
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 31 May 2019
Sec. Molecular and Cellular Oncology
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.00234
 Xiangyi Kong1†
Xiangyi Kong1† Jie Zhai1†Chengrui Yan2†Yan Song3Jing Wang1*Xiaofeng Bai4*James A. L. Brown5,6*Yi Fang1*
Jie Zhai1†Chengrui Yan2†Yan Song3Jing Wang1*Xiaofeng Bai4*James A. L. Brown5,6*Yi Fang1*FOXN3 (forkhead box N3; CHES1: check point suppressor 1) belongs to the forkhead box (FOX) protein family. FOXN3 displays transcriptional inhibitory activity, and is involved in cell cycle regulation and tumorigenesis. FOXN3 is a tumor suppresser and alterations in FOXN3 are found in of a variety of cancers including melanoma, osteosarcoma, and hepatocellular carcinoma. While the roles of FOXN3 role in some cancers have been explored, its role in breast cancer remains unclear. Here we describe current state of knowledge of FOXN3 functions, and focus on its roles (known and potential) in breast cancer.
The Forkhead proteins (FOX) belongs to a subgroup of “helical-fold-helical” proteins (1). At the molecular level FOX proteins act as monomers directing transcription (through DNA binding), acting as a platform to coordinate transcription factors (2). Through this activity FOX proteins influence nucleosome location and structure, promoting gene activation (3). The DNA binding domain of the FOX proteins (“forkhead box”) is a highly conserved 100 amino acids region composed of three α helices (H1, H2, and H3) and two characteristic loop structures. These form a characteristic wing-like helix structure (“wing-shaped helix”), where the H3 motif represents the main recognition and DNA binding site (4, 5). Based on homology of DNA binding domains, it has been determined that the FOX protein has over 100 family members in different species of eukaryotic cells, including 17 subfamilies (families A-Q) (Tables 1, 2) (6–8).

Table 1. Forty-eight FOX family proteins are classified into 19 subfamilies.
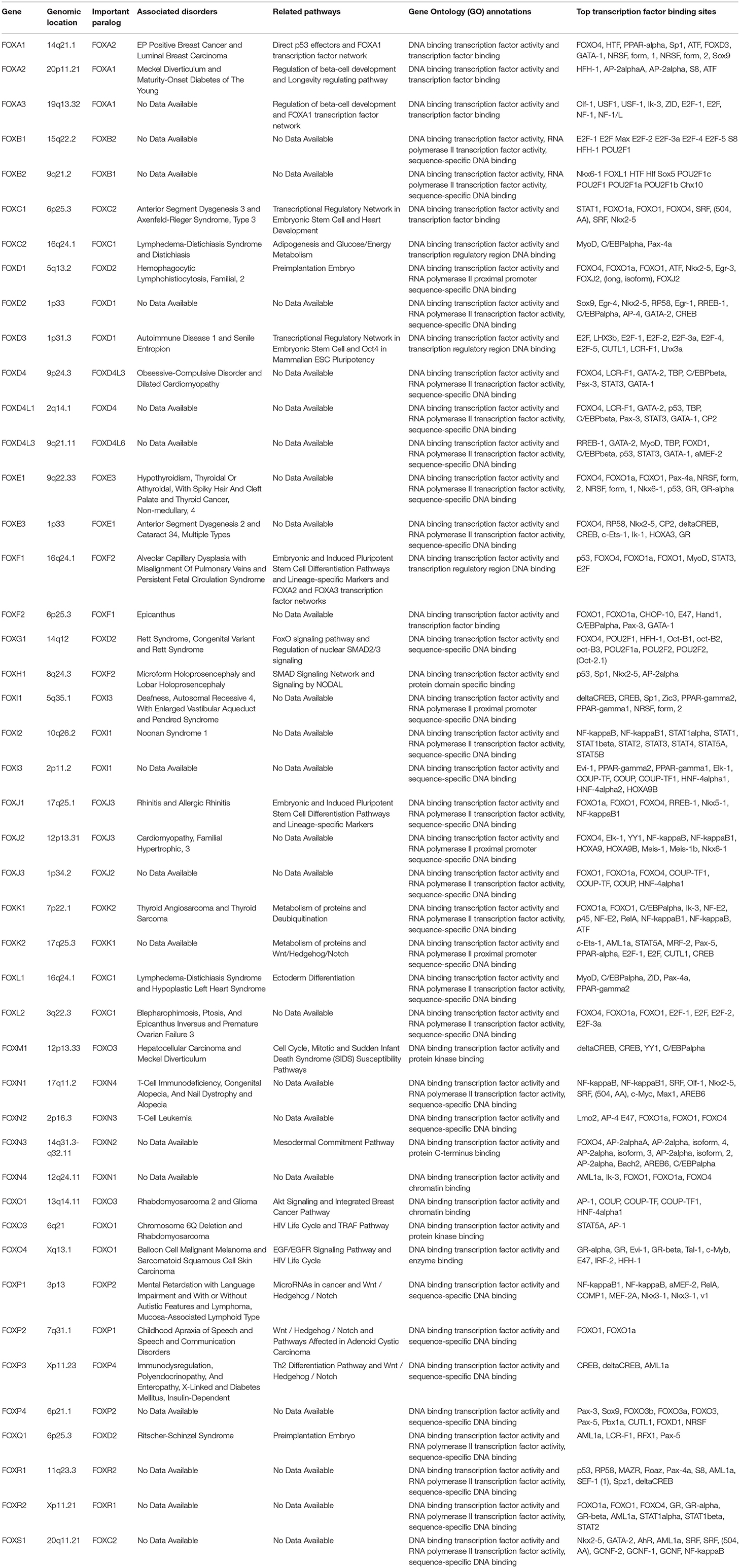
Table 2. FOX family members' gene card information.
As transcriptional regulators, the FOX protein family regulates the development of tissues, embryos as well as carbohydrate and lipid metabolism (9–11). Furthermore, FOX proteins are involved in regulating biological processes including the cell cycle (5, 12), homeostasis and immunization (13). Dysregulation of FOX family members can cause developmental malformation of tissues and organs, metabolic diseases, and importantly is associated with the development of tumors (14, 15). Down-regulation of the FOXO subfamily (regulating signal transduction pathways) is found in multiple tumors (including breast, prostate, glioblastoma, hematological malignancy, alveolar Rhabdomyosarcoma), leading to aberrant apoptosis, cell cycle, and DNA repair defects (16, 17). As many FOX proteins directly interact with kinases in multiple signaling pathways, they are attractive therapeutic targets for many diseases. Here we focus on the FOXN subfamily due to that the FOXN genes remain an understudied forkhead subfamily with little known about the transcriptional activity of the proteins within this group.
FOXN3 is among the few FOX family members that binds both a FORKHEAD (FKH) and a FORKHEAD-LIKE (FHL) sequence. A nice series of dendograms has been presented by Nakagawa et al. (18) (Figure 1). Rogers et al. has very recently (27 February, 2019) solved the crystal structures of FOXN3 bound to both FKH and FHL sites, respectively, and made very interesting observations: the same protein domain binds two different DNA sequences because the FOX domain recognizes the totality (sequence and backbone) of the element, and can bend it substantially (19). However, the DNA structure is different in the two complexes. These structures reveal how a single transcription factor binds two unrelated DNA sequences and the importance of DNA shape in the mechanism of bispecific recognition. Also, from the discovery, we could understand how the vast majority (around 80%) of precipitated chromatin in the ChIP-Seq experiment did not have either a FKH (around 18%) or a FHL (around 2–3%) sequence: FOXN3 mostly binds DNA through interaction with a partner (19).
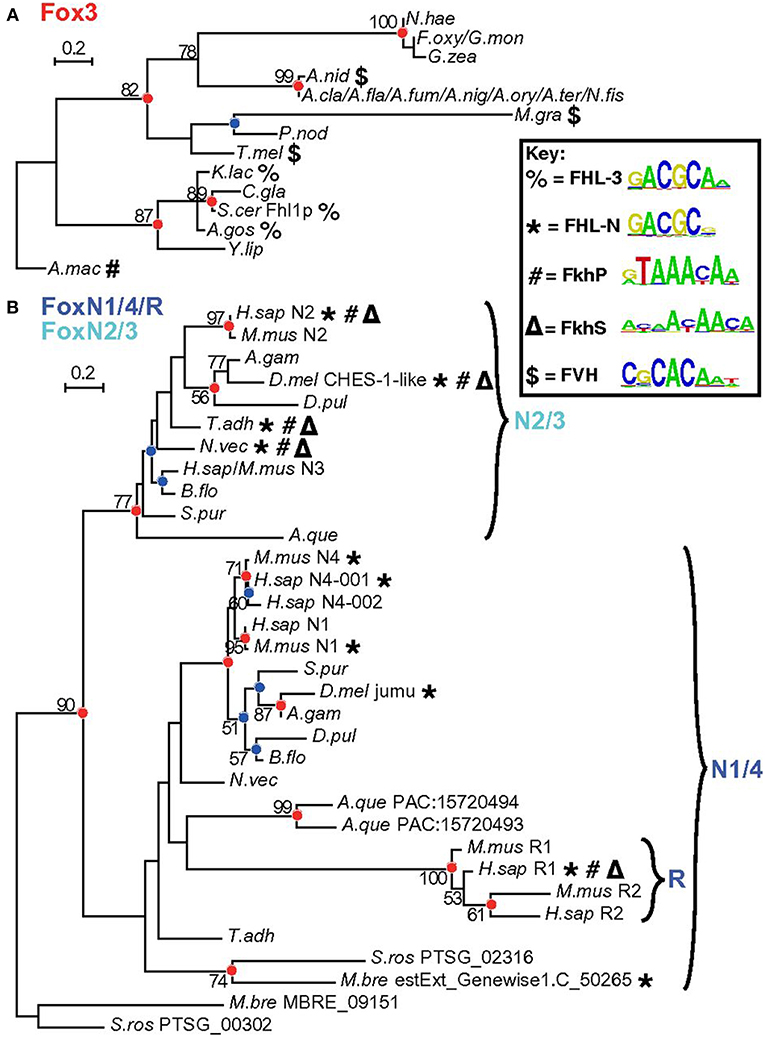
Figure 1. Detailed analysis of Fox3 and FoxN subfamilies. Maximum likelihood (ML) phylogenetic trees for Fox domains from a broader range of species for fungal Fox3 (A) and holozoan FoxN/R (B) clades. Red and blue circles indicate corresponding nodes. Bold symbols represent binding capacity for different motif classes. Reprinted from Nakagawa et al. (18). Copyright® 2013 National Academy of Sciences.
As a subfamily of the FOX family transcription factors, the FOXN protein family consists of six members: FOXN1 (Figure 2A), FOXN2 (HTLF) (Figure 2B), FOXN3 (CHES1) (Figure 2C), FOXN4 (Figure 2D), FOXN5 (FOXR1) (Figure 2E), and FOXN6 (FOXR2) (Figure 2F) (20). FOXN1 (WHN) regulates the growth of the keratinized epithelial cells and the differentiation of thymic epithelial cells required for T cell development (21, 22). In addition, FOXN1 is regulated by Wnt glycoproteins, important for regulating keratinocyte growth and differentiation of thymic epithelium (23).
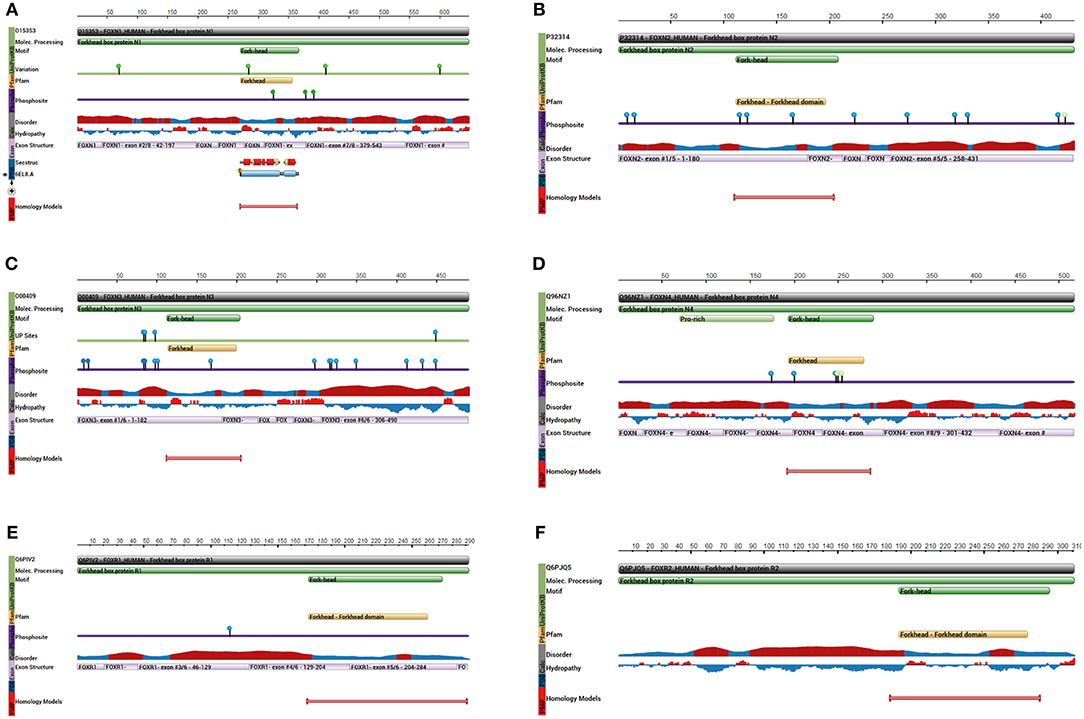
Figure 2. Key protein features for the indicated FOXN proteins. (A) The protein feature view for the FOXN1 protein. (B) The protein feature view for FOXN2 protein. (C) The protein feature view for FOXN3 protein. (D) The protein feature view for FOXN4 protein. (E) The protein feature view for FOXN5 protein. (F) The protein feature view for FOXN6 protein. All protein structures generated using the Protein Data Bank (RCSB PDB; http://www.rcsb.org/pdb/protein/).
FOXN2 (human T-cell leukemia factor: HTLF) was identified as binding to the long-chain terminal repeat sequence of the human T-cell leukemia virus (HTLV-ILTR), participating in HTLV-ILT transcriptional regulation (24).
FOXN3 is the primary focus of this review, and will be discussed extensively below in the following sections.
FOXN4 is involved in the formation of amacrine cells and horizontal cells in the retina, and can upregulate the expression of specific retinal factors (25). The chimeric expression of FOXN4 with its mutants may serve as a trigger, thereby ensuring the diversity of the neurons in the V2 domain of the ventral spinal cord. For instance, FOXN4lacZ/lacZ is an important mutant type of FOXN4. Li etc. showed that during murine lung development the FOXn4 transcription factor is expressed in proximal airways by a subpopulation of postmitotic epithelial cells which are distinct from basal and ciliated cells and of which only a small fraction are Clara cells. FOXN4lacZ/lacZ causes dilated alveoli, thinned alveolar walls and reduced septa in the distal lung but no overt gross alterations in proximal airways. The alveolar defects in mutants may result from decreased PDGFA signaling and reduced surfactant SFTPB expression (26).
Additionally, FOXN4 can regulate differentiation of the multi-center cells (MCC), and is involved in the development of multiple organelles including nucleus and mitochondria (25).
FOXN5 (FOXR1) was originally reported as a candidate tumor suppressor gene, with mutations leading to abnormal structural changes closely associated with the occurrence of neuroblastoma (27). FOXN5 is a Protein Coding gene. Gene Ontology (GO) annotations related to this gene include DNA binding transcription factor activity and RNA polymerase II transcription factor activity, sequence-specific DNA binding.
FOXN6 (FOXR2) regulates the proliferation of the hepatocellular carcinoma cells, and dysregulation promotes tumor formation in chronic hepatocellular carcinoma (HCC). It is believed that FOXN6 is a new promising therapeutic target for HCC (28). They reported that FOXN6 is frequently up-regulated in 25/42 (59.5%) of HCC specimens compared with neighboring non-cancerous tissues in messenger RNA (mRNA) level and further confirmed by immunohistochemistry analysis in protein level. Moreover, FOXN6 overexpression facilitated the development of tumor xenografts in nude mice model (28).
FOXN3 (CHES1, PRO1635, or C14orf116) was originally isolated by Pati et al. in yeast cells with multiple checkpoint mutations (29). FOXN3 suppresses a number of DNA damage-activated checkpoint mutations in S. cerevisiae, including mec1, rad9, rad24, dun1, and rad53. FOXN3 suppression of sensitivity to DNA damage is specific for checkpoint-defective strains, in contrast to DNA repair-defective strains. Presence of FOXN3 but not a control vector resulted in G2 delay after UV irradiation in checkpoint-defective strains, with kinetics, nuclear morphology, and cycloheximide resistance similar to those of a wild-type strain. FOXN3 can also suppress the lethality, UV sensitivity, and G2 checkpoint defect of a mec1 null mutation. In contrast to this activity, FOXN3 had no measurable effect on the replication checkpoint as assayed by hydroxyurea sensitivity of a mec1 strain (29). The FOXN3 gene (in humans located at 14q31.3 ~ q32. 11), is 463bp long and contains 7 exons. The protein symbol for FOXN3 gene is O00409-FOXN3_HUMAN with a size of 490 amino acids, which interacts through its C-terminus with the C-terminus of SNW1/SKIP. The structure of FOXN3 is shown in Figure 2C. FOXN3 is the only member of the FOXN family with no transcriptional binding domain (lacking transcriptional activation domains), while its carboxyl terminal serves as the transcriptional repressor (30, 31). Fusing the carboxyl terminal of FOXN3 onto a heterologous DNA binding domain allows continuous suppression of target genes (32).
In humans FOXN3 is widely expressed in many organs (including the liver, pancreas, kidneys, lungs and bone marrow), where FOXN3 plays indispensable roles in the development of these tissues (32, 33). This was found to be true in other vertebrates, where Schmidt et al. confirmed in animal models of Xenopus laevis and carp larvae that FOXN3 is critical for the development of new cartilage in the brain, and influences the development of muscle morphology indirectly (34). FOXN3 was found in studies on the regulation of fasting blood glucose, with patients carrying the SNP (rs8004664) found to have gene overexpression under fasting state. This up-regulation of FOXN3 leads to increased inhibition of the Myc gene (by FOXN3), and stimulates FOXN3 regulation of blood glucose levels. This role lead to FOXN3 being defined as a pathomechanical regulator of the fasting blood glucose (11). A paradigm-shifting study on FOXN3 intron polyadenylation in leukemia that appeared in August 2018 in Nature discussed FOXN3 intron polyadenylation, and confirms that MYC is a direct repressive target of FOXN3 (35). The tumor-suppressor gene (TSG) MGA is targeted by phenocopy truncating mutations in chronic lymphocytic leukemia (CLL) and solid cancers. MGA negatively regulates the MYC transcriptional program and represses genes with MYC- and E2F-binding sites in a Polycomb-dependent manner (35). Expression of MGA from constructs validated MGA intronic polyadenylation detected in CLL cells and confirmed the repressive effect of MGA on MYC target gene expression in malignant B cells (35). Notably, on genes with binding sites for both MYC and E2F, MGA IPA acts as dominant-negative regulator of full-length MGA as it significantly induced the expression of 5 out of 6 genes in cells that endogenously express fulllength MGA. In addition, the IPA isoform of the transcriptional repressor FOXN3 derepressed its oncogenic targets MYC and PIM2 (35).
FOXN3 functions in the DNA damage response, where FOXN3 is responsible for S phase cell cycle arrest (in drosophila) (36–38) Upon DNA damage FOXN3 can induce quiescence, allowing damaged cells to persist (33). In yeast cells with cell cycle checkpoint defect (such as mutations of mec1, rad24, dun1, rad9, and rad53), FOXN3 can bind to the Sin protein in the Sin3/Rpd3 HDAC (histone deacetylation) complex to inhibit deacetylation of histones. This facilitates DNA damage repair and a G2/M phase arrest, thereby compensating for the cell cycle checkpoint defects (39). Checkpoints are eukaryotic DNA damage-inducible cell cycle arrests at G1 and G2. Checkpoint suppressor 1 suppresses multiple yeast checkpoint mutations including mec1, rad9, rad53, and dun1 by activating a MEC1-independent checkpoint pathway. Alternative splicing is observed at the locus, resulting in distinct isoforms. The main functions of FOXN3 are shown in Figure 3.
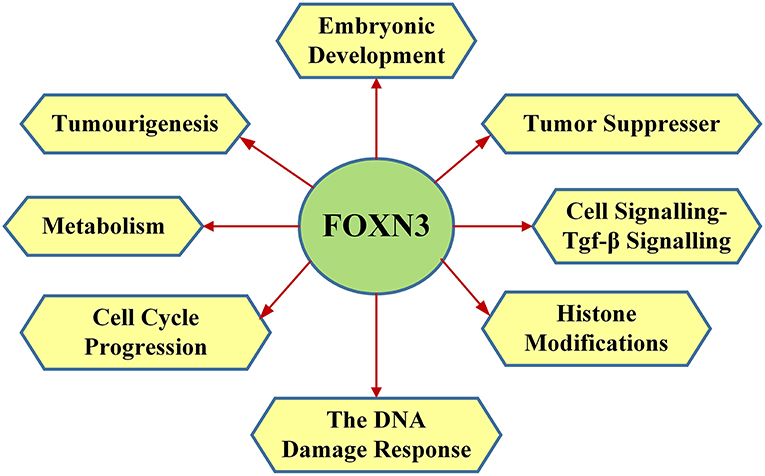
Figure 3. The main functions of FOXN3 including embryonic development, tumor suppresser, cell signalling-TGF-β signaling, histone modifications, the DNA damage response, cell cycle progression, metabolism, and tumourigenesis.
The transforming growth factor β (TGF-β) 1 superfamily members represent multifunctional cell signaling proteins that include TGF-β, bone morphogenetic protein (BMP), activin, inhibin, and growth differentiation factors. As such, the TGF-β1 family regulates many key cellular processes during growth and development (Figure 4). The TGF-β signaling pathway sequentially activates Smad 2 and Smad 3 by binding to two cell surface receptors (serine and threonine kinases), so that they can be translocated together with Smad4 to the nucleus, prompting the binding between Smad heterodimer and Smad binding element (SBE) and acting on the promoter of the target gene together with other nuclear factors (40). The Smad proteins also interact with other nuclear factors (such as the Ski, Sno, and nuclear hormone receptors), thereby regulating the transduction of TGF-β mediated signaling.
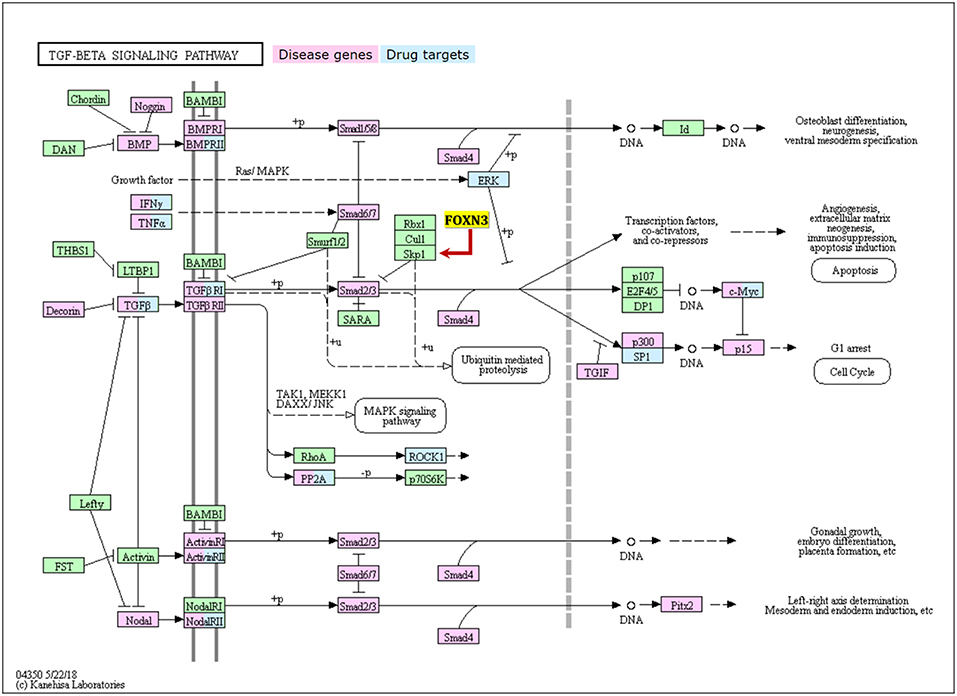
Figure 4. The TGF-β signalling pathway with showing key disease genes, drug targets, and the regulation relationship with FOXN3.
Mutations in Ski and Sno lead to oncogenic transformations, by blocking the transduction of the TGF-β signaling (41). The SKIP protein, a highly conserved transcription adaptor protein, is a nuclear hormone receptor co-activator. It was named initially for its recognition of the two-hybrid screening protein interacting with the V-Ski proto-oncogene. The protein can regulate the proliferation and differentiation of cells, and is involved in the transduction of multiple signaling pathways (42). A study in liver and melanoma malignancy cells (where FOXN3 expression has been reported to be decreased compared to corresponding normal tissue) showed that FOXN3 protein can act on the SKIP through its COOH terminal, repressing SKIP mediated TGF-β signaling (42).
In the TGF-β/smad cell signaling pathway, the Ski protein, as an inhibitor of the Smad2/3 protein transcription complex, inhibits the promoter of TGF-β by interacting with the Smad2/3 proteins, thereby inhibiting TGF-β/smad signal transduction and attenuating the anti-tumor effects of the TGF-β signaling pathway (43).
SKIP is an activator of Smad, in contrast to repressive Ski, competing with Ski to up-regulate the TGF-β signaling (44, 45). It has been proposed that this function of SKIP is a tumor-suppressive regulation of the TGF-β signaling pathway (45). c-Abl is a non-receptor-type tyrosine kinase that plays an important role in cell proliferation, migration, apoptosis, and fibrosis. c-Abl phosphorylates SKI-interacting protein (SKIP), a nuclear cofactor of the transcription factor Smad3. The c-Abl inhibitor imatinib suppressed TGF-β-induced expression of Smad3 targets as well as SKIP/Smad3 interaction (45). TGF-β-stimulation induced tyrosine phosphorylation of SKIP, and this phosphorylation was suppressed by imatinib. Tyr292, Tyr430, and Tyr433 residues in SKIP were shown to be involved in c-Abl-mediated phosphorylation. Phosphomimetic glutamic acid substitution at Tyr292 in SKIP enhanced, whereas its phospho-dead phenylalanine substitution attenuated TGF-β-induced SKIP/Smad3 interaction (45). Moreover, the phosphomimetic mutant of SKIP augmented transcriptional activity of Smad3. These results suggest that c-Abl phosphorylates SKIP mainly at Tyr292 and promotes SKIP/Smad3 interaction for the full activation of TGF-β/Smad3 signaling (45). What's more, SKIP can act on Core Binding Factor 1 (CBF1), thus inhibiting transcription in the Notch-mediated signaling pathway required for tumor growth (46). A recent work found that the FOXN3 regulated TGF-β/smad cell signaling pathway modulating the proliferation and differentiation of hematopoietic stem cells, and closely associated with the development of leukemic cells (47). However, it remains elusive whether FOXN3 mediates or causes the malignant transformation (through the TGF-β/smad cells signaling pathways).
A meta-analysis of the FOXN3 gene (using the oncomine database) demonstrated that among 15 different malignancies (including liver cancer, lung cancer, colon cancer, prostate cancer, laryngeal cancer, glioblastoma multiform, and lymphoma) there was a very marked down-regulation of the FOXN3 gene. Taken together, this supports the hypothesis that the regulatory network including FON3 is indeed valid, even across diverse cancer types, and could serve as a guide for future strategies aimed at combating colorectal cancer metastasis. Also, this suggests that FOXN3 may be an important tumor suppressor gene (48, 49).
Nuclear paraspeckle assembly transcript 1 (NEAT1), located on chromosome 11q13.1, is a lncRNA that encodes two transcriptional variants, namely NEAT1-1 (3.7 kb) and NEAT1-2 (23 kb). Previous studies have revealed that NEAT1 functions as an essential structural component of a nuclear domain called paraspeckle, which participates in gene regulation mainly by nuclear retention of proteins and RNAs (50). Cumulatively, numerous studies have identified the aberrant expression and prognostic value of NEAT1 in various types of tumors including breast cancer (51), colorectal cancer (52), liver cancer, ovarian cancer (53), cervical cancer (54), gastric cancer (55), prostate cancer (56), lung cancer (57), and papillary thyroid cancer (58), the majority of which suggest NEAT1 as an oncogene that is overexpressed in tumors compared to their respective normal tissues and promotes tumor cell progression. This lncRNA is retained in the nucleus where it forms the core structural component of the paraspeckle sub-organelles. It may act as a transcriptional regulator for numerous genes, including some genes involved in cancer progression. Diseases associated with NEAT1 include Relapsing-Remitting Multiple Sclerosis and Dengue Disease. NEAT1 could mediate TGF-β1 expression by competitively sponging miR-339-5p (59). NEAT1 induced osteosarcoma cell proliferation and cell mobility by binding to miR-339-5p and increasing TGF-β1 in osteosarcoma (59). NEAT1 can function as a ceRNA by sponging hsa-mir-139-5p (60). The authors speculated that NEAT1 can modulate TGF-β1 expression by sponging hsa-mir-139-5p in hepatocellular carcinoma. These data indicates that targeting the NEAT1/hsa-mir-139-5p/TGF-β1 axis could be a new strategy some cancers (60).
Importantly, NEAT1 was found significantly up-regulated in breast cancer tissues and cell lines (61). NEAT1 can promote the growth of breast cancer cells by interacting with miR-101, which acts as tumor suppressor in several cancers by directly targeting EZH2 (61–63). Supporting this, Zhang et. al. found that NEAT1 was highly expressed in breast cancer tissue, and the NEAT1 expression correlated to tumor size and lymph node metastasis (64). This suggests that increased NEAT1 expression can act as an oncogene (in breast cancer), promoting proliferation and metastasis of breast cancer (64).
The scaffolding protein Switch-Independent 3A (SIN3A) has been implicated in breast cancer development (65, 66). SIN3A contains paired amphipathic helix (PAH) domains, which are important for protein-protein interactions and may mediate repression by the Mad-Max complex. Diseases associated with SIN3A include Witteveen-Kolk Syndrome and Rett Syndrome. Among its related pathways are C-MYB transcription factor network and SMAD Signaling Network. GO annotations related to this gene include DNA binding transcription factor activity and transcription factor binding.
Interestingly, Li et al. reported that NEAT1 was essential for FOXN3 interactions with the SIN3A complex. NEAT1 was found to be induced by estrogen in breast cancer cells (MCF-7), where. ERα (Estrogen receptor alpha) knock-down resulted in repression of NEAT1 induction. ChIP-Seq experiments found that the FOXN3-NEAT1-SIN3A complex suppressed a group of genes, including GATA3 and TJP1 (genes regulating cell maintenance and differentiation). It was found that ERα signaling and FOXN3-NEAT1-SIN3A complex function in an inhibitor feedback loop, under mammary epithelial cell growth ERα induced NEAT1 leading to SIN3A and FOXN3 mediated inhibition of GATA3 and TJP1 expression. Conversely, when conditions favored differentiation or the maintenance, ERα signaling reduced the growth signal. This regulatory mechanism contributed to the balance or choice (between differentiation or maintenance) of mammary epithelial cells. NEAT1 is induced by estrogen and participates in transcription regulation by the FOXN3-NEAT1-SIN3A complex, transcriptional targeting of ERα by the FOXN3-NEAT1-SIN3A complex implies that there exists a negative-feedback loop in ERα+ breast cancer cells between ERα and the FOXN3-NEAT1-SIN3A complex, in which ERα transactivates NEAT1, which is assembled into the FOXN3-NEAT1-SIN3A complex, and this complex, in turn, transrepresses ERα. The balance in this regulatory mechanism is required for normal development of mammary epithelial cells. The disturbance in this balance leads to breast cancer development and metastasis. The up-regulated FOXN3-NEAT1-SIN3A complex promotes EMT and invasion of breast cancer cells in vitro and the dissemination and metastasis of breast cancer in vivo (32).
Katoh et al. employed the Array-CGH technique to elucidate the characteristics of the overall chromosomal mutation of HCC and revealed the new hepatocellular carcinoma-associated genes, FOXN3 and caspase3. By using such methods as hierarchical clustering, according to HCC, they assigned 2 subgroups according to the chromosomal variation patterns, and investigated its relationship with the relevant clinical data, further confirm that HCC can be divided into genetically similar subgroups based on the heterogeneity of genetic spectra, and that the optimal therapeutic drugs for various HCCs can be expected to be developed based on the different types of genome chromosome of HCC (67). Sun et al. detected the expressions of FOXN3 in HCC and the matched normal tissues by means of real-time quantitative PCR and Western blot, and they found that FOXN3 was significantly down-regulated in HCC. In both in vivo and in vitro experiments, FOXN3 can inhibit the proliferation of liver cancer cells significantly (68). In addition, further mechanistic studies have shown that the forkhead box region of FOXN3 can bind to the promoter of the E2F5 transcription factor and inhibit the expression of E2F5 mRNA and protein, thereby inhibiting the proliferation of HCC cells. This mechanism is expected to be a therapeutic target for liver cancer (68).
The toll-like receptor (TLR) is one of the innate immunospecific receptors, and the TLR9 signals in both in vivo and in vitro experiments could promote the metastasis of human lung cancer cells (69). Given that CpG oligo-deoxynucleotides (CpG ODNs) can activate the TLR9 signaling pathways effectively in human lung cancer cells, they used the miRNA microarray technique to detect the expression of miRNA of 95D cells treated or untreated with CpG ODNs and look for the differentially expressed miRNAs after treatment with CpG ODNs. They found that the expression level of miR-574-5p was significantly up-regulated with the increase in CpG ODNs dose and the eclipse of act time. To clarify the potential effects of miR-574-5p on human lung cancer cells under the influence of TLR9 signals, researchers used the TLR9 signaling inhibitor chloroquine and the MyD88 inhibitory peptide to block the signaling pathway in both in vivo and in vitro experiments, and found that both chloroquine and MyD88 inhibitory peptide could significantly inhibit the up-regulation of CpG ODN-induced miR-574-5p in 95D cells, further confirming that miR-574-5p is regulated by the TLR9 signaling pathway. Further in vitro and in vivo experiments have shown that miR-574-5p can promote the proliferation of the human lung cancer tumor cells. In order to determine the exact mechanisms underlying miR-574-5p in the progression of TLR9 signaling-induced human lung cancer tumors, they selected multiple possible target genes (calcoco1, Rfx4, CD96, chex1, FOXi2, dgkg, znf589, zdhhc14, ccdc88c) using Target Scan and Miranda and conducted real-time quantitative PCR analysis. They found that the expression levels of FOXN3 mRNA and protein were significantly increased in the 95D cells transfected with the miR-574-5p inhibitor; they also found in the luciferase reporter assay that FOXN3 is a target gene of miR-574-5p. It can lead to the G0/G1 phase arrest by regulating the cell cycle of the human lung cancer cells, and affect the expression of CDK2 to further regulate the proliferation of the lung cancer cells, providing a new potential therapeutic target for the treatment of cancer (70).
Given Sun's previous research results (68), they found when using the oncomine database to analyze the sample datasets in the normal colon tissue and colon adenocarcinoma tissue that compared with the normal tissues, the expression of FOXN3 was decreased in colon adenocarcinoma tissue. By using such techniques as fluorescence quantitative PCR, Western blot and immunohistochemistry, they found that the mRNA and protein levels of FOXN3 were decreased in the colon cancer tissues. In the subsequently different pretreated colon cancer mouse model experiments, Western blot revealed that FOXN3 was expressed in the normal colon tissues and tumors, demonstrating the reduction in the level of FOXN3 protein. To further explore the effect of FOXN3 in the progression of colon cancer, they overexpressed and knocked down the FOXN3 gene in colon cancer cell line, and then used such experimental methods as MTT, soft agar assay, cell migration assay, cell invasion, and flow analysis, whereby they confirmed that FOXN3 could inhibit the proliferation, migration, and invasion of colon cancer cells and inhibit cell cycle progression. To explore the potential mechanisms underlying FOXN3 inhibition of the growth, migration and invasion of colon cancer cells, they used luciferase reporter gene assay to detect the activity of FOXN3 in various signaling pathways, whereby they found that the overexpression of FOXN3 inhibited the activity of Top flash (the target gene for the β-catenin/TCF signaling pathway when treating was performed using lithium chloride). In addition, they found by silencing FOXN3 that the expressions of N-cadherin, Snail, and c-Myc (three downstream genes of the β-catenin/TCF signaling pathway) were up-regulated and they promoted the activation of the β-catenin/TCF signaling pathway. This results further confirmed that the down-regulation of FOXN3 activates the β-catenin/TCF signaling pathway, thereby promoting the growth, migration and invasion of the colon cancer tumor cells. TCF4 binds to β-catenin to initiate transcription in the nucleus (33). Through the GST pull-down assay, they found that FOXN3 and β-catenin could form a complex in the colon cancer cell lines. By using the immunoprecipitation assay in cell lines overexpressing FOXN3 and β-catenin subsequently, they found that endogenous β-catenin and FOXN3 could also form a complex. At the same time, they used the co-immunoprecipitation assay in the normal cell line 293T, and discovered that the exogenously expressed FOXN3, TCF4, and β-catenin could form complexes. Then, they used immunoprecipitation in colon cancer cell lines, and found in the FOXN3 antibody-positive results that the capability of TCF4 to form complexes with β-catenin was weaker than in the FOXN3 antibody-negative results, showing that FOXN3 inhibits the catenin/TCF signaling pathway by blocking the binding between β-catenin and TCF4. To investigate the effect of FOXN3 in the regulation of metastasis of colon cancer cells, they silenced the FOXN3 gene and injected the luciferase-labeled colon cancer cells into nude mice by way of the left ventricle. Monitoring revealed that FOXN3 was down-regulated, which promoted the metastasis of the colon cancer cells, and immunohistochemistry showed that knocking out the expression of FOXN3 promotes tumorigenesis of colon cancer cells in the colonic tissues. This study showed that FOXN3 can inhibit the progression of the colon cancer, and that restoration of the function of FOXN3 is expected to become a novel therapeutic strategy for the colon cancer (71).
In the laryngeal cancer and paracancerous mucosa specimens, gene chip data analysis showed that the FOXN3 gene was significantly down-regulated while the ADAM12 gene expression was significantly up-regulated, and subsequent real-time quantitative PCR for the validation of the mRNA expressions in 24 cases of laryngeal cancer and 17 cases of normal mucosa showed that the results were consistent with the those of detection with gene chip. As a result of the high expression of ADAM12 and the low expression of CHES1 in laryngeal carcinoma, they analyzed the presumed diagnostic values of the ratios of expressions of CHES1 and ADAM12 genes by setting the threshold of the ratio of the two genes (ADAM12/CHES1) at 0.4. After validation in 24 laryngeal cancer and 17 normal mucous membranes, they found that the ratio of the two genes could be used to distinguish between tumor and normal tissues, with the correct validation rate being as high as 70%. Therefore, it is expected that the ratio of ADAM12/CHES1 can be taken as a molecular marker to distinguish between tumor and non-tumors, and that it may become the best diagnostic indicator for laryngeal cancer (72).
The abnormal expression of FOXN3 protein is of great significance in hematological tumors. When Stefan Nagel et al. used the gene chip technique to find abnormal expressions of the FOX gene in the Hodgkin lymphoma (HL) cell lines and patients, they found that the expression levels of the FOXC1 gene and the FOXD gene were increased, and that the transcriptional levels of FOXN3, FOXO1, and FOXP1 genes were reduced. They discovered when using enrichment analysis to explore the mechanisms underlying the regulation of FOX gene expression that there was a potential activation of the JAK-STAT signaling pathway when FOXD1 and FOXN3 regulate transcription. In further studies, they found that TGF-β could inhibit the expression of FOXD1 in HL cell lines pretreated with IL13, TGF-β, and WNT5B using RQ-PCR, and that the WNT signaling pathway could reduce the expression of FOXN3. Therefore, they determined that FOXD1 and FOXN3 are affected by specific signaling pathways, and that these pathways have already shown abnormal activity in HL (73).
FOXN3 protein can inhibit the expression of the PIM2 gene and protein biosynthesis, thereby regulating cell proliferation (73); similarly, studies on three different types of lymphomas revealed that the expression of CHES1 in primary exudative lymphoma, diffuse large B-cell lymphoma and hairy cell leukemia was reduced. After overexpression of FOXN3 in lymphoma cell lines, they found through qPCR, Western blot and immunoprecipitation found that CHES1 can bind to the PIM2 gene directly to reduce the phosphorylation level of 4EBP1, the target of PIM2, leading to a decreased level of PIM2 and thus promoting tumorigenesis. This study confirmed that in lymphomas, the deletion of FOXN3 leads to increased level of the target gene PIM2, thereby weakening the inhibitory effect of FOXN3 on tumor growth. Specifically, PIM2 represents an important oncogene as well as the therapeutic target for multiple solid tumors and hematological tumors (74, 75).
In a study on the changes in the expression levels of genes of T-cell large granular lymphocyte leukemia (T-LGL), they used such research methods as multicolor fluorescence in situ hybridization, gene sequencing and LM-PCR and found that there was an increase in FOXN3 gene expression in a T-LGL patient with gene amplification in 14q, indicating that sub-genomic rearrangement alters the gene expression in patients with T-LGL, and that it is associated with the pathogenesis and survival pattern of T-LGL (76).
In a related study on the T-cell leukemia tumor suppressor network, the FOXN3 gene was down-regulated in T-lymphocytic leukemia (T-ALL) significantly. Analysis of the target genes revealed that the homologous gene ZHX1 could be directly activated by FOXN3. In T-ALL, a decreased expression of FOXN3 can lead to decrease in the level of ZHX1, while ZHX1 is mainly expressed in the normal T cells. Its homologous gene ZHX2 is more vigorously expressed in the normal B cells, and ZHX1 and ZHX2 represent the TS gene in both T and B cell malignancies, respectively. Generally speaking, the TS gene regulates proliferation and apoptosis of cells via synergy with other TS genes or oncogenes, indicating that FOXN3 and ZHX1 form a regulatory network in the development of T cells and the genesis of leukemia. This also shows that FOXN3 can interact with other transcription factors or transcriptional regulators to inhibit transcription indirectly, jointly regulating proliferation and differentiation of cells (77).
Zhang et al. used the high-resolution aCGH technique (resolution, 2kb) to scan the bone marrow leukemia cell genome in 24 patients with acute myeloid leukemia-M5(AML-M5), and found 2 (8.3%) patients had deletion of chromosome 14 involving the FOXN3 gene. Then, they employed qRT-PCR to detect the expression levels of FOXN3 in the bone marrow of 97 cases of acute leukemia [78 AML cases, and 19 acute lymphocytic leukemia (ALL) 19 cases] and 16 normal humans, and found that the expression levels of FOXN3 in patients with ALL and AML were decreased to varying degrees, especially in AML patients in whom the expression level of FOXN3 was significantly different from that in the normal controls. Thus, it can be concluded that FOXN3 may serve as an anti-oncogene in AML (78).
FOXN3 is mostly down-regulated in most solid tumors and hematological malignancies, and up-regulated in only a small number of diseases. However, the specific functions of FOXN3 in the cells of mammals, including humans, and its mechanisms of action in tumor cells have not yet been clearly confirmed. In particular, the mechanisms of action of FOXN3 in AML is still elusive, and whether FOXN3 mediates the malignancy transformations of hematopoietic stem cells through the TGF-beta/smad cell signaling pathway is still unknown. We now speculate that FOXN3, as a tumor suppressor gene, is involved in the development of leukemia. The deletion or mutation of the FOXN3 gene may lead to a decreased level of FOXN3 expression and affect the formation of the FOXN3-SKIP complex, thereby resulting in the relatively increased inhibition of the smad2/3 transcription complex by Ski. Inhibition of the negative regulation of the TGF-beta-mediated signaling pathway on cell proliferation leads to imbalanced proliferation and differentiation of the hematopoietic stem cells and induces leukemia. In the future, it is expected to further elucidate the molecular biology and cytogenetic basis of a low expression of FOXN3 in patients with AML. In addition, the mechanisms of the targeted protein of the FOXN3 transcription factor may be discovered through further studies, which will give insight into the novel tumor suppressor gene for the complex network of the development of leukemia.
XK, JZ, CY, YS, JW, XB, JB, and YF wrote and edited the paper. XK, JB, and YF final approval of the paper.
This study was supported by grants from Natural Science Foundation of China (No. 81872160), Beijing Municipal Natural Science Foundation (key program, No. 7191009), Capital Public Health Education, Beijing Science and Technology Program (No. Z171100000417028), and Chinese Academy of Medical Sciences Initiative for Innovative Medicine (No. 2017-I2M-3-020). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank our colleagues of Department of Breast Surgical Oncology, China National Cancer Center/Cancer Hospital, Chinese Academy of Medical and Peking Union Medical College.
FOXN3, forkhead box N3; CHES1, check point suppressor 1; FOX, forkhead box; HTLF, human T-cell leukemia factor; ILTR, long-chain terminal repeat; PDGFA, platelet-derived growth factor A; SFTPB, surfactant-associated protein B; MCC, multi-center cell; GO, Gene Ontology; HCC, hepatocellular carcinoma; mRNA, messenger RNA; TGF-β, transforming growth factor β; BMP, bone morphogenetic protein; SBE, Smad binding element; SKIP, Ski-interacting protein; CBF1, Core Binding Factor 1; NEAT1, nuclear paraspeckle assembly transcript 1; SIN3A, Switch-Independent 3A; PAH, paired amphipathic helix; ERα, Estrogen receptor alpha; TLR, toll-like receptor; CpG ODNs, CpG oligodeoxynucleotides; HL, Hodgkin lymphoma; T-LGL, T-cell large granular lymphocyte leukemia; AML, acute myeloid leukemia; ALL, acute lymphocytic leukemia.
1. Kaufmann E, Knochel W. Five years on the wings of fork head. Mech Dev. (1996) 57:3–20. doi: 10.1016/0925-4773(96)00539-4
2. Ikeda T, Uno M, Honjoh S, Nishida E. The MYST family histone acetyltransferase complex regulates stress resistance and longevity through transcriptional control of DAF-16/FOXO transcription factors. EMBO Rep. (2017) 18:e201743907. doi: 10.15252/embr.201743907
3. Nazario-Toole AE, Robalino J, Okrah K, Corrada-Bravo H, Mount SM, Wu LP. The splicing factor RNA-binding fox protein 1 mediates the cellular immune response in drosophila melanogaster. J Immunol. (2018) 201:1154–64. doi: 10.4049/jimmunol.1800496
4. Zhang W, Duan N, Song T, Li Z, Zhang C, Chen X. The emerging roles of forkhead box (FOX) proteins in osteosarcoma. J Cancer. (2017) 8:1619–28. doi: 10.7150/jca.18778
5. Bicknell KA. Forkhead (FOX) transcription factors and the cell cycle: measurement of DNA binding by FoxO and FoxM transcription factors. Methods Mol Biol. (2005) 296:247–62. doi: 10.1385/1-59259-857-9:247
6. Ostrow AZ, Kalhor R, Gan Y, Villwock SK, Linke C, Barberis M, et al. Conserved forkhead dimerization motif controls DNA replication timing and spatial organization of chromosomes in S. cerevisiae. Proc Natl Acad Sci USA. (2017) 114:E2411–9. doi: 10.1073/pnas.1612422114
7. Wang J, Li W, Zhao Y, Kang, Fu W, Zheng X, et al. Members of FOX family could be drug targets of cancers. Pharmacol Ther. (2018) 181:183–96. doi: 10.1016/j.pharmthera.2017.08.003
8. Munoz-Pedreros A, Yanez J, Norambuena HV, Zuniga A. Diet, dietary selectivity and density of South American grey fox, Lycalopex griseus, in Central Chile. Integr Zool. (2018) 13:46–57. doi: 10.1111/1749-4877.12260
9. Polonen IJ, Partanen KH, Jalava TK, Toivonen VF. Effect of dietary glycine and benzoate level on benzoate metabolism in mink (Mustela vision), blue fox (Alopex lagopus), and raccoon dog (Nyctereutes procyonoides). J Anim Sci. (2000) 78:976–86. doi: 10.2527/2000.784976x
10. Saavedra-Garcia P, Nichols K, Mahmud Z, Fan LY, Lam EW. Unravelling the role of fatty acid metabolism in cancer through the FOXO3-FOXM1 axis. Mol Cell Endocrinol. (2018) 462:82–92. doi: 10.1016/j.mce.2017.01.012
11. Karanth S, Zinkhan EK, Hill JT, Yost HJ, Schlegel A. FOXN3 regulates hepatic glucose utilization. Cell Rep. (2016) 15:2745–55. doi: 10.1016/j.celrep.2016.05.056
12. Barger CJ, Zhang W, Hillman J, Stablewski AB, Higgins MJ, Vanderhyden BC, et al. Genetic determinants of FOXM1 overexpression in epithelial ovarian cancer and functional contribution to cell cycle progression. Oncotarget. (2015) 6:27613–27. doi: 10.18632/oncotarget.4546
13. Fu SH, Yeh LT, Chu CC, Yen BL, Sytwu HK. New insights into Blimp-1 in T lymphocytes: a divergent regulator of cell destiny and effector function. J Biomed Sci. (2017) 24:49. doi: 10.1186/s12929-017-0354-8
14. Szafranski P, Gambin T, Dharmadhikari AV, Akdemir KC, Jhangiani SN, Schuette J, et al. Pathogenetics of alveolar capillary dysplasia with misalignment of pulmonary veins. Hum Genet. (2016) 135:569–86. doi: 10.1007/s00439-016-1655-9
15. Szafranski P, Herrera C, Proe LA, Coffman B, Kearney DL, Popek E, et al. Narrowing the FOXF1 distant enhancer region on 16q24.1 critical for ACDMPV. Clin Epigenetics. (2016) 8:112. doi: 10.1186/s13148-016-0278-2
16. Park SY, Piao Y, Thomas C, Fuller GN, de Groot JF. Cdc2-like kinase 2 is a key regulator of the cell cycle via FOXO3a/p27 in glioblastoma. Oncotarget. (2016) 7:26793–805. doi: 10.18632/oncotarget.8471
17. Ushmorov A, Wirth T. FOXO in B-cell lymphopoiesis and B cell neoplasia. Semin Cancer Biol. (2018) 50:132–41. doi: 10.1016/j.semcancer.2017.07.008
18. Nakagawa S, Gisselbrecht SS, Rogers JM, Hartl DL, Bulyk ML. DNA-binding specificity changes in the evolution of forkhead transcription factors. Proc Natl Acad Sci USA. (2013) 110:12349–54. doi: 10.1073/pnas.1310430110
19. Rogers JM, Waters CT, Seegar TCM, Jarrett SM, Hallworth AN, Blacklow SC, et al. Bispecific Forkhead transcription factor foxN3 recognizes two distinct motifs with different DNA shapes. Mol Cell. (2019) S1097-2765(19)30039-5. doi: 10.1016/j.molcel.2019.01.019
20. Schuff M, Rossner A, Donow C, Knochel W. Temporal and spatial expression patterns of FoxN genes in Xenopus laevis embryos. Int J Dev Biol. (2006) 50:429–34. doi: 10.1387/ijdb.052126ms
21. Bukowska J, Kopcewicz M, Kur-Piotrowska A, Szostek-Mioduchowska AZ, Walendzik K, Gawronska-Kozak B. Effect of TGFbeta1, TGFbeta3 and keratinocyte conditioned media on functional characteristics of dermal fibroblasts derived from reparative (Balb/c) and regenerative (Foxn1 deficient; nude) mouse models. Cell Tissue Res. (2018) 374:149–63. doi: 10.1007/s00441-018-2836-8
22. Kur-Piotrowska A, Bukowska J, Kopcewicz MM, Dietrich M, Nynca J, Slowinska M, et al. Foxn1 expression in keratinocytes is stimulated by hypoxia: further evidence of its role in skin wound healing. Sci Rep. (2018) 8:5425. doi: 10.1038/s41598-018-23794-5
23. Chen Y, Liu X, Liu Y, Wang Y, Wang H, Lu C, et al. Decreased Wnt4 expression inhibits thymoma development through downregulation of FoxN1. J Thorac Dis. (2017) 9:1574–83. doi: 10.21037/jtd.2017.05.28
24. Rho HK, McClay DR. The control of foxN2/3 expression in sea urchin embryos and its function in the skeletogenic gene regulatory network. Development. (2011) 138:937–45. doi: 10.1242/dev.058396
25. Campbell EP, Quigley IK, Kintner C. Foxn4 promotes gene expression required for the formation of multiple motile cilia. Development. (2016) 143:4654–64. doi: 10.1242/dev.143859
26. Li S, Xiang M. Foxn4 influences alveologenesis during lung development. Dev Dyn. (2011) 240:1512–7. doi: 10.1002/dvdy.22610
27. Katoh M, Katoh M. Germ-line mutation of Foxn5 gene in mouse lineage. Int J Mol Med. (2004) 14:463–7. doi: 10.3892/ijmm.14.3.463
28. Wang X, He B, Gao Y, Li Y. FOXR2 contributes to cell proliferation and malignancy in human hepatocellular carcinoma. Tumour Biol. (2016) 37:10459–67. doi: 10.1007/s13277-016-4923-3
29. Pati D, Keller C, Groudine M, Plon SE. Reconstitution of a MEC1-independent checkpoint in yeast by expression of a novel human fork head cDNA. Mol Cell Biol. (1997) 17:3037–46. doi: 10.1128/MCB.17.6.3037
30. Schuff M, Rossner A, Wacker SA, Donow C, Gessert S, Knochel W. FoxN3 is required for craniofacial and eye development of Xenopus laevis. Dev Dyn. (2007) 236:226–39. doi: 10.1002/dvdy.21007
31. Samaan G, Yugo D, Rajagopalan S, Wall J, Donnell R, Goldowitz D, et al. Foxn3 is essential for craniofacial development in mice and a putative candidate involved in human congenital craniofacial defects. Biochem Biophys Res Commun. (2010) 400:60–5. doi: 10.1016/j.bbrc.2010.07.142
32. Li W, Zhang Z, Liu X, Cheng X, Zhang Y, Han X, et al. The FOXN3-NEAT1-SIN3A repressor complex promotes progression of hormonally responsive breast cancer. J Clin Invest. (2017) 127:3421–40. doi: 10.1172/JCI94233
33. Dai Y, Wang M, Wu H, Xiao M, Liu H, Zhang D. Loss of FOXN3 in colon cancer activates beta-catenin/TCF signaling and promotes the growth and migration of cancer cells. Oncotarget. (2017) 8:9783–93. doi: 10.18632/oncotarget.14189
34. Schmidt J, Piekarski N, Olsson L. Cranial muscles in amphibians: development, novelties and the role of cranial neural crest cells. J Anat. (2013) 222:134–46. doi: 10.1111/j.1469-7580.2012.01541.x
35. Lee SH, Singh I, Tisdale S, Abdel-Wahab O, Leslie CS, Mayr C. Widespread intronic polyadenylation inactivates tumour suppressor genes in leukaemia. Nature. (2018) 561:127–31. doi: 10.1038/s41586-018-0465-8
36. Grassi D, Franz H, Vezzali R, Bovio P, Heidrich S, Dehghanian F, et al. Neuronal activity, TGFbeta-signaling and unpredictable chronic stress modulate transcription of Gadd45 family members and DNA methylation in the hippocampus. Cereb Cortex. (2017) 27:4166–81. doi: 10.1093/cercor/bhx095
37. Busygina V, Kottemann MC, Scott KL, Plon SE, Bale AE. Multiple endocrine neoplasia type 1 interacts with forkhead transcription factor CHES1 in DNA damage response. Cancer Res. (2006) 66:8397–403. doi: 10.1158/0008-5472.CAN-06-0061
38. Scott KL, Plon SE. Loss of Sin3/Rpd3 histone deacetylase restores the DNA damage response in checkpoint-deficient strains of Saccharomyces cerevisiae. Mol Cell Biol. (2003) 23:4522–31. doi: 10.1128/MCB.23.13.4522-4531.2003
39. Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. (2011) 471:74–9. doi: 10.1038/nature09803
40. Cote JA, Lessard J, Pelletier M, Marceau S, Lescelleur O, Fradette J, et al. Role of the TGF-beta pathway in dedifferentiation of human mature adipocytes. FEBS Open Bio. (2017) 7:1092–101. doi: 10.1002/2211-5463.12250
41. Zeglinski MR, Hnatowich M, Jassal DS, Dixon IM. SnoN as a novel negative regulator of TGF-beta/Smad signaling: a target for tailoring organ fibrosis. Am J Physiol Heart Circ Physiol. (2015) 308:H75–82. doi: 10.1152/ajpheart.00453.2014
42. Scott KL, Plon SE. CHES1/FOXN3 interacts with Ski-interacting protein and acts as a transcriptional repressor. Gene. (2005) 359:119–26. doi: 10.1016/j.gene.2005.06.014
43. Kocic J, Villar V, Krstic A, Santibanez JF. SKIP Downregulation Increases TGF-beta1-induced matrix metalloproteinase-9 production in transformed keratinocytes. Scientifica. (2012) 2012:861647. doi: 10.6064/2012/861647
44. Villar V, Kocic J, Santibanez JF. Skip regulates TGF- beta 1-induced extracellular matrix degrading proteases expression in human PC-3 Prostate cancer cells. Prostate Cancer. (2013) 2013:398253. doi: 10.1155/2013/398253
45. Kuki K, Yamaguchi N, Iwasawa S, Takakura Y, Aoyama K, Yuki R, et al. Enhancement of TGF-beta-induced Smad3 activity by c-Abl-mediated tyrosine phosphorylation of its coactivator SKI-interacting protein (SKIP). Biochem Biophys Res Commun. (2017) 490:1045–51. doi: 10.1016/j.bbrc.2017.06.163
46. Lee HJ, Kim MY, Park HS. Phosphorylation-dependent regulation of Notch1 signaling: the fulcrum of Notch1 signaling. BMB Rep. (2015) 48:431–7. doi: 10.5483/BMBRep.2015.48.8.107
47. Kirmizitas A, Meiklejohn S, Ciau-Uitz A, Stephenson R, Patient R. Dissecting BMP signaling input into the gene regulatory networks driving specification of the blood stem cell lineage. Proc Natl Acad Sci USA. (2017) 114:5814–21. doi: 10.1073/pnas.1610615114
48. Mudduluru G, Abba M, Batliner J, Patil N, Scharp M, Lunavat TR, et al. A systematic approach to defining the microRNA landscape in metastasis. Cancer Res. (2015) 75:3010–9. doi: 10.1158/0008-5472.CAN-15-0997
49. Robertson E, Perry C, Doherty R, Madhusudan S. Transcriptomic profiling of forkhead box transcription factors in adult glioblastoma multiforme. Cancer Genomics Proteomics. (2015) 12:103–12.
50. Li S, Li J, Chen C, Zhang R, Wang K. Pan-cancer analysis of long non-coding RNA NEAT1 in various cancers. Genes Dis. (2018) 5:27–35. doi: 10.1016/j.gendis.2017.11.003
51. Arshi A, Sharifi FS, Khorramian Ghahfarokhi M, Faghih Z, Doosti A, Ostovari S, et al. Expression analysis of MALAT1, GAS5, SRA, and NEAT1 lncRNAs in breast cancer tissues from young women and women over 45 years of age. Mol Ther Nucleic Acids. (2018) 12:751–7. doi: 10.1016/j.omtn.2018.07.014
52. Zhang M, Weng W, Zhang Q, Wu Y, Ni S, Tan C, et al. The lncRNA NEAT1 activates Wnt/beta-catenin signaling and promotes colorectal cancer progression via interacting with DDX5. J Hematol Oncol. (2018) 11:113. doi: 10.1186/s13045-018-0656-7
53. Yong W, Yu D, Jun Z, Yachen D, Weiwei W, Midie X, et al. Long noncoding RNA NEAT1, regulated by LIN28B, promotes cell proliferation and migration through sponging miR-506 in high-grade serous ovarian cancer. Cell Death Dis. (2018) 9:861. doi: 10.1038/s41419-018-0908-z
54. Xie Q, Lin S, Zheng M, Cai Q, Tu Y. Long noncoding RNA NEAT1 promoted the growth of cervical cancer cells via sponging miR-9-5p. Biochem Cell Biol. (2018) 10:1–9. doi: 10.1139/bcb-2018-0111
55. Wang CL, Wang D, Yan BZ, Fu JW, Qin L. Long non-coding RNA NEAT1 promotes viability and migration of gastric cancer cell lines through up-regulation of microRNA-17. Eur Rev Med Pharmacol Sci. (2018) 22:4128–37. doi: 10.26355/eurrev_201807_15405
56. Li X, Wang X, Song W, Xu H, Huang R, Wang Y, et al. Oncogenic properties of NEAT1 in prostate cancer cells depend on the CDC5L-AGRN transcriptional regulation circuit. Cancer Res. (2018) 78:4138–49. doi: 10.1158/0008-5472.CAN-18-0688
57. Qi L, Liu F, Zhang F, Zhang S, Lv L, Bi Y, et al. lncRNA NEAT1 competes against let-7a to contribute to non-small cell lung cancer proliferation and metastasis. Biomed Pharmacother. (2018) 103:1507–15. doi: 10.1016/j.biopha.2018.04.053
58. Sun W, Lan X, Zhang H, Wang Z, Dong W, He L, et al. NEAT1_2 functions as a competing endogenous RNA to regulate ATAD2 expression by sponging microRNA-106b-5p in papillary thyroid cancer. Cell Death Dis. (2018) 9:380. doi: 10.1038/s41419-018-0418-z
59. Zhang L, Lu XQ, Zhou XQ, Liu QB, Chen L, Cai F. NEAT1 induces osteosarcoma development by modulating the miR-339-5p/TGF-beta1 pathway. J Cell Physiol. 234:5097–105. doi: 10.1002/jcp.27313
60. Tu J, Zhao Z, Xu M, Lu X, Chang L, Ji J. NEAT1 upregulates TGF-beta1 to induce hepatocellular carcinoma progression by sponging hsa-mir-139-5p. J Cell Physiol. (2018) 233:8578–87. doi: 10.1002/jcp.26524
61. Qian K, Liu G, Tang Z, Hu Y, Fang Y, Chen Z, et al. The long non-coding RNA NEAT1 interacted with miR-101 modulates breast cancer growth by targeting EZH2. Arch Biochem Biophys. (2017) 615:1–9. doi: 10.1016/j.abb.2016.12.011
62. Zheng Y, Yin L, Chen H, Yang S, Pan C, Lu S, et al. miR-376a suppresses proliferation and induces apoptosis in hepatocellular carcinoma. FEBS Lett. (2012) 586:2396–403. doi: 10.1016/j.febslet.2012.05.054
63. Fellenberg J, Sahr H, Kunz P, Zhao Z, Liu L, Tichy D, et al. Restoration of miR-127-3p and miR-376a-3p counteracts the neoplastic phenotype of giant cell tumor of bone derived stromal cells by targeting COA1, GLE1 and PDIA6. Cancer Lett. (2016) 371:134–41. doi: 10.1016/j.canlet.2015.10.039
64. Zhang M, Wu WB, Wang ZW, Wang XH. lncRNA NEAT1 is closely related with progression of breast cancer via promoting proliferation and EMT. Eur Rev Med Pharmacol Sci. (2017) 21:1020–6.
65. Bansal N, David G, Farias E, Waxman S. Emerging roles of epigenetic regulator sin3 in cancer. Adv Cancer Res. (2016) 130:113–35. doi: 10.1016/bs.acr.2016.01.006
66. Hurst DR. Metastasis suppression by BRMS1 associated with SIN3 chromatin remodeling complexes. Cancer Metastasis Rev. (2012) 31:641–51. doi: 10.1007/s10555-012-9363-y
67. Katoh H, Ojima H, Kokubu A, Saito S, Kondo T, Kosuge T, et al. Genetically distinct and clinically relevant classification of hepatocellular carcinoma: putative therapeutic targets. Gastroenterology. (2007) 133:1475–86. doi: 10.1053/j.gastro.2007.08.038
68. Sun J, Li H, Huo Q, Cui M, Ge C, Zhao F, et al. The transcription factor FOXN3 inhibits cell proliferation by downregulating E2F5 expression in hepatocellular carcinoma cells. Oncotarget. (2016) 7:43534–45. doi: 10.18632/oncotarget.9780
69. Ren T, Xu L, Jiao S, Wang Y, Cai Y, Liang Y, et al. TLR9 signaling promotes tumor progression of human lung cancer cell in vivo. Pathol Oncol Res. (2009) 15:623–30. doi: 10.1007/s12253-009-9162-0
70. Li Q, Li X, Guo Z, Xu F, Xia J, Liu Z, et al. MicroRNA-574-5p was pivotal for TLR9 signaling enhanced tumor progression via down-regulating checkpoint suppressor 1 in human lung cancer. PLoS ONE. (2012) 7:e48278. doi: 10.1371/journal.pone.0048278
71. Miravet S, Piedra J, Miro F, Itarte E, Garcia de Herreros A, Dunach M. The transcriptional factor Tcf-4 contains different binding sites for beta-catenin and plakoglobin. J Biol Chem. (2016) 291:18854. doi: 10.1074/jbc.A116.110248
72. Markowski J, Tyszkiewicz T, Jarzab M, Oczko-Wojciechowska M, Gierek T, Witkowska M, et al. Metal-proteinase ADAM12, kinesin 14 and checkpoint suppressor 1 as new molecular markers of laryngeal carcinoma. Eur Arch Otorhinolaryngol. (2009) 266:1501–7. doi: 10.1007/s00405-009-1019-3
73. Nagel S, Meyer C, Kaufmann M, Drexler HG, MacLeod RA. Deregulated FOX genes in hodgkin lymphoma. Genes Chromosomes Cancer. (2014) 53:917–33. doi: 10.1002/gcc.22204
74. Ren C, Yang T, Qiao P, Wang L, Han X, Lv S, et al. PIM2 interacts with tristetraprolin and promotes breast cancer tumorigenesis. Mol Oncol. (2018) 12:690–704. doi: 10.1002/1878-0261.12192
75. Hospital MA, Jacquel A, Mazed F, Saland E, Larrue C, Mondesir J, et al. RSK2 is a new Pim2 target with pro-survival functions in FLT3-ITD-positive acute myeloid leukemia. Leukemia. (2018) 32:597–605. doi: 10.1038/leu.2017.284
76. Gupta A, Nagilla P, Le HS, Bunney C, Zych C, Thalamuthu A, et al. Comparative expression profile of miRNA and mRNA in primary peripheral blood mononuclear cells infected with human immunodeficiency virus (HIV-1). PLoS ONE. (2011) 6:e22730. doi: 10.1371/journal.pone.0022730
77. Nagel S, Pommerenke C, Meyer C, Kaufmann M, MacLeod RAF, Drexler HG. Identification of a tumor suppressor network in T-cell leukemia. Leuk Lymphoma. (2017) 58:2196–207. doi: 10.1080/10428194.2017.1283029
Keywords: FOXN3, forkhead, function, breast, cancer, malignancy, regulation, review
Citation: Kong X, Zhai J, Yan C, Song Y, Wang J, Bai X, Brown JAL and Fang Y (2019) Recent Advances in Understanding FOXN3 in Breast Cancer, and Other Malignancies. Front. Oncol. 9:234. doi: 10.3389/fonc.2019.00234
Received: 12 December 2018; Accepted: 15 March 2019;
Published: 31 May 2019.
Edited by:
Juan José Lasarte, University of Navarra, SpainReviewed by:
Sabarish Ramachandran, Texas Tech University Health Sciences Center, United StatesCopyright © 2019 Kong, Zhai, Yan, Song, Wang, Bai, Brown and Fang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Wang, d3d3ampqMTIzNEB2aXAuc2luYS5jb20=; Xiaofeng Bai, YmFpeGlhb2ZlbmcxOTczQDE2My5jb20=; James A. L. Brown, amFtZXMuYnJvd25AbnVpZ2Fsd2F5Lmll; Yi Fang, ZmFuZ3lpMDUwMUB2aXAuc2luYS5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.