces cytokine secretion from human epithelial cells and alveolar macrophages. Clin Exp Immunol. (1999) 115:6–12. doi: 10.1046/j.1365-2249.1999.00776.x
339. Dosman JA, Fukushima Y, Senthilselvan A, Kirychuk SP, Lawson JA, Pahwa P, et al. Respiratory response to endotoxin and dust predicts evidence of inflammatory response in volunteers in a swine barn. Am J Ind Med. (2006) 49:761–6. doi: 10.1002/ajim.20339
340. Larsson KA, Eklund AG, Hansson LO, Isaksson BM, Malmberg PO. Swine dust causes intense airways inflammation in healthy subjects. Am J Respir Crit Care Med. (1994) 150:973–7. doi: 10.1164/ajrccm.150.4.7921472
341. Basinas I, Schlunssen V, Heederik D, Sigsgaard T, Smit LA, Samadi S, et al. Sensitisation to common allergens and respiratory symptoms in endotoxin exposed workers: a pooled analysis. Occup Environ Med. (2012) 69:99–106. doi: 10.1136/oem.2011.065169
342. Eduard W, Douwes J, Omenaas E, Heederik D. Do farming exposures cause or prevent asthma? Results from a study of adult Norwegian farmers. Thorax. (2004) 59:381–6. doi: 10.1136/thx.2004.013326
Keywords: Th2 cells, infants, LPS (lipopolysaccharide), dendritic cells, IL-12 cytokine, T-bet, IL-2 cytokine, GM-CSF
Citation: León B (2023) Understanding the development of Th2 cell-driven allergic airway disease in early life. Front. Allergy 3:1080153. doi: 10.3389/falgy.2022.1080153
Received: 25 October 2022; Accepted: 21 December 2022;
Published: 10 January 2023.
Edited by:
Kirsi Jarvinen-Seppo, University of Rochester, United StatesReviewed by:
Stephane Lajoie, Johns Hopkins University, United StatesElia Tait Wojno, University of Washington, United States
© 2023 León. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beatriz León Ymxlb25AdWFiLmVkdQ==
Specialty Section: This article was submitted to Allergens, a section of the journal Frontiers in Allergy