 Elie Haddad1
Elie Haddad1 Manfred Hoenig2* on behalf of the EBMT-IEWP
Manfred Hoenig2* on behalf of the EBMT-IEWP- 1CHU Sainte-Justine, Department of Pediatrics, Microbiology, Immunology and Infectious Diseases, University of Montreal, Montreal, QC, Canada
- 2Department of Pediatrics, University Medical Center Ulm, Ulm, Germany
Severe Combined Immunodeficiencies (SCID) are a heterogeneous group of monogenetic diseases. We describe the typical clinical presentation of patients with SCID as well as basic principles in diagnosis and therapy by hematopoietic stem cell transplantation. Therapeutic strategies may differ between subtypes and the inherent reduced capacity or inablility to reject a graft have to be considered.
Introduction
SCID is defined by a severely impaired T-lymphocyte development responsible for the absence of adaptive immune response and profound T-lymphocytopenia. SCID diagnostic criteria proposed by the Primary immune Deficiency Treatment Consortium (PIDTC) help to distinguish typical SCID from leaky SCID and Omenn syndrome (Table 1) (1). There is a cohort of patients who do not fulfill the criteria given in Table 1 but can present with identical clinical features and have the same need for a rapid diagnosis and therapy. It is a matter of ongoing debate but not subject of this review whether these entities can be called “SCID.” As there is no other published definition for SCID this review will keep to the definition proposed by the PIDTC.
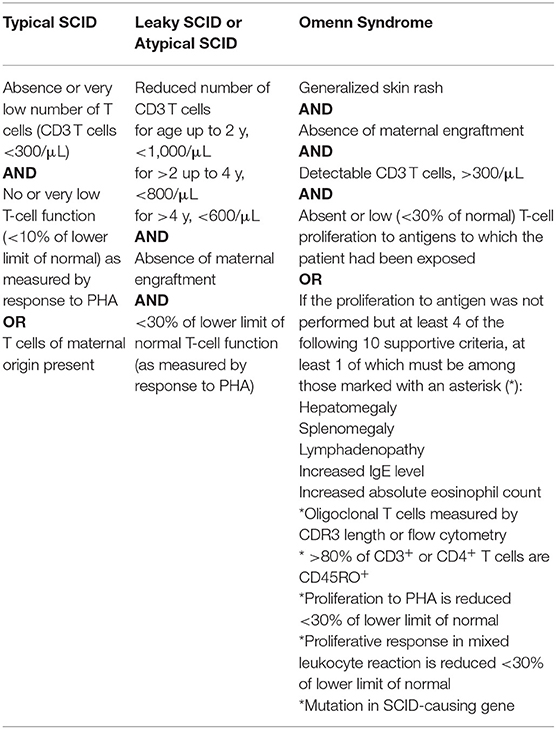
Table 1. SCID definition according to the primary immune deficiency treatment consortium published by Shearer et al. (1).
Genetic Heterogeneity
SCID is a heterogeneous genetic disorder (see Table 2 for genes list). In the most recent prospective cohort of SCID in North America, only 7% of SCID patients don't show any mutation of known SCID-causing genes (3).
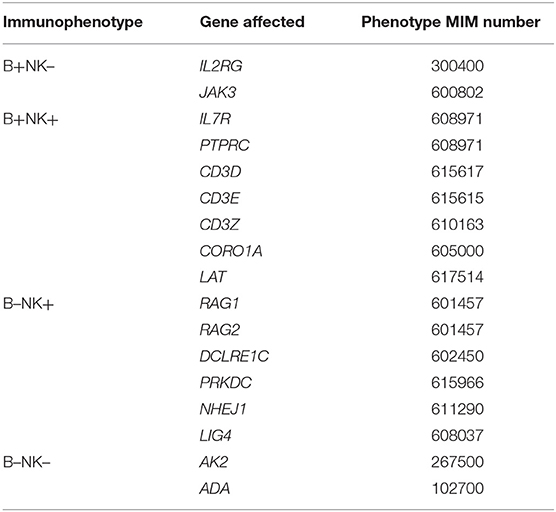
Table 2. Genetic SCID entities according to the IUIS classification (2).
Immunophenotype
SCID entities are also classified by immunophenotype, depending on the presence of B-lymphocytes and NK cells, which are usually associated with specific gene defects (genotypes) (Table 2). This immunophenotypic categorization does not replace molecular identification, which is key for prognosis (4).
Clinical Presentation
The clinical presentation of patients with SCID is variable. Usually, patients are asymptomatic at birth and present within their first weeks or months of life with infections and/or failure to thrive (FTT) (5). Some patients may also present with signs of immune dysregulation caused by autologous (Omenn syndrome) or allogeneic (maternal GvHD) T cells. Some genetic defects cause non-immunological and disease specific features. In the absence of allogenic hematopoietic stem cell transplantation (HSCT) or gene therapy (GT), patients ultimately die generally within the first year of life. Some patients may also present with leaky-SCID, usually caused by hypomorphic mutations in classical SCID causing-genes, responsible for a less severe phenotype with infections and autoimmunity.
Recurrent and Severe Infections
Patients are classically not infected at birth, but in the absence of diagnosis, they will eventually present with severe, atypical and/or recurrent life threatening infections. Opportunistic infections can suggest a defect of specific cellular immunity but non-specific bacterial infections are also frequently observed in SCID patients (5). Airway infections due to viral (Respiratory Syncytial Virus, Myxovirus, Adenovirus) and opportunistic microorganisms such as Pneumocystis jirovecii (PJP) can cause severe pneumonias requiring mechanical ventilation. Intestinal infections with Rota-, Noro or Adenovirus can lead to severe chronic diarrhea. Cytomegalovirus (CMV) infection can be particularly severe and diffuse with pulmonary, liver, cardiac, intestinal, retinal, and/or central nervous system involvement.
Live vaccines with attenuated microorganisms, such as Bacille Calmette-Guérin (BCG) or Rotavirus, may lead to atypical infections and can have an unwanted diagnostic value. A chronic swelling or ulceration at the BCG injection site or a chronic diarrhea after oral Rotavirus vaccination should lead to further diagnostic steps and the formal exclusion of a T-lymphocyte deficiency.
Specific Features Due to the Affected Gene
Patients with Adenosine-Deaminase (ADA)-deficiency may present with alveolar proteinosis (6) and/or bone abnormalities (cupping of osteochondral junctions) detectable on the chest radiograph. Some patients with DNA repair defects associated with SCID, such as Ligase-4 deficiency or Cernunnos/XLF may present with microcephalia and delayed neurological development.
Signs of Immunodysregulation: Maternal Graft vs. Host Disease (GvHD) and Omenn Syndrome
T-lymphocytes transferred from the mother during pregnancy can proliferate, expand, and cause skin lesions and more rarely liver manifestations such as in GvHD following allogeneic HSCT (7). In most cases however, maternal T-lymphocytes are detected in patient's peripheral blood without any clinical signs. An increased risk for post-transplant acute GvHD has been demonstrated for patients with maternal T cells detectable at diagnosis (8).
In some cases, “hypomorphic mutations” of SCID-causing genes encode for proteins with some residual function, allowing for the development of few oligoclonal autoreactive T-lymphocytes, which proliferate and invade peripheral tissues and may be responsible for a so-called Omenn syndrome (definition see Table 1) with a generalized exanthema, lymphadenopathy, alopecia, hepato-/ splenomegaly, intractable diarrhea, and eosinophilia (9). Omenn syndrome and rarely maternal GvHD require immunosuppressive drugs.
New-Born Screening by Quantification of T-cell Receptor Excision Circles (TREC)
Active infection and age > 3.5 months at HSCT are associated with poor survival after HSCT (4, 10, 11), underlining the importance of new-born screening (NBS) to diagnose patients as early as possible and to prevent infections (12, 13) (see review on Universal newborn screening for SCID). Screening programs have been currently established in all 50 US-states, 4 Canadian Provinces, Israel, Switzerland and Germany, and will be introduced in various European countries within next years.
Basal and Specific Diagnostic Measures
In the absence of NBS, SCID should be suspected in presence of profound lymphopenia (14). However, blood cell count may have not been performed or the lymphopenia may be missed or absent (see below) and unfortunately, SCID diagnosis is then classically suspected on the clinical history of recurrent infections and/or failure to thrive (FTT). Present maternal T-lymphocytes or expansion of autoreactive autologous T-lymphocytes may lead to normal (or rarely increased) lymphocyte counts. Therefore, a normal lymphocyte count should not delay the analysis of lymphocyte subpopulations that is usually key for the diagnosis (1) and for the SCID categorization (see above). A lack of naïve T-lymphocytes (e.g., CD3+CD4+CD45RA+CCR7+) or more specifically of recent thymic emigrant cells (CD4+CD45RA+CD31+) is an important laboratory finding suggestive of thymic dysfunction. Lack of or diminished proliferation to PHA is considered an important diagnostic criterion (1), although it is sometimes difficult to perform when T-lymphocyte counts are very low. A skewed TCR-repertoire is a strong hint toward peripheral oligoclonal expansion of either maternal or autologous autoreactive T-lymphocytes.
Immunoglobulin levels should be interpreted with caution since IgG are of maternal origin during the first months of life, IgA can be physiologically absent, IgM can sometimes be normal, and increased IgE can be observed despite a complete absence of circulating B-lymphocytes and is a typical component of Omenn's syndrome.
Thoracic radiography can also be helpful to the diagnosis by showing, in addition to the absence of thymic shadow, characteristic features of ADA deficiency (15).
It is very important to look for and rule out a DiGeorge syndrome or any other thymic disorder, which can be responsible for a SICD phenotype, because the therapeutic strategy may involve thymic transplantation. The observation of the classical features associated with Del22q11 or CHARGE syndrome (for coloboma, heart defects, choanal atresia, growth retardation, genital, and ear abnormalities) together with a T-B+NK+ phenotype are important diagnostic criteria although it may be sometimes very difficult to distinguish primary thymic disorder from “classical” SCID in the absence of SCID-causing gene identification.
Therapeutic Principles and Options
When SCID is suspected, the patient should be referred to an experienced center to perform the necessary diagnostic procedures (see above) and implement the first therapeutic measures as soon as possible (16). Infections have to be carefully investigated (including invasive measures such as biopsies or bronchoalveolar lavage) to provide specific therapy. In parallel, supportive care with Trimethoprim/sulfamethoxazole as prophylaxis for PJP, Ig replacement therapy, and patient isolation to avoid additional infections should be instituted. Some centers use Fluconazole to prevent candidiasis. Screening for CMV infection should be systematically performed (e.g., by a PCR in blood). Since breastfeeding is the major source of CMV transmission in SCID patients, many centers recommend to stop breastfeeding as soon as SCID diagnosis is suspected and allow it only when the mother is shown negative for CMV serology. There are considerable differences in institutional protocols for supportive care after diagnosis until HSCT for patients with SCID (17) and there is an urgent need for evidence-based strategies to limit patient infection.
HSCT or gene therapy (GT) (when feasible) represent the definitive treatment of SCID. In the absence of such therapy, patients usually die, except ADA-deficient SCID for which enzyme replacement by PEG-ADA may provide sufficient T-lymphocyte function for prolonged survival. Nevertheless, PEG-ADA is best given as a short bridging therapy before definitive treatment by HSCT or GT (18).
HSCT for SCID
Overall survival after HCT was between 65 and 70% in big multicenter retrospective cohorts (4, 11, 19, 20) and between 85 and 90% in more recent prospective cohorts (21, 22). The factor with most impact on survival was the donor category with matched sibling donor (MSD) being better than all other donor categories (4, 11, 19, 20, 23). In a multivariate analysis including 571 patients with non-MSD HSCT, the variables associated with survival were: age and infection at HSCT, genotype, FTT, and race/ethnicity (4). Interestingly, in this study that was the first to assess the impact of genotype, survival in patients with RAG mutations was significantly better than in patients with DCLRE1C mutations (both being T-B-NK+ phenotypes).
The unique aspect of HSCT for SCID is that it can be performed without any conditioning regimen (CR). Nevertheless, the role of CR in HSCT for SCID remains very controversial. The theoretical advantages of the absence of CR are the absence of chemotherapy-induced toxicity and the lower incidence of GvHD, likely due to the absence of stress of recipient epithelial cells. However, in the absence of CR there is little chance for donor myeloid engraftment and only donor T-lymphocyte progenitors will engraft (or mature donor T-lymphocytes from the graft depending on the intensity of T-lymphocyte depletion). On the other hand, CR that contain myeloablative agents usually allow for stem cell engraftment, which is associated with a better T- and B-lymphocyte reconstitution (4, 11, 23–26). No significant difference in survival was observed when no-CR or immunosuppression (IS) only (IS-only) was compared with busulfan-containing reduced intensity (RIC) or myeloablative (MAC) in retrospective cohorts (4, 11, 20). However, RIC/MAC were associated with a better T- and B-lymphocyte reconstitution (4, 11) while no-CR/IS-only was associated with less acute GvHD (4). Most importantly, T- and B-lymphocyte reconstitution have been shown to be associated with a better long term prognosis (4, 11, 25, 27). Patients with absent (RAG1, RAG2, DCLRE1C) or non-functional B-lymphocytes (JAK3, IL2RG) usually need CR in order to have a normal B-lymphocyte function post-HSCT (28–30). In the context of active infection at transplantation, it has been shown that CR was associated with a poor survival (11). In this situation, a 2-step procedure has been proposed: a first HCT without CR and then, once the infection is cleared, a second HCT with CR to allow for stem cell engraftment.
It is logical to propose a CR for SCID patients with RAG and DCLRE1C deficiency because of residual NK cell function and in order to empty thymic and marrow niches that inhibit donor progenitor engraftment (25). However, the context of DCLRE1C deficit and other DNA-repair defects represent a very difficult situation since CR has been associated with severe long-term toxicity (31). For all those patients, a chemotherapy-free myeloablative strategy, such as anti-stem cell monoclonal antibody, are urgently needed (see review on “Conditioning Perspectives for Primary Immunodeficiencies.”
Usually, HSCT with a MSD are performed without CR with a survival >90%. However, in about 25% of cases (29), patients require a long-term Ig replacement therapy because of the absence of donor B-lymphocyte engraftment. Interestingly, more and more centers are using a form of reduced toxicity CR in MSD in order to induce donor B-lymphocyte engraftment to avoid this need for long-term Ig replacement therapy.
Conclusion, Challenges, New Hopes
The first challenge is the assessment of the best CR (product, dosing) for patients diagnosed by NBS and planned to be transplanted at a young age, in order to reduce the chemotherapy-induced toxicity. A prospective randomized study assessing the best dose of Busulfan is presently ongoing in North America (CSIDE study, NCT03619551). There is also a new hope with the ongoing study testing anti-CD117 antibody as a non-toxic myeloablative agent (NCT02963064). Another challenge is the reduction of the rate of infection in patients diagnosed by NBS, which is still too high at 42% (22). There is still no consensus regarding the methods of isolation and infection prophylaxis for these patients, and studies assessing how to best prevent infections prior to HSCT are urgently needed. A further challenge is the lack of prospective and retrospective studies of HSCT for each given SCID genotype, in order to define the best strategy for each genotype. Clear-cut biomarkers to clearly define which patients need a re-transplantation in situation of poor T-lymphocyte reconstitution post-HSCT are also missing. Indeed, although CD4+ and CD4+CD45RA+ cell counts as soon as 6 months post-HSCT were shown to be statistically associated with long-term immune reconstitution and long-term survival (4), the majority of patients with low CD4+ and CD4+CD45RA+ cell counts still survived and displayed a good long-term immune reconstitution, making those criteria insufficient to decide to re-transplant or not for a given patient (26). Furthermore, clinical studies assessing the quality of life and the sequelae of very long-term (>20 years) survivors of HSCT for SCID are missing. The collaboration of North-American and European Consortium should allow to perform the majority of the above mentioned studies which require a sufficient number of patients.
The emergence of techniques for the genetic manipulation of autologous hematopoietic stem cells in the 1990s opened a novel therapeutic window for permanent cure of SCID patients. The major advantage of this strategy is the avoidance of side effects caused by allogeneic immunological reactions such as rejection or GvHD. For more information on this subject see the review “Autologous stem cell-based gene therapy for inherited disorders: state-of-the-art and future prospects.”
Author Contributions
EH and MH have discussed and written the review article and have approved the final version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Shearer WT, Dunn E, Notarangelo LD, Dvorak CC, Puck JM, Logan BR, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the Primary Immune Deficiency Treatment Consortium experience. J Allergy Clin Immunol. (2014) 133:1092–8. doi: 10.1016/j.jaci.2013.09.044
2. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International union of immunological societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
3. Dvorak CC, Haddad E, Buckley RH, Cowan MJ, Logan B, Griffith LM, et al. The genetic landscape of severe combined immunodeficiency in the United States and Canada in the current era (2010-2018). J Allergy Clin Immunol. (2019) 143:405–7. doi: 10.1016/j.jaci.2018.08.027
4. Haddad E, Logan BR, Griffith LM, Buckley RH, Parrott RE, Prockop SE, et al. SCID genotype and 6-month posttransplant CD4 count predict survival and immune recovery. Blood. (2018) 132:1737–49. doi: 10.1182/blood-2018-03-840702
5. Stephan JL, Vlekova V, Le Deist F, Blanche S, Donadieu J, De Saint-Basile G, et al. Severe combined immunodeficiency: a retrospective single-center study of clinical presentation and outcome in 117 patients. J Pediatr. (1993) 123:564–72. doi: 10.1016/S0022-3476(05)80951-5
6. Dhanju R, Min W, Ackerley C, Cimpean L, Palaniyar N, Roifman CM, et al. Pulmonary alveolar proteinosis in adenosine deaminase-deficient mice. J Allergy Clin Immunol. (2014) 133:1471.e1461–4. doi: 10.1016/j.jaci.2013.11.029
7. Muller SM, Ege M, Pottharst A, Schulz AS, Schwarz K, Friedrich W. Transplacentally acquired maternal T lymphocytes in severe combined immunodeficiency: a study of 121 patients. Blood. (2001) 98:1847–51. doi: 10.1182/blood.V98.6.1847
8. Wahlstrom J, Patel K, Eckhert E, Kong D, Horn B, Cowan MJ, et al. Transplacental maternal engraftment and posttransplantation graft-versus-host disease in children with severe combined immunodeficiency. J Allergy Clin Immunol. (2017) 139:628–33.e610. doi: 10.1016/j.jaci.2016.04.049
9. Villa A, Notarangelo LD, Roifman CM. Omenn syndrome: inflammation in leaky severe combined immunodeficiency. J Allergy Clin Immunol. (2008) 122:1082–6. doi: 10.1016/j.jaci.2008.09.037
10. Myers LA, Patel DD, Puck JM, Buckley RH. Hematopoietic stem cell transplantation for severe combined immunodeficiency in the neonatal period leads to superior thymic output and improved survival. Blood. (2002) 99:872–8. doi: 10.1182/blood.V99.3.872
11. Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. N Engl J Med. (2014) 371:434–46. doi: 10.1056/NEJMoa1401177
12. Brown L, Xu-Bayford J, Allwood Z, Slatter M, Cant A, Davies EG, et al. Neonatal diagnosis of severe combined immunodeficiency leads to significantly improved survival outcome: the case for newborn screening. Blood. (2011) 117:3243–6. doi: 10.1182/blood-2010-08-300384
13. Kwan A, Abraham RS, Currier R, Brower A, Andruszewski K, Abbott JK, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. (2014) 312:729–38. doi: 10.1001/jama.2014.9132
14. Gossage DL, Buckley RH. Prevalence of lymphocytopenia in severe combined immunodeficiency. N Engl J Med. (1990) 323:1422–3. doi: 10.1056/NEJM199011153232014
15. Jindal AK, Rawat A. Cupping at the ends of ribs is not always rickets. BMJ Case Rep. (2017) 2017:bcr-2017-220642. doi: 10.1136/bcr-2017-220642
17. Dergousoff BA, Vayalumkal JV, Wright NAM. Survey of infection control precautions for patients with severe combined immune deficiency. J Clin Immunol. (2019). doi: 10.1007/s10875-019-00671-y. [Epub ahead of print].
18. Gaspar HB, Aiuti A, Porta F, Candotti F, Hershfield MS, Notarangelo LD. How I treat ADA deficiency. Blood. (2009) 114:3524–32. doi: 10.1182/blood-2009-06-189209
19. Antoine C, Muller S, Cant A, Cavazzana-Calvo M, Veys P, Vossen J, et al. Long-term survival and transplantation of haemopoietic stem cells for immunodeficiencies: report of the European experience 1968-99. Lancet. (2003) 361:553–60. doi: 10.1016/S0140-6736(03)12513-5
20. Gennery AR, Slatter MA, Grandin L, Taupin P, Cant AJ, Veys P, et al. (2010). Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: entering a new century, do we do better? J Allergy Clin Immunol. 126:602–10.e601–11. doi: 10.1016/j.jaci.2010.06.015
21. Dvorak CC, Cowan MJ, Logan BR, Notarangelo LD, Griffith LM, Puck JM, et al. The natural history of children with severe combined immunodeficiency: baseline features of the first fifty patients of the primary immune deficiency treatment consortium prospective study 6901. J Clin Immunol. (2013) 33:1156–64. doi: 10.1007/s10875-013-9917-y
22. Heimall J, Logan BR, Cowan MJ, Notarangelo LD, Griffith LM, Puck JM, et al. Immune reconstitution and survival of 100 SCID patients post-hematopoietic cell transplant: a PIDTC natural history study. Blood. (2017) 130:2718–27. doi: 10.1182/blood-2017-05-781849
23. Cavazzana-Calvo M, Carlier F, Le Deist F, Morillon E, Taupin P, Gautier D, et al. Long-term T-cell reconstitution after hematopoietic stem-cell transplantation in primary T-cell-immunodeficient patients is associated with myeloid chimerism and possibly the primary disease phenotype. Blood. (2007) 109:4575–81. doi: 10.1182/blood-2006-07-029090
24. Mazzolari E, Forino C, Guerci S, Imberti L, Lanfranchi A, Porta F, et al. Long-term immune reconstitution and clinical outcome after stem cell transplantation for severe T-cell immunodeficiency. J Allergy Clin Immunol. (2007) 120:892–9. doi: 10.1016/j.jaci.2007.08.007
25. Neven B, Leroy S, Decaluwe H, Le Deist F, Picard C, Moshous D, et al. Long-term outcome after hematopoietic stem cell transplantation of a single-center cohort of 90 patients with severe combined immunodeficiency. Blood. (2009) 113:4114–24. doi: 10.1182/blood-2008-09-177923
26. Ege MJ, Schuetz C, Jacobsen EM, Muller-Langer SM, Furlan I, Sirin M, et al. Late thymic deficiency after HLA-haploidentical hematopoietic stem cell transplantation for severe combined immunodeficiency. J Allergy Clin Immunol. (2019) 143:1623–6.e1613. doi: 10.1016/j.jaci.2018.11.027
27. Haddad E, Landais P, Friedrich W, Gerritsen B, Cavazzana-Calvo M, Morgan G, et al. Long-term immune reconstitution and outcome after HLA-nonidentical T-cell-depleted bone marrow transplantation for severe combined immunodeficiency: a European retrospective study of 116 patients. Blood. (1998) 91:3646–53.
28. Buckley RH, Win CM, Moser BK, Parrott RE, Sajaroff E, Sarzotti-Kelsoe M. Post-transplantation B cell function in different molecular types of SCID. J Clin Immunol. (2013) 33:96–110. doi: 10.1007/s10875-012-9797-6
29. Haddad E, Leroy S, Buckley RH. B-cell reconstitution for SCID: should a conditioning regimen be used in SCID treatment? J Allergy Clin Immunol. (2013) 131:994–1000. doi: 10.1016/j.jaci.2013.01.047
30. Abd Hamid IJ, Slatter MA, Mckendrick F, Pearce MS, Gennery AR. Long-term outcome of hematopoietic stem cell transplantation for IL2RG/JAK3 SCID: a cohort report. Blood. (2017) 129:2198–201. doi: 10.1182/blood-2016-11-748616
Keywords: severe combined immunodeficiency, SCID, clinical presentation, infections, hematopoietic stem cell transplantation, HSCT
Citation: Haddad E and Hoenig M (2019) Hematopoietic Stem Cell Transplantation for Severe Combined Immunodeficiency (SCID). Front. Pediatr. 7:481. doi: 10.3389/fped.2019.00481
Received: 26 September 2019; Accepted: 01 November 2019;
Published: 19 November 2019.
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Michael Daniel Keller, Children's National Health System, United StatesMario Abinun, Newcastle upon Tyne Hospitals NHS Foundation Trust, United Kingdom
Copyright © 2019 Haddad and Hoenig. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manfred Hoenig, bWFuZnJlZC5ob2VuaWcmI3gwMDA0MDt1bmlrbGluaWstdWxtLmRl