
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurosci. , 24 September 2018
Sec. Neurodegeneration
Volume 12 - 2018 | https://doi.org/10.3389/fnins.2018.00668
 Xin Yi Choo1,2,3†
Xin Yi Choo1,2,3† Jeffrey R. Liddell1,3†
Jeffrey R. Liddell1,3† Mikko T. Huuskonen5†
Mikko T. Huuskonen5† Alexandra Grubman1,2,4Diane Moujalled1
Alexandra Grubman1,2,4Diane Moujalled1 Jessica Roberts1
Jessica Roberts1 Kai Kysenius3,4Lauren Patten1
Kai Kysenius3,4Lauren Patten1 Hazel Quek6
Hazel Quek6 Lotta E. Oikari6Clare Duncan1Simon A. James4,7
Lotta E. Oikari6Clare Duncan1Simon A. James4,7 Lachlan E. McInnes8
Lachlan E. McInnes8 David J. Hayne8Paul S. Donnelly8
David J. Hayne8Paul S. Donnelly8 Eveliina Pollari5
Eveliina Pollari5 Suvi Vähätalo5Katarína Lejavová5Mikko I. Kettunen5
Suvi Vähätalo5Katarína Lejavová5Mikko I. Kettunen5 Tarja Malm5
Tarja Malm5 Jari Koistinaho5,9
Jari Koistinaho5,9 Anthony R. White1,6*†
Anthony R. White1,6*† Katja M. Kanninen1,5*†
Katja M. Kanninen1,5*†Background: Neuroinflammation and biometal dyshomeostasis are key pathological features of several neurodegenerative diseases, including Alzheimer’s disease (AD). Inflammation and biometals are linked at the molecular level through regulation of metal buffering proteins such as the metallothioneins. Even though the molecular connections between metals and inflammation have been demonstrated, little information exists on the effect of copper modulation on brain inflammation.
Methods: We demonstrate the immunomodulatory potential of the copper bis(thiosemicarbazone) complex CuII(atsm) in an neuroinflammatory model in vivo and describe its anti-inflammatory effects on microglia and astrocytes in vitro.
Results: By using a sophisticated in vivo magnetic resonance imaging (MRI) approach, we report the efficacy of CuII(atsm) in reducing acute cerebrovascular inflammation caused by peripheral administration of bacterial lipopolysaccharide (LPS). CuII(atsm) also induced anti-inflammatory outcomes in primary microglia [significant reductions in nitric oxide (NO), monocyte chemoattractant protein 1 (MCP-1), and tumor necrosis factor (TNF)] and astrocytes [significantly reduced NO, MCP-1, and interleukin 6 (IL-6)] in vitro. These anti-inflammatory actions were associated with increased cellular copper levels and increased the neuroprotective protein metallothionein-1 (MT1) in microglia and astrocytes.
Conclusion: The beneficial effects of CuII(atsm) on the neuroimmune system suggest copper complexes are potential therapeutics for the treatment of neuroinflammatory conditions.
Chronic neuroinflammation, manifested by increased activation of glial cells and excessive production of pro-inflammatory mediators, is one of the hallmark pathological features of several neurodegenerative diseases, including AD, multiple sclerosis, PD, and ALS. During neurodegeneration, chronic stimulation results in unresolved inflammation and creates a neurotoxic environment that exacerbates degeneration in the brain. The view that the brain is an immune-privileged site has been challenged by studies demonstrating the presence of bidirectional crosstalk between the CNS and the immune system (Louveau et al., 2015). It is now clear that during peripheral immune activation, the innate immune system signals to the brain, thereby inducing pro-inflammatory cytokine production and global gene expression changes in the brain (Thomson et al., 2014), as well as defects in spatial learning and memory (Jurgens et al., 2012; Eimerbrink et al., 2015; Elmore et al., 2015). Therapeutic approaches targeted to the modulation of both central and peripheral inflammation during neurodegeneration have shown beneficial outcomes in animal models and in human patients (Heneka et al., 2015; Venigalla et al., 2016).
Copper is required for many critical neuronal processes and functions, including differentiation, cell signaling, and synaptic transmission (D’Ambrosi and Rossi, 2015; Maureira et al., 2015; Hatori et al., 2016). Alterations to biometal homeostasis are implicated in several neurodegenerative diseases. For example, in PD, copper levels are diminished in the affected substantia nigra brain region (Genoud et al., 2017). In AD, X-ray fluorescence (XRF) analyses have shown that amyloid plaques are enriched for copper and zinc (Miller et al., 2006), yet several studies have demonstrated that many important regions such as the neocortex are in fact copper deficient in AD patient brains (Schrag et al., 2011; Rembach et al., 2013; Graham et al., 2014). Therefore, both the sequestration of copper in plaques and loss of copper-dependent functions have been proposed to contribute to AD pathology. Given the importance of biometal homeostasis on neuronal function, therapeutic targeting of abnormal biometal balance has been clinically demonstrated to be a potential therapeutic approach for neurodegeneration (Faux et al., 2010).
Modulating brain copper homeostasis is beneficial in animal models of neurodegenerative diseases. Copper bis(thiosemicarbazones) (btscs) are stable, lipophilic neutral copper(II) complexes that are capable of crossing cell membranes and the blood–brain barrier (Donnelly et al., 2008; Torres et al., 2016). Changes to the backbone of the ligand change the CuII/I reduction potential and lead to changes in the propensity of Cu-btscs to release Cu(I) inside cells (Donnelly et al., 2012). Metal delivery and metal redistribution with these complexes reduced disease-associated cognitive decline and improved learning and memory in AD model mice (Crouch et al., 2009), preserved motor function and survival in ALS mice (Soon et al., 2011; McAllum et al., 2013; Roberts et al., 2014), and improved both motor and cognitive functions in mouse models of PD (Hung et al., 2012). The copper bis(thiosemicarbazone) CuII(atsm) is currently under clinical investigation in ALS and PD patients (NCT02870634, NCT03136809, and NCT03204929). In addition, it has been shown that other copper modulating compounds also improve learning and memory in AD model mice (Malm et al., 2007; Adlard et al., 2011), while dietary treatment of AD model mice with copper (Bayer et al., 2003), and crossing AD model mice with ATP7b mutant mice (Phinney et al., 2003), which display elevated copper levels, both result in extended lifetime and reduced production of amyloid beta (Aβ). The ability of the metal complexes CuII(gtsm) and CuII(atsm) to improve cognition in AD and motor performance in ALS has been attributed to reduction of Aβ oligomers and tau phosphorylation (Crouch et al., 2009), as well as the ability to improve the copper content of the metallo-protein Cu/Zn superoxide dismutase (SOD1) (Roberts et al., 2014) and reduce nitrosative damage (Soon et al., 2011). However, the observed reductions in the activation status of microglia and astrocytes in ALS and stroke mice treated with the copper complex CuII(atsm) (Soon et al., 2011; Roberts et al., 2014; Huuskonen et al., 2017) suggest a potential anti-inflammatory effect of copper delivery in vivo, that has not been fully explored.
Inflammation regulates the expression of critical metal ion transport proteins in the brain (Terwel et al., 2011; Pelizzoni et al., 2013; Raha et al., 2013; Urrutia et al., 2013; Zhang et al., 2014; Michaelis et al., 2015), and several inflammatory proteins and transcription factors are dependent on metal ions for their activity. Inflammation also regulates metal buffering proteins such as the metallothioneins (Liu et al., 2014; Zhou et al., 2014; Luo et al., 2015). Metallothioneins are present in elevated levels in neurodegenerative diseases including AD and PD (Duguid et al., 1989; Adlard et al., 1998; Carrasco et al., 2006; Hidalgo et al., 2006; Michael et al., 2011) influence inflammatory responses in neurodegenerative diseases (Potter et al., 2007; Chung et al., 2009; Kim et al., 2012), and their deficiency is reported to enhance inflammatory cell responses (Potter et al., 2007). Even though the molecular links between metals and inflammation have been demonstrated, little information exists on the effect of copper modulation on brain inflammation.
In this study, we investigated the immunomodulatory potential of the neuroprotective copper agent CuII(atsm) in a neuroinflammatory model in vivo and examined the effects of the copper complex on microglia and astrocytes in vitro. We demonstrate that CuII(atsm) reduces brain inflammation caused by peripheral administration of bacterial LPS. We demonstrate that CuII(atsm) has cell-specific inflammation-reducing actions in vitro, and report that its anti-inflammatory actions are associated with increased expression of Mt1, encoding the neuroprotective protein metallothionein-1 (MT1). The observed anti-inflammatory actions could account for some of the previously observed neurotherapeutic effects of copper delivery.
Four-month-old C57BL/6J male mice (n = 5–6/group) were used in this study. Male mice were used, because previous studies suggest that brain copper levels depend more on strain and age rather than sex (Maynard et al., 2002). Animals were housed in normal conditions with 12-h light/dark cycle and water and food available ad libitum. Animal handling and experimentation were performed in accordance with the national regulation of the usage and welfare of laboratory animals and approved by the Animal Experiment Committee in State Provincial Office of Southern Finland.
Lipopolysaccharide from Escherichia coli (clone 055:B5, Sigma-Aldrich, St. Louis, MO, United States) was dissolved to 1 mg/ml in sterile PBS and intraperitoneally administered to 4-month-old mice at a dose of 1 mg/kg. Two hours after LPS administration, mice were treated with 60 mg/kg CuII(atsm) dissolved in SSV solution containing 0.9% (w/v) NaCl, 0.5% (w/v) sodium carboxymethylcellulose, 0.5% (v/v) benzyl alcohol, and 0.4% (v/v) Tween 80, or SSV alone by oral gavage. CuII(atsm) was prepared according to published procedures (Gingras et al., 1962). 23.5 h after LPS administration, mice were injected with MPIO-VCAM-1 (conjugate) that was prepared according to Montagne et al. (2012) by coating MyOne Tosyl Activated Dynabeads (Thermo Fisher Scientific, Waltham, MA, United States) with 0.1 M sodium borate buffer (pH 9.5) and conjugating with VCAM-1 antibody (40 μg/1 mg of beads, BD Biosciences, San Jose, CA, United States) in 3 M ammonium sulfate at 37°C for 48 h . The conjugate was blocked with 0.5% (w/v) bovine serum albumin (BSA) in PBS-Tween for 24 h at 37°C and stored in 0.1% (w/v) BSA in PBS-Tween at 4°C. Each animal received an intravenous dose of the MPIO-VCAM-1 conjugate containing 1.0 mg/kg of Fe 30 min prior to imaging.
Magnetic resonance imaging was performed using a Biospec 9.4 T/12 cm system (Bruker, Germany) using a volume coil transmit/surface coil receive pair (Rapid Biomed, Germany) to produce 3D T2∗ weighted images (spatial resolution of 78 μm × 78 μm × 78 μm, TE/TR 12 ms/200 ms, flip angle of 25°, acquisition time 52 min). Four consecutive slices were used to construct minimum intensity projections. Systematic variation in signal intensity was removed prior to analysis by normalization to overall brain signal intensity. This approach allowed reliable detection of hypointense regions due to contrast agent. Data are the mean percentages of VCAM-1 signal area in nine consecutive minimum intensity projections per animal relative to control, beginning from the first slice where the hippocampus is visible. The VCAM-1 signal area was determined in oval regions of interest encompassing the coronal brain section within each image, and images were manually thresholded by a blinded investigator.
For neonatal astrocyte-enriched cultures, newborn C57Bl/6J mice were decapitated, and the brains removed and placed into ice-cold preparation buffer (containing 68 mM NaCl, 2.7 mM KCl, 110 μM KH2PO4, 84.5 μM Na2HPO4, 29 mM sucrose, 2.8 mM glucose, 20 U/mL penicillin, and 34.4 pM streptomycin). Brains were sequentially passed through 250 and 135 μm gauze and centrifuged at 500 ×g for 5 min. Cell pellets were resuspended in growth medium consisting of high glucose DMEM (Life Technologies, Thermo Fischer Scientific) containing 10% (v/v) FBS (Bovogen, VIC, Australia), 20 U/mL penicillin, and 34.4 pM streptomycin (Life Technologies) and plated at a density of 150,000 cells/cm2. Cells were maintained at 37°C with 10% CO2. The growth medium was replaced every 7 days, and experiments were performed after 14 DIV.
Astrocyte cultures were prepared as described previously (Pihlaja et al., 2011) with some modifications. Briefly, cortices were isolated from 6- to 8-week-old mice, the tissue suspended in Hank’s Balanced Salt Solution (Lonza, Allendale, NJ, United States) and centrifuged at 500 ×g for 5 min. After the addition of 0.25% trypsin-EDTA (Life Technologies, Great Island, NY, United States), the suspension was incubated at 37°C for 30 min with occasional shaking. After the addition of fresh culture medium to neutralize the effect of trypsin, the suspension was centrifuged at 500 ×g for 5 min. The cell suspension was added on top of Percoll (Sigma) and centrifuged. The layer of glial cells was washed once with fresh culture medium. The cells were plated onto poly-L-lysine pre-coated flask in DMEM/F12 media containing 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin–streptomycin, and G5 supplement (Life Technologies, Great Island, NY, United States). Microglia were removed by shaking the plates before the experiments. Cells were used for experiments at passages 4–8.
Microglia were harvested from astrocyte-enriched cultures plated at 65,000 cells/cm2 in six-well plates or T175 flasks after 18 DIV. Adherent glial cells were washed in PBS and incubated at 37°C with 1:5 dilution of 10× Trypsin-EDTA (Sigma-Aldrich) in high glucose DMEM until astrocyte detachment was complete (∼25 min). Astrocyte-conditioned media was added to adherent microglia in 6 well plates. Microglia in T175 flasks were scraped, counted and re-plated at a density of 65,000 cells/cm2. The media was changed to IMDM phenol red free media (Life Technologies) containing 10% (v/v) FBS, 20 U/mL penicillin, 34.4 pM streptomycin, and 2 mM L-glutamine (Life Technologies) the following day and cells were allowed to rest for a further 2 days prior to experiments.
Cells were stimulated with 15 ng/ml IFNγ and 10 ng/ml TNFα (Peprotech, Stockholm, Sweden), or 20 μg/ml LPS (Sigma-Aldrich) for 24–48 h, as indicated. CuII(atsm) (10 mM), prepared fresh in DMSO, was diluted in cell culture media and added to cells at concentrations from 0.2 to 2 μM for the indicated times. Minocycline (Sigma Aldrich) was used at 20 μM. Control groups were treated with the vehicle solutions of the treatment groups.
Cell viability was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Amresco Inc., OH, United States). The MTT assay was performed by addition of 0.5 mg/ml (final concentration) MTT to the cell media. Cells were incubated at 37°C until blue crystals were visible by eye (approximately 30–60 min). The media was removed and replaced with 100 μl of DMSO per well to dissolve the crystals. One hundred microliter aliquots were distributed to 96-well plate wells and the absorbance at 585 nm was determined using an EnSpire® Multimode Plate Reader (Perkin Elmer, Waltham, MA, United States). MTT reduction was blank corrected and expressed as a percentage of untreated controls.
Cell death was determined by measuring the enzymatic activity of extracellular LDH released from cells due to loss of membrane integrity indicating cell death using a Cytotoxicity Detection Kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions. Briefly, media samples were combined with reaction mixture and absorbance measured at 492 nm using an EnSpire® Multimode Plate Reader. Extracellular LDH activity was expressed as percentage of total cellular LDH activity determined from untreated cells lysed in culture media containing 1% triton X-100.
Nitric oxide concentrations in media were determined using a Nitric Oxide Assay Kit (Abcam, England, United Kingdom) or a Griess Reagent Kit (Thermo Fisher Scientific) according to the manufacturer’s protocols. Briefly, standards were prepared by serial dilution of the nitrite standard solution to final concentrations between 0 and 30 μM. 100 or 240 μl, for each respective kit, of freshly collected samples and standards (in duplicate) in 96-well microplate (Greiner Bio-One, Frickenhausen, Germany) were reacted with Griess reagents 1 and 2 (50 or 30 μl of each reagent, for each respective kit). Following 15 min of color development at RT, absorbance at 540 nm was determined using an EnSpire® Multimode Plate Reader. Standard curves were constructed, and sample nitrite concentrations were calculated from standard curves. Results were expressed as percentage NO released relative to TNFα- and IFNγ-stimulated positive controls.
Total RNA was isolated from cells using the MagMaxTM Total RNA isolation kit or from frozen tissue samples using TRIzol® (both from Invitrogen, Thermo Fisher Scientific), according to the manufacturer’s instructions. RNA concentrations were measured using the Qubit® 2.0 fluorometer (Life Technologies) or NanoDrop Spectrophotometer (Thermo Fisher Scientific). RNA (200 ng–2 μg) was reverse transcribed using random hexamers and High Capacity cDNA kit (Life Technologies) or Maxima reverse transcriptase (Fermentas, Thermo Fisher Scientific). The relative expression levels of mRNA were measured according to the manufacturer’s protocol by qRT-PCR (StepOnePlus; Life Technologies, LightCycler 480; Roche, Basel, Switzerland, or QuantStudio 6; Thermo Fisher Scientific) using TaqMan chemistry and specific assays-on-demand target mixes (Arg1 Mm00475988_m1, Tgfb1 Mm01178820_m1, Ccl2 Mm00441242_m1, Tnf Mm00443260_g1 or Mm00443258_m1, Mt1 Mm00496660_g1, Il1a Mm00439620_m1, Il1b Mm00434228_m1, Il6 Mm00446190_m1, Nos2 Mm00440502_m1, Ptgs2 Mm00478374_m1; Life Technologies). The expression levels were obtained by normalizing the target gene to the geometric mean of GAPDH and beta-actin (Mm99999915_g1 and Mm02619580_g1, respectively; Life Technologies), and presented as fold change in the expression using the 2-ΔΔCt method, where Ct is the threshold cycle value.
Monocyte chemoattractant protein 1 (MCP-1/CCL2) and interleukin 6 (IL-6) concentrations were quantified by ELISA using the Mouse CCL2/JE/MCP-1 DuoSet and Mouse IL-6 DuoSet (R&D systems, Minneapolis, MN, United States), respectively, according to the manufacturer’s instructions. Results were expressed as percentages relative to the LPS or TNF-α and IFNγ-stimulated positive controls.
Inductively coupled plasma mass spectrometry analysis of metal levels was performed as reported previously (Grubman et al., 2014b). Briefly, cell pellets collected for metal analysis were digested overnight in concentrated nitric acid (Aristar, BDH, Kilsyth, VIC, Australia), after which samples were heated for 20 min at 90°C. The volume of each sample was reduced to approximately 40–50 μl then 1 ml of 1% (v/v) nitric acid diluent was added to the samples. Some samples were analyzed using laser ablation ICP-MS in micro-droplet format as described previously (Yang et al., 2005; Hare et al., 2016). Briefly, cell cultures and certified element copper standard (100014-1; Choice Analytical, Australia) were prepared in phospho-safe extraction buffer (Merck). One microliter of each sample was pipetted onto a glass slide and left to air-dry overnight. Droplet residues were ablated off the slide surface using laser ablation and analysed using Iolite software (Paul et al., 2015; Hare et al., 2017). Measurements were made using an Agilent 7700x series ICP-MS instrument or a NWR-213 laser ablation unit (Electro Scientific Industries, Portland, OR, United States) hyphenated to an Agilent 8800 ICP-QQQ-MS. The concentrations of copper were calculated as μg of metal per mg of protein based on the protein concentration of the sample, as determined by BCA protein assay (Thermo Fisher Scientific).
The distribution of copper was mapped at the XFM beamline (Paterson et al., 2011) at the Australian Synchrotron on cryofrozen freeze-dried cell culture samples grown on SiN X-ray transparent membranes, as described previously (Grubman et al., 2014a). An incident beam of 12.7 keV X-rays was used to induce K-shell ionization of copper, while providing adequate separation of the Rayleigh and Compton peaks from the elemental fluorescence of interest. The incident beam was focused to a ∼1.5 μm spot (full-width at half maximum) using a Kirkpatrick–Baez mirror pair and specimens were fly-scanned through X-ray focus. The resulting XRF was collected in event mode using the low latency, 384-channel Maia XRF detector (Ryan et al., 2014) situated in the backscatter geometry. Full XRF spectra were used to reconstruct copper maps of the specimen using a virtual pixel size of 0.8 μm (corresponding to the sample interval), giving a pixel transit time of 25 μs. Reference foils of Mn and Pt (Micromatter, Canada), were scanned in the same geometry at regular intervals throughout the measurement and used as references to establish elemental quantitation. Deconvolution of the Maia data was performed using GeoPIXE v6.6 software (Kirkham et al., 2011).
Results were analyzed by ANOVA followed by Holm–Sidak’s multiple comparison test using GraphPad Prism software. Statistical significance was assumed if p < 0.05. Data presented in graphs represent the experimental mean ± SEM of the indicated number of independent experiments, each derived from multiple technical replicates.
Peripheral inflammation induced by LPS triggers an inflammatory response that is characterized by increased expression of pro-inflammatory cytokines in the brain and plasma (Thomson et al., 2014). Furthermore, brain expression of VCAM-1, which plays a major role in the trafficking of leukocytes across the blood–brain barrier, is upregulated in response to systemic LPS (O’Sullivan et al., 2010; Thomson et al., 2014). To study the effect of a copper bis(thiosemicarbazone) complex on peripheral inflammation induced by systemic LPS, we employed a sophisticated in vivo MRI approach to measure brain inflammation that detects brain VCAM-1 expression in live animals. For this purpose, microsized particles of iron oxide targeting VCAM-1 (MPIO-VCAM-1) were intravenously administered to mice 30 min prior to imaging by MRI (Figure 1A). The MRI imaging revealed a robust, close to 100% increase in the expression of VCAM-1 at 24 h after administration of 1 mg/kg LPS (Figures 1B,C).
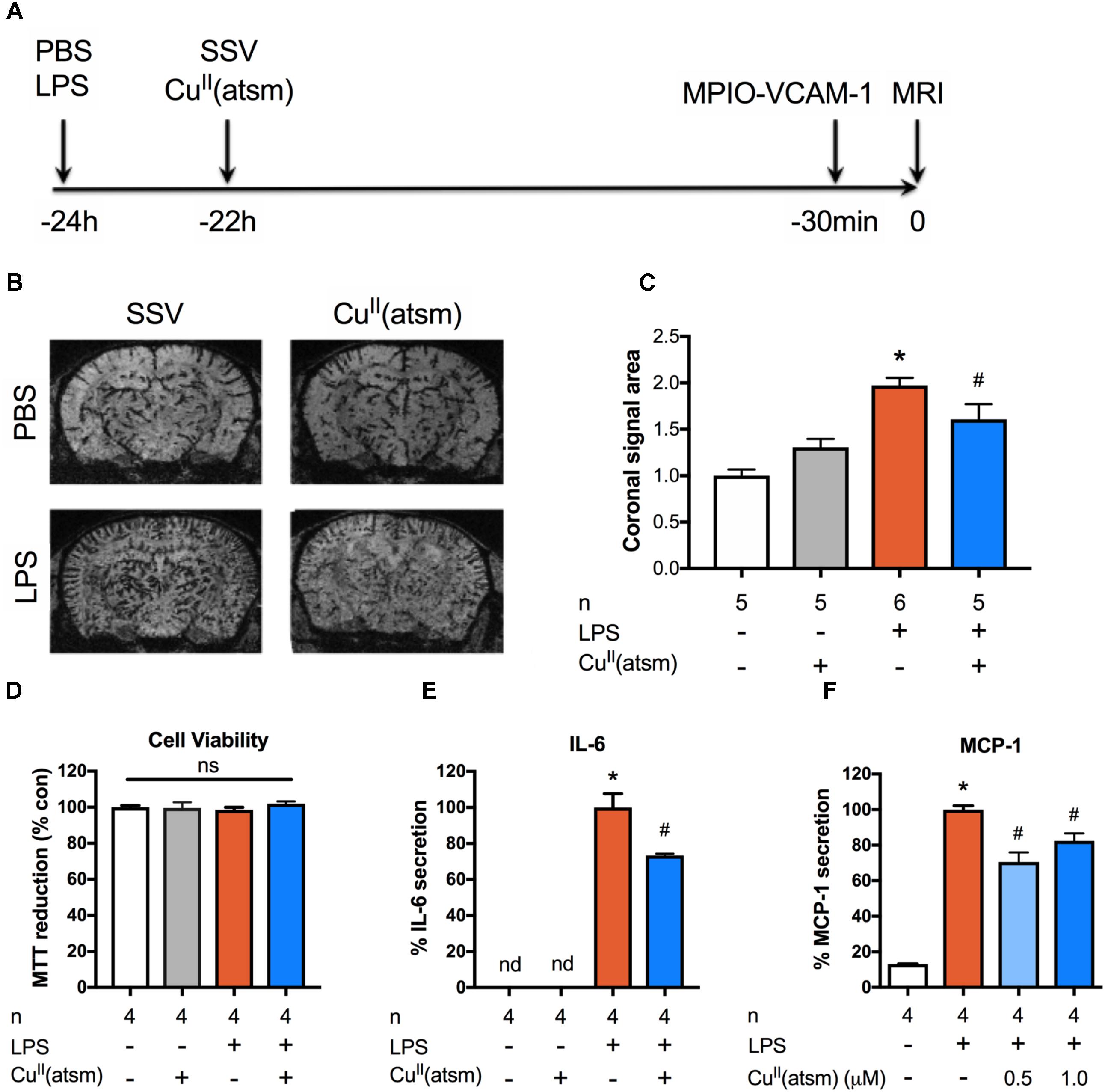
FIGURE 1. Copper delivery reduces inflammation induced by administration of LPS. (A) Timeline demonstrating time points for administration of the indicated compounds to adult mice and in vivo imaging of inflammation by MRI. (B) Representative MRI images acquired 30 min after intravenous administration of MPIO-VCAM-1 (24 h after LPS and 22 h after CuII(atsm) or SSV) showing signal voids corresponding to MPIO binding. (C) Quantitative analysis of the volume of signal void in the brain induced by MPIO-VCAM-1 following treatment with LPS and CuII(atsm). (D–F) Cultured adult astrocytes were stimulated with 20 μg/ml LPS with or without 0.5 μM CuII(atsm) for 48 h and (D) MTT reduction, (E) IL-6 secretion, and (F) MCP-1 secretion were measured 48 h later. Sample sizes per group (n) as indicated. ∗p < 0.05 compared to control, #p < 0.05 compared to LPS-treated group. ns, not significant. nd, not detected.
To investigate whether the copper complex, CuII(atsm), possesses anti-inflammatory activity in vivo, mice were treated with a single dose of CuII(atsm) by oral gavage after peripheral administration of LPS. As the inflammatory response in brain peaks 1 h after administration of LPS (Qin et al., 2007), mice were treated with CuII(atsm) 2 h after LPS administration to investigate the effect of CuII(atsm) on an ongoing inflammatory response. Mice were administered 60 mg/kg CuII(atsm) as we have demonstrated this to be sufficient to induce neuroprotective effects in vivo (Huuskonen et al., 2017). MPIO-VCAM-1 imaging revealed a significant, ∼20% reduction in the level of brain VCAM-1 expression in the CuII(atsm)-treated mice compared to control, SSV-treated mice at 24 h after LPS injection (Figure 1C). These data demonstrate the efficacy of the neuroprotective agent CuII(atsm) in reducing acute cerebrovascular inflammation (measured as VCAM-1 expression) caused by systemic administration of LPS in vivo.
Given that our in vivo findings during acute neuroinflammation suggest that copper may regulate inflammatory pathways, we next investigated inflammation in vitro utilizing adult astrocytes co-stimulated with LPS and CuII(atsm) for 48 h. Neither 20 μg/ml LPS or 0.5 μM CuII(atsm) alone or combined were toxic after 48 h (Figure 1D). This concentration of CuII(atsm) is consistent with our previous pharmacokinetic studies which demonstrate CuII(atsm) concentration reaches 0.6 μM in the mouse CNS following a single administration of 30 mg/kg (Hung et al., 2012). As expected, secretion of the cytokine IL-6 was strongly induced by LPS treatment in adult astrocytes (Figure 1E). This was significantly attenuated by co-treatment with CuII(atsm). MCP-1 (CCL-2) is a chemoattractant cytokine, the suppression of which is known to be protective during neuroinflammation (Madrigal and Caso, 2014). To assess the effects of copper delivery on LPS-induced production of MCP-1, we next measured secreted MCP-1 (Figure 1F). LPS treatment induced a ∼10-fold increase in MCP-1 secretion from adult astrocytes, which was reduced by co-treatment with CuII(atsm).
Although LPS is useful to investigate acute inflammatory insults, and cultured cells robustly respond to it, LPS is unlikely to directly stimulate brain cells in vivo due to poor blood–brain barrier penetration (Banks and Robinson, 2010). Therefore, we further studied the anti-inflammatory potential of CuII(atsm) in cultured microglia and astrocytes stimulated with endogenous mediators of inflammation.
Modulation of microglial activity is a viable therapeutic approach for diseases with a strong neuroinflammatory component. To determine whether copper bis(thiosemicarbazone) complexes modulate the inflammatory phenotype of microglia, we used a treatment paradigm where primary microglia were stimulated with the pro-inflammatory cytokines IFNγ and TNFα during co-treatment with CuII(atsm). The maximum non-toxic doses of CuII(atsm) for cultured microglia based on MTT and LDH assays were chosen for these experiments (Supplementary Figure S1). The inflammation-modulating activity of CuII(atsm) was also compared to that of the well-known anti-inflammatory agent, minocycline (Malm et al., 2010). We first tested the effects of non-toxic concentrations (Figure 2A) of CuII(atsm) on IFNγ/TNFα-induced NO production. The amount of NO was increased 2.5-fold after the 48 h IFNγ/TNFα stimulation (Figure 2B). Co-treatment with 0.5 μM CuII(atsm) significantly suppressed the IFNγ/TNFα-induced NO production by ∼50%, whereas CuII(atsm) alone did not affect NO secretion.
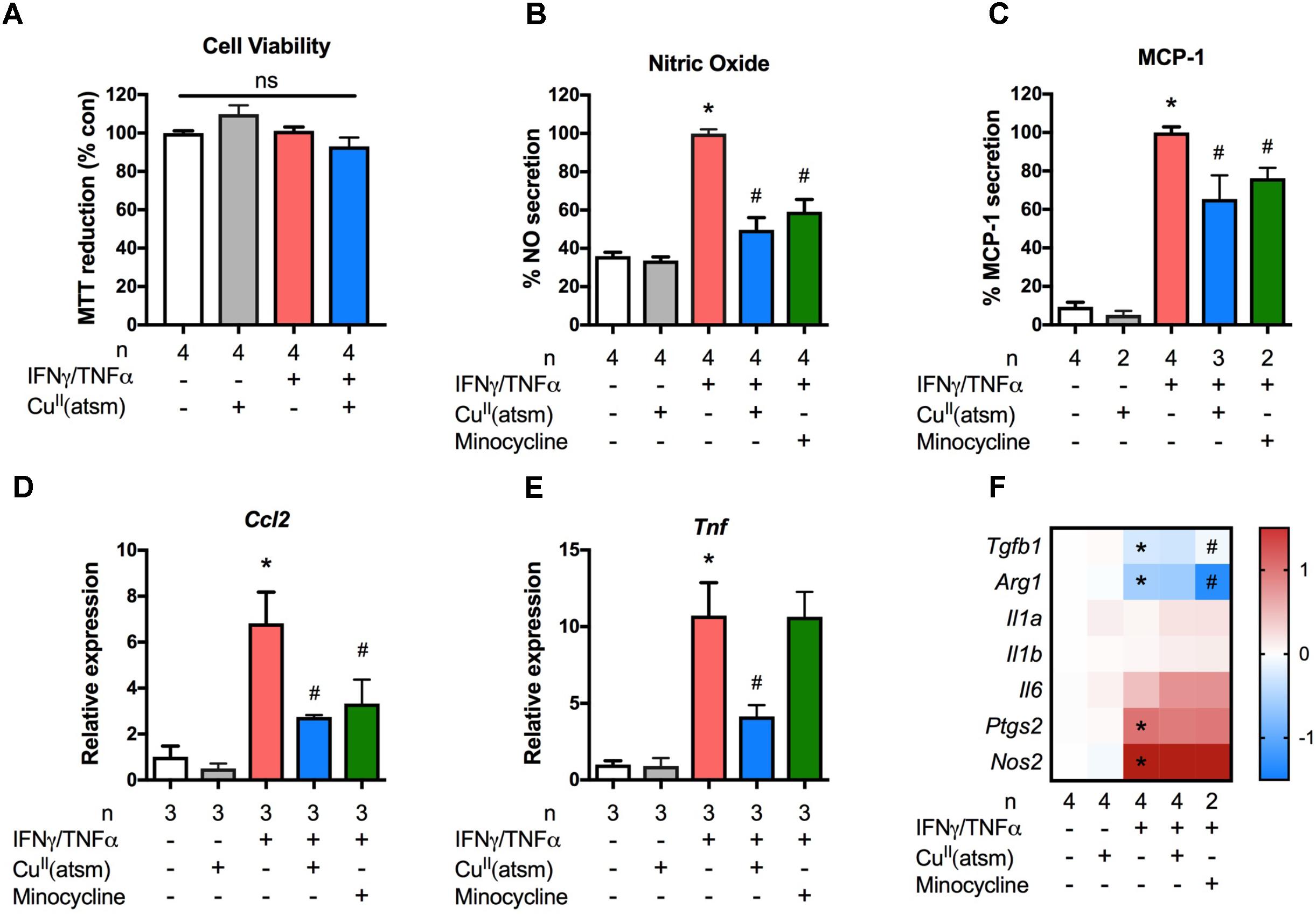
FIGURE 2. Copper delivery dampens microglial activation during inflammation. Primary microglia were stimulated with 15 ng/ml IFNγ and 10 ng/ml TNFα with or without 0.5 μM CuII(atsm). (A) MTT reduction was measured to determine the cell viability 24 h later. Primary microglia were stimulated with 15 ng/ml IFNγ and 10 ng/ml TNFα with or without 0.5 μM CuII(atsm) or 20 μM minocycline for 24 h. (B) Nitric oxide release into the media was measured 24 h later. (C) The secretion of MCP-1 into the media was measured by ELISA. qRT-PCR was used to measure microglial mRNA expression levels of (D) Ccl2, (E) Tnf, and (F) the indicated genes. (F) Presented as log fold-increase over control. Sample sizes per group (n) as indicated. ∗p < 0.05 compared to control, #p < 0.05 compared to IFNγ/TNFα-treated group. ns, not significant.
We next measured secreted MCP-1 (Figure 2C) and the mRNA levels of the MCP-1 gene, Ccl2, (Figure 2D) in primary microglial cultures. As expected, treatment with IFNγ/TNFα increased the secretion of MCP-1 into the media and the mRNA level of Ccl2. When compared to IFNγ/TNFα stimulation, co-treatment with CuII(atsm) reduced the MCP-1 protein secreted into the media by 40% (Figure 2C). This reduction was comparable to that conferred by minocycline (Figure 2C). Similarily, CuII(atsm) treatment was effective at reducing the mRNA expression of Ccl2 (Figure 2D). Co-treatment with CuII(atsm) also reduced the mRNA expression of Tnf encoding TNFα (Figure 2E).
Further analysis of inflammatory gene expression revealed these effects of CuII(atsm) to be quite selective. In contrast to Ccl2 and Tnf, CuII(atsm) did not significantly alter the expression of Il1a, Il1b, Il6, Ptgs2, or Nos2 (Figure 2F). The latter is intruiging, as this gene codes for inducible NO synthase, and would be expected to be decreased by CuII(atsm) in light of the attenuated NO levels (Figure 2B). As expected, IFNγ/TNFα significantly decreased expression of two anti-inflammatory genes, Tgfb1 and Arg1 (Figure 2F). While CuII(atsm) did not alter this response, minocycline increased expression of Tgfb1 and further decreased expression of Arg1.
To determine whether the observed anti-inflammatory effects of copper delivery were specific for microglia only, we next employed a similar treatment paradigm for murine astrocytes with non-toxic concentrations (Figures 3A,B). In comparison to neonatal astrocytes stimulated with IFNγ/TNFα, co-treatment with CuII(atsm) resulted in significantly reduced secretion of MCP-1 (Figure 3C) and NO (Figure 3D) into the culture media. This was accompanied by a corresponding attenuation of IFNγ/TNFα-induced Ccl2 (Figure 3E), Nos2 (Figure 3F), and Tnf (Figure 3G) expression by CuII(atsm). Analysis of other inflammatory genes revealed IFNγ/TNFα increased expression of Tgfb1 and Arg1 in astrocytes, which was either attenuated or exacerbated by CuII(atsm) co-treatment, respectively (Figure 3H). IFNγ/TNFα treatment increased expression of Il1a, Il1b, and Il6, but not Ptgs2 (Figure 3H). Co-treatment with CuII(atsm) did not diminish expression of these genes, and actually increased expression of Il1a.
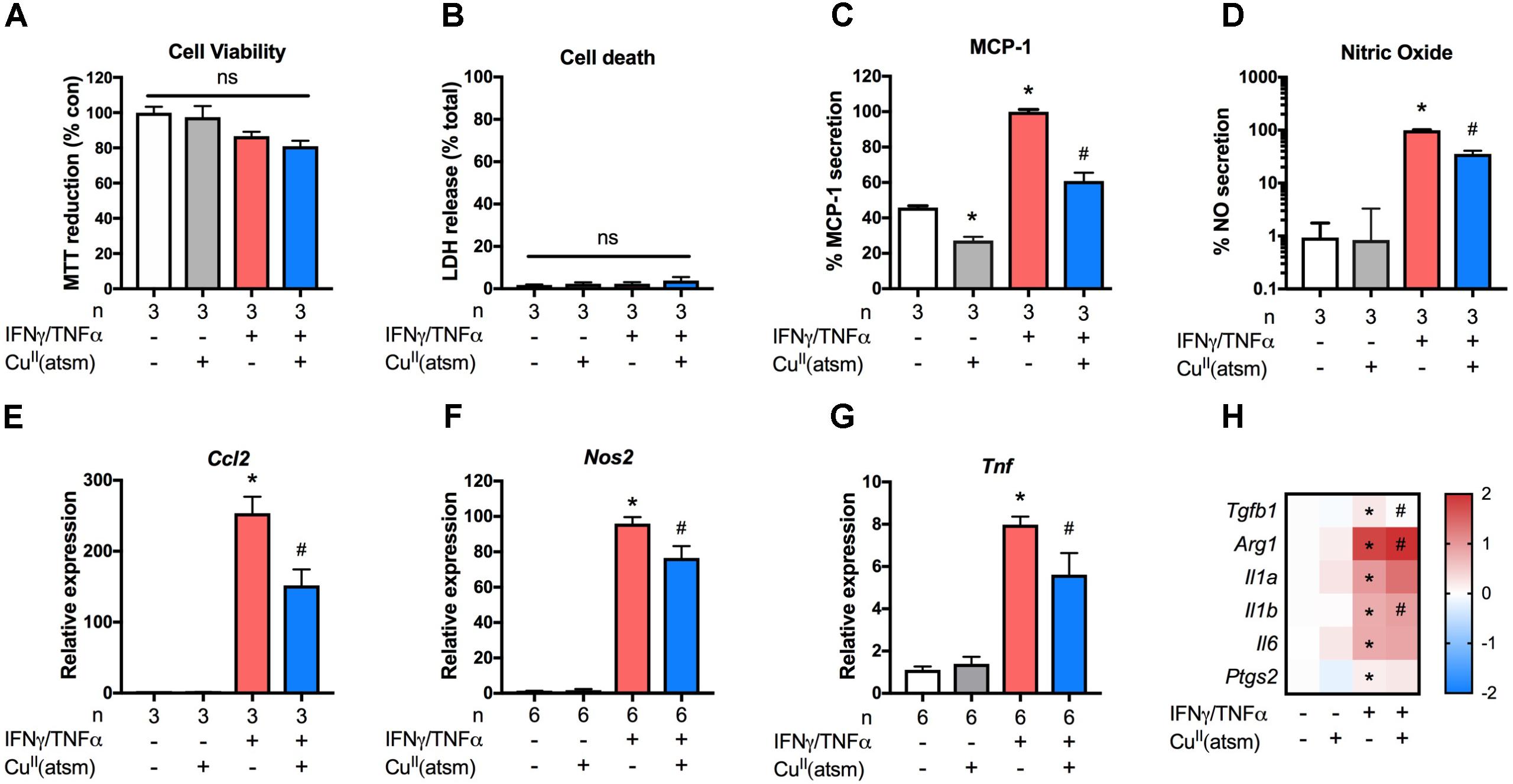
FIGURE 3. Anti-inflammatory effects of copper delivery in astrocytes. Neonatal astrocytes were stimulated for 48 h with 15 ng/ml IFNγ and 10 ng/ml TNFα with or without 2μM CuII(atsm) for the final 24 h (A–H). (A) MTT reduction and (B) LDH release was measured to determine the cell viability and cell death, respectively, after treatment. (C) MCP-1 secretion and (D) nitric oxide release were measured after treatment. qRT-PCR was used to measure astrocyte mRNA expression levels of (E) Ccl2, (F) Nos2, (G) Tnf, and (H) the indicated genes. (H) Presented as log fold-increase over control. Sample sizes per group (n) as indicated, or 6–7/treatment group for (H). ∗p < 0.05 compared to control, #p < 0.05 compared to IFNγ/TNFα-treated group. ns, not significant.
Metal accumulation in response to treatment with CuII(atsm) was examined to determine the effect of the compound on metal delivery in primary microglia and neonatal astrocytes under basal and inflammatory conditions. Treatment of either astrocytes or microglia with CuII(atsm) caused a significant increase in cellular copper levels as measured by ICP-MS (Figures 4A,C). IFNγ/TNFα alone did neither alter copper levels as compared to basal levels nor alter copper content when co-treated with CuII(atsm) relative to CuII(atsm) alone.
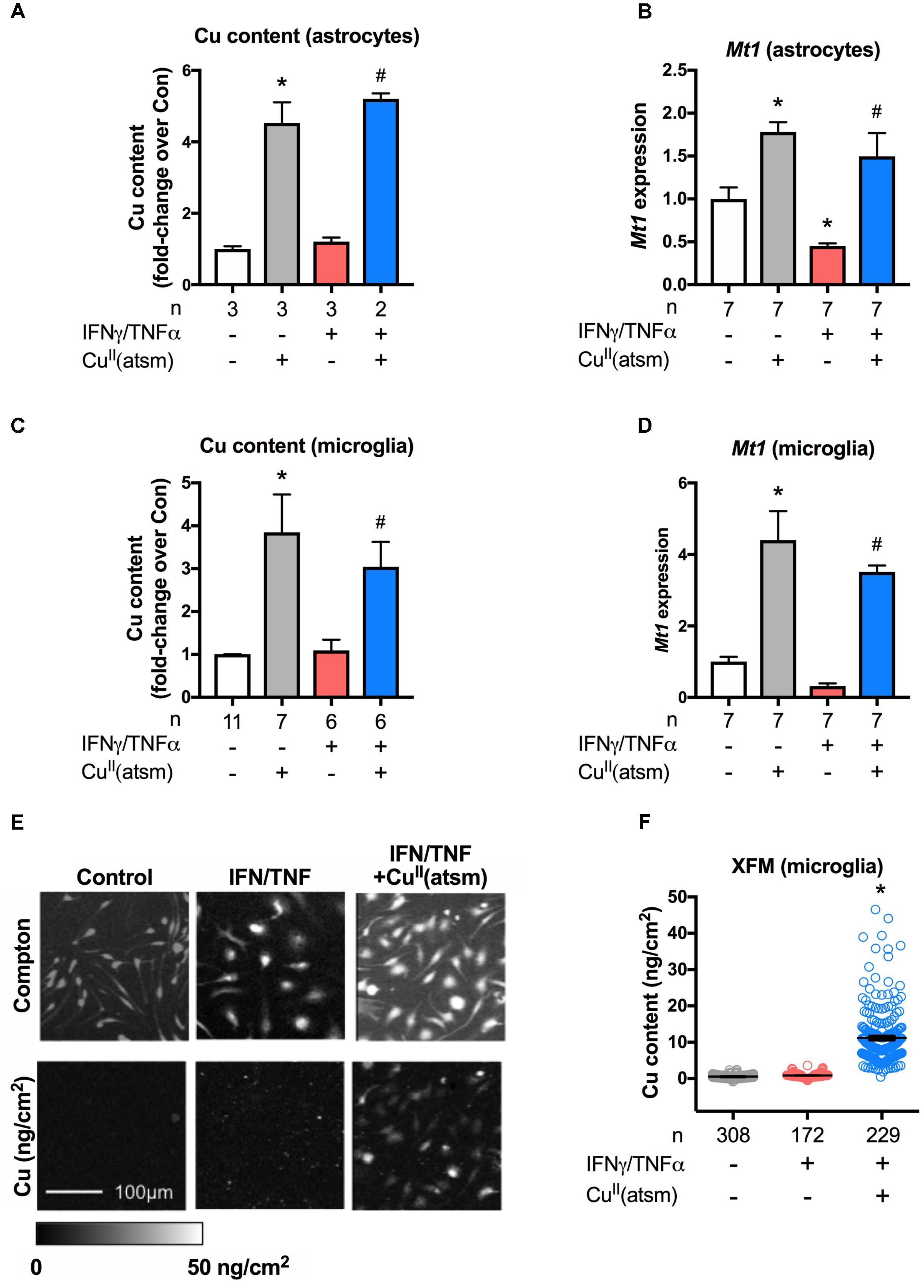
FIGURE 4. CuII(atsm) delivers copper into cells. (A,B) Neonatal astrocytes were treated for 48 h with 15 ng/ml IFNγ and 10 ng/ml TNFα with or without 2 μM CuII(atsm) for the final 24 h. (C,D) Microglia were treated for 24 h and with 15 ng/ml IFNγ and 10 ng/ml TNFα with or without 0.5 μM CuII(atsm). Cellular copper content of (A) astrocytes and (C) microglia was measured by ICP-MS. mRNA expression of Mt1 was measured by qRT-PCR in (B) astrocytes and (D) microglia. Sample sizes per group (n) as indicated. (E) Representative images of microglia analyzed for cellular copper content by XFM (reflected by elemental areal density). Scale bar 100 μm. (F) Quantification of XFM-determined copper levels per cell in microglia treated with 15 ng/ml IFNγ and 10 ng/ml TNFα with or without 0.5 μM CuII(atsm). Cells per treatment group (n) as indicated. ∗p < 0.05 compared to control, #p < 0.05 compared to IFNγ/TNFα-treated group.
Metallothionein-1 is a critical metal-binding protein that is involved in the regulation of metals and the modulation of anti-inflammatory and anti-oxidative pathways (Huh et al., 2007). To determine whether MT1 is associated with the anti-inflammatory effects induced by copper delivery, Mt1 expression was investigated. In contrast to reports indicating that Mt1 expression is increased by pro-inflammatory conditions (De et al., 1990; Manso et al., 2011; Everhardt Queen et al., 2016), here Mt1 expression was decreased when either astrocytes or microglia were treated with IFNγ/TNFα alone (Figure 4B; Supplementary Figure S2). Treatment of astrocytes or microglia with CuII(atsm) increased Mt1 expression (Figures 4B,D). This expression was unchanged by co-treatment with IFNγ/TNFα As Mt1 expression is well known to be strongly regulated by copper levels via metal regulatory transcription factor 1 (Juarez-Rebollar et al., 2017), these results indicate that CuII(atsm) is delivering bioavailable copper into cultured glial cells. That the presence or absence of IFNγ/TNFα did not alter CuII(atsm)-mediated copper content or Mt1 expression of either astrocytes or microglia indicates that inflammatory conditions do not alter the accumulation of copper by glial cells, at least in culture.
Finally, XFM was used next to investigate the delivery and subcellular localisation of copper into primary microglia. Inelastic scatter (Compton) of incident photons was used to define the boundaries of cells in order to quantify metal levels for each individual cell (Figure 4E). We observed a dramatic increase in cellular copper in response to IFNγ/TNFα plus CuII(atsm) treatment, with little overlap in copper content per cell as compared to cells treated in absence of CuII(atsm) (Figure 4F). Copper appears to be diffusely localized throughout microglia, with no discernable nuclear exclusion or punctate localisation. In microglia treated with CuII(atsm), copper levels per cell are somewhat heterogeneous, with some cells accumulating much more copper than others. Together, these results demonstrate that CuII(atsm) efficiently delivers copper into cells under both basal and inflammatory conditions.
Modulation of copper and inflammation are both beneficial during neurodegeneration (Soon et al., 2011; Hung et al., 2012; Heneka et al., 2013; Roberts et al., 2014; Spangenberg and Green, 2016), yet the direct inflammation-modulating effects of copper compounds have remained relatively unexplored. Here we demonstrate, for the first time, that a bis(thiosemicarbazone) copper complex has anti-inflammatory effects in cultured microglia and astrocytes, and moderates inflammation in mice subjected to acute peripheral inflammation.
The role of copper in neuroinflammatory conditions, including AD, is not clear. Multiple studies have shown that increases in free copper levels impairs the clearance of Aβ and worsens behavioral outcome in animal models of AD (Kitazawa et al., 2009; Singh et al., 2013; Kitazawa et al., 2016). Copper triggers activation of cultured microglia, causing them to release the pro-inflammatory cytokine TNFα and NO into the culture media (Hu et al., 2014; Yu et al., 2015). Accordingly, sequestering systemic copper via D-penicillamine or tetrathiomolybdate reduces systemic oxidative stress in AD patients (Squitti et al., 2002), and inhibits acute LPS-induced peripheral inflammatory reactions in vivo (Wei et al., 2011). However, these chelator studies did not assess inflammation in the brain, where in contrast, intracellular copper deficiency elicits a neurodegenerative phenotype in mice and promotes the activation of microglia (Zucconi et al., 2007). Together, these studies suggest that too much or too little copper is detrimental and that intracellular, physiological copper concentrations are essential for the prevention of neuroinflammation. In line with this, neuroinflammatory conditions, including AD, have profoundly disturbed copper homeostasis. Copper-induced Aβ deposition occurs concomitantly with a reduction in the amount of intracellular, bioavailable copper, rendering the AD brain copper deficient (Schrag et al., 2011; Choo et al., 2013). In contrast to the inflammatory effects of free copper treatments, we find that the copper bis(thiosemicarbazone) complex applied in the current study moderates peripherally induced neuroinflammation in vivo and attenuates inflammation in cultured glial cells. This is in line with our previous findings that modulation of copper by cell permeable copper-delivering compounds is potently neuroprotective in various in vivo models of neurodegeneration (Crouch et al., 2009; Adlard et al., 2011; Soon et al., 2011; Hung et al., 2012; McAllum et al., 2013; Roberts et al., 2014), and that these protective effects are accompanied by modulation of acute and chronic neuroinflammation in vivo (Soon et al., 2011; Roberts et al., 2014; Huuskonen et al., 2017). The discrepancy between free copper and these complexes is likely due to the specific actions of these copper complexes, redistributing copper ions, and circumventing the endogenous copper trafficking machinery to restore copper homeostasis. The strong neuroprotective effects of these complexes demonstrates the potential therapeutic utility of our approach: CuII(atsm) is currently under investigation in clinical trials for the treatment of PD and ALS (NCT02870634, NCT03136809, and NCT03204929).
We demonstrated by MRI imaging that an acute inflammatory insult induces a robust cerebrovascular inflammatory phenotype, which is dampened by treatment with CuII(atsm). Our findings corroborate earlier reports whereby the expression of VCAM-1 in the brain is upregulated in response to systemic LPS (O’Sullivan et al., 2010; Thomson et al., 2014). VCAM-1 is an adhesion molecule overexpressed by the activated cerebral vasculature during inflammation and is considered a promising marker for molecular imaging of neuroinflammation in CNS disorders (Gauberti et al., 2014). For the first time, we report that peripheral LPS-induced brain VCAM-1 expression is modulated by a copper complex. It has been shown that superoxide dismutase 1 (SOD1/Cu-Zn SOD) plays an important role in scavenging the superoxide anions generated by inflammatory challenge and limiting VCAM-1-mediated propagation of the injury (Ishigami et al., 2011). Since CuII(atsm) is known to increase SOD1 copper content in animal models of ALS (Roberts et al., 2014), we hypothesize that the antioxidant reactions raised by CuII(atsm) may contribute to reducing VCAM-1 expression after systemic LPS challenge. Interestingly, administration of copper-bound anti-inflammatory agents significantly reduces endothelial expression of VCAM-1 after acute injury in rabbits (Puranik et al., 2016). By contrast, an in vitro study has shown that treatment of human aortic endothelial cells with cupric sulfate increases VCAM-1 expression, indicative of increased inflammation (Wei et al., 2014). These apparently conflicting results can be reconciled by the fact that the latter study did not assess the effect of copper under inflammatory conditions, used supra-physiological concentrations of copper, and used free copper as opposed to a membrane-permeable copper delivery agent, which as discussed above does not allow for tight control of metal delivery, or a direct comparison to be made to our data. While examination of the attenuation of inflammation by CuII(atsm) in vivo in the current study is limited to VCAM-1 expression, our previous studies demonstrate that CuII(atsm) robustly attenuates the neuroinflammation associated with acute and chronic neurodegenerative insults (Soon et al., 2011; Roberts et al., 2014; Huuskonen et al., 2017). Nevertheless, future studies will be useful to examine additional markers of inflammation both in the CNS and periphery to further elucidate the effects of CuII(atsm) in acute models of inflammation.
Inflammation in the CNS is regulated by microglia and to a lesser extent astrocytes. Our in vitro data support the notion that copper delivery is beneficial in these glial cells through the demonstration that under inflammatory conditions, treatment with CuII(atsm) reduced secretion of NO and IL-6, reduced the expression of the gene encoding the classical pro-inflammatory molecule TNFα, and reduced both the expression of the gene encoding MCP-1 and the secretion of the protein into the culture media. However, no changes were observed in the expression of several other inflammatory genes. This may indicate that the anti-inflammatory effects of CuII(atsm) are somewhat selective. Interestingly, NO secretion by microglia is attenuated by CuII(atsm), but not the elevated expression of Nos2. This suggests post-transcriptional modifications are responsible for decreased NO secretion. Indeed, a direct interact of copper with NO species has been reported for microglia (Rossi-George and Guo, 2016). Such interactions could also influence other inflammatory mediators, and this remains to be determined. An alternative option for lack of change in expression is that more wide-ranging anti-inflammatory effects were not able to be observed under the conditions utilized in this study but may occur under different conditions. In support of this, minocycline has been reported to attenuate the expression of many inflammatory genes, however several were examined here and were found to be unaltered by minocycline treatment, including Nos2, Il6, Il1b, and Ptgs2 (Matsui et al., 2010; Piotrowska et al., 2017). That CuII(atsm) consistently induced more robust anti-inflammatory effects than minocycline supports the strong anti-inflammatory action of CuII(atsm).
Arginase-1 and TGB-β are strongly induced in microglia and macrophages during anti-inflammatory conditions and are often used as a marker of anti-inflammatory/alternatively activated cells (Jha et al., 2016). However, mounting evidence suggests that this bipolar classification of microglia is not reflective of the complexity of dynamic microglial transcriptional fingerprints in the physiological and pathological brain (Ransohoff, 2016; Keren-Shaul et al., 2017). Our results are consistent with this, with microglia exhibiting decreased expression of both Arg1 and Tgfb1 in response to IFNγ/TNFα-induced inflammation, whereas both are increased in response to the same stimuli in cultured astrocytes. It is important to note that our astrocyte cultures contain a substantial subpopulation of microglia. The phenotype of both microglia and astrocytes are strongly influenced by communication with neighboring cells. For example, MCP-1 released by astrocytes is known to activate microglia, cause their pro-inflammatory phenotype switch, and to enhance their migratory ability (He et al., 2016), and the presence of even a small number of microglia is capable of altering the phenotype of cultured astrocytes (Liddelow et al., 2017). Given this extensive crosstalk between microglia and astrocytes, this mixed glial culture perhaps represents a more physiological model than either isolated microglia or astrocytes. Regardless, with the exception of Arg1 and Tgfb1, that similar anti-inflammatory results were obtained in both cultured astrocytes and microglia further strengthens the validity of these effects and the likelihood of translation in vivo.
How does CuII(atsm) trigger the anti-inflammatory effects demonstrated herein? CuII(atsm) can deliver bioavailable copper into the CNS (Crouch et al., 2009; Roberts et al., 2014). Conditions consistent with oxidative stress promote the dissociation of copper from CuII(atsm) (Sirois et al., 2018). This produces a targeted effect proven to be beneficial in animal models of conditions such as stroke and ALS, where neuroinflammation is strongly related to increased oxidative stress (Roberts et al., 2014; Huuskonen et al., 2017). Copper-induced increases in microglial and astrocytic Mt1 expression have been reported by others (Agullo et al., 1998; Bulcke and Dringen, 2015) and are now confirmed by the current report. This increase indicates copper dissociation from CuII(atsm). In addition to the previously reported mechanisms of action of CuII(atsm) connected to increased antioxidant scavenging (Soon et al., 2011; Hung et al., 2012; Roberts et al., 2014), we show here that MT1 may also mediate the protective effects of CuII(atsm). Existing reports demonstrate that metallothioneins, metal-buffering proteins with numerous protective functions, modulate anti-inflammatory pathways (Inoue et al., 2009). Mt1 expression is also upregulated in ALS patients and model mice (Sillevis Smitt et al., 1994), in AD patients and in mice modeling the disease, specifically in cells surrounding amyloid plaques (Carrasco et al., 2006; Kim et al., 2012) and in astrocytes (Kim et al., 2012). In vitro studies have demonstrated that in addition to preventing amyloid neurotoxicity directly, MT1 also suppresses Aβ-dependent microglial activation and reduces the neurotoxicity of Aβ-stimulated microglia (Kim et al., 2012). Furthermore, in vivo studies have reported that over-expression of MT1 modulates key phenotypes of AD model mice in a gender- and age-related manner (Kim et al., 2012), and extends lifespan of ALS model mice (Tokuda et al., 2014). These studies demonstrate the critical role of MT1 during neuroinflammatory conditions, which may be involved in the therapeutic action of CuII(atsm). While the in vitro results suggest inflammation does not alter CuII(atsm) or copper accumulation, our previous studies indicate the accumulation of copper from CuII(atsm) is enhanced under disease conditions (Donnelly et al., 2012; Roberts et al., 2014). Hence, it remains to be determined whether the pharmacokinetics of CuII(atsm) in vivo are altered under acute inflammatory conditions.
To have clinical utility, anti-inflammatory therapeutics must be able to inhibit ongoing inflammation. For the in vivo analyses in the current study, as 1 h is reportedly sufficient for LPS to generate a robust inflammatory response (Qin et al., 2007), the effects of CuII(atsm) administered 2 h after LPS are likely to indicate the attenuation of ongoing inflammation, rather than preventing the onset of inflammation. Similarly, while inflammation was induced in microglia simultaneously with CuII(atsm) treatment, primary astrocytes were treated both simultaneously and 24 h following induction of inflammation. Both paradigms elicited similar results demonstrating CuII(atsm) attenuates prevailing inflammation in vitro. Furthermore, our previous studies in animal models of several neurodegenerative conditions also demonstrate that CuII(atsm) elicits robust protective effects even when administered post-symptom onset (Hung et al., 2012; McAllum et al., 2013; Huuskonen et al., 2017). Thus CuII(atsm) can ameliorate ongoing inflammation.
Available treatment options for neuroinflammatory diseases are inadequate and those available have limited efficacy. Discovering novel approaches that modulate neuroinflammation are therefore of immense therapeutic interest for these disorders. Here, we demonstrated robust inflammation-reducing effects of a cell permeable copper complex in both in vivo and in vitro models of neuroinflammation. Given our newly reported insights into its neuroimmune role, this study demonstrates that copper delivery is a strong candidate for the development of therapies for neuroinflammatory conditions.
Animal handling and experimentation were performed in accordance with the national regulation of the usage and welfare of laboratory animals and approved by the Animal Experiment Committee in State Provincial Office of Southern Finland and the University of Melbourne Animal Ethics Committee (ethics ID. 1312831).
XC, JL, AG, MH, DM, JR, KK, LP, HQ, LO, CD, SJ, LM, DH, EP, SV, KL, MK, and JR performed experimental work, data analysis, and contributed to manuscript preparation. XC, AG, MH, LM, PD, TM, JR, JK, AW, and KK developed the concept of the project and contributed to the manuscript preparation.
This study was supported by the Academy of Finland, Sigrid Juselius Foundation and Tekes: Finnish Funding Agency for the Innovation. AW was supported by an Australian National Health and Medical Research Council Senior Research Fellowship. AG was supported by a Melbourne Neuroscience Institute Fellowship, a Melbourne University Early Career Grant, and NHMRC-ARC Dementia Research Development Fellowship. XC was supported by a Henry and Rachael Ackman Travelling Scholarships from The University of Melbourne. JL was supported by an Australian National Health and Medical Research Council Peter Doherty Fellowship. MH was supported by Kuopio University Foundation at the University of Eastern Finland. Part of this work was conducted at the XFM beamline of the Australian Synchrotron, and supported by the Multi-modal Australian ScienceS Imaging and Visualisation Environment (MASSIVE) (www.massive.org.au).
Collaborative Medicinal Development LLC has licensed intellectual property related to this subject from The University of Melbourne, where the inventors include PD and AW.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Mirka Tikkanen, Laila Kaskela, Riikka Lampinen, and Martin D. de Jonge for expert technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2018.00668/full#supplementary-material
FIGURE S1 | Toxicity analysis of CuII(atsm) in primary microglial cultures. Primary microglia were treated with the indicated concentrations of CuII(atsm) for 24 h. (A) MTT reduction and (B) LDH release were measured after 24 h. N = 2–5/group. ∗p < 0.05 compared to control.
FIGURE S2 | Inflammation decreases metallothionein-1 expression in microglia. Microglia were treated for 24 h with or without 15 ng/ml IFNγ and 10 ng/ml TNFα. mRNA expression of Mt1 was measured by qRT-PCR. N = 7/group. ∗p < 0.05 compared to control.
Aβ, amyloid beta; AD, Alzheimer’s disease; ALS, amyotrophic lateral sclerosis; BCA, bichinchoninic acid; CNS, central nervous system; CuII(gtsm), glyoxalbis(N(4)-methyl-3-thiosemicarbazonato)copper(II); CuII(atsm), diacetylbis(N(4)-methyl-3-thiosemicarbazonato)copper(II); DIV, days in vitro; DMSO, dimethyl sulfoxide; ELISA, enzyme-linked immunosorbent assay; FBS, fetal bovine serum; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; ICP-MS, inductively coupled plasma mass spectrometry; IFNγ, interferon gamma; IL, interleukin; iNOS, inducible nitric oxide synthase; LPS, lipopolysaccharide; MCP-1/CCL2, monocyte chemoattractant protein 1; MPIO-VCAM-1, microparticles of iron conjugated to vascular cell adhesion molecule 1 antibody; MRI, magnetic resonance imaging; MT1, metallothionein-1; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NO, nitric oxide; Nrf2, nuclear factor erythroid 2-like 2; PBS, phosphate-buffered saline; PD, Parkinson’s disease; PDTC, pyrrolidine dithiocarbamate; PFA, paraformaldehyde; qRT-PCR, quantitative real-time polymerase chain reaction; RT, room temperature; SSV, standard suspension vehicle; TNFα, tumor necrosis factor α; XFM, X-ray fluorescence microscope.
Adlard, P. A., Bica, L., White, A. R., Nurjono, M., Filiz, G., Crouch, P. J., et al. (2011). Metal ionophore treatment restores dendritic spine density and synaptic protein levels in a mouse model of alzheimer’s disease. PLoS One 6:e17669. doi: 10.1371/journal.pone.0017669
Adlard, P. A., West, A. K., and Vickers, J. C. (1998). Increased density of metallothionein I/II-immunopositive cortical glial cells in the early stages of alzheimer’s disease. Neurobiol. Dis. 5, 349–356. doi: 10.1006/nbdi.1998.0203
Agullo, L., Garcia, A., and Hidalgo, J. (1998). Metallothionein-I + II induction by zinc and copper in primary cultures of rat microglia. Neurochem. Int. 33, 237–242. doi: 10.1016/S0197-0186(98)00022-9
Banks, W. A., and Robinson, S. M. (2010). Minimal penetration of lipopolysaccharide across the murine blood-brain barrier. Brain Behav. Immun. 24, 102–109. doi: 10.1016/j.bbi.2009.09.001
Bayer, T. A., Schafer, S., Simons, A., Kemmling, A., Kamer, T., Tepest, R., et al. (2003). Dietary cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid abeta production in APP23 transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 100, 14187–14192. doi: 10.1073/pnas.2332818100
Bulcke, F., and Dringen, R. (2015). Copper oxide nanoparticles stimulate glycolytic flux and increase the cellular contents of glutathione and metallothioneins in cultured astrocytes. Neurochem. Res. 40, 15–26. doi: 10.1007/s11064-014-1458-0
Carrasco, J., Adlard, P., Cotman, C., Quintana, A., Penkowa, M., Xu, F., et al. (2006). Metallothionein-I and -III expression in animal models of alzheimer disease. Neuroscience 143, 911–922. doi: 10.1016/j.neuroscience.2006.08.054
Choo, X. Y., Alukaidey, L., White, A. R., and Grubman, A. (2013). Neuroinflammation and copper in alzheimer’s disease. Int. J. Alzheimers Dis. 2013, 145345. doi: 10.1155/2013/145345
Chung, R. S., Leung, Y. K., Butler, C. W., Chen, Y., Eaton, E. D., Pankhurst, M. W., et al. (2009). Metallothionein treatment attenuates microglial activation and expression of neurotoxic quinolinic acid following traumatic brain injury. Neurotox. Res. 15, 381–389. doi: 10.1007/s12640-009-9044-y
Crouch, P. J., Hung, L. W., Adlard, P. A., Cortes, M., Lal, V., Filiz, G., et al. (2009). Increasing cu bioavailability inhibits abeta oligomers and tau phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 106, 381–386. doi: 10.1073/pnas.0809057106
D’Ambrosi, N., and Rossi, L. (2015). Copper at synapse: release, binding and modulation of neurotransmission. Neurochem. Int. 90, 36–45. doi: 10.1016/j.neuint.2015.07.006
De, S. K., McMaster, M. T., and Andrews, G. K. (1990). Endotoxin induction of murine metallothionein gene expression. J. Biol. Chem. 265, 15267–15274.
Donnelly, P. S., Caragounis, A., Du, T., Laughton, K. M., Volitakis, I., Cherny, R. A., et al. (2008). Selective intracellular release of copper and zinc ions from bis(thiosemicarbazonato) complexes reduces levels of alzheimer disease amyloid-beta peptide. J. Biol. Chem. 283, 4568–4577. doi: 10.1074/jbc.M705957200
Donnelly, P. S., Liddell, J. R., Lim, S., Paterson, B. M., Cater, M. A., Savva, M. S., et al. (2012). An impaired mitochondrial electron transport chain increases retention of the hypoxia imaging agent diacetylbis(4-methylthiosemicarbazonato)copperII. Proc. Natl. Acad. Sci. U.S.A. 109, 47–52. doi: 10.1073/pnas.1116227108
Duguid, J. R., Bohmont, C. W., Liu, N. G., and Tourtellotte, W. W. (1989). Changes in brain gene expression shared by scrapie and alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 86, 7260–7264. doi: 10.1073/pnas.86.18.7260
Eimerbrink, M. J., White, J. D., Pendry, R. J., Hodges, S. L., Sadler, L. N., Wiles, J. D., et al. (2015). Administration of the inverse benzodiazepine agonist MRK-016 rescues acquisition and memory consolidation following peripheral administration of bacterial endotoxin. Behav. Brain Res. 288, 50–53. doi: 10.1016/j.bbr.2015.03.048
Elmore, M. R., Lee, R. J., West, B. L., and Green, K. N. (2015). Characterizing newly repopulated microglia in the adult mouse: impacts on animal behavior, cell morphology, and neuroinflammation. PLoS One 10:e0122912. doi: 10.1371/journal.pone.0122912
Everhardt Queen, A., Moerdyk-Schauwecker, M., McKee, L. M., Leamy, L. J., and Huet, Y. M. (2016). Differential expression of inflammatory cytokines and stress genes in male and female mice in response to a lipopolysaccharide challenge. PLoS One 11:e0152289. doi: 10.1371/journal.pone.0152289
Faux, N. G., Ritchie, C. W., Gunn, A., Rembach, A., Tsatsanis, A., Bedo, J., et al. (2010). PBT2 rapidly improves cognition in alzheimer’s disease: additional phase II analyses. J. Alzheimers Dis. 20, 509–516. doi: 10.3233/JAD-2010-1390
Gauberti, M., Montagne, A., Quenault, A., and Vivien, D. (2014). Molecular magnetic resonance imaging of brain-immune interactions. Front. Cell. Neurosci. 8:389. doi: 10.3389/fncel.2014.00389
Genoud, S., Roberts, B. R., Gunn, A. P., Halliday, G. M., Lewis, S. J. G., Ball, H. J., et al. (2017). Subcellular compartmentalisation of copper, iron, manganese, and zinc in the Parkinson’s disease brain. Metallomics 9, 1447–1455. doi: 10.1039/c7mt00244k
Gingras, B. A., Suprunchuk, T., and Bayley, C. H. (1962). The preparation of some thiosemicarbazones and their copper complexes: part III < br / >. Can. J. Chem. 40, 1053–1054. doi: 10.1139/v62-161
Graham, S. F., Nasaruddin, M. B., Carey, M., Holscher, C., McGuinness, B., Kehoe, P. G., et al. (2014). Age-associated changes of brain copper, iron, and zinc in alzheimer’s disease and dementia with lewy bodies. J. Alzheimers Dis. 42, 1407–1413. doi: 10.3233/JAD-140684
Grubman, A., James, S. A., James, J., Duncan, C., Volitakis, I., Hickey, J. L., et al. (2014a). X-ray fluorescence imaging reveals subcellular biometal disturbances in a childhood neurodegenerative disorder. Chem. Sci. 5, 2503–2516. doi: 10.1039/C4SC00316K
Grubman, A., Lidgerwood, G. E., Duncan, C., Bica, L., Tan, J. L., Parker, S. J., et al. (2014b). Deregulation of subcellular biometal homeostasis through loss of the metal transporter, Zip7, in a childhood neurodegenerative disorder. Acta Neuropathol. Commun. 2:25. doi: 10.1186/2051-5960-2-25
Hare, D., Fryer, F., Bence, P., Bishopa, D., and Doble, P. (2016). Characterisation of matrix-based polyatomic interference formation in laser ablation-inductively coupled plasma-mass spectrometry using dried micro-droplet ablation and its relevance for bioimaging. Anal. Methods 8, 7552–7556. doi: 10.1039/C6AY02545E
Hare, D. J., Kysenius, K., Paul, B., Knauer, B., Hutchinson, R. W., O’Connor, C., et al. (2017). Imaging metals in brain tissue by laser ablation - inductively coupled plasma - mass spectrometry (LA-ICP-MS). J. Vis. Exp. 119:55042. doi: 10.3791/55042
Hatori, Y., Yan, Y., Schmidt, K., Furukawa, E., Hasan, N. M., Yang, N., et al. (2016). Neuronal differentiation is associated with a redox-regulated increase of copper flow to the secretory pathway. Nat. Commun. 7:10640. doi: 10.1038/ncomms10640
He, M., Dong, H., Huang, Y., Lu, S., Zhang, S., Qian, Y., et al. (2016). Astrocyte-derived CCL2 is associated with M1 activation and recruitment of cultured microglial cells. Cell. Physiol. Biochem. 38, 859–870. doi: 10.1159/000443040
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Hidalgo, J., Penkowa, M., Espejo, C., Martinez-Caceres, E. M., Carrasco, J., Quintana, A., et al. (2006). Expression of metallothionein-I, -II, and -III in Alzheimer disease and animal models of neuroinflammation. Exp. Biol. Med. 231, 1450–1458. doi: 10.1177/153537020623100902
Hu, Z., Yu, F., Gong, P., Qiu, Y., Zhou, W., Cui, Y., et al. (2014). Subneurotoxic copper(II)-induced NF-kappaB-dependent microglial activation is associated with mitochondrial ROS. Toxicol. Appl. Pharmacol. 276, 95–103. doi: 10.1016/j.taap.2014.01.020
Huh, S., Lee, K., Yun, H. S., Paik, D. J., Kim, J. M., and Youn, J. (2007). Functions of metallothionein generating interleukin-10-producing regulatory CD4 + T cells potentiate suppression of collagen-induced arthritis. J. Microbiol. Biotechnol. 17, 348–358.
Hung, L. W., Villemagne, V. L., Cheng, L., Sherratt, N. A., Ayton, S., White, A. R., et al. (2012). The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of parkinson’s disease. J. Exp. Med. 209, 837–854. doi: 10.1084/jem.20112285
Huuskonen, M. T., Tuo, Q. Z., Loppi, S., Dhungana, H., Korhonen, P., McInnes, L. E., et al. (2017). The copper bis(thiosemicarbazone) complex CuII(atsm) is protective against cerebral ischemia through modulation of the inflammatory milieu. Neurotherapeutics 14, 519–532. doi: 10.1007/s13311-016-0504-9
Inoue, K., Takano, H., Shimada, A., and Satoh, M. (2009). Metallothionein as an anti-inflammatory mediator. Mediators Inflamm. 2009:101659. doi: 10.1155/2009/101659
Ishigami, N., Isoda, K., Adachi, T., Niida, T., Kujiraoka, T., Hakuno, D., et al. (2011). Deficiency of CuZn superoxide dismutase promotes inflammation and alters medial structure following vascular injury. J. Atheroscler. Thromb. 18, 1009–1017. doi: 10.5551/jat.9324
Jha, M. K., Lee, W. H., and Suk, K. (2016). Functional polarization of neuroglia: implications in neuroinflammation and neurological disorders. Biochem. Pharmacol. 103, 1–16. doi: 10.1016/j.bcp.2015.11.003
Juarez-Rebollar, D., Rios, C., Nava-Ruiz, C., and Mendez-Armenta, M. (2017). Metallothionein in brain disorders. Oxid. Med. Cell. Longev. 2017:5828056. doi: 10.1155/2017/5828056
Jurgens, H. A., Amancherla, K., and Johnson, R. W. (2012). Influenza infection induces neuroinflammation, alters hippocampal neuron morphology, and impairs cognition in adult mice. J. Neurosci. 32, 3958–3968. doi: 10.1523/JNEUROSCI.6389-11.2012
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of alzheimer’s disease. Cell 169, 1276.e–1290.e. doi: 10.1016/j.cell.2017.05.018
Kim, J. H., Nam, Y. P., Jeon, S. M., Han, H. S., and Suk, K. (2012). Amyloid neurotoxicity is attenuated by metallothionein: dual mechanisms at work. J. Neurochem. 121, 751–762. doi: 10.1111/j.1471-4159.2012.07725.x
Kirkham, R., Dunn, P., Kuczewski, A., Siddons, D., Dodanwela, R., Moorhead, G., et al. (2011). The maia spectroscopy detector system: engineering for integrated pulse capture, low-latency scanning and real-time processing. AIP Conf. Proc. 1234, 240–243.
Kitazawa, M., Cheng, D., and Laferla, F. M. (2009). Chronic copper exposure exacerbates both amyloid and tau pathology and selectively dysregulates cdk5 in a mouse model of AD. J. Neurochem. 108, 1550–1560. doi: 10.1111/j.1471-4159.2009.05901.x
Kitazawa, M., Hsu, H. W., and Medeiros, R. (2016). Copper exposure perturbs brain inflammatory responses and impairs clearance of amyloid-beta. Toxicol. Sci. 152, 194–204. doi: 10.1093/toxsci/kfw081
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Liu, Y., Liu, H., Chen, W., Yang, T., and Zhang, W. (2014). EOLA1 protects lipopolysaccharide induced IL-6 production and apoptosis by regulation of MT2A in human umbilical vein endothelial cells. Mol. Cell. Biochem. 395, 45–51. doi: 10.1007/s11010-014-2110-7
Louveau, A., Harris, T. H., and Kipnis, J. (2015). Revisiting the mechanisms of CNS immune privilege. Trends Immunol. 36, 569–577. doi: 10.1016/j.it.2015.08.006
Luo, K., Long, H., Xu, B., and Luo, Y. (2015). Metallothionein ameliorates burn sepsis partly via activation of akt signaling pathway in mice: a randomized animal study. World J. Emerg. Surg. 10:53. doi: 10.1186/s13017-015-0044-3
Madrigal, J. L., and Caso, J. R. (2014). The chemokine (C-C motif) ligand 2 in neuroinflammation and neurodegeneration. Adv. Exp. Med. Biol. 824, 209–219. doi: 10.1007/978-3-319-07320-0_15
Malm, T., Koistinaho, M., Muona, A., Magga, J., and Koistinaho, J. (2010). The role and therapeutic potential of monocytic cells in alzheimer’s disease. Glia 58, 889–900. doi: 10.1002/glia.20973
Malm, T. M., Iivonen, H., Goldsteins, G., Keksa-Goldsteine, V., Ahtoniemi, T., Kanninen, K., et al. (2007). Pyrrolidine dithiocarbamate activates akt and improves spatial learning in APP/PS1 mice without affecting beta-amyloid burden. J. Neurosci. 27, 3712–3721. doi: 10.1523/JNEUROSCI.0059-07.2007
Manso, Y., Adlard, P. A., Carrasco, J., Vasak, M., and Hidalgo, J. (2011). Metallothionein and brain inflammation. J. Biol. Inorg. Chem. 16, 1103–1113. doi: 10.1007/s00775-011-0802-y
Matsui, T., Svensson, C. I., Hirata, Y., Mizobata, K., Hua, X. Y., and Yaksh, T. L. (2010). Release of prostaglandin E(2) and nitric oxide from spinal microglia is dependent on activation of p38 mitogen-activated protein kinase. Anesth. Analg. 111, 554–560. doi: 10.1213/ANE.0b013e3181e3a2a2
Maureira, C., Letelier, J. C., Alvarez, O., Delgado, R., and Vergara, C. (2015). Copper enhances cellular and network excitabilities, and improves temporal processing in the rat hippocampus. Eur. J. Neurosci. 42, 3066–3080. doi: 10.1111/ejn.13104
Maynard, C. J., Cappai, R., Volitakis, I., Cherny, R. A., White, A. R., Beyreuther, K., et al. (2002). Overexpression of alzheimer’s disease amyloid-beta opposes the age-dependent elevations of brain copper and iron. J. Biol. Chem. 277, 44670–44676. doi: 10.1074/jbc.M204379200
McAllum, E. J., Lim, N. K., Hickey, J. L., Paterson, B. M., Donnelly, P. S., Li, Q. X., et al. (2013). Therapeutic effects of cu(II)(atsm) in the SOD1-G37R mouse model of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 586–590. doi: 10.3109/21678421.2013.824000
Michael, G. J., Esmailzadeh, S., Moran, L. B., Christian, L., Pearce, R. K., and Graeber, M. B. (2011). Up-regulation of metallothionein gene expression in parkinsonian astrocytes. Neurogenetics 12, 295–305. doi: 10.1007/s10048-011-0294-5
Michaelis, M., Nieswandt, B., Stegner, D., Eilers, J., and Kraft, R. (2015). STIM1, STIM2, and Orai1 regulate store-operated calcium entry and purinergic activation of microglia. Glia 63, 652–663. doi: 10.1002/glia.22775
Miller, L. M., Wang, Q., Telivala, T. P., Smith, R. J., Lanzirotti, A., and Miklossy, J. (2006). Synchrotron-based infrared and X-ray imaging shows focalized accumulation of cu and zn co-localized with beta-amyloid deposits in alzheimer’s disease. J. Struct. Biol. 155, 30–37. doi: 10.1016/j.jsb.2005.09.004
Montagne, A., Gauberti, M., Macrez, R., Jullienne, A., Briens, A., Raynaud, J. S., et al. (2012). Ultra-sensitive molecular MRI of cerebrovascular cell activation enables early detection of chronic central nervous system disorders. Neuroimage 63, 760–770. doi: 10.1016/j.neuroimage.2012.07.018
O’Sullivan, J. B., Ryan, K. M., Harkin, A., and Connor, T. J. (2010). Noradrenaline reuptake inhibitors inhibit expression of chemokines IP-10 and RANTES and cell adhesion molecules VCAM-1 and ICAM-1 in the CNS following a systemic inflammatory challenge. J. Neuroimmunol. 220, 34–42. doi: 10.1016/j.jneuroim.2009.12.007
Paterson, D., de Jonge, M. D., Howard, D., Lewis, W., McKinlay, J., Starritt, A., et al. (2011). The X-ray fluorescence microscopy beamline at the australian synchrotron. AIP Conf. Proc. 1365, 219–222. doi: 10.1063/1.3625343
Paul, B., Hare, D., Bishop, D., Paton, C., Nguyen, V. T., Cole, N., et al. (2015). Visualising mouse neuroanatomy and function by metal distribution using laser ablation-inductively coupled plasma-mass spectrometry imaging. Chem. Sci. 6, 5383–5393. doi: 10.1039/C5SC02231B
Pelizzoni, I., Zacchetti, D., Campanella, A., Grohovaz, F., and Codazzi, F. (2013). Iron uptake in quiescent and inflammation-activated astrocytes: a potentially neuroprotective control of iron burden. Biochim. Biophys. Acta 1832, 1326–1333. doi: 10.1016/j.bbadis.2013.04.007
Phinney, A. L., Drisaldi, B., Schmidt, S. D., Lugowski, S., Coronado, V., Liang, Y., et al. (2003). In vivo reduction of amyloid-beta by a mutant copper transporter. Proc. Natl. Acad. Sci. U.S.A. 100, 14193–14198. doi: 10.1073/pnas.2332851100
Pihlaja, R., Koistinaho, J., Kauppinen, R., Sandholm, J., Tanila, H., and Koistinaho, M. (2011). Multiple cellular and molecular mechanisms are involved in human abeta clearance by transplanted adult astrocytes. Glia 59, 1643–1657. doi: 10.1002/glia.21212
Piotrowska, A., Popiolek-Barczyk, K., Pavone, F., and Mika, J. (2017). Comparison of the expression changes after botulinum toxin type A and minocycline administration in lipopolysaccharide-stimulated rat microglial and astroglial cultures. Front. Cell. Infect. Microbiol. 7:141. doi: 10.3389/fcimb.2017.00141
Potter, E. G., Cheng, Y., Knight, J. B., Gordish-Dressman, H., and Natale, J. E. (2007). Basic science; metallothionein I and II attenuate the thalamic microglial response following traumatic axotomy in the immature brain. J. Neurotrauma 24, 28–42. doi: 10.1089/neu.2006.0056.R1
Puranik, R., Bao, S., Bonin, A. M., Kaur, R., Weder, J. E., Casbolt, L., et al. (2016). A novel class of copper(II)- and zinc(II)-bound non-steroidal anti-inflammatory drugs that inhibits acute inflammation in vivo. Cell Biosci. 6:9. doi: 10.1186/s13578-016-0076-8
Qin, L., Wu, X., Block, M. L., Liu, Y., Breese, G. R., Hong, J. S., et al. (2007). Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55, 453–462. doi: 10.1002/glia.20467
Raha, A. A., Vaishnav, R. A., Friedland, R. P., Bomford, A., and Raha-Chowdhury, R. (2013). The systemic iron-regulatory proteins hepcidin and ferroportin are reduced in the brain in alzheimer’s disease. Acta Neuropathol. Commun. 1:55. doi: 10.1186/2051-5960-1-55
Ransohoff, R. M. (2016). A polarizing question: do M1 and M2 microglia exist? Nat. Neurosci. 19, 987–991. doi: 10.1038/nn.4338
Rembach, A., Doecke, J. D., Roberts, B. R., Watt, A. D., Faux, N. G., Volitakis, I., et al. (2013). Longitudinal analysis of serum copper and ceruloplasmin in alzheimer’s disease. J. Alzheimers Dis. 34, 171–182. doi: 10.3233/JAD-121474
Roberts, B. R., Lim, N. K., McAllum, E. J., Donnelly, P. S., Hare, D. J., Doble, P. A., et al. (2014). Oral treatment with cu(II)(atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosci. 34, 8021–8031. doi: 10.1523/JNEUROSCI.4196-13.2014
Rossi-George, A., and Guo, C. J. (2016). Copper disrupts S-nitrosothiol signaling in activated BV2 microglia. Neurochem. Int. 99, 1–8. doi: 10.1016/j.neuint.2016.05.011
Ryan, C., Siddons, D., Kirkham, R., Li, Z., de Jonge, M., Paterson, D., et al. (2014). Maia X-ray fluorescence imaging: capturing detail in complex natural samples. J. Phys. Conf. Ser. 499:012002. doi: 10.1088/1742-6596/499/1/012002
Schrag, M., Mueller, C., Oyoyo, U., Smith, M. A., and Kirsch, W. M. (2011). Iron, zinc and copper in the alzheimer’s disease brain: a quantitative meta-analysis. some insight on the influence of citation bias on scientific opinion. Prog. Neurobiol. 94, 296–306. doi: 10.1016/j.pneurobio.2011.05.001
Sillevis Smitt, P. A., Mulder, T. P., Verspaget, H. W., Blaauwgeers, H. G., Troost, D., and de Jong, J. M. (1994). Metallothionein in amyotrophic lateral sclerosis. Biol. Signals 3, 193–197. doi: 10.1159/000109545
Singh, I., Sagare, A. P., Coma, M., Perlmutter, D., Gelein, R., Bell, R. D., et al. (2013). Low levels of copper disrupt brain amyloid-beta homeostasis by altering its production and clearance. Proc. Natl. Acad. Sci. U.S.A. 110, 14771–14776. doi: 10.1073/pnas.1302212110
Sirois, J. J., Padgitt-Cobb, L., Gallegos, M. A., Beckman, J. S., Beaudry, C. M., and Hurst, J. K. (2018). Oxidative release of copper from pharmacologic copper bis(thiosemicarbazonato) compounds. Inorg. Chem. 57, 8923–8932. doi: 10.1021/acs.inorgchem.8b00853
Soon, C. P., Donnelly, P. S., Turner, B. J., Hung, L. W., Crouch, P. J., Sherratt, N. A., et al. (2011). Diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. J. Biol. Chem. 286, 44035–44044. doi: 10.1074/jbc.M111.274407
Spangenberg, E. E., and Green, K. N. (2016). Inflammation in alzheimer’s disease: lessons learned from microglia-depletion models. Brain Behav. Immun. 61, 1–11. doi: 10.1016/j.bbi.2016.07.003
Squitti, R., Rossini, P. M., Cassetta, E., Moffa, F., Pasqualetti, P., Cortesi, M., et al. (2002). D-penicillamine reduces serum oxidative stress in alzheimer’s disease patients. Eur. J. Clin. Invest. 32, 51–59. doi: 10.1046/j.1365-2362.2002.00933.x
Terwel, D., Loschmann, Y. N., Schmidt, H. H., Scholer, H. R., Cantz, T., and Heneka, M. T. (2011). Neuroinflammatory and behavioural changes in the Atp7B mutant mouse model of wilson’s disease. J. Neurochem. 118, 105–112. doi: 10.1111/j.1471-4159.2011.07278.x
Thomson, C. A., McColl, A., Cavanagh, J., and Graham, G. J. (2014). Peripheral inflammation is associated with remote global gene expression changes in the brain. J. Neuroinflammation 11:73. doi: 10.1186/1742-2094-11-73
Tokuda, E., Okawa, E., Watanabe, S., and Ono, S. (2014). Overexpression of metallothionein-I, a copper-regulating protein, attenuates intracellular copper dyshomeostasis and extends lifespan in a mouse model of amyotrophic lateral sclerosis caused by mutant superoxide dismutase-1. Hum. Mol. Genet. 23, 1271–1285. doi: 10.1093/hmg/ddt517
Torres, J. B., Andreozzi, E. M., Dunn, J. T., Siddique, M., Szanda, I., Howlett, D. R., et al. (2016). PET imaging of copper trafficking in a mouse model of alzheimer disease. J. Nucl. Med. 57, 109–114. doi: 10.2967/jnumed.115.162370
Urrutia, P., Aguirre, P., Esparza, A., Tapia, V., Mena, N. P., Arredondo, M., et al. (2013). Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 126, 541–549. doi: 10.1111/jnc.12244
Venigalla, M., Sonego, S., Gyengesi, E., Sharman, M. J., and Munch, G. (2016). Novel promising therapeutics against chronic neuroinflammation and neurodegeneration in alzheimer’s disease. Neurochem. Int. 95, 63–74. doi: 10.1016/j.neuint.2015.10.011
Wei, H., Frei, B., Beckman, J. S., and Zhang, W. J. (2011). Copper chelation by tetrathiomolybdate inhibits lipopolysaccharide-induced inflammatory responses in vivo. Am. J. Physiol. Heart Circ. Physiol. 301, H712–H720. doi: 10.1152/ajpheart.01299.2010
Wei, H., Zhang, W. J., Leboeuf, R., and Frei, B. (2014). Copper induces–and copper chelation by tetrathiomolybdate inhibits–endothelial activation in vitro. Redox Rep. 19, 40–48. doi: 10.1179/1351000213Y.0000000070
Yang, L., Sturgeon, R. E., and Mester, Z. (2005). Quantitation of trace metals in liquid samples by dried-droplet laser ablation inductively coupled plasma mass spectrometry. Anal. Chem. 77, 2971–2977. doi: 10.1021/ac048275a
Yu, F., Gong, P., Hu, Z., Qiu, Y., Cui, Y., Gao, X., et al. (2015). Cu(II) enhances the effect of alzheimer’s amyloid-beta peptide on microglial activation. J. Neuroinflammation 12:122. doi: 10.1186/s12974-015-0343-3
Zhang, Z., Hou, L., Song, J. L., Song, N., Sun, Y. J., Lin, X., et al. (2014). Pro-inflammatory cytokine-mediated ferroportin down-regulation contributes to the nigral iron accumulation in lipopolysaccharide-induced Parkinsonian models. Neuroscience 257, 20–30. doi: 10.1016/j.neuroscience.2013.09.037
Zhou, S., Wang, Y., Tan, Y., Cai, X., Cai, L., Cai, J., et al. (2014). Deletion of metallothionein exacerbates intermittent hypoxia-induced oxidative and inflammatory injury in aorta. Oxid. Med. Cell. Longev. 2014:141053. doi: 10.1155/2014/141053
Keywords: microglia, astrocyte, inflammation, neurodegeneration, copper
Citation: Choo XY, Liddell JR, Huuskonen MT, Grubman A, Moujalled D, Roberts J, Kysenius K, Patten L, Quek H, Oikari LE, Duncan C, James SA, McInnes LE, Hayne DJ, Donnelly PS, Pollari E, Vähätalo S, Lejavová K, Kettunen MI, Malm T, Koistinaho J, White AR and Kanninen KM (2018) CuII(atsm) Attenuates Neuroinflammation. Front. Neurosci. 12:668. doi: 10.3389/fnins.2018.00668
Received: 14 February 2018; Accepted: 05 September 2018;
Published: 24 September 2018.
Edited by:
Wendy Noble, King’s College London, United KingdomReviewed by:
Jessica L. Teeling, University of Southampton, United KingdomCopyright © 2018 Choo, Liddell, Huuskonen, Grubman, Moujalled, Roberts, Kysenius, Patten, Quek, Oikari, Duncan, James, McInnes, Hayne, Donnelly, Pollari, Vähätalo, Lejavová, Kettunen, Malm, Koistinaho, White and Kanninen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anthony R. White, dG9ueS53aGl0ZUBxaW1yYmVyZ2hvZmVyLmVkdS5hdQ== Katja M. Kanninen, a2F0amEua2FubmluZW5AdWVmLmZp
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.