 Elvira Akhmetzyanova
Elvira Akhmetzyanova Konstantin Kletenkov1
Konstantin Kletenkov1 Yana Mukhamedshina
Yana Mukhamedshina Albert Rizvanov
Albert Rizvanov
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Syst. Neurosci. , 27 August 2019
Volume 13 - 2019 | https://doi.org/10.3389/fnsys.2019.00037
This article is part of the Research Topic Frontiers in Systems Neuroscience – Editors’ Pick 2021 View all 10 articles
Microglial cells, which are highly plastic, immediately respond to any change in the microenvironment by becoming activated and shifting the phenotype toward neurotoxicity or neuroprotection. The polarization of microglia/macrophages after spinal cord injury (SCI) seems to be a dynamic process and can change depending on the microenvironment, stage, course, and severity of the posttraumatic process. Effective methods to modulate microglia toward a neuroprotective phenotype in order to stimulate neuroregeneration are actively sought for. In this context, available approaches that can selectively impact the polarization of microglia/macrophages regulate synthesis of trophic factors and cytokines/chemokines in them, and their phagocytic function and effects on the course and outcome of SCI are discussed in this review.
Spinal cord injury (SCI) is characterized by numerous pathologic reactions that involve every cell type of the central nervous system (CNS). The activation of microglial cells, which are the first to respond to nervous tissue damage, is one of the essential events of posttraumatic reactions (Gensel and Zhang, 2015). Activated microglia can synthesize not only trophic biomolecules such as neurotrophins, glutamate transporters, and antioxidants, but also effectors such as nitric oxide (NO) and pro-inflammatory cytokines that can be potentially neurotoxic (Persson et al., 2005; Lai and Todd, 2006; Hellwig et al., 2013). In addition to synthesis of many biomolecules, a phagocytic function of microglia is critically important also due to its essential for the removal of degenerating/lost neurons and neuroglial cells, and rearrangement or destruction of synaptic connections (Chen and Trapp, 2016; Jin and Yamashita, 2016; Wolf et al., 2017). Previous studies have shown that the activation of microglia is not a single phenomenon and that there are several different “states of activation” when microglia can have a selective neurotoxic or neuroprotective effect. Given the diversity of microglia functions, the M1/M2 paradigm is a simplified model that reflects two opposite effects on inflammatory responses. However, one should take into consideration that the microglia microenvironment in vivo is diverse and its phenotype may rarely shift directly to the other state.
To date, several states of microglia polarization have been described: they are classic activation (M1), alternative activation (M2a), alternative type II activation (M2b), and acquired deactivation (M2c). A number of investigators question whether microglia can acquire an M3 phenotype (Malyshev and Malyshev, 2015; Walker and Lue, 2015). A number of principal studies have identified which markers are specific for classically or alternatively activated microglia (Martinez et al., 2006; Martinez et al., 2013).
M1 microglia are capable of producing active oxygen species that promote a respiratory burst, as well as produce cytokines such as tumor necrosis factor-α (TNF-α), IL-1β, IL-6, and IL-12, thereby mediating inflammatory tissue damage (Liu et al., 2018). M1 microglia are involved in secondary damage after SCI, producing proinflammatory molecules and the formation of a glial scar, which, in turn, creates an environment at the site of injury that is adverse for neuroprotection. Therefore, this phenotype is commonly referred to as neurotoxic (Shechter and Schwartz, 2013; Fan et al., 2016). The phagocytic activity was shown to be inhibited in M1 polarization (Durafourt et al., 2012); at the same time, M1 microglia regulate synaptic pruning and labeling synapses for phagocytosis (Schafer et al., 2012).
Alternative activation is subdivided into two subcategories: M2a and M2b. M2à microglia are considered to respond to IL-4 and IL-13; to have an increased phagocytic activity; to produce an insulin-like growth factor-1, trophic polyamines, and anti-inflammatory cytokines such as IL-10; and to express G-CSF, GM-CSF, and CD209 (Martinez and Gordon, 2014; Franco and Fernandez-Suarez, 2015; Peferoen et al., 2015). The microglia of this type can eliminate cellular debris and stimulate tissue regeneration. M2b microglia are induced by ligation of immunoglobulin Fc-gamma-receptors that results in IL-12 expression, increased IL-10 secretion, and HLA-DR expression. This phenotype is also characterized by active phagocytosis and an increased expression of CD32 and CD64, which are detected in the cerebral microglia in Alzheimer’s disease (Peress et al., 1993). M2c (acquired deactivation) polarization can be caused by the anti-inflammatory cytokine IL-10 or glucocorticoids, an increased expression of transforming growth factor (TGF), sphingosine kinase (SPHK1), and CD163, a membrane-bound receptor for haptoglobin/hemoglobin complexes (Wilcock, 2014). The polarization of microglia/macrophages toward the M2 phenotype occurs to resolve inflammation and degeneration as a whole; thus, this phenotype is characterized as neuroprotective. It is worth noting, however, that although the M2 phenotype of microglia/macrophages plays a positive role in neuroregeneration processes, it has an absolutely opposite role in the case of neoplastic processes in the CNS and has a pro-tumor action (Wu and Watabe, 2017).
A similar pattern of polarization is involved in peripheral macrophages that actively migrate after injury when the blood–brain barrier is damaged. It should be noted that most researchers do not distinguish between microglia and macrophages, subsuming them into the same cell population and using pan markers for their identification. This might be due to the lack of highly specific markers for resident microglia and macrophages migrating toward a site of injury (Franco and Fernandez-Suarez, 2015; Martin et al., 2017).
It has been previously shown that microglia are activated within the first 24 h after SCI. In the acute period, polarization shifts primarily toward M1 microglia, which release proinflammatory cytokines and chemokines. This results in progression of inflammatory processes after primary mechanical injury (Lee et al., 2009; Nakajima et al., 2012). Shortly thereafter (2–3 days post-injury, dpi), blood monocytes that subsequently differentiate into macrophages phenotypically and morphologically indistinguishable from activated microglia migrate toward the site of injury (Donnelly and Popovich, 2008; Beck et al., 2010). The appearance of M2 microglia/macrophages and their secretion of anti-inflammatory cytokines and chemokines results in inhibiting excessive inflammatory reactions around the site of injury and stimulating regeneration of damaged spinal cord tissues (Gratchev et al., 2008; Varnum and Ikezu, 2012; Shechter and Schwartz, 2013; Weisser et al., 2013). M2 microglia/macrophages are shown to possess an increased phagocytic activity that promotes clearance of posttraumatic debris, leading to accelerated demyelination and resolution of the initial traumatic events (Redondo-Castro et al., 2013; Lampron et al., 2015; Orihuela et al., 2016; Akhmetzyanova et al., 2018).
The primary phase of microglia/macrophage activation peaks on 7 dpi; microglia are reactivated after 14 dpi and then peak on 60 dpi and remain for up to 180 dpi (Beck et al., 2008; Conta and Stelzner, 2008; Kigerl et al., 2009; Bellver-Landete et al., 2019). M1 and M2 microglia/macrophages co-exist at the injury site within the 1st week after SCI, with M1 cells prevailing. However, other researchers have demonstrated that there were no M2 cells and the population of M1 cells significantly decreased after 28 dpi (Kigerl et al., 2009; Francos-Quijorna et al., 2016). These results confirm data on the population of ED1+ phagocytic macrophages/microglia, which peak by day 7 after SCI, significantly decreasing by 28 dpi and abruptly increasing again by 90 dpi (Beck et al., 2010). At the same time, Bellver-Landete et al. (2019) showed that activated, proliferating microglia play an important role in the healing process, having a positive effect on tissue sparing and functional recovery after SCI, and this effect persists for 5 weeks after SCI.
These phases of microglia/macrophage activation in SCI can be paralleled with changes observed in the population of macrophages when other tissues and organs are damaged. For example, at the end of the remodeling phase when the main healing processes are completed, macrophages are deactivated, and inflammation resolves. The behavior of microglia/macrophages whose number reduces significantly though variably by 2–4 weeks after SCI is possibly the same. With effective healing, the level of macrophages in non-nervous tissues returns to normal within several weeks after injury in parallel with its healing. On the contrary, wounds that do not heal within 3 months result in a stable activation of macrophages that is a distinctive feature of chronicity (Sindrilaru et al., 2011). In turn, we observed a similar picture during the second phase of microglia/macrophage activation that seems to be triggered by ongoing neurodegeneration in response to which re-activation of these cells prevents a subsequent loss of function.
The polarization of microglia/macrophages after SCI seems to be a dynamic process and can be altered depending on the microenvironment, the stage of the posttraumatic process, and its severity (Kigerl et al., 2009; David and Kroner, 2011). This phenomenon has been demonstrated in several studies and has shown that the behavior of microglia/macrophages depended on the factors of activation, in particular, the type of cells that activated them and the specific activating molecule (Nakajima and Kohsaka, 2002; Nakajima and Kohsaka, 2004; Stout and Suttles, 2004; Shaked et al., 2005; Lai and Todd, 2006; Nakajima et al., 2006; Menzies et al., 2010). An effective method to modulate microglia toward a neuroprotective phenotype in order to stimulate neuroregeneration is actively sought for in addition to investigation into the factors of activation. For this purpose, new approaches are being developed and different biomolecules potentially possessing a selective effect on the polarization of microglia/macrophages regulate their synthesis of trophic factors, cytokines/chemokines, and a phagocytic function tested. The latter can be achieved by affecting the signaling pathways that control microglia activation and polarization, discussed in the following section.
It is now increasingly evident that there are various ways of activation for microglia that determine the generation of cells with divergent abilities (Figure 1). Toll-like receptors (TLRs) are a class of transmembrane receptors involved in the activation of cell-mediated immune response. Out of more than 10 TLRs, identified in both rodents and humans, microglia express at least 9 TLRs along with their adapter proteins (Laflamme et al., 2001; Bsibsi et al., 2002; Dalpke et al., 2002; Olson and Miller, 2004; Zhang et al., 2013). Previous studies have demonstrated TLR-dependent microglia activation in neurodegenerative disorders and different types of CNS injury (Heneka et al., 2005; Fernandez-Lizarbe et al., 2009; Song et al., 2011; Yao et al., 2013). A classical/canonical activation of the nuclear factor κB (NF-κB) signaling, which is essential for both acute and chronic inflammatory responses, is initiated by TLRs, as well as other cell surface receptors, including those for IL-1 and TNF (Shih et al., 2015; Noort et al., 2015). The activated NF-κB allows translocation to the nucleus that results in production of anti-inflammatory cytokines, release of reactive oxygen species (ROS), and microglia modulation toward the M1 phenotype (Pan et al., 2010; Taetzsch et al., 2015; Zhang et al., 2017). The activation of NF-κB transcription factors also plays a key role in neurogenesis, synaptic plasticity, and protection of neurons (O’Riordan et al., 2006; Ahn et al., 2008; Koo et al., 2010). Therefore, NF-κB should be selectively inhibited in microglia and possibly in astrocytes in order to neutralize its neurotoxic role and maintain the neuroprotective one (Brambilla et al., 2005; Crosio et al., 2011; Frakes et al., 2014).
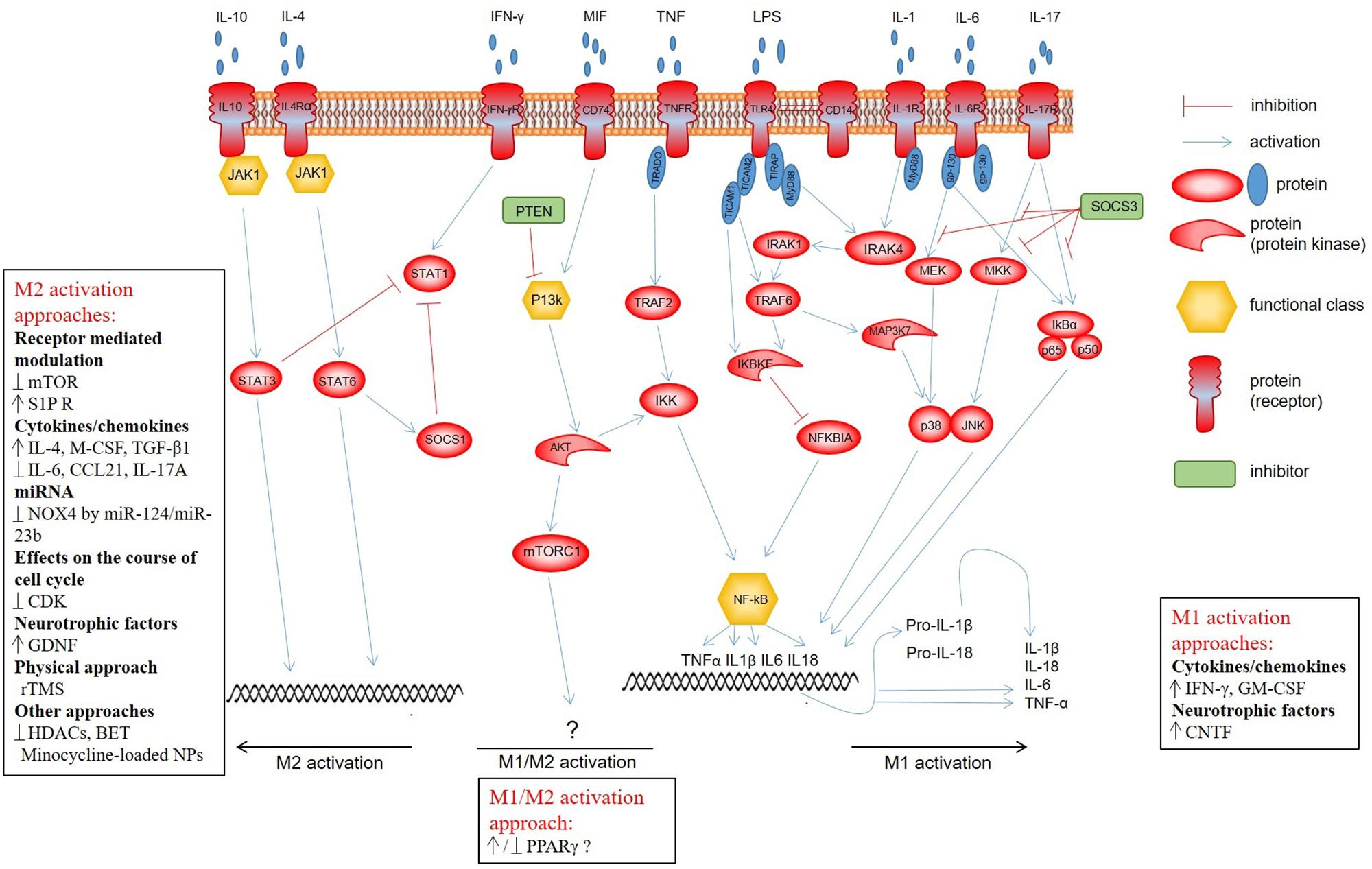
Figure 1. Microglial signaling pathways determining development of cells with divergent abilities. Classical activation of the NF-κB signaling is initiated by TLRs, as well as other cell surface receptors, including specific IL-1 and TNF, and provides M1 polarization of microglia. PI3K/Akt/mTOR is triggered through the CD74 receptor, whose activation is promoted by MIF. There is quite contradictory evidence that this pathway affects the shift of the microglia/macrophage phenotype toward M1 or M2. Anti-inflammatory cytokines IL-10 and IL-4 induce STAT3 and STAT6 phosphorylation, respectively, via JAK1, which promotes polarization toward the M2 phenotype. The activation of STAT1, in turn, leads to polarization toward a neurotoxic M2 phenotype of microglia. Normally, there is a balance between the activation of STAT1 and STAT3/STAT6 that strictly regulates the microglia polarization and activity.
In the presence of inflammation, microglia are activated by means of phosphorylation of p38 mitogen-activated protein kinase (p38/MAPK) and extracellular signal–regulated kinases (ERKs), thereby enhancing phagocytosis, chemotaxis, and the expression of proinflammatory cytokines (Wang et al., 2011; Fan et al., 2017). At the same time, the phosphorylation of p38/MAPK inhibited ULK1 kinase activity and reduced autophagy, allowing the full induction of the inflammatory process during microglia activation (He et al., 2018). The activation of glial cells and the p38/MAPK signaling pathway was demonstrated to be involved in the development of a chronic neuropathic pain that affects up to 80% of patients with SCI (Finnerup et al., 2001; Crown et al., 2008; Detloff et al., 2008). Therefore, p38/MAPK inhibitors are intensively used to reduce activation of the spinal microglia, to prevent/reverse the neuropathic pain symptoms and neuroinflammation in general (Rojewska et al., 2014; Cheng et al., 2015; Kim et al., 2016; Taves et al., 2016).
A phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) signaling pathway that is involved in neuropathic pain progression, as well as astrocyte and microglia activation, is known. Its inhibition reduces the ability of microglial cells to migrate and their number in the site of neurodegeneration (Guo et al., 2017). PI3K/Akt/mTOR is triggered through the CD74 receptor, whose activation is promoted by a macrophage migration inhibitory factor (MIF). The use of MIF suppresses the microglia M1 activation and mitigates the severity of secondary injury around the lesion site in the murine dorsal hemisection model of SCI (Emmetsberger and Tsirka, 2012). At the same time, there is quite contradictory evidence that this pathway affects the shift of the microglia/macrophage phenotype toward M1 or M2 stages (Wang G. et al., 2015; Wang et al., 2016). Therefore, the role of the PI3K/Akt/mTOR pathway in microglia activation and neuroregeneration as a whole following SCI is still controversial (Kanno et al., 2012; Chen et al., 2016). Some researchers relate this to the possibility of isoform-specific cross-talk between PI3K, Akt, and mTORC (Vergadi et al., 2017).
There are natural (phosphatase and tensin homolog deleted on chromosome 10, PTEN) and artificial (ZSTK474, NVP-BEZ235, LY294002, PI828, etc.) inhibitors of the PI3K/Akt/mTOR signaling pathway. PTEN is a lipid and protein phosphatase that has dual substrate specificity and serves as the main negative regulator of PI3K and the PI3K/Akt/mTOR signaling pathway by converting phosphatidylinositol (3,4,5)-trisphosphate (PIP3) into phosphatidylinositol (4,5)-biphosphate (P1P2). In a model of chronic peripheral nerve injury, the PTEN gene overexpression resulted from its delivery with an adenoviral vector (Ad5-PTEN) in the spinal cord. It significantly reduced activation of microglia and astrocytes and prevented a neuropathic pain (Huang et al., 2015). At the same time, such microglia modulation in neurotrauma therapy can negatively affect regeneration as the PTEN expression has been shown to be able to attenuate neuroprotection and lead to an impairment of axonal growth in particular (Zukor et al., 2013; Ohtake et al., 2014; Yin et al., 2018). Thus, the system regulation at the level of this enzyme is a quite dangerous process leading to disarrangement of oppositely directed processes.
A Janus tyrosine kinase (JAK)/signal transducer and activator of transcription (STAT) signaling pathway is one of the most important cascades triggered in response to many modulators of inflammation. Most studies focused on activation of the JAK/STAT3 signaling pathway in the case of neoplastic activity of microglia/macrophages (Zhang et al., 2009; Zhang et al., 2011; Oliva et al., 2012; Koscsó et al., 2013; Popiolek-Barczyk and Mika, 2016). As for microglia/macrophage modulation after SCI, anti-inflammatory cytokines IL-10 and IL-4 induce STAT3 and STAT6 phosphorylation, respectively, via JAK1, and promote polarization toward the M2 phenotype (Koscsó et al., 2013; Wang et al., 2014; Cianciulli et al., 2015; Popiolek-Barczyk and Mika, 2016). Activation of STAT1 and NF-κB transcription factors leads, in turn, to polarization toward a neurotoxic phenotype of microglia/macrophages. There is normally a balance between the activation of STAT1 and STAT3/STAT6 that strictly regulates the microglia/macrophage polarization and activity.
STAT3 is recognized as the main mediator of IL-6 and IL-17 functions (Camporeale and Poli, 2012). There are two main types of IL6 signalization: pro-inflammatory and anti-inflammatory. In microglia, the IL-6 pro-inflammatory signaling pathway is carried out through gp-130, which acts as an antagonist sequestering IL-6 (Hodes et al., 2016). Results obtained by Ma et al. (2010) indicate that SOCS3 can function as a negative regulator of NF-κB, p38 MAPK, and JNK signaling; moreover, an important role for SOCS3 in the regulation of IL-17 and IL-6/R-dependent induction of IL-6 was elucidated. Guerrero et al. (2012) demonstrated that classical microglia activation in mouse SCI could be inhibited by IL-6 blockade. Redondo-Castro et al. (2013) used glibenclamide, an inhibitor of ATP-sensitive potassium channels KATP to relieve a neuropathic pain in rats with SCI. The inhibition of IL-17, as a mediator of microglia activation, when injecting hyperforin enabled microglia polarization toward the M2 phenotype (Ma et al., 2018). Although studies to inhibit individual mediators of microglia activation demonstrate certain positive changes in some neurodegenerative disorders, they cannot completely neutralize the neurotoxic potential of these cells that is related to their possible activation by means of a common regulator in a signaling cascade.
Supplementary Table 1 contains published data available on the different approaches to modulation of microglia/macrophages in vitro and in vivo.
Peroxisome proliferator-activated receptor (PPARγ) is a key regulator of the microglia/macrophage M2 phenotype. It is a nuclear receptor capable of modulating inflammatory processes and controlling lipid and lipoprotein metabolism as well as glucose homeostasis (Chinetti et al., 2003; McTigue, 2008). PPARγ was shown to be immediately induced in monocyte differentiation into macrophages (Chinetti et al., 1998). In addition, activation of PPARγ signaling can suppress an inflammatory response by inhibiting NF-κB (Zolezzi et al., 2017). Therefore, PPARγ is described as the main anti-inflammatory regulator of macrophages (Ahmadian et al., 2013). Han et al. (2017) demonstrated that 6-Shogaol, a pungent constituent extracted from Zingiber officinale Roscoe can enhance PPARγ expression. Also, the addition of 6-Shogaol to an in vitro microglia culture could reduce the lipopolysaccharide-induced (LPS) expression of proinflammatory factors TNF-α, IL1β, IL6, and PGE2 as measured using ELISA. The use of a PPARγ agonist, rosiglitazone, was shown in vitro with PCR and flow cytometry to lead to monocyte modulation toward the M2 phenotype (Bouhlel et al., 2007). Additionally, in a murine brain injury model intravenously injected PPARγ activator, malibatol A, an anti-oxidant extracted from Hopea hainanensis, could shift the microglia phenotype toward M2 (Pan et al., 2015). Wen et al. (2018) compared the effects of intraperitoneal injections of the PPARγ agonist, rosiglitazone, and the PPARγ antagonist, GW9662, in a lateral fluid percussion injury model in mice. Using ELISA, real-time PCR and immunohistochemistry, they observed that 72 h after injury, expression levels of proinflammatory cytokines TNF-α, IL-1β, and IL-6 were significantly higher and that of anti-inflammatory IL-10 was lower in the group treated with GW9662. Further in vitro experiments in a primary culture of mouse microglia were conducted and results demonstrated that rosiglitazone increased the expression of M2 markers (CD206 and YM-1) and decreased that of M1 markers (TNF-α, IL-6, IL-1β, and IL-10). Interesting results were obtained by Park et al. (2007) using pioglitazone in a rat model of SCI. They showed that intraperitoneal injection of pioglitazone causes a decrease in the number of reactive macrophages, attenuates myelin loss, and improves functional recovery from SCI. The results of this study were confirmed by McTigue et al. (2007), where the rat electromagnetic SCI model again showed pioglitazone’s ability to reduce the number of activated phagocytic microglia by 7 dpi.
Neuropeptides Y (NPY), Y1 receptor activators, suppress the innate immune response by reducing the release of interleukin-1β and NO, migration, and phagocytosis of activated microglial cells (Farzi et al., 2015). NPY were demonstrated to significantly restrain the microglia activation by inhibiting LPS-induced Fc-receptor-mediated phagocytosis (Ferreira et al., 2012). Macrophage antigen complex-1 receptor (MAC1R), a molecule mediating the macrophage activation in response to various stimuli, plays an important role in phagocytosis (Le Cabec et al., 2002). In fact, MAC1R is a key receptor for both toxins and classic acute-phase reactants such as fibrinogen, which activate microglia/macrophages manifesting in enhanced phagocytic activity and a release of ROS (Adams et al., 2007; Pei et al., 2007). The investigation of the role that NPY and MAC1R play in regulation of phagocytic activity of microglial cells is promising and requires further research.
Rapamycin, an inhibitor of the mTOR receptor in mammals, is involved in numerous cellular processes such as neuroprotection in neurodegenerative disorders (Ravikumar et al., 2004; Malagelada et al., 2010) and neuroregeneration after cerebral injury and SCI (Erlich et al., 2007; Kanno et al., 2012). Rapamycin inhibits the mTOR pathway by preventing the activation of p70S6K protein kinase (Schmelzle and Hall, 2000). Rapamycin neuroprotective properties are due to its ability to stimulate autophagy (Ravikumar et al., 2004; Malagelada et al., 2010). It is also involved in suppression of microglia activation and reduction of inflammation in the CNS by selective inhibition of the mTORC1 complex (Russo et al., 2009). In vivo studies using rat SCI model showed the ability of rapamycin to attenuate microglial activation and neuroinflammation processes by reducing the number of M1 cells and, as a result, TNF-α production (Chen et al., 2013; Song et al., 2015). However, Eldahan et al. (2018) caution against the use of rapamycin as a therapeutic intervention for SCI due to its toxic effects and exacerbation of cardiovascular dysfunction.
FTY720 is an agonist of the S1P receptor and a derivative of ISP-1 (myriocin), a metabolite of the Chinese fungus Iscaria sinclairii, as well as a sphingosine structural analog. This is a novel immunomodulator that promotes transplantability in numerous models by inhibiting lymphocytes. FTY720 plays the role of a switch in the polarization of microglia from M1 to M2 through the STAT3 protein activation that has been established in a white matter ischemic injury model (Qin et al., 2017). The results obtained also provide evidence that FTY720 has a protective effect against structural damage to the nodes of Ranvier and demyelination after hypoperfusion. It should be noted that, in some cases, FTY720 was effective in treating SCI but did not affect the activation of microglia/macrophages (Norimatsu et al., 2012; Wang J. et al., 2015).
Cytokines play an important role in posttraumatic processes; in particular, microglia cells can influence healing by controlling levels of some of them. Activating and blocking agents to modulate inflammatory processes for most of these cytokines are under development. For example, IL-4 is considered the strongest polarizing cytokine for M2a microglia response. In the IL-4R-deficient mice SCI model, there was a decrease in the production of anti-inflammatory cytokines such as arginase, IL-1, and CCL2, which indicates the predominance of M1 microglia (Fenn et al., 2014). A single intraspinal injection of IL-4 48 h after SCI was shown to be sufficient to switch the microglia phenotype toward M2 and, what is more important, it was associated with improved functional recovery in mice with an SCI (Francos-Quijorna et al., 2016). Interestingly, in response to exposure, IL-4 can modulate the morphology of microglia in vitro from amoeboid (activated) to a more ramified (quiescent) one associated with a more activated phenotype; at the same time, IFN-γ and GM-CSF have the opposite effect (Rostam et al., 2017).
IL-6 is a key factor triggering inflammation after SCI; it also promotes microglia M1 activation (Bethea and Dietrich, 2002). An IL-6 blocking agent—monoclonal anti-mouse receptor antibody IL-6 (MR16-1)—administered into the site of SCI in mice promoted alternative M2 microglia activation and resulted in improved tissue integrity as well as an increased number of myelinated fibers (Guerrero et al., 2012). Moreover, inhibition of the EGFR/MAPK pathway that suppresses microglia activation and associated cytokine production decreases neuroinflammation-related secondary damage and thereby provides neuroprotection in rats after SCI (Qu et al., 2012). It is thought that EGFR can be a therapeutic target and inhibitors C225 and AG1478 have the potential to be used in the treatment of SCI (Qu et al., 2012). Chemokine CCL21 neutralization was also shown to reduce microglia M1 activation and to cause neuronal hyperexcitability of lateral posterior thalamic nuclei (Zhao et al., 2007).
IL-6 is a key cytokine accelerating the IL-17 production (Camporeale and Poli, 2012). IL-17 is a well-known proinflammatory cytokine associated with M1 activation of microglia (Kim and Moalem-Taylor, 2011; Zong et al., 2014). The inhibition of IL-17 as a mediator of microglia activation after hyperforin injection promoted microglia polarization toward the M2 phenotype in a murine acute cerebral mechanic trauma model (Ma et al., 2018). TGF-β1 is a polypeptide component of a transforming cytokine factor. It was shown in an in vivo murine stroke model that cerebro-ventricular injections of TGF-β1 promoted microglia M2 activation as well as improvement of functional recovery in mice (Taylor et al., 2017).
Lately, special emphasis is being given to the role that miRNA plays in the pathogenesis of many diseases including diseases of the CNS. It has been shown that miRNA administration can be an effective therapeutic approach to the management of neurodegenerative processes. MiRNAs regulate the expression of a great number of genes by stimulating RNA interference pathway degradation or by preventing the translation of target genes. High miR-124 levels were reported in resident cerebral and spinal microglia, as well as their activation in vitro and in vivo to promote a decreased miR-124 expression (Ponomarev et al., 2011). miR-124 is considered to regulate the activity of microglia/macrophages by down-regulating the expression of CCAAT-enhancer-binding protein-α, a transcription factor regulating the differentiation of myeloid cells. Therefore, high miR-124 levels are thought to be required to maintain microglia in a quiescent state. Willemen et al. (2012) demonstrated that intrathecally injected miR-124 promoted the maintenance of microglia in this quiescent state and alleviated chronic posttraumatic processes in the spinal cord of rats with hyperalgesia. Im et al. (2012) reported similar results when injecting miR-23b intrathecally to mice in a neuropathic pain model. Based on their results, a return of miR-23b to normal levels decreased the expression of inflammatory proteins, reduced the number of Iba1+ cells in the spinal cord tissue, and alleviated a neuropathic pain resulting from SCI.
It was found that exposure on a cell cycle course can also modulate cell phenotype. There were changes in cell cycle course following SCI and effects of systemic administration of delayed (24 h) flavopiridol, an inhibitor of major cyclin-dependent kinases, on functional recovery and histopathology in a rat SCI model (Wu et al., 2012). The treatment with flavopiridol attenuated the number of Iba-1+-microglia in the intact tissue and, as a result, increased the myelinated area of the white matter. Moreover, flavopiridol attenuated the expression of Iba-1 and glactin-3, associated with microglia M1 activation and astrocyte reactivity by reducing the GFAP, NG2, and CHL1 expression.
Although there are numerous approaches to modulate microglia in the CNS, the search for new approaches to effective polarization strictly toward a neuroprotective phenotype and introduction of results into clinical practice is still relevant. Neurotrophic factors are molecules that increase the potential of nervous system cells to proliferate, survive, migrate, and differentiate. For instance, a ciliary neurotrophic factor (CNTF) exerts a positive effect on reactive M1 microglia, promoting their survival and activation after intracerebral injection to mice in vivo (Kahn et al., 1995). However, Rocha et al. (2012) demonstrated that, on the contrary, a glial cell line-derived neurotrophic factor (GDNF) inhibited the activation of reactive M1 microglia in vitro (Rocha et al., 2012).
Selective modulation of microglia with recombinant adenoviruses carrying the GDNF gene in vivo seems promising. Zhuravleva et al. (2016) demonstrated that microglia transduction with an adenovirus encoding for GDNF (Ad5-GDNF) promoted a reduced phagocytic activity of these cells. We have conducted a study to evaluate effects of Ad5-GDNF transduction on the morphology and phenotype of microglial cells as well as effects of transplanting these cells on posttraumatic processes in the rat spinal cord. It was shown that microglia transduction with Ad5-GDNF down-regulated expression of CD45, but their transplantation into the site of a rat SCI did not increase the area of intact tissue as compared to similar transplantation of Ad5-EGFP microglia with fair phagocytic activity (Akhmetzyanova et al., 2018).
Repetitive transcranial magnetic stimulation of the motor cortex was shown to reduce microglia M1 activation after SCI and alleviated symptoms of neuropathic pain and allodynia (Kim et al., 2013). A method of physical exposure to alleviate SCI consequences is promising as it is non-invasive and has few side effects. However, this approach to treatment is just gaining ground and is not yet fully understood. Therefore a comprehensive assessment of modulation mechanisms triggered by a physical exposure is required.
Lipopolysaccharide is an essential molecular component of the outer membrane of gram-negative bacteria and is recognized by an immune system as an invasion marker of bacterial pathogens. LPS is more often used to induce a potent immunological response and activate microglia/macrophages. It was found that pre-conditioning of microglia with LPS 48 h prior to transplantation enabled M2 polarization in mouse SCI (Hayakawa et al., 2014). The results obtained were evaluated by measuring the expression of mRNA markers of M1 (iNOS, CD86, and CD16) and M2 (arginase1 and CD206) microglia.
Histone deacetylases (HDACs) are proteins targeted to remove acetyl groups from lysine residues of target proteins. HDAC3 is most commonly found in the brain and is a regulator of inflammatory processes (Broide et al., 2007). HDAC3-deficient macrophages have a reduced ability to activate the expression of inflammatory genes in response to LPS stimulation (Chen et al., 2012). At the same time, it was found that HDAC3 is an epigenomic brake in macrophage alternative (M2) activation (Mullican et al., 2011). Malvaez et al. (2013) used protein mass spectrometry in the study in vitro to detect global molecular changes in resident microglia exposed to RGFP966, a selective HDAC3 inhibitor, by investigating a signaling pathway through which RGFP966 regulated an inflammation. They observed that RGFP966 could inhibit TLR and STAT3/5 signaling pathways of microglia M1 activation and that this resulted in an anti-inflammatory microglia response manifesting as a reduced expression of proinflammatory cytokines such as IL-6 and TNF-α (Xia et al., 2017). This is confirmed by another in vitro study, where it was shown in a primary culture that treatment with HDAC inhibitors promoted suppression of the innate immune activation of microglia (Kannan et al., 2013). A similar study demonstrated that HDAC3 arrest with the same selective inhibitor RGFP966 facilitated the shift toward an anti-inflammatory microglia response that resulted in gaining a neuroprotective phenotype by these cells and improved functional recovery in an SCI model in vivo (Kuboyama et al., 2017). Bromodomain and extraterminal (BET) proteins are readers of histone acetylation labels, thereby affecting the transcription of genes and thus playing an important role in regulation the expression of pro-inflammatory cytokine expression (Belkina et al., 2013). Sánchez-Ventura et al. (2019) investigated the influence of BET inhibitor JQ1 in polarizing microglia on bone-marrow-derived macrophage in vitro and in vivo in SCI mice and showed that JQ1 promotes polarization of microglia toward the M2 phenotype, reducing the expression of pro-inflammatory cytokines IL-6, IL-1β, and TNF-α and increasing the expression of anti-inflammatory cytokines Arg1 and CD206.
It was shown that an early administration of minocycline, a known anti-inflammatory agent, inhibiting poly (ADP-ribose) polymerase-1 (PARP-1), which both promotes cell death and inhibits microglia activation and an inflammation in general, can reduce a degree of neuronal hyperexcitability for up to 4 weeks after SCI (Alano et al., 2006; Tan, 2009). In addition, minocycline-loaded polymeric nanoparticles (NPs) injected into the site of an SCI can selectively target activated microglial cells and modulate their phenotypes toward the anti-inflammatory one by inhibiting PARP-1 and matrix metalloproteinases 2 and 9, which improves the course of secondary traumatic processes in a murine SCI model. The treatment with minocycline-loaded NPs resulted in a reduced activation and decreased proliferation of microglia around the site of injury. As a result, the decreased number of cells with a phagocytic phenotype switched toward quiescent microglia with a low CD68 staining level. The treatment with these particles appeared effective for 15 post-injury days and was related to a prolonged anti-inflammatory stimulus associated with microglia activation (Papa et al., 2013). Another study demonstrated that the administration of minocycline-loaded NPs in an acute period following trauma in a murine SCI model could effectively modulate resident microglial cells from M1 to M2 phenotype, which reduced a proinflammatory response, restored the nervous tissue integrity, and improved behavior test scores for up to 63 post-injury days (Papa et al., 2016).
Although there are many studies aimed at elucidating mechanisms of microglia/macrophage modulation, their phenotype, and role in various pathologies, currently, no effective methods to modulate microglia toward a neuroprotective phenotype in order to stimulate neuroregeneration are employed in clinical practice. In addition, there is an urgent need to develop a highly specific panel of markers for resident microglia and macrophages migrating to a site of pathology, as well as complete elucidation of every external (specifically activating molecules secreted by surrounding cells) and internal factor (signaling pathways) affecting the modulation of their phenotype.
EA was in charge of the collection of data on the approaches to modulation of microglia/macrophages and compilation of a table. KK was in charge of the collection of data on the microglial signaling pathways. YM was in charge of the data collection about the microglia phenotypes and behavior of microglia/macrophages in the area of SCI and Figure 1 drawing. AR was in charge of the article content compilation and manuscript writing.
This work was supported by a grant from the Russian Science Foundation, No. 18-75-00043 to YM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was performed in accordance with the Program of Competitive Growth of the Kazan Federal University.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnsys.2019.00037/full#supplementary-material
Adams, R. A., Bauer, J., Flick, M. J., Sikorski, S. L., Nuriel, T., Lassmann, H., et al. (2007). The fibrin-derived γ377–395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J. Exp. Med. 204, 571–582. doi: 10.1084/jem.20061931
Ahmadian, M., Suh, J. M., Hah, N., Liddle, C., Atkins, A. R., Downes, M., et al. (2013). PPARγ signaling and metabolism: the good, the bad and the future. Nat. Med. 19, 557–566. doi: 10.1038/nm.3159
Ahn, H. J., Hernandez, C. M., Levenson, J. M., Lubin, F. D., Liou, H. C., and Sweat, J. D. (2008). c-Rel, an NF-κB family transcription factor, is required for hippocampal long-term synaptic plasticity and memory formation. Learn. Mem. 15, 539–549. doi: 10.1101/lm.866408
Akhmetzyanova, E., Mukhamedshina, Y. O., Zhuravleva, M., Galieva, L., Kostennikov, A., Garanina, E., et al. (2018). Transplantation of microglia in the area of spinal cord injury in an acute period increases tissue sparing, but not functional recovery. Front. Cell. Neurosci. 12:507. doi: 10.3389/fncel.2018.00507
Alano, C. C., Kauppinen, T. M., Valls, A. V., and Swanson, R. A. (2006). Minocycline inhibits poly (ADP-ribose) polymerase-1 at nanomolar concentrations. Proc. Natl. Acad. Sci. 103, 9685–9690. doi: 10.1073/pnas.0600554103
Beck, A., Penner, R., and Fleig, A. (2008). Lipopolysaccharide-induced down-regulation of Ca2+ release-activated Ca2+ currents (ICRAC) but not Ca2+-activated TRPM4-like currents (ICAN) in cultured mouse microglial cells. J. Physiol. 586, 427–439. doi: 10.1113/jphysiol.2007.145151
Beck, K. D., Nguyen, H. X., Galvan, M. D., Salazar, D. L., Woodruff, T. M., and Anderson, A. (2010). Quantitative analysis of cellular inflammation after traumatic spinal cord injury: evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain 133, 433–447. doi: 10.1093/brain/awp322
Belkina, A. C., Nikolajczyk, B. S., and Denis, G. V. (2013). BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J. Immunol. 190, 3670–3678. doi: 10.4049/jimmunol.1202838
Bellver-Landete, V., Bretheau, F., Mailhot, B., Vallières, N., Lessard, M., Janelle, M. E., et al. (2019). Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat. Commun. 10, 518–536. doi: 10.1038/s41467-019-08446-0
Bethea, J. R., and Dietrich, D. W. (2002). Targeting the host inflammatory response in traumatic spinal cord injury. Curr. Opin. Neurol. 15, 355–360. doi: 10.1097/00019052-200206000-00021
Bouhlel, M. A., Derudas, B., Rigamonti, E., Dièvart, R., Brozek, J., Haulon, S., et al. (2007). PPARγ activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 6, 137–143. doi: 10.1016/j.cmet.2007.06.010
Brambilla, R., Bracchi-Ricard, V., Hu, W. H., Frydel, B., Bramwell, A., Karmally, S., et al. (2005). Inhibition of astroglial nuclear factor κB reduces inflammation and improves functional recovery after spinal cord injury. J. Exp. Med. 202, 145–156. doi: 10.1084/jem.20041918
Broide, R. S., Redwine, J. M., Aftahi, N., Young, W., Bloom, F. E., and Winrow, C. J. (2007). Distribution of histone deacetylases 1–11 in the rat brain. J. Mol. Neurosci. 31, 47–58. doi: 10.1007/bf02686117
Bsibsi, M., Ravid, R., Gveric, D., and van Noort, J. M. (2002). Broad expression of toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 61, 1013–1021. doi: 10.1093/jnen/61.11.1013
Camporeale, A., and Poli, V. (2012). IL-6, IL-17 and STAT3: a holy trinity in auto-immunity? Front Biosci. 17:2306–2326.
Chen, C. H., Sung, C. S., Huang, S. Y., Feng, C. W., Hung, H. C., Yang, S. N., et al. (2016). The role of the PI3K/Akt/mTOR pathway in glial scar formation following spinal cord injury. Exp. Neurol. 278, 27–41. doi: 10.1016/j.expneurol.2016.01.023
Chen, H. C., Fong, T. H., Hsu, P. W., and Chiu, W. T. (2013). Multifaceted effects of rapamycin on functional recovery after spinal cord injury in rats through autophagy promotion, anti-inflammation, and neuroprotection. J. Surg. Res. 179, e203–e210. doi: 10.1016/j.jss.2012.02.023
Chen, X., Barozzi, I., Termanini, A., Prosperini, E., Recchiuti, A., Dalli, J., et al. (2012). Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci.U.S.A. 109, 2865–2874. doi: 10.1073/pnas.1121131109
Chen, Z., and Trapp, B. D. (2016). Microglia and neuroprotection. J. Neurochem. 136, 10–17. doi: 10.1111/jnc.13062
Cheng, B., Lin, Y., Kuang, M., Fang, S., Gu, Q., Xu, J., et al. (2015). Synthesis and anti-neuroinflammatory activity of lactone benzoyl hydrazine and 2-nitro-1-phenyl-1H-indole derivatives as p38α MAPK inhibitors. Chem. Biol. Drug Des. 86, 1121–1130. doi: 10.1111/cbdd.12581
Chinetti, G., Fruchart, J. C., and Staels, B. (2003). Peroxisome proliferator-activated receptors: new targets for the pharmacological modulation of macrophage gene expression and function. Curr. Opin. Lipidol. 14, 459–468.
Chinetti, G., Griglio, S., Antonucci, M., Torra, I. P., Delerive, P., Majd, Z., et al. (1998). Activation of proliferator-activated receptors α and γ induces apoptosis of human monocyte-derived macrophages. J. Biol. Chem. 273, 25573–25580. doi: 10.1074/jbc.273.40.25573
Cianciulli, A., Dragone, T., Calvello, R., Porro, C., Trotta, T., Lofrumento, D. D., et al. (2015). IL-10 plays a pivotal role in anti-inflammatory effects of resveratrol in activated microglia cells. Int. Immunopharmacol. 24, 369–376. doi: 10.1016/j.intimp.2014.12.035
Conta, A. C., and Stelzner, D. J. (2008). Immunomodulatory effect of the purine nucleoside inosine following spinal cord contusion injury in rat. Spinal Cord 46, 39. doi: 10.1038/sj.sc.3102057
Crosio, C., Valle, C., Casciati, A., Iaccarino, C., and Carrì, M. T. (2011). Astroglial inhibition of NF-κB does not ameliorate disease onset and progression in a mouse model for amyotrophic lateral sclerosis (ALS). PLoS One 6:17187. doi: 10.1371/journal.pone.0017187
Crown, E. D., Gwak, Y. S., Ye, Z., Johnson, K. M., and Hulsebosch, C. E. (2008). Activation of p38 MAP kinase is involved in central neuropathic pain following spinal cord injury. Exp. Neurol. 213, 257–267. doi: 10.1016/j.expneurol.2008.05.025
Dalpke, A. H., Schäfer, M. K., Frey, M., Zimmermann, S., Tebbe, J., Weihe, E., et al. (2002). Immunostimulatory CpG-DNA activates murine microglia. J. Immunol. 168, 4854–4863. doi: 10.4049/jimmunol.168.10.4854
David, S., and Kroner, A. (2011). Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 12, 388. doi: 10.1038/nrn3053
Detloff, M. R., Fisher, L. C., McGaughy, V., Longbrake, E. E., Popovich, P. G., and Basso, D. M. (2008). Remote activation of microglia and pro-inflammatory cytokines predict the onset and severity of below-level neuropathic pain after spinal cord injury in rats. Exp. Neurol. 212, 337–347. doi: 10.1016/j.expneurol.2008.04.009
Donnelly, D. J., and Popovich, P. G. (2008). Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp. Neurol. 209, 378–388. doi: 10.1016/j.expneurol.2007.06.009
Durafourt, B. A., Moore, C. S., Zammit, D. A., Johnson, T. A., Zaguia, F., Guiot, M. C., et al. (2012). Comparison of polarization properties of human adult microglia and blood-derived macrophages. GLIA 60, 717–727. doi: 10.1002/glia.22298
Eldahan, K. C., Cox, D. H., Gollihue, J. L., Patel, S. P., and Rabchevsky, A. G. (2018). Rapamycin exacerbates cardiovascular dysfunction after complete high-thoracic spinal cord injury. J. Neurotrauma. 35, 842–853. doi: 10.1089/neu.2017.5184
Emmetsberger, J., and Tsirka, S. E. (2012). Microglial inhibitory factor (MIF/TKP) mitigates secondary damage following spinal cord injury. Neurobiol. Dis. 47, 295–309. doi: 10.1016/j.nbd.2012.05.001
Erlich, S., Alexandrovich, A., Shohami, E., and Pinkas-Kramarski, R. (2007). Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol. Dis. 26, 86–93. doi: 10.1016/j.nbd.2006.12.003
Fan, H., Zhang, K., Shan, L., Kuang, F., Chen, K., Zhu, K., et al. (2016). Reactive astrocytes undergo M1 microglia/macrophages-induced necroptosis in spinal cord injury. Mol. Neurodegener. 11:14. doi: 10.1186/s13024-016-0081-8
Fan, Y., Xie, L., and Chung, C. Y. (2017). Signaling pathways controlling microglia chemotaxis. Mol. Cells 40, 163–168. doi: 10.14348/molcells.2017.0011
Farzi, A., Reichmann, F., and Holzer, P. (2015). The homeostatic role of neuropeptide Y in immune function and its impact on mood and behaviour. Acta Physiol. 213, 603–627. doi: 10.1111/apha.12445
Fenn, A. M., Hall, J. C., Gensel, J. C., Popovich, P. G., and Godbout, J. P. (2014). IL-4 signaling drives a unique arginase+/IL-1β+ microglia phenotype and recruits macrophages to the inflammatory CNS: consequences of age-related deficits in IL-4Rα after traumatic spinal cord injury. J. Neurosci. 34, 8904–8917. doi: 10.1523/jneurosci.1146-14.2014
Fernandez-Lizarbe, S., Pascual, M., and Guerri, C. (2009). Critical role of TLR4 response in the activation of microglia induced by ethanol. J. Immunol. 183, 4733–4744. doi: 10.4049/jimmunol.0803590
Ferreira, R., Santos, T., Cortes, L., Cochaud, S., Agasse, F., Silva, A. P., et al. (2012). Neuropeptide Y inhibits interleukin-1 beta-induced microglia motility. J. Neurochem. 120, 93–105. doi: 10.1111/j.1471-4159.2011.07541.x
Finnerup, N. B., Johannesen, I. L., Sindrup, S. H., Bach, F. W., and Jensen, T. S. (2001). Pain and dysesthesia in patients with spinal cord injury: a postal survey. Spinal cord 39, 256–262. doi: 10.1038/sj.sc.3101161
Frakes, A. E., Ferraiuolo, L., Haidet-Phillips, A. M., Schmelzer, L., Braun, L., Miranda, C. J., et al. (2014). Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron 81, 1009–1023. doi: 10.1016/j.neuron.2014.01.013
Franco, R., and Fernandez-Suarez, D. (2015). Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 131, 65–86. doi: 10.1016/j.pneurobio.2015.05.003
Francos-Quijorna, I., Amo-Aparicio, J., Martinez-Muriana, A., and López-Vales, R. (2016). IL-4 drives microglia and macrophages toward a phenotype conducive for tissue repair and functional recovery after spinal cord injury. GLIA 64, 2079–2092. doi: 10.1002/glia.23041
Gensel, J. C., and Zhang, B. (2015). Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res. 1619, 1–11. doi: 10.1016/j.brainres.2014.12.045
Gratchev, A., Kzhyshkowska, J., Kannookadan, S., Ochsenreiter, M., Popova, A., Yu, X., et al. (2008). Activation of a TGF-β-specific multistep gene expression program in mature macrophages requires glucocorticoid-mediated surface expression of TGF-β receptor II. J. Immunol. 180, 6553–6555.
Guerrero, A. R., Uchida, K., Nakajima, H., Watanabe, S., Nakamura, M., Johnson, W. E., et al. (2012). Blockade of interleukin-6 signaling inhibits the classic pathway and promotes an alternative pathway of macrophage activation after spinal cord injury in mice. J. Neuroinflammation. 9:40. doi: 10.1186/1742-2094-9-40
Guo, J. R., Wang, H., Jin, X. J., Jia, D. L., Zhou, X., and Tao, Q. (2017). Effect and mechanism of inhibition of PI3K/Akt/mTOR signal pathway on chronic neuropathic pain and spinal microglia in a rat model of chronic constriction injury. Oncotarget 8:52923. doi: 10.18632/oncotarget.17629
Han, J., Harris, R. A., and Zhang, X. M. (2017). An updated assessment of microglia depletion: current concepts and future directions. Mol. Brain 10:25. doi: 10.1186/s13041-017-0307-x
Hayakawa, K., Okazaki, R., Morioka, K., Nakamura, K., Tanaka, S., and Ogata, T. (2014). Lipopolysaccharide preconditioning facilitates M2 activation of resident microglia after spinal cord injury. J. Neurosci. Res. 92, 1647–1658. doi: 10.1002/jnr.23448
He, Y., She, H., Zhang, T., Xu, H., Cheng, L., Yepes, M., et al. (2018). p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1. J Cell Biol. 217, 315–328. doi: 10.1083/jcb.201701049
Hellwig, S., Heinrich, A., and Biber, K. (2013). The brain’s best friend: microglial neurotoxicity revisited. Front. Cell. Neurosci. 7:71. doi: 10.3389/fncel.2013.00071
Heneka, M. T., Sastre, M., Dumitrescu-Ozimek, L., Hanke, A., Dewachter, I., Kuiperi, C., et al. (2005). Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1–42 levels in APPV717I transgenic mice. Brain 128, 1442–1453. doi: 10.1093/brain/awh452
Hodes, G. E., Ménard, C., and Russo, S. J. (2016). Integrating interleukin-6 into depression diagnosis and treatment. Neurobiol. Stress 4, 15–22. doi: 10.1016/j.ynstr.2016.03.003
Huang, Z., Luo, Q., Guo, Y., Chen, J., Xiong, G., Peng, Y., et al. (2015). Mycobacterium tuberculosis-induced polarization of human macrophage orchestrates the formation and development of tuberculous granulomas in vitro. PLoS One 10:e0129744. doi: 10.1371/journal.pone.0129744
Im, Y. B., Jee, M. K., Choi, J. I., Cho, H. T., Kwon, O. H., and Kang, S. K. (2012). Molecular targeting of NOX4 for neuropathic pain after traumatic injury of the spinal cord. Cell Death Dis. 3, E426. doi: 10.1038/cddis.2012.168
Jin, X., and Yamashita, T. (2016). Microglia in central nervous system repair after injury. J. Biochem. 159, 491–496. doi: 10.1093/jb/mvw009
Kahn, M. A., Ellison, J. A., Speight, G. J., and De Vellis, J. (1995). CNTF regulation of astrogliosis and the activation of microglia in the developing rat central nervous system. Brain Res. 685, 55–67. doi: 10.1016/0006-8993(95)00411-i
Kannan, V., Brouwer, N., Hanisch, U. K., Regen, T., Eggen, B. J., and Boddeke, H. W. (2013). Histone deacetylase inhibitors suppress immune activation in primary mouse microglia. J. Neurosci. Res. 91, 1133–1142. doi: 10.1002/jnr.23221
Kanno, H., Ozawa, H., Sekiguchi, A., Yamaya, S., Tateda, S., Yahata, K., et al. (2012). The role of mTOR signaling pathway in spinal cord injury. Cell Cycle 11, 3175–3179. doi: 10.4161/cc.21262
Kigerl, K. A., Gensel, J. C., Ankeny, D. P., Alexander, J. K., Donnelly, D. J., and Popovich, P. G. (2009). Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 29, 13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009
Kim, B. W., More, S. V., Yun, Y. S., Ko, H. M., Kwak, J. H., Lee, H., et al. (2016). A novel synthetic compound MCAP suppresses LPS-induced murine microglial activation in vitro via inhibiting NF-kB and p38 MAPK pathways. Acta Pharmacol. Sin. 37, 334–343
Kim, C. F., and Moalem-Taylor, G. (2011). Interleukin-17 contributes to neuroinflammation and neuropathic pain following peripheral nerve injury in mice. J. Pain 12, 370–383. doi: 10.1016/j.jpain.2010.08.003
Kim, J. Y., Choi, G. S., Cho, Y. W., Cho, H., Hwang, S. J., and Ahn, S. H. (2013). Attenuation of spinal cord injury-induced astroglial and microglial activation by repetitive transcranial magnetic stimulation in rats. J. Korean Med. Sci. 28, 295–299. doi: 10.3346/jkms.2013.28.2.295
Koo, J. W., Russo, S. J., Ferguson, D., Nestler, E. J., and Duman, R. S. (2010). Nuclear factor-κB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc. Natl. Acad. Sci.U.S.A. 107, 2669–2674. doi: 10.1073/pnas.0910658107
Koscsó, B., Csóka, B., Kókai, E., Németh, Z. H., Pacher, P., Virág, L., et al. (2013). Adenosine augments IL-10-induced STAT3 signaling in M2c macrophages. J. Leukoc. Biol. 94, 1309–1315. doi: 10.1189/jlb.0113043
Kuboyama, T., Wahane, S., Huang, Y., Zhou, X., Wong, J. K., Koemeter-Cox, A., et al. (2017). HDAC3 inhibition ameliorates spinal cord injury by immunomodulation. Sci. Rep. 7:8641. doi: 10.1038/s41598-017-08535-4
Laflamme, N., Soucy, G., and Rivest, S. (2001). Circulating cell wall components derived from gram-negative, not gram-positive, bacteria cause a profound induction of the gene-encoding Toll-like receptor 2 in the CNS. J. Neurochem. 79, 648–657. doi: 10.1046/j.1471-4159.2001.00603.x
Lai, A. Y., and Todd, K. G. (2006). Microglia in cerebral ischemia: molecular actions and interactions. Can. J. Physiol. Pharmacol. 84, 49–59. doi: 10.1139/Y05-143
Lampron, A., Larochelle, A., Laflamme, N., Préfontaine, P., Plante, M. M., Sánchez, M. G., et al. (2015). Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 212, 481–495. doi: 10.1084/jem.20141656
Le Cabec, V., Carréno, S., Moisand, A., Bordier, C., and Maridonneau-Parini, I. (2002). Complement receptor 3 (CD11b/CD18) mediates type I and type II phagocytosis during nonopsonic and opsonic phagocytosis, respectively. J. Immunol. 169, 2003–2009. doi: 10.4049/jimmunol.169.4.2003
Lee, K. D., Chow, W. N., Sato-Bigbee, C., Graf, M. R., Graham, R. S., Colello, R. J., et al. (2009). FTY720 reduces inflammation and promotes functional recovery after spinal cord injury. J. Neurotrauma 26, 2335–2344. doi: 10.1089/neu.2008.0840
Liu, X., Wen, S., Yan, F., Liu, K., Liu, L., Wang, L., et al. (2018). Salidroside provides neuroprotection by modulating microglial polarization after cerebral ischemia. J. Neuroinflammation 15:39. doi: 10.1186/s12974-018-1081-0
Ma, L., Pan, X., Zhou, F., Liu, K., and Wang, L. (2018). Hyperforin protects against acute cerebral ischemic injury through inhibition of interleukin-17A-mediated microglial activation. Brain Res. 1678, 254–261. doi: 10.1016/j.brainres.2017.08.023
Ma, X., Reynolds, S. L., Baker, B. J., Li, X., Benveniste, E. N., and Qin, H. (2010). IL-17 enhancement of the IL-6 signaling cascade in astrocytes. J. Immunol. 184, 4898–4906. doi: 10.4049/jimmunol.1000142
Malagelada, C., Jin, Z. H., Jackson-Lewis, V., Przedborski, S., and Greene, L. A. (2010). Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 30, 1166–1175. doi: 10.1523/JNEUROSCI.3944-09.2010
Malvaez, M., McQuown, S. C., Rogge, G. A., Astarabadi, M., Jacques, V., Carreiro, S., et al. (2013). HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc. Natl. Acad. Sci. U.S.A. 110, 2647–2652. doi: 10.1073/pnas.1213364110
Malyshev, I., and Malyshev, Y. (2015). Current concept and update of the macrophage plasticity concept: intracellular mechanisms of reprogramming and M3 macrophage “switch” phenotype. Biomed Res. Int. 2015:341308. doi: 10.1155/2015/341308
Martin, E., El-Behi, M., Fontaine, B., and Delarasse, C. (2017). Analysis of microglia and monocyte-derived macrophages from the central nervous system by flow cytometry. J. Vis. Exp. 22:55781. doi: 10.3791/55781
Martinez, F. O., and Gordon, S. (2014). The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime Rep. 6:13. doi: 10.12703/P6-13
Martinez, F. O., Gordon, S., Locati, M., and Mantovani, A. (2006). Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J. Immunol. 177, 7303–7311. doi: 10.4049/jimmunol.177.10.7303
Martinez, F. O., Helming, L., Milde, R., Varin, A., Melgert, B. N., Draijer, C., et al. (2013). Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: similarities and differences. Blood 121, 57–69. doi: 10.1182/blood-2012-06-436212
McTigue, D. M. (2008). Potential therapeutic targets for PPAR after spinal cord injury. PPAR Res. 2008:517162. doi: 10.1155/2008/517162
McTigue, D. M., Tripathi, R., Wei, P., and Lash, A. T. (2007). The PPAR gamma agonist pioglitazone improves anatomical and locomotor recovery after rodent spinal cord injury. Exp. Neurol. 205, 396–406. doi: 10.1016/j.expneurol.2007.02.009
Menzies, F. M., Henriquez, F. L., Alexander, J., and Roberts, C. W. (2010). Sequential expression of macrophage anti-microbial/inflammatory and wound healing markers following innate, alternative and classical activation. Clin. Exp. Immunol. 160, 369–379. doi: 10.1111/j.1365-2249.2009.04086.x
Mullican, S. E., Gaddis, C. A., Alenghat, T., Nair, M. G., Giacomin, P. R., Everett, L. J., et al. (2011). Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev. 25, 2480–2488. doi: 10.1101/gad.175950.111
Nakajima, H., Uchida, K., Guerrero, A. R., Watanabe, S., Sugita, D., Takeura, N., et al. (2012). Transplantation of mesenchymal stem cells promotes an alternative pathway of macrophage activation and functional recovery after spinal cord injury. J. Neurotrauma. 29, 1614–1625. doi: 10.1089/neu.2011.2109
Nakajima, K., and Kohsaka, S. (2002). Neuroprotective Roles of Microglia in the Central Nervous system, in Microglia in the Regenerating and Degenerating Central Nervous System. Berlin: Springer, 188–208.
Nakajima, K., and Kohsaka, S. (2004). Microglia: neuroprotective and neurotrophic cells in the central nervous system. Curr. Drug Targets Cardiovasc. Haematol. Disord. 4, 65–84. doi: 10.2174/1568006043481284
Nakajima, K., Matsushita, Y., Tohyama, Y., Kohsaka, S., and Kurihara, T. (2006). Differential suppression of endotoxin-inducible inflammatory cytokines by nuclear factor kappa B (NFκB) inhibitor in rat microglia. Neurosci. Lett. 401, 199–202. doi: 10.1016/j.neulet.2006.03.014
Noort, A. R., Tak, P. P., and Tas, S. W. (2015). Non-canonical NF-κB signaling in rheumatoid arthritis: Dr Jekyll and Mr Hyde? Arthritis Res. Ther. 17:15. doi: 10.1186/s13075-015-0527-3
Norimatsu, Y., Ohmori, T., Kimura, A., Madoiwa, S., Mimuro, J., Seichi, A., et al. (2012). FTY720 improves functional recovery after spinal cord injury by primarily nonimmunomodulatory mechanisms. Am. J. Pathol. 180, 1625–1635. doi: 10.1016/j.ajpath.2011.12.012
Ohtake, Y., Park, D., Abdul-Muneer, P. M., Li, H., Xu, B., Sharma, K., et al. (2014). The effect of systemic PTEN antagonist peptides on axon growth and functional recovery after spinal cord injury. Biomaterials 35, 4610–4626. doi: 10.1016/j.biomaterials.2014.02.037
Oliva, A. A., Kang, Y., Sanchez-Molano, J., Furones, C., and Atkins, C. M. (2012). STAT3 signaling after traumatic brain injury. J. Neurochem. 120, 710–720. doi: 10.1111/j.1471-4159.2011.07610.x
Olson, J. K., and Miller, S. D. (2004). Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 173, 3916–3924. doi: 10.4049/jimmunol.173.6.3916
Orihuela, R., McPherson, C. A., and Harry, G. J. (2016). Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 173, 649–665. doi: 10.1111/bph.13139
O’Riordan, K. J., Huang, I. C., Pizzi, M., Spano, P., Boroni, F., Egli, R., et al. (2006). Regulation of nuclear factor κB in the hippocampus by group I metabotropic glutamate receptors. J. Neurosci. 26, 4870–4879. doi: 10.1523/JNEUROSCI.4527-05.2006
Pan, J., Jin, J. L., Ge, H. M., Yin, K. L., Chen, X., Han, L. J., et al. (2015). Malibatol A regulates microglia M1/M2 polarization in experimental stroke in a PPARγ-dependent manner. J. Neuroinflammation 12:51. doi: 10.1186/s12974-015-0270-3
Pan, W., Yu, C., Hsuchou, H., and Kastin, A. J. (2010). The role of cerebral vascular NFκB in LPS-induced inflammation: differential regulation of efflux transporter and transporting cytokine receptors. Cell. Physiol. Biochem. 25, 623–630. doi: 10.1159/000315081
Papa, S., Caron, I., Erba, E., Panini, N., De Paola, M., Mariani, A., et al. (2016). Early modulation of pro-inflammatory microglia by minocycline loaded nanoparticles confers long lasting protection after spinal cord injury. Biomaterials 75, 13–24. doi: 10.1016/j.biomaterials.2015.10.015
Papa, S., Rossi, F., Ferrari, R., Mariani, A., De Paola, M., Caron, I., et al. (2013). Selective nanovector mediated treatment of activated proinflammatory microglia/macrophages in spinal cord injury. ACS Nano 7, 9881–9895. doi: 10.1021/nn4036014
Park, S. W., Yi, J. H., Miranpuri, G., Satriotomo, I., Bowen, K., Resnick, D. K., et al. (2007). Thiazolidinedione class of peroxisome proliferator-activated receptor γ agonists prevents neuronal damage, motor dysfunction, myelin loss, neuropathic pain, and inflammation after spinal cord injury in adult rats. J. Pharmacol. Exp. Ther. 320, 1002–1012. doi: 10.1124/jpet.106.113472
Peferoen, L. A., Vogel, D. Y., Ummenthum, K., Breur, M., Heijnen, P. D., Gerritsen, W. H., et al. (2015). Activation status of human microglia is dependent on lesion formation stage and remyelination in multiple sclerosis. J. Neuropathol. Exp. Neurol. 74, 48–63. doi: 10.1097/NEN.0000000000000149
Pei, Z., Pang, H., Qian, L. I., Yang, S., Wang, T., Zhang, W., et al. (2007). MAC1 mediates LPS-induced production of superoxide by microglia: the role of pattern recognition receptors in dopaminergic neurotoxicity. GLIA 55, 1362–1373. doi: 10.1002/glia.20545
Peress, N. S., Fleit, H. B., Perillo, E., Kuljis, R., and Pezzullo, C. (1993). Identification of FcγRI, II and III on normal human brain ramified microglia and on microglia in senile plaques in Alzheimer’s disease. J. Neuroimmunol. 48, 71–79. doi: 10.1016/0165-5728(93)90060-c
Persson, M., Brantefjord, M., Hansson, E., and Rönnbäck, L. (2005). Lipopolysaccharide increases microglial GLT-1 expression and glutamate uptake capacity in vitro by a mechanism dependent on TNF-α. Glia 51, 111–120. doi: 10.1002/glia.20191
Ponomarev, E. D., Veremeyko, T., Barteneva, N., Krichevsky, A. M., and Weiner, H. L. (2011). MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-α–PU. Nat. Med. 17, 64. doi: 10.1038/nm.2266
Popiolek-Barczyk, K., and Mika, J. (2016). Targeting the microglial signaling pathways: new insights in the modulation of neuropathic pain. Curr. Med. Chem. 23, 2908–2928. doi: 10.2174/0929867323666160607120124
Qin, C., Fan, W. H., Liu, Q., Shang, K., Murugan, M., Wu, L. J., et al. (2017). Fingolimod protects against ischemic white matter damage by modulating microglia toward M2 polarization via STAT3 pathway. Stroke 48, 3336–3346. doi: 10.1161/STROKEAHA.117.018505
Qu, W. S., Tian, D. S., Guo, Z. B., Fang, J., Zhang, Q., Yu, Z. Y., et al. (2012). Inhibition of EGFR/MAPK signaling reduces microglial inflammatory response and the associated secondary damage in rats after spinal cord injury. J. Neuroinflammation 9:178. doi: 10.1186/1742-2094-9-178
Ravikumar, B., Vacher, C., Berger, Z., Davies, J. E., Luo, S., Oroz, L. G., et al. (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595. doi: 10.1038/ng1362
Redondo-Castro, E., Hernández, J., Mahy, N., and Navarro, X. (2013). Phagocytic microglial phenotype induced by glibenclamide improves functional recovery but worsens hyperalgesia after spinal cord injury in adult rats. Eur. J. Neurosci. 38, 3786–3798. doi: 10.1111/ejn.12382
Rocha, S. M., Cristovão, A. C., Campos, F. L., Fonseca, C. P., and Baltazar, G. (2012). Astrocyte-derived GDNF is a potent inhibitor of microglial activation. Neurobiol. Dis. 47, 407–415. doi: 10.1016/j.nbd.2012.04.014
Rojewska, E., Popiolek-Barczyk, K., Jurga, A. M., Makuch, W., Przewlocka, B., and Mika, J. (2014). Involvement of pro-and antinociceptive factors in minocycline analgesia in rat neuropathic pain model. J. Neuroimmunol. 277, 57–66. doi: 10.1016/j.jneuroim.2014.09.020
Rostam, H. M., Reynolds, P. M., Alexander, M. R., Gadegaard, N., and Ghaemmaghami, A. M. (2017). Image based machine learning for identification of macrophage subsets. Sci. Rep. 7:3521. doi: 10.1038/s41598-017-03780-z
Russo, C. D., Lisi, L., Tringali, G., and Navarra, P. (2009). Involvement of mTOR kinase in cytokine-dependent microglial activation and cell proliferation. Biochem. Pharmacol. 78, 1242–1251. doi: 10.1016/j.bcp.2009.06.097
Sánchez-Ventura, J., Amo-Aparicio, J., Navarro, X., and Penas, C. (2019). BET protein inhibition regulates cytokine production and promotes neuroprotection after spinal cord injury. J. Neuroinflammation 16:124. doi: 10.1186/s12974-019-1511-7
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Schmelzle, T., and Hall, M. N. (2000). TOR, a central controller of cell growth. Cell 103, 253–262. doi: 10.1016/s0092-8674(00)00117-3
Shaked, I., Tchoresh, D., Gersner, R., Meiri, G., Mordechai, S., Xiao, X., et al. (2005). Protective autoimmunity: interferon-γ enables microglia to remove glutamate without evoking inflammatory mediators. J. Neurochem. 92, 997–1009. doi: 10.1111/j.1471-4159.2004.02954.x
Shechter, R., and Schwartz, M. (2013). Harnessing monocyte-derived macrophages to control central nervous system pathologies: no longer ‘if’ but ‘how’. J. Pathol. 229, 332–346. doi: 10.1002/path.4106
Shih, R. H., Wang, C. Y., and Yang, C. M. (2015). NF-kappaB signaling pathways in neurological inflammation: a mini review. Front. Mol. Neurosci. 8:77. doi: 10.3389/fnmol.2015.00077
Sindrilaru, A., Peters, T., Wieschalka, S., Baican, C., Baican, A., Peter, H., et al. (2011). An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Invest. 121, 985–997. doi: 10.1172/JCI44490
Song, M., Jin, J., Lim, J. E., Kou, J., Pattanayak, A., Rehman, J. A., et al. (2011). TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J. Neuroinflammation 8:92. doi: 10.1186/1742-2094-8-92
Song, Y., Xue, H., Liu, T. T., Liu, J. M., and Chen, D. (2015). Rapamycin plays a neuroprotective effect after spinal cord injury via anti-inflammatory effects. J. Biochem. Mol. Toxicol. 29, 29–34. doi: 10.1002/jbt.21603
Stout, R. D., and Suttles, J. (2004). Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J. Leukoc. Biol. 76, 509–513. doi: 10.1189/jlb.0504272
Taetzsch, T., Levesque, S., McGraw, C., Brookins, S., Luqa, R., Bonini, M. G., et al. (2015). Redox regulation of NF-κB p50 and M1 polarization in microglia. GLIA 63, 423–440. doi: 10.1002/glia.22762
Tan, A. M. (2009). Early microglial inhibition preemptively mitigates chronic pain development after experimental spinal cord injury. J. Rehab. Res. Dev. 46, 123–133.
Taves, S., Berta, T., Liu, D. L., Gan, S., Chen, G., Kim, Y. H., et al. (2016). Spinal inhibition of p38 MAP kinase reduces inflammatory and neuropathic pain in male but not female mice: sex-dependent microglial signaling in the spinal cord. Brain Behav. Immun. 55, 70–81. doi: 10.1016/j.bbi.2015.10.006
Taylor, R. A., Chang, C. F., Goods, B. A., Hammond, M. D., Mac Grory, B., Ai, Y., et al. (2017). TGF-β1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J. Clin. Invest. 127, 280–292. doi: 10.1172/JCI88647
Varnum, M. M., and Ikezu, T. (2012). The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch. Immunol. Ther. Exp. 60, 251–266. doi: 10.1007/s00005-012-0181-2
Vergadi, E., Ieronymaki, E., Lyroni, K., Vaporidi, K., and Tsatsanis, C. (2017). Akt signaling pathway in macrophage activation and M1/M2 polarization. J. Immunol. 198, 1006–1014. doi: 10.4049/jimmunol.1601515
Walker, D. G., and Lue, L. F. (2015). Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res. Ther. 7:56. doi: 10.1186/s13195-015-0139-9
Wang, G., Shi, Y., Jiang, X., Leak, R. K., Hu, X., Wu, Y., et al. (2015). HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3β/PTEN/Akt axis. Proc. Natl. Acad. Sci. 112, 2853–2858. doi: 10.1073/pnas.1501441112
Wang, J., Wang, J., Lu, P., Cai, Y., Wang, Y., Hong, L., et al. (2015). Local delivery of FTY720 in PCL membrane improves SCI functionbal recovery by reducing reactive astrogliosis. Biomaterials 62, 76–87. doi: 10.1016/j.biomaterials.2015.04.060
Wang, N., Liang, H., and Zen, K. (2014). Molecular mechanisms that influence the macrophage M1–M2 polarization balance. Front. Immunol. 5:614. doi: 10.3389/fimmu.2014.00614
Wang, P., He, Y., Li, D., Han, R., Liu, G., Kong, D., et al. (2016). Class I PI3K inhibitor ZSTK474 mediates a shift in microglial/macrophage phenotype and inhibits inflammatory response in mice with cerebral ischemia/reperfusion injury. J. Neuroinflammation 13:192. doi: 10.3389/fimmu.2014.00614
Wang, Y. P., Wu, Y., Li, L. Y., Zheng, J., Liu, R. G., Zhou, J. P., et al. (2011). Aspirin-triggered lipoxin A 4 attenuates LPS-induced pro-inflammatory responses by inhibiting activation of NF-κB and MAPKs in BV-2 microglial cells. J. Neuroinflammation 8:95. doi: 10.1186/s12974-016-0660-1
Weisser, S. B., McLarren, K. W., Kuroda, E., and Sly, L. M. (2013). Generation and characterization of murine alternatively activated macrophages. Basic Cell Cult. Protoc. 2013, 225–239. doi: 10.1007/978-1-62703-128-8_14
Wen, L., You, W., Wang, H., Meng, Y., Feng, J., and Yang, X. (2018). Polarization of microglia to the M2 phenotype in a peroxisome proliferator-activated receptor gamma–dependent manner attenuates axonal injury induced by traumatic brain injury in mice. J. Neurotrauma 35, 2330–2340. doi: 10.1089/neu.2017.5540
Wilcock, D. M. (2014). Neuroinflammatory phenotypes and their roles in Alzheimer’s disease. Neurodegener. Dis. 13, 183–185. doi: 10.1007/s11307-017-1099-1
Willemen, H. L., Huo, X. J., Mao-Ying, Q. L., Zijlstra, J., Heijnen, C. J., and Kavelaars, A. (2012). MicroRNA-124 as a novel treatment for persistent hyperalgesia. J. Neuroinflammation 9:143. doi: 10.1186/1742-2094-9-143
Wolf, S. A., Boddeke, H. W. G. M., and Kettenmann, H. (2017). Microglia in physiology and disease. Annu. Rev. Physiol. 79, 619–643. doi: 10.1146/annurev-physiol-022516-034406
Wu, J., Stoica, B. A., Dinizo, M., Pajoohesh-Ganji, A., Piao, C., and Faden, A. I. (2012). Delayed cell cycle pathway modulation facilitates recovery after spinal cord injury. Cell Cycle 11, 1782–1795. doi: 10.4161/cc.20153
Wu, S. Y., and Watabe, K. (2017). The roles of microglia/macrophages in tumor progression of brain cancer and metastatic disease. Front. Biosci. 22:1805. doi: 10.2741/4573
Xia, M., Zhao, Q., Zhang, H., Chen, Y., Yuan, Z., Xu, Y., et al. (2017). Proteomic analysis of HDAC3 selective inhibitor in the regulation of inflammatory response of primary microglia. Neural Plast 2017:6237351. doi: 10.1155/2017/6237351
Yao, L., Kan, E. M., Lu, J., Hao, A., Dheen, S. T., Kaur, C., et al. (2013). Toll-like receptor 4 mediates microglial activation and production of inflammatory mediators in neonatal rat brain following hypoxia: role of TLR4 in hypoxic microglia. J. Neuroinflammation 10:23. doi: 10.1186/1742-2094-10-23
Yin, H., Shen, L., Xu, C., and Liu, J. (2018). Lentivirus-mediated overexpression of miR-29a promotes axonal regeneration and functional recovery in experimental spinal cord injury via PI3K/Akt/mTOR pathway. Neurochem. Res. 43, 2038–2046. doi: 10.1007/s11064-018-2625-5
Zhang, F., Zhong, R., Li, S., Fu, Z., Cheng, C., Cai, H., et al. (2017). Acute hypoxia induced an imbalanced M1/M2 activation of microglia through NF-κB signaling in Alzheimer’s disease mice and wild-type littermates. Front. Aging Neurosci. 9:282. doi: 10.3389/fnagi.2017.00282
Zhang, L., Alizadeh, D., Van Handel, M., Kortylewski, M., Yu, H., and Badie, B. (2009). Stat3 inhibition activates tumor macrophages and abrogates glioma growth in mice. GLIA 57, 1458–1467. doi: 10.1002/glia.20863
Zhang, L., Liu, W., Alizadeh, D., Zhao, D., Farrukh, O., Lin, J., et al. (2011). S100B attenuates microglia activation in gliomas: possible role of STAT3 pathway. GLIA 59, 486–498. doi: 10.1002/glia.21118
Zhang, Y. K., Liu, J. T., Peng, Z. W., Fan, H., Yao, A. H., Cheng, P., et al. (2013). Different TLR4 expression and microglia/macrophage activation induced by hemorrhage in the rat spinal cord after compressive injury. J. Neuroinflammation 10:112. doi: 10.1186/1742-2094-10-112
Zhao, P., Waxman, S. G., and Hains, B. C. (2007). Modulation of thalamic nociceptive processing after spinal cord injury through remote activation of thalamic microglia by cysteine–cysteine chemokine ligand 21. J. Neurosci. 27, 8893–8902. doi: 10.1523/jneurosci.2209-07.2007
Zhuravleva, M., Rizvanov, A., and Mukhamedshina, Y. (2016). Effect of GDNF on morphology, proliferation, and phagocytic activity of rat neonatal cortex isolated microglia. Bionanoscience 6, 379–383. doi: 10.1007/s12668-016-0247-4
Zolezzi, J. M., Santos, M. J., Bastías-Candia, S., Pinto, C., Godoy, J. A., and Inestrosa, N. C. (2017). PPARs in the central nervous system: roles in neurodegeneration and neuroinflammation. Biol. Rev. 92, 2046–2069. doi: 10.1111/brv.12320
Zong, S., Zeng, G., Fang, Y., Peng, J., Tao, Y., Li, K., et al. (2014). The role of IL-17 promotes spinal cord neuroinflammation via activation of the transcription factor STAT3 after spinal cord injury in the rat. Mediators Inflamm. 2014:786947. doi: 10.1155/2014/786947
Keywords: microglia, phenotypes, modulation, spinal cord injury, neuroregeneration
Citation: Akhmetzyanova E, Kletenkov K, Mukhamedshina Y and Rizvanov A (2019) Different Approaches to Modulation of Microglia Phenotypes After Spinal Cord Injury. Front. Syst. Neurosci. 13:37. doi: 10.3389/fnsys.2019.00037
Received: 20 April 2019; Accepted: 29 July 2019;
Published: 27 August 2019.
Edited by:
Preston E. Garraghty, Indiana University Bloomington, United StatesReviewed by:
John Gensel, University of Kentucky, United StatesCopyright © 2019 Akhmetzyanova, Kletenkov, Mukhamedshina and Rizvanov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yana Mukhamedshina, eWFuYS5rLXotbkBtYWlsLnJ1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.