
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol. , 10 December 2019
Sec. Hematologic Malignancies
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.01374
 Benjamin Fournier1†Estelle Balducci2†Nicolas Duployez3Emmanuelle Clappier4Wendy Cuccuini4Chloé Arfeuille5Aurélie Caye-Eude5Eric Delabesse6Elodie Bottollier-Lemallaz Colomb7Karin Nebral8Marie-Lorraine Chrétien9Coralie Derrieux10Aurélie Cabannes-Hamy11Florent Dumezy3Pascaline Etancelin12Odile Fenneteau13Jamile Frayfer14Antoine Gourmel15Marie Loosveld16Gérard Michel17Nathalie Nadal18Dominique Penther12Isabelle Tigaud19Elise Fournier3Bettina Reismüller20Andishe Attarbaschi20Marina Lafage-Pochitaloff21†André Baruchel1,22*†
Benjamin Fournier1†Estelle Balducci2†Nicolas Duployez3Emmanuelle Clappier4Wendy Cuccuini4Chloé Arfeuille5Aurélie Caye-Eude5Eric Delabesse6Elodie Bottollier-Lemallaz Colomb7Karin Nebral8Marie-Lorraine Chrétien9Coralie Derrieux10Aurélie Cabannes-Hamy11Florent Dumezy3Pascaline Etancelin12Odile Fenneteau13Jamile Frayfer14Antoine Gourmel15Marie Loosveld16Gérard Michel17Nathalie Nadal18Dominique Penther12Isabelle Tigaud19Elise Fournier3Bettina Reismüller20Andishe Attarbaschi20Marina Lafage-Pochitaloff21†André Baruchel1,22*†Background: B-cell acute lymphoblastic leukemia associated with t(5;14)(q31;q32); IGH-IL3 is an exceptional cause of eosinophilia. The IGH enhancer on 14q32 is juxtaposed to the IL3 gene on 5q31, leading to interleukin-3 overproduction and release of mature eosinophils in the blood. Clinical, biological and outcome data are extremely scarce in the literature. Except for eosinophilia, no relevant common feature has been highlighted in these patients. However, it has been classified as a distinct entity in the World Health Organization classification.
Cases Presentation: Eight patients with t(5;14)(q31;q32) treated by French or Austrian protocols were retrospectively enrolled. Array comparative genomic hybridization, multiplex ligation-dependent probe amplification or genomic PCR search for IKZF1 deletion were performed in 7. Sixteen patients found through an exhaustive search in the literature were also analyzed.
For those 24 patients, median age at diagnosis is 14.3 years with a male predominance (male to female ratio = 5). Eosinophilia-related symptoms are common (neurologic in 26%, thromboembolic in 26% or pulmonary in 50%). Median white blood cells count is high (72 × 109/L) and linked to eosinophilia (median: 32 × 109/L). Peripheral blasts are present at a low level or absent (median: 0 × 109/L; range: 0–37 × 109/L). Bone marrow morphology is marked by a low blast infiltration (median: 42%). We found an IKZF1 deletion in 5 out of 7 analyzable patients Outcome data are available for 14 patients (median follow-up: 28 months): 8 died and 6 are alive in complete remission.
Some of these features are concordant with those seen in patients with other IGH-rearranged B-cell acute lymphoblastic leukemias: young age at onset, male sex, low blast count, high incidence of IKZF1 deletion and intermediate prognosis.
Conclusion: Based on shared epidemiological and biological features, B-cell acute lymphoblastic leukemia with t(5;14)(q31;q32) is a peculiar subset of IGH-rearranged B-cell acute lymphoblastic leukemia with an intermediate prognosis and particular clinical features related to eosinophilia.
B-cell acute lymphoblastic leukemia (B-ALL) with translocation t(5;14)(q31;q32); IGH-IL3 is an exceptional cause of massive eosinophilia recognized by the World Health Organization (WHO) classification of hematological malignancies as a distinct entity (1, 2). The translocation t(5;14)(q31;q32) juxtaposes the IGH enhancer located on 14q32 to the IL3 gene on 5q31. The subsequent production of interleukin-3 (IL3) induces the maturation and release of eosinophils in the blood stream (3–5). The relationship between IL3 and eosinophilia is also supported by the constant eosinophilia in all patients treated with recombinant IL3 for Blackfan-Diamond anemia in one study (6). The IL3-receptor has been hypothesized to be present on blast cells and to induce JAK-STAT-dependent cell survival and proliferation in an autocrine manner (4, 7). The other part of the translocation involves the IGH gene, which is frequently rearranged via chromosomal translocations in mature B-cell neoplasms such as Burkitt leukemia/lymphoma, mantle cell or follicular lymphomas with MYC, CCND1, or BCL2 as partner genes, respectively, whose expression is upregulated (8). IGH translocations are also reported in B-ALL and usually involve other partners such as members of the CEBP gene family, ID4, CRLF2 or EPOR (9–13). Due to the rarity of t(5;14)(q31;q32) associated B-ALL, little is known regarding clinical and molecular features. To further characterize this very rare entity, we have analyzed in detail 8 new patients and combined these data to those of 16 patients from the literature over 35 years.
Patient #1 and patient #2 were first diagnosed in two of our centers in the same period. Consequently, we screened the database of FRALLE 2000 French ALL treatment study (2000–2010) and launched a call within the GFCH (Groupe Francophone de Cytogénétique Hématologique) for patients with B-ALL presenting with t(5;14)(q31;q32). Four other cases were retrieved including a recently relapsed case previously reported at diagnosis (14) and one published very recently (15). All these 6 patients were diagnosed between 2004 and 2016 and were treated according to national protocols (FRALLE 2000 for the 4 pediatric patients, GRAALL 05 and 14 for the 2 adult patients). Given the rarity of the disease, we seized the opportunity to include 2 additional patients diagnosed and treated in Austria in 1999 and 2010 according to the Associazione Italiana di Ematologia Oncologia Pediatrica and the Berlin-Frankfurt-Münster Acute Lymphoblastic Leukemia 2000 [AIEOP-BFM ALL 2000 (16)] and 2009 protocols.
Karyotypes were analyzed on bone marrow samples at diagnosis and relapse according to the International System for Human Cytogenetic Nomenclature (ISCN, 2016). Whenever possible, at least 20 metaphases were analyzed.
Fluorescence in situ hybridization (FISH) was performed on cytogenetic preparations using an IGH dual-color break-apart probe (Vysis, Abbott Molecular Inc.) according to the manufacturer's instructions.
IKZF1 deletions were detected using both multiplex ligation-dependent probe amplification (MLPA) (P335-B2 kit; MRC Holland) and genomic fluorescent multiplex PCR (17). MRD monitoring was based on Ig-TCR rearrangements (18, 19) except in patient #4 (IGH-IL3 genomic PCR quantification).
a-CGH (SurePrint G3 Human CGH Microarray Kit, 4 × 180K; Agilent Technologies, USA) or SNP-array (CytoScan™ HD array; ThermoFisher Scientific) was performed in all cases (except for patient #8) according to manufacturers' instructions. Data were analyzed with the DNA Analytics software v4.0.85 and the Chromosome Analysis Suite version 3.1 (ChAS; Affymetrix) software package and annotated using the human genome version 19 (hg19) of the UCSC Genome Browser, excluding copy number variants (CNVs) arising from B-cell and T-cell antigen receptor gene rearrangements and known CNVs reported in public databases.
The study was approved by the local ethics committee (Comité Local d'Ethique pour la recherche clinique des HUPSSD Avicenne-Jean Verdier-René Muret (CLEA), December 13th, 2018, CLEA-2018-58). According to French legislation, due to the retrospective observational design of the study, informed consent from patient was waived.
Firstly, we report here on our 8 patients selected for the presence of the specific t(5;14)(q31;q32). Clinical characteristics are detailed in Table 1, main biological features in Table 2 and outcome in Table S1. Patients #3 and #7 were previously reported (14, 15), but are yet further described here because of availability of new relevant biological data and because of occurrence of events during longer follow-up.
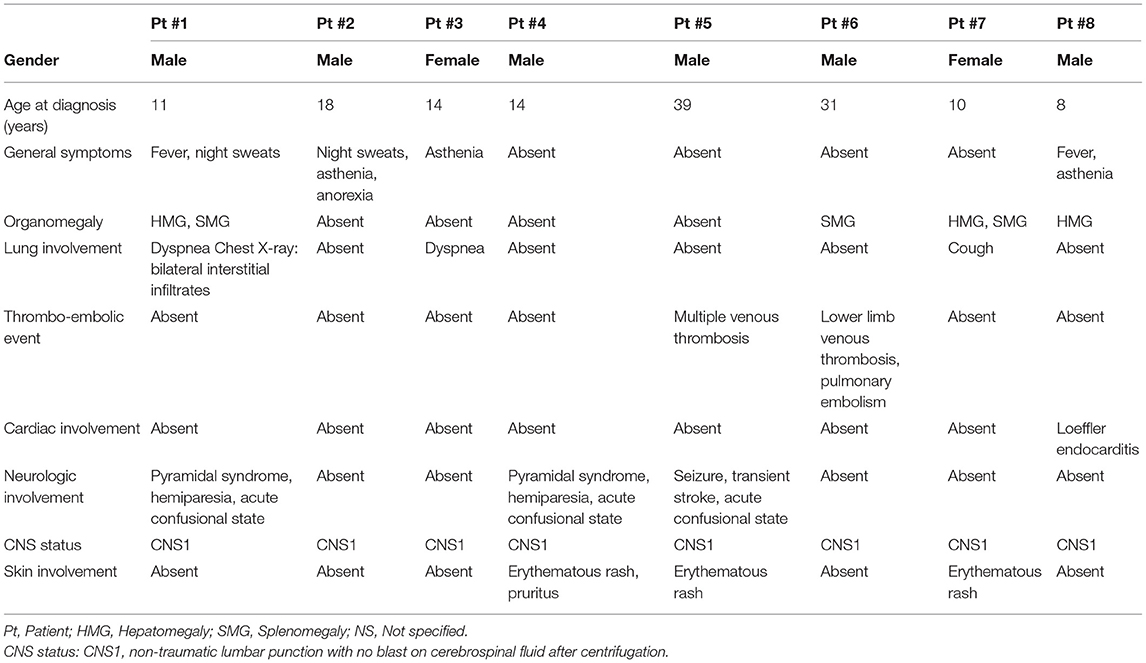
Table 1. Clinical characteristics at onset of the 8 newly reported patients.
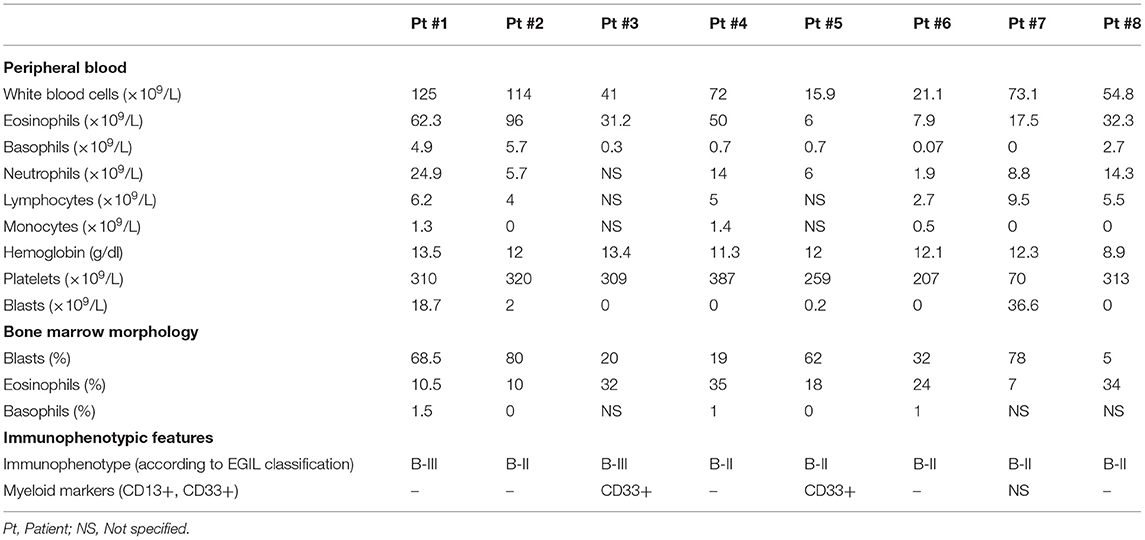
Table 2. Biological characteristics at onset of the 8 newly reported patients.
We chose to highlight hereunder the main clinical features of these patients according to age at onset: pediatric/adolescent onset (8–18 years old) and young adult onset (21–39 years old). We then analyzed them together with the 16 patients from the literature in a third section.
Six patients were children or adolescent (8, 10, 11, 14, 14, and 18 year-old) including one with Down syndrome (patient #7). Two patients presented severe neurologic involvement (encephalopathy with pyramidal syndrome and acute confusion). Magnetic resonance imaging (MRI) showed scattered involvement of white matter and myelitis in one case, multiple cortical and subcortical ischemic lesions in another. Symptoms disappeared after the first courses of chemotherapy. No thrombo-embolic event was recorded. Peripheral blast cells count was increased in 2 (36.6 and 18.7 × 109/L) but was low in others (range: 0–2 × 109/L) and even absent in 10. Peripheral eosinophilia was increased (range: 17.5–96 × 109/L). All patients reached complete remission after induction therapy. All patients are in first (n = 3), second (n = 1), or third (n = 1) continuous complete remission (CCR) for more than 3 years, except for the patient with Down syndrome who died from disease progression after a second relapse.
Two patients were young adults (31 and 39 years of age). Both presented with severe thrombo-embolic events (lower limb venous thrombosis with pulmonary embolism and multiple venous thrombosis with transient strokes, respectively). No encephalopathy was documented. Multiple but asymptomatic subcortical lesions on MRI were present in 1 patient.
Peripheral blast cells count was 0 and 0.2 × 109/L, respectively. Eosinophilia was much lower (7.9 and 6 × 109/L). Prognosis was poor: none reached complete remission after induction therapy, one is in second complete remission (CR2) 1 month after hematopoietic stem cell transplantation and one is deceased after relapse.
We next pooled and analyzed our 8 newly described patients with 16 patients found through an extensive search in the literature, to enlighten the specific features of this very rare entity (20–35). Consequently, the following data concern 24 patients (recognizing nevertheless that not all features are constantly available for each patient of the literature) (Table 3).

Table 3. Main characteristics of 24 patients at onset of t(5;14)(q31;q32) B-ALL -our patients (n = 8) and patients from literature (n = 16).
Median age at onset was 14.3 years (range: 4–60 years); median age for pediatric (n = 16) and adult patients over 18-year-old (n = 8) was 11 and 32 years, respectively. Nineteen out of 24 patients (79%) were above 10 years of age.
A strong male predominance was observed (20 males vs. 4 females).
Eosinophilia-related symptoms were common at onset. Ten out of 20 (50%) patients had lung involvement (dyspnea, cough, chest pain, pleural effusions with eosinophilia, interstitial alterations on chest X-ray). Four patients presented multiple venous thromboses (pulmonary embolism in 3 patients and cerebral venous thrombosis in 1), 1 patient had arterial thrombosis of the lower limb [i.e., 5 out of 19 (26%)]. Nine out of 19 (47%) patients had cardiac involvement, e.g., newly acquired ECG abnormalities, global cardiac failure (n = 1), Loeffler myocarditis on MRI (n = 3), restrictive cardiomyopathy (n = 1), or ventricular hypertrophy (n = 1), leading to death due to ventricular fibrillation in one patient. Five out of 19 (26%) patients had neurologic symptoms (decreased consciousness, cerebral palsy, generalized seizure, and transient ischemic attack). MRI data were available for 3 of our patients (described in text above) and in Kobayashi et al. (34). No patient had blasts in the cerebrospinal fluid at initial presentation. Five out of 19 (26%) patients had an erythematous urticaria-like skin involvement.
Regarding biologic parameters, median white blood cell count was high (72 × 109/L), mainly due to eosinophilia (median: 32 × 109/L; range: 6–111 × 109/L). Basophil count was increased (median: 0.8 × 109/L; range: 0–7 × 109/L). Hemoglobin and platelets were mainly normal (median: 12 g/dL, range 7–13.7 g/dL, and 171 × 109/L, range 60–532 × 109/L, respectively). Peripheral blasts were present at a very low count or even absent in 9 patients (median: 0 × 109/L; range 0–36.6 × 109/L). Of note, in these 9 patients, blast count was determined only on blood smears except for 2 patients who had flow cytometry analysis [patient #6 and reference (35)]. An only partial lymphoblastic infiltration in the bone marrow was frequent (median 42%; range: 5–80%). Two patients had a prolonged interval between blood eosinophilia with normal bone marrow morphology and the appearance of blast cells in the bone marrow [12 months for patient #5, 6 months in Knuutila et al. (26)].
Flow cytometry data were available for 12 patients classified patients in BI (n = 1), BII (n = 7), or BIII (n = 4) subsets according to EGIL classification. Myeloid markers (CD13 and/or CD33) were positive in 4/13 patients (31%).
Regarding cytogenetics, detailed karyotypes are provided in Table S2 and array-based Comparative Genomic Hybridization (a-CGH) in Table S3. All but 2 patients had an abnormal karyotype harboring the t(5;14)(q31;q32): concerning patient #6 and in Yu et al. (35), the translocation was proven by genomic IGH-IL3 PCR or next generation sequencing, respectively. t(5;14)(q31;q32) was the sole cytogenetic finding in 13 patients, 9 having additional abnormalities. The number of abnormal metaphases was low (range: 1–22), except for one case where all metaphases were abnormal.
a-CGH abnormalities found in at least 2 patients included deletion of SLX4IP (n = 4), VPREB1 (n = 3), CDKN2A (n = 3), PAX5 (n = 2), MEF2C (n = 2), ARHGAP24 (n = 2), and ETV6 (n = 2).
An IKZF1 deletion was searched only in our newly described patients and detected in a large proportion [5/7 (71%), Table S4]. The isochromosome 7q in Knuutila et al. (26) also leads to the loss of IKZF1.
Outcome and remission status were both available for 14 out of 24 patients [Figure S1: our 8 patients (detailed in Table S1), 6 from literature (20–22, 28, 30) with additional information provided by Toboso and Campos (33)]. Median follow-up was 28 months (range: 4 days−13 years). Early treatment response was characterized by a high induction failure proportion: only 9 out 14 patients were in complete remission (CR) after first induction, 4 more reached CR1 after a further course. Minimal residual disease (MRD), only available for our newly described patients, was high for 4 patients after first induction (range: 0.1–10%). Eight out of 14 patients died [cardiac involvement secondary to eosinophilia (n = 1) (20), uncontrolled disease and/or sepsis (n = 7)].
t(5;14)(q31;q32); IGH-IL3 B-ALL is an exceptional hence poorly described subset of B-ALL, which is however described as a distinct entity in the WHO classification. None was reported despite the use of extensive FISH screening for IGH-translocation in 3,302 patients from UK protocols (9). By combining 8 new patients (spanning over 15 years of French and Austrian protocols) to 16 patients from the literature (20–35), we could highlight new striking features.
(1) Most patients are adolescents or young adults (median age: 14.3 years). (2) Male to female ratio is highly biased (5 to 1). (3) Eosinophilia-related symptoms are common and severe. It is the expected consequence of the underlying translocation: IL-3 overproduction induces maturation and release of eosinophils in the blood from the bone marrow. (4) Lymphoblastic infiltration of the bone marrow is partial and explains that peripheral blasts are almost undetectable in a vast proportion of patients. This partial infiltration may also account for normal to slightly decreased hemoglobin and platelet count. In cases with <20% of infiltrating blast cells, the specific translocation permitted a diagnosis of t(5;14)(q31;q32) associated B-ALL (2). The frequent CNS-related symptoms with no ALL blast infiltration suggests a direct eosinophil toxicity on neuronal, glial and endothelial cells caused either by eosinophilic proteins release or eosinophil cells infiltration (36, 37). Depending on the damaged cell type, symptoms may include signs of encephalopathy or focal symptoms due to local thrombosis and multiple infarcts (38). (5) IKZF1 deletions are frequently detected. (6) Response to treatment is poor: induction failure and high levels of MRD are frequently observed, and may account for the intermediate prognosis.
These particularities suggest new diagnostic strategies, notably because (a) peripheral blasts are often absent (even by flow cytometry analysis) and (b) the translocation t(5;14)(q31;q32) may be missed owing to the low rate of abnormal metaphases on standard karyotype and metaphase FISH. It is worth noting that the 2 patients with a prolonged interval between blood eosinophilia and the appearance of blast cells in the bone marrow could fulfill criteria of idiopathic hypereosinophilic syndrome (39). Persistent and/or massive eosinophilia should thus be investigated by repeated bone marrow aspirations, including cytogenetic investigations and IGH-FISH on cytogenetic pellet on a sufficient number of metaphases and interphase nuclei. FISH analysis on bone marrow smears (patients #2 and #6) or on leukemic cytometer-sorted cells (34) (patient #8) can be used. As suggested in (35, 40), a bone marrow biopsy may reveal focal clusters of blasts missed by bone marrow aspiration. Next generation sequencing may be an alternative to detect IGH-IL3 rearrangement (35). Finally, careful monitoring of eosinophils should be performed during and after treatment as increased eosinophils may suggest a relapse.
Of note, t(5;14)(q31;q32); IGH-IL3 B-ALL shares remarkable features with other IGH-rearranged B-ALL (5–11% of B-ALL) (9, 10, 12, 41) (Table 4). (1) IGH-rearranged B-ALL also affects especially adolescents and young adults [median age of onset: 25 years (12) or 16 years (9)]. (2) A male predominance is present even if male to female ratio is lower (1.44) (12). (3) In both entities, a majority of patients with low blast count is observed (9, 10, 41). (4) A high incidence of IKZF1 deletion (40%) is frequent in patients with IGH-rearranged B-ALL (9). (5) Only 6 out 14 evaluable patients with t(5;14)(q31;q32); IGH-IL3 B-ALL are alive which mirrors the 27–30% overall survival observed in 3 studies (9, 12, 41).
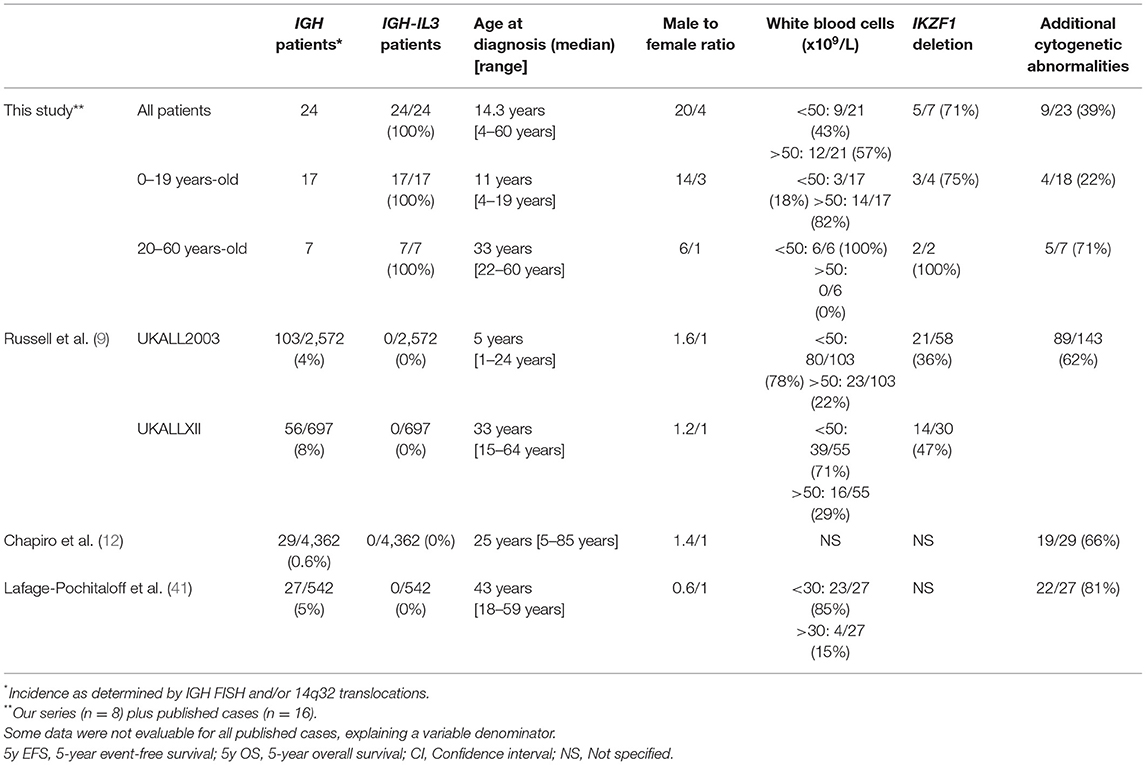
Table 4. Comparison of IGH-rearranged B-ALL from literature to t(5;14)(q31;q32); IGH-IL3 B-ALL (our series plus published cases).
In conclusion, t(5;14)(q31;q32); IGH-IL3 B-ALL constitutes a very rare subset of IGH-rearranged B-ALL. The IL3 partner gene is responsible for the specific symptomatology and increased awareness of this B-ALL may lead to a correct diagnosis of B-ALL when facing a severe symptomatic eosinophilia. International collaboration should provide new insights into its biology and bring new treatment strategies.
The datasets generated for this study are available on request to the corresponding author.
The studies involving human participants were reviewed and approved by Comité Local d'Ethique pour la recherche clinique des HUPSSD Avicenne-Jean Verdier-René Muret (CLEA), December 13th, 2018, CLEA-2018-58. Written informed consent from the participants' legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
BF, EB, AA, ML-P, and AB: conception of the study. All authors: collection and assembly of data, final approval of manuscript, and accountable for all aspects of the work. BF, EB, ML-P, and AB: data analysis and interpretation and manuscript writing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank M. V. Larsen, D. G. Toboso, and C. B. Campos for providing additional data on their patients, respectively published in Larsen et al. (27) and Toboso and Campos (33). EB, WC, NN, DP, IT, and ML-P are members of the Groupe Francophone de Cytogénétique Hématologique.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.01374/full#supplementary-material
1. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. (2009) 114:937–51. doi: 10.1182/blood-2009-03-209262
2. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Beau MML, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
3. Esnault S, Kelly EA. Essential mechanisms of differential activation of eosinophils by IL-3 compared to GM-CSF and IL-5. Crit Rev Immunol. (2016) 36:429–44. doi: 10.1615/CritRevImmunol.2017020172
4. Meeker TC, Hardy D, Willman C, Hogan T, Abrams J. Activation of the interleukin-3 gene by chromosome translocation in acute lymphocytic leukemia with eosinophilia. Blood. (1990) 76:285–9. doi: 10.1182/blood.V76.2.285.285
5. Grimaldi JC, Meeker TC. The t(5;14) chromosomal translocation in a case of acute lymphocytic leukemia joins the interleukin-3 gene to the immunoglobulin heavy chain gene. Blood. (1989) 73:2081–5. doi: 10.1182/blood.V73.8.2081.2081
6. Gillio AP, Faulkner LB, Alter BP, Reilly L, Klafter R, Heller G, et al. Treatment of diamond-blackfan anemia with recombinant human interleukin- 3 [see comments]. Blood. (1993) 82:744–51. doi: 10.1182/blood.V82.3.744.744
7. Liu K, Zhu M, Huang Y, Wei S, Xie J, Xiao Y. CD123 and its potential clinical application in leukemias. Life Sci. (2015) 122:59–64. doi: 10.1016/j.lfs.2014.10.013
8. Willis TG, Dyer MJS. The role of immunoglobulin translocations in the pathogenesis of B-cell malignancies. Blood. (2000) 96:808–22. doi: 10.1182/blood.V96.3.808
9. Russell LJ, Enshaei A, Jones L, Erhorn A, Masic D, Bentley H, et al. IGH@ translocations are prevalent in teenagers and young adults with acute lymphoblastic leukemia and are associated with a poor outcome. J Clin Oncol. (2014) 32:1453–62. doi: 10.1200/JCO.2013.51.3242
10. Moorman AV, Schwab C, Ensor HM, Russell LJ, Morrison H, Jones L, et al. IGH@ translocations, CRLF2 deregulation, and microdeletions in adolescents and adults with acute lymphoblastic leukemia. J Clin Oncol. (2012) 30:3100–8. doi: 10.1200/JCO.2011.40.3907
11. Iacobucci I, Mullighan CG. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol. (2017) 35:975–83. doi: 10.1200/JCO.2016.70.7836
12. Chapiro E, Radford-Weiss I, Cung HA, Dastugue N, Nadal N, Taviaux S, et al. Chromosomal translocations involving the IGH@ locus in B-cell precursor acute lymphoblastic leukemia: 29 new cases and a review of the literature. Cancer Genet. (2013) 206:162–73. doi: 10.1016/j.cancergen.2013.04.004
13. Moorman AV. New and emerging prognostic and predictive genetic biomarkers in B-cell precursor acute lymphoblastic leukemia. Haematologica. (2016) 101:407–16. doi: 10.3324/haematol.2015.141101
14. Girodon F, Bergoin E, Favre B, Martha SA, Mugneret F, Couillault G, et al. Hypereosinophilia in acute B-lineage lymphoblastic leukaemia. Br J Haematol. (2005) 129:568. doi: 10.1111/j.1365-2141.2005.05469.x
15. Derrieux C, Freynet N, Frayfer J, Delabesse E, Clappier E, Defasque S, et al. A case of B-cell precursor acute lymphoblastic leukemia with IL3-IGH rearrangement revealed by thromboembolism and marked eosinophilia. Leuk Lymphoma. (2018) 59:2489–92. doi: 10.1080/10428194.2018.1430796
16. Conter V, Bartram CR, Valsecchi MG, Schrauder A, Panzer-Grümayer R, Möricke A, et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood. (2010) 115:3206–14. doi: 10.1182/blood-2009-10-248146
17. Clappier E, Grardel N, Bakkus M, Rapion J, De Moerloose B, Kastner P, et al. IKZF1 deletion is an independent prognostic marker in childhood B-cell precursor acute lymphoblastic leukemia, and distinguishes patients benefiting from pulses during maintenance therapy: results of the EORTC Children's Leukemia Group study 58951. Leukemia. (2015) 29:2154–61. doi: 10.1038/leu.2015.134
18. Brüggemann M, Schrauder A, Raff T, Pfeifer H, Dworzak M, Ottmann OG, et al. Standardized MRD quantification in European ALL trials: proceedings of the second international symposium on MRD assessment in Kiel, Germany, 18–20 september 2008. Leukemia. (2010) 24:521–35. doi: 10.1038/leu.2009.268
19. Velden VHJ van der, Cazzaniga G, Schrauder A, Hancock J, Bader P, Panzer-Grumayer ER, et al. Analysis of minimal residual disease by Ig/TCR gene rearrangements: guidelines for interpretation of real-time quantitative PCR data. Leukemia. (2007) 21:604–11. doi: 10.1038/sj.leu.2404586
20. Tono-oka T, Sato Y, Matsumoto T, Ueno N, Ohkawa M, Shikano T, et al. Hypereosinophilic syndrome in acute lymphoblastic leukemia with a chromosome translocation [t(5q;14q)]. Med Pediatr Oncol. (1984) 12:33–7. doi: 10.1002/mpo.2950120109
21. Baumgarten E, Wegner RD, Fengler R, Ludwig WD, Schulte-Overberg U, Domeyer C, et al. Calla-positive acute leukaemia with t(5q;14q) translocation and hypereosinophilia–a unique entity? Acta Haematol. (1989) 82:85–90. doi: 10.1159/000205289
22. Hogan TF, Koss W, Murgo AJ, Amato RS, Fontana JA, VanScoy FL. Acute lymphoblastic leukemia with chromosomal 5;14 translocation and hypereosinophilia: case report and literature review. J Clin Oncol. (1987) 5:382–90. doi: 10.1200/JCO.1987.5.3.382
23. McConnell TS, Foucar K, Hardy WR, Saiki J. Three-way reciprocal chromosomal translocation in an acute lymphoblastic leukemia patient with hypereosinophilia syndrome. J Clin Oncol. (1987) 5:2042–4. doi: 10.1200/JCO.1987.5.12.2042
24. Chen Z, Morgan R, Sandberg AA. Non-random involvement of chromosome 5 in ALL. Cancer Genet Cytogenet. (1992) 61:106–7. doi: 10.1016/0165-4608(92)90381-H
25. Heerema NA, Palmer CG, Weetman R, Bertolone S. Cytogenetic analysis in relapsed childhood acute lymphoblastic leukemia. Leukemia. (1992) 6:185–92.
26. Knuutila S, Alitalo R, Ruutu T. Power of the MAC (morphology-antibody-chromosomes) method in distinguishing reactive and clonal cells: report of a patient with acute lymphatic leukemia, eosinophilia, and t(5;14). Genes Chromosomes Cancer. (1993) 8:219–23. doi: 10.1002/gcc.2870080403
27. Larsen MV, Karstoft K, Andersen MK. [Acute lymphatic leukaemia with eosinophilia in a younger man returning from a travel in the tropics–case report]. Ugeskr Laeger. (2011) 173:1363–4.
28. Gallego M, Coccé M, Felice M, Rossi J, Eandi S, Sciuccati G, et al. A new case of t(5;14)(q31;q32) in a pediatric acute lymphoblastic leukemia presenting with hypereosinophilia. Atlas Genet Ctyogenet Oncol Haematol. (2012) 16:183–4. doi: 10.4267/2042/46952
29. George S, Kumar P, Quarta G, Shankar A, Hough R, Samarasinghe S. Loeffler myocarditis in pre-B acute lymphoblastic leukaemia with t(5;14)(q31;q32). Br J Haematol. (2012) 157:517. doi: 10.1111/j.1365-2141.2012.09098.x
30. Kaneko H, Shimura K, Yoshida M, Ohkawara Y, Ohshiro M, Tsutsumi Y, et al. Acute lymphoblastic leukemia with eosinophilia lacking peripheral blood leukemic cell: a rare entity. Indian J Hematol Blood Transfus. (2014) 30:80–3. doi: 10.1007/s12288-013-0255-2
31. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. (2014) 371:1005–15. doi: 10.1056/NEJMoa1403088
32. Delabesse E, Thomas C, Radford I, Valensi F, Macintyre E. Leucémie, origine d'un syndrome hyperéosinophilique d'apparence idiopathique. Hématologie. (1995) 1:423–6.
33. Toboso DG, Campos CB. Peripheral eosinophilia as the first manifestation of B-cell acute lymphoblastic leukemia with t(5;14)(q31;q32). Blood. (2017) 130:380. doi: 10.1182/blood-2016-12-754812
34. Kobayashi K, Mizuta S, Yamane N, Ueno H, Yoshida K, Kato I, et al. Paraneoplastic hypereosinophilic syndrome associated with IL3-IgH positive acute lymphoblastic leukemia. Pediatr Blood Cancer. (2019) 66:e27449. doi: 10.1002/pbc.27449
35. Yu H, Wertheim G, Shankar S, Paessler M, Aplenc R, Pillai V. Marked eosinophilia masking B lymphoblastic leukemia. Am J Hematol. (2016) 91:543–4. doi: 10.1002/ajh.24266
36. Wang JG, Mahmud SA, Thompson JA, Geng JG, Key NS, Slungaard A. The principal eosinophil peroxidase product, HOSCN, is a uniquely potent phagocyte oxidant inducer of endothelial cell tissue factor activity: a potential mechanism for thrombosis in eosinophilic inflammatory states. Blood. (2006) 107:558–65. doi: 10.1182/blood-2005-05-2152
37. Ackerman SJ, Loegering DA, Venge P, Olsson I, Harley JB, Fauci AS, et al. Distinctive cationic proteins of the human eosinophil granule: major basic protein, eosinophil cationic protein, and eosinophil-derived neurotoxin. J Immunol. (1983) 131:2977–82.
38. Moore PM, Harley JB, Fauci AS. Neurologic dysfunction in the idiopathic hypereosinophilic syndrome. Ann Intern Med. (1985) 102:109–14. doi: 10.7326/0003-4819-102-1-109
39. Reiter A, Gotlib J. Myeloid neoplasms with eosinophilia. Blood. (2017) 129:704–14. doi: 10.1182/blood-2016-10-695973
40. Wang SA, Hasserjian RP, Tam W, Tsai AG, Geyer JT, George TI, et al. Bone marrow morphology is a strong discriminator between chronic eosinophilic leukemia, not otherwise specified and reactive idiopathic hypereosinophilic syndrome. Haematologica. (2017) 102:1352–60. doi: 10.3324/haematol.2017.165340
Keywords: acute lymphoblastic leukemia, cytogenetics and molecular genetics, clinical and molecular epidemiology, eosinophilia, IKZF1 rearrangement
Citation: Fournier B, Balducci E, Duployez N, Clappier E, Cuccuini W, Arfeuille C, Caye-Eude A, Delabesse E, Bottollier-Lemallaz Colomb E, Nebral K, Chrétien M-L, Derrieux C, Cabannes-Hamy A, Dumezy F, Etancelin P, Fenneteau O, Frayfer J, Gourmel A, Loosveld M, Michel G, Nadal N, Penther D, Tigaud I, Fournier E, Reismüller B, Attarbaschi A, Lafage-Pochitaloff M and Baruchel A (2019) B-ALL With t(5;14)(q31;q32); IGH-IL3 Rearrangement and Eosinophilia: A Comprehensive Analysis of a Peculiar IGH-Rearranged B-ALL. Front. Oncol. 9:1374. doi: 10.3389/fonc.2019.01374
Received: 01 September 2019; Accepted: 21 November 2019;
Published: 10 December 2019.
Edited by:
Anjali Mishra, Sidney Kimmel Cancer Center, United StatesReviewed by:
Lisa Jane Russell, Newcastle University, United KingdomCopyright © 2019 Fournier, Balducci, Duployez, Clappier, Cuccuini, Arfeuille, Caye-Eude, Delabesse, Bottollier-Lemallaz Colomb, Nebral, Chrétien, Derrieux, Cabannes-Hamy, Dumezy, Etancelin, Fenneteau, Frayfer, Gourmel, Loosveld, Michel, Nadal, Penther, Tigaud, Fournier, Reismüller, Attarbaschi, Lafage-Pochitaloff and Baruchel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: André Baruchel, andre.baruchel@aphp.fr
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.