 Rosemary M. Clark
Rosemary M. Clark Mariana Brizuela
Mariana Brizuela Catherine A. Blizzard
Catherine A. Blizzard Tracey C. Dickson
Tracey C. Dickson
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Cell. Neurosci. , 28 September 2018
Sec. Cellular Neurophysiology
Volume 12 - 2018 | https://doi.org/10.3389/fncel.2018.00328
This article is part of the Research Topic Motor System and Motor Diseases: from Molecules to Circuits View all 28 articles
Cortical interneurons play a crucial role in regulating inhibitory-excitatory balance in brain circuits, filtering synaptic information and dictating the activity of pyramidal cells through the release of GABA. In the fatal motor neuron (MN) disease, amyotrophic lateral sclerosis (ALS), an imbalance between excitation and inhibition is an early event in the motor cortex, preceding the development of overt clinical symptoms. Patients with both sporadic and familial forms of the disease exhibit reduced cortical inhibition, including patients with mutations in the copper/zinc superoxide-dismutase-1 (SOD1) gene. In this study, we investigated the influence of the familial disease-causing hSOD1-G93A ALS mutation on cortical interneurons in neuronal networks. We performed whole-cell patch-clamp recordings and neurobiotin tracing from GFP positive interneurons in primary cortical cultures derived from Gad67-GFP::hSOD1G93A mouse embryos. Targeted recordings revealed no overt differences in the passive properties of Gad67-GFP::hSOD1G93A interneurons, however the peak outward current was significantly diminished and cells were less excitable compared to Gad67-GFP::WT controls. Post hoc neurite reconstruction identified a significantly increased morphological complexity of the Gad67-GFP::hSOD1G93A interneuron neurite arbor compared to Gad67-GFP::WT controls. Our results from the SOD1 model suggest that cortical interneurons have electrophysiological and morphological alterations that could contribute to attenuated inhibitory function in the disease. Determining if these phenomena are driven by the network or represent intrinsic alteration of the interneuron may help explain the emergence of inhibitory susceptibility and ultimately disrupted excitability, in ALS.
Amyotrophic lateral sclerosis (ALS) is the most common and severe form of motor neuron (MN) disease. It is clinically characterized by selective loss of the upper and lower MNs in the primary motor cortex and spinal cord, resulting in progressive motor system failure and death within 3–5 years of diagnosis (Talbot, 2014; Brown and Al-Chalabi, 2017). This currently incurable disease is clinically heterogeneous and its etiology remains unknown. However, accumulating evidence from several clinical and experimental studies suggests the disease pathogenesis may center on altered regulation of MN excitability (Turner and Kiernan, 2012; Clark et al., 2015; Geevasinga et al., 2016).
In both sporadic and familial forms of ALS, patients have been found to present with neurophysiological alterations described as hyperexcitability (Vucic and Kiernan, 2006; Vucic et al., 2008; Geevasinga et al., 2015). Originating in the motor cortex and preceding detectable lower MN dysfunction and symptom onset (Menon et al., 2015), it is proposed that hyperexcitability enhances the susceptibility of MNs to cell death through glutamatergic excitotoxicity (Blizzard et al., 2015; Eisen et al., 2017). In ALS, hyperexcitability likely results from dysfunctional inhibition exerted by GABAergic interneurons, as well as intrinsic changes to sodium (Na+) and potassium (K+) channel function on MNs (Geevasinga et al., 2016; Do-Ha et al., 2018).
Evidence for ion channel dysfunction is highlighted by a convergent hyperexcitability phenotype in patient-derived MNs, including TARDBP, C9ORF72 and superoxide-dismutase-1 (SOD1) mutation carriers (Wainger et al., 2014; Devlin et al., 2015). TDP-43 and SOD1 ALS rodent models identify changes in the excitability of MNs prior to symptoms (Fogarty et al., 2015; Handley et al., 2017). However, more recent studies recognize progressive alterations in the number and excitability of interneurons throughout the disease course in TDP-43 and SOD1 models (Zhang et al., 2016; Clark et al., 2017; Kim et al., 2017). Clinical imaging studies indicate loss of inhibitory activity is a common and early feature of cortical hyperexcitability (Menon et al., 2015), and a key determinant of clinical disease progression (Shibuya et al., 2016). As such, there is a growing body of evidence to suggest dysregulated inhibition, presumably mediated by cortical interneurons, may drive an excitatory/inhibitory imbalance in ALS.
Here, we focused on the potential for the familial hSOD1G93A mutation to influence firing properties and morphology of cortical interneurons in Gad67-GFP::hSOD1G93A cultures. Structural alteration of pyramidal neurons is demonstrated in hSOD1G93A studies (Jara et al., 2012; Fogarty et al., 2016; Saba et al., 2016), but few studies report changes to the morphological fine structure of interneurons (Clark et al., 2017). Additionally, mutant SOD1 is theorized to mediate non-cell autonomous pathogenicity through perturbed function of multiple cell types early in development (Kuo et al., 2004; van Zundert et al., 2008; Martin et al., 2013; Wainger et al., 2014; Devlin et al., 2015). As such, we hypothesize that the hSOD1G93A mutation can perturb the excitability and neurite structure of cortical interneurons in culture.
All procedures were approved by the Animal Ethics Committee of the University of Tasmania (#A0013586) and conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. Gad67-GFP knock-in transgenic mice (Tamamaki et al., 2003) express green fluorescent protein (GFP) under the interneuron-specific Gad67 promoter. High copy number hSOD1G93A mice were maintained and fully backcrossed to the C57BL/6 background (Gurney et al., 1994; B6.Cg-Tg(SOD1G93A)1Gur.J1, Jackson Laboratories, Bar Harbor, ME, USA).
Primary cortical cultures were prepared by carefully dissecting individual mouse embryo neocortices to enrich for neurons as previously described (Brizuela et al., 2015, 2017), with some modifications. Cells were plated at 3.5 × 104 cells/mm2 for up to 12 days and genotyped for GFP (Brizuela et al., 2015) and the hSOD1G93A mutation (Leitner et al., 2009). Whole-cell voltage and current clamp recordings were performed as previously described (Brizuela et al., 2015), with minor modifications. Glass capillaries (impedance 7–9 Ω) were filled with intracellular solution containing neurobiotin for post hoc cell identification. Voltage responses to current injection were recorded from the cell’s resting potential (applying 25 pA steps for 200 ms from −100 pA to 500 pA). Recordings were filtered at 5 kHz and sampled at 10 kHz and terminated when access resistance was ≥15 MΩ. Voltage-gated sodium and potassium currents were investigated as previously described (Brizuela et al., 2015). Data was analyzed using the programs Igor (WaveMetrics, USA) and Axograph (Axograph Scientific, Australia).
Cultures were processed for immunocytochemistry as previously described (Brizuela et al., 2015). Primary antibodies (rat anti-GFP, 1:3,000, Nacalai tesque, RRID: AB_10013361). Secondary antibodies (Alexa Fluor anti-rat 488, 1:1,000, Molecular Probes, RRID: AB_2534074). Neurobiotin-filled interneurons were labeled with streptavidin-546 and imaged using a UltraView Spinning disc confocal microscope (Perkin Elmer) with Velocity Software (Velocity v6 3.0, 2013, Perkin Elmer).
Cell processes were traced through Z-stack series (5 μm, 0.5 μm intervals) with Neurolucida™ (MBF Bioscience, VT, USA) and assessed using branched structure and sholl analyses with Neurolucida Explorer 11 (MBF Bioscience). Interneurons that met electrophysiology inclusion criteria were morphometrically assessed.
All statistical analysis was performed in GraphPad Prism (Version 6.0c, GraphPad Software La Jolla, CA, USA). Unless otherwise stated, comparisons utilized Mann-Whitney tests, after applying d’Agostino and Pearson’s normality test, results expressed as median with interquartile range. Sholl analysis and current-frequency relationships used two-way ANOVA. Data was considered significant at *p < 0.05.
Previous studies have demonstrated that mutant hSOD1G93A can perturb neuronal excitability during development, which may contribute to cellular vulnerability in disease (Kuo et al., 2004; van Zundert et al., 2008; Martin et al., 2013; Wainger et al., 2014; Devlin et al., 2015). Here, we focus on investigating cortical interneuron excitability in neuronal culture by examining firing patterns and membrane properties of interneurons in Gad67-GFP::hSOD1G93A cultures.
We used whole-cell recordings from multipolar cortical interneurons to characterize the effect of the hSOD1G93A mutation on cortical interneuron excitability in the presence of synaptic currents. Selection of GFP-positive neurons ensured studied cells were interneurons, with neurobiotin labeling used for post hoc identification (Figure 1A). We found the hSOD1G93A mutation significantly affected firing properties of cortical interneurons, decreasing the number of action potentials (APs; Figure 1B) in response to 200 ms depolarizing current step injections at 350pA (Figure 1C; 8.675 ± 2.752pA for hSOD1G93A, 27.42 ± 3.888pA for WT; p < 0.05, two-way ANOVA, Bonferroni post hoc; F(8,47) = 2.305, p = 0.0357, interaction between genotype and frequency in two-way ANOVA). There was no significant difference in passive electrophysiological properties, including the resting membrane potential (RMP; Figure 1D; −66.5 mv, −67.30 to −63.82 for WT (n = 16) v −63.73, −68.75 to −58.71 for hSOD1G93A (n = 11), p > 0.05), capacitance (Figure 1E; 32.50 pF, 27.17–38.20 for WT (n = 16) v 32.00, 25.39–35.89 for hSOD1G93A (n = 11), p > 0.05) and input resistance (Figure 1F; 463 MΩ, 394.1–652.1 for WT (n = 16) v 378.0, 255.9–513.4 for hSOD1G93A (n = 11), p > 0.05). Further alterations in excitability were not detected through investigation of AP characteristics, including the spike threshold (Figure 1G; −35.24 mv, −41.59 to −31.07 for WT (n = 16) v −33.43, −36.52 to −29.64 for hSOD1G93A (n = 11), p > 0.05), the minimal stimulus required to initiate an AP (Figure 1H; rheobase, 150.0 pA, 156.2–186.1 for WT (n = 16) v 187.5, 158.6–206.4 for hSOD1G93A (n = 11), p > 0.05) and the AP duration (Figure 1I; 5.504 ms, 4.649–7.086 for WT (n = 16) v 6.066, 4.849–7.563 for hSOD1G93A (n = 11), p > 0.05). However, investigation of voltage-dependent currents identified a significant decrease in the peak outward current (Figure 1J; 1.644 nA, 1.524–2.442 for WT (n = 16) v 1.151, 0.6636–1.624 for hSOD1G93A (n = 11), p < 0.05), while there was no significant difference in the peak inward current (Figure 1K; −1.575 nA, −2.691 to −1.207 for WT (n = 16) v −1.598, −2.214 to −1.273 for hSOD1G93A (n = 11), p > 0.05). Taken together, this data suggests that cortical interneurons are less excitable and have decreased peak outward currents in the presence of the hSOD1G93A mutation.
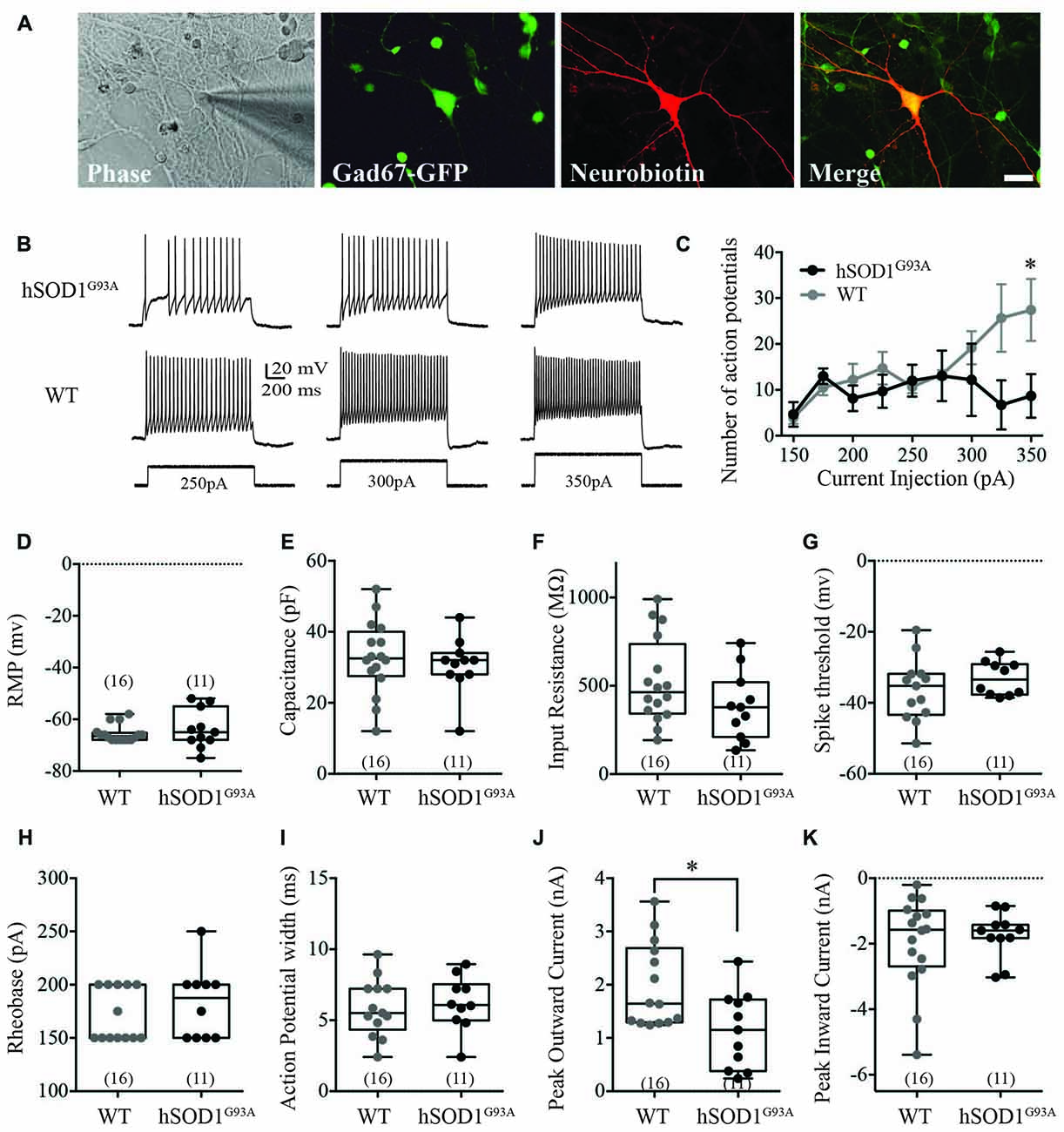
Figure 1. Electrophysiological characterization of Gad67-GFP positive hSOD1G93A cortical interneurons in vitro. Primary neuronal cultures were prepared from E15.5 Gad67-GFP::hSOD1G93A embryos as described in “Methods” section. (A) Whole cell patch-clamped interneurons were positive for Gad67-GFP and post hoc labeled for neurobiotin-streptavidin-546. (B,C) Representative voltage traces (B) and current-spike frequency relationship (C) measured from GFP-positive interneurons in 12 DIV cortical culture (Gad67-GFP::hSOD1G93A, n = 11 cells from five cultures; Gad67-GFP::WT, n = 16 cells from five cultures; *p < 0.05, two-way ANOVA), error bars show mean ± SEM. (D–K) The active and passive electrophysiological properties of GFP-positive interneurons, including: the resting membrane potential (RMP; D), capacitance (E), input resistance (F), threshold to fire (G), rheobase (H), action potential (AP) width (I), peak inward current (J) and significantly decreased peak outward current (K; Gad67-GFP::hSOD1G93A, n = 11 cells from five cultures; Gad67-GFP::WT, n = 16 cells from five cultures; *p < 0.05, Mann-Whitney test). Box-and-whisker plots show the interquartile range.
GABAergic interneurons have dynamic axonal structures that can alter in response to changes in activity, to modify post-synaptic targets (Flores and Méndez, 2014). Given the role of interneuron morphology in shaping inhibition during development (Huang, 2009; Le Magueresse and Monyer, 2013), we determined whether there was evidence of changes to the interneuron neurite arbor in Gad67-GFP::hSOD1G93A cultures.
Interneuron morphology was characterized by tracing the three-dimensional structure of neurobiotin-filled GFP-positive interneuron processes in WT and hSOD1G93A cultures. Initial examination revealed a distinct increase in the complexity of hSOD1G93A interneurons (Figure 2A). Assessing the characteristics of the interneuron neurite field with sholl analysis (intersections per concentric shell placed at 10 μm radiating outward from the soma), we identified a significant increase in the number of processes on hSOD1G93A interneurons at 110–150 μm from the cell soma compared to WT (p < 0.05, two-way ANOVA, Bonferroni post hoc; F(1,849) = 24.77, p = 0.0001, main effect of genotype in two-way ANOVA; see Figure 2B). The distance from the soma also independently influenced the neurite arbor complexity (F(40,849) = 10.58, p = 0.0001, main effect of distance in two-way ANOVA). To investigate the increased complexity of hSOD1G93A interneurons we next used branched structural analyses. As shown in Figure 2C, the path length was significantly increased in hSOD1G93A interneurons (2363 μm, 1914–3020 for WT (n = 20) v 3380, 2673–4929 for hSOD1G93A (n = 11), p < 0.05). However, there was also a significant increase in total branch numbers (Figure 2D; 91.00, 76.65–121.0 for WT (n = 20) v 117.0, 105.1–166.7 for hSOD1G93A (n = 11), p < 0.05) and the highest order of branches on hSOD1G93A interneurons (Figure 2E; 10.00, 9.77–12.83 for WT (n = 20) v 14.0, 12.16–19.30 for hSOD1G93A (n = 11), p < 0.05). These results suggest that the presence of the hSOD1G93A mutation can result in early inhibitory arbor structural remodeling in cortical networks.
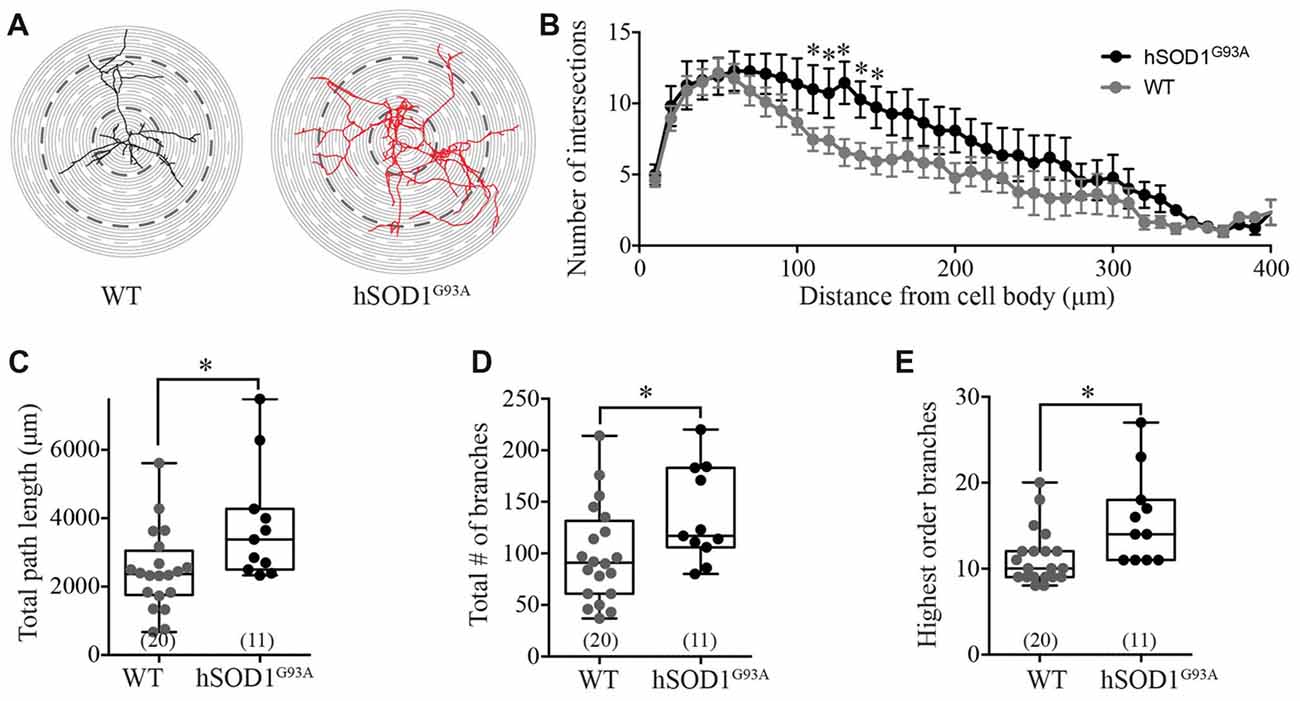
Figure 2. Morphological characterization of Gad67-GFP positive hSOD1G93A cortical interneurons in vitro. (A) Representative images of patched interneurons reconstructed from post hoc neurobiotin-streptavidin labeling in Gad67-GFP::hSOD1G93A and Gad67-GFP::WT cultures. Each concentric circle represents 10 μm, and each dashed line represents 50 μm from the cell soma. (B) Sholl analysis denoting the morphological complexity of GFP-positive interneurons as measured by the average number of neurites intersecting with concentric circles placed at 10 μm intervals from the cell soma (Gad67-GFP::hSOD1G93A, n = 11 cells from five cultures; Gad67-GFP::WT, n = 20 cells from five cultures; *p < 0.05, two-way ANOVA), error bars show mean ± SEM. (C–E) Histograms quantifying significantly increased total neurite path length (μm; C), total branch number (D) and average number of branches (E) of Gad67-GFP::hSOD1G93A interneurons compared to Gad67-GFP::WT controls (Gad67-GFP::hSOD1G93A, n = 11 cells from 5 cultures; Gad67-GFP::WT, n = 20 cells from five cultures; *p < 0.05, Mann-Whitney test). Box-and-whisker plots show the interquartile range.
This study is the first investigation of changes in the excitability and neurite arborization of cortical interneurons in neocortical embryonic cultures from a SOD1 rodent model. The principal finding is that excitability of cultured cortical interneurons may be altered by the hSOD1G93A mutation, and that changes to the complexity of the neurite structure accompany this effect in vitro. Specifically, attenuation of excitability is supported by differences in the current-voltage relationship between WT and hSOD1G93A mouse interneurons, and by the observation of reduced peak outward currents in hSOD1G93A interneurons. Interestingly, outward potassium currents are affected in MNs derived from SOD1 ALS patients (Wainger et al., 2014), and by mutant SOD1 oligomers (Zhang et al., 2017), which could account for the changes in the hSOD1G93A cortical interneurons.
However, a number of studies highlight increased excitability of cortical pyramidal neuron populations both in vivo and in vitro (Pieri et al., 2009; Fogarty et al., 2015), which could drive changes in the morphological development of cortical interneurons. The inhibitory neurite arbor can undergo activity-dependent structural remodeling (Bartolini et al., 2013; Babij and De Marco Garcia, 2016) and the level of network activity can produce subtle but significant changes to interneuron morphology (Schuemann et al., 2013). Importantly, in networks deprived of activity, cortical interneurons have been shown to extend collaterals beyond their normal projection range (Marik et al., 2010). While the data presented here are correlational not causal, altered structural and electrophysiological properties of cortical interneurons provide evidence that hSOD1G93A could disturb the inhibitory/excitatory balance in developing neuronal networks, early in the disease pathogenesis.
Previous studies highlight progressive and differential interneuron involvement in the hSOD1G93A mouse model (Clark et al., 2017; Kim et al., 2017). In particular, Kim et al support altered interneuron excitability in hSOD1G93A models, finding progressive hyperexcitability with disease progression in vivo (Kim et al., 2017). In the current study we find evidence for intrinsically hypoexcitability. These data support a role for the hSOD1G93A mutation in the early perturbation of inhibitory neuron populations in the disease. However, to determine the effect on functional inhibition, future studies should establish if synaptic excitability is changed to compensate for abnormal firing properties which could normalize firing rates (Turrigiano, 2008). In addition, further experimentation will be required to delineate if this is the cause or consequence of perturbed excitatory neuron excitability in the disease.
All data generated or analyzed in this study are available from the corresponding author upon request.
RC performed cortical culturing, genotyping, immunocytochemistry, data analysis and was a major contributor in writing the manuscript. MB performed electrophysiology and data analysis. CB, MB and TD contributed to experimental design, drafting and editing of manuscript. All authors read and approved the final manuscript.
This work was supported by the Stanford Family Motor Neuron Disease Collaboration Grant (#D0023678) and the Betty Laidlaw Motor Neuron Disease Research Grant (#BLP17), Motor Neuron Disease Research Institute of Australia; the Tasmanian Masonic Centenary Medical Research Foundation (#D0019780); Motor Neuron Disease Research Institute of Australia PhD Scholarship (C0021191); the Select Foundation Fellowship (#D0020742); Australian Research Council Discovery Early Career Research Award (#DE170101514) and the Broadreach Holdings Post-doctoral Medical Research Fellowship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SG and the handling Editor declared their shared affiliation.
We authors would like to thank Profs. John Bekkers (Australian National University) and Nobuaki Tamamaki (Kyoto University) for access to Gad67-GFP mice. We thank our colleagues Dr. Kaylene Young and Dr. Kimberley Pitman, and in particular Dr. Katherine Lewis for manuscript feedback.
Babij, R., and De Marco Garcia, N. (2016). Neuronal activity controls the development of interneurons in the somatosensory cortex. Front. Biol. 11, 459–470. doi: 10.1007/s11515-016-1427-x
Bartolini, G., Ciceri, G., and Marin, O. (2013). Integration of GABAergic interneurons into cortical cell assemblies: lessons from embryos and adults. Neuron 79, 849–864. doi: 10.1016/j.neuron.2013.08.014
Blizzard, C. A., Southam, K. A., Dawkins, E., Lewis, K. E., King, A. E., Clark, J. A., et al. (2015). Identifying the primary site of pathogenesis in amyotrophic lateral sclerosis—vulnerability of lower motor neurons to proximal excitotoxicity. Dis. Model. Mech. 8, 215–224. doi: 10.1242/dmm.018606
Brizuela, M., Blizzard, C. A., Chuckowree, J. A., Dawkins, E., Gasperini, R. J., Young, K. M., et al. (2015). The microtubule-stabilizing drug Epothilone D increases axonal sprouting following transection injury in vitro. Mol. Cell. Neurosci. 66, 129–140. doi: 10.1016/j.mcn.2015.02.006
Brizuela, M., Blizzard, C. A., Chuckowree, J. A., Pitman, K. A., Young, K. M., and Dickson, T. (2017). Mild traumatic brain injury leads to decreased inhibition and a differential response of calretinin positive interneurons in the injured cortex. J. Neurotrauma 34, 2504–2517. doi: 10.1089/neu.2017.4977
Brown, R. H., and Al-Chalabi, A. (2017). Amyotrophic lateral sclerosis. N. Engl. J. Med. 377, 162–172. doi: 10.1056/NEJMra1603471
Clark, R. M., Blizzard, C., and Dickson, T. (2015). Inhibitory dysfunction in amyotrophic lateral sclerosis: future therapeutic opportunities. Neurodegener. Dis. Manag. 5, 511–525. doi: 10.2217/nmt.15.49
Clark, R. M., Blizzard, C. A., Young, K. M., King, A. E., and Dickson, T. C. (2017). Calretinin and Neuropeptide Y interneurons are differentially altered in the motor cortex of the SOD1G93A mouse model of ALS. Sci. Rep. 7:44461. doi: 10.1038/srep44461
Devlin, A. C., Burr, K., Borooah, S., Foster, J. D., Cleary, E. M., Geti, I., et al. (2015). Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat. Commun. 6:5999. doi: 10.1038/ncomms6999
Do-Ha, D., Buskila, Y., and Ooi, L. (2018). Impairments in motor neurons, interneurons and astrocytes contribute to hyperexcitability in ALS: underlying mechanisms and paths to therapy. Mol. Neurobiol. 55, 1410–1418. doi: 10.1007/s12035-017-0392-y
Eisen, A., Braak, H., Del Tredici, K., Lemon, R., Ludolph, A. C., and Kiernan, M. C. (2017). Cortical influences drive amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 88, 917–924. doi: 10.1136/jnnp-2017-315573
Flores, C. E., and Méndez, P. (2014). Shaping inhibition: activity dependent structural plasticity of GABAergic synapses. Front. Cell. Neurosci. 8:327. doi: 10.3389/fncel.2014.00327
Fogarty, M. J., Mu, E. W., Noakes, P. G., Lavidis, N. A., and Bellingham, M. C. (2016). Marked changes in dendritic structure and spine density precede significant neuronal death in vulnerable cortical pyramidal neuron populations in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 4:77. doi: 10.1186/s40478-016-0347-y
Fogarty, M. J., Noakes, P. G., and Bellingham, M. C. (2015). Motor cortex layer V pyramidal neurons exhibit dendritic regression, spine loss, and increased synaptic excitation in the presymptomatic hSOD1G93A mouse model of amyotrophic lateral sclerosis. J. Neurosci. 35, 643–647. doi: 10.1523/JNEUROSCI.3483-14.2015
Geevasinga, N., Menon, P., Nicholson, G. A., Ng, K., Howells, J., Kril, J. J., et al. (2015). Cortical function in asymptomatic carriers and patients with C9orf72 amyotrophic lateral sclerosis. JAMA Neurol 72, 1268–1274. doi: 10.1001/jamaneurol.2015.1872
Geevasinga, N., Menon, P., Özdinler, P. H., Kiernan, M. C., and Vucic, S. (2016). Pathophysiological and diagnostic implications of cortical dysfunction in ALS. Nat. Rev. Neurol. 12, 651–661. doi: 10.1038/nrneurol.2016.140
Gurney, M., Pu, H., Chiu, A., Dal Canto, M., Polchow, C., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264, 1772–1775. doi: 10.1126/science.8209258
Handley, E. E., Pitman, K. A., Dawkins, E., Young, K. M., Clark, R. M., Jiang, T. C., et al. (2017). Synapse dysfunction of layer V pyramidal neurons precedes neurodegeneration in a mouse model of TDP-43 proteinopathies. Cereb. Cortex 27, 3630–3647. doi: 10.1093/cercor/bhw185
Huang, Z. J. (2009). Activity-dependent development of inhibitory synapses and innervation pattern: role of GABA signalling and beyond. J. Physiol. 587, 1881–1888. doi: 10.1113/jphysiol.2008.168211
Jara, J. H., Villa, S. R., Khan, N. A., Bohn, M. C., and Ozdinler, P. H. (2012). AAV2 mediated retrograde transduction of corticospinal motor neurons reveals initial and selective apical dendrite degeneration in ALS. Neurobiol. Dis. 47, 174–183. doi: 10.1016/j.nbd.2012.03.036
Kim, J., Hughes, E. G., Shetty, A. S., Arlotta, P., Goff, L. A., Bergles, D. E., et al. (2017). Changes in the excitability of neocortical neurons in a mouse model of amyotrophic lateral sclerosis are not specific to corticospinal neurons and are modulated by advancing disease. J. Neurosci. 37, 9037–9053. doi: 10.1523/JNEUROSCI.0811-17.2017
Kuo, J. J., Schonewille, M., Siddique, T., Schults, A. N., Fu, R., Bär, P. R., et al. (2004). Hyperexcitability of cultured spinal motoneurons from presymptomatic ALS mice. J. Neurophysiol. 91, 571–575. doi: 10.1152/jn.00665.2003
Le Magueresse, C., and Monyer, H. (2013). GABAergic interneurons shape the functional maturation of the cortex. Neuron 77, 388–405. doi: 10.1016/j.neuron.2013.01.011
Leitner, M., Menzies, S., and Lutz, C. (2009). Working with ALS mice: Guidelines for Preclinical Testing and Colony Management. Bar Harbor, ME: Prize4Life and The Jackson Laboratory.
Marik, S. A., Yamahachi, H., McManus, J. N., Szabo, G., and Gilbert, C. D. (2010). Axonal dynamics of excitatory and inhibitory neurons in somatosensory cortex. PLoS Biol. 8:e1000395. doi: 10.1371/journal.pbio.1000395
Martin, E., Cazenave, W., Cattaert, D., and Branchereau, P. (2013). Embryonic alteration of motoneuronal morphology induces hyperexcitability in the mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 54, 116–126. doi: 10.1016/j.nbd.2013.02.011
Menon, P., Kiernan, M. C., and Vucic, S. (2015). Cortical hyperexcitability precedes lower motor neuron dysfunction in ALS. Clin. Neurophysiol. 126, 803–809. doi: 10.1016/j.clinph.2014.04.023
Pieri, M., Carunchio, I., Curcio, L., Mercuri, N. B., and Zona, C. (2009). Increased persistent sodium current determines cortical hyperexcitability in a genetic model of amyotrophic lateral sclerosis. Exp. Neurol. 215, 368–379. doi: 10.1016/j.expneurol.2008.11.002
Saba, L., Viscomi, M. T., Caioli, S., Pignataro, A., Bisicchia, E., Pieri, M., et al. (2016). Altered functionality, morphology and vesicular glutamate transporter expression of cortical motor neurons from a presymptomatic mouse model of amyotrophic lateral sclerosis. Cereb. Cortex 26, 1512–1528. doi: 10.1093/cercor/bhu317
Schuemann, A., Klawiter, A., Bonhoeffer, T., and Wierenga, C. J. (2013). Structural plasticity of GABAergic axons is regulated by network activity and GABAA receptor activation. Front. Neural Circuits 7:113. doi: 10.3389/fncir.2013.00113
Shibuya, K., Park, S. B., Geevasinga, N., Menon, P., Howells, J., Simon, N. G., et al. (2016). Motor cortical function determines prognosis in sporadic ALS. Neurology 87, 513–520. doi: 10.1212/wnl.0000000000002912
Talbot, K. (2014). Amyotrophic lateral sclerosis: cell vulnerability or system vulnerability? Epidemiology 224, 45–51. doi: 10.1111/joa.12107
Tamamaki, N., Yanagawa, Y., Tomioka, R., Miyazaki, J., Obata, K., and Kaneko, T. (2003). Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J. Comp. Neurol. 467, 60–79. doi: 10.1002/cne.10905
Turner, M. R., and Kiernan, M. C. (2012). Does interneuronal dysfunction contribute to neurodegeneration in amyotrophic lateral sclerosis? Amyotroph. Lateral Scler. 13, 245–250. doi: 10.3109/17482968.2011.636050
Turrigiano, G. G. (2008). The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435. doi: 10.1016/j.cell.2008.10.008
van Zundert, B., Peuscher, M. H., Hynynen, M., Chen, A., Neve, R. L., Brown, R. H. Jr., et al. (2008). Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J. Neurosci. 28, 10864–10874. doi: 10.1523/JNEUROSCI.1340-08.2008
Vucic, S., and Kiernan, M. C. (2006). Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain 129, 2436–2446. doi: 10.1093/brain/awl172
Vucic, S., Nicholson, G. A., and Kiernan, M. C. (2008). Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 131, 1540–1550. doi: 10.1093/brain/awn071
Wainger, B. J., Kiskinis, E., Mellin, C., Wiskow, O., Han, S. S., Sandoe, J., et al. (2014). Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 7, 1–11. doi: 10.1016/j.celrep.2014.03.019
Zhang, Y., Ni, W., Horwich, A. L., and Kaczmarek, L. K. (2017). An ALS-associated mutant SOD1 rapidly suppresses KCNT1 (Slack) Na+-activated K+ channels in aplysia neurons. J. Neurosci. 37, 2258–2265. doi: 10.1523/JNEUROSCI.3102-16.2017
Keywords: excitability, structure, interneuron, cortex, SOD1 G93A mutant
Citation: Clark RM, Brizuela M, Blizzard CA and Dickson TC (2018) Reduced Excitability and Increased Neurite Complexity of Cortical Interneurons in a Familial Mouse Model of Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 12:328. doi: 10.3389/fncel.2018.00328
Received: 25 July 2018; Accepted: 10 September 2018;
Published: 28 September 2018.
Edited by:
Paul G. Mermelstein, University of Minnesota Twin Cities, United StatesReviewed by:
Steven M. Graves, University of Minnesota Twin Cities, United StatesCopyright © 2018 Clark, Brizuela, Blizzard and Dickson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tracey C. Dickson, dHJhY2V5LmRpY2tzb25AdXRhcy5lZHUuYXU=
† These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.