 Thomas N. Seyfried1*
Thomas N. Seyfried1* Purna Mukherjee1
Purna Mukherjee1 Mehmet S. Iyikesici2Abdul Slocum3
Mehmet S. Iyikesici2Abdul Slocum3 Miriam Kalamian4
Miriam Kalamian4 Jean-Pierre Spinosa5Christos Chinopoulos6
Jean-Pierre Spinosa5Christos Chinopoulos6- 1Biology Department, Boston College, Chestnut Hill, MA, United States
- 2Medical Oncology, Kemerburgaz University Bahcelievler Medical Park Hospital, Istanbul, Turkey
- 3Medical Oncology, Chemo Thermia Oncology Center, Istanbul, Turkey
- 4Dietary Therapies LLc, Hamilton, MT, United States
- 5Gynecologic and Breast Oncology, Clinique Cecil, Lausanne, Switzerland
- 6Department of Medical Biochemistry, Semmelweis University, Budapest, Hungary
Breast cancer remains as a significant cause of morbidity and mortality in women. Ultrastructural and biochemical evidence from breast biopsy tissue and cancer cells shows mitochondrial abnormalities that are incompatible with energy production through oxidative phosphorylation (OxPhos). Consequently, breast cancer, like most cancers, will become more reliant on substrate level phosphorylation (fermentation) than on oxidative phosphorylation (OxPhos) for growth consistent with the mitochondrial metabolic theory of cancer. Glucose and glutamine are the prime fermentable fuels that underlie therapy resistance and drive breast cancer growth through substrate level phosphorylation (SLP) in both the cytoplasm (Warburg effect) and the mitochondria (Q-effect), respectively. Emerging evidence indicates that ketogenic metabolic therapy (KMT) can reduce glucose availability to tumor cells while simultaneously elevating ketone bodies, a non-fermentable metabolic fuel. It is suggested that KMT would be most effective when used together with glutamine targeting. Information is reviewed for suggesting how KMT could reduce systemic inflammation and target tumor cells without causing damage to normal cells. Implementation of KMT in the clinic could improve progression free and overall survival for patients with breast cancer.
Introduction
Breast cancer persists as a significant cause of morbidity and mortality in woman. According to the American Cancer Society, the number of new cases and deaths from breast cancer in US woman is estimated to be 268,600 and 41,760, respectively, for 2019 (1). Indeed, breast cancer alone will account for 30% of all female cancers. Although the incidence of breast cancer is lower in black women than in white women, the death rate is 41% higher in blacks than in whites possibly due in part to diet and lifestyle (2). The failure to effectively manage malignant breast cancer, and most malignant cancers for that matter, comes in large part from a misunderstanding on the origin of cancer. Although cancer has long been considered a genetic disease based on the somatic mutation theory (3–5), recent revelations have raised serious concerns that question the validity of this theory. Major concerns include:
1 The absence of gene and chromosomal mutations in some cancers (6–9). Indeed, Greenman et al. found no mutations following extensive sequencing in 73/210 cancers (3), while Parsons et al. found no mutations in the P53, the PI3K, and the RB1 pathways in the Br20P tissue sample of a glioblastoma patient (10). Such samples should not exist according to the somatic mutation theory.
2 The presence and clonal expansion of numerous so-called driver gene mutations in a broad range of normal human tissues including breast tissue (11–15). It is not clear how the somatic mutation theory can account for malignant tumors that have no mutations or for normal cells and tissues that express driver mutations.
3 The absence of breast cancer and most other cancers in chimpanzees despite having about 98.5% gene and protein sequence identity with humans even at the BRCA1 locus (16–19). Indeed, breast cancer has never been documented in a female chimpanzee suggesting that diet and lifestyle issues, rather than genetic mutations, are largely responsible for the disease (18, 20).
4 The nuclear/cytoplasm transfer experiments showing that normal cells and tissues can be produced from tumorigenic nuclei as long as the tumorigenic nuclei are localized in cytoplasm containing normal mitochondria (21). Furthermore, recent studies show that normal mitochondria can down-regulate multiple oncogenic pathways and growth behavior in metastatic breast cancer cells (22, 23). These findings show that normal mitochondrial function can suppress tumorigenesis regardless of the gene or chromosomal abnormalities that might be present in the tumor nucleus. Viewed collectively, these findings suggest that the somatic mutations found in breast cancer and in most other cancers are not the primary cause of the disease. It is therefore unlikely that therapeutic strategies based on the somatic mutation theory will have major impact on the management of most cancers including breast cancer.
The Mitochondrial Metabolic Theory of Cancer
Emerging evidence indicates that most if not all cancers display deranged energy metabolism (24–35). It is well-documented that the tumor cells found in most cancerous tissues including breast cancer tissue, have abnormalities in the number, structure, and function of their mitochondria (26, 27, 29, 33, 36–42). These abnormalities would compromise efficient energy production through oxidative phosphorylation (OxPhos). Figure 1 documents ultrastructural abnormalities in breast cancer mitochondria that are linked to abnormalities in proteins of the electron transport chain (43). In addition to abnormalities in mitochondrial membranes, breast cancer cells also express abnormalities in mitochondrial-associated membranes (MAM), that would further reduce energy production through OxPhos (44, 45). Consequently, increased fermentation metabolism would be necessary to compensate for OxPhos deficiency in order to maintain sufficient energy for breast cancer viability and growth.
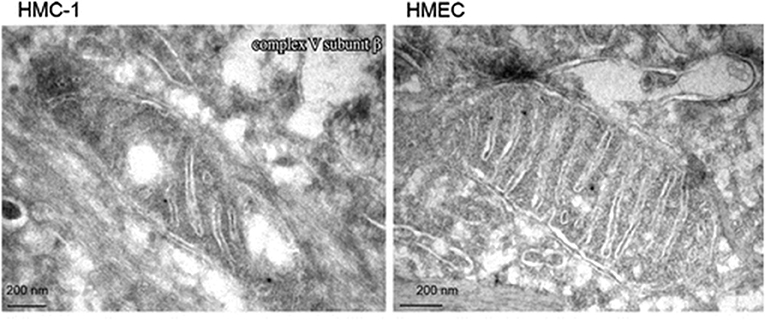
Figure 1. Electron microscopy of primary breast cancer cells (human mammary carcinoma HMC-1) and human epithelial mammary cell control line (HEMC). Abnormal mitochondrial morphology in the HMC-1 cell showing loss of invaginations and vacuoles. These abnormalities in mitochondria ultrastructure were linked to abnormalities in the electron transport chain and are in general agreement with those from other studies of breast cancer mitochondria (37, 40, 42). Reprinted with permission from Putignani et al. (43).
Aerobic Fermentation of Glucose and Glutamine in Cancer Cells
Glucose and glutamine are the major fermentable fuels used by cancer cells with impaired OxPhos (39, 46). Glucose is fermented to lactic acid through glycolysis in the cell cytoplasm, while glutamine is fermented to succinic acid through glutaminolysis in the tricarboxylic acid (TCA) cycle. Notably, both fermentation processes can generate energy in the presence or absence of oxygen through substrate level phosphorylation (SLP) at the pyruvate kinase reaction in glycolysis and at the succinate ligase reaction in glutaminolysis, respectively (46). Warburg first described the aerobic fermentation of glucose as a major phenotype of most cancers (47–51). This metabolic phenotype has become known as the Warburg effect (52). In contrast to the Pasteur effect, where fermentation is suppressed in the presence of oxygen, the Warburg effect involves robust glucose-derived lactic acid fermentation even in the presence of 100% oxygen (aerobic fermentation).
Glutaminolysis
In addition to aerobic fermentation in the cytoplasm, glutaminolysis can also support high-energy phosphate synthesis in the mitochondria through the sequential conversion of glutamine-glutamate-alpha-ketoglutarate-succinyl CoA-succinate (Figure 2) (46). ATP synthesis through the succinate-CoA ligase reaction in the TCA cycle can compensate for reduced ATP synthesis through either glycolysis or OxPhos. We recently proposed that most of the ATP synthesized in tumor cells would come from mitochondrial substrate level phosphorylation (mSLP) at the succinate ligase reaction (46). Evidence showing that mSLP can compensate for OxPhos deficiency in cancer is emerging (57–60). mSLP could also compensate for minimal energy production through glycolysis due to the predominance of the glycolytic pyruvate kinase M2 (PKM2) isoform, which produces less ATP than the PKM1 isoform (61). The PKM2 isoform is predominant in many cancers including breast cancer (62, 63). As Q is the single-letter designation of glutamine, the aerobic fermentation of glutamine through mSLP was recently defined as the Q effect; which has been recognized as the missing link in the mitochondrial metabolic theory of cancer (32, 46, 64, 65). Unfortunately, the glutaminolysis pathway and the role of glutamine and mSLP was unknown to Warburg (46). We find it remarkable that nearly all of the major reviews or previous studies on cancer energy metabolism have not addressed or possibly even recognized the role of SUCL activity and mSLP, as a compensatory energy mechanism for deficient OxPhos. We consider mSLP as the key mechanism that underlies energy production in tumor cells with defective respiration (46).

Figure 2. The glutaminolysis pathway. The succinyl-CoA ligase reaction, metabolizing succinyl-CoA to succinate, produces high-energy phosphates (ATP) in the absence of oxidative phosphorylation through the process of substrate-level phosphorylation in the mitochondrial matrix. Provision of succinyl-CoA by the α-ketoglutarate dehydrogenase complex is crucial for maintaining the function of succinyl-CoA ligase thus preventing the adenine nucleotide translocase from reversing. Succinate contributes to inflammation and stabilizes Hif-1a, a key transcription factor that contributes to the aerobic fermentation (53–56).
Aerobic fermentation (Warburg effect) is a common metabolic phenotype in breast cancer regardless of histopathological type, grade, or gene expression profile (24, 66–74). In addition to glucose, glutamine is the other major fuel necessary for breast cancer cells (73, 75–78). The elevated utilization of glucose and glutamine becomes necessary to sustain the viability of OxPhos-impaired cancer cells through SLP in the cytoplasm and mitochondria, respectively. The Q-effect provides a rational explanation for the high glutamine use in cells with compromised OxPhos (46). The aerobic fermentation of glucose and glutamine in cancer cells would be the expected consequence of impaired OxPhos, as fermentation can compensate for insufficient respiration. Some have suggested that the function of the Warburg-effect is to provide a growth advantage for tumor cells. This is a teleological explanation, i.e., design with a purpose or intelligent design, which should not be part of modern biological thought (79–81). Cells do not make choices or have preferences, but simply respond to conditions in their internal and external environments according to evolutionarily designed metabolic programs. As proliferation, rather than quiescence, is the default state of metazoan cells (8, 82), unbridled proliferation becomes the consequence when fermentation gradually replaces respiration in cancer cells (21, 83, 84). Indeed, unbridled proliferation was the dominant growth phenotype of all organisms that existed on the planet before oxygen entered the atmosphere some 2.5 billion years ago (83). Hence, glucose and glutamine become the drivers of cancer cell fermentation and growth.
Role of Reactive Oxygen Species (ROS) and Oncogenes in the Origin and Progression of Cancer
Impaired OxPhos together with compensatory fermentation leads to the accumulation of reactive oxygen species (ROS) (85–87). ROS are carcinogenic and mutagenic, and are largely responsible for the genomic instability and mutations seen in tumor cells (85, 87–93). In other words, the mutations seen in tumor cells arise as a consequence of impaired energy metabolism (32, 64). Oncogenes such as Hif-1alpha, Myc, Ras, BRAF, etc., facilitate the dependence of tumor cells on glucose and glutamine while defects in the tumor suppressor genes p53 and pRb will compromise OxPhos function thus causing further dependency on fermentation for growth (28, 46, 84, 94–99). These genes mutations are linked to breast cancer and other cancers through mitochondrial dysfunction. Compromised OxPhos would require compensatory glucose and glutamine fermentation to maintain membrane pump activity and tumor cell viability (64, 100). A mutation in the p53 gene was found to increase glucose consumption and the Warburg effect, whereas the retinoblastoma Rb protein was found to increase glutamine metabolism through an effect on the E2F-3 transcription factor (101, 102). Mutations in these and in other oncogenes, and tumor suppressor genes, have been detected in breast cancer cells (103, 104). It is interesting that Her2 signaling in breast tumor promotes glycolysis and glucose utilization with lactate accumulation (105). Moreover, the ErbB2 targeting antibody, trastuzumab (Herceptin), inhibits glycolysis via downregulation of HSF1 and LDH-A in ErbB2-positive cancer cells. The results of Ding suggest that ErbB2 not only promotes glycolysis, but also inhibits mitochondrial oxidative phosphorylation by decreasing ETC activities (106). Mutations in the BRCA1 gene, which is known to play an important role in maintaining metabolic homeostasis, have also been linked to elevated glycolysis and other metabolic disturbances (107, 108). As no known inherited breast cancer gene is 100% penetrant, germline mutations are considered secondary risk factors and cannot therefore be considered primary causes of cancer. A penetrance of 100% is necessary for any tumor gene to be considered a primary cause of cancer. OxPhos impairment with compensatory glucose and glutamine fermentation is the common underlying phenotype of most if not all cancers.
Origin of Metastasis
Metastasis involves the dissemination of cancer cells from the primary tumor to surrounding tissues and to distant organs and is the primary cause of cancer morbidity and mortality (109). Most tumors that fail to show local invasion or metastasis are considered benign and do not pose serious risk despite expressing a majority of the so called “Hallmarks of Cancer” (110). Tumor cell metastasis involves a stereotypic cascade of biological phenomenon including local invasion, intravasation into the blood, survival in the circulation, immune suppression, extravasation from the blood, and growth in a distant organ or tissue (111). Identification of the unique features of the metastatic cell can expand understanding of metastasis and facilitate rational therapeutic strategies based on knowledge of cell biology and biochemistry.
Two major theories have been advanced to explain metastasis that include the epithelial-mesenchymal transition (EMT), and the macrophage fusion hybrid hypothesis. The EMT is the dominant explanation and is based on the somatic mutation theory of cancer. The EMT proposes that an initial series of random mutations disturb cell-cell interaction causing a normal epithelial cell to transform into an invasive mesenchymal cell. Further additional random mutations cause the mesenchymal cell to intravasate, and subsequently extravasate into the parenchyma at some distant organ site (4, 112). Once established at the distant site, the metastatic tumor cells transform back to an epithelial phenotype via the so called mesenchymal epithelial transition (MET) (4, 112). No explanation has been presented, however, as to how the multiple random gene mutations responsible for the initial events of the metastatic cascade could be reversed or suppressed during the MET (4). Despite its linkage to phenomenology, the EMT is considered the dominant explanation for breast cancer metastasis (113).
In contrast to the EMT, the macrophage fusion hybrid hypothesis posits that metastasis arises from macrophages either directly or indirectly from the fusion of macrophages with neoplastic cells (109, 114–117). The metastatic breast cancer cell, like many metastatic cancer cells, expresses characteristics of myeloid cells and macrophages (109, 114, 118–125). These characteristics include phagocytosis, fusogenicity, and expression of multiple myeloid/macrophage biomarkers. Fusion hybridization could also better account for the large degree of cellular and genetic heterogeneity seen in metastatic breast cancer than can the EMT (126, 127). A macrophage origin of metastasis is also more consistent with Paget's “seed soil” hypothesis than is an origin based on EMT (109, 128, 129). Macrophages (seeds) are known to infiltrate organs (soil) non-randomly (109). As glutamine is a major fuel for macrophages (130, 131), targeting the availability of glutamine together with glucose becomes a rational, yet largely overlooked, therapeutic strategy for managing metastatic cancer (60).
Current Standard of Care (SOC) for Breast Cancer
Current standard management of breast cancer requires histological diagnosis obtained by core needle biopsy prior to surgical procedure (132). Therapies for breast cancer depend largely on the stage, grade, and the biology of the disease. Surgery and radiotherapy (breast and axilla) are used for local disease control, staging, and tumor extirpation. It is important to mention, however, that the extent of surgery and radiotherapy are not linked to a survival advantage (133). Chemotherapy, hormone therapy (tamoxifen, aromatase inhibitor, or one followed by the other), targeted therapies, trastuzumab (Herceptin) and pertuzumab (Perjeta), or combinations of these are designed to destroy local residual cancer cells and latent metastasis, while also reducing the risk of recurrence. Mechanical interventions (core needle biopsies and surgery) can cause wound-induced inflammatory oncotaxis, which influences the natural history of breast cancer (129). Data from several studies show that biopsies and surgery can cause inflammatory oncotaxis thus increasing tissue angiogenesis and spread of tumor cells (134–140). Invasive and metastatic behavior would be expected for breast cancer cells with myeloid properties (123). Some have hypothesized that growth factor stimulation in response to intraoperative tissue damage, can increase HER2 receptor activation in incompletely resected pre-invasive breast cancer thus increasing risk for tumor cell proliferation and spread (141). Procedures that might elevate blood glucose or insulin levels should be avoided, as glucose is known to accelerate breast cancer development (142–147). Products used in anesthesia might also increase blood glucose and insulin levels (148). The type of anesthesia and analgesia used during and after surgery should be carefully monitored for possible influence on glucose and insulin metabolism (149). Glucocorticoids, which are given to some breast cancer patients, can also elevate blood glucose levels thus supporting glucose-dependent aerobic fermentation. In addition to elevating blood glucose, glucocorticoids can also block estrogen-induced apoptosis to further stimulate breast cancer growth (150). Hence, risk for tumor spread can be associated with accepted procedures used in breast cancer management.
Ketogenic Metabolic Therapy
Ketogenic metabolic therapy (KMT) is emerging as an effective complementary or alternative therapeutic strategy for managing a broad range of malignant cancers including breast cancer (151–160). Calorie restriction and low-carbohydrate high-fat ketogenic diets (KD) reduce the glucose needed to propell the Warburg effect while also elevating ketone bodies (34). Cancer cells cannot effectively use ketone bodies or fatty acids for ATP synthesis through OxPhos due to defects in the number, structure, and function of their mitochondria (34, 46). Moreover, ketone bodies and fatty acids cannot be fermented, and thus cannot effectively replace glucose and glutamine as an alternative energy source for cancer (46). Bartmann et al. showed that the major ketone body, beta-hydroxybutyrate, could not stimulate breast tumor growth in vitro (161). Hence, KMT becomes a putative therapeutic strategy for managing most cancers including breast cancer (34).
There have been reports, however, suggesting that some cancers, including breast cancer, can oxidize ketone bodies and fatty acids for growth (162–165). The uptake of ketone bodies or fatty acids together with oxygen consumption in tumor cells is not proof that the ketone bodies or fatty acids can be used to generate energy through OxPhos (34, 46, 166). Indeed, Kuok et al. recently showed that palmitate could increase oxygen consumption rate (OCR) by stimulating ATP usage and insulin secretion rather than by increasing beta-oxidation (167). Fatty acids are potent swelling and uncoupling agents that can stimulate insulin secretion and glucose/glutamine consumption thus making it appear as if tumor cells can metabolize fatty acids for energy (166, 168–170). In other words, fatty acids can stimulate utilization of glucose and glutamine. Many tumor cells including breast cancer cells will store fatty acids as lipid droplets (166, 171, 172). Lipid droplet storage is considered a protection mechanism from the lethal effects of saturated fatty acids in cells that cannot metabolize fats for energy (171–174). If tumor cells could use fatty acids for growth, then water-only fasting and calorie restricted ketogenic diets should accelerate tumor growth, as these dietary changes elevate free fatty acids in the blood (175, 176). This is clearly not the case. Also, palmitic acid cannot support in vitro tumor cell growth in the absence of glucose and glutamine. Any disruption of the mitochondrial proton motive gradient will provide ATP for the F1-F0 ATP synthase thus hydrolyzing ATP rather than synthesizing ATP (46). mSLP will provide ATP for F1-F0-ATP synthase in an effort to maintain a moderate mitochondrial membrane potential and prevent reversal. Based on the foundational biological principle that structure determines function (43, 168, 177, 178), ketone bodies and fatty acids cannot serve as major respiratory fuels for tumor cells containing defects in mitochondrial structure and function (46, 84). Hence, it would be helpful to include evidence of normal mitochondria ultrastructure and electron transport chain activities in reports indicating that ketone bodies and fatty acids are fuels for OxPhos-generated ATP synthesis in cancer cells.
Depletion of fermentable fuels from KMT will facilitate catastrophic tumor cell death. Ketogenic diet appears to work better when used with glutamine targeting and is consumed in restricted amounts (60). The simultaneous targeting of glycolysis and glutaminolysis is now emerging as a potential therapeutic strategy for managing a broad range of cancers including breast cancer (60, 179, 180). KMT reduces circulating levels of glucose and insulin that are needed for rapid tumor growth (176). Excessive consumption of ketogenic diets, however, can provoke tumor growth by causing insulin insensitivity and glucose elevation (176, 181). Hence, KMT becomes a logical therapeutic strategy for managing breast cancer when used correctly.
The microenvironment of many tumors is hypoxic, acidotic, and enriched with glucose and glutamine. Under KMT, this pro-tumorigenic microenvironment becomes less inflamed (84, 182, 183). Restricted KDs and calorie restriction are antiinvasive, antiangiogenic, anti-inflammatory, and capable of killing tumor cells through a pro-apoptotic mechanism (181–188). Metabolism of the major circulating ketone body, D-beta-hydroxybutyrate, reduces reactive oxygen species production through the mitochondrial Co-enzyme Q couple in normal cells, while simultaneously elevating oxidative stress in tumor cells (34, 189–191). Implementation of KMT prior to any surgical procedure could also benefit patients (152). KMT should decrease the need for dexamethasone pretreatment, a problematic therapy that can inadvertently increase availability of glucose to the tumor cells while also inhibiting chemotherapy-induced apoptosis (192–194). It is well-known that glucose and hyperglycemia contribute to rapid breast cancer growth (146, 195, 196). Consequently, KMT could reduce inflammation and glucose systemically, thus enhancing the anti-tumorigenic properties of the microenvironment.
Therapeutic ketosis is linked to reduced blood glucose levels and to elevated ketone body levels within normal physiological ranges (Figure 3). Reduction of carbohydrate intake after breast cancer diagnosis in women reduces risk of recurrence (198). Evidence shows that therapeutic ketosis can act synergistically with several drugs and procedures to enhance cancer management while improving both progression-free and overall survival (34, 154, 199, 200). For example, hyperbaric oxygen therapy (HBOT) increases oxidative stress on tumor cells especially when used alongside therapies that reduce blood glucose and elevate blood ketones (201). By reducing blood glucose, KMT would also reduce the immunosuppressive effects of lactic acid in the tumor microenvironment (202). Recent studies show that therapeutic ketosis can facilitate drug delivery through the blood-brain barrier (60, 203). This would be important in helping to target breast cancer cells that metastasize to the brain. Also, it has been reported that caloric restriction reduces leaky tumor blood vessels by increasing neovascular smooth muscle (183). This could account, in part, for a better drug delivery to solid tumors under KMT. The anti-malarial drug, chloroquine neutralizes lysosomal pH reducing phagocytosis and autophagy, and thus depriving invasive and metastatic tumor cells from obtaining glucose and glutamine (34, 204, 205). Chloroquine can also inhibit mitochondrial diaphorases that oxidize NADPH to NAD+, which in turn would reduce mSLP (46, 206). The glutamine dehydrogenase inhibitor, epigallocatechin gallate (EGCG) is also proposed to target glutamine metabolism through an effect on glutamate dehydrogenase (207). The glutaminase inhibitor, 6-diazo-5-oxo-L-norleucine (DON), is a powerful glutamine-targeting drug that can work synergistically with a restricted KD for managing brain cancer and metastasis (60, 208, 209). Hence, KMT can, (a), target the multiple drivers of rapid tumor growth, (b), facilitate drug delivery to the tumor tissue, (c), work synergistically with glutamine-targeting drugs, and, (d), enhance the metabolic efficiency in normal healthy cells. To our knowledge, there are currently no cancer therapies that can target these multiple drivers of tumor growth while simultaneously protecting normal cells.
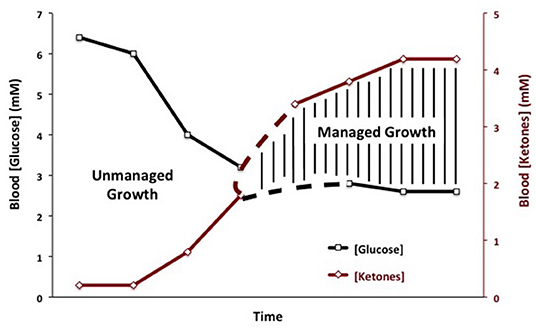
Figure 3. Linkage of plasma glucose and ketone body levels to cancer management. The glucose and ketone (beta-hydroxybutyrate) values are within normal physiological ranges for humans that are under water-only fasting. This state is considered the zone of metabolic management for most cancers. The zone of metabolic management is obtained gradually, as circulating levels of glucose fall and ketones rise. the Glucose Ketone Index (GKI) tracks the transition to therapeutic ketosis. The dashed lines highlight individual variability that could exist in reaching a therapeutic GKI. GKI values approaching 1.0 and below are considered potentially therapeutic. [Reprinted from Meidenbauer et al. (197), and distributed under a Creative Commons license].
The Glucose Ketone Index
The Glucose Ketone Index (GKI) is single number representing the glucose-to-ketone ratio (expressed in mmol/L), and was developed as a guide for evaluating therapeutic efficacy of KMT. The GKI serves as a proxy for the degree of metabolic stress placed on tumor cells through the reduction of circulating glucose and elevation of ketone bodies (beta-hydroxybutyrate, acetoacetate) (34, 197). A GKI value of 1.0 or below has been suggested as the therapeutic goal for cancer management (Figure 4). However, therapeutic GKI values can be difficult to achieve for many cancer patients. For example, tumor burden, toxic treatment protocols, and emotional and physical stress can combine to elevate blood glucose and insulin levels thus preventing the patient from achieving therapeutic GKI values (34, 84, 210). Further refinement of existing KMT therapies along with introduction of new therapies may serve to mitigate this obstacle.
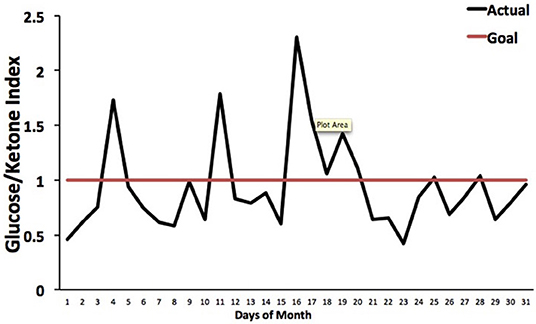
Figure 4. Tracking an individual's GKI using the Glucose Ketone Index Calculator. Index values of 1.0 or below are considered best for managing cancer growth. Individual glucose and ketone values are shown, along with the corresponding GKI values. The GKI values are plotted over the course of a month, whereas the target GKI value (1.0) is plotted as a single line. Tumor management is predicted to be better (slower growth) within the metabolic target zone than outside of the zone. [Reprinted from Meidenbauer et al. (197), and distributed under a Creative Commons license].
KMT as a Complementary or Alternative to SOC
KMT has been used together with low dose chemotherapy and other treatments to manage tumor progression in a woman with stage IV triple negative breast cancer (154). The woman responded well to the combined treatment, and initially reported as a complete therapeutic response. Although overall survival exceeded the median expected for her stage and grade, she eventually succumbed to her cancer. A failure to continue with the KMT protocol was considered responsible in part for her tumor recurrence (154). Nevertheless, a subsequent clinical study showed that a ketogenic diet combined with carboplatin/paclitaxel, hyperthermia, and hyperbaric oxygen therapy significantly improved progression-free and overall survival in patients with advanced non-small cell lung cancer (211). Currently, neoadjuvant therapies are commonly prescribed for a subset of invasive or late-stage cancers, including breast cancers. KMT initiated soon after diagnosis and prior to surgery may prove to be a non-toxic adjunct treatment capable of downgrading the aggressive and invasive nature of the cancer, thereby increasing the efficacy of subsequent treatments (212). It is our view that improvements in selection, dosage, timing, and scheduling of drugs, diet, and procedures will offer benefits in survival and quality of life to patients with advanced metastatic breast cancer when used as a complementary or alternative therapeutic strategy alongside the SOC (Figure 5). The Press-Pulse therapeutic strategy for cancer management was based on the concept of Arens and West, who described how the simultaneous occurrence of “press-pulse” disturbances was responsible for the extinction of organic populations during prior evolutionary epochs (213). A similar concept can be used to show how tumors can be slowly degraded. Optimization of dosing, timing, and scheduling of KMT used together with synergistic drugs and procedures will facilitate the eradication of breast tumor cells with minimal patient toxicity. This therapeutic strategy can serve as a framework for the design of clinical trials for the non-toxic management of most cancers (34). Further details are described in Figure 5.
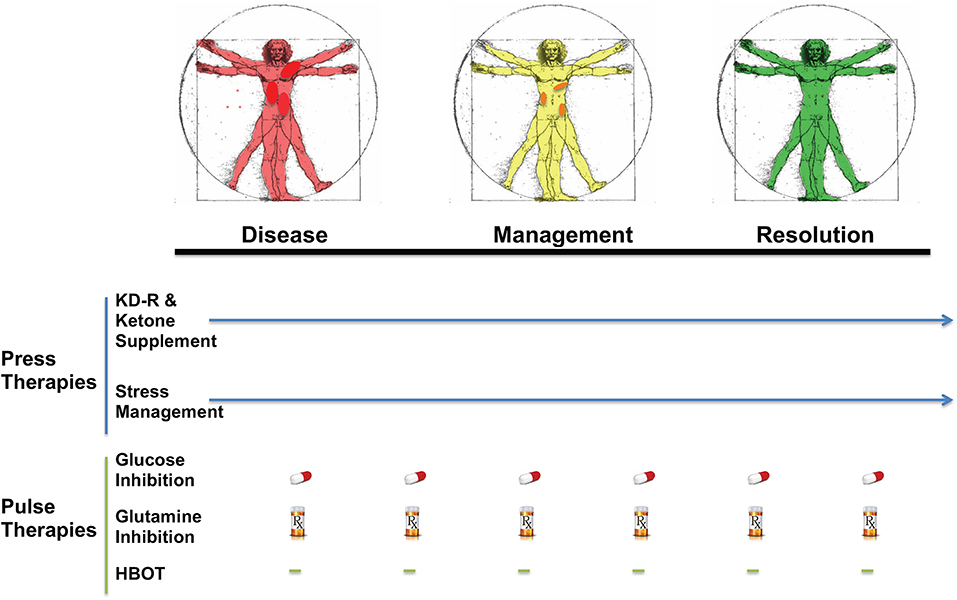
Figure 5. Breast Cancer Management with Press-Pulse Therapeutic Strategy. Arens and West considered the simultaneous occurrence of “Press-Pulse” disturbances as the mechanism responsible for the mass extinction of organic populations during prior evolutionary epochs (213). We described how this concept could be adopted as a therapeutic strategy for the management and possible eradication of cancer (34). This therapeutic strategy considers all cancer, including breast cancer, as a single disease that can be managed by transitioning the energy metabolism of normal cells from glucose to non-fermentable ketone bodies, while simultaneously restricting the availability of fermentable fuels (glucose and glutamine) to tumor cells (34). The reduction in blood glucose levels will also reduce insulin and insulin-like growth factor 1, which are known to drive rapid tumor growth (184, 214, 215). This metabolic therapeutic strategy exploits the dependency of tumor cells on glucose and glutamine fermentation and their inability to metabolize ketone bodies for energy due to defects in the number, structure and function of the tumor mitochondria. In essence, the press-pulse therapy pits the metabolic demands of the mutated tumor cells against those of the normal cells, which evolved to adapt and survive under the extremes of nutrient stress (216). Collections of random mutations will prevent tumor cells from adapting to nutrient stress, thus leading to their extinction according to evolutionary theory (81). As a cancer diagnosis can increase emotional stress and blood glucose, stress management techniques together with exercise could improve general health while reducing glucose availability to the tumor. The press therapies would work synergistically with acute pulse therapies to further restrict glucose and glutamine metabolism. HBOT would work synergistically with the press therapies to increase oxidative stress selectively in tumor cells. The timing (spacing) between the various pulse therapies is designed to stress tumor cell metabolism while minimizing toxicity to normal body cells (34). This therapeutic strategy will target the fermentation metabolism common to most breast tumor cells, thus degrading tumor burden gradually with little or no toxicity. The color change from red (diseased with darker red spots indicative of metastatic lesions), to yellow (with reduced metastasis), to green (resolution) in the Vitruvian man indicates the gradual metabolic management and possible resolution of the breast cancer. The pill and Rx symbols are suggestive of drugs taken orally and/or intravenously (prescription) that would be effective in targeting simultaneously glycolysis and glutaminolysis. Pulse therapies would be eliminated with evidence of tumor management or resolution, while press therapies could continue under modifications or adjustments (arrow). Optimization of timing, dosing, and scheduling of the press-pulse treatments will facilitate eradication of tumor cells with minimal patient toxicity. This therapeutic strategy is a framework for future clinical trials. HBOT, hyperbaric oxygen therapy; KD-R, calorie restricted ketogenic diet. The figure is reprinted with modifications, as described previously (34) and distributed under a Creative Commons license.
Conclusions
Breast cancer, like many cancers, are dependent on fermentation metabolism for growth. Substrate level phosphorylation drives the fermentation metabolism of tumor cells using glucose and glutamine as major fuels in the cytoplasm and mitochondria, respectively. The protracted replacement of respiration with fermentation in cancer cells leads to unbridled cell proliferation. Fusion hybridization of epithelial-derived cancer stem cells with glutamine-dependent tissue macrophages is an alternative explanation to the epithelial-mesenchymal-transition for the origin of metastatic breast cancer cells. The aim of our review is to illustrate that the restriction of glucose and glutamine together with elevation of non-fermentable ketone bodies offers a complementary or alternative therapeutic strategy to the SOC for the non-toxic management of breast cancer. It is our view that KMT could improve progression-free and overall survival for most breast cancer patients.
Author Contributions
TS conceived and wrote the review. MI, AS, MK, and J-PS contributed clinical information and edited the review. PM and CC contributed scientific information to the review.
Conflict of Interest
MK was employed by Dietary Therapies LLC.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Foundation for Metabolic Cancer Therapies, CrossFit Inc., the Nelson and Claudia Peltz Foundation, Joseph C. Maroon, the George Yu Foundation, Kenneth Rainin Foundation, and the Boston College Research Expense Fund for their support.
Abbreviations
SLP, Substrate level phosphorylation; KMT, Ketogenic metabolic therapy; OxPhos, Oxidative phosphorylation; EMT, Epithelial mesenchymal transition; GKI, Glucose Ketone Index; SUCL, succinate-CoA ligase.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. (2019) 69:7–34. doi: 10.3322/caac.21551
2. Desantis CE, Miller KD, Goding Sauer A, Jemal A, Siegel RL. Cancer statistics for African Americans, 2019. CA Cancer J Clin. (2019) 69:211–33. doi: 10.3322/caac.21555
3. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature. (2007) 446:153–8. doi: 10.1038/nature05610
4. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
5. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LAJr, Kinzler KW. Cancer genome landscapes. Science. (2013) 339:1546–58. doi: 10.1126/science.1235122
6. Bayreuther K. Chromosomes in primary neoplastic growth. Nature. (1960) 186:6–9. doi: 10.1038/186006a0
7. Pitot HC. Some biochemical aspects of malignancy. Ann Review Biochem. (1966) 35:335–68. doi: 10.1146/annurev.bi.35.070166.002003
8. Soto AM, Sonnenschein C. The somatic mutation theory of cancer: growing problems with the paradigm? Bioessays. (2004) 26:1097–107. doi: 10.1002/bies.20087
9. Baker SG. A cancer theory kerfuffle can lead to new lines of research. J Natl Cancer Inst. (2015) 107:dju405. doi: 10.1093/jnci/dju405
10. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. (2008) 321:1807–12. doi: 10.1126/science.1164382
11. Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science. (2015) 349:1483–9. doi: 10.1126/science.aab4082
12. Chanock SJ. The paradox of mutations and cancer. Science. (2018) 362:893–4. doi: 10.1126/science.aav5697
13. Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, et al. Somatic mutant clones colonize the human esophagus with age. Science. (2018) 362:911–7. doi: 10.1126/science.aau3879
14. Yizhak K, Aguet F, Kim J, Hess JM, Kubler K, Grimsby J, et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science. (2019) 364:eaaw0726. doi: 10.1126/science.aaw0726
15. Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H, Takeuchi Y, et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature. (2019) 565:312–7. doi: 10.1038/s41586-018-0811-x
16. Huttley GA, Easteal S, Southey MC, Tesoriero A, Giles GG, Mccredie MR, et al. Adaptive evolution of the tumour suppressor BRCA1 in humans and chimpanzees. Aus Breast Cancer Family Study Nat Genet. (2000) 25:410–3. doi: 10.1038/78092
17. Puente XS, Velasco G, Gutierrez-Fernandez A, Bertranpetit J, King MC, Lopez-Otin C. Comparative analysis of cancer genes in the human and chimpanzee genomes. BMC Genomics. (2006) 7:15. doi: 10.1186/1471-2164-7-15
18. Varki NM, Varki A. On the apparent rarity of epithelial cancers in captive chimpanzees. Philos Trans R Soc Lond B Biol Sci. (2015) 370:20140225. doi: 10.1098/rstb.2014.0225
19. Lowenstine LJ, Mcmanamon R, Terio KA. Comparative pathology of aging great apes: bonobos, chimpanzees, gorillas, and orangutans. Vet Pathol. (2016) 53:250–76. doi: 10.1177/0300985815612154
20. Kopp W. How western diet and lifestyle drive the pandemic of obesity and civilization diseases. Diabetes Metab Syndr Obes. (2019) 12:2221–36. doi: 10.2147/DMSO.S216791
21. Seyfried TN. Cancer as a mitochondrial metabolic disease. Front Cell Dev Biol. (2015) 3:43. doi: 10.3389/fcell.2015.00043
22. Kaipparettu BA, Ma Y, Park JH, Lee TL, Zhang Y, Yotnda P, et al. Crosstalk from non-cancerous mitochondria can inhibit tumor properties of metastatic cells by suppressing oncogenic pathways. PLoS ONE. (2013) 8:e61747. doi: 10.1371/journal.pone.0061747
23. Chang JC, Chang HS, Wu YC, Cheng WL, Lin TT, Chang HJ, et al. Mitochondrial transplantation regulates antitumour activity, chemoresistance and mitochondrial dynamics in breast cancer. J Exp Clin Cancer Res. (2019) 38:30. doi: 10.1186/s13046-019-1028-z
24. Isidoro A, Casado E, Redondo A, Acebo P, Espinosa E, Alonso AM, et al. Breast carcinomas fulfill the Warburg hypothesis and provide metabolic markers of cancer prognosis. Carcinogenesis. (2005) 26:2095–104. doi: 10.1093/carcin/bgi188
25. Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci USA. (2005) 102:719–24. doi: 10.1073/pnas.0408894102
26. Ayyasamy V, Owens KM, Desouki MM, Liang P, Bakin A, Thangaraj K, et al. Cellular model of warburg effect identifies tumor promoting function of UCP2 in breast cancer and its suppression by genipin. PLoS ONE. (2011) 6:e24792. doi: 10.1371/journal.pone.0024792
27. Owens KM, Kulawiec M, Desouki MM, Vanniarajan A, Singh KK. Impaired OXPHOS complex III in breast cancer. PLoS ONE. (2011) 6:e23846. doi: 10.1371/journal.pone.0023846
28. Hu Y, Lu W, Chen G, Wang P, Chen Z, Zhou Y, et al. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. (2012) 22:399–412. doi: 10.1038/cr.2011.145
29. Putignani L, Raffa S, Pescosolido R, Rizza T, Del Chierico F, Leone L, et al. Preliminary evidences on mitochondrial injury and impaired oxidative metabolism in breast cancer. Mitochondrion. (2012) 12:363–9. doi: 10.1016/j.mito.2012.02.003
30. Seyfried TN. Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer. Hoboken, NJ: John Wiley & Sons (2012).
31. Verschoor ML, Ungard R, Harbottle A, Jakupciak JP, Parr RL, Singh G. Mitochondria and cancer: past, present, and future. Biomed Res Int. (2013) 2013:612369. doi: 10.1155/2013/612369
32. Seyfried TN, Flores RE, Poff AM, D'agostino DP. Cancer as a metabolic disease: implications for novel therapeutics. Carcinogenesis. (2014) 35:515–27. doi: 10.1093/carcin/bgt480
33. Srinivasan S, Guha M, Dong DW, Whelan KA, Ruthel G, Uchikado Y, et al. Disruption of cytochrome c oxidase function induces the warburg effect and metabolic reprogramming. Oncogene. (2016) 35:1585–95. doi: 10.1038/onc.2015.227
34. Seyfried TN, Yu G, Maroon JC, D'agostino DP. Press-pulse: a novel therapeutic strategy for the metabolic management of cancer. Nutr Metab. (2017) 14:19. doi: 10.1186/s12986-017-0178-2
35. Da Silva I, Da Costa Vieira R, Stella C, Loturco E, Carvalho AL, Veo C, et al. Inborn-like errors of metabolism are determinants of breast cancer risk, clinical response and survival: a study of human biochemical individuality. Oncotarget. (2018) 9:31664–81. doi: 10.18632/oncotarget.25839
36. Rouiller C. Physiological and pathological changes in mitochondrial morphology. Int Rev Cytol. (1960) 9:227–92. doi: 10.1016/S0074-7696(08)62748-5
37. Roddy HJ, Silverberg SG. Ultrastructural analysis of apocrine carcinoma of the human breast. Ultrastruct Pathol. (1980) 1:385–93. doi: 10.3109/01913128009141441
38. Gadaleanu V, Craciun C. Malignant oncocytoma of the breast. Zentralbl Allg Pathol. (1987) 133:279–83.
39. Pelicano H, Xu RH, Du M, Feng L, Sasaki R, Carew JS, et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J Cell Biol. (2006) 175:913–23. doi: 10.1083/jcb.200512100
40. Elliott RL, Jiang XP, Head JF. Mitochondria organelle transplantation: introduction of normal epithelial mitochondria into human cancer cells inhibits proliferation and increases drug sensitivity. Breast Cancer Res Treat. (2012) 136:347–54. doi: 10.1007/s10549-012-2283-2
41. Zeczycki TN, Whelan J, Hayden WT, Brown DA, Shaikh SR. Increasing levels of cardiolipin differentially influence packing of phospholipids found in the mitochondrial inner membrane. Biochem Biophys Res Commun. (2014) 450:366–71. doi: 10.1016/j.bbrc.2014.05.133
42. Jogalekar MP, Serrano EE. Morphometric analysis of a triple negative breast cancer cell line in hydrogel and monolayer culture environments. PeerJ. (2018) 6:e4340. doi: 10.7717/peerj.4340
43. Putignani L, Raffa S, Pescosolido R, Aimati L, Signore F, Torrisi MR, et al. Alteration of expression levels of the oxidative phosphorylation system. (OXPHOS) in breast cancer cell mitochondria. Breast Cancer Res Treat. (2008) 110:439–52. doi: 10.1007/s10549-007-9738-x
44. Arismendi-Morillo G, Castellano-Ramirez A, Seyfried TN. Ultrastructural characterization of the Mitochondria-associated membranes abnormalities in human astrocytomas: functional and therapeutics implications. Ultrastruct Pathol. (2017) 41:234–44. doi: 10.1080/01913123.2017.1300618
45. Morciano G, Marchi S, Morganti C, Sbano L, Bittremieux M, Kerkhofs M, et al. Role of mitochondria-associated ER membranes in calcium regulation in cancer-specific settings. Neoplasia. (2018) 20:510–23. doi: 10.1016/j.neo.2018.03.005
46. Chinopoulos C, Seyfried TN. Mitochondrial substrate level phosphorylation as energy source for glioblastoma: review and hypothesis. ASN Neuro. (2018) 10:1–27. doi: 10.1177/1759091418818261
47. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. (1927) 8:519–30. doi: 10.1085/jgp.8.6.519
49. Warburg O. On the origin of cancer cells. Science. (1956) 123:309–14. doi: 10.1126/science.123.3191.309
51. Warburg O. Revidsed lindau lectures: the prime cause of cancer and prevention - parts 1 & 2, In: Burk D, editor. Meeting of the Nobel-Laureates. Lindau: K.Triltsch (1969). p. 1–9.
53. Selak MA, Armour SM, Mackenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. (2005) 7:77–85. doi: 10.1016/j.ccr.2004.11.022
54. Tannahill GM, Curtis AM, Adamik J, Palsson-Mcdermott EM, Mcgettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. (2013) 496:238–42. doi: 10.1038/nature11986
55. Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. (2014) 515:431–5. doi: 10.1038/nature13909
56. Semenza GL. Hypoxia-inducible factors: coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J. (2017) 36:252–9. doi: 10.15252/embj.201695204
57. Gao C, Shen Y, Jin F, Miao Y, Qiu X. Cancer stem cells in small cell lung cancer cell line H446: higher dependency on oxidative phosphorylation and mitochondrial substrate-level phosphorylation than non-stem cancer cells. PLoS ONE. (2016) 11:e0154576. doi: 10.1371/journal.pone.0154576
58. Chen Q, Kirk K, Shurubor YI, Zhao D, Arreguin AJ, Shahi I, et al. Rewiring of glutamine metabolism is a bioenergetic adaptation of human cells with mitochondrial DNA mutations. Cell Metab. (2018) 27:1007–25 e1005. doi: 10.1016/j.cmet.2018.03.002
59. Flores RE, Brown AK, Taus L, Khoury J, Glover F, Kami K, et al. Mycoplasma infection and hypoxia initiate succinate accumulation and release in the VM-M3 cancer cells. Biochim Biophys Acta. (2018) 1859:975–83. doi: 10.1016/j.bbabio.2018.03.012
60. Mukherjee P, Augur ZM, Li M, Hill C, Greenwood B, Domin MA, et al. Therapeutic benefit of combining calorie-restricted ketogenic diet and glutamine targeting in late-stage experimental glioblastoma. Commun Biol. (2019) 2:200. doi: 10.1038/s42003-019-0455-x
61. Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. (2010) 329:1492–9. doi: 10.1126/science.1188015
62. Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. (2013) 155:397–409. doi: 10.1016/j.cell.2013.09.025
63. Yu M, Chen S, Hong W, Gu Y, Huang B, Lin Y, et al. Prognostic role of glycolysis for cancer outcome: evidence from 86 studies. J Cancer Res Clin Oncol. (2019) 145:967–99. doi: 10.1007/s00432-019-02847-w
64. Seyfried TN, Shelton LM. Cancer as a metabolic disease. Nutr Metab. (2010) 7:7. doi: 10.1186/1743-7075-7-7
65. Seyfried TN. Is mitochondrial glutamine fermentation a missing link in the metabolic theory of cancer? In: Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer. Hoboken, NJ: John Wiley & Sons (2012). p. 133–144. doi: 10.1002/9781118310311.ch8
66. Merchant TE, Gierke LW, Meneses P, Glonek T. 31P magnetic resonance spectroscopic profiles of neoplastic human breast tissues. Cancer Res. (1988) 48:5112–8.
67. Semenza GL, Artemov D, Bedi A, Bhujwalla Z, Chiles K, Feldser D, et al. The metabolism of tumours: 70 years later. Novartis Found Symp. (2001) 240:251–60. doi: 10.1002/0470868716.ch17
68. Isidoro A, Martinez M, Fernandez PL, Ortega AD, Santamaria G, Chamorro M, et al. Alteration of the bioenergetic phenotype of mitochondria is a hallmark of breast, gastric, lung and oesophageal cancer. Biochem J. (2004) 378:17–20. doi: 10.1042/bj20031541
69. Robey IF, Stephen RM, Brown KS, Baggett BK, Gatenby RA, Gillies RJ. Regulation of the warburg effect in early-passage breast cancer cells. Neoplasia. (2008) 10:745–56. doi: 10.1593/neo.07724
70. Palaskas N, Larson SM, Schultz N, Komisopoulou E, Wong J, Rohle D, et al. 18F-fluorodeoxy-glucose positron emission tomography marks MYC-overexpressing human basal-like breast cancers. Cancer Res. (2011) 71:5164–74. doi: 10.1158/0008-5472.CAN-10-4633
71. Pelicano H, Zhang W, Liu J, Hammoudi N, Dai J, Xu RH, et al. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: role of mTOR pathway and therapeutic potential. Breast Cancer Res. (2014) 16:434. doi: 10.1186/s13058-014-0434-6
72. Madonna MC, Fox DB, Crouch BT, Lee J, Zhu C, Martinez AF, et al. Optical imaging of glucose uptake and mitochondrial membrane potential to characterize Her2 breast tumor metabolic phenotypes. Mol Cancer Res. (2019) 17:1545–55. doi: 10.1158/1541-7786.MCR-18-0618
73. Reis LMD, Adamoski D, Ornitz Oliveira Souza R, Rodrigues Ascencao CF, Sousa De Oliveira KR, Correa-Da-Silva F, et al. Dual inhibition of glutaminase and carnitine palmitoyltransferase decreases growth and migration of glutaminase inhibition-resistant triple-negative breast cancer cells. J Biol Chem. (2019) 294:9342–57. doi: 10.1074/jbc.RA119.008180
74. Rosso M, Lapyckyj L, Besso MJ, Monge M, Reventos J, Canals F, et al. Characterization of the molecular changes associated with the overexpression of a novel epithelial cadherin splice variant mRNA in a breast cancer model using proteomics and bioinformatics approaches: identification of changes in cell metabolism and an increased expression of lactate dehydrogenase B. Cancer Metab. (2019) 7:5. doi: 10.1186/s40170-019-0196-9
75. Katt WP, Ramachandran S, Erickson JW, Cerione RA. Dibenzophenanthridines as inhibitors of glutaminase C and cancer cell proliferation. Mol Cancer Ther. (2012) 11:1269–78. doi: 10.1158/1535-7163.MCT-11-0942
76. Van Geldermalsen M, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D, et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene. (2016) 35:3201–8. doi: 10.1038/onc.2015.381
77. Demas DM, Demo S, Fallah Y, Clarke R, Nephew KP, Althouse S, et al. Glutamine metabolism drives growth in advanced hormone receptor positive breast cancer. Front Oncol. (2019) 9:686. doi: 10.3389/fonc.2019.00686
78. Gwangwa MV, Joubert AM, Visagie MH. Effects of glutamine deprivation on oxidative stress and cell survival in breast cell lines. Biol Res. (2019) 52:15. doi: 10.1186/s40659-019-0224-9
79. Mayr E. The Growth of Biological Thought: Diversity, Evolution, and Inheritance. Cambridge: Belknap Harvard (1982).
81. Seyfried TN. Nothing in cancer biology makes sense except in the light of evolution In: Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer. Hoboken, NJ: John Wiley & Sons (2012). p. 261–75. doi: 10.1002/9781118310311.ch15
82. Sonnenschein C, Soto AM. Somatic mutation theory of carcinogenesis: why it should be dropped and replaced. Mol Carcinog. (2000) 29:205–211. doi: 10.1002/1098-2744(200012)29:4>205::AID-MC1002<3.0.CO;2-W
83. Szent-Gyorgyi A. The living state and cancer. Proc Natl Acad Sci USA. (1977) 74:2844–7. doi: 10.1073/pnas.74.7.2844
84. Seyfried TN, Shelton L, Arismendi-Morillo G, Kalamian M, Elsakka A, Maroon J, et al. Provocative question: should ketogenic metabolic therapy become the standard of care for glioblastoma? Neurochem Res. (2019) 44:2392–404. doi: 10.1007/s11064-019-02795-4
85. Klaunig JE, Kamendulis LM, Hocevar BA. Oxidative stress and oxidative damage in carcinogenesis. Toxicol Pathol. (2010) 38:96–109. doi: 10.1177/0192623309356453
86. Iglesias P, Salas A, Costoya JA. The maintenance of mitochondrial genetic stability is crucial during the oncogenic process. Commun Integ Biol. (2012) 5:34–8. doi: 10.4161/cib.18160
87. Galadari S, Rahman A, Pallichankandy S, Thayyullathil F. Reactive oxygen species and cancer paradox: to promote or to suppress? Free Radic Biol Med. (2017) 104:144–64. doi: 10.1016/j.freeradbiomed.2017.01.004
88. Desler C, Lykke A, Rasmussen LJ. The effect of mitochondrial dysfunction on cytosolic nucleotide metabolism. J Nucleic Acids. (2010) 2010:701518. doi: 10.4061/2010/701518
89. Ralph SJ, Rodriguez-Enriquez S, Neuzil J, Saavedra E, Moreno-Sanchez R. The causes of cancer revisited: mitochondrial malignancy and ROS-induced oncogenic transformation - why mitochondria are targets for cancer therapy. Mol Aspects Med. (2010) 31:145–70. doi: 10.1016/j.mam.2010.02.008
90. Valle A, Oliver J, Roca P. Role of uncoupling proteins in cancer. Cancers. (2010) 2:567–91. doi: 10.3390/cancers2020567
91. Degtyareva NP, Heyburn L, Sterling J, Resnick MA, Gordenin DA, Doetsch PW. Oxidative stress-induced mutagenesis in single-strand DNA occurs primarily at cytosines and is DNA polymerase zeta-dependent only for adenines and guanines. Nucleic Acids Res. (2013) 41:8995–9005. doi: 10.1093/nar/gkt671
92. Bartesaghi S, Graziano V, Galavotti S, Henriquez NV, Betts J, Saxena J, et al. Inhibition of oxidative metabolism leads to p53 genetic inactivation and transformation in neural stem cells. Proc Natl Acad Sci USA. (2015) 112:1059–64. doi: 10.1073/pnas.1413165112
93. Rodic S, Vincent MD. Reactive oxygen species. (ROS) are a key determinant of cancer's metabolic phenotype. Int J Cancer. (2018) 142:440–8. doi: 10.1002/ijc.31069
94. Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the warburg effect in carcinogenesis. J Biol Chem. (2002) 277:23111–5. doi: 10.1074/jbc.M202487200
95. Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, et al. p53 regulates mitochondrial respiration. Science. (2006) 312:1650–3. doi: 10.1126/science.1126863
96. Lu W, Pelicano H, Huang P. Cancer metabolism: is glutamine sweeter than glucose? Cancer Cell. (2010) 18:199–200. doi: 10.1016/j.ccr.2010.08.017
97. Yang D, Wang MT, Tang Y, Chen Y, Jiang H, Jones TT, et al. Impairment of mitochondrial respiration in mouse fibroblasts by oncogenic H-RASQ61L. Cancer Biol Ther. (2010) 9:122–33. doi: 10.4161/cbt.9.2.10379
98. Nicolay BN, Danielian PS, Kottakis F, Lapek JD Jr, Sanidas I, Miles WO, et al. Proteomic analysis of pRb loss highlights a signature of decreased mitochondrial oxidative phosphorylation. Genes Dev. (2015) 29:1875–89. doi: 10.1101/gad.264127.115
99. Szeliga M, Albrecht J. Opposing roles of glutaminase isoforms in determining glioblastoma cell phenotype. Neurochem Int. (2015) 88:6–9. doi: 10.1016/j.neuint.2014.11.004
100. Seyfried TN. Energetics of normal cells and cancer cells In: Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer. Hoboken, NJ: John Wiley & Sons (2012). p. 47–72. doi: 10.1002/9781118310311.ch4
101. Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, et al. Tumour-associated mutant p53 drives the warburg effect. Nat Commun. (2013) 4:2935. doi: 10.1038/ncomms3935
102. Reynolds MR, Lane AN, Robertson B, Kemp S, Liu Y, Hill BG, et al. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene. (2014) 33:556–66. doi: 10.1038/onc.2012.635
103. Osborne C, Wilson P, Tripathy D. Oncogenes and tumor suppressor genes in breast cancer: potential diagnostic and therapeutic applications. Oncologist. (2004) 9:361–77. doi: 10.1634/theoncologist.9-4-361
104. Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. (2016) 7:11479. doi: 10.1038/ncomms11479
105. Zhang D, Tai LK, Wong LL, Chiu LL, Sethi SK, Koay ES. Proteomic study reveals that proteins involved in metabolic and detoxification pathways are highly expressed in HER-2/neu-positive breast cancer. Mol Cell Proteomics. (2005) 4:1686–96. doi: 10.1074/mcp.M400221-MCP200
106. Ding Y, Liu Z, Desai S, Zhao Y, Liu H, Pannell LK, et al. Receptor tyrosine kinase ErbB2 translocates into mitochondria and regulates cellular metabolism. Nat Commun. (2012) 3:1271. doi: 10.1038/ncomms2236
107. Privat M, Radosevic-Robin N, Aubel C, Cayre A, Penault-Llorca F, Marceau G, et al. BRCA1 induces major energetic metabolism reprogramming in breast cancer cells. PLoS ONE. (2014) 9:e102438. doi: 10.1371/journal.pone.0102438
108. Cuyas E, Fernandez-Arroyo S, Alarcon T, Lupu R, Joven J, Menendez JA. Germline BRCA1 mutation reprograms breast epithelial cell metabolism towards mitochondrial-dependent biosynthesis: evidence for metformin-based starvation strategies in BRCA1 carriers. Oncotarget. (2016) 7:52974–92. doi: 10.18632/oncotarget.9732
109. Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Crit Rev Oncogene. (2013) 18:43–73. doi: 10.1615/CritRevOncog.v18.i1-2.40
110. Lazebnik Y. What are the hallmarks of cancer? Nat Rev Cancer. (2010) 10:232–3. doi: 10.1038/nrc2827
111. Fidler IJ. The pathogenesis of cancer metastasis: the seed and soil hypothesis revisited. Nat Rev Cancer. (2003) 3:453–8. doi: 10.1038/nrc1098
113. Kar R, Jha NK, Jha SK, Sharma A, Dholpuria S, Asthana N, et al. A NOTCH Deeper into the epithelial-to-mesenchymal transition. (EMT) program in breast cancer. Genes. (2019) 10:961. doi: 10.3390/genes10120961
114. Pawelek JM. Cancer-cell fusion with migratory bone-marrow-derived cells as an explanation for metastasis: new therapeutic paradigms. Fut Oncol. (2008) 4:449–52. doi: 10.2217/14796694.4.4.449
115. Pawelek JM, Chakraborty AK. The cancer cell–leukocyte fusion theory of metastasis. Adv Cancer Res. (2008) 101:397–444. doi: 10.1016/S0065-230X(08)00410-7
116. Powell AE, Anderson EC, Davies PS, Silk AD, Pelz C, Impey S, et al. Fusion between Intestinal epithelial cells and macrophages in a cancer context results in nuclear reprogramming. Cancer Res. (2011) 71:1497–505. doi: 10.1158/0008-5472.CAN-10-3223
117. Gast CE, Silk AD, Zarour L, Riegler L, Burkhart JG, Gustafson KT, et al. Cell fusion potentiates tumor heterogeneity and reveals circulating hybrid cells that correlate with stage and survival. Sci Adv. (2018) 4:eaat7828. doi: 10.1126/sciadv.aat7828
118. Marin-Padilla M. Erythrophagocytosis by epithelial cells of a breast carcinoma. Cancer. (1977) 39:1085–1089. doi: 10.1002/1097-0142(197703)39:3>1085::AID-CNCR2820390312<3.0.CO;2-U
119. Ruff MR, Pert CB. Small cell carcinoma of the lung: macrophage-specific antigens suggest hemopoietic stem cell origin. Science. (1984) 225:1034–6. doi: 10.1126/science.6089338
120. Ruff MR, Pert CB. Origin of human small cell lung cancer. Science. (1985) 229:680. doi: 10.1126/science.229.4714.680
121. Calvo F, Martin PM, Jabrane N, De Cremoux P, Magdelenat H. Human breast cancer cells share antigens with the myeloid monocyte lineage. Br J Cancer. (1987) 56:15–9. doi: 10.1038/bjc.1987.145
122. Kobayashi M, Sugimoto T, Okabayashi T, Okamoto K, Namikawa T, Tochika N, et al. Localization of thymidine phosphorylase in breast cancer tissue. Med Mol Morphol. (2005) 38:112–7. doi: 10.1007/s00795-005-0282-7
123. Huysentruyt LC, Seyfried TN. Perspectives on the mesenchymal origin of metastatic cancer. Cancer Metastasis Rev. (2010) 29:695–707. doi: 10.1007/s10555-010-9254-z
124. Schramm HM. Should EMT of cancer cells be understood as epithelial-myeloid transition? J Cancer. (2014) 5:125–32. doi: 10.7150/jca.8242
125. Rowan DJ, Logunova V, Van Tuinen P, Olteanu H, Peterson JF. Circulating breast carcinoma cells mimicking therapy-related acute myeloid leukemia. Int J Surg Pathol. (2017) 25:87–93. doi: 10.1177/1066896916664986
126. Wu JM, Fackler MJ, Halushka MK, Molavi DW, Taylor ME, Teo WW, et al. Heterogeneity of breast cancer metastases: comparison of therapeutic target expression and promoter methylation between primary tumors and their multifocal metastases. Clin Cancer Res. (2008) 14:1938–46. doi: 10.1158/1078-0432.CCR-07-4082
127. Weigelt B, Reis-Filho JS. Histological and molecular types of breast cancer: is there a unifying taxonomy? Nat Rev Clin Oncol. (2009) 6:718–30. doi: 10.1038/nrclinonc.2009.166
128. Paget S. The distribution of secondary growths in cancer of the breast. Lancet. (1889) 1:571–3. doi: 10.1016/S0140-6736(00)49915-0
129. Kawaguchi T. Organ preference of cancer metastasis and metastasis-related cell adhesion molecules including carbohydrates. Cardiovasc Hematol Disord Drug Targets. (2016) 15:164–86. doi: 10.2174/1871529X15666151102102551
130. Newsholme P. Why is L-glutamine metabolism important to cells of the immune system in health, postinjury, surgery or infection? J Nutr. (2001) 131:2515S−22S. doi: 10.1093/jn/131.9.2515S
131. Ren W, Xia Y, Chen S, Wu G, Bazer FW, Zhou B, et al. Glutamine metabolism in macrophages: a novel target for obesity/type 2 diabetes. Adv Nutr. (2019) 10:321–30. doi: 10.1093/advances/nmy084
132. Wallis M, Tardivon A, Helbich T, Schreer I, European Society of Breast I. Guidelines from the European society of breast imaging for diagnostic interventional breast procedures. Eur Radiol. (2007) 17:581–8. doi: 10.1007/s00330-006-0408-x
133. Kurian AW, Lichtensztajn DY, Keegan TH, Nelson DO, Clarke CA, Gomez SL. Use of and mortality after bilateral mastectomy compared with other surgical treatments for breast cancer in California, 1998-2011. JAMA. (2014) 312:902–14. doi: 10.1001/jama.2014.10707
134. Hofer SO, Molema G, Hermens RA, Wanebo HJ, Reichner JS, Hoekstra HJ. The effect of surgical wounding on tumour development. Eur J Surg Oncol. (1999) 25:231–43. doi: 10.1053/ejso.1998.0634
135. Tagliabue E, Agresti R, Carcangiu ML, Ghirelli C, Morelli D, Campiglio M, et al. Role of HER2 in wound-induced breast carcinoma proliferation. Lancet. (2003) 362:527–33. doi: 10.1016/S0140-6736(03)14112-8
136. Baum M. Does the act of surgery provoke activation of latent metastases in early breast cancer? Breast Cancer Res. (2004) 6:160–1. doi: 10.1186/bcr902
137. Demicheli R, Retsky MW, Hrushesky WJ, Baum M, Gukas ID. The effects of surgery on tumor growth: a century of investigations. Ann Oncol. (2008) 19:1821–8. doi: 10.1093/annonc/mdn386
138. Walter ND, Rice PL, Redente EF, Kauvar EF, Lemond L, Aly T, et al. Wound healing after trauma may predispose to lung cancer metastasis: review of potential mechanisms. Am J Respir Cell Mol Biol. (2011) 44:591–6. doi: 10.1165/rcmb.2010-0187RT
139. Hobson J, Gummadidala P, Silverstrim B, Grier D, Bunn J, James T, et al. Acute inflammation induced by the biopsy of mouse mammary tumors promotes the development of metastasis. Breast Cancer Res Treat. (2013) 139:391–401. doi: 10.1007/s10549-013-2575-1
140. Szalayova G, Ogrodnik A, Spencer B, Wade J, Bunn J, Ambaye A, et al. Human breast cancer biopsies induce eosinophil recruitment and enhance adjacent cancer cell proliferation. Breast Cancer Res Treat. (2016) 157:461–74. doi: 10.1007/s10549-016-3839-3
141. Singer CF, Hudelist G, Fuchs EM, Kostler W, Fink-Retter A, Gschwantler-Kaulich D, et al. Incomplete surgical resection of ductal carcinomas in situ results in activation of ERBB2 in residual breast cancer cells. Endocr Relat Cancer. (2009) 16:73–83. doi: 10.1677/ERC-08-0065
142. Muti P, Quattrin T, Grant BJ, Krogh V, Micheli A, Schunemann HJ, et al. Fasting glucose is a risk factor for breast cancer: a prospective study. Cancer Epidemiol Biomarkers Prev. (2002) 11:1361–8.
143. Krone CA, Ely JT. Controlling hyperglycemia as an adjunct to cancer therapy. Integr Cancer Ther. (2005) 4:25–31. doi: 10.1177/1534735404274167
144. Rapp K, Schroeder J, Klenk J, Ulmer H, Concin H, Diem G, et al. Fasting blood glucose and cancer risk in a cohort of more than 140,000 adults in Austria. Diabetologia. (2006) 49:945–52. doi: 10.1007/s00125-006-0207-6
145. Alokail MS, Al-Daghri N, Abdulkareem A, Draz HM, Yakout SM, Alnaami AM, et al. Metabolic syndrome biomarkers and early breast cancer in Saudi women: evidence for the presence of a systemic stress response and/or a pre-existing metabolic syndrome-related neoplasia risk? BMC Cancer. (2013) 13:54. doi: 10.1186/1471-2407-13-54
146. Lende TH, Austdal M, Bathen TF, Varhaugvik AE, Skaland I, Gudlaugsson E, et al. Metabolic consequences of perioperative oral carbohydrates in breast cancer patients - an explorative study. BMC Cancer. (2019) 19:1183. doi: 10.1186/s12885-019-6393-7
147. Li W, Zhang X, Sang H, Zhou Y, Shang C, Wang Y, et al. Effects of hyperglycemia on the progression of tumor diseases. J Exp Clin Cancer Res. (2019) 38:327. doi: 10.1186/s13046-019-1309-6
148. Windelov JA, Pedersen J, Holst JJ. Use of anesthesia dramatically alters the oral glucose tolerance and insulin secretion in C57Bl/6 mice. Physiol Rep. (2016) 4:e12824. doi: 10.14814/phy2.12824
149. Guo NL, Zhang JX, Wu JP, Xu YH. Isoflurane promotes glucose metabolism through up-regulation of miR-21 and suppresses mitochondrial oxidative phosphorylation in ovarian cancer cells. Biosci Rep. (2017) 37:BSR20170818. doi: 10.1042/BSR20170818
150. Fan P, Siwak DR, Abderrahman B, Agboke FA, Yerrum S, Jordan VC. Suppression of nuclear factor- κb by glucocorticoid receptor blocks estrogen-induced apoptosis in estrogen-deprived breast cancer cells. Mol Cancer Ther. (2019) 18:1684–95. doi: 10.1158/1535-7163.MCT-18-1363
151. Gluschnaider U, Hertz R, Ohayon S, Smeir E, Smets M, Pikarsky E, et al. Long-chain fatty acid analogues suppress breast tumorigenesis and progression. Cancer Res. (2014) 74:6991–7002. doi: 10.1158/0008-5472.CAN-14-0385
152. Branca JJ, Pacini S, Ruggiero M. Effects of pre-surgical vitamin d supplementation and ketogenic diet in a patient with recurrent breast cancer. Anticancer Res. (2015) 35:5525–32.
153. Hyde PN, Lustberg MB, Miller VJ, Lafountain RA, Volek JS. Pleiotropic effects of nutritional ketosis: conceptual framework for ketoadaptation as a breast cancer therapy. Cancer Treat Res Comm. (2017) 12:32–9. doi: 10.1016/j.ctarc.2017.06.001
154. Iyikesici MS, Slocum AK, Slocum A, Berkarda FB, Kalamian M, Seyfried TN. Efficacy of metabolically supported chemotherapy combined with ketogenic diet, hyperthermia, and hyperbaric oxygen therapy for stage IV triple-negative breast cancer. Cureus. (2017) 9:e1445. doi: 10.7759/cureus.1445
155. Klement RJ, Bandyopadhyay PS, Champ CE, Walach H. Application of Bayesian evidence synthesis to modelling the effect of ketogenic therapy on survival of high grade glioma patients. Theor Biol Med Model. (2018) 15:12. doi: 10.1186/s12976-018-0084-y
156. Cohen CW, Fontaine KR, Arend RC, Gower BA. A ketogenic diet is acceptable in women with ovarian and endometrial cancer and has no adverse effects on blood lipids: a randomized, controlled trial. Nutr Cancer. (2019) 27:1–11. doi: 10.1080/01635581.2019.1645864
157. Khodabakhshi A, Akbari ME, Mirzaei HR, Mehrad-Majd H, Kalamian M, Davoodi SH. Feasibility, safety, and beneficial effects of MCT-based ketogenic diet for breast cancer treatment: a randomized controlled trial study. Nutr Cancer. (2019) 9:1–8. doi: 10.1080/01635581.2019.1650942
158. Licha D, Vidali S, Aminzadeh-Gohari S, Alka O, Breitkreuz L, Kohlbacher O, et al. Untargeted metabolomics reveals molecular effects of ketogenic diet on healthy and tumor xenograft mouse models. Int J Mol Sci. (2019) 20:3873. doi: 10.3390/ijms20163873
159. Mitchell T, Clarke L, Goldberg A, Bishop KS. Pancreatic cancer cachexia: the role of nutritional interventions. Healthcare. (2019) 7:89. doi: 10.3390/healthcare7030089
160. Weber DD, Aminzadeh-Gohari S, Tulipan J, Catalano L, Feichtinger RG, Kofler B. Ketogenic diet in the treatment of cancer - Where do we stand? Mol Metab. (2019) 118:668–88. doi: 10.1016/j.molmet.2019.06.026
161. Bartmann C, Janaki Raman SR, Floter J, Schulze A, Bahlke K, Willingstorfer J, et al. Beta-hydroxybutyrate. (3-OHB) can influence the energetic phenotype of breast cancer cells, but does not impact their proliferation and the response to chemotherapy or radiation. Cancer Metab. (2018) 6:8. doi: 10.1186/s40170-018-0180-9
162. Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. (2010) 9:3506–14. doi: 10.4161/cc.9.17.12731
163. Zheng J. Energy metabolism of cancer: glycolysis versus oxidative phosphorylation. Oncol Lett. (2012) 4:1151–7. doi: 10.3892/ol.2012.928
164. Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. (2017) 541:41–5. doi: 10.1038/nature20791
165. Rodrigues LM, Uribe-Lewis S, Madhu B, Honess DJ, Stubbs M, Griffiths JR. The action of beta-hydroxybutyrate on the growth, metabolism and global histone H3 acetylation of spontaneous mouse mammary tumours: evidence of a beta-hydroxybutyrate paradox. Cancer Metab. (2017) 5:4. doi: 10.1186/s40170-017-0166-z
166. Giudetti AM, De Domenico S, Ragusa A, Lunetti P, Gaballo A, Franck J, et al. A specific lipid metabolic profile is associated with the epithelial mesenchymal transition program. Biochim Biophys Acta Mol Cell Biol Lipids. (2019) 1864:344–57. doi: 10.1016/j.bbalip.2018.12.011
167. Kuok IT, Rountree AM, Jung SR, Sweet IR. Palmitate is not an effective fuel for pancreatic islets and amplifies insulin secretion independent of calcium release from endoplasmic reticulum. Islets. (2019) 11:51–64. doi: 10.1080/19382014.2019.1601490
168. Lehninger AL. The Mitochondrion: Molecular Basis of Structure and Function. New York, NY: W.A. Benjamin, INC (1964).
169. Samudio I, Fiegl M, Andreeff M. Mitochondrial uncoupling and the warburg effect: molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. (2009) 69:2163–6. doi: 10.1158/0008-5472.CAN-08-3722
170. Vozza A, Parisi G, De Leonardis F, Lasorsa FM, Castegna A, Amorese D, et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc Natl Acad Sci USA. (2014) 111:960–5. doi: 10.1073/pnas.1317400111
171. Hardy S, El-Assaad W, Przybytkowski E, Joly E, Prentki M, Langelier Y. Saturated fatty acid-induced apoptosis in MDA-MB-231 breast cancer cells. a role for cardiolipin. J Biol Chem. (2003) 278:31861–70. doi: 10.1074/jbc.M300190200
172. Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr, Ory DS, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci USA. (2003) 100:3077–82. doi: 10.1073/pnas.0630588100
173. Kamili A, Roslan N, Frost S, Cantrill LC, Wang D, Della-Franca A, et al. TPD52 expression increases neutral lipid storage within cultured cells. J Cell Sci. (2015) 128:3223–38. doi: 10.1242/jcs.167692
174. Ta NL, Seyfried TN. Influence of serum and hypoxia on incorporation of [(14)C]-D-glucose or [(14)C]-L-glutamine into lipids and lactate in murine glioblastoma cells. Lipids. (2015) 50:1167–84. doi: 10.1007/s11745-015-4075-z
175. Mahoney LB, Denny CA, Seyfried TN. Caloric restriction in C57BL/6J mice mimics therapeutic fasting in humans. Lipids Health Dis. (2006) 5:13. doi: 10.1186/1476-511X-5-13
176. Meidenbauer JJ, Ta N, Seyfried TN. Influence of a ketogenic diet, fish-oil, and calorie restriction on plasma metabolites and lipids in C57BL/6J mice. Nutr Metab. (2014) 11:23. doi: 10.1186/1743-7075-11-23
177. Kiebish MA, Han X, Cheng H, Chuang JH, Seyfried TN. Cardiolipin and electron transport chain abnormalities in mouse brain tumor mitochondria: lipidomic evidence supporting the Warburg theory of cancer. J Lipid Res. (2008) 49:2545–56. doi: 10.1194/jlr.M800319-JLR200
178. Cogliati S, Enriquez JA, Scorrano L. Mitochondrial cristae: where beauty meets functionality. Trends Biochem Sci. (2016) 41:261–73. doi: 10.1016/j.tibs.2016.01.001
179. Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. (2019) 366:1013–21. doi: 10.1126/science.aav2588
180. Reckzeh ES, Karageorgis G, Schwalfenberg M, Ceballos J, Nowacki J, Stroet MCM, et al. Inhibition of glucose transporters and glutaminase synergistically impairs tumor cell growth. Cell Chem Biol. (2019) 26:1214–28 e1225. doi: 10.1016/j.chembiol.2019.06.005
181. Zhou W, Mukherjee P, Kiebish MA, Markis WT, Mantis JG, Seyfried TN. The calorically restricted ketogenic diet, an effective alternative therapy for malignant brain cancer. Nutr Metab. (2007) 4:5. doi: 10.1186/1743-7075-4-5
182. Mulrooney TJ, Marsh J, Urits I, Seyfried TN, Mukherjee P. Influence of Caloric Restriction on Constitutive Expression of NF-kappaB in an Experimental Mouse Astrocytoma. PLoS ONE. (2011) 6:e18085. doi: 10.1371/journal.pone.0018085
183. Urits I, Mukherjee P, Meidenbauer J, Seyfried TN. Dietary restriction promotes vessel maturation in a mouse astrocytoma. J Oncol. (2012) 264039:10. doi: 10.1155/2012/264039
184. Mukherjee P, El-Abbadi MM, Kasperzyk JL, Ranes MK, Seyfried TN. Dietary restriction reduces angiogenesis and growth in an orthotopic mouse brain tumour model. Br J Cancer. (2002) 86:1615–21. doi: 10.1038/sj.bjc.6600298
185. Mukherjee P, Mulrooney TJ, Marsh J, Blair D, Chiles TC, Seyfried TN. Differential effects of energy stress on AMPK phosphorylation and apoptosis in experimental brain tumor and normal brain. Mol Cancer. (2008) 7:37. doi: 10.1186/1476-4598-7-37
186. Shelton LM, Huysentruyt LC, Mukherjee P, Seyfried TN. Calorie restriction as an anti-invasive therapy for malignant brain cancer in the VM mouse. ASN Neuro. (2010) 2:e00038. doi: 10.1042/AN20100002
187. Simone BA, Palagani A, Strickland K, Ko K, Jin L, Lim MK, et al. Caloric restriction counteracts chemotherapy-induced inflammation and increases response to therapy in a triple negative breast cancer model. Cell Cycle. (2018) 17:1536–44. doi: 10.1080/15384101.2018.1471314
188. Wallace TC, Bultman S, D'adamo C, Daniel CR, Debelius J, Ho E, et al. Personalized nutrition in disrupting cancer - proceedings from the 2017 american college of nutrition annual meeting. J Am Coll Nutr. (2019) 38:1–14. doi: 10.1080/07315724.2018.1500499
189. Veech RL. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids. (2004) 70:309–19. doi: 10.1016/j.plefa.2003.09.007
190. D'agostino DP, Olson JE, Dean JB. Acute hyperoxia increases lipid peroxidation and induces plasma membrane blebbing in human U87 glioblastoma cells. Neuroscience. (2009) 159:1011–22. doi: 10.1016/j.neuroscience.2009.01.062
191. Veech RL, Todd King M, Pawlosky R, Kashiwaya Y, Bradshaw PC, Curtis W. The great controlling nucleotide coenzymes. IUBMB Life. (2019) 71:565–79. doi: 10.1002/iub.1997
192. Wu W, Chaudhuri S, Brickley DR, Pang D, Karrison T, Conzen SD. Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer Res. (2004) 64:1757–64. doi: 10.1158/0008-5472.CAN-03-2546
193. Champ CE, Palmer JD, Volek JS, Werner-Wasik M, Andrews DW, Evans JJ, et al. Targeting metabolism with a ketogenic diet during the treatment of glioblastoma multiforme. J Neuro Oncol. (2014) 117:125–31. doi: 10.1007/s11060-014-1362-0
194. Rieger J, Bahr O, Maurer GD, Hattingen E, Franz K, Brucker D, et al. ERGO: a pilot study of ketogenic diet in recurrent glioblastoma. Int J Oncol. (2014) 44:1843–52. doi: 10.3892/ijo.2014.2382
195. De Beer JC, Liebenberg L. Does cancer risk increase with HbA1c, independent of diabetes? Br J Cancer. (2014) 110:2361–8. doi: 10.1038/bjc.2014.150
196. Wu K, Yu X, Huang Z, Zhu D, Yi X, Wu YL, et al. Targeting of PP2Cdelta By a small molecule C23 inhibits high glucose-induced breast cancer progression in vivo. Antioxid Redox Signal. (2019) 30:1983–98. doi: 10.1089/ars.2017.7486
197. Meidenbauer JJ, Mukherjee P, Seyfried TN. The glucose ketone index calculator: a simple tool to monitor therapeutic efficacy for metabolic management of brain cancer. Nutr Metab. (2015) 12:12. doi: 10.1186/s12986-015-0009-2
198. Emond JA, Pierce JP, Natarajan L, Gapuz LR, Nguyen J, Parker BA, et al. Risk of breast cancer recurrence associated with carbohydrate intake and tissue expression of IGFI receptor. Cancer Epidemiol Biomark Prev. (2014) 23:1273–9. doi: 10.1158/1055-9965.EPI-13-1218
199. Klement RJ. Beneficial effects of ketogenic diets for cancer patients: a realist review with focus on evidence and confirmation. Med Oncol. (2017) 34:132. doi: 10.1007/s12032-017-0991-5
200. Santos JG, Souza Da Cruz WM, Schonthal AH, Salazar MD, Fontes CA, Qiuirico-Santos T, et al. Efficacy of a ketogenic diet with concomitant intranasal perillyl alcohol as a novel strategy for the therapy of recurrent glioblastoma. Onc Lett. (2018) 15:1263–70. doi: 10.3892/ol.2017.7362
201. Poff AM, Ari C, Seyfried TN, D'agostino DP. The ketogenic diet and hyperbaric oxygen therapy prolong survival in mice with systemic metastatic cancer. PLoS ONE. (2013) 8:e65522. doi: 10.1371/journal.pone.0065522
202. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol. (2013) 191:1486–95. doi: 10.4049/jimmunol.1202702
203. Denny CA, Heinecke KA, Kim YP, Baek RC, Loh KS, Butters TD, et al. Restricted ketogenic diet enhances the therapeutic action of N-butyldeoxynojirimycin towards brain GM2 accumulation in adult sandhoff disease mice. J Neurochem. (2010) 113:1525–35. doi: 10.1111/j.1471-4159.2010.06733.x
204. Al-Bari MA. Chloroquine analogues in drug discovery: new directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J Antimicrob Chemother. (2015) 70:1608–21. doi: 10.1093/jac/dkv018
205. Ye H, Chen M, Cao F, Huang H, Zhan R, Zheng X. Chloroquine, an autophagy inhibitor, potentiates the radiosensitivity of glioma initiating cells by inhibiting autophagy and activating apoptosis. BMC Neurol. (2016) 16:178. doi: 10.1186/s12883-016-0700-6
206. Hrabak A, Sefrioui H, Vercruysse V, Temesi A, Bajor T, Vray B. Action of chloroquine on nitric oxide production and parasite killing by macrophages. Eur J Pharmacol. (1998) 354:83–90. doi: 10.1016/S0014-2999(98)00427-0
207. Yang C, Ko B, Hensley CT, Jiang L, Wasti AT, Kim J, et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol Cell. (2014) 56:414–24. doi: 10.1016/j.molcel.2014.09.025
208. Shelton LM, Huysentruyt LC, Seyfried TN. Glutamine targeting inhibits systemic metastasis in the VM-M3 murine tumor model. Inter J Cancer. (2010) 127:2478–85. doi: 10.1002/ijc.25431
209. Lemberg KM, Vornov JJ, Rais R, Slusher BS. We're not DON Yet: optimal dosing and prodrug delivery of 6-Diazo-5-oxo-L-norleucine. Mol Cancer Ther. (2018) 17:1824–32. doi: 10.1158/1535-7163.MCT-17-1148
210. Kalamian M. KETO for CANCER: Ketogenic Metabolic Therapy as a Targeted Nutritional Strategy. White River Junction, VT: Chelsea Green (2017).
211. Iyikesici MS. Feasibility study of metabolically supported chemotherapy with weekly carboplatin/paclitaxel combined with ketogenic diet, hyperthermia and hyperbaric oxygen therapy in metastatic non-small cell lung cancer. Int J Hyperthermia. (2019) 36:446–55. doi: 10.1080/02656736.2019.1589584
212. Elsakka AMA, Bary MA, Abdelzaher E, Elnaggar M, Kalamian M, Mukherjee P, et al. Management of glioblastoma multiforme in a patient treated with ketogenic metabolic therapy and modified standard of care: A 24-month follow-up. Front Nutr. (2018) 5:20. doi: 10.3389/fnut.2018.00020
213. Arens NC, West ID. Press-pulse: a general theory of mass extinction? Paleobiology. (2008) 34:456–71. doi: 10.1666/07034.1
214. Mukherjee P, Abate LE, Seyfried TN. Antiangiogenic and proapoptotic effects of dietary restriction on experimental mouse and human brain tumors. Clin Cancer Res. (2004) 10:5622–9. doi: 10.1158/1078-0432.CCR-04-0308
215. Marsh J, Mukherjee P, Seyfried TN. Akt-dependent proapoptotic effects of dietary restriction on late-stage management of a phosphatase and tensin homologue/tuberous sclerosis complex 2-deficient mouse astrocytoma. Clin Cancer Res. (2008) 14:7751–62. doi: 10.1158/1078-0432.CCR-08-0213
Keywords: glycolysis, survival, glutaminolysis, non-toxic, fermentation, metastasis, inflammation
Citation: Seyfried TN, Mukherjee P, Iyikesici MS, Slocum A, Kalamian M, Spinosa J-P and Chinopoulos C (2020) Consideration of Ketogenic Metabolic Therapy as a Complementary or Alternative Approach for Managing Breast Cancer. Front. Nutr. 7:21. doi: 10.3389/fnut.2020.00021
Received: 10 October 2019; Accepted: 21 February 2020;
Published: 11 March 2020.
Edited by:
Ornella Ida Selmin, University of Arizona, United StatesReviewed by:
Charikleia Stefanaki, National and Kapodistrian University of Athens, GreeceSharon Ross, National Cancer Institute (NCI), United States
Copyright © 2020 Seyfried, Mukherjee, Iyikesici, Slocum, Kalamian, Spinosa and Chinopoulos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas N. Seyfried, dGhvbWFzLnNleWZyaWVkJiN4MDAwNDA7YmMuZWR1