 Richie D. Barclay
Richie D. Barclay Nicholas A. Burd
Nicholas A. Burd Christopher Tyler
Christopher Tyler Neale A. Tillin
Neale A. Tillin Richard W. Mackenzie1*
Richard W. Mackenzie1*- 1Department of Life Sciences, University of Roehampton, London, United Kingdom
- 2Division of Nutritional Sciences, Department of Kinesiology and Community Health, University of Illinois, Urbana, IL, United States
Sarcopenia is defined as the combined loss of skeletal muscle strength, function, and/or mass with aging. This degenerative loss of muscle mass is associated with poor quality of life and early mortality humans. The loss of muscle mass occurs due to acute changes in daily muscle net protein balance (NPB). It is generally believed a poor NPB occurs due to reduced muscle protein synthetic responses to exercise, dietary amino acid availability, or an insensitivity of insulin to suppress breakdown. Hence, aging muscles appear to be resistant to the anabolic action of exercise and protein (amino acids or hormonal) when compared to their younger counterparts. The mechanisms that underpin anabolic resistance to anabolic stimuli (protein and resistance exercise) are multifactorial and may be partly driven by poor lifestyle choices (increased sedentary time and reduced dietary protein intake) as well as an inherent dysregulated mechanism in old muscles irrespective of the environmental stimuli. The insulin like growth factor 1 (IGF-1), Akt /Protein Kinase B and mechanistic target of rapamycin (mTOR) pathway is the primary driver between mechanical contraction and protein synthesis and may be a site of dysregulation between old and younger people. Therefore, our review aims to describe and summarize the differences seen in older muscle in this pathway in response to resistance exercise (RE) and describe approaches that researchers have sought out to maximize the response in muscle. Furthermore, this review will present the hypothesis that inositol hexakisphosphate kinase 1 (IP6K1) may be implicated in IGF-1 signaling and thus sarcopenia, based on recent evidence that IGF-1 and insulin share some intracellular bound signaling events and that IP6K1 has been implicated in skeletal muscle insulin resistance.
Setting the Scene
Sarcopenia is defined as the combined loss of skeletal muscle mass, function and strength (1) and it can progress at a rate of approximately 0.8% skeletal muscle loss per year from the 5th decade in adult life (2). Sarcopenia is diagnosed using a battery of clinical assessments and globally effects 1 in 10 adults above the age of 60 (3). Given that sarcopenia costs the American Health Service an estimated $18.5 billion annually (4) and the United Kingdom's National Health Service £4592 for 11% of the total aging population which costs £2.5 billion annually (5), it is important that we develop a greater understanding of the cellular pathways that drive this disease and how interventions such as exercise and dietary protein are used to delay this processes implicated in its progression.
Human skeletal muscle is of high plasticity and is in a constant state of remodeling. Skeletal muscle remodeling occurs due to the dynamic balance between muscle protein synthesis (MPS) and muscle protein breakdown rates (MPB). The daily difference between MPS and MPB defines net protein balance (NPB), which is a key regulator of overall skeletal muscle mass. A positive NPB is generally indicative of a positive remodeling response that can be hypertrophic [i.e., increase fiber cross sectional area (6)] or non-hypertrophic [i.e., increased metabolic quality (7, 8)] in nature, whereas a reduced NPB reflects an overt phenotype being negative by inducing a loss of muscle mass or poor metabolic quality (9). Changes in MPB are small in normal aging, whilst changes in MPS seem to be larger in amplitude and more obvious in response to the main anabolic stimuli to muscle tissue. As such, the measurement of MPS is the primary focus in human metabolic research (10).
Protein ingestion stimulates an increase in MPS; however, a decrease in habitual physical activity, which is often observed with aging and/or injury, can induce anabolic resistance of MPS to protein ingestion (11) (Figure 1). For example, 7 days of unilateral leg immobilization in young men caused significant decreases in quadriceps cross sectional area (CSA) compared to the control limb and leucine supplementation did not reduce the loss in CSA (13). Similarly, 14 days immobilization in young men caused decreased CSA and an amino acid infusion in varying doses showed decreased post prandial MPS in the immobilized leg vs. the control limb (14). To reach the same myofibrillar protein synthetic response in muscle, older individuals need to consume more relative amounts of protein than younger individuals. In vivo, the stimulation of myofibrillar protein synthesis rates is dependent on intracellular molecular signaling pathways that become activated in response to extracellular cues. The mTOR complex 1 (mTORC1) pathway appears to play an important role in stimulating postprandial / post-exercise myofibrillar protein synthesis rates (15), and activation of this pathway can occur in two ways; firstly, through mechanical contraction (i.e., resistance exercise) which causes a release of skeletal muscle IGF-1 (8) and secondly via amino acid/protein intake (16). Furthermore, resistance exercise (RE) plus protein ingestion increases mTORC1 phosphorylation to a greater extent than protein or RE alone (17, 18), and likely intracellular redistribution of mTORC1 toward the sarcolemma as well (19).
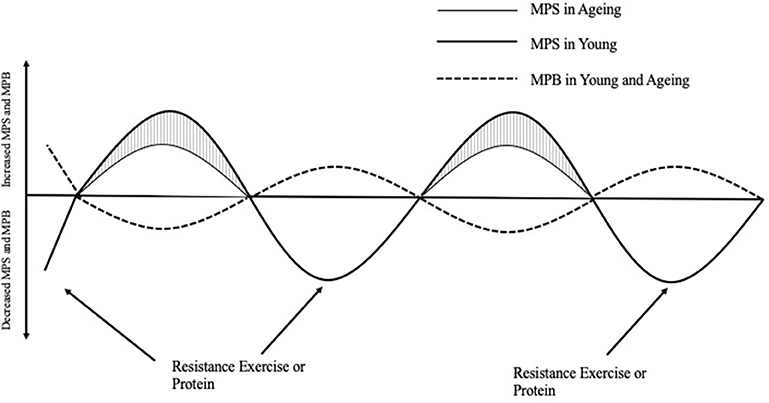
Figure 1. The response of muscle protein synthesis (MPS) and breakdown (MPB) on net protein balance after acute resistance exercise or protein ingestion in young and aging populations [Adapted from Breen and Phillips (12)]. In the morning after an overnight fast, muscle protein breakdown exceeds muscle protein synthesis such that net protein balance is negative. After a bout of resistance exercise or the ingestion of protein, young people respond greater in their myofibrillar protein synthesis response compared to aging people, which is appears to be the major attenuating factor to decreased NPB leading to skeletal muscle protein loss over time. MPS, Muscle protein synthesis; MPB, Muscle protein breakdown.
As humans age, the muscle's ability to respond to both exercise and dietary protein diminishes leading to reduced NPB, and particularly in the myofibrillar protein fraction (11, 20, 21). The insulin like growth factor 1 (IGF-1), Akt/Protein Kinase B–mTOR pathway is acutely stimulated to promote ribosomal biogenesis and translation to form new myofibril proteins using these elongated ribosomes across the mRNA, which allows for the remodeling of skeletal muscle (22, 23). This occurs through amino acid sensed stimulation of phosphatidylinositol 3-phosphate (PtdIns3P) and localization of mTOR to the lysosome surface where mTOR positive lysosomes can translocate to the cell periphery which creates the most anabolic environment for myofibrillar remodeling (24).
Inositol hexakisphosphate kinase 1 (IP6K1) has recently been shown to inhibit Akt308 activity in IGF-1 stimulated hepatic cell lines (25) as well as skeletal muscle contraction having the ability to reverse this negative effect by reducing IP6K1 content (26). Normal IGF-1 signaling and Akt activity are vital for maintaining anabolic sensitivity (27). Taken together, it is hypothesized that IP6K1 may have a role in the onset of sarcopenia, and this review aims to discuss the possible role it has in anabolic resistance in aging skeletal muscle. Moreover, this review will describe and explain the molecular regulation of MPS and the role of RE and dietary protein has on the Akt-mTOR pathway in anabolic resistant, aging muscle.
Molecular Regulation of Muscle Protein Synthesis (MPS) in Response to Exercise and Nutrition
MPS is regulated by an intrinsic cell signaling response which is activated by various external cues, such as dietary amino acids and RE. These cues drive the molecular regulation that augments MPS rates which facilitates the protein remodeling response in skeletal muscle, which is generally considered to be a hypertrophic response when studied during recovery from RE. The muscle protein remodeling response after endurance exercise is likely more aimed at non-hypertrophic remodeling. What is noteworthy is that it is still unclear how different anabolic signaling pathways coordinate the synthesis of specific muscle protein sub-fractions such as myofibrillar or mitochondrial proteins throughout the postprandial or post-exercise period. In addition, the time course of activation for these signaling transduction pathways is still unclear. The signaling response to RE and protein is described in detail below both in healthy and anabolic resistant phenotypes.
Insulin Like Growth Factor-1 Activation
IGF-1, owing its name to high homogeneity to insulin, is a small peptide structurally bound by 70 amino acids (28). It is secreted by many tissues including the liver and skeletal muscle. Once IGF-1 enters the blood stream, there is not a specific tissue target (adipose, skeletal muscle, brain, cardiac muscle) although it is agreed that IGF-1 secreted from the liver (the largest contributor to circulating IGF-1), will not act on those tissues that have capabilities of producing the hormone themselves, such as skeletal muscle (29–33). At rest, circulating IGF-1 is relatively stable and young adults have significantly greater concentrations compared to older individuals (34). It has recently been suggested that large, but transient, increases in exercise-induced plasma IGF-1 concentrations are not required for activation of intracellular muscle signaling and the subsequent skeletal muscle adaptive response (35, 36). For example, Morton et al. (35) observed no relationship between circulating anabolic hormones (IGF-1) in plasma and strength gains in resistance trained young men after 12 weeks of low or high load RE. This suggests that intrinsic secretion of muscle IGF-1, not circulating plasma IGF-1, is a key determinant for switching on anabolic pathways (37). This is supported by Morton et al. (37) who suggested muscle androgen receptor content and not circulating hormones was associated with changes in lean body mass following 12 weeks of RE in previously trained men. In muscle, IGF-1 is stimulated by mechanical loading and contraction to which IGF receptor (IGFR) is activated in the cell to allow for membrane bound protein signaling pathways to become active. IGF-1 is secreted from muscle fibers into the extracellular matrix (ECM) to which it is bound by IGF binding proteins (IGFBPs). Given, the half-life of IGF-1 is just 5–10 min (38), these pools of IGFBPs must be local to the ECM (39–41). Upon binding to IGFBPs, IGF-1 activates its receptor to which intracellular signaling processes driving MPS can occur.
IGF-1 enters the cell via IGFR, it triggers phosphoinositide 3-kinase (P13-K) to generate hosphatidylinositol (4, 5)-bisphosphate (PIP2), leading to the production of hosphatidylinositol 3,4,5-trisphosphate (PIP3) (27, 42, 43). PIP3 is then free to bind to phosphoinositide-dependent kinase-1 (PDK1) which binds to the pleskstrin homology (PH) domain of Akt, allowing for translocation to the cell membrane preceding phosphorylation at Akt308 (27, 43).
Akt-mTOR Signaling in Response to Protein and Resistance Exercise
Akt is a threonine and serine protein kinase which has three isoforms; Akt1, Akt2, and Akt3 (44). Akt1 has been linked to cell survival and may inhibit apoptosis (45). The same isoform is also implicated in MPS (46). Akt2 is the predominant form present in skeletal muscle and is heavily involved in glucose uptake and MPS via Akt308 and Akt473 (47) whilst Akt3 is vital for brain development and cell death (48). Diez et al. (49) described the activity of specific isoforms on Akt308 phosphorylation and despite Akt1 resulting in over half of total Akt expression, inhibition of Akt1 and Akt3 through use of shRNA techniques had no significant effect on total phosphorylated Akt308. In contrast to the little effect of Akt1 and Akt3 inhibition had on total phosphorylated Akt308, inhibition of Akt2 was related to reduction in total Akt and phosphorylated Akt308 in all conditions respective to control. Mutant mice lacking the Akt2 gene have significantly reduced total and phosphorylated Akt compared to mice lacking the Akt3 gene (49). Furthermore, Akt2 mutants had impaired glucose homeostasis, and growth deficiencies which may have been partially due to significantly blunted Ribosomal protein S6 kinase beta-1 (p70s6k) phosphorylation. Akt activation by mTOR complex 2 (mTORC2) uses PIP3, the product of P13K, to bind to the pleskstrin homology (PH) domain of mSin1 which localized mTORC2 to the cell membrane and relieves inhibition of mTOR which allows for Akt473 phosphorylation (50). Phosphorylation of both serine threonine residues are vital for maximal Akt activity and disruption of either site will have a knock-on effect downstream (51). In healthy phenotype, this mechanism occurs quickly and of high magnitude in response to RE (52) whilst this response is blunted in metabolically unhealthy or aging (53). Interference at any stage of this sequential reaction can have a profound effect on downstream signaling and metabolism. For example, inhibition of insulin receptor substrate 1 (IRS-1) in genetically modified mice had little effect on muscle atrophy, however when IGFR and IRS-1 are both disrupted, mice showed marked atrophy but had good glucose tolerance, measured by intravenous glucose tolerance test in gastrocnemius and soleus muscle which may have been due to increased glucose transporter 1 (GLUT-1) and glucose transporter 4 (GLUT-4) (42).
Downstream, Akt308 activates mTOR complex 1 (mTORC1) at mTOR2448 via the tuberous sclerosis complex (TSC). The TSC is phosphorylated by Akt which then disassociates the TSC from the small GTPase Ras homolog enriched in brain (Rheb) to localize to the lysosome where Rheb and mTORC1 can interact and phosphorylate mTORC1 (54). Similar to Akt, mTOR is a serine/threonine kinase and is made up of 5 components; Raptor, mLST, mTOR, PRAS40, and Deptor. All five of these components play a key role in mTORC1 signaling that subsequently results in MPS via downstream protein phosphorylation. Following RE and dietary protein ingestion, mTORC1 activity is upregulated which allows for subsequent membrane bound proteins to increase MPS (55). Phosphorylation and activity of mTORC1 at mTOR2448 and mTOR2481 are directly correlated with MPS (17, 56). mTORC2481 is a marker of intact mTORC2 and phosphorylation of this site is an indicator that the actin cytoskeleton and Akt473 are metabolically active (57). RE and dietary protein ingestion actively promotes skeletal muscle remodeling, at least partly, through acute activation of the Akt-mTORC1 pathway, which prevent MPB pathways such as autophagy (8, 27, 58, 59). Acute increases in Akt-mTORC1 signaling increase MPS to prevent breakdown in skeletal muscle, albeit MPS blunted in older humans compared to younger counterparts (8, 59). However, it has been established that targeted exercise prescription aimed at encouraging the recruitment of type II muscle fibers with the more volume (6 sets > 3 sets) can restore the post-exercise MPS response to a more youthful-state (60). To prevent negative protein balance and therefore muscle atrophy, RE and increased protein intake can be used as a tool to prevent progressive loss of muscle mass and function in aging adults (12, 21).
Downstream of mTORC1, two key proteins are activated to regulate muscle mass. Previous efforts have shown that mTORC1 signaling to p70s6k and 4E-BP1 are both required for an optimal amount and quality of muscle mass during hypertrophic remodeling (61). In particular, 4E-BP1 phosphorylation can support hypertrophy, but without p70s6k phosphorylation the quality of muscle is poor and leads to impaired force production due to formation of protein aggregates (61). In humans, it was demonstrated that p70s6k phosphorylation 6 h post exercise is a strong predictor of skeletal muscle mass using a small sample size, which suggested that disruption of these anabolic signaling proteins can have a profound effect on muscle plasticity (62). However, Mitchell et al. (63) showed that the predictive nature of p70s6k phosphorylation immediately resistance exercise for muscle mass gain is less apparent when using a larger sample size. Such a finding is consistent with the heterogeneity of skeletal muscle adaptive potential in humans (64, 65), and re-underlines the challenges associated with emphasizing the value of a single point snapshot of phosphorylation after exercise as the “holy grail” in terms of predicting the extent of skeletal muscle adaptations with progressive resistance exercise training in humans.
Causes of Anabolic Resistance
There are several factors likely contributing toward the anabolic resistance of aging muscles to RE or protein ingestion (66), many of which have been highlighted above, including elevated mTORC1 phosphorylation in the post absorptive state (67). This systemic activation of mTORC1 in the post absorptive state inhibits autophagy in muscle, a protein breakdown pathway, which leads to the build-up of excess, unused ribosomes that have been synthesized as a result of mTORC1 phosphorylation and ultimately leading to reduced structural integrity of the ECM of muscle which causes insulin resistance (68, 69). It is thought that this is due to an insensitivity to anabolic stimuli whereby mTORC1 is elevated at rest and causing a systematic upregulation of MPS, similar to insulin resistance (67). A similar adaptation is seen in obese muscle that is resistant to anabolic stimuli (70); therefore, this suggests that a major difference between unhealthy and healthy muscle is post absorptive mTORC1 activity. TSC1 deficient mice, showing prolonged and chronic elevated mTORC1, developed late onset myopathy, showed vacuolated and basophilic fibers, intracellular inclusions, and abnormally large myonuclei. Furthermore, extracellular binding proteins were impaired in TSC1 deficient mice which may be an attenuating factor to myopathy. In older adults after an acute bout of RE, p70s6k and 4E-BP1 do not increase to the same magnitude as young people (53). This decreased activation of key proteins may be caused by upstream signaling pathways being disrupted, however is it not possible to turnover new proteins without a distinct activation of p70s6k and 4E-BP1. Drummond et al. (53) saw increases in 4E-BP1 phosphorylation at 1 and 6 h in young men however the increase was maintained for only 1 h in older adults. Kumar et al. (21) saw a similar effect in response to several different bouts of RE in a scaled protocol for one rep max percentage, older individuals had a significantly reduced FSR at all intensities compared to young and this response was coupled with a decreased p70s6k phosphorylation. With the evidence above in mind, it is clear that elevated post absorptive mTORC1 activity is a negative adaptation, however the reasons to which this occurs are currently not known.
Recently a NAD+ dependent deacetylase called Sirtuin 1 (SIRT1) has gained some attention due to its emerging role in increased cell longevity (71). This protein is implicated in a number of pathways including cellular energy production, mitochondrial biogenesis via AMPK and PGC-1α while reducing inflammation through the inhibition of NF-kB (71–73). SIRT1 acts negatively on Akt and mTORC1 in the basal state, a finding previously shown to be a major attenuating factor to the normal response to anabolic stimuli (70). Furthermore, aging is associated with an increase in SIRT1 muscle content, however anabolic resistant skeletal muscle in aging populations display less SIRT1 vs. active counterparts, a finding that may contribute to increased post-absorptive Akt-mTORC1 activity (74, 75). Furthermore, caloric restriction also appears to increase SIRT1 muscle content, adding support for the combination of caloric restriction, RE, and leucine rich protein sources as a strategy for improving anabolic resistance in aging populations (71).
Habitual diet must be considered as a potential attenuating factor of sarcopenia given the reduced response older adults have to protein ingestion and the profound effect this has on skeletal muscle remodeling. Mediterranean diets rich in protein, fats and vegetables have been shown to reduce the risk of sarcopenia through its anti-inflammatory effects and high anti-oxidant content (76). Some research suggests that individuals consuming a diet high in protein also present with increased serum IGF-1 concentrations (77, 78). Yet more recently, older adults consuming a protein rich diet (1.4 g/kg/day) showed no difference in circulating IGF-1 levels vs. a low protein group (0.8 g/kg/day) (79). Whilst older adults clearly require greater quantities of protein to gain the desired MPS response, the evidence suggests that the IGF-1 pathway is not responsive to their habitual diet. A recent study on old mice (26 months old) suggested that increased expression of local skeletal muscle IGF-1 isoforms (IGF-1 Ea and IGF-1 Eb) counteracted sarcopenia, which included increased SIRT1 muscle content, autophagy and PGC-1α, without inducing any negative effects in other tissues compared to wild type mice (80). No study to the authors knowledge has investigated the local skeletal muscle isoforms of IGF-1 and the effect of acute or habitual diet on its concentration and secretion, and therefore requires exploration. Yet, as previously discussed, local skeletal muscle IGF-1 seems to play an important factor to skeletal muscle remodeling and that this effect is greater that any contribution to IGF-1 produced and secreted by other tissue types.
Counteracting Anabolic Resistance With Nutrition and Exercise
With the aim of counteracting anabolic resistance, studies have investigated how MPS can be maximized in older adults. Moore et al. (81) compared young and older adults myofibrillar protein synthetic response to graded intakes of whey protein and it was suggested that young and older adults could reach a similar response in their muscle however the quantity of protein required in g/kg lean body mass (LBM) in a single meal was far greater [0.6 g/kg LBM vs. 0.25 g/kg LBM (Figure 2)] in the older population. One explanation for this reduced ability of aging muscles to “sense” dietary amino acids in circulation is likely related to impairments in molecular signaling (e.g., decreased amino acid transports, elevated mTORC1, and IP6K1). Due to impaired molecular signaling intrinsic to muscle, older adults require greater amounts of leucine to have a similar response as younger individuals (81–83). To maximize MPS in aging adults, Kumar et al. (21) suggested that MPS was at its peak following 6 sets of 8 repetitions at 60% 1RM although this was significantly less compared to their young counterparts. This is important for the prescription of exercise protocols in research to ensure MPS is stimulated optimally and also important for exercise prescription in older adults aiming to prevent muscle atrophy and improve muscle function.
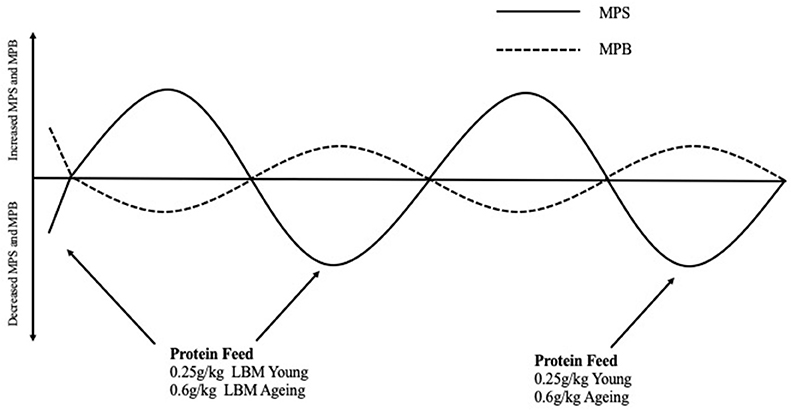
Figure 2. Normal fluctuations in Muscle Protein Synthesis and Muscle Protein Breakdown rates throughout the day in response to eating a protein containing meal and the effect this has on net protein balance. Protein Requirements to stimulate myofibrillar protein synthesis rates in young and aging populations are described in g/kg of lean body mass These protein meal requirements should be spread equally throughout the day (i.e., 4–5 meal times) to facilitate non-hypertrophic protein remodeling and counterbalance fasting-state protein losses that occurred in between meals (81). MPS, Muscle protein synthesis; MPB, Muscle protein breakdown; LBM, Lean body mass.
In summary, RE and higher protein diets have the ability to restore the anabolic sensitivity of MPS rates in aging adults, and either low or high load RE has the ability to stimulate a robust post-exercise muscle protein synthetic response throughout adult life. A sedentary lifestyle and reduced protein intake progressively cause a reduced response to anabolic stimuli (driven by blunted Akt-mTOR signaling), which leads to the progressive loss of muscle mass and function over time. Adults should engage with RE throughout their lives (2–3 times per week), and current evidence suggests that higher dietary protein intakes (0.6 g/kg LBM in each meal X 4–5 meal times) combined with modest caloric restriction is required to support a robust simulation in postprandial MPS rates and perhaps to maintain muscle mass and function with age. It is important to note, however, that a large-scale randomized clinical trial that combines exercise and protein intake manipulations (from RDA and beyond) is needed to confirm the value of eating protein in far excess of the RDA to support a more youthful phenotype with age. In addition, design of RE programme should consider adherence, motivation and enjoyment as these are the key factors for long term participation and ultimately skeletal muscle health.
Possible Mechanism of Anabolic Resistance Involving Inositol Hexakisphosphate Kinase 1 (IP6K1)
Inositol hexakisphosphate kinase 1 (IP6K1) is a six carbon cylitol kinase which has recently been well documented in the attenuation of insulin resistance and type 2 diabetes (25, 26, 84–87). IP6K1 synthesizes IP6 to IP7 which binds to the PH domain of Akt (Figure 3), preventing translocation to the cell membrane (25). Inhibition of IP6K1 using N2-(m-Trifluorobenzyl), N6-(p-nitrobenzyl) purine (TNP) prevents diet induced obesity in mice through increased Akt activity (88). These mice maintained lean mass and had improved insulin sensitivity, reduced blood glucose, and insulin. Similarly, in Chakraborty et al. (25) study, mice that were treated with TNP had similar traits to the above mice in response to a high fat diet. They had healthy metabolic parameters (low serum insulin and blood glucose) and increased energy expenditure compared to wild type mice. Phosphorylation of Akt308 and p70s6k was increased in TNP treated mice which increased glucose uptake and reduced fat mass whilst maintaining lean body mass, therefore having a positive effect on skeletal muscle remodeling, likely via mTORC1 and p70s6k activity. In the same study, Chakraborty et al. (25) treated mouse embryonic fibroblasts with 100 mCi[3H]myoinositol for 3 days to inhibit inositol phosphates before stimulating with or without IGF-1. Graded concentrations of IGF-1 increased IP7 cell content in wild type cell lines vs. inositol knockout (-/-inositol) cells. In the -/-inositol cells, Akt308 and Akt473 activity increased with IGF-1 administration and more importantly for our hypotheses, p70s6k activity was increased in the same way. The mTORC1 regulators TSC were also phosphorylated in IGF-1 treated -/-inositol cell lines compared to control which suggests IP6K1 could be implicated in Akt-mTORC1 signaling and therefore sarcopenia. Given this evidence it is conceivable that IP6K1 may have a role in the onset of anabolic resistance in aging phenotype via downregulated Akt, TSC and mTORC1.
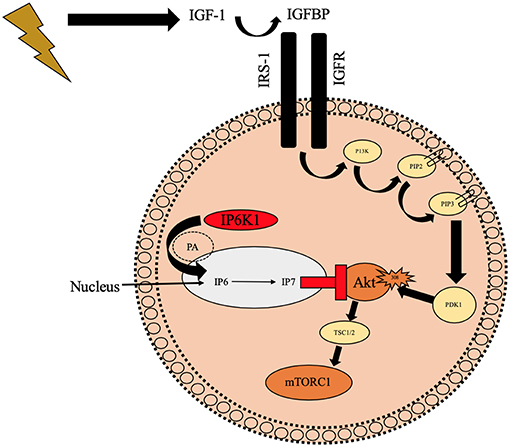
Figure 3. Schematic diagram illustrating the potential negative role of IP6K1 on Akt translocation to the cell membrane preventing phosphorylation of Akt308 which may reduce mTORC1. IP6K1 enters the nucleus via PA and it then synthesizes IP7 from IP6 which prevents Akt from translocating to the cell membrane and ultimately preventing Akt308 phosphorylation. IGFBP, Insulin like growth factor binding proteins; IGF-1, Insulin like growth factor-1; IP6K1, inositol hexakisphosphate kinase 1; IGFR, Insulin like growth factor receptor; IRS-1, Insulin receptor substrate 1; P13K, phosphoinositide 3-kinase; PIP2, hosphatidylinositol (4, 5)-bisphosphate; PIP3, hosphatidylinositol 3,4,5-trisphosphate; PDK1, phosphoinositide-dependent kinase-1; Akt, Protein kinase B; mTORC2, Mechanistic target of rapamycin; PA, Phosphotadic acid; IP6, inositol hexaphosphate; IP7, Inositol pyrophosphate;  Illustrates contraction of skeletal muscle;
Illustrates contraction of skeletal muscle;  Illustrates binding/translocationto the cell membrane;
Illustrates binding/translocationto the cell membrane;  Illustrates activation;
Illustrates activation;  Illustrates phosphorylation;
Illustrates phosphorylation;  Illustrates binding to PH domain and downregulating Akt;
Illustrates binding to PH domain and downregulating Akt;  Illustrates preventing translocation to cell membrane.
Illustrates preventing translocation to cell membrane.
Recently, our research group showed that IP6K1 was altered in adult (47 years ± 3) skeletal muscle in response to exercise (26). In this case, insulin resistant pre-diabetic individuals, whose IP6K1 was elevated at basal, were able to decrease the content in skeletal muscle following an acute high intensity interval training (HIIT) protocol (26). In this study, a HIIT intervention was superior compared to a continuous exercise protocol for increasing Akt308 activity and decreasing IP6K1 content. Given the findings of Chakraborty et al. (25), Naufahu et al. (26) and O'Neill et al. (42), it is hypothesized that elevated IP6K1 has a role in MPS via reduced Akt activity and physical inactivity. Habitual RE attenuates anabolic sensitivity via increased Akt and mTOR activity in response to anabolic stimuli (12, 89). Previous work has highlighted the negative role IP6K1 has on Akt activity whilst this negative effect is reversed with HIIT (26), thus we hypothesize that IP6K1 may be downregulated following RE in older adults.
To conclude, dysregulated Akt-mTOR signaling in response to RE and protein intake ingesting a current recommended intake (15 g per meal or 0.8 g protein/kg/day) is the primary driver of anabolic resistance and sarcopenia (90). Recent research has aimed at identifying strategies to maximize the response in the muscle in older individuals (21, 81), which include 0.6g/kg LBM protein in each meal and 60% 1RM for 6 sets of 8 repetitions. Thus far, characteristics of anabolic resistant muscle are known (i.e., elevated post absorptive mTORC1 phosphorylation, reduced SIRT1 muscle content, reduced structural integrity via reduced autophagy, and muscle atrophy); however, it is not clear why reduced protein intake and exercise cause these characteristics. With the evidence presented above, it is hypothesized that IP6K1 has a role in this diminished response to anabolic stimuli, similar to the onset of insulin resistance, and obesity. We are currently testing this hypothesis in vitro and in vivo at our University of Roehampton labs with the aim of disseminating our findings over the next 12 months.
Author Contributions
RB was responsible for writing the manuscript. NB, NT, CT, and RM were all responsible for reviewing and contributing to the manuscript. RM was the senior author in the group.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
IP6K1, Inositol hexakisphosphate kinase 1; IGF-1, Insulin like growth factor-1; Akt, Protein kinase B; mTOR, Mechanistic target of rapamycin; mTORC1, Mechanistic target of rapamycin complex 1; SIRT1, Sirtuin 1; AMPK, 5′ AMP-activated protein kinase; Peroxisome proliferator-activated receptor-γ coactivator, PGC-1α; Nuclear factor kappa-light-chain-enhancer of activated B cells, NF-kB; PB, Net protein balance; RE, Resistance exercise; MPS, Muscle protein synthesis; MPB, Muscle protein balance; CSA, Cross sectional area; IGFR, Insulin like growth factor receptor; ECM, Extracellular matrix; IGFBP, Insulin like growth factor binding proteinsl PH, Pleckstrin homology; P13K, Phosphoinositide 3-kinase; PA, Phosphotadic acid; PDK1, Phosphoinositide-dependent kinase-1; PIP3, Phosphatidylinositol 3,4,5-trisphosphate; PIP2, Phosphatidylinositol (4,5)-bisphosphate; mTORC2, Mechanistic target of rapamycin complex 2; IRS1, Insulin receptor substrate 1; GLUT-1, Glucose transporter-1; GLUT-4, Glucose transporter-4; TSC, Tuberous sclerosis complex; p70s6k, Ribosomal protein S6 kinase beta-1; 4E-BP1, 4E Binding protein 1; FSR, Fractional synthesis rate; IRM, 1 Repetition maximum; IP6, Inositol hexophosphate; IP7, Inositol pyrophosphate; TNP, N2-(m-Trifluorobenzyl), N6-(p-nitrobenzyl)purine; HIIT, High intensity interval training.
References
1. Santilli V, Bernetti A, Mangone M, Paoloni M. Clinical definition of sarcopenia. Clin Cases Min Bone Metabol. (2014) 11:177. doi: 10.11138/ccmbm/2014.11.3.177
2. Janssen I. Evolution of sarcopenia research. Appl Physiol Nutr Metabol. (2010) 35:707–12. doi: 10.1139/H10-067
3. Shafiee G, Keshtkar A, Soltani A, Ahadi Z, Larijani B, Heshmat R. Prevalence of sarcopenia in the world: a systematic review and meta-analysis of general population studies. J Diabetes Metab Disord. (2017) 16:21. doi: 10.1186/s40200-017-0302-x
4. Janssen I, Shepard DS, Katzmarzyk PT, Roubenoff R. The healthcare costs of sarcopenia in the united states. J Am Geriatr Soc. (2004) 52:80–5. doi: 10.1111/j.1532-5415.2004.52014.x
5. Pinedo-Villanueva R, Westbury LD, Syddall HE, Sanchez-Santos MT, Dennison EM, Robinson SM, et al. Health care costs associated with muscle weakness: a UK population-based estimate. Calcif Tissue Int. (2019) 104:137–44. doi: 10.1007/s00223-018-0478-1
6. Phillips SM, Hartman JW, Wilkinson SB. Dietary protein to support anabolism with resistance exercise in young men. J Am Coll Nutr. (2005) 24:134–9. doi: 10.1080/07315724.2005.10719454
7. Tipton KD, Wolfe RR. Exercise, protein metabolism, and muscle growth. Int J Sport Nutr Exerc Metab. (2001) 11:109–32. doi: 10.1123/ijsnem.11.1.109
8. Fry CS, Drummond MJ, Glynn EL, Dickinson JM, Gundermann DM, Timmerman KL, et al. Aging impairs contraction-induced human skeletal muscle mTORC1 signaling and protein synthesis. Skelet Muscle. (2011) 1:11. doi: 10.1186/2044-5040-1-11
9. Fiatarone MA, Marks EC, Ryan ND, Meredith CN, Lipsitz LA, Evans WJ. High-intensity strength training in nonagenarians: Effects on skeletal muscle. JAMA. (1990) 263:3029–34. doi: 10.1001/jama.1990.03440220053029
10. Biolo G, Ciocchi B, Lebenstedt M, Barazzoni R, Zanetti M, Platen P, et al. Short-term bed rest impairs amino acid-induced protein anabolism in humans. J Physiol. (2004) 558:381–8. doi: 10.1113/jphysiol.2004.066365
11. Wall BT, Gorissen SH, Pennings B, Koopman R, Groen BB, Verdijk LB, et al. Aging is accompanied by a blunted muscle protein synthetic response to protein ingestion. PLoS ONE. (2015) 10:e0140903. doi: 10.1371/journal.pone.0140903
12. Breen L, Phillips SM. Skeletal muscle protein metabolism in the elderly: Interventions to counteract the'anabolic resistance'of ageing. Nutr Metab. (2011) 8:68. doi: 10.1186/1743-7075-8-68
13. Backx EM, Horstman AM, Marzuca-Nassr GN, van Kranenburg J, Smeets JS, Fuchs CJ, et al. Leucine supplementation does not attenuate skeletal muscle loss during leg immobilization in healthy, young men. Nutrients. (2018) 10:635. doi: 10.3390/nu10050635
14. Glover EI, Phillips SM, Oates BR, Tang JE, Tarnopolsky MA, Selby A, et al. Immobilization induces anabolic resistance in human myofibrillar protein synthesis with low and high dose amino acid infusion. J Physiol. (2008) 586:6049–61. doi: 10.1113/jphysiol.2008.160333
15. Abou Sawan S, Vliet S, Parel JT, Beals JW, Mazzulla M, West DW, et al. Translocation and protein complex co-localization of mTOR is associated with postprandial myofibrillar protein synthesis at rest and after endurance exercise. Physiol Rep. (2018) 6:e13628 doi: 10.14814/phy2.13628
16. Manifava M, Smith M, Rotondo S, Walker S, Niewczas I, Zoncu R, et al. Dynamics of mTORC1 activation in response to amino acids. Elife. (2016) 5:e19960. doi: 10.7554/eLife.19960
17. Deldicque L, Theisen D, Francaux M. Regulation of mTOR by amino acids and resistance exercise in skeletal muscle. Eur J Appl Physiol. (2005) 94:1–10. doi: 10.1007/s00421-004-1255-6
18. Burd NA, West DW, Moore DR, Atherton PJ, Staples AW, Prior T, et al. Enhanced amino acid sensitivity of myofibrillar protein synthesis persists for up to 24 h after resistance exercise in young Men−3. J Nutr. (2011) 141:568–73. doi: 10.3945/jn.110.135038
19. Song Z, Moore DR, Hodson N, Ward C, Dent JR, O'leary MF, et al. Resistance exercise initiates mechanistic target of rapamycin (mTOR) translocation and protein complex co-localisation in human skeletal muscle. Sci Rep. (2017) 7:5028. doi: 10.1038/s41598-017-05483-x
20. Cuthbertson D, Smith K, Babraj J, Leese G, Waddell T, Atherton P, et al. Anabolic signaling deficits underlie amino acid resistance of wasting, aging muscle. FASEB J. (2005) 19:422–4. doi: 10.1096/fj.04-2640fje
21. Kumar V, Selby A, Rankin D, Patel R, Atherton P, Hildebrandt W, et al. Age-related differences in the dose–response relationship of muscle protein synthesis to resistance exercise in young and old men. J Physiol. (2009) 587:211–7. doi: 10.1113/jphysiol.2008.164483
22. Wen Y, Alimov AP, McCarthy JJ. Ribosome biogenesis is necessary for skeletal muscle hypertrophy. Exerc Sport Sci Rev. (2016) 44:110. doi: 10.1249/JES.0000000000000082
23. Latres E, Amini AR, Amini AA, Griffiths J, Martin FJ, Wei Y, et al. Insulin-like growth factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/akt/mammalian target of rapamycin (PI3K/akt/mTOR) pathway. J Biol Chem. (2005) 280:2737–44. doi: 10.1074/jbc.M407517200
24. Hong Z, Pedersen NM, Wang L, Torgersen ML, Stenmark H, Raiborg C. PtdIns3P controls mTORC1 signaling through lysosomal positioning. J Cell Biol. (2017) 216:4217. doi: 10.1083/jcb.201611073
25. Chakraborty A, Koldobskiy MA, Bello NT, Maxwell M, Potter JJ, Juluri KR, et al. Inositol pyrophosphates inhibit akt signaling, thereby regulating insulin sensitivity and weight gain. Cell. (2010) 143:897–910. doi: 10.1016/j.cell.2010.11.032
26. Naufahu J, Elliott B, Markiv A, Dunning-Foreman P, McGrady M, Howard D, et al. High intensity exercise decreases IP6K1 muscle content and improves insulin sensitivity (SI2*) in glucose intolerant individuals. J Clin Endocrinol Metab. (2018) 103:1479–90. doi: 10.1210/jc.2017-02019
27. Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, et al. The IGF-1/PI3K/akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. (2004) 14:395–403. doi: 10.1016/S1097-2765(04)00211-4
28. Rinderknecht E, Humbel RE. Primary structure of human insulin-like growth factor II. FEBS Lett. (1978) 89:283–6. doi: 10.1016/0014-5793(78)80237-3
29. Turner JD, Rotwein P, Novakofski J, Bechtel PJ. Induction of mRNA for IGF-I and-II during growth hormone-stimulated muscle hypertrophy. Am J Physiol Endocrinol Metabol. (1988) 255:E517. doi: 10.1152/ajpendo.1988.255.4.E513
30. Barton ER, Park S, James JK, Makarewich CA, Philippou A, Eletto D, et al. Deletion of muscle GRP94 impairs both muscle and body growth by inhibiting local IGF production. FASEB J. (2012) 26:3691–702. doi: 10.1096/fj.11-203026
31. Sjögren K, Liu J, Blad K, Skrtic S, Vidal O, Wallenius V, et al. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc Natl Acad Sci USA. (1999) 96:7088–92. doi: 10.1073/pnas.96.12.7088
32. Yakar S, Liu J, Stannard B, Butler A, Accili D, Sauer B, et al. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci USA. (1999) 96:7324–9. doi: 10.1073/pnas.96.13.7324
33. Cohick WS, Clemmons DR. The insulin-like growth factors. Annu. Rev. Physiol. (1993) 55:131–53. doi: 10.1146/annurev.ph.55.030193.001023
34. Huffman DM, Farias Quipildor G, Mao K, Zhang X, Wan J, Apontes P, et al. Central insulin-like growth factor-1 (IGF-1) restores whole-body insulin action in a model of age-related insulin resistance and IGF-1 decline. Aging Cell. (2016) 15:181–6. doi: 10.1111/acel.12415
35. Morton RW, Oikawa SY, Wavell CG, Mazara N, McGlory C, Quadrilatero J, et al. Neither load nor systemic hormones determine resistance training-mediated hypertrophy or strength gains in resistance-trained young men. J Appl Physiol. (2016) 121:129–38. doi: 10.1152/japplphysiol.00154.2016
36. West DW, Burd NA, Churchward-Venne TA, Camera DM, Mitchell CJ, Baker SK, et al. Sex-based comparisons of myofibrillar protein synthesis after resistance exercise in the fed state. J Appl Physiol. (2012) 112:1805–13. doi: 10.1152/japplphysiol.00170.2012
37. Morton RW, Sato K, Gallaugher MP, Oikawa SY, McNicholas PD, Fujita S, et al. Muscle androgen receptor content but not systemic hormones is associated with resistance training-induced skeletal muscle hypertrophy in healthy, young men. Front Physiol. (2018) 9:1373. doi: 10.3389/fphys.2018.01373
38. Morimoto LM, Newcomb PA, White E, Bigler J, Potter JD. Variation in plasma insulin-like growth factor-1 and insulin-like growth factor binding protein-3: genetic factors. Cancer Epidemiol Prevent Biomark. (2005) 14:1394–401. doi: 10.1158/1055-9965.EPI-04-0694
39. Jones JI, Gockerman A, Busby WH, Camacho-Hubner C, Clemmons DR. Extracellular matrix contains insulin-like growth factor binding protein-5: potentiation of the effects of IGF-I. J Cell Biol. (1993) 121:679–87. doi: 10.1083/jcb.121.3.679
40. Hede MS, Salimova E, Piszczek A, Perlas E, Winn N, Nastasi T, et al. E-peptides control bioavailability of IGF-1. PLoS ONE. (2012) 7:e51152. doi: 10.1371/journal.pone.0051152
41. Philippou A, Barton ER. Optimizing IGF-I for skeletal muscle therapeutics. Growth Hormone IGF Res. (2014) 24:157–63. doi: 10.1016/j.ghir.2014.06.003
42. O'Neill BT, Lauritzen HP, Hirshman MF, Smyth G, Goodyear LJ, Kahn CR. Differential role of insulin/IGF-1 receptor signaling in muscle growth and glucose homeostasis. Cell Rep. (2015) 11:1220–35. doi: 10.1016/j.celrep.2015.04.037
43. Schiaffino S, Mammucari C. Regulation of skeletal muscle growth by the IGF1-akt/PKB pathway: Insights from genetic models. Skelet Muscle. (2011) 1:4. doi: 10.1186/2044-5040-1-4
44. Manning BD. Insulin signaling: Inositol phosphates get into the akt. Cell. (2010) 143:861–3. doi: 10.1016/j.cell.2010.11.040
45. Faissner A, Heck N, Dobbertin A, Garwood J. DSD-1-proteoglycan/phosphacan and receptor protein tyrosine phosphatase-beta isoforms during development and regeneration of neural tissues. In: Bähr M, editor. Brain Repair. Boston, MA: Springer (2006). p. 25–53. doi: 10.1007/0-387-30128-3_3
46. Lai KM, Gonzalez M, Poueymirou WT, Kline WO, Na E, Zlotchenko E, et al. Conditional activation of akt in adult skeletal muscle induces rapid hypertrophy. Mol Cell Biol. (2004) 24:9295–304. doi: 10.1128/MCB.24.21.9295-9304.2004
47. Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest. (2003) 112:197–208. doi: 10.1172/JCI16885
48. Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, et al. Role for Akt3/protein kinase bγ in attainment of normal brain size. Mol Cell Biol. (2005) 25:1869–78. doi: 10.1128/MCB.25.5.1869-1878.2005
49. Diez H, Garrido JJ, Wandosell F. Specific roles of akt iso forms in apoptosis and axon growth regulation in neurons. PLoS ONE. (2012) 7:e32715. doi: 10.1371/journal.pone.0032715
50. Liu P, Gan W, Chin YR, Ogura K, Guo J, Zhang J, et al. PtdIns (3:4, 5) P3-dependent activation of the mTORC2 kinase complex. Cancer Discov. (2015) 5:1194–209. doi: 10.1158/2159-8290.CD-15-0460
51. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. (2007) 129:1261–74. doi: 10.1016/j.cell.2007.06.009
52. Camera DM, Edge J, Short MJ, Hawley JA, Coffey VG. Early time course of akt phosphorylation after endurance and resistance exercise. Med Sci Sports Exerc. (2010) 42:1843–52. doi: 10.1249/MSS.0b013e3181d964e4
53. Drummond MJ, Dreyer HC, Pennings B, Fry CS, Dhanani S, Dillon EL, et al. Skeletal muscle protein anabolic response to resistance exercise and essential amino acids is delayed with aging. J Appl Physiol. (2008) 104:1452–61. doi: 10.1152/japplphysiol.00021.2008
54. Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, et al. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell. (2014) 156:771–85. doi: 10.1016/j.cell.2013.11.049
55. Ogasawara R, Fujita S, Hornberger TA, Kitaoka Y, Makanae Y, Nakazato K, et al. The role of mTOR signalling in the regulation of skeletal muscle mass in a rodent model of resistance exercise. Sci. Rep. (2016) 6:31142. doi: 10.1038/srep31142
56. Bolster DR, Kubica N, Crozier SJ, Williamson DL, Farrell PA, Kimball SR, et al. Immediate response of mammalian target of rapamycin (mTOR)-mediated signalling following acute resistance exercise in rat skeletal muscle. J Physiol. (2003) 553:213–20. doi: 10.1113/jphysiol.2003.047019
57. Copp J, Manning G, Hunter T. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): Phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res. (2009) 69:1821–7. doi: 10.1158/0008-5472.CAN-08-3014
58. Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. (2007) 6:458–71. doi: 10.1016/j.cmet.2007.11.001
59. Francaux M, Demeulder B, Naslain D, Fortin R, Lutz O, Caty G, et al. Aging reduces the activation of the mTORC1 pathway after resistance exercise and protein intake in human skeletal muscle: potential role of REDD1 and impaired anabolic sensitivity. Nutrients. (2016) 8:47. doi: 10.3390/nu8010047
60. Kumar V, Atherton PJ, Selby A, Rankin D, Williams J, Smith K, et al. Muscle protein synthetic responses to exercise: effects of age, volume, and intensity. J Gerontol Ser A: Biomed Sci Med Sci. (2012) 67:1170–7. doi: 10.1093/gerona/gls141
61. Marabita M, Baraldo M, Solagna F, Ceelen JJM, Sartori R, Nolte H, et al. S6K1 is required for increasing skeletal muscle force during hypertrophy. Cell Rep. (2016) 17:501–13. doi: 10.1016/j.celrep.2016.09.020
62. Baar K, Esser K. Phosphorylation of p70S6kcorrelates with increased skeletal muscle mass following resistance exercise. Am J Physiol Cell Physiol. (1999) 276:C127. doi: 10.1152/ajpcell.1999.276.1.C120
63. Mitchell CJ, Churchward-Venne TA, West DW, Burd NA, Breen L, Baker SK, et al. Resistance exercise load does not determine training-mediated hypertrophic gains in young men. J Appl Physiol. (2012) 113:71–7. doi: 10.1152/japplphysiol.00307.2012
64. Davidsen PK, Gallagher IJ, Hartman JW, Tarnopolsky MA, Dela F, Helge JW, et al. High responders to resistance exercise training demonstrate differential regulation of skeletal muscle microRNA expression. J Appl Physiol. (2010) 110:309–17. doi: 10.1152/japplphysiol.00901.2010
65. Roberts MD, Haun CT, Mobley CB, Mumford PW, Romero MA, Roberson PA, et al. Physiological differences between low versus high skeletal muscle hypertrophic responders to resistance exercise training: current perspectives and future research directions. Front Physiol. (2018) 9:834. doi: 10.3389/fphys.2018.00834
66. Burd NA, Gorissen SH, van Loon LJ. Anabolic resistance of muscle protein synthesis with aging. Exerc Sport Sci Rev. (2013) 41:169–73. doi: 10.1097/JES.0b013e318292f3d5
67. Markofski MM, Dickinson JM, Drummond MJ, Fry CS, Fujita S, Gundermann DM, et al. Effect of age on basal muscle protein synthesis and mTORC1 signaling in a large cohort of young and older men and women. Exp Gerontol. (2015) 65:1–7. doi: 10.1016/j.exger.2015.02.015
68. Castets P, Lin S, Rion N, Di Fulvio S, Romanino K, Guridi M, et al. Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab. (2013) 17:731–44. doi: 10.1016/j.cmet.2013.03.015
69. Williams AS, Kang L, Wasserman DH. The extracellular matrix and insulin resistance. Trends Endocrinol Metabol. (2015) 26:357–66. doi: 10.1016/j.tem.2015.05.006
70. Beals JW, Sukiennik RA, Nallabelli J, Emmons RS, Van Vliet S, Young JR, et al. Anabolic sensitivity of postprandial muscle protein synthesis to the ingestion of a protein-dense food is reduced in overweight and obese young adults, 2. Am. J Clin Nutr. (2016) 104:1014–22. doi: 10.3945/ajcn.116.130385
71. Sharples AP, Hughes DC, Deane CS, Saini A, Selman C, Stewart CE. Longevity and skeletal muscle mass: the role of IGF signalling, the sirtuins, dietary restriction and protein intake. Aging Cell. (2015) 14:511–23. doi: 10.1111/acel.12342
72. Lin R, Yan D, Zhang Y, Liao X, Gong G, Hu J, et al. Common variants in SIRT1 and human longevity in a Chinese population. BMC Med Genet. (2016) 17:31. doi: 10.1186/s12881-016-0293-3
73. Grounds MD, Radley HG, Gebski BL, Bogoyevitch MA, Shavlakadze T. Implications of cross-talk between tumour necrosis factor and insulin-like growth factor-1 signalling in skeletal muscle. Clin Exp Pharmacol Physiol. (2008) 35:846–51. doi: 10.1111/j.1440-1681.2007.04868.x
74. Kilic U, Gok O, Erenberk U, Dundaroz MR, Torun E, Kucukardali Y, et al. A remarkable age-related increase in SIRT1 protein expression against oxidative stress in elderly: SIRT1 gene variants and longevity in human. PLoS ONE. (2015) 10:e0117954. doi: 10.1371/journal.pone.0117954
75. Jing XM, Wang QQ, Hao YL. Alterations of Sirt1 and mTOR activity in gastrocnemius muscle of mice with sarcopenia. Basic Clin Med. (2018) 38:922–7. doi: 10.3969/j.issn.1001-6325.2018.07.005
76. McClure R, Villani A. Mediterranean diet attenuates risk of frailty and sarcopenia: new insights and future directions. JCSM Clin Rep. (2017) 2:1–17. doi: 10.17987/jcsm-cr.v2i2.45
77. Crowe FL, Key TJ, Allen NE, Appleby PN, Roddam A, Overvad K, et al. The association between diet and serum concentrations of IGF-I, IGFBP-1, IGFBP-2, and IGFBP-3 in the European Prospective Investigation into Cancer and Nutrition. Cancer Epidemiol Prevent Biomark. (2009) 18:1333–40. doi: 10.1158/1055-9965.EPI-08-0781
78. Larsson SC, Wolk K, Brismar K, Wolk A. Association of diet with serum insulin-like growth factor I in middle-aged and elderly men. Am J Clin Nutr. (2005) 81:1163–7. doi: 10.1093/ajcn/81.5.1163
79. Wright C, Zhou J, Sayer R, Kim J, Campbell W. Effects of a high-protein diet including whole eggs on muscle composition and indices of cardiometabolic health and systemic inflammation in older adults with overweight or obesity: a randomized controlled trial. Nutrients. (2018) 10:946. doi: 10.3390/nu10070946
80. Ascenzi F, Barberi L, Dobrowolny G, Villa Nova Bacurau A, Nicoletti C, Rizzuto E, et al. Effects of IGF-1 isoforms on muscle growth and sarcopenia. Aging Cell. (2019) 18:e12954. doi: 10.1111/acel.12954
81. Moore DR, Churchward-Venne TA, Witard O, Breen L, Burd NA, Tipton KD, et al. Protein ingestion to stimulate myofibrillar protein synthesis requires greater relative protein intakes in healthy older versus younger men. J Gerontol Ser A Biol Sci Med Sci. (2015) 70:57–62. doi: 10.1093/gerona/glu103
82. Yang Y, Breen L, Burd NA, Hector AJ, Churchward-Venne TA, Josse AR, et al. Resistance exercise enhances myofibrillar protein synthesis with graded intakes of whey protein in older men. Br J Nutr. (2012) 108:1780–8. doi: 10.1017/S0007114511007422
83. Volpi E, Campbell WW, Dwyer JT, Johnson MA, Jensen GL, Morley JE, et al. Is the optimal level of protein intake for older adults greater than the recommended dietary allowance? J Gerontol Ser A Biomed Sci Med Sci. (2012) 68:677–81. doi: 10.1093/gerona/gls229
84. Jadav RS, Kumar D, Buwa N, Ganguli S, Thampatty SR, Balasubramanian N, et al. Deletion of inositol hexakisphosphate kinase 1 (IP6K1) reduces cell migration and invasion, conferring protection from aerodigestive tract carcinoma in mice. Cell. Signal. (2016) 28:1124–36. doi: 10.1016/j.cellsig.2016.04.011
85. Ghoshal S, Zhu Q, Asteian A, Lin H, Xu H, Ernst G, et al. TNP [N2-(m-trifluorobenzyl), N6-(p-nitrobenzyl) purine] ameliorates diet induced obesity and insulin resistance via inhibition of the IP6K1 pathway. Mol Metabol. (2016) 5:903–17. doi: 10.1016/j.molmet.2016.08.008
86. Mackenzie RW, Elliott BT. Akt/PKB activation and insulin signaling: a novel insulin signaling pathway in the treatment of type 2 diabetes. Diab Metab Syndrome Obesity Targ Therapy. (2014) 7:55. doi: 10.2147/DMSO.S48260
87. Zhu Q, Ghoshal S, Rodrigues A, Gao S, Asterian A, Kamenecka TM, et al. Adipocyte-specific deletion of Ip6k1 reduces diet-induced obesity by enhancing AMPK-mediated thermogenesis. J Clin Invest. (2016) 126:4273–88. doi: 10.1172/JCI85510
88. Ghoshal S, Tyagi R, Zhu Q, Chakraborty A. Inositol hexakisphosphate kinase-1 interacts with perilipin1 to modulate lipolysis. Int J Biochem Cell Biol. (2016) 78:149–55. doi: 10.1016/j.biocel.2016.06.018
89. Moore DR. Keeping older muscle “young” through dietary protein and physical activity. Adv Nutr. (2014) 5:599S−607S. doi: 10.3945/an.113.005405
Keywords: IP6K1, sarcopenia, aging, protein, resistance exercise, anabolic resistance, Akt, mTOR
Citation: Barclay RD, Burd NA, Tyler C, Tillin NA and Mackenzie RW (2019) The Role of the IGF-1 Signaling Cascade in Muscle Protein Synthesis and Anabolic Resistance in Aging Skeletal Muscle. Front. Nutr. 6:146. doi: 10.3389/fnut.2019.00146
Received: 29 March 2019; Accepted: 23 August 2019;
Published: 10 September 2019.
Edited by:
Maurizio Muscaritoli, Sapienza University of Rome, ItalyReviewed by:
Miguel Luiz Batista Júnior, University of Mogi das Cruzes, BrazilSergio Davinelli, University of Molise, Italy
Copyright © 2019 Barclay, Burd, Tyler, Tillin and Mackenzie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard W. Mackenzie, cmljaGFyZC5tYWNrZW56aWUmI3gwMDA0MDtyb2VoYW1wdG9uLmFjLnVr