 Pei-Hui Lin1*
Pei-Hui Lin1* Pu Duann2
Pu Duann2- 1Department of Surgery, Davis Heart and Lung Research Institute, The Ohio State University, Columbus, OH, United States
- 2Research and Development, Salem Veteran Affairs Medical Center, Salem, VA, United States
To excrete body nitrogen waste and regulate electrolyte and fluid balance, the kidney has developed into an energy factory with only second to the heart in mitochondrial content in the body to meet the high-energy demand and regulate homeostasis. Energy supply from the renal mitochondria majorly depends on lipid metabolism, with programed enzyme systems in fatty acid β-oxidation and Krebs cycle. Renal mitochondria integrate several metabolic pathways, including AMPK/PGC-1α, PPARs, and CD36 signaling to maintain energy homeostasis for dynamic and static requirements. The pathobiology of several kidney disorders, including diabetic nephropathy, acute and chronic kidney injuries, has been primarily linked to impaired mitochondrial bioenergetics. Such homeostatic disruption in turn stimulates a pathological adaptation, with mitochondrial enzyme system reprograming possibly leading to dyslipidemia. However, this alteration, while rescuing oncotic pressure deficit secondary to albuminuria and dissipating edematous disorder, also imposes an ominous lipotoxic consequence. Reprograming of lipid metabolism in kidney injury is essential to preserve the integrity of kidney mitochondria, thereby preventing massive collateral damage including excessive autophagy and chronic inflammation. Here, we review dyslipidemia in kidney disorders and the most recent advances on targeting mitochondrial energy metabolism as a therapeutic strategy to restrict renal lipotoxicity, achieve salutary anti-edematous effects, and restore mitochondrial homeostasis.
Introduction
The kidney is characterized by a complex anatomy, with millions of nephrons as the functional unit to excrete nitrogen waste and secure fluid homeostasis. The kidney is composed of multiple specialized cell types ensuring vital homeostasis of acid-base and electrolyte balance, blood pressure regulation, nutrient reabsorption, and hormone secretion (Hoenig and Zeidel, 2014; Duann and Lin, 2017; Yu et al., 2019). Therefore, it is one of the most metabolically active organs other than heart and skeletal muscle, with proximal tubules presenting a very high density of mitochondria required for energy consumption (Meyer et al., 1997; Wang et al., 2000). For example, the human proximal convoluted tubules (S1 and S2 combined) contain abundant large mitochondria, which occupy about 16.3% cell volume (Møller and Skriver, 1985). Notably, the mature nephron comprises distinct segments, each utilizing metabolic pathways to varying degrees depending on the specific function (Cargill and Sims-Lucas, 2020).
Complete oxidation of fatty acids (FAs), which are high-energy substrates, to CO2 and H2O gives rise to roughly 9 Kcal/g fat, while only 4 Kcal/g are generated from carbohydrates or proteins. The heart possesses metabolic flexibility and powerful catabolic capacity to use various energy substrates, mainly FAs (40–60%) and glucose (20–40%), for ATP production (Karwi et al., 2018). Instead, most proximal tubule epithelial cells (PTEC) have low metabolic flexibility toward glycolysis and rely on FAs as energy source at baseline (Bonventre and Yang, 2011). This was shown by early in vivo studies measuring ATP synthesis by tracking isotope-labeled FAs with NMR in rat kidney, which indicated that FAs are a preferred fuel (Freeman et al., 1986). However, PTEC are able to shift to anaerobic glycolysis to produce ATP required for cellular regeneration after ischemic acute kidney injury (AKI; Lan et al., 2016). In mice, glomerular podocytes display much lower mitochondrial density than in PTECs and rely primarily on anaerobic glycolysis to maintain glomerular filtration barrier and are relatively insensitive to defect in mitochondrial biogenesis during ischemia damage (Brinkkoetter et al., 2019). Instead, as lipid accumulation is commonly observed in patients with chronic kidney disease, podocytes are rather sensitive to cellular cholesterol-mediated glomerular injury (Merscher et al., 2014).
Mitochondria are pivotal for maintaining the health and function of the metabolically active kidney by providing efficient energy support through the process of oxidative phosphorylation (OXPHOS) and aerobic glycolysis. Several factors such as mitochondria biogenesis, bioenergetics, dynamics, and autophagy regulate the mitochondrial physiology (Duann and Lin, 2017). In addition, mitochondria also contribute to production of reactive oxygen species (ROS) free radicals and transduction of metabolic and stress signals (Galvan et al., 2017; Flemming et al., 2018). Persistent mitochondrial damage is a major source of oxidants. Consequently, mitochondrial fitness translates into body’s general health. Mitochondrial dysfunction is involved in various kidney diseases, such as acute kidney injury (AKI), chronic kidney disease (CKD), diabetic nephropathy (DN), and glomerulonephritis (GN; Duann and Lin, 2017; Eirin et al., 2017; Galvan et al., 2017; Flemming et al., 2018).
Dysregulated lipid metabolism with defective cholesterol/free fatty acid (FFA) metabolism leading to dyslipidemia is common in patients of several kidney diseases, including acute kidney disease, CKD, diabetic kidney disease (DKD), nephrotic syndrome, and uremia (Agrawal et al., 2017; Hager et al., 2017; Gai et al., 2019; Nishi et al., 2019; Nishi and Nangaku, 2019; Jang et al., 2020b), and may contribute to end-stage kidney disease. In this review, we summarize the recent advances in understanding lipid metabolism in the function of kidney mitochondria and the molecular mechanisms related to dyslipidemia during kidney disease progression.
Basic Lipid Biology
In biological systems, lipids include fats, sterols, phospholipids, and triacylglycerides (TAG). In the cell, lipids have numerous functions: they constitute the cell membrane as a protective barrier; form membranous compartments of intracellular organelles; provide energy source and storage; provide building blocks for hormones; and serve as secondary cellular messengers within body. FAs are carboxylic acids with a long aliphatic tail, which constitute building blocks for other lipids such as TAG and phospholipids.
Within the body, lipid metabolism comprises several inter-dependent pathways for the generation, storage, and transport of lipids, which involves plasma lipoprotein particles [chylomicrons, high density lipoproteins (HDL), low density lipoproteins (LDL), intermediate density lipoproteins (IDL), and very low density lipoproteins (VLDL)] in circulation. Dietary lipids, mainly (95%) TAG, some FFAs, and cholesterol, carried by chylomicrons into circulation, are degraded into FFAs and glycerol by lipoprotein lipase (LPL) activity on the capillaries. These FFAs are taken up by muscle, heart, and adipose and peripheral tissues like kidney; remnants of chylomicrons are subsequently cleared in the liver (Florens et al., 2016; Agrawal et al., 2017; Kronenberg, 2018). FFAs are transported by serum albumin to the liver and periphery and could be stored as TAG in kidney capillaries. Additionally, esterified cholesterol could be stored as a lipid droplet within the kidney.
Transport of Cellular FFAs
Lipid Uptake by CD36 in the Kidney
FA uptake from the extracellular milieu is the first step in their utilization. Multiple cell surface lipid transport proteins, such as cluster of differentiation 36 (CD36), scavenger receptor B1 (SR-B1), tissue-specific fatty acid transport proteins (FATPs), and plasma membrane fatty acid-binding protein (FABPpm) facilitate cellular FFA uptake (Haunerland and Spener, 2004; Su and Abumrad, 2009; Figure 1).
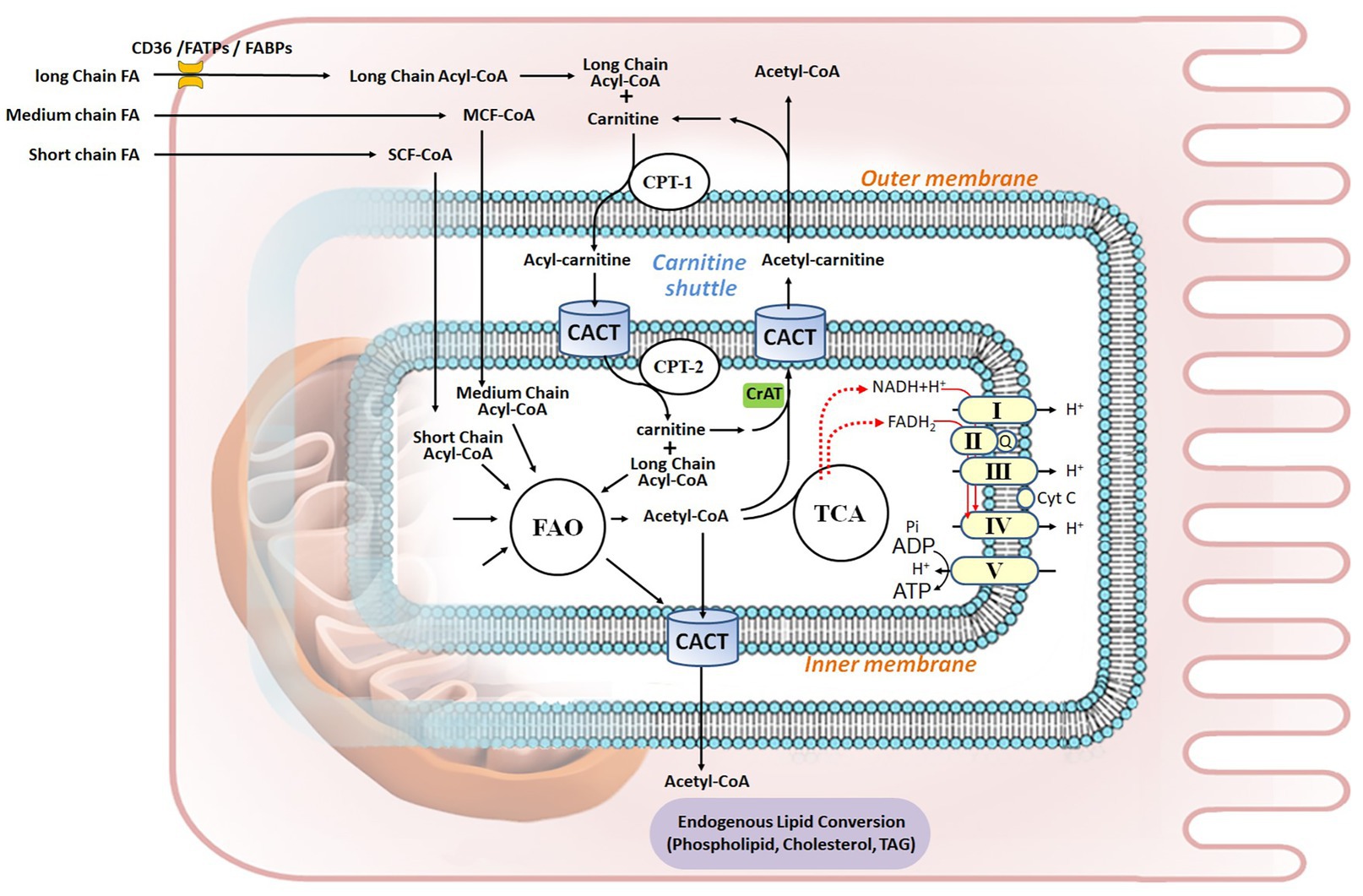
Figure 1. Fatty acid metabolism in renal proximal tubule epithelial cell. Fatty acids (FAs) are the preferred energy substrates for the kidney. Uptake of FAs from capillaries into kidney cells is facilitated by either FAT/CD36 or FABPs and FATPs. In the cytosol, FAs are activated to acyl-CoA, esterified with carnitine, and transported into the mitochondrial matrix through the carnitine shuttle, which is composed of CPT-1, CACT, and CPT-2. Medium-chain FAs and short-chain FAs can permeate the mitochondrial membranes. In the matrix, acyl-CoA undergoes FA β-oxidation (FAO), thereby generating acetyl-CoA to fuel the TCA cycle, as well as FADH2 and NADH that serve as electron donors to the five ETC complex for ATP production via oxidative phosphorylation. Acetyl-CoA can be shuttled out of mitochondria through carnitine acetyltransferase (CrAT), while it goes through integrated endogenous lipid conversion to form phospholipids, cholesterol, and triacylglycerol (TAG). Q, Coenzyme Q; Cyt C, cytochrome C.
Long-chain fatty acids (LCFAs, referring to FAs with 12 or longer carbons chains) primarily enter the cell via FA transporter CD36 [also known as Fatty acid translocase (FAT) or SR-B2]. CD36 is expressed in multiple cell types and mediates diverse functions, such as lipid uptake, inflammation, ROS production, molecular adhesion, and apoptosis. CD36 is a multifunctional receptor for many ligands, including collagen, native lipoproteins, LCFA, oxidized phospholipids, oxidized LDL, thrombospondin, and apoptotic cells (Yang et al., 2017; Wang and Li, 2019). Several post-translational modifications, including phosphorylation, palmitoylation, ubiquitylation, and glycosylation regulate CD36 stability and dimerization, and correlate its function to myocellular FA uptake (Luiken et al., 2016). In adipocytes, two palmitoyl-acyltransferases (PATs), namely DHHC4/5, modulate CD36 palmitoylation and target it to the plasma membrane lipid rafts, where it mediates FA adsorption and transport (Wang et al., 2019). Interestingly, in addition to the cell surface, CD36 also localizes to the ER, endosomes, and mitochondria (Bonen et al., 2000; Smith et al., 2011). In response to diverse signaling transduction pathways, rapid mobilization of the vesicular transport system mediates dynamic intracellular distribution of CD36 to reprogram energy utilization and control lipid metabolism (Georgiou et al., 2015; Glatz et al., 2016).
In the kidney, CD36 mediates FA uptake and lipid metabolic reprograming and functions (Yang et al., 2017). CD36 is highly expressed in mesangial cells (Ruan et al., 1999), renal proximal (Susztak et al., 2005) and distal tubular epithelial cells (Okamura et al., 2007), podocytes (Hua et al., 2015), microvascular endothelial cells, and interstitial macrophages (Rahman et al., 2008; Kennedy et al., 2013). Transgenic mice with tubular overexpression of CD36 demonstrate tubular-specific accumulation of lipids, TAG, and LCFAs (Kang et al., 2015). Other kidney CD36 substrates include oxidized phospholipids, advanced oxidation protein products (AOPPs; Li et al., 2019), and advanced glycation end products (AGEs), which promote inflammation, ER stress, and renal cells apoptosis and contribute to renal fibrosis (Okamura et al., 2009; Ruggiero et al., 2014; Pennathur et al., 2015).
Lipid Uptake by Other Transporters in the Kidney: FABPs and FATPs
FA-binding proteins (FABPs) are low molecular weight (14–15 kDa) proteins that transport LCFAs through cell membranes, transport FAs to mitochondria and peroxisomes, and function as chaperones to mediate intracellular transport. Two major FABP isoforms are expressed in human kidneys, the proximal tubule-enriched FABP1 (also known as liver type L-FABP) and the distal tubule-enriched FABP3 (Maatman et al., 1991). Urinary FABP1 level was proposed as a biomarker of acute tubulointerstitial damage (Yamamoto et al., 2007; Pelsers, 2008).
Emerging data also support proximal tubular apical expression of FA transporter-2 (FATP2, encoded by Slc27a2) and its role in luminal non-esterified FA (NEFA) reabsorption from glomerular filtrate and NEFA metabolism in mice. Silencing of FATP2 in human renal PTEC in vitro leads to increased Oil Red O staining and subsequent apoptosis following FA exposure. Moreover, tubular lipoapoptosis in lipidated albumin-injected mice decreases in Slc27a2-deficient mice. These data suggest that luminal NEFA uptake by FATP2 causes proximal tubule lipoapoptosis, which may contribute to tubular atrophy and CKD progression (Khan et al., 2018).
Fatty Acid Metabolism in Kidney Mitochondria
FA β-oxidation (FAO) may occur in both mitochondria and peroxisomes. While mitochondria majorly oxidize LCFAs, and medium-chain and short-chain FAs (MCFAs and SCFAs, referring to FAs with less than 12 carbons chains), peroxisomes oxidize specific carboxylic acids such as very long-chain FAs (VLCFAs), branched-chain FAs, fatty dicarboxylic acids, and bile acid intermediates (in the liver; Cipolla and Lodhi, 2017). Interestingly, peroxisomal FAO provides alternative metabolism of LCFAs and MCFAs in case of mitochondrial long-chain FAO deficiencies (Violante et al., 2019). Mitochondrial FAO is thus the major pathway for the degradation of FAs to sustain cellular energy homeostasis (Houten et al., 2016). This process includes six tightly-regulated steps: (i) FA esterification to acyl-CoA; (ii) mitochondrial CPT shuttle (or the carnitine shuttle); (iii) the FAO pathway; (iv) the OXPHOS pathway; (v) allosteric control of FAO; and (vi) integrated nutrient metabolism in the kidney (Figure 1).
Fatty Acid Esterification to acyl-CoA
FAs must be converted to fatty acyl-CoA by cytosolic acyl-CoA synthetases in order to enter mitochondria. Once inside the cell, MCFAs or SCFAs can freely diffuse into mitochondria. However, LCFAs need to be activated to long-chain acyl-CoA (LC acyl-CoA) and esterified with carnitine into LC-acylcarnitine to permeate the outer mitochondrial membrane (OMM) and subsequently be transported into the mitochondrial matrix (Bremer, 1983).
The Carnitine Shuttle
The carnitine shuttle, mediated by the rate-limiting enzyme carnitine palmitoyltransferase I (CPT-1, on the OMM) and the two inner mitochondrial membrane (IMM) proteins carnitine-acylcarnitine translocase (CACT) and carnitine palmitoyltransferase II (CPT-2), serves to transport the FA moiety into mitochondria. CPT-2 conducts a reverse reaction to convert LC-acylcarnitine back to LC acyl-CoA and carnitine. Carnitine is transported back to the cytoplasm by the same shuttle (Brivet et al., 1999).
The FAO Pathway
FAO is the process of breaking down a LC acyl-CoA into acetyl-CoA molecules inside the mitochondrial matrix. The term β-oxidation refers to the position of the carbon group being oxidized. The number of acetyl-CoA molecules produced depends on the initial carbon length of the FA. When LC acyl-CoA enters FAO, two carbons are cleaved to generate an acetyl-CoA and an acyl-CoA that is two carbons shorter from each β-oxidation cycle. This process continues until all of the carbons in the FA are turned into acetyl-CoA to fuel the tricarboxylic acid (TCA) cycle and generate ATP. The two redox active coenzymes – the reduced form of nicotinamide adenine dinucleotide (NADH) and the hydroquinone form of flavin adenine dinucleotide (FADH2) – produced during each β-oxidation cycle, along with those generated from TCA cycle, are used as electron donors by the electron transport chain (ETC) complex, in the redox reaction that produces ATP (the OXPHOS pathway). LCFA oxidation yields high energy: for instance, 137 ATP are generated from palmitate as opposed to 38 obtained from glucose oxidation (Nsiah-Sefaa and McKenzie, 2016).
The OXPHOS Pathway
The mitochondrial ETC/OXPHOS respiratory chain contains five complexes. Complexes I–IV transfer electrons (e−) and protons (H+) across IMM to generate an electrochemical gradient for ATP synthesis in complex V (ATP synthase). Several critical steps regulate this process. The concentration of NAD constitutes the rate-limiting process (Canto et al., 2015; Verdin, 2015). Coenzyme Q10 (CoQ10) is a component of ETC, which shuttles electrons in the respiratory chain. Moreover, the reduced form of CoQ10 is also a potent antioxidant (Ernster and Dallner, 1995; Thomas et al., 1996). CoQ deficiency could cause nephropathies (Ozaltin, 2014) and mutation in ADCK4 (CoQ8B), a protein required for stabilizing CoQ complex in podocyte, is an etiology of steroid-resistant nephrotic syndrome (SRNS or FSGS; Ashraf et al., 2013; Widmeier et al., 2020). Cardiolipin, an IMM phospholipid, plays a central structural role in cristae formation, facilitates ETC supra-complex formation for optimal OXPHOS activity, and serves as a platform to initiate apoptosis (Birk et al., 2014; O’Brien et al., 2015).
Allosteric Control of FAO
Mitochondrial bioenergetic homeostasis is subjected to allosteric regulation by the ratios of the [Acetyl CoA/CoA], [NADH/NAD+], and [FADH2/FAD+]. Therefore, FAO enzymatic activities are affected by the levels of the metabolic products of their own reactions, and a rise in [Acetyl-CoA/CoA] or [NADH/NAD+] leads to feedback inhibition of FAO (Karwi et al., 2019). For example, mice with proximal tubule-specific deletion of carnitine acetyltransferase (CrAT), an enzyme that controls inter-conversion of Acetyl-CoA/CoA and shuttles excess FA products out of the mitochondria, develop mitochondrial dysfunction, cellular apoptosis, and tubular and glomerular fibrosis (Kruger et al., 2019). Interestingly, de novo synthesis of NAD+, a central metabolic coenzyme/co-substrate involved in cellular energy metabolism, profoundly affects mitochondrial fitness in organ health and injury, including kidney (Hershberger et al., 2017; Katsyuba et al., 2018; Poyan Mehr et al., 2018; Ralto et al., 2020).
Integrated Nutrient Metabolism in the Kidney
Acetyl-CoA is a critical metabolite derived from catabolism of all major nutrient sources, such as glucose, FAs, and amino acids. Moreover, acetyl-CoA can be diverted from the TCA cycle to synthesize cholesterol, phospholipids, and TAG in the cell (Pietrocola et al., 2015; Shi and Tu, 2015). Proper integration and regulation of energy metabolism during ATP loss or excess are thus key to maintain mitochondrial health during injury and repair in renal pathophysiology (Vamecq et al., 2012; Aon et al., 2014; Fornoni et al., 2014; Szeto, 2017; Jang et al., 2020a).
Transcriptional, Epigenetic, and Post-Translational Regulation of FAO and Mitochondria Biogenesis
Several transcriptional, epigenetic, and post-translational regulators are involved in the crosstalk between peroxisomes, nucleus, and mitochondria to control the expression/functions of FAO enzymes, mitochondrial biogenesis, and energy reprograming in health and disease-stressed states (Stallons et al., 2013; Bhargava and Schnellmann, 2017; Figure 2). These key molecules include the nuclear hormone receptor, peroxisome proliferator-activated receptors (PPARs, PPARα, and PPARγ as examples; Wu et al., 2009; Corrales et al., 2018); PPARγ coactivator 1α (PGC-1α; Weinberg, 2011; Li and Susztak, 2018; Fontecha-Barriuso et al., 2020); the NAD+-dependent deacetylases sirtuins (SIRTs; Wakino et al., 2015; Hershberger et al., 2017; Morigi et al., 2018); AMP-activated protein kinase (AMPK); and nuclear respiratory factors 1 and 2 (NRF1 and NRF2; Akhtar and Siragy, 2019).
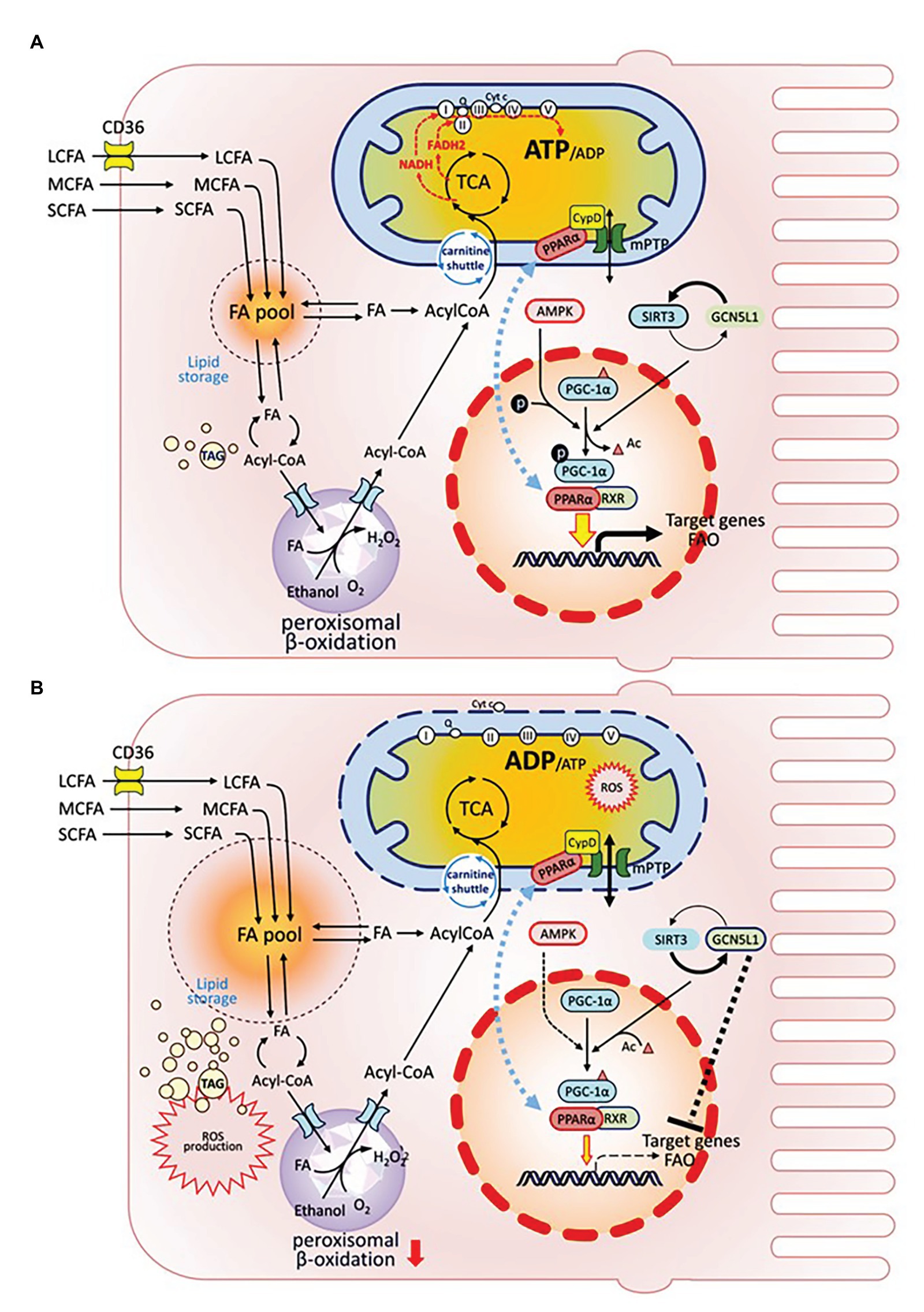
Figure 2. Organelle crosstalk regulates fatty acids metabolism in renal PTEC under healthy or injury/disease states. Intracellular FA metabolism includes catabolic and anabolic pathways. FAs are oxidized either in mitochondria or peroxisome to generate ATP (catabolism), or are stored as global triglyceride pool (anabolism). PGC-1α is the mitochondrial master regulator, which drives mitochondrial biogenesis by co-activating transcriptional factors PPAR-α and RXR to regulate the expression of target genes affecting biogenesis, OXPHOS, and FAO. PGC-1α is also extensively regulated by post-translational modifications: PGC-1α is activated via phosphorylation by AMPK; its acetylation state is regulated by the counter-balance between SIRT deacetylase and GCN5L1 acetylase. GCN5L1 activation also negatively modulates FAO target genes. Translocation of PPARα between nucleus and IMM affects PPARα activity as a transcription factor. (A) In healthy condition. (B) Under injury/disease state, impaired PGC-1α leads to defective FAO and is associated with reduced FA catabolism, increased FA pool and TAG accumulation, increased cellular ROS production, and PPARα mitochondrial translocation, which induces PPARα interaction with CypD, mPTP opening, ETC disruption, cytochrome C release, and mitochondrial damage.
PGC-1α, the mitochondrial biogenesis master regulator, is predominantly expressed in proximal tubules and interacts directly with multiple transcription factors to integrate upstream signaling events with mitochondrial biogenesis and functional capacity. Downstream transcription factors control all aspects of mitochondrial function, including biogenesis, energy production, dynamics, and protein homeostasis. PGC-1α regulates the expression of NRF1 and NRF2 to increase NAD+ biosynthesis (Tran et al., 2016) and activates genes coding for the OXPHOS system (Zoja et al., 2014). In obesity-related nephropathy models, reduced NRF2 along with suppressed expression of the key FAO enzyme long-chain acyl-CoA synthetase-1 (ACSL1) are associated with elevated renal lipid deposition, further supporting the importance of mitochondria in lipid metabolism and energy homeostasis (Chen et al., 2019).
As the PGC-1α/PPARα axis governs transcriptional regulation of FAO, it was proposed as therapeutic target in AKI and CKD (Simon and Hertig, 2015; Stadler et al., 2015). Defects in the FAO pathway, such as reduced expression of CPT-1 (Kang et al., 2015), CrAT (Kruger et al., 2019), and PPARα (Chung et al., 2018) are associated with CKD and renal fibrosis. PPARα heterodimerizes with its obligate partner, the retinoid-X-receptor (RXR), to regulate FAO and energy metabolism. FAs are natural activators of PPARα, and one of PPARα target genes is CD36 (which increases FA uptake; Portilla, 2003). Moreover, impaired expression of PPARα and specific proteins in FAO pathway are associated with lipid accumulation and fibrosis in renal tubular epithelial cells in aging rats (Chung et al., 2018). Interestingly, Jang et al. demonstrated that proximal tubular mitochondrial interaction of PPARα with cyclophilin D (CypD), a component of the IMM structural protein complex mitochondrial permeability transition pore (mPTP), could repress nuclear PPARα activity and negatively modulate FAO in cisplatin-induced AKI (Jang et al., 2020a). Several PPARα agonists have been shown to enhance FAO activity in kidney (Konig et al., 2008; Lakhia et al., 2018).
PGC-1α is extensively regulated by post-translational modifications. AMPK and SIRT positively regulate PGC-1α through phosphorylation or deacetylation, respectively (Jager et al., 2007; Canto and Auwerx, 2009). Interestingly, liver histone demethylase JMJD3 was identified as a gene-specific transcriptional partner of SIRT1 that epigenetically activates mitochondrial β-oxidation during fasting (Seok et al., 2018). The counterpart of JMJD3 in kidney remains to be uncovered. General control of amino acid synthesis-5-like 1 (GCN5L1), a protein acetylase counteracting the function of SIRT3, was recently shown to negatively modulate hepatic FAO enzyme activities via acetylation (Thapa et al., 2018). Similarly, GCN5L1-mediated hyper-acetylation and impairment of FAO enzymes might be a key pathogenic event underlying lipid overload-induced kidney injury (Lv et al., 2019).
In summary, the integrated regulation of FA metabolism at the genetic, epigenetic, and protein level is tightly associated with mitochondrial homeostasis. Under injury or disease state, deficiency in PGC-1α and associated transcription factors leads to defective FAO, enlarged FA pool and TAG accumulation, massive ROS production, increased PPARα mitochondrial translocation inducing mPTP opening and loss of mitochondrial membrane potential, cytochrome C release, and mitochondrial damage (Figure 2).
Dyslipidemia and Cellular Lipotoxicity-Mediated Kidney Injury
Dyslipidemia is an abnormal amount of lipids (e.g., TAG, cholesterol or phospholipids) in the blood. Lipids in excess, which is delivered to organs beyond their energy demands, can be stored mainly as TAG in intracellular lipid droplets (LDs), an ubiquitous organelle that serves as energy stores, dynamic membrane synthesis, and as a hub for further metabolic regulation (Walther et al., 2017). Accumulation of such lipid intermediates or final products in non-adipose tissues, along with the subsequent multi-factorial disturbance of intracellular homeostasis, could result in lipotoxicity of target tissues. Lipotoxicity thus represents a pathologic phenomenon with hallmarks of aberrant lipid accumulation, causing metabolic, inflammatory, oxidative stress in intracellular organelles, and further triggering cell damages (Su et al., 2017; Opazo-Ríos et al., 2020).
Moorhead et al. first hypothesized “lipid nephrotoxicity” in 1982, proposing that dyslipidemia may contribute to the progression of renal dysfunction (Moorhead et al., 1982). This hypothesis had gained supportive evidence in several contexts. For examples, renal lipid accumulation has been shown with high clinical prevalence in patients with CKD, including the insulin resistant obese subjects with diabetic nephropathy (Herman-Edelstein et al., 2014; Escasany et al., 2019; Opazo-Ríos et al., 2020), in nephrotic syndrome (Vaziri, 2016; Agrawal et al., 2017), focal segmental glomerulosclerosis (FSGS; Sasaki et al., 2018), and also as a consequence of acute ischemic renal injury (Zager et al., 2011). Significant alterations in renal lipid metabolism are typified as high TAG, variation in the composition of apolipoproteins and lipids, the accumulation of atherogenic particles VLDL and IDL, and decreased HDL cholesterol (Vaziri, 2006; Stadler et al., 2015; Florens et al., 2016; Kronenberg, 2018; Du and Ruan, 2019; Gai et al., 2019; Thongnak et al., 2020; Jang et al., 2020b). As discussed earlier, systematic lipid metabolism involves multi-organ crosstalk, ultimately also affecting kidney function. Therefore, dyslipidemia and lipid nephrotoxicity could be not only a consequence but also a cause of kidney disease (Florens et al., 2016; Agrawal et al., 2017; Kronenberg, 2018; Czumaj et al., 2019; Nishi et al., 2019).
Excess fat could be derived from either dysfunctional capacity of adipose lipid storage, or from diet-induced hyperlipidemia (high plasma albumin-bound FFAs and cholesterol), or in the condition of renal dysfunction (as commonly exemplified by renal mass reduction in animal model) and defective insulin signaling. Excess kidney ectopic fat deposition and lipid overload in intracellular organelles could lead to ER stress (Zhao et al., 2008), mitochondria dysfunction (Vamecq et al., 2012; Szeto et al., 2016), and lysosomal stress (Yamamoto et al., 2017, 2020). These alterations could change cellular protective mechanisms such as autophagy, mitophagy, lipophagy and contribute to apoptosis and cell damage. These observations thus support the notion of dyslipidemia contributes to the progression of renal injury, and lipid-lowering therapies or shielding mitochondria could provide beneficial effects on lipotoxicity-mediated kidney injury (Izquierdo-Lahuerta et al., 2016; Su et al., 2017).
Dyslipidemia could appear in various forms with different causes and consequences. In lipid-mediated podocyte damage, FFAs and their metabolism affect function and survival of podocytes (Sieber and Jehle, 2014). Dyslipidemia is also a common feature, rather than a complication, of nephrotic syndrome. Excessive urinal protein loss results in hypoproteinemia, in turn leading to low serum oncotic pressure, and even edematous change in severe cases. To rescue the oncotic pressure deficit, the body initiates a reactive hepatic protein synthesis, including lipoproteins (Attman and Alaupovic, 1990; Merscher et al., 2014; Vaziri, 2016; Agrawal et al., 2017). Additionally, reduced plasma levels of lipoprotein lipase results in decreased lipid catabolism. Elevated serum levels of LDL and IDL are filtered through glomeruli and lead to lipiduria, which manifests with fatty casts containing oval fat bodies in the urine sediment (Cavanaugh and Perazella, 2019).
Mutations affecting cholesterol metabolism in the process of lipid trafficking, storage, influx, or efflux, could mediate glomerular injury (Merscher et al., 2014). For example, Tangier disease (OMIM #205400) or DKD caused by mutations in ATP-binding cassette A1 (ABCA1) gene result in reduced HDL in circulation, albuminuria, podocyte phenotype with esterified cholesterol accumulation and dysfunctional mitochondria due to cardiolipin hyperoxidation (Ducasa et al., 2019a,b). For the topics on glomerular diseases-related renal lipotoxicity and mitochondrial dysfunction, please refer to the comprehensive review in the same special issue (Ge et al., 2020).
Furthermore, excess of FFAs leads to TAG accumulation and renal tubular toxicity (Johnson et al., 2005; Scerbo et al., 2017). Increased LCFA-bound albumin induces altered redox balance, high tubular cell apoptosis, and kidney fibrosis (Ruggiero et al., 2014). As lipoprotein abnormalities also correlate with high risk of both cardiovascular and kidney diseases, these modified lipoproteins could be accounted as actual mediators of uremic toxicity (Florens et al., 2016). LDL and oxidized (ox)-LDL uptake by mesangial cells lead to cell proliferation and glomerular matrix expansion, while uptake by PTE results in tubulointerstitial lesions with remarks of heightened expression of extracellular matrix proteins (Nosadini and Tonolo, 2011). HDL is a key player in reverse cholesterol transport to shuttle cholesterol from peripheral cells, such as macrophages, to the liver, therefore relieving the cholesterol burden of these cells. HDL thus exerts its anti-oxidant function through preventing LDL oxidation by ROS and protecting against the adverse effects of ox-LDL on the endothelium. Reduced levels and dysfunction of HDL, which could be due to perturbed HDL proteome composition, are common in CKD patients (Vaziri, 2006; Yamamoto et al., 2012; Agrawal et al., 2017; Kronenberg, 2018; Rysz et al., 2020).
Deficiency of FA metabolism and lipid overload are the main drivers in the progression of both glomerular and tubular kidney diseases. Lipid accumulation, particularly in ischemic proximal tubules, may result in persistent energy depletion with FFA-induced mitochondrial dysfunction, which could play an important role in the AKI to CKD transition (Szeto, 2017). Conversely, mitochondrial protection prevents high-fat diet-induced glomerular and tubular lesions (Szeto et al., 2016).
The pathophysiological changes underlying hyperlipidemia may involve energy shortage from impaired mitochondrial biogenesis or ATP energetics, and systemic oxidative stress due to excessive ROS production accompanied by ER stress and influx of inflammatory cytokines. Without timely intervention, these changes could eventually lead to apoptosis and kidney fibrosis (Agrawal et al., 2017; Du and Ruan, 2019). Less is known about the molecular mechanism of some toxic lipid intermediates (“metabolic poison”) derived from deficiency or decreased expression of FAO-related enzymes in kidney disease development (Stadler et al., 2015; Su et al., 2017); and future research may elucidate this process.
Targeting Mitochondrial Energy Metabolism and Lipotoxicity in Kidney Diseases
Lipid-lowering therapies in kidney diseases have been studied for many years, although statins is still the first choice of conventional hypolipidemia strategies for its effect on HMGCoA inhibition to block cholesterol synthesis. Cumulative pharmacological efforts have advanced the field to develop classic and novel lipid modifying therapies in kidney diseases, as extensively reviewed recently (Ferro et al., 2018; Sudhakaran et al., 2018; Filippatos et al., 2019; Rosenson et al., 2019; Heine et al., 2020; Opazo-Ríos et al., 2020). These include effective and well-tolerated drugs targeting various lipid synthesis, uptake, trafficking and metabolism pathways. Recent years, compounds that specifically target mitochondria have emerged as promising therapeutic options for patients with renal disease. Here, we discuss molecules targeting mitochondrial lipid metabolism and mitochondrial dysfunction pathways, including pharmacological agents promoting mitochondrial FAO, mitochondrial biogenesis, and ATP synthesis, as well as mitochondrial antioxidants (regulating ROS metabolism) and cardiolipin stabilizers.
Mitochondrial FAO-Promoting Agents
Carnitine and acetyl-L-carnitine are nonessential nutrients, as kidney of healthy subjects normally produce sufficient carnitine from daily food intake/metabolism and preserve its excretion well. However, carnitine could be used as dietary supplements to help with carnitine shuttle of FAO in conditions of “primary carnitine deficiency” (children with genetic disorder of carnitine transporter OCTN2 encoded by the SLC22A5 gene; Frigeni et al., 2017) or adults with secondary carnitine deficiencies due to chronic renal failure (Ames, 2010).
PPARα is crucially involved in energy and metabolic homeostasis. Fibrates (fibric acid derivatives, including fenofibrate and the enhanced medication-pemafibrate) are a class of PPARα agonists that lowers blood TAG through decreasing VLDL production by liver and promoting the removal of TAG from blood. Fibrates also moderately increase blood HDL cholesterol. Mechanistically, the PPARα agonists activate PPARα, promote peroxisomal and mitochondrial FAO, initiate cellular cascade to upregulate lipoprotein lipase, and ultimately cause more efficient catabolism of VLDL and TAG (Lakhia et al., 2018; Cheng et al., 2019; Yamashita et al., 2020).
CD36 mediates the internalization of lipids such as LCFAs, oxLDL, and oxidized phospholipid in both proximal tubule cells and podocytes. CD36 signaling is involved in FA-induced glomerular injury (Hua et al., 2015). The ApoA-I mimetic 5A peptide is a CD36 antagonist, which was shown to reduce glomerular injury and tubulointerstitial fibrosis in mouse CKD models of subtotal nephrectomy with angiotensin II infusion or unilateral ureteral obstruction (Souza et al., 2016). 5A peptide was shown to form HDL-like particles to promote ABCA1-dependent cholesterol efflux (Islam et al., 2018) and thus may effectively treat patients with cardiovascular disease.
The herbal alkaloid Berberine (BBR) is used as a supplemental medicine and has shown clinical benefit in reduction of LDL and TAG in diabetic and hypertensive patients (Koppen et al., 2017). BBR has wide spectrum pharmacological effects through its various action of mechanisms such as increasing LDL-receptor mediated hepatic clearance of LDL cholesterol (Wang et al., 2014), protection of lipid-induced apoptosis by promoting FAO in PTEC (Sun et al., 2018), supporting PGC1α-regulated mitochondrial energy homeostasis in CKD model of db/db mice and cultured podocytes (Qin et al., 2019a), and podocyte protection via inhibition of mitochondrial fission and dysfunction (Qin et al., 2019b).
Mitochondrial Bioenergetics and Biogenesis-Promoting Agents
Niacin (vitamin B-3) was the first identified lipid-lowering drug in patients at late 1950s and currently used as an adjunct therapy to help the control of cholesterol. Niacin, at pharmacological dose, increases circulating HDL level to improve cholesterol clearance in peripheral tissues and also changes the composition and metabolism of ApoA-I and ApoA-II (Shepherd et al., 1979). The HDL boost effect of niacin is through different molecular mechanisms. First, niacin stabilizes surface ABCA1 expression and ApoA-I lipidation. Second, niacin inhibits surface expression of the hepatic HDL receptor β-ATP synthase, and thus increases HDL blood availability (Zhang et al., 2008). Third, niacin inhibits the hepatic TAG biosynthesis enzyme “diacylglycerol acyltransferase-2 (DGAT2)” to reduce TAG synthesis and leads to the subsequent VLDL/LDL destabilization (Ganji et al., 2004). The mechanisms of DGAT inhibition and TAG metabolism are active research area as more pharmacological drugs designs centering on the two DGAT enzymes (DGAT1 and DGAT2), which apparently have distinct and overlapping functions (Chitraju et al., 2019). Niacin was later found to be an important precursor of cofactor NAD+, which promotes SIRT/PGC-1α activity and thus modulates mitochondrial energy homeostasis, biogenesis, and lipid metabolism (Kirkland and Meyer-Ficca, 2018; Romani et al., 2019). Moreover, niacin provides vascular benefits through NAD+/SIRT mediated mechanism during endothelial lipotoxicity (Hughes-Large et al., 2014).
The AMPK/SIRT/PGC-1α axis is crucial for mitochondrial biogenesis (Duann and Lin, 2017). Agents modulating this process include metformin. Metformin, the most commonly prescribed drug for the treatment of type 2 diabetes as a glucose-lowering and insulin-sensitizing agent, is a biguanide drug that also actives the energy sensor AMPK. In animal nephropathy models, several pathologies were observed including reduced phosphorylation of acetyl-CoA carboxylase (ACC), a target of AMPK and the major enzyme in the control of FAO rate; decreased expressions of CPT1 and enzymes in mitochondrial biogenesis; and increased lipid accumulation and expression of pro-inflammatory cytokines and tubulointerstitial fibrosis. Metformin reduces renal fibrosis by improving AMPK-mediated phosphorylation of ACC and FA energy metabolism (Lee et al., 2018).
Mitochondria-Targeted Anti-oxidants
Lipid-mediated mitochondrial oxidative stress is common in many kidney diseases. The selective mitochondria-targeted antioxidants, such as MitoQ and MitoTEMPO, have been developed to mitigate mitochondrial oxidative stress. These small molecule agents could be delivered and concentrated at mitochondria matrix to function as ROS scavenger (Kezic et al., 2016). They are chimeric molecules of a lipophilic cation triphenylphosphonium (TPP+) conjugated with an antioxidant moiety such as ubiquinone (MitoQ; Kelso et al., 2001) or piperidine nitroxides (TEMPOL and TEMPO; Trnka et al., 2008).
MitoTEMPO could be uptaken and accumulated in energized mitochondria matrix several 100-fold to modulate coenzyme Q (CoQ) pool within mitochondria (Trnka et al., 2008). In a diabetic db/db mouse model, 7-week of CoQ10 (0.1% in food) oral administration significantly reduced the levels of serum creatinine and blood glucose and albumin-to-creatinine ratio, in accordance with renal morphological restoration (Sun et al., 2019). CoQ10 ameliorates DN-induced mitochondrial dysfunction and oxidative stress through its activation of mitophagy-mediated glomerular mitochondria homeostasis both in vivo and in vitro. In this study, MitoTEMPO (3 mg/kg/day) restored mitophagy and alleviated kidney dysfunction in glomeruli of db/db mice in a similar manner as CoQ10 treatment (Sun et al., 2019). In a mouse sub-total nephrectomy-induced renal fibrosis CKD model, MitoTEMPO rescued impaired renal function and alleviated renal fibrosis by reducing inflammation cytokines, mitochondrial dysfunction, ER stress, and profibrotic factors (Liu et al., 2018).
Additionally, in a clinically relevant murine model of abdominal sepsis (cecal ligation and puncture, CLP), a single delayed high dose of MitoTEMPO (10 mg/kg, given at 6 h post-CLP) could reverse renal mitochondrial dysfunction and attenuated sepsis-induced AKI by 18 h. MitoTEMPO decreased mitochondrial superoxide level, protected ETC respiration, improved renal microcirculation and glomerular filtration rate. Importantly, MitoTEMPO treatment significantly increased 96-h survival rate from 40% in untreated mice to 80% (Patil et al., 2014). The beneficial effect of MitoTEMPO is still under debate as it failed to exert long-term benefits in a later CLP-AKI study (Rademann et al., 2017). However, in a rat puromycin aminonucleoside (PAN)-induced glomerular damage model, a model mimicking children minimal-change nephrotic syndrome (MCNS), a 10-day MitoTEMPO treatment (1 day prior to PAN-injury and continued for 9 additional days) reduced the level of urinary protein, urinary lipid peroxidation and the expression of oxidative stress markers in glomeruli and plasma; although the overall renal function seemed not significantly improved as measure of creatinine clearance (Fujii et al., 2020). In summary, more research is warranted to validate renoprotective effects of MitoTEMPO.
MitoQ is a mitochondria targeted antioxidant of CoQ analogue, which could be accumulated in mitochondria up to 1,000-fold. In a type 1 monogenic diabetes of the young [MODY, the Ins2Akita (Akita)] mouse model, oral administration of MitoQ over a 12-week period prevented diabetic nephropathy (Chacko et al., 2010). MitoQ treatment did not alter the glycaemic status of diabetic animals. However, MitoQ significantly decreased urinary albumin levels in diabetic mice. MitoQ offered benefits in prevention of diabetes-induced tubular dysfunction and protection of glomerular function as measured by radioactive tracer clearance capacity. Moreover, MitoQ decreased pathogenic glomerular GBM thickening and reduced interstitial fibrosis through prevention of EMT (epithelial-to-mesenchymal transition) process in Akita mice (Chacko et al., 2010). Recently, in a diabetic db/db mouse model, Ward et al. confirmed the renoprotective effects of MitoQ treatment through daily intragastric gavage over a period of 12-week. MitoQ improved renal function, decreased glomerular hyperfiltration, albuminuria, and prevented interstitial fibrosis (Ward et al., 2017). In a mouse ischemia-reperfusion induced AKI (IRI) model, administration of MitoQ prior to the onset of ischemia was shown to reduce oxidative damage and severity of renal IRI (Dare et al., 2015). Despite the great success of mitochondria-targeting antioxidants in preclinical studies, their clinical effects on CKD patients remain to be verified. However, MitoQ supplementation was linked to restoration of endothelial function and reduces aortic arterial stiffness in aging humans, thus offers potential promise in vascular treatment in CKD patients (Rossman et al., 2018).
Cardiolipin-Targeting Peptides
In mice, a long-term (28 weeks) high fat diet (HFD) caused mitochondrial dysfunction and structural alterations, such as reduction in size and loss of matrix density and IMM cristae, in renal cells including proximal tubular cells, podocytes and glomerular endothelial cells. The mitochondrial injury led to ER stress, lipid droplets accumulation, autophagy, apoptosis, and subsequent inflammation, proteinuria, and fibrosis (Szeto et al., 2016). The mitochondrial injury could be due to loss and/or peroxidation of cardiolipin, the major structural and functional regulator of IMM cristae. Such mitochondrial injury could be prevented with cardiolipin-stabilizing tetrapeptide SS31 (namely, Elamipretide, MTP-13, or Bendavia), which reduces HFD-induced lipid accumulation, toxic ROS production, regulates cytochrome C activity, and restores AMPK signaling (Szeto et al., 2016; Szeto, 2017). The mitochondria protective effect of SS31 after ischemia-AKI prevents prolonged inflammation and arrests CKD transition (Szeto et al., 2017). Elamipretide is on a phase 2a clinical trial in patients with atherosclerotic renal artery stenosis during stent revascularization, with promising results (NCT01755858; Saad et al., 2017) and was shown to improve mitochondria function in the human failing heart (Chatfield et al., 2019). The clinical effects of Elamipretide on kidney disease, however, require further investigations.
Concluding Remarks
Mitochondria are the “powerhouse” of the high-energy demanding kidney cells. Crosstalk between mitochondria, nucleus, endoplasmic reticulum, and peroxisomes impacts numerous cellular functions. Mitochondrial bioenergetics, adaptation of energy metabolism, and mitochondrial biogenesis during physiology or stress conditions are tightly linked to body lipid homeostasis, as well as health and disease states of kidney. Dysfunctional mitochondria could lead to dyslipidemia, microvasculature damage, inflammation, kidney fibrosis, or even kidney failure. The evolving knowledge of the molecular mechanisms modulating mitochondrial energy homeostasis and lipid metabolism suggest that normalizing renal cell mitochondrial function and energy balance could be an important preventative strategy against dyslipidemia and could provide new drug targets in kidney diseases.
Author Contributions
All authors contributed to the conception and drafting the work and critically reviewed and revised for the intellectual accuracy of the contents. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by an intramural Fund from The Ohio State University (to P-HL).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Agrawal, S., Zaritsky, J. J., Fornoni, A., and Smoyer, W. E. (2017). Dyslipidaemia in nephrotic syndrome: mechanisms and treatment. Nat. Rev. Nephrol. 14:70. doi: 10.1038/nrneph.2017.175
Akhtar, S., and Siragy, H. M. (2019). Pro-renin receptor suppresses mitochondrial biogenesis and function via AMPK/SIRT-1/PGC-1alpha pathway in diabetic kidney. PLoS One 14:e0225728. doi: 10.1371/journal.pone.0225728
Ames, B. N. (2010). Optimal micronutrients delay mitochondrial decay and age-associated diseases. Mech. Ageing Dev. 131, 473–479. doi: 10.1016/j.mad.2010.04.005
Aon, M. A., Bhatt, N., and Cortassa, S. C. (2014). Mitochondrial and cellular mechanisms for managing lipid excess. Front. Physiol. 5:282. doi: 10.3389/fphys.2014.00282
Ashraf, S., Gee, H. Y., Woerner, S., Xie, L. X., Vega-Warner, V., Lovric, S., et al. (2013). ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J. Clin. Invest. 123, 5179–5189. doi: 10.1172/JCI69000
Attman, P. O., and Alaupovic, P. (1990). Pathogenesis of hyperlipidemia in the nephrotic syndrome. Am. J. Nephrol. 10, 69–75. doi: 10.1159/000168197
Bhargava, P., and Schnellmann, R. G. (2017). Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 13, 629–646. doi: 10.1038/nrneph.2017.107
Birk, A. V., Chao, W. M., Bracken, C., Warren, J. D., and Szeto, H. H. (2014). Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 171, 2017–2028. doi: 10.1111/bph.12468
Bonen, A., Luiken, J. J., Arumugam, Y., Glatz, J. F., and Tandon, N. N. (2000). Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. J. Biol. Chem. 275, 14501–14508. doi: 10.1074/jbc.275.19.14501
Bonventre, J. V., and Yang, L. (2011). Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Invest. 121, 4210–4221. doi: 10.1172/JCI45161
Bremer, J. (1983). Carnitine-metabolism and functions. Physiol. Rev. 63, 1420–1480. doi: 10.1152/physrev.1983.63.4.1420
Brinkkoetter, P. T., Bork, T., Salou, S., Liang, W., Mizi, A., Özel, C., et al. (2019). Anaerobic glycolysis maintains the glomerular filtration barrier independent of mitochondrial metabolism and dynamics. Cell Rep. 27, 1551.e5–1566.e5. doi: 10.1016/j.celrep.2019.04.012
Brivet, M., Boutron, A., Slama, A., Costa, C., Thuillier, L., Demaugre, F., et al. (1999). Defects in activation and transport of fatty acids. J. Inherit. Metab. Dis. 22, 428–441. doi: 10.1023/A:1005552106301
Canto, C., and Auwerx, J. (2009). PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 20, 98–105. doi: 10.1097/MOL.0b013e328328d0a4
Canto, C., Menzies, K. J., and Auwerx, J. (2015). NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 22, 31–53. doi: 10.1016/j.cmet.2015.05.023
Cargill, K., and Sims-Lucas, S. (2020). Metabolic requirements of the nephron. Pediatr. Nephrol. 35, 1–8. doi: 10.1007/s00467-018-4157-2
Cavanaugh, C., and Perazella, M. A. (2019). Urine sediment examination in the diagnosis and management of kidney disease: core curriculum 2019. Am. J. Kidney Dis. 73, 258–272. doi: 10.1053/j.ajkd.2018.07.012
Chacko, B. K., Reily, C., Srivastava, A., Johnson, M. S., Ye, Y., Ulasova, E., et al. (2010). Prevention of diabetic nephropathy in Ins2(+/)−(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochem. J. 432, 9–19. doi: 10.1042/BJ20100308
Chatfield, K. C., Sparagna, G. C., Chau, S., Phillips, E. K., Ambardekar, A. V., Aftab, M., et al. (2019). Elamipretide improves mitochondrial function in the failing human heart. JACC Basic Transl. Sci. 4, 147–157. doi: 10.1016/j.jacbts.2018.12.005
Chen, Y., He, L., Yang, Y., Chen, Y., Song, Y., Lu, X., et al. (2019). The inhibition of Nrf2 accelerates renal lipid deposition through suppressing the ACSL1 expression in obesity-related nephropathy. Ren. Fail. 41, 821–831. doi: 10.1080/0886022X.2019.1655450
Cheng, H. S., Tan, W. R., Low, Z. S., Marvalim, C., Lee, J. Y. H., and Tan, N. S. (2019). Exploration and development of PPAR modulators in health and disease: an update of clinical evidence. Int. J. Mol. Sci. 20:5055. doi: 10.3390/ijms20205055
Chitraju, C., Walther, T. C., and Farese, R. V. Jr. (2019). The triglyceride synthesis enzymes DGAT1 and DGAT2 have distinct and overlapping functions in adipocytes. J. Lipid Res. 60, 1112–1120. doi: 10.1194/jlr.M093112
Chung, K. W., Lee, E. K., Lee, M. K., Oh, G. T., Yu, B. P., and Chung, H. Y. (2018). Impairment of PPARalpha and the fatty acid oxidation pathway aggravates renal fibrosis during aging. J. Am. Soc. Nephrol. 29, 1223–1237. doi: 10.1681/ASN.2017070802
Cipolla, C. M., and Lodhi, I. J. (2017). Peroxisomal dysfunction in age-related diseases. Trends Endocrinol. Metab. 28, 297–308. doi: 10.1016/j.tem.2016.12.003
Corrales, P., Izquierdo-Lahuerta, A., and Medina-Gomez, G. (2018). Maintenance of kidney metabolic homeostasis by PPAR gamma. Int. J. Mol. Sci. 19:2063. doi: 10.3390/ijms19072063
Czumaj, A., Sledzinski, T., Carrero, J. J., Stepnowski, P., Sikorska-Wisniewska, M., Chmielewski, M., et al. (2019). Alterations of fatty acid profile may contribute to dyslipidemia in chronic kidney disease by influencing hepatocyte metabolism. Int. J. Mol. Sci. 20:2470. doi: 10.3390/ijms20102470
Dare, A. J., Bolton, E. A., Pettigrew, G. J., Bradley, J. A., Saeb-Parsy, K., and Murphy, M. P. (2015). Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol. 5, 163–168. doi: 10.1016/j.redox.2015.04.008
Du, X. G., and Ruan, X. Z. (2019). Lipid metabolism disorder and renal fibrosis. Adv. Exp. Med. Biol. 1165, 525–541. doi: 10.1007/978-981-13-8871-2_26
Duann, P., and Lin, P. H. (2017). Mitochondria damage and kidney disease. Adv. Exp. Med. Biol. 982, 529–551. doi: 10.1007/978-3-319-55330-6_27
Ducasa, G. M., Mitrofanova, A., and Fornoni, A. (2019a). Crosstalk between lipids and mitochondria in diabetic kidney disease. Curr. Diab. Rep. 19:144. doi: 10.1007/s11892-019-1263-x
Ducasa, G. M., Mitrofanova, A., Mallela, S. K., Liu, X., Molina, J., Sloan, A., et al. (2019b). ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. J. Clin. Invest. 129, 3387–3400. doi: 10.1172/jci125316
Eirin, A., Lerman, A., and Lerman, L. O. (2017). The emerging role of mitochondrial targeting in kidney disease. Handb. Exp. Pharmacol. 240, 229–250. doi: 10.1007/164_2016_6
Ernster, L., and Dallner, G. (1995). Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. Acta 1271, 195–204. doi: 10.1016/0925-4439(95)00028-3
Escasany, E., Izquierdo-Lahuerta, A., and Medina-Gomez, G. (2019). Underlying mechanisms of renal lipotoxicity in obesity. Nephron 143, 28–32. doi: 10.1159/000494694
Ferro, C. J., Mark, P. B., Kanbay, M., Sarafidis, P., Heine, G. H., Rossignol, P., et al. (2018). Lipid management in patients with chronic kidney disease. Nat. Rev. Nephrol. 14, 727–749. doi: 10.1038/s41581-018-0072-9
Filippatos, T. D., Liontos, A., Christopoulou, E. C., and Elisaf, M. S. (2019). Novel hypolipidaemic drugs: mechanisms of action and main metabolic effects. Curr. Vasc. Pharmacol. 17, 332–340. doi: 10.2174/1570161116666180209112351
Flemming, N. B., Gallo, L. A., and Forbes, J. M. (2018). Mitochondrial dysfunction and signaling in diabetic kidney disease: oxidative stress and beyond. Semin. Nephrol. 38, 101–110. doi: 10.1016/j.semnephrol.2018.01.001
Florens, N., Calzada, C., Lyasko, E., Juillard, L., and Soulage, C. O. (2016). Modified lipids and lipoproteins in chronic kidney disease: a new class of uremic toxins. Toxins (Basel) 8:376. doi: 10.3390/toxins8120376
Fontecha-Barriuso, M., Martin-Sanchez, D., Martinez-Moreno, J. M., Monsalve, M., Ramos, A. M., Sanchez-Nino, M. D., et al. (2020). The role of PGC-1alpha and mitochondrial biogenesis in kidney diseases. Biomolecules 10:347. doi: 10.3390/biom10020347
Fornoni, A., Merscher, S., and Kopp, J. B. (2014). Lipid biology of the podocyte—new perspectives offer new opportunities. Nat. Rev. Nephrol. 10, 379–388. doi: 10.1038/nrneph.2014.87
Freeman, D. M., Chan, L., Yahaya, H., Holloway, P., and Ross, B. D. (1986). Magnetic resonance spectroscopy for the determination of renal metabolic rate in vivo. Kidney Int. 30, 35–42. doi: 10.1038/ki.1986.147
Frigeni, M., Balakrishnan, B., Yin, X., Calderon, F. R. O., Mao, R., Pasquali, M., et al. (2017). Functional and molecular studies in primary carnitine deficiency. Hum. Mutat. 38, 1684–1699. doi: 10.1002/humu.23315
Fujii, Y., Matsumura, H., Yamazaki, S., Shirasu, A., Nakakura, H., Ogihara, T., et al. (2020). Efficacy of a mitochondrion-targeting agent for reducing the level of urinary protein in rats with puromycin aminonucleoside-induced minimal-change nephrotic syndrome. PLoS One 15:e0227414. doi: 10.1371/journal.pone.0227414
Gai, Z., Wang, T., Visentin, M., Kullak-Ublick, G. A., Fu, X., and Wang, Z. (2019). Lipid accumulation and chronic kidney disease. Nutrients 11:722. doi: 10.3390/nu11040722
Galvan, D. L., Green, N. H., and Danesh, F. R. (2017). The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 92, 1051–1057. doi: 10.1016/j.kint.2017.05.034
Ganji, S. H., Tavintharan, S., Zhu, D., Xing, Y., Kamanna, V. S., and Kashyap, M. L. (2004). Niacin noncompetitively inhibits DGAT2 but not DGAT1 activity in HepG2 cells. J. Lipid Res. 45, 1835–1845. doi: 10.1194/jlr.M300403-JLR200
Ge, M., Fontanesi, F., Merscher, S., and Fornoni, A. (2020). The vicious cycle of renal lipotoxicity and mitochondrial dysfunction. Front. Physiol. 11:732. doi: 10.3389/fphys.2020.00732
Georgiou, D. K., Dagnino-Acosta, A., Lee, C. S., Griffin, D. M., Wang, H., Lagor, W. R., et al. (2015). Ca2+ binding/permeation via calcium channel, CaV1.1, regulates the intracellular distribution of the fatty acid transport protein, CD36, and fatty acid metabolism. J. Biol. Chem. 290, 23751–23765. doi: 10.1074/jbc.M115.643544
Glatz, J. F., Nabben, M., Heather, L. C., Bonen, A., and Luiken, J. J. (2016). Regulation of the subcellular trafficking of CD36, a major determinant of cardiac fatty acid utilization. Biochim. Biophys. Acta 1861, 1461–1471. doi: 10.1016/j.bbalip.2016.04.008
Hager, M. R., Narla, A. D., and Tannock, L. R. (2017). Dyslipidemia in patients with chronic kidney disease. Rev. Endocr. Metab. Disord. 18, 29–40. doi: 10.1007/s11154-016-9402-z
Haunerland, N. H., and Spener, F. (2004). Fatty acid-binding proteins—insights from genetic manipulations. Prog. Lipid Res. 43, 328–349. doi: 10.1016/j.plipres.2004.05.001
Heine, G. H., Eller, K., Stadler, J. T., Rogacev, K. S., and Marsche, G. (2020). Lipid-modifying therapy in chronic kidney disease: pathophysiological and clinical considerations. Pharmacol. Ther. 207:107459. doi: 10.1016/j.pharmthera.2019.107459
Herman-Edelstein, M., Scherzer, P., Tobar, A., Levi, M., and Gafter, U. (2014). Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 55, 561–572. doi: 10.1194/jlr.P040501
Hershberger, K. A., Martin, A. S., and Hirschey, M. D. (2017). Role of NAD(+) and mitochondrial sirtuins in cardiac and renal diseases. Nat. Rev. Nephrol. 13, 213–225. doi: 10.1038/nrneph.2017.5
Hoenig, M. P., and Zeidel, M. L. (2014). Homeostasis, the milieu interieur, and the wisdom of the nephron. Clin. J. Am. Soc. Nephrol. 9, 1272–1281. doi: 10.2215/cjn.08860813
Houten, S. M., Violante, S., Ventura, F. V., and Wanders, R. J. (2016). The biochemistry and physiology of mitochondrial fatty acid beta-oxidation and its genetic disorders. Annu. Rev. Physiol. 78, 23–44. doi: 10.1146/annurev-physiol-021115-105045
Hua, W., Huang, H. Z., Tan, L. T., Wan, J. M., Gui, H. B., Zhao, L., et al. (2015). CD36 mediated fatty acid-induced podocyte apoptosis via oxidative stress. PLoS One 10:e0127507. doi: 10.1371/journal.pone.0127507
Hughes-Large, J. M., Pang, D. K., Robson, D. L., Chan, P., Toma, J., and Borradaile, N. M. (2014). Niacin receptor activation improves human microvascular endothelial cell angiogenic function during lipotoxicity. Atherosclerosis 237, 696–704. doi: 10.1016/j.atherosclerosis.2014.10.090
Islam, R. M., Pourmousa, M., Sviridov, D., Gordon, S. M., Neufeld, E. B., Freeman, L. A., et al. (2018). Structural properties of apolipoprotein A-I mimetic peptides that promote ABCA1-dependent cholesterol efflux. Sci. Rep. 8:2956. doi: 10.1038/s41598-018-20965-2
Izquierdo-Lahuerta, A., Martínez-García, C., and Medina-Gómez, G. (2016). Lipotoxicity as a trigger factor of renal disease. J. Nephrol. 29, 603–610. doi: 10.1007/s40620-016-0278-5
Jager, S., Handschin, C., St-Pierre, J., and Spiegelman, B. M. (2007). AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. U. S. A. 104, 12017–12022. doi: 10.1073/pnas.0705070104
Jang, H. S., Noh, M. R., Jung, E. M., Kim, W. Y., Southekal, S., Guda, C., et al. (2020a). Proximal tubule cyclophilin D regulates fatty acid oxidation in cisplatin-induced acute kidney injury. Kidney Int. 97, 327–339. doi: 10.1016/j.kint.2019.08.019
Jang, H. S., Noh, M. R., Kim, J., and Padanilam, B. J. (2020b). Defective mitochondrial fatty acid oxidation and lipotoxicity in kidney diseases. Front. Med. (Lausanne) 7:65. doi: 10.3389/fmed.2020.00065
Johnson, A. C., Stahl, A., and Zager, R. A. (2005). Triglyceride accumulation in injured renal tubular cells: alterations in both synthetic and catabolic pathways. Kidney Int. 67, 2196–2209. doi: 10.1111/j.1523-1755.2005.00325.x
Kang, H. M., Ahn, S. H., Choi, P., Ko, Y. A., Han, S. H., Chinga, F., et al. (2015). Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 21, 37–46. doi: 10.1038/nm.3762
Karwi, Q. G., Jorg, A. R., and Lopaschuk, G. D. (2019). Allosteric, transcriptional and post-translational control of mitochondrial energy metabolism. Biochem. J. 476, 1695–1712. doi: 10.1042/BCJ20180617
Karwi, Q. G., Uddin, G. M., Ho, K. L., and Lopaschuk, G. D. (2018). Loss of metabolic flexibility in the failing heart. Front. Cardiovasc. Med. 5:68. doi: 10.3389/fcvm.2018.00068
Katsyuba, E., Mottis, A., Zietak, M., De Franco, F., van der Velpen, V., Gariani, K., et al. (2018). De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature 563, 354–359. doi: 10.1038/s41586-018-0645-6
Kelso, G. F., Porteous, C. M., Coulter, C. V., Hughes, G., Porteous, W. K., Ledgerwood, E. C., et al. (2001). Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J. Biol. Chem. 276, 4588–4596. doi: 10.1074/jbc.M009093200
Kennedy, D. J., Chen, Y., Huang, W., Viterna, J., Liu, J., Westfall, K., et al. (2013). CD36 and Na/K-ATPase-alpha1 form a proinflammatory signaling loop in kidney. Hypertension 61, 216–224. doi: 10.1161/HYPERTENSIONAHA.112.198770
Kezic, A., Spasojevic, I., Lezaic, V., and Bajcetic, M. (2016). Mitochondria-targeted antioxidants: future perspectives in kidney ischemia reperfusion injury. Oxid. Med. Cell. Longev. 2016:2950503. doi: 10.1155/2016/2950503
Khan, S., Cabral, P. D., Schilling, W. P., Schmidt, Z. W., Uddin, A. N., Gingras, A., et al. (2018). Kidney proximal tubule lipoapoptosis is regulated by fatty acid transporter-2 (FATP2). J. Am. Soc. Nephrol. 29, 81–91. doi: 10.1681/asn.2017030314
Kirkland, J. B., and Meyer-Ficca, M. L. (2018). Niacin. Adv. Food Nutr. Res. 83, 83–149. doi: 10.1016/bs.afnr.2017.11.003
Konig, B., Koch, A., Giggel, K., Dordschbal, B., Eder, K., and Stangl, G. I. (2008). Monocarboxylate transporter (MCT)-1 is up-regulated by PPARalpha. Biochim. Biophys. Acta 1780, 899–904. doi: 10.1016/j.bbagen.2008.03.002
Koppen, L. M., Whitaker, A., Rosene, A., and Beckett, R. D. (2017). Efficacy of berberine alone and in combination for the treatment of hyperlipidemia: a systematic review. J. Evid. Based Complementary Altern. Med. 22, 956–968. doi: 10.1177/2156587216687695
Kronenberg, F. (2018). HDL in CKD-the devil is in the detail. J. Am. Soc. Nephrol. 29, 1356–1371. doi: 10.1681/ASN.2017070798
Kruger, C., Nguyen, T. T., Breaux, C., Guillory, A., Mangelli, M., Fridianto, K. T., et al. (2019). Proximal tubular cell-specific ablation of carnitine acetyltransferase causes tubular disease and secondary glomerulosclerosis. Diabetes 68, 819–831. doi: 10.2337/db18-0090
Lakhia, R., Yheskel, M., Flaten, A., Quittner-Strom, E. B., Holland, W. L., and Patel, V. (2018). PPARalpha agonist fenofibrate enhances fatty acid beta-oxidation and attenuates polycystic kidney and liver disease in mice. Am. J. Physiol. Ren. Physiol. 314, F122–F131. doi: 10.1152/ajprenal.00352.2017
Lan, R., Geng, H., Singha, P. K., Saikumar, P., Bottinger, E. P., Weinberg, J. M., et al. (2016). Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J. Am. Soc. Nephrol. 27, 3356–3367. doi: 10.1681/ASN.2015020177
Lee, M., Katerelos, M., Gleich, K., Galic, S., Kemp, B. E., Mount, P. F., et al. (2018). Phosphorylation of acetyl-CoA carboxylase by AMPK reduces renal fibrosis and is essential for the anti-fibrotic effect of metformin. J. Am. Soc. Nephrol. 29, 2326–2336. doi: 10.1681/ASN.2018010050
Li, S. Y., and Susztak, K. (2018). The role of peroxisome proliferator-activated receptor gamma coactivator 1alpha (PGC-1alpha) in kidney disease. Semin. Nephrol. 38, 121–126. doi: 10.1016/j.semnephrol.2018.01.003
Li, X., Zhang, T., Geng, J., Wu, Z., Xu, L., Liu, J., et al. (2019). Advanced oxidation protein products promote lipotoxicity and tubulointerstitial fibrosis via CD36/beta-catenin pathway in diabetic nephropathy. Antioxid. Redox Signal. 31, 521–538. doi: 10.1089/ars.2018.7634
Liu, Y., Wang, Y., Ding, W., and Wang, Y. (2018). Mito-TEMPO alleviates renal fibrosis by reducing inflammation, mitochondrial dysfunction, and endoplasmic reticulum stress. Oxid. Med. Cell. Longev. 2018:5828120. doi: 10.1155/2018/5828120
Luiken, J. J., Chanda, D., Nabben, M., Neumann, D., and Glatz, J. F. (2016). Post-translational modifications of CD36 (SR-B2): implications for regulation of myocellular fatty acid uptake. Biochim. Biophys. Acta 1862, 2253–2258. doi: 10.1016/j.bbadis.2016.09.004
Lv, T., Hu, Y., Ma, Y., Zhen, J., Xin, W., and Wan, Q. (2019). GCN5L1 controls renal lipotoxicity through regulating acetylation of fatty acid oxidation enzymes. J. Physiol. Biochem. 75, 597–606. doi: 10.1007/s13105-019-00711-6
Maatman, R. G., Van Kuppevelt, T. H., and Veerkamp, J. H. (1991). Two types of fatty acid-binding protein in human kidney. Isolation, characterization and localization. Biochem. J. 273, 759–766. doi: 10.1042/bj2730759
Merscher, S., Pedigo, C. E., and Mendez, A. J. (2014). Metabolism, energetics, and lipid biology in the podocyte—cellular cholesterol-mediated glomerular injury. Front. Endocrinol. (Lausanne) 5:169. doi: 10.3389/fendo.2014.00169
Meyer, C., Nadkarni, V., Stumvoll, M., and Gerich, J. (1997). Human kidney free fatty acid and glucose uptake: evidence for a renal glucose-fatty acid cycle. Am. J. Phys. 273, E650–E654. doi: 10.1152/ajpendo.1997.273.3.E650
Møller, J. C., and Skriver, E. (1985). Quantitative ultrastructure of human proximal tubules and cortical interstitium in chronic renal disease (hydronephrosis). Virchows Arch. A Pathol. Anat. Histopathol. 406, 389–406. doi: 10.1007/BF00710231
Moorhead, J. F., Chan, M. K., El-Nahas, M., and Varghese, Z. (1982). Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease. Lancet 2, 1309–1311. doi: 10.1016/s0140-6736(82)91513-6
Morigi, M., Perico, L., and Benigni, A. (2018). Sirtuins in renal health and disease. J. Am. Soc. Nephrol. 29, 1799–1809. doi: 10.1681/ASN.2017111218
Nishi, H., Higashihara, T., and Inagi, R. (2019). Lipotoxicity in kidney, heart, and skeletal muscle dysfunction. Nutrients 11:1664. doi: 10.3390/nu11071664
Nishi, H., and Nangaku, M. (2019). Podocyte lipotoxicity in diabetic kidney disease. Kidney Int. 96, 809–812. doi: 10.1016/j.kint.2019.07.013
Nosadini, R., and Tonolo, G. (2011). Role of oxidized low density lipoproteins and free fatty acids in the pathogenesis of glomerulopathy and tubulointerstitial lesions in type 2 diabetes. Nutr. Metab. Cardiovasc. Dis. 21, 79–85. doi: 10.1016/j.numecd.2010.10.002
Nsiah-Sefaa, A., and McKenzie, M. (2016). Combined defects in oxidative phosphorylation and fatty acid β-oxidation in mitochondrial disease. Biosci. Rep. 36:e00313. doi: 10.1042/BSR20150295
O’Brien, E. S., Nucci, N. V., Fuglestad, B., Tommos, C., and Wand, A. J. (2015). Defining the apoptotic trigger: the interaction of cytochrome C and cardiolipin. J. Biol. Chem. 290, 30879–30887. doi: 10.1074/jbc.M115.689406
Okamura, D. M., Lopez-Guisa, J. M., Koelsch, K., Collins, S., and Eddy, A. A. (2007). Atherogenic scavenger receptor modulation in the tubulointerstitium in response to chronic renal injury. Am. J. Physiol. Ren. Physiol. 293, F575–F585. doi: 10.1152/ajprenal.00063.2007
Okamura, D. M., Pennathur, S., Pasichnyk, K., Lopez-Guisa, J. M., Collins, S., Febbraio, M., et al. (2009). CD36 regulates oxidative stress and inflammation in hypercholesterolemic CKD. J. Am. Soc. Nephrol. 20, 495–505. doi: 10.1681/ASN.2008010009
Opazo-Ríos, L., Mas, S., Marín-Royo, G., Mezzano, S., Gómez-Guerrero, C., Moreno, J. A., et al. (2020). Lipotoxicity and diabetic nephropathy: novel mechanistic insights and therapeutic opportunities. Int. J. Mol. Sci. 21:2632. doi: 10.3390/ijms21072632
Ozaltin, F. (2014). Primary coenzyme Q10 (CoQ 10) deficiencies and related nephropathies. Pediatr. Nephrol. 29, 961–969. doi: 10.1007/s00467-013-2482-z
Patil, N. K., Parajuli, N., MacMillan-Crow, L. A., and Mayeux, P. R. (2014). Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: mitochondria-targeted antioxidant mitigates injury. Am. J. Physiol. Ren. Physiol. 306, F734–F743. doi: 10.1152/ajprenal.00643.2013
Pelsers, M. M. (2008). Fatty acid-binding protein as marker for renal injury. Scand. J. Clin. Lab. Investig. Suppl. 241, 73–77. doi: 10.1080/00365510802150133
Pennathur, S., Pasichnyk, K., Bahrami, N. M., Zeng, L., Febbraio, M., Yamaguchi, I., et al. (2015). The macrophage phagocytic receptor CD36 promotes fibrogenic pathways on removal of apoptotic cells during chronic kidney injury. Am. J. Pathol. 185, 2232–2245. doi: 10.1016/j.ajpath.2015.04.016
Pietrocola, F., Galluzzi, L., Bravo-San Pedro, J. M., Madeo, F., and Kroemer, G. (2015). Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 21, 805–821. doi: 10.1016/j.cmet.2015.05.014
Portilla, D. (2003). Energy metabolism and cytotoxicity. Semin. Nephrol. 23, 432–438. doi: 10.1016/S0270-9295(03)00088-3
Poyan Mehr, A., Tran, M. T., Ralto, K. M., Leaf, D. E., Washco, V., Messmer, J., et al. (2018). De novo NAD(+) biosynthetic impairment in acute kidney injury in humans. Nat. Med. 24, 1351–1359. doi: 10.1038/s41591-018-0138-z
Qin, X., Jiang, M., Zhao, Y., Gong, J., Su, H., Yuan, F., et al. (2019a). Berberine protects against diabetic kidney disease via promoting PGC-1α-regulated mitochondrial energy homeostasis. Br. J. Pharmacol. 177, 3646–3661. doi: 10.1111/bph.14935
Qin, X., Zhao, Y., Gong, J., Huang, W., Su, H., Yuan, F., et al. (2019b). Berberine protects glomerular podocytes via inhibiting Drp1-mediated mitochondrial fission and dysfunction. Theranostics 9, 1698–1713. doi: 10.7150/thno.30640
Rademann, P., Weidinger, A., Drechsler, S., Meszaros, A., Zipperle, J., Jafarmadar, M., et al. (2017). Mitochondria-targeted antioxidants SkQ1 and MitoTEMPO failed to exert a long-term beneficial effect in murine polymicrobial sepsis. Oxid. Med. Cell. Longev. 2017:6412682. doi: 10.1155/2017/6412682
Rahman, E. U., Ruan, X. Z., Chana, R. S., Brunskill, N. J., Gaya, J., Powis, S. H., et al. (2008). Mesangial matrix-activated monocytes express functional scavenger receptors and accumulate intracellular lipid. Nephrol. Dial. Transplant. 23, 1876–1885. doi: 10.1093/ndt/gfm901
Ralto, K. M., Rhee, E. P., and Parikh, S. M. (2020). NAD(+) homeostasis in renal health and disease. Nat. Rev. Nephrol. 16, 99–111. doi: 10.1038/s41581-019-0216-6
Romani, M., Hofer, D. C., Katsyuba, E., and Auwerx, J. (2019). Niacin: an old lipid drug in a new NAD(+) dress. J. Lipid Res. 60, 741–746. doi: 10.1194/jlr.S092007
Rosenson, R. S., Hegele, R. A., and Koenig, W. (2019). Cholesterol-lowering agents. Circ. Res. 124, 364–385. doi: 10.1161/CIRCRESAHA.118.313238
Rossman, M. J., Santos-Parker, J. R., Steward, C. A. C., Bispham, N. Z., Cuevas, L. M., Rosenberg, H. L., et al. (2018). Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension 71, 1056–1063. doi: 10.1161/HYPERTENSIONAHA.117.10787
Ruan, X. Z., Varghese, Z., Powis, S. H., and Moorhead, J. F. (1999). Human mesangial cells express inducible macrophage scavenger receptor. Kidney Int. 56, 440–451. doi: 10.1046/j.1523-1755.1999.00587.x
Ruggiero, C., Elks, C. M., Kruger, C., Cleland, E., Addison, K., Noland, R. C., et al. (2014). Albumin-bound fatty acids but not albumin itself alter redox balance in tubular epithelial cells and induce a peroxide-mediated redox-sensitive apoptosis. Am. J. Physiol. Ren. Physiol. 306, F896–F906. doi: 10.1152/ajprenal.00484.2013
Rysz, J., Gluba-Brzozka, A., Rysz-Gorzynska, M., and Franczyk, B. (2020). The role and function of HDL in patients with chronic kidney disease and the risk of cardiovascular disease. Int. J. Mol. Sci. 21:601. doi: 10.3390/ijms21020601
Saad, A., Herrmann, S. M. S., Eirin, A., Ferguson, C. M., Glockner, J. F., Bjarnason, H., et al. (2017). Phase 2a clinical trial of mitochondrial protection (elamipretide) during stent revascularization in patients with atherosclerotic renal artery stenosis. Circ. Cardiovasc. Interv. 10:e005487. doi: 10.1161/CIRCINTERVENTIONS.117.005487
Sasaki, M., Yasuno, T., Ito, K., Matsunaga, A., Hisano, S., Abe, Y., et al. (2018). Focal segmental glomerulosclerosis with heterozygous apolipoprotein E5 (Glu3Lys). CEN Case Rep. 7, 225–228. doi: 10.1007/s13730-018-0331-4
Scerbo, D., Son, N. H., Sirwi, A., Zeng, L., Sas, K. M., Cifarelli, V., et al. (2017). Kidney triglyceride accumulation in the fasted mouse is dependent upon serum free fatty acids. J. Lipid Res. 58, 1132–1142. doi: 10.1194/jlr.M074427
Seok, S., Kim, Y. C., Byun, S., Choi, S., Xiao, Z., Iwamori, N., et al. (2018). Fasting-induced JMJD3 histone demethylase epigenetically activates mitochondrial fatty acid beta-oxidation. J. Clin. Invest. 128, 3144–3159. doi: 10.1172/JCI97736
Shepherd, J., Packard, C. J., Patsch, J. R., Gotto, A. M. Jr., and Taunton, O. D. (1979). Effects of nicotinic acid therapy on plasma high density lipoprotein subfraction distribution and composition and on apolipoprotein a metabolism. J. Clin. Invest. 63, 858–867. doi: 10.1172/JCI109385
Shi, L., and Tu, B. P. (2015). Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr. Opin. Cell Biol. 33, 125–131. doi: 10.1016/j.ceb.2015.02.003
Sieber, J., and Jehle, A. W. (2014). Free fatty acids and their metabolism affect function and survival of podocytes. Front. Endocrinol. (Lausanne) 5:186. doi: 10.3389/fendo.2014.00186
Simon, N., and Hertig, A. (2015). Alteration of fatty acid oxidation in tubular epithelial cells: from acute kidney injury to renal fibrogenesis. Front. Med. (Lausanne) 2:52. doi: 10.3389/fmed.2015.00052
Smith, B. K., Jain, S. S., Rimbaud, S., Dam, A., Quadrilatero, J., Ventura-Clapier, R., et al. (2011). FAT/CD36 is located on the outer mitochondrial membrane, upstream of long-chain acyl-CoA synthetase, and regulates palmitate oxidation. Biochem. J. 437, 125–134. doi: 10.1042/BJ20101861
Souza, A. C., Bocharov, A. V., Baranova, I. N., Vishnyakova, T. G., Huang, Y. G., Wilkins, K. J., et al. (2016). Antagonism of scavenger receptor CD36 by 5A peptide prevents chronic kidney disease progression in mice independent of blood pressure regulation. Kidney Int. 89, 809–822. doi: 10.1016/j.kint.2015.12.043
Stadler, K., Goldberg, I. J., and Susztak, K. (2015). The evolving understanding of the contribution of lipid metabolism to diabetic kidney disease. Curr. Diab. Rep. 15:40. doi: 10.1007/s11892-015-0611-8
Stallons, L. J., Funk, J. A., and Schnellmann, R. G. (2013). Mitochondrial homeostasis in acute organ failure. Curr. Pathobiol. Rep. 1, 169–177. doi: 10.1007/s40139-013-0023-x
Su, X., and Abumrad, N. A. (2009). Cellular fatty acid uptake: a pathway under construction. Trends Endocrinol. Metab. 20, 72–77. doi: 10.1016/j.tem.2008.11.001
Su, H., Wan, C., Lei, C. T., Zhang, C. Y., Ye, C., Tang, H., et al. (2017). Lipid deposition in kidney diseases: interplay among redox, lipid mediators, and renal impairment. Antioxid. Redox Signal. 28, 1027–1043. doi: 10.1089/ars.2017.7066
Sudhakaran, S., Bottiglieri, T., Tecson, K. M., Kluger, A. Y., and McCullough, P. A. (2018). Alteration of lipid metabolism in chronic kidney disease, the role of novel antihyperlipidemic agents, and future directions. Rev. Cardiovasc. Med. 19, 77–88. doi: 10.31083/j.rcm.2018.03.908
Sun, J., Chen, X., Liu, T., Jiang, X., Wu, Y., Yang, S., et al. (2018). Berberine protects against palmitate-induced apoptosis in tubular epithelial cells by promoting fatty acid oxidation. Med. Sci. Monit. 24, 1484–1492. doi: 10.12659/MSM.908927
Sun, J., Zhu, H., Wang, X., Gao, Q., Li, Z., and Huang, H. (2019). CoQ10 ameliorates mitochondrial dysfunction in diabetic nephropathy through mitophagy. J. Endocrinol. doi: 10.1530/JOE-18-0578 [Epub ahead of print]
Susztak, K., Ciccone, E., McCue, P., Sharma, K., and Bottinger, E. P. (2005). Multiple metabolic hits converge on CD36 as novel mediator of tubular epithelial apoptosis in diabetic nephropathy. PLoS Med. 2:e45. doi: 10.1371/journal.pmed.0020045
Szeto, H. H. (2017). Pharmacologic approaches to improve mitochondrial function in AKI and CKD. J. Am. Soc. Nephrol. 28, 2856–2865. doi: 10.1681/ASN.2017030247
Szeto, H. H., Liu, S., Soong, Y., Alam, N., Prusky, G. T., and Seshan, S. V. (2016). Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int. 90, 997–1011. doi: 10.1016/j.kint.2016.06.013
Szeto, H. H., Liu, S., Soong, Y., Seshan, S. V., Cohen-Gould, L., Manichev, V., et al. (2017). Mitochondria protection after acute ischemia prevents prolonged upregulation of IL-1β and IL-18 and arrests CKD. J. Am. Soc. Nephrol. 28, 1437–1449. doi: 10.1681/ASN.2016070761
Thapa, D., Wu, K., Stoner, M. W., Xie, B., Zhang, M., Manning, J. R., et al. (2018). The protein acetylase GCN5L1 modulates hepatic fatty acid oxidation activity via acetylation of the mitochondrial beta-oxidation enzyme HADHA. J. Biol. Chem. 293, 17676–17684. doi: 10.1074/jbc.AC118.005462
Thomas, S. R., Neuzil, J., and Stocker, R. (1996). Cosupplementation with coenzyme Q prevents the prooxidant effect of alpha-tocopherol and increases the resistance of LDL to transition metal-dependent oxidation initiation. Arterioscler. Thromb. Vasc. Biol. 16, 687–696. doi: 10.1161/01.ATV.16.5.687
Thongnak, L., Pongchaidecha, A., and Lungkaphin, A. (2020). Renal lipid metabolism and lipotoxicity in diabetes. Am. J. Med. Sci. 359, 84–99. doi: 10.1016/j.amjms.2019.11.004
Tran, M. T., Zsengeller, Z. K., Berg, A. H., Khankin, E. V., Bhasin, M. K., Kim, W., et al. (2016). PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531, 528–532. doi: 10.1038/nature17184
Trnka, J., Blaikie, F. H., Smith, R. A., and Murphy, M. P. (2008). A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radic. Biol. Med. 44, 1406–1419. doi: 10.1016/j.freeradbiomed.2007.12.036
Vamecq, J., Dessein, A. F., Fontaine, M., Briand, G., Porchet, N., Latruffe, N., et al. (2012). Mitochondrial dysfunction and lipid homeostasis. Curr. Drug Metab. 13, 1388–1400. doi: 10.2174/138920012803762792
Vaziri, N. D. (2006). Dyslipidemia of chronic renal failure: the nature, mechanisms, and potential consequences. Am. J. Physiol. Ren. Physiol. 290, F262–F272. doi: 10.1152/ajprenal.00099.2005
Vaziri, N. D. (2016). Disorders of lipid metabolism in nephrotic syndrome: mechanisms and consequences. Kidney Int. 90, 41–52. doi: 10.1016/j.kint.2016.02.026
Verdin, E. (2015). NAD(+) in aging, metabolism, and neurodegeneration. Science 350, 1208–1213. doi: 10.1126/science.aac4854
Violante, S., Achetib, N., van Roermund, C. W. T., Hagen, J., Dodatko, T., Vaz, F. M., et al. (2019). Peroxisomes can oxidize medium‐ and long-chain fatty acids through a pathway involving ABCD3 and HSD17B4. FASEB J. 33, 4355–4364. doi: 10.1096/fj.201801498R
Wakino, S., Hasegawa, K., and Itoh, H. (2015). Sirtuin and metabolic kidney disease. Kidney Int. 88, 691–698. doi: 10.1038/ki.2015.157
Walther, T. C., Chung, J., and Farese, R. V. Jr. (2017). Lipid droplet biogenesis. Annu. Rev. Cell Dev. Biol. 33, 491–510. doi: 10.1146/annurev-cellbio-100616-060608
Wang, J., Hao, J. W., Wang, X., Guo, H., Sun, H. H., Lai, X. Y., et al. (2019). DHHC4 and DHHC5 facilitate fatty acid uptake by palmitoylating and targeting CD36 to the plasma membrane. Cell Rep. 26, 209.e5–221.e5. doi: 10.1016/j.celrep.2018.12.022
Wang, Z., Heshka, S., Gallagher, D., Boozer, C. N., Kotler, D. P., and Heymsfield, S. B. (2000). Resting energy expenditure-fat-free mass relationship: new insights provided by body composition modeling. Am. J. Physiol. Endocrinol. Metab. 279, E539–E545. doi: 10.1152/ajpendo.2000.279.3.E539
Wang, J., and Li, Y. (2019). CD36 tango in cancer: signaling pathways and functions. Theranostics 9, 4893–4908. doi: 10.7150/thno.36037
Wang, Y., Yi, X., Ghanam, K., Zhang, S., Zhao, T., and Zhu, X. (2014). Berberine decreases cholesterol levels in rats through multiple mechanisms, including inhibition of cholesterol absorption. Metabolism 63, 1167–1177. doi: 10.1016/j.metabol.2014.05.013
Ward, M. S., Flemming, N. B., Gallo, L. A., Fotheringham, A. K., McCarthy, D. A., Zhuang, A., et al. (2017). Targeted mitochondrial therapy using MitoQ shows equivalent renoprotection to angiotensin converting enzyme inhibition but no combined synergy in diabetes. Sci. Rep. 7:15190. doi: 10.1038/s41598-017-15589-x
Weinberg, J. M. (2011). Mitochondrial biogenesis in kidney disease. J. Am. Soc. Nephrol. 22, 431–436. doi: 10.1681/ASN.2010060643
Widmeier, E., Yu, S., Nag, A., Chung, Y. W., Nakayama, M., Fernández-Del-Río, L., et al. (2020). ADCK4 deficiency destabilizes the coenzyme Q complex, which is rescued by 2,4-Dihydroxybenzoic acid treatment. J. Am. Soc. Nephrol. 31, 1191–1211. doi: 10.1681/ASN.2019070756
Wu, J., Chen, L., Zhang, D., Huo, M., Zhang, X., Pu, D., et al. (2009). Peroxisome proliferator-activated receptors and renal diseases. Front. Biosci. (Landmark Ed.) 14, 995–1009. doi: 10.2741/3291
Yamamoto, T., Noiri, E., Ono, Y., Doi, K., Negishi, K., Kamijo, A., et al. (2007). Renal L-type fatty acid—binding protein in acute ischemic injury. J. Am. Soc. Nephrol. 18, 2894–2902. doi: 10.1681/ASN.2007010097
Yamamoto, T., Takabatake, Y., Minami, S., Sakai, S., Fujimura, R., Takahashi, A., et al. (2020). Eicosapentaenoic acid attenuates renal lipotoxicity by restoring autophagic flux. Autophagy, 1–14. doi: 10.1080/15548627.2020.1782034
Yamamoto, T., Takabatake, Y., Takahashi, A., Kimura, T., Namba, T., Matsuda, J., et al. (2017). High-fat diet-induced lysosomal dysfunction and impaired autophagic flux contribute to lipotoxicity in the kidney. J. Am. Soc. Nephrol. 28, 1534–1551. doi: 10.1681/ASN.2016070731
Yamamoto, S., Yancey, P. G., Ikizler, T. A., Jerome, W. G., Kaseda, R., Cox, B., et al. (2012). Dysfunctional high-density lipoprotein in patients on chronic hemodialysis. J. Am. Coll. Cardiol. 60, 2372–2379. doi: 10.1016/j.jacc.2012.09.013
Yamashita, S., Masuda, D., and Matsuzawa, Y. (2020). Pemafibrate, a new selective PPARalpha modulator: drug concept and its clinical applications for dyslipidemia and metabolic diseases. Curr. Atheroscler. Rep. 22:5. doi: 10.1007/s11883-020-0823-5
Yang, X., Okamura, D. M., Lu, X., Chen, Y., Moorhead, J., Varghese, Z., et al. (2017). CD36 in chronic kidney disease: novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 13, 769–781. doi: 10.1038/nrneph.2017.126
Yu, A. S. L., Chertow, G. M., Luyckx, V., Marsden, P. A., Skorecki, K., and Taal, M. W. (2019). Brenner and Rector’s the kidney. 11th Edn. Vol. 2. Philadelphia: Elsevier.
Zager, R. A., Johnson, A. C., and Becker, K. (2011). Acute unilateral ischemic renal injury induces progressive renal inflammation, lipid accumulation, histone modification, and “end-stage” kidney disease. Am. J. Physiol. Ren. Physiol. 301, F1334–F1345. doi: 10.1152/ajprenal.00431.2011
Zhang, L. H., Kamanna, V. S., Zhang, M. C., and Kashyap, M. L. (2008). Niacin inhibits surface expression of ATP synthase beta chain in HepG2 cells: implications for raising HDL. J. Lipid Res. 49, 1195–1201. doi: 10.1194/jlr.M700426-JLR200
Zhao, H. L., Sui, Y., Guan, J., He, L., Zhu, X., Fan, R. R., et al. (2008). Fat redistribution and adipocyte transformation in uninephrectomized rats. Kidney Int. 74, 467–477. doi: 10.1038/ki.2008.195
Keywords: fatty acid β-oxidation, energy metabolism, lipotoxicity, podocyte, proximal tubule cells, oxidative stress, homeostasis, fibrosis
Citation: Lin P-H and Duann P (2020) Dyslipidemia in Kidney Disorders: Perspectives on Mitochondria Homeostasis and Therapeutic Opportunities. Front. Physiol. 11:1050. doi: 10.3389/fphys.2020.01050
Edited by:
Giuliano Ciarimboli, University of Münster, GermanyReviewed by:
Michele Visentin, University Hospital Zürich, SwitzerlandMelinda Coughlan, Monash University, Australia
Copyright © 2020 Lin and Duann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pei-Hui Lin, cGVpLWh1aS5saW5Ab3N1bWMuZWR1;cGxpbjE5OTlAeWFob28uY29t