 Victoria M. Pak1*
Victoria M. Pak1* S.-Hakki Onen2,3
S.-Hakki Onen2,3 Donald L. Bliwise4Nancy G. Kutner5
Donald L. Bliwise4Nancy G. Kutner5 Katherine L. Russell1
Katherine L. Russell1 Fannie Onen6,7
Fannie Onen6,7- 1Nell Hodgson Woodruff School of Nursing, Emory University, Atlanta, GA, United States
- 2Centre de Sommeil, Hôpital de la Croix-Rousse, Lyon, France
- 3INSERM U128, Université de Lyon, Lyon, France
- 4Department of Neurology, Emory University, Atlanta, GA, United States
- 5Department of Rehabilitation Medicine, Emory University, Atlanta, GA, United States
- 6CHU Bichat–Claude-Bernard, AP-HP, Service de Gériatrie, Paris, France
- 7CESP & INSERM 1178 Université Paris Sud, Paris, France
Mild cognitive impairment (MCI) and Alzheimer’s disease (AD) affect a high proportion of the elderly population with an increasing prevalence. Sleep disturbances are frequent in those with MCI and AD. This review summarizes existing research on sleep disturbances and neuroinflammation in MCI and AD. Although strong evidence supports various pathways linking sleep and AD pathology, the temporal direction of this central relationship is not yet known. Improved understanding of sleep disturbance and neuroinflammation in MCI and AD may aid in the identification of targets for their prevention.
Introduction
Alzheimer’s disease (AD) is a leading public health problem (Ballard et al., 2011) affecting 24 million people worldwide (Ballard et al., 2011; Reitz and Mayeux, 2014). Descriptive evidence implies that sleep may play a crucial role in optimal daytime cognitive functioning, attention, executive functioning, and memory (Diekelmann and Born, 2010; Killgore, 2010). Disturbed sleep is associated with cognitive disorders (da Silva, 2015), and early observational data from institutionalized AD patients showed worse nocturnal sleep among those with more severe dementia (Bliwise et al., 1995). Epidemiological research among older adults has shown that both short and long sleepers have worse cognitive function (Potvin et al., 2012; Devore et al., 2014) and greater cognitive decline in contrast to adults sleeping 7 or 8 h per night (Keage et al., 2012; Benito-León et al., 2013). Thus, a U-shaped association between sleep duration and cognitive outcomes has been suggested (Yaffe et al., 2014). Both cross-sectional (Muangpaisan et al., 2008; Naismith et al., 2010; Guarnieri et al., 2012) and longitudinal studies (Westerberg et al., 2012), have shown that sleep disturbance is linked to mild cognitive impairment (MCI) and AD.
Patients with dementias, especially AD, often have disrupted nocturnal sleep. When monitored in overnight sleep studies (Vitiello et al., 1990; Montplaisir et al., 1998), they have earlier bedtimes and much more variable wake-up times than those without AD (Bliwise et al., 1992). Approximately 25–44% of patients with AD reported having sleep disturbances, although sleep was measured via self-report (Guarnieri et al., 2012). Nocturnal sleep disturbance is more frequent in dementia patients with Lewy bodies than in patients with AD, although nocturnal sleep disturbances in AD are more associated with advanced disease (Bliwise et al., 2011). There may also be a higher prevalence of sleep apnea in AD patients, though this remains a controversial area (Bliwise, 1996; Bliwise et al., 2019). Nevertheless, a potential causal role for sleep apnea in impaired cognition in the aged could be exacerbated by sleep fragmentation, inflammation, oxidative stress, and hypoxemia, all of which are features of disturbed respiration during sleep (Daulatzai, 2012, 2013). In this review, we examine relevant existing human research on neuroinflammation and sleep disturbance in MCI and AD.
Importance of Exploring Microglia, Neuroinflammation, and Sleep in MCI and AD
We propose a model in which disturbed sleep may play a crucial causal role for cognitive decline in old age. This places neuroinflammation as the most important mediating variable underlying many of the associations noted to date. Sleep disturbance may be a major risk factor for AD and is associated with increased inflammation (Hansson et al., 2006; Alvarez et al., 2007). The amyloid cascade hypothesis proposed by Hardy and Higgins (1992) postulates that the accumulation of insoluble amyloid β (Aβ) is the primary driver of AD pathology. However, research has demonstrated neuroinflammatory responses connected to Aβ deposition common to both sleep disturbance and AD.
Microglia are a type of glial cell that is common in the central nervous system (CNS) and functions as a primary line of immune defense (Nayak et al., 2014). They manage immunosurveillance and mediate inflammation, both of which are suggested to be important in AD (Olsson et al., 2013). Activated microglia appear to have a beneficial effect in response to brain injury, although uncontrolled microglial activation that is part of aging may lead to neuronal dysfunction and cognitive decline (Ojo et al., 2015). Upon exposure to Aβ, microglia and astrocytes release cytokines, interleukins, nitric oxide, and other potentially cytotoxic molecules (McGeer and McGeer, 2010; Krabbe et al., 2013; Heneka et al., 2015). Twenty-four-hour sleep deprivation followed by 24-h recovery was associated with elevated levels of IL-6 and microglial activation in the mouse hippocampus, although not in the cortex (Zhu et al., 2012). Chronic sleep restriction and low-level microglial activation may promote neuroinflammation and increase the brain’s vulnerability to insult (Atienza et al., 2018). Xie et al. (2019) have shown that chronic sleep fragmentation can induce dysfunction of intracellular protein degradation and activate microglia predominately in the hippocampus, which in turn may induce this pro-inflammatory response (Xie et al., 2019). Rodent models of chronic sleep deprivation have reported microglial morphological changes, including enlargement of the cell body and increased expression of pro-inflammatory cytokines (Hsu et al., 2003; Wisor et al., 2011). Thus, microglial dysfunction and neuroinflammation may contribute to the pathophysiology of sleep disorders.
Additionally, intrinsic molecular clock control within microglia regulates sleep–wake cycle-dependent changes in synaptic strengths (Hayashi et al., 2013). This intrinsic circadian control within microglia is an example of a peripheral clock of the circadian sleep-wake system. These are centrally directed from the suprachiasmatic nucleus (SCN), which controls metabolomic changes and synchronizes tissues within the body in response to internal and external “zeitgebers” such as light (Musiek, 2017). These cellular circadian rhythms are often expressed through a transcriptional–translational feedback loop (TTFL) in which the transcriptional factors CLOCK and BMAL1 heterodimerize and bind to E-box sequences in target gene promoters to drive rhythmic expression of Period (Per1-3) and Cryptochrome (Cry1-2) proteins. These Per and Cry proteins then form a complex to return to the nucleus and inhibit CLOCK- and BMAL1-mediated gene expression, completing the feedback loop (Eckel-Mahan and Sassone-Corsi, 2013; Gachon et al., 2017). Studies in mice show that when Bmal1 or Clock is deleted or deficient, they produce arrhythmic circadian cycles in the SCN (Eckel-Mahan and Sassone-Corsi, 2013; Krishnaiah et al., 2017). This process may be disrupted in AD patients, as differences in the phase of these clock gene rhythms have been observed in the brains of individuals with AD when compared to controls (Cermakian et al., 2011; Weissova et al., 2016). Additionally, Ni et al. (2019) show that reduced expression of BMAL1 (a clock gene) is responsible for increased expression of an inflammatory type of microglia in APP knockout mice (Ni et al., 2019). This circadian rhythm dysfunction links AD and sleep disturbance pathology (Musiek, 2017).
Recent research has demonstrated the importance of a missense mutation in the triggering receptor expressed on myeloid cells 2 (TREM2) in increasing one’s risk for AD (Guerreiro et al., 2013; Jonsson et al., 2013; Wang et al., 2015). The R47H variant of the gene confers a 2–4.5-fold increased risk of developing late-onset AD. Behind the APOE ε4 gene, these results place Trem2 as the strongest associated risk gene for AD (Guerreiro et al., 2013; Jonsson et al., 2013). In studies of mice with Aβ accumulation, deletion of Trem2 significantly reduced the number of plaque-associated macrophages (Jay et al., 2015). Deletion of Trem2 also led to decreases in astrocytosis and declines in inflammatory cytokines IL-1β, IL-6, and TNF-α (Jay et al., 2017). Additionally, studies have shown that microglia and macrophages lacking TREM2 have a reduced ability to clear Aβ and apoptotic cells from the brain (Savage et al., 2015). In addition to TREM2, research has explored other microglia-specific receptors and their role in immune response in AD. CX3CL1 is an inhibitory chemokine that binds to CX3CR1 on the surface of microglia, regulating microglial activity and migration in injury conditions (Limatola and Ransohoff, 2014). CX3CL1 may also dose-dependently reduce expression of nitric oxide, IL-6, and TNF-α, and suppress neuronal death induced by microglial activation. Disrupting the receptor can lead to system inflammation and microglial neurotoxicity (Jung et al., 2000; Zujovic et al., 2000; Mizuno et al., 2003; Cardona et al., 2006). Plasma levels of CX3CL1 have been shown to be elevated in patients with AD and more so in patients with MCI, possibly due to increased inflammation preceding AD pathology (Kim et al., 2008). Additionally, postmortem analysis shows significantly lower levels of CX3CL1 in the hippocampus of AD brains compared to controls without dementia (Cho et al., 2011). Microglia, particularly those that have been activated, exhibit GABAB receptors (Kuhn et al., 2004). This is important as GABA decreases the release of pro-inflammatory cytokines IL-6 and TNF-α by activating astrocytes and microglia through this receptor (Lee et al., 2011).
Astrocytes reside in the extracellular space (ECS) of the CNS and are also involved in the clearance of metabolites from the brain. They are responsible for clearing Aβ from parenchyma through the recently described glymphatic system (Jessen et al., 2015). There is a 60% increase in the ECS during sleep, allowing increased flow of interstitial fluid from para-arterial to para-venous space that doubles the rate of Aβ clearance at night (Cedernaes et al., 2017; Mander et al., 2016). In order for this to occur, low adrenergic input is required to the area. The locus coeruleus (LC), best known for its role in alertness and attention and as a center of noradrenergic neurons, acts as the main driver of metabolite clearance in the brain by reducing its adrenergic input (Mander et al., 2016). Any activation of the LC during the night, which occurs in obstructive sleep apnea (OSA), may affect its role in sleep-mediated memory consolidation (Twigg et al., 2010; Rosenzweig et al., 2016). Conversely, normal activation of adrenergic neurons in the LC during the day is neuroprotective, as noradrenaline (NA) is anti-inflammatory and anti-oxidative (Heneka et al., 2015). NA is also responsible for reducing TNF-α expression in microglia, interferon γ-induced expression of class II antigens in astrocytes, and cytokine induction of type 2 nitric oxide synthase expression in astrocytes and microglia (Feinstein et al., 2016). The number of neurons in the LC naturally declines with age, but this occurs more significantly in individuals with AD (Robinson, 1975; Heneka et al., 2015). In fact, this reduction in LC neurons is significantly correlated with the number of plaques, the number of neurofibrillary tangles, and the severity of dementia (Bondareff et al., 1981, 1987). It is hypothesized that increased damage to the LC in AD pathology results from hyperphosphorylated tau, which appears before tau lesions or Aβ accumulation in the cortex (Braak et al., 2011).
Melatonin, a major hormone controlling circadian rhythm and the sleep–wake cycle, is decreased significantly in the cerebrospinal fluid (CSF) and blood of individuals with AD (Zhou et al., 2003; Wu and Swaab, 2005). Additionally, reductions of MT2, a melatonin receptor, are found in the hippocampus of AD patients (Savaskan et al., 2005, 2002). In addition to its role in the sleep–wake cycle, melatonin inhibits tau phosphorylation (Deng et al., 2005) and reduces the release of APP from neurons (Lahiri, 1999), both hallmarks of AD. Melatonin also plays an anti-inflammatory role by inhibiting the activation of nuclear factor kappa B (NF-κB) (Tamura et al., 2009) and reducing adherence of leukocytes and neutrophils to endothelium, preventing vascular permeability (Lotufo et al., 2006). Melatonin is released from the pineal gland, which can be reduced in volume or undergo calcification in AD (Bumb et al., 2013, 2014). When Aβ interacts with toll-like receptors in the pineal gland, subsequent release of pro-inflammatory factors inhibit synthesis of melatonin through the NF-κB pathway (Cecon et al., 2015). These pineal gland volume and function changes in AD are also significantly associated with sleep disturbances and insomnia (Matsuoka et al., 2017).
TNF-α plays an important role as a pro-inflammatory cytokine and is elevated in the brains and plasma of individuals with AD (Chang et al., 2017). TNF-α binds to TNFR1 or TNFR2 to activate a cytokine cascade. Overexpression of TNFR1 in the hippocampi of mice resulted in activation of Aβ-induced apoptosis, while mice lacking the receptor exhibited reduced microglial activation and improved cognition performance (He et al., 2007; Li et al., 2004). As noted above, microglia produce TNF-α in response to Aβ. Perpetuating this cycle, TNF-α can then promote Aβ production through upregulation of β-secretase (Liao et al., 2004). IL-1β is another pro-inflammatory cytokine involved in Aβ plaque development that is found in increased levels in the hippocampus and prefrontal cortex of individuals with AD (Cacabelos et al., 1994). IL-6 is a cytokine whose overproduction can lead to neuroinflammation (Rothaug et al., 2016). Elevated levels in midlife predict cognitive decline 10 years later and are elevated in the CSF and plasma of individuals with AD (Blum-Degena et al., 1995; Singh-Manoux et al., 2014). In addition to its release by glial cells in response to Aβ, activation of IL-6 receptors enhance APP transcription and expression (Chong, 1997; Ringheim et al., 1998). Elevated levels of these pro-inflammatory cytokines have also been linked to sleep disturbances, such as excessive daytime sleepiness (EDS), obstructive sleep apnea, and narcolepsy (Vgontzas et al., 1997; Okun et al., 2004). Studies simulating acute sleep loss in humans have shown TNF-α, IL-6, and cellular adhesion molecules to be dysregulated (Vgontzas et al., 2004; Frey et al., 2007; Chennaoui et al., 2011). Even modest sleep restriction is associated with secretion of IL-6 in both men and women and TNF-α in men (Mullington et al., 2016).
It is believed that impaired permeability of the blood–brain barrier (BBB) may be related to neuroinflammation and sleep disruption (Atienza et al., 2018). Specifically, chronic restriction of either total sleep time (He et al., 2014) or REM sleep results in a reduction in BBB integrity (Montagne et al., 2015). Sleep fragmentation may lead to age-related disruption of the BBB’s ability to block proinflammatory compounds (Atienza et al., 2018). Increased BBB permeability in aging may augment low-grade inflammation on cerebral integrity and mediate the relationship between disrupted sleep, dysfunctional adiposity, and impaired cognition (Atienza et al., 2018). These results suggest that disturbed sleep may play a causative role in neuroinflammation and subsequent cognitive decline.
Existing human studies on microglia and neuroinflammation are limited in that they do not measure sleep explicitly (Schuitemaker et al., 2009; Olsson et al., 2013) but may hold relevance for possible causal pathways linking poor sleep to cognitive decline. CSF YKL-40, a glycoprotein and biomarker of glial inflammation, is associated with a cerebral structural signature distinct from that related to p-tau neurodegeneration at the earliest stages of cognitive decline due to AD (Gispert et al., 2016). In order to investigate if microglial markers could differentiate between AD and controls, and secondly between stable MCI and those progressing to AD, the Olsson et al. study quantified YKL-40 and sCD14 in CSF from 96 AD patients, 65 healthy controls, and 170 patients with MCI from baseline and over 5.7 years (Olsson et al., 2013). YKL-40, and not sCD14, was significantly elevated in AD compared with healthy controls (p = 0.003). Furthermore, according to the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) (Olsson et al., 2013), YKL-40 and sCD14 were increased in MCI patients who converted to vascular dementia (VaD) (p = 0.029 and p = 0.008), but not to AD. However, when stratified according to CSF levels of tau and Aβ42, YKL-40 was elevated in those with an AD-indicative profile compared with stable MCI with a normal profile (p = 0.037). In addition, YKL-40 and sCD14 were very stable in AD patients with strong correlation between time-points (r = 0.94, p = 3.4 × 10−25; r = 0.77, p = 2.0 × 10−11) and the cortical damage marker T-tau (Olsson et al., 2013). Thus, microglial markers appear stable for monitoring CNS inflammation and microglia activation in clinical trials (Olsson et al., 2013). Moreover, YKL-40 differentiates between AD and controls, between stable MCI to AD, and those that convert to AD and VaD. In order to clarify the role of disturbed sleep and its impact on neuroinflammation, it will be important to measure sleep explicitly in future studies.
Studies conducted in human subjects have demonstrated that neuroinflammation, which includes microglia activation and increases in the levels of pro-inflammatory cytokines in the brain, may lead to cognitive function decline (Schuitemaker et al., 2009). Schuitemaker et al. (2009) conducted a study examining levels of pro-inflammatory cytokines in patients with MCI and probable AD. In this study, 145 subjects had probable AD and 67 had MCI. Patients were diagnosed with MCI according to Petersen et al. (1999): failing in one or more cognitive domains or a greater cognitive decline than that expected for the person’s age or education but not interfering notably with daily activities (Petersen et al., 1999). Patients were diagnosed with probable AD according to McKhann et al. (1984): the gradual worsening of specific cognitive functions such as language, motor skills, and perception (McKhann et al., 1984). The MCI patients were further classified as having either a low-risk or high-risk for AD according to their Aβ42 and tau levels in CSF, with cut-off values from previously published work (Schoonenboom et al., 2005). High risk CSF consisted of Aβ42 ≤ 494 pg/mL and tau > 356 pg/mL and low risk CSF consisted of Aβ42 > 494 pg/mL and tau > 356 pg/mL. CSF and serum CRP levels were significantly higher in MCI compared to AD patients after adjustment for age, APOE ε4 genotype, and cardiovascular diseases. This difference remained present in patients with a low-risk biomarker profile for AD after adjustment for the abovementioned covariates. CSF IL-6 levels were also significantly higher in MCI patients with a low-risk CSF profile (Schuitemaker et al., 2009). Again, these studies have not assessed sleep per se but do suggest that associations between cognitive impairment and sleep noted in the literature may well be reflecting such mediational effects.
Sleep Disturbances in Alzheimer’S
The adverse effects of sleep on the production and clearance of proteins have been seen in neurodegenerative diseases such as AD (Ju et al., 2017; Olsson et al., 2018; Shokri-Kojori et al., 2018). Sleep has a role in regulating neuronal activity that affects the release of glymphatic proteins, Aβ, and τ (Huang et al., 2012). The brain relies on the glymphatic clearance pathway to remove these waste materials (Xie et al., 2013). Studies have shown positive associations between sleep deprivation, OSA, and increased efflux of these proteins in the brain. Spira et al. (2013) found that older adults with shorter sleep duration have an increase in Aβ (Spira et al., 2013). Another study conducted with healthy volunteers revealed that a night of total sleep deprivation led to a physiological increase in cerebral Aβ42, an isoform of Aβ (Ooms et al., 2014). A 2-year prospective longitudinal study conducted in community-dwelling healthy older adults found that OSA was associated with markers of increased amyloid burden (Sharma et al., 2018). A study of OSA patients from the Korean Genome and Epidemiology Study revealed significant Pittsburgh compound B (PiB) deposition compared to controls at the right posterior cingulate gyrus and the right temporal cortex; there was no area of higher uptake in the controls compared with the OSA group (Yun et al., 2017). The study found that OSA accelerates amyloid deposition, contributing to the development or progression of AD (Yun et al., 2017).
Clarifying the underlying causes of sleep disturbances in AD can help to improve quality of life and understanding of disease progression. A prior study by Lee et al. (2007) investigating whether reports of daytime sleepiness in AD were correlated with lower functional status showed that EDS is linked to lower quality of life in AD subjects (Lee et al., 2007). EDS has been reported to range from 44.5 (Guarnieri et al., 2012) to 52.1% (Park et al., 2011) in AD patients. Despite this high prevalence, there is a lack of research on the mechanistic causes of sleepiness in AD, which will have important implications for improving quality of life and disease progression.
Recent research reveals a correlation between self-reported EDS and Aβ deposition in cognitively normal subjects (Carvalho et al., 2018; Spira et al., 2018). Spira et al. (2018) showed that EDS is associated with more than 2.5 times the odds of Aβ deposition at follow-up 15.7 years later in 124 cognitively normal subjects (Spira et al., 2018). However, they did not look at objectively measured sleepiness and did not adequately control for variables such as sleep quality, which may be linked to higher CSF Aβ (Ooms et al., 2014); sleep duration, which may influence Aβ accumulation (Spira et al., 2013); or sleep apnea, which is linked to sleepiness and may influence Aβ (Spira et al., 2014; Bu et al., 2015). Another study of 2,172 subjects found that baseline EDS is associated with a longitudinal increase in Aβ accumulation [measured by Pittsburgh compound B positron emission tomography (PiB-PET)] in elderly persons without dementia (Carvalho et al., 2018). One study of 1,041 non-demented participants over 65 years found that increased daytime sleepiness and sleep inadequacy was a risk factor for development of dementia, which occurred in 78 participants over the following 3 years (Tsapanou et al., 2015). Additionally, increased Aβ deposition in the brainstems of patients with more frequent nocturnal awakenings is associated with cognitive impairment (You et al., 2019). Prior studies have confirmed this link between poor sleep and/or sleep disturbance and cognitive impairment (Blackwell et al., 2006; Tworoger et al., 2006; Xu et al., 2011; Yaffe et al., 2011). Aβ deposition as a result of less adequate sleep, more sleep problems, and greater daytime somnolence has been reported in AD-sensitive brain regions and is associated with greater AD pathology (Sprecher et al., 2015, 2017). This relationship between sleep and Aβ accumulation has also been demonstrated in subjects with pre-clinical AD (Ju et al., 2013). A cross-sectional study of 145 cognitively normal individuals (half of which had a parental history of late-onset AD) measured sleep quality using wrist actigraphy and Aβ levels in CSF samples (Ju et al., 2013). 22% of the sample met their definition of “preclinical” AD, in which CSF Aβ42 levels were 500 pg/mL or less. This group experienced worse sleep quality and more frequent napping (Ju et al., 2013).
The apolipoprotein (APOE) ε4 allele significantly increases one’s risk for developing AD. One copy of the allele can increase risk of developing AD by nearly threefold, while two alleles may increase one’s risk by 8–15 times (Corder et al., 1993). APOE ε4 has also been linked to sleep disturbances. For example, Koo et al. (2019) showed that carriers of the gene more frequently experienced disrupted sleep as compared to non-carriers (Koo et al., 2019). Sleep quality measured by polysomnography (PSG) and wrist actigraphy was also shown to be lower in individuals with APOE ε4 as compared to non-carriers (Drogos et al., 2016). Additionally, APOE ε4 has been linked to an increased risk of developing sleep apnea (Kadotani et al., 2001; Gottlieb et al., 2004).
Sleep Disturbances in Mci
Sleep disturbances are not restricted to those with AD but are also prevalent in patients with MCI (da Silva, 2015). MCI is viewed as a transitional stage between normal decline of cognitive aging to dementia, which is a general term for loss of memory and other cognitive abilities severe enough to interfere with daily life (Muangpaisan et al., 2008). The specific transition between normal aging and MCI can be subtle and the distinction between MCI and very early dementia is challenging (da Silva, 2015). Patients affected with this condition have a higher conversion rate to AD, with an estimated average rate of 10–15% annually (Muangpaisan et al., 2008). MCI patients may show intermediate levels of electroencephalographically (EEG) defined sleep instability relative to AD patients (Maestri et al., 2015). Sleep disturbance is prevalent and may be predictive of cognitive decline in older adults and in those with neurodegenerative disorders (da Silva, 2015). Though these studies suggest linkage between sleep disturbance and MCI, the underlying mechanisms of sleep disturbances remain unclear.
A prior small case-control study of 16 older adults and 8 MCI subjects found that amnesiac MCI (aMCI) patients had lower delta and theta power during non-REM sleep and spent less time in slow-wave sleep. This is concerning since the sleep changes common in aMCI may interfere with sleep-dependent memory consolidation (Westerberg et al., 2012). Brayet et al. (2016) reported slower EEG frequencies during REM, particularly for amnestic- relative to non-amnestic MCI (Brayet et al., 2016). Additionally, at least one study has indicated that the associations between disrupted sleep and impaired cognition in MCI may be, at least in part, moderated by APOE genotype (Hita-Yanez et al., 2012).
Integration of Evidence: Neuroinflammation as a Mediator of the Association of Sleep Disturbance With AD
We propose neuroinflammation as the mediator between AD and sleep disturbance. AD and sleep disturbance, as described in Figure 1. Evidence has accumulated linking sleep disturbance to Aβ deposits in the brain, particularly in regions that Aβ is typically found in AD. Aβ also serves as a target of inflammatory response, attracting microglia that release cytokines, interleukins, and other potentially cytotoxic molecules. Other pathological changes in melatonin and NA release, both serving roles in sleep-wake neural circuitry and functioning in anti-inflammatory responses, reduce the brain’s ability to respond to inflammation generated by Aβ deposition. Sleep disturbances also independently promote a rise in inflammatory cytokines and interleukins and are separately linked to cognitive decline and AD. Fewer studies, however, have examined all three outcomes – AD, sleep disturbance, and inflammatory markers – within a human population. Most have addressed only one or two of these relationships at a time.
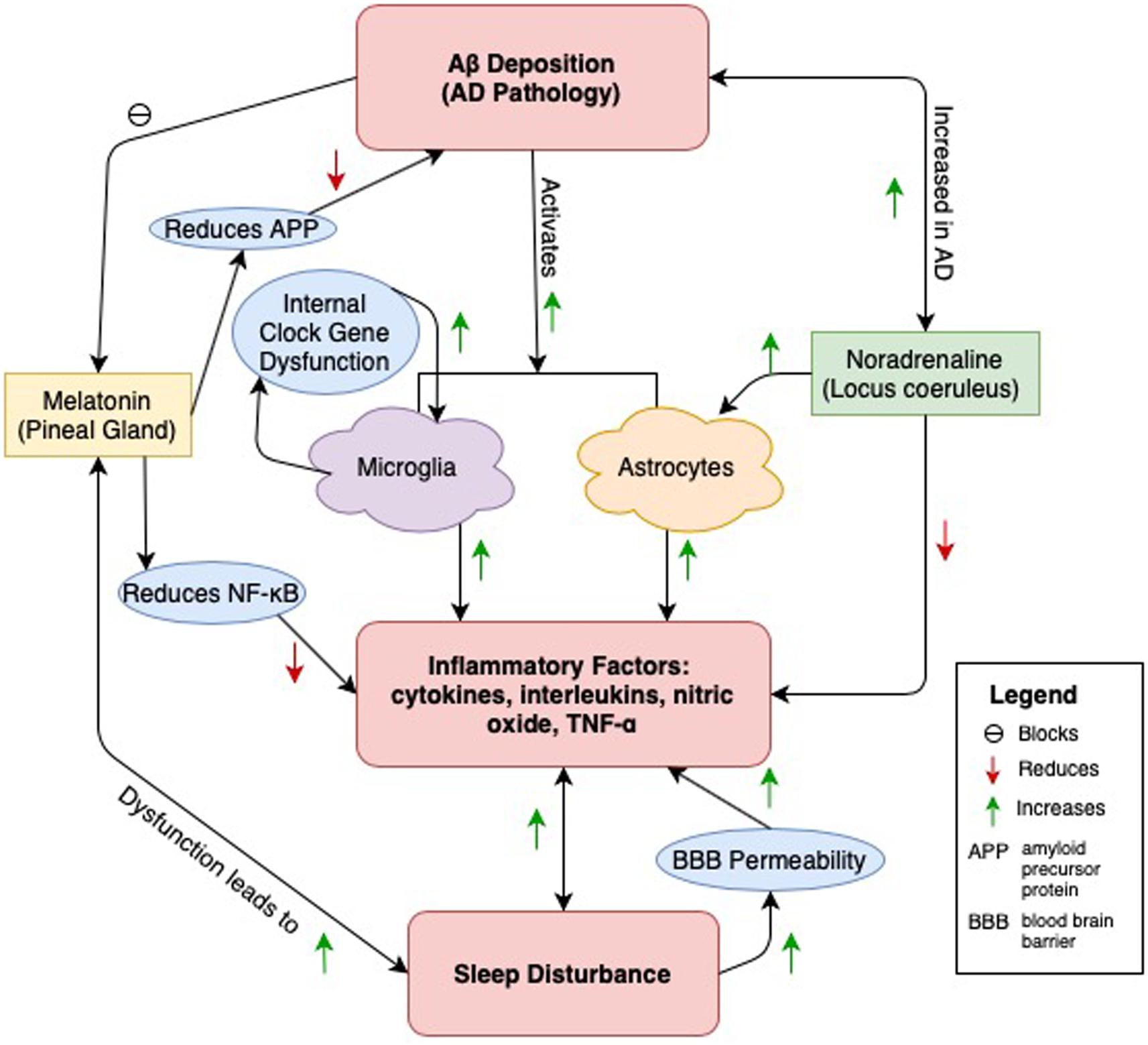
Figure 1. A central pathway through which sleep disturbance and AD pathology is connected is through neuroinflammation caused by Aβ deposition (Heneka et al., 2015). Microglia and astrocytes are both activated and release inflammatory factors in response to Aβ (Krabbe et al., 2013). Additionally, disruption of internal clock gene function in microglia causes an increase in their release (Ni et al., 2019). AD pathology may damage neural pathways and lead to calcification within the pineal gland (Bumb et al., 2013, 2014). This leads to a reduction in the amount of melatonin which in turn causes sleep disturbance (Matsuoka et al., 2017). This relationship is bidirectional, as sleep disturbance is linked to disruptions of melatonin release (Wu and Swaab, 2005). Melatonin has anti-inflammatory properties, reducing the release of these factors by attenuating the release of NF-κB (Cecon et al., 2015). Additionally, melatonin reduces amyloid precursor protein (APP), thus protecting against Aβ creation (Lahiri, 1999). However, Aβ, by binding toll-like receptors of the pineal gland, may block melatonin release, leading to sleep disturbance in AD (Cecon et al., 2015). Noradrenaline, released from the locus coeruleus (LC) also plays an anti-inflammatory role, as the neurotransmitter can reduce the release of inflammatory factors (Feinstein et al., 2016). Low adrenergic input from the LC to the extracellular space (ECS, location of astrocytes) during sleep is required to allow Aβ and other metabolite clearance to occur (Mander et al., 2016). The LC-noradrenergic system activates with cortico-hippocampal neuronal replay during NREM EEG slow oscillations, suggesting a prominent role in sleep dependent memory consolidation (Rosenzweig et al., 2016; Twigg et al., 2010). Thus, inappropriate activation of the LC may compromise its role in memory consolidation during sleep and reduce Aβ clearance, leading to AD pathology. Finally, sleep disturbance has been independently linked to the release of inflammatory factors and increased blood brain barrier (BBB) permeability (He et al., 2014; Montagne et al., 2015). Sleep disturbance is correlated with increased Aβ deposition and may be occurring through any of the above pathways (Ju et al., 2017; Ooms et al., 2014; Spira et al., 2013).
Two studies have examined all three outcomes within mouse models. Ni et al. (2019) studied APP knock-in (APP-KI) and wild-type (WT) mice injected with an agonist for REV-ERB designed to reduce expression of BMAL1, a central clock gene (Ni et al., 2019). This disruption of CLOCK/BMAL1-driven transcriptional loops impaired microglia in APP-KI mice. This led to NF-κB activation, increasing expression of pro-inflammatory genes (TNF-α, IL-1β, and IL-6) (Ni et al., 2019). This study demonstrates that clock gene disturbance in microglia is involved in early onset of AD by inducing chronic neuroinflammation (Ni et al., 2019). Thus, disturbed sleep and its subsequent triggering of inflammatory responses may play a critical role in AD pathology development. Cecon et al. (2015) also studied these outcomes in a mouse model. They treated rat pineal glands with Aβ1–40 or Aβ1–42 before inducing melatonin synthesis with NA (Cecon et al., 2015). They found that Aβ interfered with melatonin binding. Additionally, 52 inflammatory genes were upregulated in response to Aβ. Through the NF-κB pathway, these inflammatory factors inhibited synthesis of melatonin, reducing production of the hormone by 75% (Cecon et al., 2015). This study demonstrates a way in which Aβ, a hallmark of AD, may interfere with sleep regulation through disruption of melatonin via inflammatory pathways.
We have included a summary of the key human and animal studies discussed above in Table 1. It is unclear whether sleep disturbance leads to AD through inflammation or whether AD pathology leads to inflammation and subsequent sleep disturbance. The amyloid cascade hypothesis has provided a platform for researchers to target Aβ in hopes of developing effective AD drugs. Given that Aβ deposition triggers inflammation in the brain, it is hypothesized that treatment of Aβ would solve both problems. However, clinical trials in this area have been lacking. Others have instead focused on the brain’s inflammatory response to Aβ, specifically whether non-steroidal anti-inflammatory drugs (NSAIDs) may reduce AD risk or stop progression of the disease. These studies have also been inconclusive (Miguel-Álvarez et al., 2015). Largely missing from the literature are human studies examining all three outcomes: AD pathology, sleep disturbance, and inflammatory markers. Additionally, randomized studies examining the effect of treating sleep disturbance on AD risk would aid in determining the temporal relationship between AD and sleep (Irwin and Vitiello, 2019).
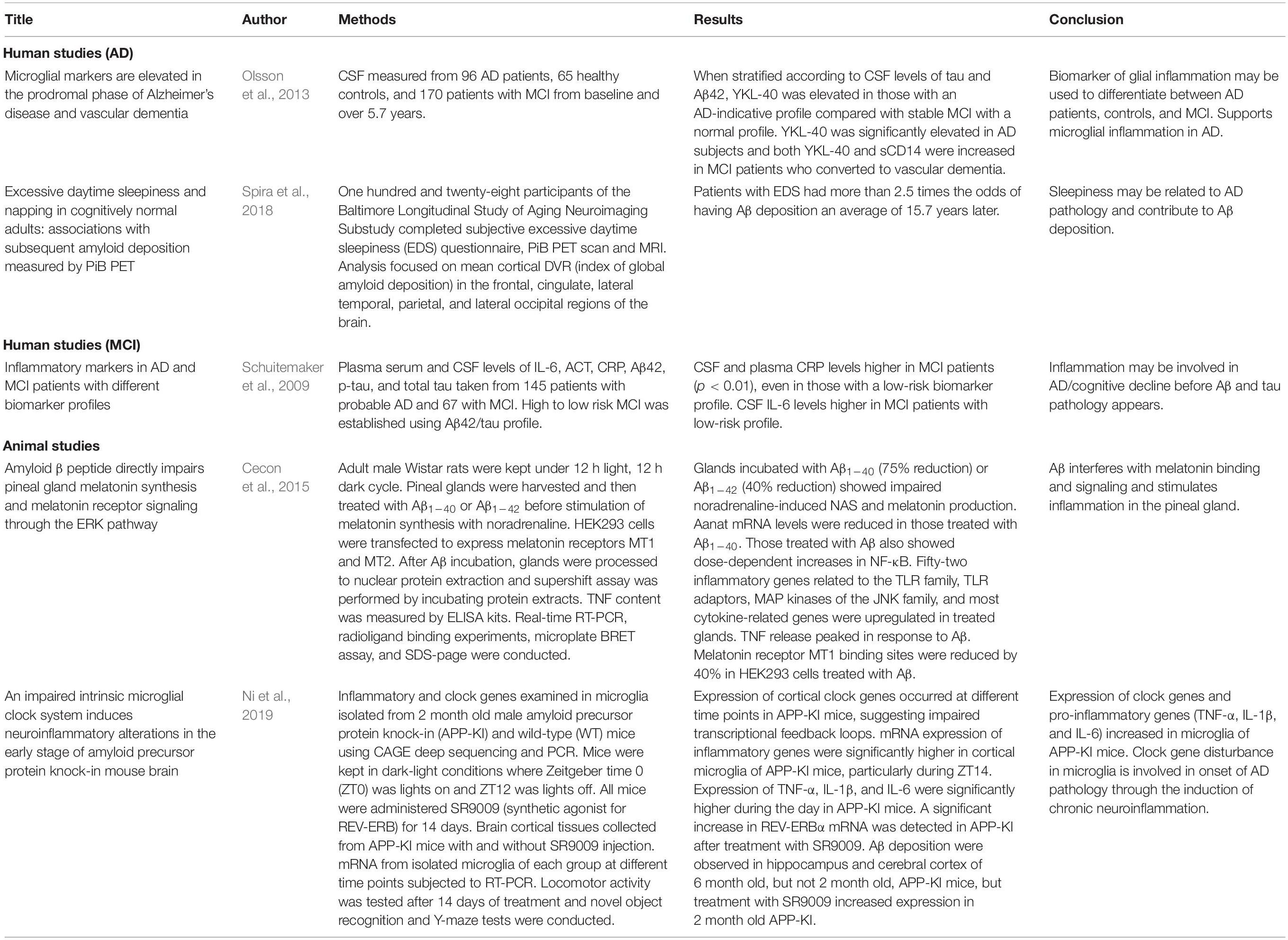
Table 1. Key studies examining AD, MCI, sleep disturbance and/or inflammation.
Conclusion
Sleep disturbances are common in both MCI and AD. There is a need to better evaluate the role of neuroinflammation associated with sleep disturbance in the progression of MCI and AD. There are methodological limitations of existing studies that must be acknowledged. Results are supported by a small number of studies, have small sample sizes, often do not differentiate between different subgroups of MCI (amnesiac versus non-amnesiac), and do not consistently address confounders, including several common comorbidities in older adults such as depression or OSA, which could also negatively impact sleep quality and AD biomarkers. Large scale human studies with more comprehensive and objective sleep measures are needed. Future studies should consider the important biological/metabolic signatures associated with sleep disturbance and neuroinflammation, and subsequent cognitive decline with age. From a clinical perspective, a better understanding of the associations between sleep disturbances, cognitive integrity, and brain integrity in older adults may allow for more targeted preventive strategies that promote healthy aging.
Author Contributions
VP, S-HO, DB, NK, KR, and FO wrote and edited the manuscript.
Funding
This research work was supported by R00NR014675-03 NIH Pathway to Independence Award (Pak).
Conflict of Interest
DB was a consultant for Merck, Eisai, and Ferring in the last 12 months.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alvarez, A., Cacabelos, R., Sanpedro, C., Garcia-Fantini, M., and Aleixandre, M. (2007). Serum TNF-alpha levels are increased and correlate negatively with free IGF-I in Alzheimer disease. Neurobiol. Aging 28, 533–536. doi: 10.1016/j.neurobiolaging.2006.02.012
Atienza, M., Ziontz, J., and Cantero, J. L. (2018). Low-grade inflammation in the relationship between sleep disruption, dysfunctional adiposity, and cognitive decline in aging. Sleep Med. Rev. 42, 171–183. doi: 10.1016/j.smrv.2018.08.002
Ballard, C., Gauthier, S., Corbett, A., Brayne, C., Aarsland, D., and Jones, E. (2011). Alzheimer’s disease. Lancet 377, 1019–1031. doi: 10.1016/s0140-6736(10)61349-9
Benito-León, J., Louis, E. D., and Bermejo-Pareja, F. (2013). Cognitive decline in short and long sleepers: a prospective population-based study (NEDICES). J. Psychiatr. Res. 47, 1998–2003. doi: 10.1016/j.jpsychires.2013.09.007
Blackwell, T., Yaffe, K., Ancoli-Israel, S., Schneider, J. L., Cauley, J. A., Hillier, T. A., et al. (2006). Poor sleep is associated with impaired cognitive function in older women: the study of osteoporotic fractures. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 61, 405–410. doi: 10.1093/gerona/61.4.405
Bliwise, D. L. (1996). Is sleep apnea a cause of reversible dementia in old age? J. Am. Geriatr. Soc. 44, 1407–1409. doi: 10.1111/j.1532-5415.1996.tb01421.x
Bliwise, D. L., Hughes, M., McMahon, P. M., and Kutner, N. (1995). Observed sleep/wakefulness and severity of dementia in an Alzheimer’s disease special care unit. J. Gerontol. A Biol. Sci. Med. Sci. 50, M303–M306. doi: 10.1093/gerona/50a.6.m303
Bliwise, D. L., Mercaldo, N. D., Avidan, A. Y., Boeve, B. F., Greer, S. A., and Kukull, W. A. (2011). Sleep disturbance in dementia with Lewy bodies and Alzheimer’s disease: a multicenter analysis. Dement. Geriatr. Cogn. Disord. 31, 239–246. doi: 10.1159/000326238
Bliwise, D. L., Tinklenberg, J. R., and Yesavage, J. A. (1992). Timing of sleep and wakefulness in Alzheimer’s disease patients residing at home. Biol. Psychiatry 31, 1163–1165. doi: 10.1016/0006-3223(92)90162-S
Bliwise, D. L., Trotti, L., Wood-Siverio, C., and Hu, W. (2019). APOE4, but not desaturation index, is associated with dementia severity in a memory clinic population. Sleep 42, A285–A286.
Blum-Degena, D., Müller, T., Kuhn, W., Gerlach, M., Przuntek, H., and Riederer, P. (1995). Interleukin-1β and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci. Lett. 202, 17–20. doi: 10.1016/0304-3940(95)12192-7
Bondareff, W., Mountjoy, C., and Roth, M. (1981). Selective loss of neurones of origin of adrenergic projection to cerebral cortex (nucleus locus coeruleus) in senile dementia. Lancet 317, 783–784. doi: 10.1016/s0140-6736(81)92657-x
Bondareff, W., Mountjoy, C., Roth, M., Rossor, M., Iversen, L., Reynolds, G., et al. (1987). Neuronal degeneration in locus ceruleus and cortical correlates of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1, 256–262. doi: 10.1097/00002093-198701040-00005
Braak, H., Thal, D. R., Ghebremedhin, E., and Del Tredici, K. (2011). Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969. doi: 10.1097/nen.0b013e318232a379
Brayet, P., Petit, D., Frauscher, B., Gagnon, J. F., Gosselin, N., Gagnon, K., et al. (2016). Quantitative EEG of rapid-eye-movement sleep: a marker of amnestic mild cognitive impairment. Clin. EEG Neurosci. 47, 134–141. doi: 10.1177/1550059415603050
Bu, X. L., Liu, Y. H., Wang, Q. H., Jiao, S. S., Zeng, F., Yao, X. Q., et al. (2015). Serum amyloid-beta levels are increased in patients with obstructive sleep apnea syndrome. Sci. Rep. 5:13917. doi: 10.1038/srep13917
Bumb, J. M., Brockmann, M. A., Groden, C., and Nolte, I. (2013). Microstructural analysis of pineal volume using trueFISP imaging. World J. Radiol. 5, 166–172.
Bumb, J. M., Schilling, C., Enning, F., Haddad, L., Paul, F., Lederbogen, F., et al. (2014). Pineal gland volume in primary insomnia and healthy controls: a magnetic resonance imaging study. J. Sleep Res. 23, 276–282. doi: 10.1111/jsr.12125
Cacabelos, R., Alvarez, X., Fernandez-Novoa, L., Franco, A., Mangues, R., Pellicer, A., et al. (1994). Brain interleukin-1 beta in Alzheimer’s disease and vascular dementia. Methods Find. Exp. Clin. Pharmacol. 16, 141–151.
Cardona, A. E., Pioro, E. P., Sasse, M. E., Kostenko, V., Cardona, S. M., Dijkstra, I. M., et al. (2006). Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 9, 917–924. doi: 10.1038/nn1715
Carvalho, D. Z., St Louis, E. K., Knopman, D. S., Boeve, B. F., Lowe, V. J., Roberts, R. O., et al. (2018). Association of excessive daytime sleepiness with longitudinal beta-amyloid accumulation in elderly persons without dementia. JAMA Neurol. 75, 672–680. doi: 10.1001/jamaneurol.2018.0049
Cecon, E., Chen, M., Marçola, M., Fernandes, P. A., Jockers, R., and Markus, R. P. (2015). Amyloid β peptide directly impairs pineal gland melatonin synthesis and melatonin receptor signaling through the ERK pathway. FASEB J. 29, 2566–2582. doi: 10.1096/fj.14-265678
Cedernaes, J., Osorio, R. S., Varga, A. W., Kam, K., Schiöth, H. B., and Benedict, C. (2017). Candidate mechanisms underlying the association between sleep-wake disruptions and Alzheimer’s disease. Sleep Med. Rev. 31, 102–111. doi: 10.1016/j.smrv.2016.02.002
Cermakian, N., Waddington Lamont, E., Boudreau, P., and Boivin, D. B. (2011). Circadian clock gene expression in brain regions of Alzheimer’s disease patients and control subjects. J. Biol. Rhythms 26, 160–170. doi: 10.1177/0748730410395732
Chang, R., Yee, K.-L., and Sumbria, R. K. (2017). Tumor necrosis factor α Inhibition for Alzheimer’s disease. J. Cent. Nerv. Syst. Dis. 9, 1179573517709278.
Chennaoui, M., Sauvet, F., Drogou, C., Van Beers, P., Langrume, C., Guillard, M., et al. (2011). Effect of one night of sleep loss on changes in tumor necrosis factor alpha (TNF-α) levels in healthy men. Cytokine 56, 318–324. doi: 10.1016/j.cyto.2011.06.002
Cho, S.-H., Sun, B., Zhou, Y., Kauppinen, T. M., Halabisky, B., Wes, P., et al. (2011). CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 286, 32713–32722. doi: 10.1074/jbc.m111.254268
Chong, Y. (1997). Effect of a carboxy-terminal fragment of the Alzheimer’s amyloid precursor protein on expression of proinflammatory cytokines in rat glial cells. Life Sci. 61, 2323–2333. doi: 10.1016/s0024-3205(97)00936-3
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
da Silva, R. A. (2015). Sleep disturbances and mild cognitive impairment: a review. Sleep Sci. 8, 36–41. doi: 10.1016/j.slsci.2015.02.001
Daulatzai, M. A. (2012). Quintessential risk factors: their role in promoting cognitive dysfunction and Alzheimer’s disease. Neurochem. Res. 37, 2627–2658. doi: 10.1007/s11064-012-0854-6
Daulatzai, M. A. (2013). Neurotoxic saboteurs: straws that break the Hippo’s (Hippocampus) back drive cognitive impairment and Alzheimer’s disease. Neurotox. Res. 24, 407–459. doi: 10.1007/s12640-013-9407-2
Deng, Y.-Q., Xu, G.-G., Duan, P., Zhang, Q., and Wang, J.-Z. (2005). Effects of melatonin on wortmannin-induced tau hyperphosphorylation. Acta Pharmacol. Sin. 26, 519–526. doi: 10.1111/j.1745-7254.2005.00102.x
Devore, E. E., Grodstein, F., Duffy, J. F., Stampfer, M. J., Czeisler, C. A., and Schernhammer, E. S. (2014). Sleep duration in midlife and later life in relation to cognition. J. Am. Geriatr. Soc. 62, 1073–1081. doi: 10.1111/jgs.12790
Drogos, L. L., Gill, S. J., Tyndall, A. V., Raneri, J. K., Parboosingh, J. S., Naef, A., et al. (2016). Evidence of association between sleep quality and APOE ε4 in healthy older adults: a pilot study. Neurology 87, 1836–1842. doi: 10.1212/wnl.0000000000003255
Eckel-Mahan, K., and Sassone-Corsi, P. (2013). Metabolism and the circadian clock converge. Physiol. Rev. 93, 107–135. doi: 10.1152/physrev.00016.2012
Feinstein, D. L., Kalinin, S., and Braun, D. (2016). Causes, consequences, and cures for neuroinflammation mediated via the locus coeruleus: noradrenergic signaling system. J. Neurochem. 139, 154–178. doi: 10.1111/jnc.13447
Frey, D. J., Fleshner, M., and Wright, K. P. Jr. (2007). The effects of 40 hours of total sleep deprivation on inflammatory markers in healthy young adults. Brain Behav. Immun. 21, 1050–1057. doi: 10.1016/j.bbi.2007.04.003
Gachon, F., Loizides-Mangold, U., Petrenko, V., and Dibner, C. (2017). Glucose homeostasis: regulation by peripheral circadian clocks in rodents and humans. Endocrinology 158, 1074–1084. doi: 10.1210/en.2017-00218
Gispert, J. D., Monte, G. C., Falcon, C., Tucholka, A., Rojas, S., Sanchez-Valle, R., et al. (2016). CSF YKL-40 and pTau181 are related to different cerebral morphometric patterns in early AD. Neurobiol. Aging 38, 47–55. doi: 10.1016/j.neurobiolaging.2015.10.022
Gottlieb, D., DeStefano, A., Foley, D., Mignot, E., Redline, S., Givelber, R., et al. (2004). APOE ε4 is associated with obstructive sleep apnea/hypopnea: the Sleep Heart Health Study. Neurology 63, 664–668. doi: 10.1212/01.wnl.0000134671.99649.32
Guarnieri, B., Adorni, F., Musicco, M., Appollonio, I., Bonanni, E., Caffarra, P., et al. (2012). Prevalence of sleep disturbances in mild cognitive impairment and dementing disorders: a multicenter Italian clinical cross-sectional study on 431 patients. Dement. Geriatr. Cogn. Disord. 33, 50–58. doi: 10.1159/000335363
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127.
Hansson, O., Zetterberg, H., Buchhave, P., Londos, E., Blennow, K., and Minthon, L. (2006). Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 5, 228–234. doi: 10.1016/S1474-4422(06)70355-6
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–186.
Hayashi, Y., Koyanagi, S., Kusunose, N., Okada, R., Wu, Z., Tozaki-Saitoh, H., et al. (2013). The intrinsic microglial molecular clock controls synaptic strength via the circadian expression of cathepsin S. Sci. Rep. 3:2744.
He, J., Hsuchou, H., He, Y., Kastin, A. J., Wang, Y., and Pan, W. (2014). Sleep restriction impairs blood-brain barrier function. J. Neurosci. 34, 14697–14706. doi: 10.1523/JNEUROSCI.2111-14.2014
He, P., Zhong, Z., Lindholm, K., Berning, L., Lee, W., Lemere, C., et al. (2007). Deletion of tumor necrosis factor death receptor inhibits amyloid β generation and prevents learning and memory deficits in Alzheimer’s mice. J. Cell Biol. 178, 829–841. doi: 10.1083/jcb.200705042
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405.
Hita-Yanez, E., Atienza, M., Gil-Neciga, E., and Cantero, J. L. (2012). Disturbed sleep patterns in elders with mild cognitive impairment: the role of memory decline and ApoE epsilon4 genotype. Curr. Alzheimer Res. 9, 290–297. doi: 10.2174/156720512800107609
Hsu, J. C., Lee, Y. S., Chang, C. N., Chuang, H. L., Ling, E. A., and Lan, C. T. (2003). Sleep deprivation inhibits expression of NADPH-d and NOS while activating microglia and astroglia in the rat hippocampus. Cells Tissues Organs 173, 242–254. doi: 10.1159/000070380
Huang, Y., Potter, R., Sigurdson, W., Santacruz, A., Shih, S., Ju, Y.-E., et al. (2012). Effects of age and amyloid deposition on Aβ dynamics in the human central nervous system. Arch. Neurol. 69, 51–58. doi: 10.1001/archneurol.2011.235
Irwin, M. R., and Vitiello, M. V. (2019). Implications of sleep disturbance and inflammation for Alzheimer’s disease dementia. Lancet Neurol. 18, 296–306. doi: 10.1016/s1474-4422(18)30450-2
Jay, T. R., Hirsch, A. M., Broihier, M. L., Miller, C. M., Neilson, L. E., Ransohoff, R. M., et al. (2017). Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci. 37, 637–647. doi: 10.1523/jneurosci.2110-16.2016
Jay, T. R., Miller, C. M., Cheng, P. J., Graham, L. C., Bemiller, S., Broihier, M. L., et al. (2015). TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 212, 287–295. doi: 10.1084/jem.20142322
Jessen, N. A., Munk, A. S. F., Lundgaard, I., and Nedergaard, M. (2015). The glymphatic system: a beginner’s guide. Neurochem. Res. 40, 2583–2599.
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116.
Ju, Y.-E. S., McLeland, J. S., Toedebusch, C. D., Xiong, C., Fagan, A. M., Duntley, S. P., et al. (2013). Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 70, 587–593.
Ju, Y.-E. S., Ooms, S. J., Sutphen, C., Macauley, S. L., Zangrilli, M. A., Jerome, G., et al. (2017). Slow wave sleep disruption increases cerebrospinal fluid amyloid-β levels. Brain 140, 2104–2111. doi: 10.1093/brain/awx148
Jung, S., Aliberti, J., Graemmel, P., Sunshine, M. J., Kreutzberg, G. W., Sher, A., et al. (2000). Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 20, 4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000
Kadotani, H., Kadotani, T., Young, T., Peppard, P. E., Finn, L., Colrain, I. M., et al. (2001). Association between apolipoprotein e? 4 and sleep-disordered breathing in adults. JAMA 285, 2888–2890.
Keage, H. A. D., Banks, S., Yang, K. L., Morgan, K., Brayne, C., and Matthews, F. E. (2012). What sleep characteristics predict cognitive decline in the elderly? Sleep Med. 13, 886–892. doi: 10.1016/j.sleep.2012.02.003
Killgore, W. D. S. (2010). Effects of sleep deprivation on cognition. Prog. Brain Res. 185, 105–129. doi: 10.1016/b978-0-444-53702-7.00007-5
Kim, T.-S., Lim, H.-K., Lee, J. Y., Kim, D.-J., Park, S., Lee, C., et al. (2008). Changes in the levels of plasma soluble fractalkine in patients with mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 436, 196–200. doi: 10.1016/j.neulet.2008.03.019
Koo, K. Y. G., Schweizer, T. A., Fischer, C. E., and Munoz, D. G. (2019). Abnormal sleep behaviours across the spectrum of Alzheimer’s disease severity: influence of Apoe genotypes and Lewy Bodies. Curr. Alzheimer Res. 16, 243–250. doi: 10.2174/1567205016666190103161034
Krabbe, G., Halle, A., Matyash, V., Rinnenthal, J. L., Eom, G. D., Bernhardt, U., et al. (2013). Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One 8:e60921. doi: 10.1371/journal.pone.0060921
Krishnaiah, S. Y., Wu, G., Altman, B. J., Growe, J., Rhoades, S. D., Coldren, F., et al. (2017). Clock regulation of metabolites reveals coupling between transcription and metabolism. Cell Metab. 25, 961–974.e4. doi: 10.1016/j.cmet.2017.03.019
Kuhn, S. A., van Landeghem, F. K., Zacharias, R., Färber, K., Rappert, A., Pavlovic, S., et al. (2004). Microglia express GABAB receptors to modulate interleukin release. Mol. Cell. Neurosci. 25, 312–322. doi: 10.1016/j.mcn.2003.10.023
Lahiri, D. K. (1999). Melatonin affects the metabolism of the β-amyloid precursor protein in different cell types. J. Pineal Res. 26, 137–146. doi: 10.1111/j.1600-079x.1999.tb00575.x
Lee, J. H., Bliwise, D. L., Ansari, F. P., Goldstein, F. C., Cellar, J. S., Lah, J. J., et al. (2007). Daytime sleepiness and functional impairment in Alzheimer disease. Am. J. Geriatr. Psychiatry 15, 620–626. doi: 10.1097/JGP.0b013e3180381521
Lee, M., Schwab, C., and Mcgeer, P. L. (2011). Astrocytes are GABAergic cells that modulate microglial activity. Glia 59, 152–165. doi: 10.1002/glia.21087
Li, R., Yang, L., Lindholm, K., Konishi, Y., Yue, X., Hampel, H., et al. (2004). Tumor necrosis factor death receptor signaling cascade is required for amyloid-β protein-induced neuron death. J. Neurosci. 24, 1760–1771. doi: 10.1523/jneurosci.4580-03.2004
Liao, Y.-F., Wang, B.-J., Cheng, H.-T., Kuo, L.-H., and Wolfe, M. S. (2004). Tumor necrosis factor-α, interleukin-1β, and interferon-γ stimulate γ-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J. Biol. Chem. 279, 49523–49532. doi: 10.1074/jbc.m402034200
Limatola, C., and Ransohoff, R. M. (2014). Modulating neurotoxicity through CX3CL1/CX3CR1 signaling. Front. Cell. Neurosci. 8:229. doi: 10.3389/fncel.2014.00229
Lotufo, C. M., Yamashita, C. E., Farsky, S. H., and Markus, R. P. (2006). Melatonin effect on endothelial cells reduces vascular permeability increase induced by leukotriene B4. Eur. J. Pharmacol. 534, 258–263. doi: 10.1016/j.ejphar.2006.01.050
Maestri, M., Carnicelli, L., Tognoni, G., Di Coscio, E., Giorgi, F. S., Volpi, L., et al. (2015). Non-rapid eye movement sleep instability in mild cognitive impairment: a pilot study. Sleep Med. 16, 1139–1145. doi: 10.1016/j.sleep.2015.04.027
Mander, B. A., Winer, J. R., Jagust, W. J., and Walker, M. P. (2016). Sleep: a novel mechanistic pathway, biomarker, and treatment target in the pathology of Alzheimer’s disease? Trends Neurosci. 39, 552–566. doi: 10.1016/j.tins.2016.05.002
Matsuoka, T., Imai, A., Fujimoto, H., Kato, Y., Shibata, K., Nakamura, K., et al. (2017). Reduced pineal volume in alzheimer disease: a retrospective cross-sectional MR imaging study. Radiology 286, 239–248. doi: 10.1148/radiol.2017170188
McGeer, E. G., and McGeer, P. L. (2010). Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: a field in its infancy. J. Alzheimers Dis. 19, 355–361. doi: 10.3233/jad-2010-1219
McKhann, G., Drachman, D., Folstein, M., Katzman, R., Price, D., and Stadlan, E. M. (1984). Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group∗ under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–939. doi: 10.1212/wnl.34.7.939
Miguel-Álvarez, M., Santos-Lozano, A., Sanchis-Gomar, F., Fiuza-Luces, C., Pareja-Galeano, H., Garatachea, N., et al. (2015). Non-steroidal anti-inflammatory drugs as a treatment for Alzheimer’s disease: a systematic review and meta-analysis of treatment effect. Drugs Aging 32, 139–147. doi: 10.1007/s40266-015-0239-z
Mizuno, T., Kawanokuchi, J., Numata, K., and Suzumura, A. (2003). Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 979, 65–70. doi: 10.1016/s0006-8993(03)02867-1
Montagne, A., Barnes, S. R., Sweeney, M. D., Halliday, M. R., Sagare, A. P., Zhao, Z., et al. (2015). Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302. doi: 10.1016/j.neuron.2014.12.032
Montplaisir, J., Petit, D., Gauthier, S., Gaudreau, H., and Decary, A. (1998). Sleep disturbances and eeg slowing in Alzheimer’s disease. Sleep Res. Online 1, 147–151.
Muangpaisan, W., Intalapaporn, S., and Assantachai, P. (2008). Neuropsychiatric symptoms in the community-based patients with mild cognitive impairment and the influence of demographic factors. Int. J. Geriatr. Psychiatry 23, 699–703. doi: 10.1002/gps.1963
Mullington, J. M., Abbott, S. M., Carroll, J. E., Davis, C. J., Dijk, D.-J., Dinges, D. F., et al. (2016). Developing biomarker arrays predicting sleep and circadian-coupled risks to health. Sleep 39, 727–736. doi: 10.5665/sleep.5616
Musiek, E. S. (2017). Circadian rhythms in AD pathogenesis: a critical appraisal. Curr. Sleep Med. Rep. 3, 85–92. doi: 10.1007/s40675-017-0072-5
Naismith, S. L., Rogers, N. L., Hickie, I. B., Mackenzie, J., Norrie, L. M., and Lewis, S. J. (2010). Sleep well, think well: sleep-wake disturbance in mild cognitive impairment. J. Geriatr. Psychiatry Neurol. 23, 123–130. doi: 10.1177/0891988710363710
Nayak, D., Roth, T. L., and McGavern, D. B. (2014). Microglia development and function. Annu. Rev. Immunol. 32, 367–402. doi: 10.1146/annurev-immunol-032713-120240
Ni, J., Wu, Z., Meng, J., Saito, T., Saido, T. C., Qing, H., et al. (2019). An impaired intrinsic microglial clock system induces neuroinflammatory alterations in the early stage of amyloid precursor protein knock-in mouse brain. J. Neuroinflammation 16:173.
Ojo, J. O., Rezaie, P., Gabbott, P. L., and Stewart, M. G. (2015). Impact of age-related neuroglial cell responses on hippocampal deterioration. Front. Aging Neurosci. 7:57. doi: 10.3389/fnagi.2015.00057
Okun, M., Giese, S., Lin, L., Einen, M., Mignot, E., and Coussons-Read, M. (2004). Exploring the cytokine and endocrine involvement in narcolepsy. Brain Behav. Immun. 18, 326–332. doi: 10.1016/j.bbi.2003.11.002
Olsson, B., Hertze, J., Lautner, R., Zetterberg, H., Nagga, K., Hoglund, K., et al. (2013). Microglial markers are elevated in the prodromal phase of Alzheimer’s disease and vascular dementia. J. Alzheimers Dis. 33, 45–53. doi: 10.3233/jad-2012-120787
Olsson, M., Ärlig, J., Hedner, J., Blennow, K., and Zetterberg, H. (2018). Sleep deprivation and cerebrospinal fluid biomarkers for Alzheimer’s disease. Sleep 41:zsy025. doi: 10.1093/sleep/zsy025
Ooms, S., Overeem, S., Besse, K., Rikkert, M. O., Verbeek, M., and Claassen, J. A. (2014). Effect of 1 night of total sleep deprivation on cerebrospinal fluid beta-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol. 71, 971–977. doi: 10.1001/jamaneurol.2014.1173
Park, M., Shah, R. C., Fogg, L. F., and Wyatt, J. K. (2011). Daytime sleepiness in mild Alzheimer’s disease with and without parkinsonian features. Sleep Med. 12, 397–402. doi: 10.1016/j.sleep.2010.09.006
Petersen, R. C., Smith, G. E., Waring, S. C., Ivnik, R. J., Tangalos, E. G., and Kokmen, E. (1999). Mild cognitive impairment: clinical characterization and outcome. Arch. Neurol. 56, 303–308.
Potvin, O., Lorrain, D., Forget, H., Dubé, M., Grenier, S., Préville, M., et al. (2012). Sleep quality and 1-year incident cognitive impairment in community-dwelling older adults. Sleep 35, 491–499. doi: 10.5665/sleep.1732
Reitz, C., and Mayeux, R. (2014). Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 88, 640–651. doi: 10.1016/j.bcp.2013.12.024
Ringheim, G. E., Szczepanik, A. M., Petko, W., Burgher, K. L., Zu Zhu, S., and Chao, C. C. (1998). Enhancement of beta-amyloid precursor protein transcription and expression by the soluble interleukin-6 receptor/interleukin-6 complex. Mol. Brain Res. 55, 35–44. doi: 10.1016/s0169-328x(97)00356-2
Robinson, D. (1975). Changes in monoamine oxidase and monoamines with human development and aging. Fed. Proc. 34, 103–107.
Rosenzweig, I., Glasser, M., Crum, W. R., Kempton, M. J., Milosevic, M., McMillan, A., et al. (2016). Changes in neurocognitive architecture in patients with obstructive sleep apnea treated with continuous positive airway pressure. EBioMedicine 7, 221–229. doi: 10.1016/j.ebiom.2016.03.020
Rothaug, M., Becker-Pauly, C., and Rose-John, S. (2016). The role of interleukin-6 signaling in nervous tissue. Biochim. Biophys. Acta 1863, 1218–1227. doi: 10.1016/j.bbamcr.2016.03.018
Savage, J. C., Jay, T., Goduni, E., Quigley, C., Mariani, M. M., Malm, T., et al. (2015). Nuclear receptors license phagocytosis by trem2+ myeloid cells in mouse models of Alzheimer’s disease. J. Neurosci. 35, 6532–6543. doi: 10.1523/jneurosci.4586-14.2015
Savaskan, E., Ayoub, M. A., Ravid, R., Angeloni, D., Fraschini, F., Meier, F., et al. (2005). Reduced hippocampal MT2 melatonin receptor expression in Alzheimer’s disease. J. Pineal Res. 38, 10–16. doi: 10.1111/j.1600-079x.2004.00169.x
Savaskan, E., Olivieri, G., Meier, F., Brydon, L., Jockers, R., Ravid, R., et al. (2002). Increased melatonin 1a-receptor immunoreactivity in the hippocampus of Alzheimer’s disease patients. J. Pineal Res. 32, 59–62. doi: 10.1034/j.1600-079x.2002.00841.x
Schoonenboom, S., Visser, P. J., Mulder, C., Lindeboom, J., Van Elk, E.-J., Van Kamp, G. J., et al. (2005). Biomarker profiles and their relation to clinical variables in mild cognitive impairment. Neurocase 11, 8–13. doi: 10.1080/13554790490896785
Schuitemaker, A., Dik, M. G., Veerhuis, R., Scheltens, P., Schoonenboom, N. S., Hack, C. E., et al. (2009). Inflammatory markers in AD and MCI patients with different biomarker profiles. Neurobiol. Aging 30, 1885–1889. doi: 10.1016/j.neurobiolaging.2008.01.014
Sharma, R. A., Varga, A. W., Bubu, O. M., Pirraglia, E., Kam, K., Parekh, A., et al. (2018). Obstructive sleep apnea severity affects amyloid burden in cognitively normal elderly. A longitudinal study. Am. J. Respir. Crit. Care Med. 197, 933–943.
Shokri-Kojori, E., Wang, G.-J., Wiers, C. E., Demiral, S. B., Guo, M., Kim, S. W., et al. (2018). β-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc. Natl. Acad. Sci. U.S.A. 115, 4483–4488. doi: 10.1073/pnas.1721694115
Singh-Manoux, A., Dugravot, A., Brunner, E., Kumari, M., Shipley, M., Elbaz, A., et al. (2014). Interleukin-6 and C-reactive protein as predictors of cognitive decline in late midlife. Neurology 83, 486–493. doi: 10.1212/wnl.0000000000000665
Spira, A. P., An, Y., Wu, M. N., Owusu, J. T., Simonsick, E. M., Bilgel, M., et al. (2018). Excessive daytime sleepiness and napping in cognitively normal adults: associations with subsequent amyloid deposition measured by PiB PET. Sleep 41:zsy152. doi: 10.1093/sleep/zsy152
Spira, A. P., Gamaldo, A. A., An, Y., Wu, M. N., Simonsick, E. M., Bilgel, M., et al. (2013). Self-reported Sleep and β-Amyloid Deposition in Community-Dwelling Older AdultsSelf-reported Sleep and β-Amyloid DepositionSelf-reported Sleep and β-Amyloid Deposition. JAMA Neurol. 70, 1537–1543. doi: 10.1001/jamaneurol.2013.4258
Spira, A. P., Yager, C., Brandt, J., Smith, G. S., Zhou, Y., Mathur, A., et al. (2014). Objectively measured sleep and beta-amyloid burden in older adults: a pilot study. SAGE Open Med. 2:2050312114546520. doi: 10.1177/2050312114546520
Sprecher, K. E., Bendlin, B. B., Racine, A. M., Okonkwo, O. C., Christian, B. T., Koscik, R. L., et al. (2015). Amyloid burden is associated with self-reported sleep in nondemented late middle-aged adults. Neurobiol. Aging 36, 2568–2576. doi: 10.1016/j.neurobiolaging.2015.05.004
Sprecher, K. E., Koscik, R. L., Carlsson, C. M., Zetterberg, H., Blennow, K., Okonkwo, O. C., et al. (2017). Poor sleep is associated with CSF biomarkers of amyloid pathology in cognitively normal adults. Neurology 89, 445–453. doi: 10.1212/wnl.0000000000004171
Tamura, E. K., Cecon, E., Monteiro, A. W. A., Silva, C. L. M., and Markus, R. P. (2009). Melatonin inhibits LPS-induced NO production in rat endothelial cells. J. Pineal Res. 46, 268–274. doi: 10.1111/j.1600-079x.2008.00657.x
Tsapanou, A., Gu, Y., Manly, J., Schupf, N., Tang, M.-X., Zimmerman, M., et al. (2015). Daytime sleepiness and sleep inadequacy as risk factors for dementia. Dement. Geriatr. Cogn. Disord. Extra 5, 286–295. doi: 10.1159/000431311
Twigg, G. L., Papaioannou, I., Jackson, M., Ghiassi, R., Shaikh, Z., Jaye, J., et al. (2010). Obstructive sleep apnea syndrome is associated with deficits in verbal but not visual memory. Am. J. Respir. Crit. Care Med. 182, 98–103. doi: 10.1164/rccm.200901-0065oc
Tworoger, S. S., Lee, S., Schernhammer, E. S., and Grodstein, F. (2006). The association of self-reported sleep duration, difficulty sleeping, and snoring with cognitive function in older women. Alzheimer Dis. Assoc. Disord. 20, 41–48. doi: 10.1097/01.wad.0000201850.52707.80
Vgontzas, A. N., Papanicolaou, D. A., Bixler, E. O., Kales, A., Tyson, K., and Chrousos, G. P. (1997). Elevation of plasma cytokines in disorders of excessive daytime sleepiness: role of sleep disturbance and obesity. J. Clin. Endocrinol. Metab. 82, 1313–1316. doi: 10.1210/jcem.82.5.3950
Vgontzas, A. N., Zoumakis, E., Bixler, E. O., Lin, H.-M., Follett, H., Kales, A., et al. (2004). Adverse effects of modest sleep restriction on sleepiness, performance, and inflammatory cytokines. J. Clin. Endocrinol. Metab. 89, 2119–2126. doi: 10.1210/jc.2003-031562
Vitiello, M. V., Prinz, P. N., Williams, D. E., Frommlet, M. S., and Ries, R. K. (1990). Sleep disturbances in patients with mild-stage Alzheimer’s disease. J. Gerontol. 45, M131–M138.
Wang, Y., Cella, M., Mallinson, K., Ulrich, J. D., Young, K. L., Robinette, M. L., et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. doi: 10.1016/j.cell.2015.01.049
Weissova, K., Bartoš, A., Sládek, M., Novakova, M., and Sumová, A. (2016). Moderate changes in the circadian system of Alzheimer’s disease patients detected in their home environment. PLoS One 11:e0146200. doi: 10.1371/journal.pone.0146200
Westerberg, C. E., Mander, B. A., Florczak, S. M., Weintraub, S., Mesulam, M. M., Zee, P. C., et al. (2012). Concurrent impairments in sleep and memory in amnestic mild cognitive impairment. J. Int. Neuropsychol. Soc. 18, 490–500. doi: 10.1017/s135561771200001x
Wisor, J. P., Schmidt, M. A., and Clegern, W. C. (2011). Cerebral microglia mediate sleep/wake and neuroinflammatory effects of methamphetamine. Brain Behav. Immun. 25, 767–776. doi: 10.1016/j.bbi.2011.02.002
Wu, Y. H., and Swaab, D. F. (2005). The human pineal gland and melatonin in aging and Alzheimer’s disease. J. Pineal Res. 38, 145–152. doi: 10.1111/j.1600-079x.2004.00196.x
Xie, L., Kang, H., Xu, Q., Chen, M. J., Liao, Y., Thiyagarajan, M., et al. (2013). Sleep drives metabolite clearance from the adult brain. Science 342, 373–377. doi: 10.1126/science.1241224
Xie, Y., Ba, L., Wang, M., Deng, S. Y., Chen, S. M., Huang, L. F., et al. (2019). Chronic sleep fragmentation shares similar pathogenesis with neurodegenerative diseases: Endosome-autophagosome-lysosome pathway dysfunction and microglia-mediated neuroinflammation. CNS Neurosci. Ther. 26, 215–227. doi: 10.1111/cns.13218
Xu, L., Jiang, C. Q., Lam, T. H., Liu, B., Jin, Y. L., Zhu, T., et al. (2011). Short or long sleep duration is associated with memory impairment in older Chinese: the Guangzhou Biobank Cohort Study. Sleep 34, 575–580. doi: 10.1093/sleep/34.5.575
Yaffe, K., Falvey, C. M., and Hoang, T. (2014). Connections between sleep and cognition in older adults. Lancet Neurol. 13, 1017–1028. doi: 10.1016/S1474-4422(14)70172-3
Yaffe, K., Laffan, A. M., Harrison, S. L., Redline, S., Spira, A. P., Ensrud, K. E., et al. (2011). Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA 306, 613–619.
You, J. C., Jones, E., Cross, D. E., Lyon, A. C., Kang, H., Newberg, A. B., et al. (2019). Association of β-amyloid burden with sleep dysfunction and cognitive impairment in elderly individuals with cognitive disorders. JAMA Netw. Open 2:e1913383. doi: 10.1001/jamanetworkopen.2019.13383
Yun, C.-H., Lee, H.-Y., Lee, S. K., Kim, H., Seo, H. S., Bang, S., et al. (2017). Amyloid burden in obstructive sleep apnea. J. Alzheimers Dis. 59, 21–29.
Zhou, J. N., Liu, R. Y., Kamphorst, W., Hofman, M. A., and Swaab, D. F. (2003). Early neuropathological Alzheimer’s changes in aged individuals are accompanied by decreased cerebrospinal fluid melatonin levels. J. Pineal Res. 35, 125–130. doi: 10.1034/j.1600-079x.2003.00065.x
Zhu, B., Dong, Y., Xu, Z., Gompf, H. S., Ward, S. A., Xue, Z., et al. (2012). Sleep disturbance induces neuroinflammation and impairment of learning and memory. Neurobiol. Dis. 48, 348–355. doi: 10.1016/j.nbd.2012.06.022
Keywords: neuroinflammation, sleep, mild cognitive impairment, Alzheimer’s disease, aging
Citation: Pak VM, Onen S-H, Bliwise DL, Kutner NG, Russell KL and Onen F (2020) Sleep Disturbances in MCI and AD: Neuroinflammation as a Possible Mediating Pathway. Front. Aging Neurosci. 12:69. doi: 10.3389/fnagi.2020.00069
Received: 19 September 2019; Accepted: 26 February 2020;
Published: 08 May 2020.
Edited by:
Daniel Ortuño-Sahagún, Universidad de Guadalajara, MexicoReviewed by:
Luca Steardo, Magna Græcia University of Catanzaro, ItalyMarisol Godínez-Rubí, Universidad de Guadalajara, Mexico
Copyright © 2020 Pak, Onen, Bliwise, Kutner, Russell and Onen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Victoria M. Pak, dmljdG9yaWEubS5wYWtAZW1vcnkuZWR1