 Mateo Mejia Saldarriaga
Mateo Mejia Saldarriaga Amir Steinberg2
Amir Steinberg2 Adam Binder
Adam Binder
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol. , 03 December 2019
Sec. Hematologic Malignancies
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.01303
This article is part of the Research Topic Molecular and Immunological Advances in Hematological Malignancies View all 11 articles
We report the case of a patient with B-Cell Acute Lymphoblastic Leukemia (ALL) who was found to harbor a gene fusion involving the CCDC6 and RET genes. Although the RET mutations have been identified before in other malignancies, and it is thought to represent a driver mutation in these neoplasms, it has yet to be described in ALL. The identification of known fusion genes conferring activating tyrosine kinase activity in neoplasms can suggest potential therapeutic role of tyrosine kinase inhibitors (TKI), an approach that has been exploited in several other fusion genes.
Sincethe discovery of a recurrent translocation in patients with chronic myeloid leukemia, t(9:22)(q34;q11), later identified as the BCR-ABL fusion gene, the role of gene fusions involving tyrosine kinases have been recognized as important oncogenic drivers.In ALL, the t(9:22) translocation is the most frequent cytogenetic abnormality. BCR-ABL+ ALL patients comprise a distinct clinical subgroup (referred as Ph+ ALL), with a more severe clinical presentation, historically poorer prognosis, and more prevalent in adults than children (1). Therapeutic targeting of the BCR-ABL fusion gene with TKI has improved treatment response and overall survival for patients with Ph+ ALL. We describe here the case of a 55 year-old female who presented with pancytopenia, lymphadenopathy, and was found to have ALL with a CCDC6-RET fusion. Understanding the role of the different genomic alterations present in ALL is of crucial importance, as they have prognostic implications and are evolving therapeutic targets. The first targeted therapy in ALL was against the BCR-ABL fusion protein, which improved overall response and survival in “fit” patients, but also allowed a promising alternative treatment strategy for elderly or frail patients (2, 3). More recently, a subgroup of patients were noticed to share the same gene expression profile as Ph-positive, but lack the BCR-ABL translocation. These patients have several different genomic alterations, and are possibly targeted using different TKI's (4). RET gene fusions and point mutations are known driver mutation in several neoplasms, including papillary thyroid cancer (PTC), non-small cell lung cancer (NSCLC), acute myeloid leukemia, and sporadic medullary thyroid cancer. RET gene fusions are the main oncogenic driver in patients with multiple endocrine type 2 (5). RET gene fusions have not previously been described in patients with ALL (6, 7).
A 55 year-old female patient with past medical history of type 2 diabetes, hypothyroidism and opioid dependence presented to the ED with 2 weeks of right flank pain radiating to her right groin. Her initial vital signs were stable, and she was afebrile. Her physical exam was remarkable for bruising in her lower extremities and a hemorrhagic bulla under her tongue. Palpation of her back revealed right costovertebral angle tenderness. Lymph node exam demonstrated a few bilateral shotty inguinal lymph nodes.
The patient's initial blood work was remarkable for pancytopenia (white blood cell count 2.1 ×106/uL, Hb 112 g/L, platelets 0.03 ×109/uL), with normal renal function and elevated Alkaline Phosphatase, AST and ALT (265, 481, and 39 U/L, respectively). Lactate dehydrogenase (LDH) was severely elevated 17,216 U/L, with normal coagulation time, fibrinogen and uric acid. A bone marrow flow cytometry revealed a population of blasts that represented 77% of the sample. The blast were positive for CD9, CD19, CD10 (variable), CD20, cCD22, CD34, CD45, cCD79a, HLA-DR, and cTdT. The blasts were negative for: CD2, cCD3, CD4, CD5, CD7, CD8, CD1a, CD11b, CD11c, CD13, CD14, CD25, CD33, CD56, CD64, CD91, CD117, CD123, CD163, cIgM, cMPO, Kappa, and Lambda. These findings were compatible with B-lymphoblastic leukemia. BCR/ABL was negative by PCR, and cytogenetics were abnormal with 71,74,XXXX,XXX,XX,-3,+4,+5,+6,+6,+6,-7,-7,+8,-9,-9,+10,-10,+11,-12,-12,-13,-14,-14,+15,+15,-16,-16,-17,-18,-18,+19,+20,+20,-20,+21,+22,+22,+22,del(22)(q11.2),x2[cp15]/46,XX [5].
Patient completed induction chemotherapy with R-HyperCVAD, with complete remission as determined by bone marrow biopsy at the end of the first cycle. Course was complicated by neutropenic fever after the first cycle. FoundatioOne®Heme was sent as part of her diagnostic work up and revealed the following genomic alterations: CCDC6-RET gene fusion, FLT3 D835H and D835V, CDKN2A/B loss, DNMT3A truncation in intron 14, KMT2C S793* (encoding MLL3) and TP53 K132R. The patient finished induction with negative minimal residual disease (MRD) by flow cytometry and resolution of cytogenetic abnormalities observed at diagnosis, and underwent myeloablative allogenic bone marrow transplant from peripheral blood donor, with cyclophosphamide and total body irradiation conditioning regimen.
Her initial post-transplant course was uncomplicated initially; however, the patient developed acute graft vs. host disease involving the skin, with a good response to steroids. Ruxolitinib was introduced later as steroid sparing agent. To date, 500 days since transplant, she continues to be in remission, with no MRD in last assessment 1 year post-transplant. Patient authorized and consented use of her information and publication of the case.
The detection of new potential driver mutations in different neoplasms has been boosted by the introduction of several comprehensive genomic profiling assays available for commercial use. These assays both identify known driver mutations as well as novel genomic alterations.
Activating mutations involving tyrosine kinase receptors and other molecules involved in intracellular signal amplifications/transduction are known to be important drivers of oncogenesis. Moreover, the presence of the same mutation across different neoplasm, often represents an important pharmacological target. CCDC6-RET and other common fusion partners leads to homodimerization through coiled-coil interactions in the fusion partner protein, preserving the tyrosine kinase activity in the RET intracellular domain and causing ligand independent activation (9). RET autophosphorylation lead to activation of several intracellular transduction cascades involved in cellular proliferation, including MAPK, AKT, PKC, JAK-STAT, PKA and PI3K (5).
RET gene fusions were first identified in patients with PTC, followed by a subset of patients with NSCLC (10). This mutation has been targeted recently with TKIs, offering a new line of treatment for these patients (Figure 1) (11). CCDC6-RET translocations are the most common type of RET fusion present in PTC (12). These mutations are considered to have a considerable role in oncogenesis as they are usually mutually exclusive with other alterations such as BRAF and RAS in PTC (13), a hypothesis that is supported by clinical response to TKIs (5). More recently, RET gene fusions, including CCDC6 have been identified in NSCLC (10), resulting in clinical trials for TKIs in this subgroup (11, 14).
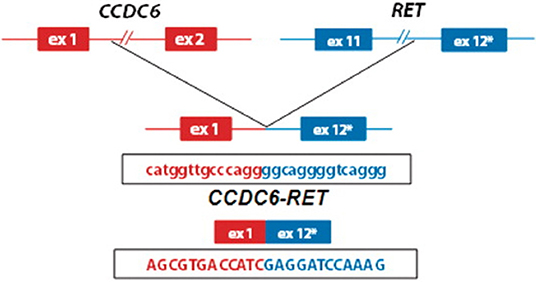
Figure 1. Schematic representation of the CCDC6-RET fusion gene identified in patients with papilary thyroid cancer (8) (used with permission, copyright Nucleic Acids Research, Oxford University Press).
Several multi-kinase inhibitors with RET inhibition are available and have been approved for several different indications. Vandetanib (14), Lenvatinib (15), Alectinib (16), and Cabozatinib (11) have been used in trials of NSCLC with rearranged RET. Ponatinib (9), Sunitinib (7), Sorafenib (17), and Axitinib (5) are molecules that inhibit multiple tyrosine kinase proteins, but also exhibit multitarget RET inhibition, and have been used in different clinical settings. Newer RET specific molecules (BLU-667, LOXO-292) are currently being developed and evaluated in different clinical scenarios (5).
Despite the evidence for the central role of RET as a driver mutation, different agents inhibiting RET in NSCLC and PTC have showed different degrees of overall response rate, ranging from 0 to 50% (5). Several hypotheses for this heterogeneous effect exist, including significant differences in potency (as reflected by IC50), different types of interaction with the ATP-binding pocket of RET, and differences in non-RET tyrosine kinase inhibition. The latter mechanism is especially of interest given the preclinical evidence of resistance to RET inhibition through EGFR-pathway activation in NSCLC, suggesting a potential rational for multiple tyrosine kinase inhibition (18).
Additionally, the RET fusion partner gene might impact the expression of RET despite not involving the tyrosine kinase domain. Reports of different degrees of RET expression varies, with a higher degree of RET expression in KIF5B-RET when compared to CCDC6-RET. Additionally, KIF5B-RET has been associated with multikinase activation independent of the RET-tyrosine kinase (19). Evidence of heterogeneous clinical activity of Vandetanib across different RET fusion partners has been observed in clinically in phase 2 trials in NSCLC (14, 20).
The previously described additional mechanism in RET mutations might explain the significant gap observed in patients with actionable NSCLC such as EGFR, ALK, ROS1, and RET, where the response rate and progression free survival is roughly half for RET inhibitors (5).
As described in NSCLC, dependence on other pathways could also potentially lead to resistance of RET inhibition. In this case, other associated mutations such as FLT3, DNMT3A, MLL, and TP53 act as oncogenes or tumor suppressor genes could further contribute to resistance, as they involve other tyrosine kinase proteins (FLT3), important epigenetic (DNTM3A, MLL3) and DNA damage-response elements (TP53). Off note, FLT3 D835 mutations have been described before in pediatric ALL (21), and is one of the most frequent mutations in acute myeloid leukemia (AML). FLT3 is a well-recognized target in AML and TKIs such as midostaurin and gilteritinib are approved to target this mutation (22). DNMT3A mutations have been described in 5% of 57 patient with ALL, predominantly of T-cell origin, and are thought to lead to worse outcomes, although there is no information in large cohorts (23). Although these mutations are known to be present in ALL, the interactions with CCDC6-RET is unknown and their overall prognostic implication is unclear.
This is the first report of CCDC6-RET rearrangement in ALL to date. The increased use of genome wide comprehensive assays is an important tool to discover novel genomic alterations and genomic alterations common in certain malignancies in unexpected contexts. These findings may be suitable for targeted therapies. The isolation of CCDC6-RET fusion raise the possibility of directed RET inhibition with selective or multitarget TKI in this disease. Drugs such as ponatinib and sorafenib are already used for patients with underlying hematologic malignancies, with a well-studied safety profile; particularly ponatinib in Ph+ ALL. Fortunately, our patient continues to be in remission post-transplant, but this could be a treatment option in the future should she relapse. Comprehensive genomic profiling including interrogation of RET rearrangement and point mutations should be conducted in ALL to identify the prevalence and clinical implications of this new finding. Further characterization of RET activation and resistance mechanism are required to assess for better strategies to improve clinical outcomes in this subset of malignancies.
Analysis and data gathering was purely retrospective and with consent by the patient.
ES is employed by Foundation Medicine, Inc. Part of this case was presented as an online abstract at American Society of Hematology annual meeting 2018 at San Diego, California.
AB and AS participated directly in the clinical care of the patient. All authors participated actively in textual elaboration.
ES is an employee at Fundation Medicine.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Schlieben S, Borkhardt A, Reinisch I, Ritterbach J, Janssen JW, Ratei R, et al. Incidence and clinical outcome of children with BCR/ABL-positive acute lymphoblastic leukemia (ALL. A prospective RT-PCR study based on 673 patients enrolled in the German pediatric multicenter therapy trials ALL-BFM-90 and CoALL-05-92. Leukemia. (1996) 10:957–63.
2. Thomas DA, Faderl S, Cortes J, O'Brien S, Giles FJ, Kornblau SM, et al. Treatment of Philadelphia chromosome-positive acute lymphocytic leukemia with hyper-CVAD and imatinib mesylate. Blood. (2004) 103:4396–407. doi: 10.1182/blood-2003-08-2958
3. Rousselot P, Coudé MM, Gokbuget N, Gambacorti Passerini C, Hayette S, Cayuela JM, et al. Dasatinib and low-intensity chemotherapy in elderly patients with Philadelphia chromosome–positive ALL. Blood. (2016) 128:774–82. doi: 10.1182/blood-2016-02-700153
4. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. (2014) 371:1005–15. doi: 10.1056/NEJMoa1403088
5. Drilon A, Hu ZI, Y Lai GG, Tan DSW. Targeting RET-driven cancers: lessons from evolving preclinical and clinical landscapes. Nat Rev Clin Oncol. (2018) 15:151–67. doi: 10.1038/nrclinonc.2017.175
6. Nelson KN, Peiris MN, Meyer AN, Siari A, Donoghue DJ. Receptor tyrosine kinases: translocation partners in hematopoietic disorders. Trends Mol Med. (2017) 23:59–79. doi: 10.1016/j.molmed.2016.11.002
7. Schram AM, Chang MT, Jonsson P, Drilon A. Fusions in solid tumours: Diagnostic strategies, targeted therapy, and acquired resistance. Nat Rev Clin Oncol. (2017) 14:735–48. doi: 10.1038/nrclinonc.2017.127
8. Chmielecki J, Peifer M, Jia P, Socci ND, Hutchinson K, Viale A, et al. Targeted next-generation sequencing of DNA regions proximal to a conserved GXGXXG signaling motif enables systematic discovery of tyrosine kinase fusions in cancer. Nucleic Acids Res. (2010) 38:6985–96. doi: 10.1093/nar/gkq579
9. De Falco V, Buonocore P, Muthu M, Torregrossa L, Basolo F, Billaud M, et al. Ponatinib (AP24534) is a novel potent inhibitor of oncogenic RET mutants associated with thyroid cancer. J Clin Endocrinol Metab. (2013) 98:E811–9. doi: 10.1210/jc.2012-2672
10. Wang R, Hu H, Pan Y, Li Y, Ye T, Li C, et al. RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol. (2012) 30:4352–9. doi: 10.1200/JCO.2012.44.1477
11. Drilon A, Rekhtman N, Arcila M, Wang L, Ni A, Albano M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol. (2016) 17:1653–60. doi: 10.1016/S1470-2045(16)30562-9
12. Santoro M, Carlomagno F. Central role of RET in thyroid cancer. Cold Spring Harb Perspect Biol. (2013) 5:a009233. doi: 10.1101/cshperspect.a009233
13. Mitsutake N, Miyagishi M, Mitsutake S, Akeno N, Mesa C Jr, Knauf JA, et al. BRAF mediates RET/PTC-induced mitogen-activated protein kinase activation in thyroid cells: functional support for requirement of the RET/PTC-RAS-BRAF pathway in papillary thyroid carcinogenesis. Endocrinology. (2006) 147:1014–9. doi: 10.1210/en.2005-0280
14. Lee S-H, Lee J-K, Ahn M-J, Kim DW, Sun JM1, Keam B, et al. Vandetanib in pretreated patients with advanced non-small cell lung cancer-harboring RET rearrangement: a phase II clinical trial. Ann Oncol. (2017) 28:292–7. doi: 10.1093/annonc/mdw559
15. Velcheti V. Phase 2 study of lenvatinib (LN) in patients (Pts) with RET fusion-positive adenocarcinoma of the lung. Ann Oncol. (2016) 27:416–54. doi: 10.1093/annonc/mdw383
16. Lin JJ, Kennedy E, Sequist LV, Brastianos PK, Goodwin KE, Stevens S, et al. Clinical activity of alectinib in advanced ret-rearranged non-small cell lung cancer. J Thorac Oncol. (2016) 11:2027–32. doi: 10.1016/j.jtho.2016.08.126
17. Plaza-Menacho I, Mologni L, Sala E, Gambacorti-Passerini C, Magee AI, Links TP, et al. Sorafenib functions to potently suppress RET tyrosine kinase activity by direct enzymatic inhibition and promoting RET lysosomal degradation independent of proteasomal targeting. J Biol Chem. (2007) 282:29230–40. doi: 10.1074/jbc.M703461200
18. Bronte G, Ulivi P, Verlicchi A, Cravero P, Delmonte A, Crinò L. Targeting RET-rearranged non-small-cell lung cancer: future prospects. Lung Cancer. (2019) 10:27–36. doi: 10.2147/LCTT.S192830
19. Das TK, Cagan RL. KIF5B-RET Oncoprotein signals through a multi-kinase signaling hub. Cell Rep. (2017) 20:2368–83. doi: 10.1016/j.celrep.2017.08.037
20. Yoh K, Seto T, Satouchi M, Nishio M, Yamamoto N, Murakami H, et al. Vandetanib in patients with previously treated RET-rearranged advanced non-small-cell lung cancer (LURET): an open-label, multicentre phase 2 trial. Lancet Respir Med. (2017) 5:42–50. doi: 10.1016/S2213-2600(16)30322-8
21. Ding L-W, Sun Q-Y, Tan K-T, Chien W, Mayakonda A, Yeoh AEJ, et al. Mutational landscape of pediatric acute lymphoblastic leukemia. Cancer Res. (2017) 77:390–400. doi: 10.1158/0008-5472.CAN-16-1303
22. Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. (2017) 377:454–64. doi: 10.1056/NEJMoa1614359
Keywords: acute lymphoblastic leukemia, RET, CCDC6-RET, novel mutation, FoundationOneHeme
Citation: Mejia Saldarriaga M, Steinberg A, Severson EA and Binder A (2019) A Case of CCDC6-RET Fusion Mutation in Adult Acute Lymphoblastic Leukemia (ALL), a Known Activating Mutation Reported in ALL. Front. Oncol. 9:1303. doi: 10.3389/fonc.2019.01303
Received: 25 April 2019; Accepted: 11 November 2019;
Published: 03 December 2019.
Edited by:
Ritu Gupta, All India Institute of Medical Sciences, IndiaReviewed by:
Deepshi Thakral, All India Institute of Medical Sciences, IndiaCopyright © 2019 Mejia Saldarriaga, Steinberg, Severson and Binder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mateo Mejia Saldarriaga, bWF0ZW9tZWppYXNAaG90bWFpbC5jb20=; Adam Binder, YWRhbS5iaW5kZXJAamVmZmVyc29uLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.