 Qiang Li
Qiang Li Xiaohui He
Xiaohui He Yuanhang Ren
Yuanhang Ren Chuan Xiong
Chuan Xiong Xin Jin
Xin Jin Lianxin Peng
Lianxin Peng Wenli Huang
Wenli Huang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 19 June 2020
Sec. Fungi and Their Interactions
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.01382
In this present study, we assembled and analyzed the mitogenomes of two asymbiotic and six ectomycorrhizal Amanita species based on next-generation sequencing data. The size of the eight Amanita mitogenomes ranged from 37,341 to 137,428 bp, and we considered introns to be one of the main factors contributing to the size variation of Amanita. The introns of the cox1 gene experienced frequent gain/loss events in Amanita; and the intron position class cox1P386 was lost in the six ectomycorrhizal Amanita species. In addition, ectomycorrhizal Amanita species had more repetitive sequences and fewer intergenic sequences than asymbiotic Amanita species in their mitogenomes. Large-scale gene rearrangements were detected in the Amanita species we tested, including gene displacements and inversions. On the basis of the combined mitochondrial gene set, we reconstructed the phylogenetic relationships of 66 Basidiomycetes. The six ectomycorrhizal Amanita species were of single origin, and the two saprophytic Amanita species formed two distinct clades. This study is the first to elucidate the functions of the mitogenome in the evolution and ecological adaptation of Amanita species.
The genus of Amanita, belonging to Agaricales, Basidiomycetes, is a group of macrofungi. Amanita species are widely distributed around the world and have a variety of lifestyles (Wolfe et al., 2012a). Most of Amanita species form ectomycorrhizal associations with host plants, with some saprotrophic representatives (Wolfe et al., 2012b; De Mares et al., 2015). All Amanita species were derived from asymbiotic ancestors (De Mares et al., 2015). Amanita species has become one of the model organisms for the study of the life history, genetics, and evolution of ectomycorrhizal and saprotrophic fungi. Kohler et al. (2015) found that Amanita may lose some genes encoding plant cell wall–degrading enzymes in ectomycorrhizal life. Hess et al. (2014, 2018) further confirmed these results and proved that ectomycorrhizal symbiosis may be accompanied by an increased transposable element (TE) activity. Some ectomycorrhizal Amanita species have obtained carbohydrate metabolism genes through horizontal transfer (De Mares et al., 2015). The features and evolution of mitochondrial genomes (mitogenomes) in Amanita species remain unknown, however, which limits our comprehensive understanding of the evolution of Amanita species.
Amanita is a genus with a rich biodiversity, and approximately 600 species have been described in the genus (Sanchez-Ramirez et al., 2015). Consumers appreciate some species in this genus as delicious edible fungi, but some Amanita species are lethal, causing hundreds of cases of food poisoning every year worldwide (Cai et al., 2016; Ye and Liu, 2018). Similar and confusing morphological characteristics make it difficult to accurately distinguish between lethal and edible Amanita species based only on their morphologies (Garcia et al., 2015; Sun et al., 2018). The introduction of molecular markers, including internal transcribed spacers, RNA polymerase II, β-tubulin gene, and translation elongation factor 1-α gene (tef1-α), has promoted the classification and identification of Amanita species (Thongbai et al., 2016, 2017). The rapid rate of evolution, independent origin from the nuclear genome, and several available molecular markers has made the mitogenome a powerful tool for species classification and population genetics studies (Li et al., 2018b, c). Until now, however, no complete mitogenome has been published for the genus of Amanita.
Mitogenomes reportedly have been derived from alphabacteria through endosymbiosis by the ancestors of eukaryotes (Lang et al., 1999). By evolving with the host organisms, the features, gene arrangement, repeat sequences, introns, and protein-coding genes (PCGs) of mitogenomes have been considered to be useful information to reveal the evolution, environmental adaptation, and phylogeny of eukaryotic organisms (Boore, 1999; Poliseno et al., 2017; Qian et al., 2018; Sankoff et al., 1992). With the rapid development of the next-generation sequencing technology, increasing numbers of mitogenomes have been obtained, including animals, plants, and fungi. The mitogenome of fungi, however, has been less studied than that of animals, especially Basidiomycetes. Thus far, less than 100 Basidiomycetes mitogenomes were available in public databases [National Center for Biotechnology Information (NCBI)1 ], which has limited our comprehensive understanding of the genetics of the world’s largest mushroom-forming fungal group. Even relative to the total number of available nuclear genomes2, the number of Basidiomycetes mitogenomes was far behind. According to limited reports, the mitogenomes of Basidiomycetes were among the most variable mitogenomes. Their genome size, gene arrangement, content of repeat sequence, and intron classes varied greatly among different Basidiomycetes species as well as between closely related species (Kanzi et al., 2016; Sandor et al., 2018). The Basidiomycetes mitogenomes, however, were found to contain 14 conserved PCGs for energy metabolism (atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, and nad6) and one rps3 gene for transcriptional regulation, which we called the core PCGs of Basidiomycetes (Li et al., 2018d, 2019a). In addition, we also detected two rRNA genes and 20–36 tRNA genes in the Basidiomycete mitogenomes.
In the present study, we assembled and annotated mitogenomes of eight Amanita species, including six ectomycorrhizal species (Amanita basii, Amanita muscaria, Amanita bisporigera, Amanita phalloides, Amanita brunnescens, and Amanita pseudoporphyria) and two asymbiotic species (Amanita inopinata and Amanita thiersii). The aims of this study are (1) to reveal the features of Amanita mitogenomes and the similarities or variations between the ectomycorrhizal and asymbiotic mitogenomes in gene content, genome size, gene order, and repeat sequence; (2) to reveal the dynamic changes of introns in ectomycorrhizal and asymbiotic Amanita species. Mitogenomes of the eight Amanita species further our understanding of the evolutionary biology, genetics, and taxonomy of this important macrofungal genus.
We downloaded the raw sequencing data of A. basii, A. muscaria, A. bisporigera, A. phalloides, A. brunnescens, A. inopinata, and A. thiersii from the NCBI Sequence Read Archive (SRA) database (Hess et al., 2014, 2018; Kohler et al., 2015; Pulman et al., 2016). We obtained sequencing data for A. pseudoporphyria from our 100 mushroom genome project (Li H. et al., 2018). The species information and SRA accession numbers used for mitogenome assembly could be found in Supplementary Table S1. We conducted a series of quality control steps to obtain clean reads from the raw sequencing data. First, we removed adapter reads in the raw sequences using the AdapterRemoval v2 (Schubert et al., 2016), and then we filtered low-quality sequences using ngsShoRT (Chen C. et al., 2014). We used MITObim V1.9 (Hahn et al., 2013) to assemble the eight Amanita mitogenomes with the obtained clean reads. We annotated the obtained complete mitogenomes of the eight Amanita species according to our previously described methods (Li et al., 2018a). Briefly, we initially annotated the rRNA genes, tRNA genes, intron, and PCGs of the eight mitogenomes using the MITOS (Bernt et al., 2013) and MFannot (Valach et al., 2014), both of which are based on the genetic code 4. We then modified or predicted the PCGs using the NCBI Open Reading Frame Finder3 and further annotated the PCGs by BLASTP searches against the NCBI non-redundant protein sequence database (Bleasby and Wootton, 1990). We also used tRNAscan-SE v1.3.1 (Lowe and Chan, 2016) to identify or predict tRNA genes in the eight Amanita mitogenomes. We used OGDraw v1.2 (Lohse et al., 2013) to draw graphical maps of the eight complete mitogenomes.
We used DNASTAR Lasergene v7.14 to calculate base compositions of the eight Amanita mitogenomes. We assessed strand asymmetries of the eight mitogenomes according to the following formulas: AT skew = (A−T) /(A + T), and GC skew = (G−C)/(G + C) (Wang et al., 2017). Pairwise genetic distances between each pair of the 15 core PCGs (atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, and rps3) were calculated using the MEGA v6.06 (Caspermeyer, 2016) with the Kimura-2-parameter (K2P) substitution model. Collinearity analysis of the eight Amanita mitogenome was conducted using Mauve (Darling et al., 2004), and color blocks of the same color represented homologous regions between different mitogenomes.
To identify whether there were interspersed repeats or intragenomic duplications of large fragments throughout the eight mitogenomes, we conducted BLASTN searches of each mitogenome against itself using discontiguous megablast with general parameters (Johnson et al., 2008). We used Tandem Repeats Finder (Benson, 1999) with default parameters to detect tandem repeats (>10 bp in length) in the eight mitogenomes. We also used REPuter (Kurtz et al., 2001) to identify forward (direct), reverse, complemented, and palindromic (revere complemented) repeats in the eight mitogenomes. We manually calculated the content of non-overlapping repeat sequence identified by the three methods.
We classified introns in cox1 genes of the eight Amanita mitogenomes into different position classes (Pcls) according to the method described by Ferandon et al. (2010). Each Pcl was constituted by introns inserted at the same position in the coding region of the cox1 gene. The same Pcl from different species usually had a high sequence similarity and contained orthologous intronic opening reading frames (ORFs). Pcls were named according to the method described by Zhang and Zhang (2019).
To investigate the phylogenetic relationships of species in the Basidiomycota phylum, we constructed a phylogenetic tree of 67 species based on the combined mitochondrial gene set (15 core PCGs + 2 rRNA genes) (Li et al., 2018d). Annulohypoxylon stygium from the Ascomycota phylum was set as the outgroup (Deng et al., 2018). We used MAFFT v7.037 software (Katoh et al., 2017) to align individual mitochondrial genes and then used SequenceMatrix v1.7.8 (Vaidya et al., 2011) to concatenate these alignments into a combined mitochondrial gene set. We used the PartitionFinder 2.1.1 (Lanfear et al., 2017) to determine best-fit models of evolution and partitioning schemes for the gene set. We used Bayesian inference (BI) and maximum likelihood (ML) methods to construct phylogenetic trees. We conducted BI analysis using the MrBayes v3.2.6 (Ronquist et al., 2012) and performed ML analysis with RAxML v 8.0.0 (Stamatakis, 2014). When conducted BI analysis, two independent BI runs with four chains (three heated and one cold) were each conducted simultaneously over 2 × 106 generations. Each run was sampled every 100 generations. Stationarity was assumed to have been reached when the estimated sample size (ESS) was greater than 100, and the potential scale reduction factor (PSRF) approached 1.0. The first 25% of the samples were discarded as burn-in, and the remaining trees were used to calculate Bayesian posterior probabilities (BPP) and construct a 50% majority-rule consensus tree.
The complete mitogenomes of A. basii, A. muscaria, A. bisporigera, A. phalloides, A. brunnescens, A. pseudoporphyria, A. inopinata, and A. thiersii were deposited in the GenBank database under the accession numbers MK993555, MK993559, MK993556, MK993560, MK993557, MK993554, MK993558, and MK993561, respectively.
The size of mitogenomes from eight Amanita species ranged from 37,341 to 137,428 bp, with an average size of 73,728 bp (Figure 1). The GC content of the eight mitogenomes ranged from 23.4 to 27.56%, and the average GC content was 24.63%. Of the eight mitogenomes we assembled, only three AT skews were positive, including A. phalloides, A. inopinata, and A. thiersii. The GC skews of all eight mitogenomes we tested were positive (Supplementary Table S1).
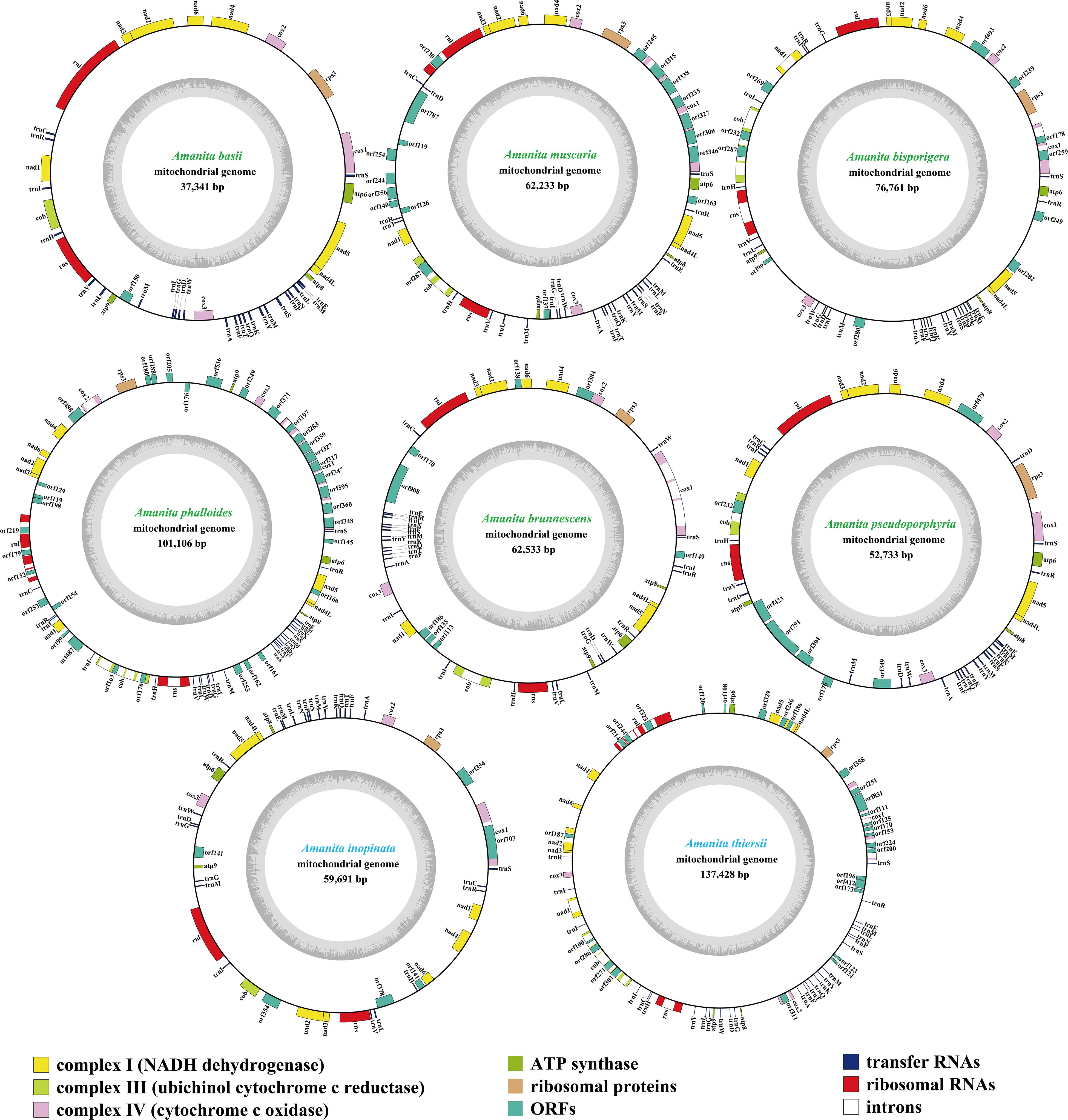
Figure 1. Circular maps of the mitogenomes of eight Amanita species. Genes are represented by different colored blocks. Colored blocks outside each ring indicate that the genes are on the direct strand, whereas colored blocks within the ring indicate that the genes are located on the reverse strand. The Amanita species with blue fonts are asymbiotic, while the species with green fonts are ectomycorrhizal.
We detected 16–49 PCGs in the eight Amanita mitogenomes. A. phalloides contained the largest number of non-intronic PCGs among the eight Amanita species detected, with 34 (Supplementary Table S1); A. basii contained the least number of non-intronic PCGs, with only 16. All eight mitogenomes contained a set of core PCGs, including 14 core PCGs for energy metabolism and 1 rps3 gene for transcriptional regulation (Supplementary Table S2). In addition, A. phalloides, A. brunnescens, A. pseudoporphyria, A. inopinata, and A. thiersii contained 2, 2, 3, 1, and 3 non-intronic genes coded DNA polymerases, respectively. A. basii, A. muscaria, A. bisporigera, A. phalloides, A. brunnescens, A. pseudoporphyria, A. inopinata, and A. thiersii contained 1, 10, 7, 17, 6, 3, 4, and 6 non-intronic genes with unknown functions, respectively. We detected 8, 4, 15, 1, 1, and 19 intronic ORFs detected in the mitogenomes of A. muscaria, A. bisporigera, A. phalloides, A. pseudoporphyria, A. inopinata, and A. thiersii, respectively. These intronic ORFs encoded homing endonucleases from different families, including the GIY-YIG homing nuclease and LAGLIDADG endonuclease.
All eight Amanita mitogenomes contained two rRNA genes, namely, the large subunit ribosomal RNA (rnl) and the small subunit ribosomal RNA (rns) (Supplementary Table S2). The rns genes of A. bisporigera, A. phalloides, and A. thiersii all contained one intron, whereas the other rns genes did not contain any intron. The length of the rns gene varied in different Amanita species. A. thiersii had the longest rns gene, reaching 2,074 bp, whereas the rns gene of A. inopinata was the shortest, with only 1,896 bp. We detected 1, 4, and 4 introns in the rnl genes of A. muscaria, A. phalloides, and A. thiersii, respectively. A. inopinata had the longest rnl gene (3,552 bp), whereas A. pseudoporphyria had the shortest (3,367 bp).
We detected between 25 and 28 tRNA genes in the eight Amanita mitogenomes, which encoded 20 standard amino acids (Supplementary Table S2). All eight mitogenomes contained two tRNAs that coded for serine and leucine with different anticodons and three tRNAs that coded for methionine with the same anticodons. We also detected additional tRNA genes, including the trnD, trnG, trnI, and trnR, in the eight mitogenomes. The length of individual tRNAs ranged from 71 to 88 bp, with these length variations mainly resulting from the expansion or contraction of extra arms. The length expansions of extra arms in the trns genes contributed to their lengths becoming the longest among all the tRNAs.
By comparing the eight mitogenomes with themselves through a BLASTN search, we identified 5, 11, 64, 51, 56, 18, 8, and 34 repeat sequences in the mitogenomes of A. basii, A. muscaria, A. bisporigera, A. phalloides, A. brunnescens, A. pseudoporphyria, A. inopinata, and A. thiersii, respectively (Supplementary Table S3). The length of the individual repeat sequences ranged from 34 to 2,875 bp, with pair-wise nucleotide similarities ranging from 67.15 to 100%. The longest repeat sequences were located in the intergenic region between orf99 and cox3 and between the orf280 and trnA genes in the A. bisporigera mitogenome. A. bisporigera contained the highest proportion of repetitive sequences, accounting for 36.62% of the entire mitogenome. Repeated sequences accounted for 2.53, 3.39, 10.26, 9.82, 12.36, 1.22, and 2.15% of the mitogenomes of A. basii, A. muscaria, A. phalloides, A. brunnescens, A. pseudoporphyria, A. inopinata, and A. thiersii, respectively.
We detected a total of 7, 15, 46, 56, 43, 7, 24, and 300 tandem repeats in the A. basii, A. muscaria, A. bisporigera, A. phalloides, A. brunnescens, A. pseudoporphyria, A. inopinata, and A. thiersii mitogenomes, respectively (Supplementary Table S4). The longest tandem sequence was located between trnG and atp8 in the A. thiersii mitogenome, with a length of 258 bp. Most of the tandem repeat sequences were repeated two to four times in the eight Amanita mitogenomes, with the highest replication number (35) observed in the A. thiersii mitogenome. Tandem repeat sequences accounted for 1.31, 1.23, 3.31, 2.83, 3.53, 0.66, 1.66, and 14.83% of the A. basii, A. muscaria, A. bisporigera, A. phalloides, A. brunnescens, A. pseudoporphyria, A. inopinata, and A. thiersii mitogenomes, respectively. Using the REPuter, we identified 42 forward and eight reverse repeats in the mitogenome of A. thiersii, accounting for 3.44% of the entire mitogenome (Supplementary Table S5). Repeats identified by REPuter accounted for 2.92, 1.91, 3.84, 3.27, 4.50, 3.47, and 2.46 of the A. basii, A. muscaria, A. bisporigera, A. phalloides, A. brunnescens, A. pseudoporphyria, and A. inopinata mitogenomes, respectively.
Phylogenetic analysis using ML and BI methods based on the combined mitochondrial gene set (14 core PCGs + rps3 + 2 rRNA genes) using the GTR + I + G substitution model yielded identical and well-supported tree topologies (Figure 2). All major clades within the trees were well supported (BPP ≥0.96; bootstrap (BS) ≥99). On the basis of phylogenetic analysis, the 66 Basidiomycota species could be divided into 13 major clades, corresponding to the orders Agaricales, Boletales, Russulales, Polyporales, Cantharellales, Tremellales, Trichosporonales, Microstromatales, Ustilaginales, Tilletiales, Microbotryales, Sporidiobolales, and Pucciniales (Supplementary Table S6). The Amanita genus was divided into four groups, wherein the first included A. thiersii, the second group included A. inopinata, the third group was recovered as [A. pseudoporphyria + A. brunnescens + (A. phalloides + A. bisporigera)], and the forth group was recovered as (A. muscaria + A. basii). The phylogenetic analyses indicated that A. phalloides was a sister species to A. bisporigera, and A. muscaria was a sister species to A. basii. A. thiersii and A. inopinata were differentiated earlier from other Amanita species.
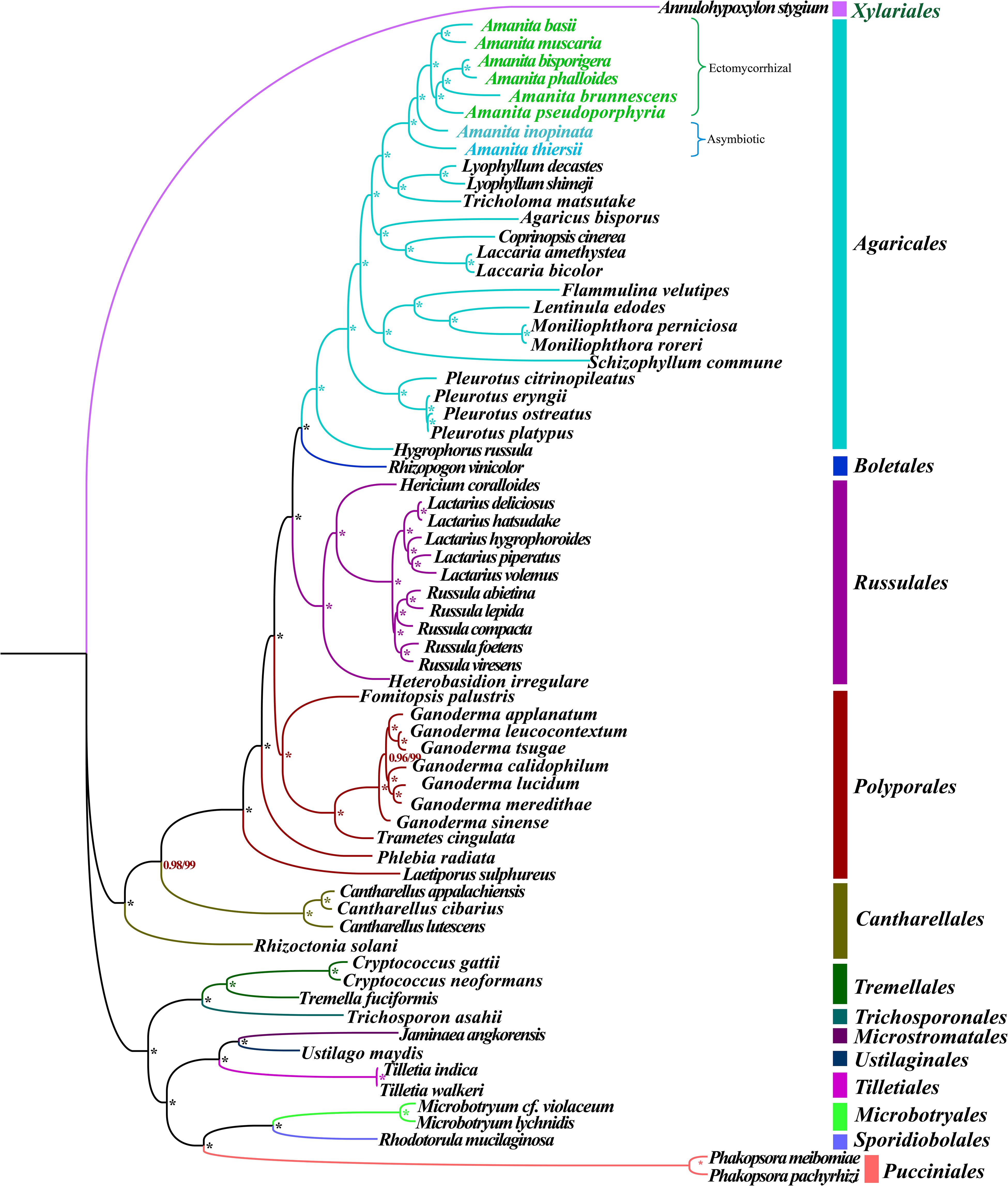
Figure 2. Molecular phylogeny of 66 Basidiomycete species based on BI and ML analysis of 15 PCGs and two rRNA genes. Support values are BPP (before slash) and BS values (after slash). The asterisk indicates that the BPP value is 1 and the BS value is 100 of the branch. Species and NCBI accession numbers for genomes used in the phylogenetic analysis are provided in Supplementary Table S6.
We frequently observed large-scale gene rearrangements at the level of families, even genera, from the Agaricales order (Li et al., 2018b, 2019b; Figure 3). The mitochondrial gene arrangements of the Amanita species were distinctly different from that of any other Agaricales species. Among the eight mitogenomes we tested, A. basii, A. muscaria, A. bisporigera, and A. pseudoporphyria had identical mitochondrial gene orders. Relative to the gene order in the four ectomycorrhizal Amanita species above, large-scale gene rearrangement events were found in the mitogenome of A. phalloides, A. brunnescens, A. inopinata, and A. thiersii. The displacement of cox3 and atp9 genes occurred in the mitogenome of A. phalloides. The mitogenome of A. brunnescens involved the displacement of cox3 gene and the inversion of atp6 and nad5 with nad4L and atp8 genes. Mitogenomes of A. inopinata, and A. thiersii involved multiple gene relocations, including nad4L, nad5, atp6, and cox3. The results indicated that the mitochondrial gene arrangement of Amanita species was diverse.
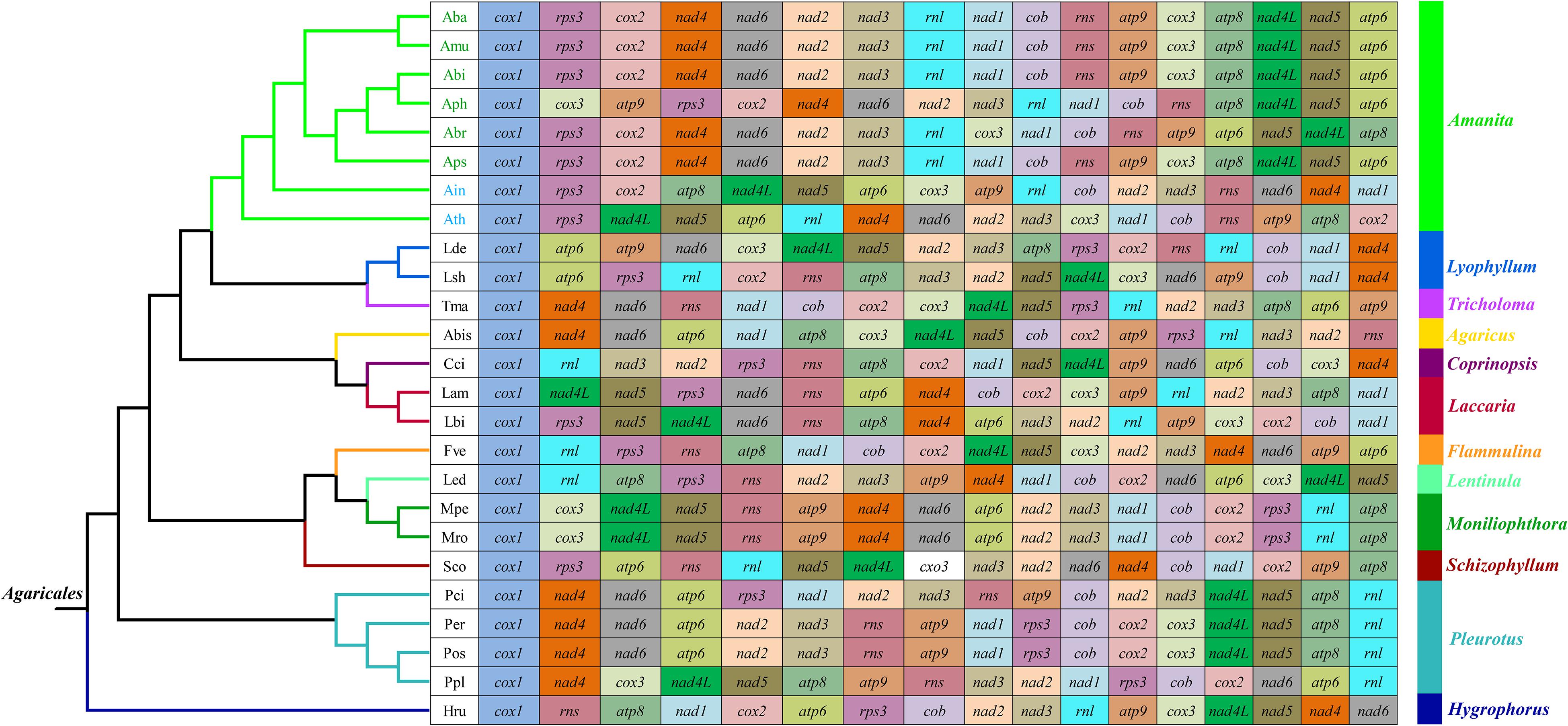
Figure 3. Mitochondrial gene arrangement analyses of 25 species from the genus of Agaricales. The phylogenetic positions of 25 Agaricales species were established using the BI method and ML method based on 15 concatenated mitochondrial core proteins and two rRNA genes. Genes are represented with different color blocks. All genes are shown in order of occurrence in the mitochondrial genome, starting from cox1. Fourteen core PCGs genes, one rps3 gene, and two rRNA genes were included in the gene arrangement analysis. Species and NCBI accession number used for gene arrangement analysis in the present study are listed in Supplementary Table S6.
We detected 20 homologous regions among the eight Amanita mitogenomes (Figure 4). The size variation of homologous regions and the expansion or contraction of regions between homologous sequences promoted the size dynamics of Amanita mitogenomes. Different Amanita species contained varied amounts and groups of homologous regions. We detected homologous region N only in A. basii, A. inopinata, and A. pseudoporphyria mitogenomes; we detected homologous region S only in A. bisporigera, A. phalloides, and A. thiersii; and we detected homologous region T only in A. brunnescens and A. phalloides species. Mitogenomes of A. basii, A. muscaria, A. bisporigera, and A. pseudoporphyria exhibited a high degree of collinearity. We detected rearrangements of homologous regions in A. phalloides, A. brunnescens, A. inopinata, and A. thiersii species, of which the two asymbiotic species (i.e., A. inopinata, and A. thiersii) had experienced large-scale rearrangements of homologous regions.
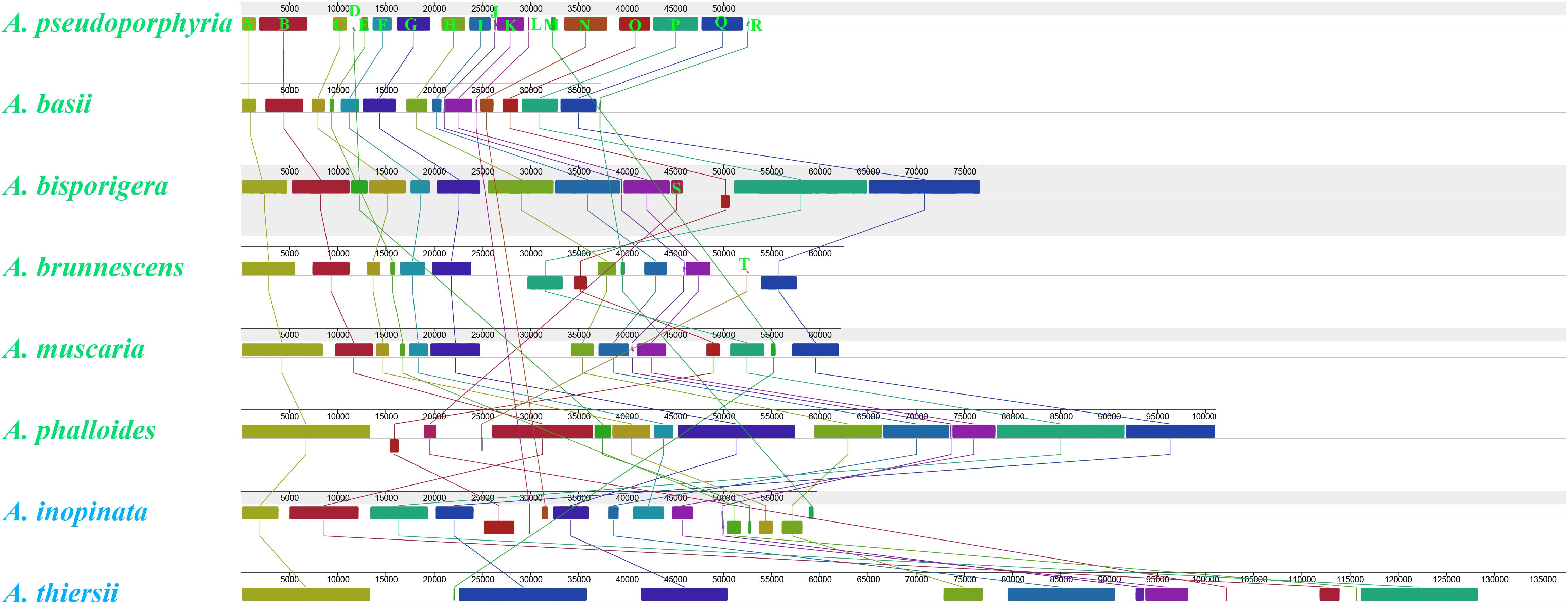
Figure 4. Colinearity analysis of eight Amanita mitogenomes using Mauve. Twenty homologous regions were detected among the eight mitogenomes. The sizes and relative positions of the homologous regions varied among mitogenomes. The Amanita species with blue fonts are asymbiotic, while the species with green fonts are ectomycorrhizal.
Among the 15 core PCGs we tested, 9 core PCGs varied in length among the eight Amanita species (Figure 5). The largest length variation was observed in the rps3 gene; no two Amanita species contained the same length of rps3 gene. As for the GC content, atp9 had the highest GC content and atp8 had the lowest content among the 15 core PCGs detected. The GC content of core PCGs varied among the different species, which indicated that the core PCGs underwent frequent base variations. According to the second parity rule, each base in the complementary DNA chain exists at a roughly equal frequency if there are no variations or selection biases (Chen H. et al., 2014). In the present study, most AT skews of core PCGs for energy metabolism were negative, except for that of the atp8 genes from A. brunnescens and A. muscaria. The AT skew of rps3 gene used for transcriptional regulation was positive. The GC skew of the core PCGs was variable: atp8 gene contained a negative GC skew; atp9 gene contained a positive GC skew; and atp6 gene was positive in A. basii, A. muscaria, and A. thiersii, but negative in A. bisporigera, A. pseudoporphyria, and A. inopinata.
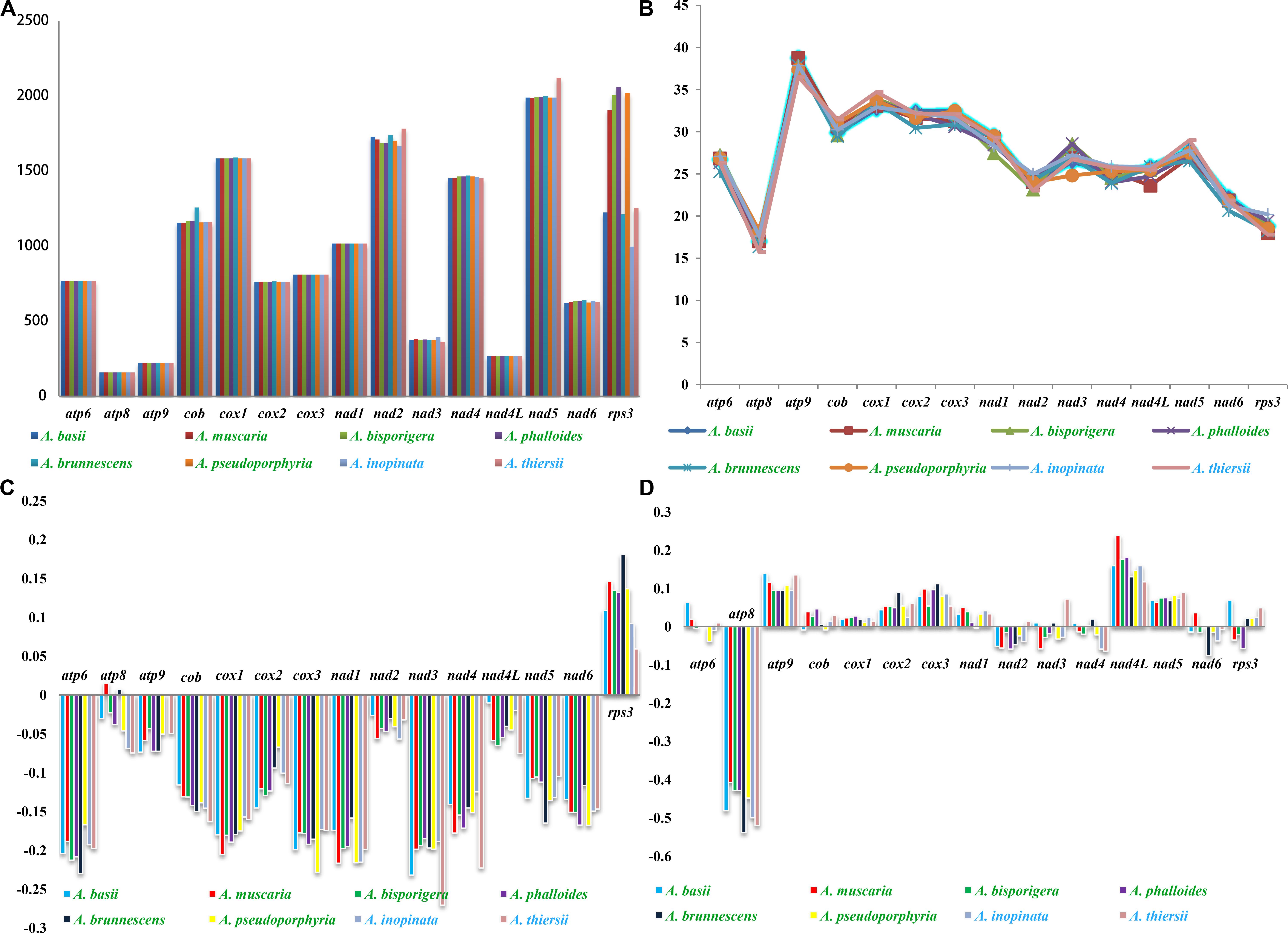
Figure 5. Variation in the length and base composition of each of the 15 PCGs among the eight Amanita mitogenomes. The Amanita species with blue fonts are asymbiotic, while the species with green fonts are ectomycorrhizal. (A) Length variation of PCGs; (B) GC content of the PCGs; (C) AT skew; (D) GC skew.
Among the 15 core PCGs detected, the average K2P genetic distance of atp9 and nad4L was the smallest, indicating that the two genes were highly conserved in Amanita species (Figure 6). The K2P genetic distances of nad2, nad3, and rps3 were the highest, indicating that these genes had larger variation between different species. In addition, the K2P genetic distance of nad3 gene varied significantly among different pairs of species, indicating the rich diversity of the nad3 gene.
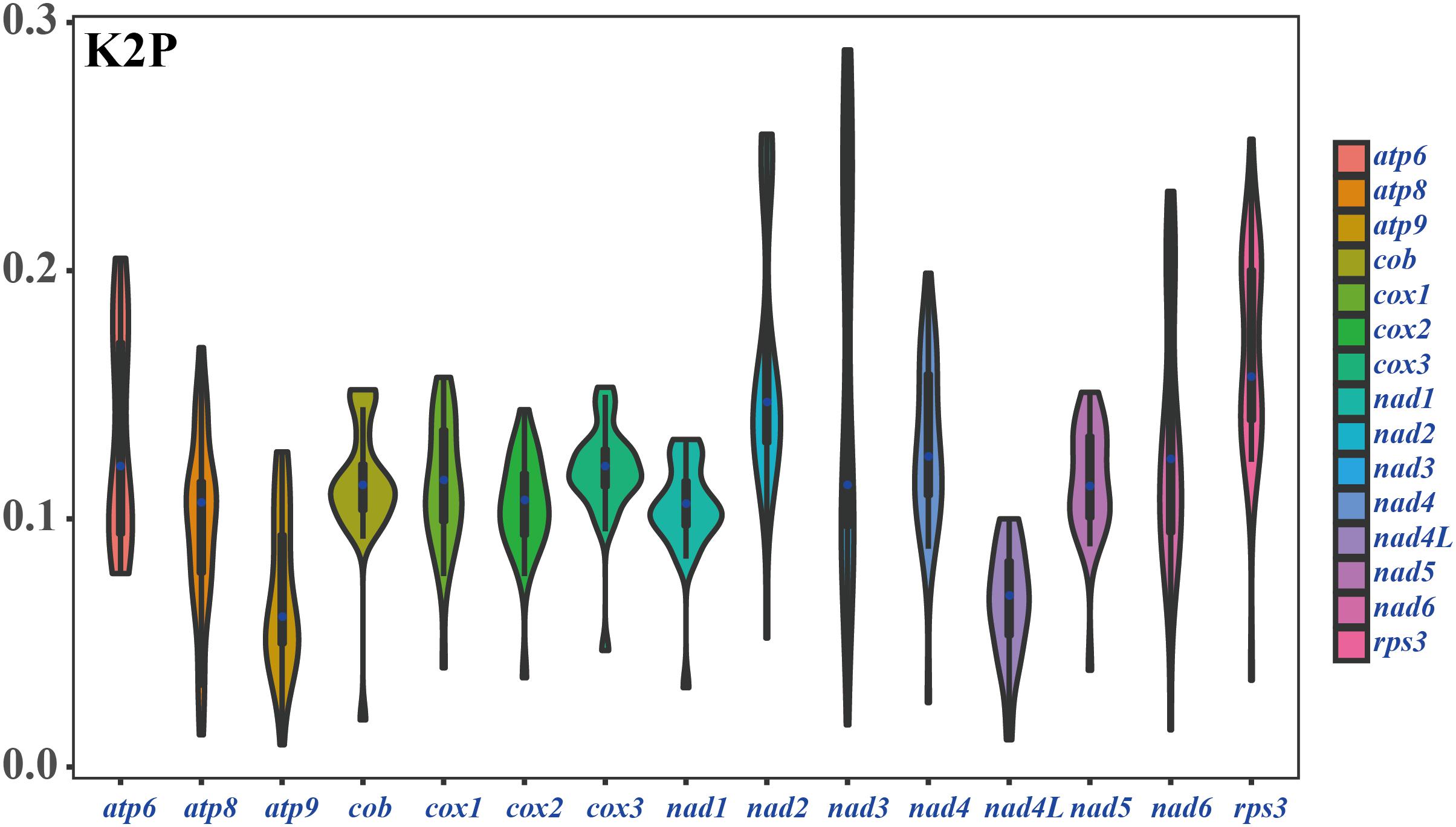
Figure 6. K2P genetic distance of 15 PCGs conserved in the eight Amanita mitogenomes.
We detected a total of 63 introns in the eight Amanita species, which belonged to two groups: group I and group II. These introns were distributed in cox1, cox2, nad1, nad2, nadd5, cob, rns, and rnl genes of mitogenomes. The number of introns in each species ranged from 0 to 22. A. thiersii contained the largest number of introns, whereas A. basii did not contain any introns. The Pearson correlation coefficient between intron sequences and mitogenome sizes was 0.97, with a p-value of 1 × 10–5, indicating that the variations of introns promoted dynamic changes in the size of Amanita mitogenomes. The cox1 gene contained the most introns in Amanita, and 39.68% of introns were harbored in the cox1 gene. Only one intron from the cox1 gene belonged to the group II.
The introns in the cox1 gene could be classified into different Pcls according to their insertion positions in the protein-coding region (Ferandon et al., 2010). We considered introns belonging to the same Pcls to be orthologous introns, with high sequence similarities. We considered introns from different Pcls to be non-homologous and they contained low sequence similarities. In the present study, we divided 25 introns in the cox1 gene of Amanita into 16 Pcls (Figure 7). Among these Pcls, P615 and P1305 were the most common in Amanita, which were distributed in 3 of 8 Agaricales species. Pcls P386, P807, P867, and P1107 were distributed in 2 of the 8 Amanita species. The other Pcls were rare introns in Amanita species, which were distributed in only 1 of the 8 Amanita species. We detected Pcls P709 and P725 in distant species Lyophyllum shimeji (Li et al., 2019a), detected Pcl P206 in Laccaria bicolor (Li et al., 2020), and detected Pcl P234 in distant species Agaricus bisporus (Ferandon et al., 2010). The number and classes of Pcls from different Amanita species varied, which indicated that frequent intron loss/gain events occurred in the evolution of Amanita species. The two asymbiotic Amanita species contained Pcl P380, which the ectomycorrhizal Amanita species did not possess, suggesting that ectomycorrhizal Amanita species may have lost this intron classes during evolution.
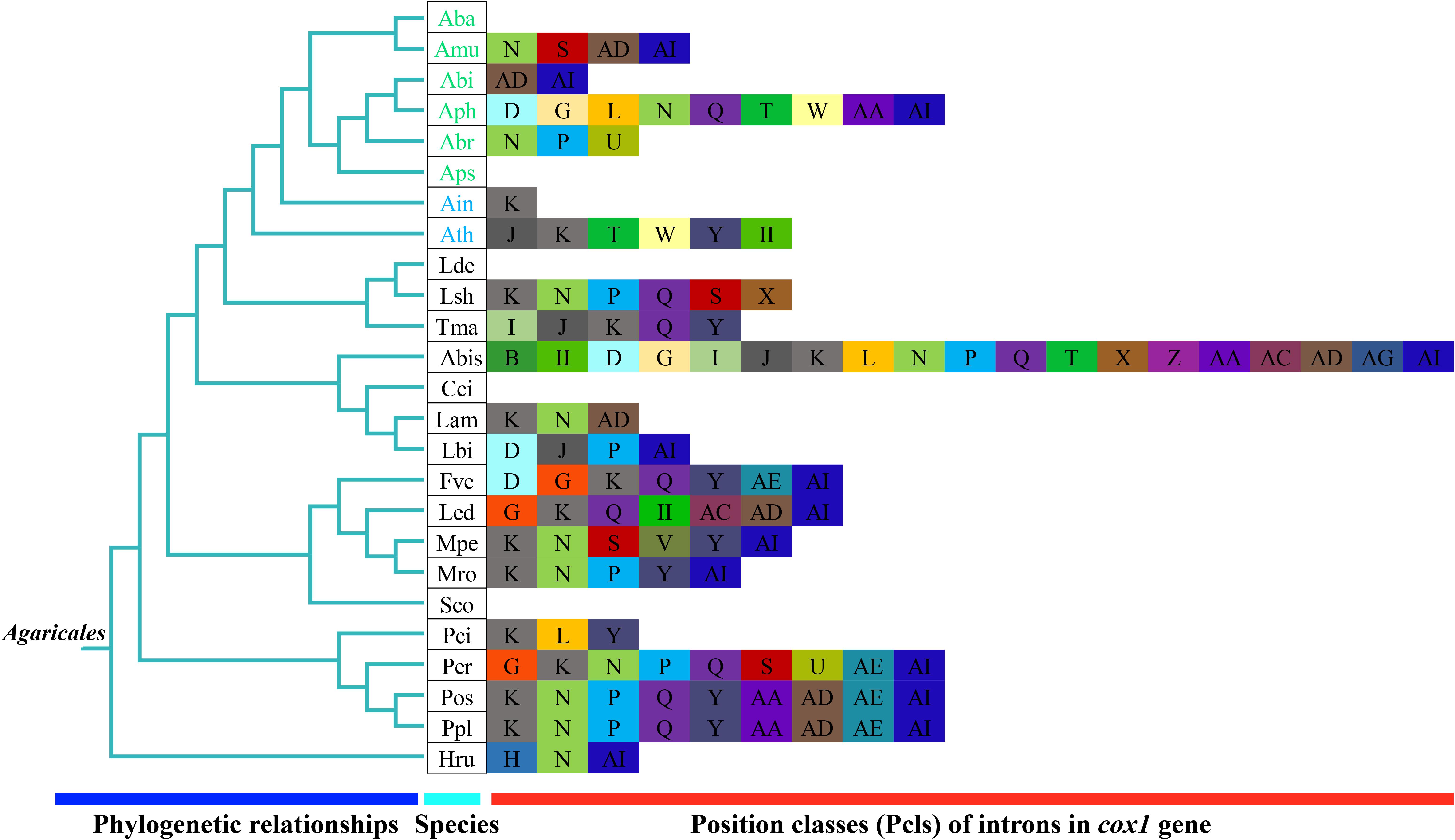
Figure 7. Position class (Pcl) information of cox1 gene of the 25 Agaricales species. The same Pcl (orthologous intron) is represented by the same letter. The phylogenetic positions of 25 Agaricales species were established using the BI method and ML method based on 15 concatenated mitochondrial core proteins and two rRNA genes. The II in the figure shows the intron belongs to the group II intron. Pcls were named according to the method described by Zhang and Zhang (2019). Species IDs are given in Supplementary Table S6.
Until now, the mitogenome of A. thiersii was the largest among all the published Agaricales mitogenomes, and the mitogenome of A. basii was the smallest among the sequenced mitogenomes in Agaricales. The largest mitogenome in Amanita was 3.68 times that of the smallest mitogenome, which indicated the great variation of the Amanita mitogenome. These results extended our understanding of the mitogenome in Amanita and in Agaricales (Ferandon et al., 2010; Stajich et al., 2010). We identified dynamic changes in the introns as the primary factors contributing to size variations in Amanita. These results were consistent with previous studies (Kanzi et al., 2016; Li et al., 2019a). In addition to the quantitative variations of introns, we found that intron classes also underwent great variations in Amanita, which promoted additional variations in the organization and sizes among Amanita mitogenomes. Comparative mitogenome analysis revealed that the frequent gain/loss, as well as the possible horizontal transfer of introns, occurred in the mitogenomes of Amanita we detected, which enabled Amanita to become one of the most varied fungal group in Agaricales.
In this study, we compared the features of mitogenomes from two saprophytic and six ectomycorrhizal Amanita species. We found that there were no obvious differences in genome size, GC content, tRNA, and rRNA between saprophytic and ectomycorrhizal Amanita species. We synthetically analyzed the repeats obtained by three methods and found that the ectomycorrhizal Amanita species had a high proportion of repeat sequences relative to saprophytic species. Previous studies also have found that ectomycorrhizal Amanita species had increased TE activities (Hess et al., 2014). The results showed that ectomycorrhizal Amanita species tended to have abundant repetitive elements in both nuclear genome and mitogenome, which may be related to ectomycorrhizal lifestyle adaptation. In addition, we found that ectomycorrhizal Amanita species had a relatively low proportion of intergenic regions, which made their mitogenomes more compact than saprophytic mitogenomes.
Most of the mitochondrial genes in eukaryotes were transferred to the nuclear genome during evolution (Adams and Palmer, 2003). Species from the Basidiomycota phylum, however, have retained the 14 core PCGs for energy metabolism and 1 rps3 gene for transcriptional regulation (Bullerwell et al., 2000; Costa et al., 2012). In the present study, we found that all eight Amanita species contained the 15 core PCGs, and their length and base composition varied between different Amanita species. In addition, the K2P genetic distances of different PCGs also varied, which indicated the different evolution rates of core PCGs in Amanita. In addition to these core PCGs, some PCGs with unknown function were found in the eight Amanita mitogenomes. In general, the functions of Amanita mitogenomes were diverse and fascinating. Further studies are needed to reveal and identify the role of Amanita mitogenomes in growth, development, environmental adaptation, and stress resistance.
Large-scale gene rearrangements were detected in the eight Amanita mitogenomes we tested, involving gene displacement and gene inversion. This was the first time that large-scale gene rearrangements were detected in Amanita species. Previous studies have shown that the accumulation of repeat sequences in fungal mitogenome promoted the mitochondrial gene rearrangement in fungi (Aguileta et al., 2014). In the present study, however, we found that A. thiersii and A. inopinata, which experienced large-scale mitochondrial rearrangements, contained fewer repetitive sequences than the other mitogenomes. Therefore, the mechanism of mitochondrial gene rearrangement in fungi needs to be further examined (Lavrov et al., 2002). A. bisporigera and its sister species A. phalloides showed significant differences in gene arrangement, indicating the variability and complexity of Amanita gene arrangements. The mitochondrial gene order of A. thiersii and A. inopinata, two asymbiotic Amanita species, varied greatly from other Amanita species, which indicated that they differentiated from other Amanita species in the early stages. The arrangement of mitochondrial genes in Amanita provided useful information for revealing the evolution of Amanita species.
Mitochondrial genes have become a powerful tool for the study of phylogeny, taxonomy, and population genetics of animals because of their distinct advantages (Johri et al., 2019; Li et al., 2015). Thus far, less than 100 Basidiomycete mitogenomes have been published, which is far less than the number of nuclear genomes available. In the present study, we first reported the eight Amanita mitogenomes based on the next-generation sequencing reads. On the basis of the combined mitochondrial gene set, we reconstructed the phylogenetic relationships of 66 Basidiomycetes and obtained tree topologies with a high support rate, which indicated that mitochondrial genes were a powerful tool for analyzing the phylogenetic relationships of fungi. From the evolutionary tree, we found that the ectomycorrhizal Amanita species were of single origin, and one monophyletic clade encompassed all ectomycorrhizal Amanita species. However, saprophytic Amanita species formed two distinct clades, which was not consistent with previous studies (Wolfe et al., 2012b). This phylogenetic difference may be caused by different genetic characteristics of mitochondrial and nuclear genomes (Saarma et al., 2009). More Basidiomycete mitogenomes need to be acquired to reveal the origin, evolution, and phylogenetic relationships of Basidiomycete species or other fungi.
The datasets generated for this study can be found in the complete mitogenomes of A. basii, A. muscaria, A. bisporigera, A. phalloides, A. brunnescens, A. pseudoporphyria, A. inopinata, and A. thiersii were deposited in the GenBank database under the accession numbers MK993555, MK993559, MK993556, MK993560, MK993557, MK993554, MK993558, and MK993561, respectively.
QL, YR, and WH conceived and designed the experiments. QL, XH, CX, and XJ performed the experiments. QL, XJ, and LP analyzed the data. QL, LP, and WH contributed reagents, materials, and analysis tools. QL wrote the manuscript. All authors contributed to the article and approved the submitted version. QL and XH revised the manuscript.
This study was funded by the Sichuan Distinguished Youth Fund (2019JDJQ0034) and Talent Fund of Sichuan Academy of Agricultural Sciences (2019QYXK003).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01382/full#supplementary-material
Adams, K. L., and Palmer, J. D. (2003). Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol. Phylogenet. Evol. 29, 380–395. doi: 10.1016/s1055-7903(03)00194-5
Aguileta, G., de Vienne, D. M., Ross, O. N., Hood, M. E., Giraud, T., Petit, E., et al. (2014). High variability of mitochondrial gene order among fungi. Genome Biol. Evol. 6, 451–465. doi: 10.1093/gbe/evu028
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bernt, M., Donath, A., Juhling, F., Externbrink, F., Florentz, C., Fritzsch, G., et al. (2013). MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319. doi: 10.1016/j.ympev.2012.08.023
Bleasby, A. J., and Wootton, J. C. (1990). Construction of validated, non-redundant composite protein sequence databases. Protein Eng. 3, 153–159. doi: 10.1093/protein/3.3.153
Boore, J. L. (1999). Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780. doi: 10.1093/nar/27.8.1767
Bullerwell, C. E., Burger, G., and Lang, B. F. (2000). A novel motif for identifying rps3 homologs in fungal mitochondrial genomes. Trends Biochem. Sci. 25, 363–365. doi: 10.1016/s0968-0004(00)01612-1
Cai, Q., Cui, Y. Y., and Yang, Z. L. (2016). Lethal Amanita species in China. Mycologia 108, 993–1009. doi: 10.3852/16-008
Caspermeyer, J. (2016). MEGA evolutionary software re-engineered to handle today’s big data demands. Mol. Biol. Evol. 33:1887.
Chen, C., Khaleel, S. S., Huang, H., and Wu, C. H. (2014). Software for pre-processing Illumina next-generation sequencing short read sequences. Source Code Biol. Med. 9:8.
Chen, H., Sun, S., Norenburg, J. L., and Sundberg, P. (2014). Mutation and selection cause codon usage and bias in mitochondrial genomes of ribbon worms (Nemertea). PLoS One 9:e85631. doi: 10.1371/journal.pone.0085631
Costa, G. G., Cabrera, O. G., Tiburcio, R. A., Medrano, F. J., Carazzolle, M. F., Thomazella, D. P., et al. (2012). The mitochondrial genome of Moniliophthora roreri, the frosty pod rot pathogen of cacao. Fungal Biol. 116, 551–562. doi: 10.1016/j.funbio.2012.01.008
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
De Mares, M. C., Hess, J., Floudas, D., Lipzen, A., Choi, C., Kennedy, M., et al. (2015). Horizontal transfer of carbohydrate metabolism genes into ectomycorrhizal Amanita. New Phytol. 205, 1552–1564. doi: 10.1111/nph.13140
Deng, Y., Hsiang, T., Li, S., Lin, L., Wang, Q., Chen, Q., et al. (2018). Comparison of the mitochondrial genome sequences of six annulohypoxylon stygium isolates suggests short fragment insertions as a potential factor leading to larger genomic size. Front. Microbiol. 9:2079. doi: 10.3389/fmicb.2018.02079
Ferandon, C., Moukha, S., Callac, P., Benedetto, J. P., Castroviejo, M., and Barroso, G. (2010). The Agaricus bisporus cox1 gene: the longest mitochondrial gene and the largest reservoir of mitochondrial group I introns. PLoS One 5:e14048. doi: 10.1371/journal.pone.0014048
Garcia, J., Costa, V. M., Carvalho, A., Baptista, P., de Pinho, P. G., Bastos, M. D., et al. (2015). Amanita phalloides poisoning: mechanisms of toxicity and treatment. Food Chem. Toxicol. 86, 41–55. doi: 10.1016/j.fct.2015.09.008
Hahn, C., Bachmann, L., and Chevreux, B. (2013). Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads–a baiting and iterative mapping approach. Nucleic Acids Res. 41:e129. doi: 10.1093/nar/gkt371
Hess, J., Skrede, I., De Mares, M. C., Hainaut, M., Henrissat, B., and Pringle, A. (2018). Rapid divergence of genome architectures following the origin of an ectomycorrhizal symbiosis in the genus Amanita. Mol. Biol. Evol. 35, 2786–2804.
Hess, J., Skrede, I., Wolfe, B. E., LaButti, K., Ohm, R. A., Grigoriev, I. V., et al. (2014). Transposable element dynamics among asymbiotic and Ectomycorrhizal Amanita fungi. Genome Biol. Evol. 6, 1564–1578. doi: 10.1093/gbe/evu121
Johnson, M., Zaretskaya, I., Raytselis, Y., Merezhuk, Y., McGinnis, S., and Madden, T. L. (2008). NCBI BLAST: a better web interface. Nucleic Acids Res. 36, W5–W9.
Johri, P., Marinov, G. K., Doak, T. G., and Lynch, M. (2019). Population genetics of Paramecium mitochondrial genomes: recombination, mutation spectrum, and efficacy of selection. Genome Biol. Evol. 11, 1398–1416. doi: 10.1093/gbe/evz081
Kanzi, A. M., Wingfield, B. D., Steenkamp, E. T., Naidoo, S., and van der Merwe, N. A. (2016). Intron derived size polymorphism in the mitochondrial genomes of closely related Chrysoporthe species. PLoS One 11:e0156104. doi: 10.1371/journal.pone.0156104
Katoh, K., Rozewicki, J., and Yamada, K. D. (2017). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kohler, A., Kuo, A., Nagy, L. G., Morin, E., Barry, K. W., Buscot, F., et al. (2015). Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists (vol 47, pg 410, 2015). Nat. Genet. 47, 410–415. doi: 10.1038/ng.3223
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., and Calcott, B. (2017). PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773.
Lang, B. F., Gray, M. W., and Burger, G. (1999). Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 33, 351–397. doi: 10.1146/annurev.genet.33.1.351
Lavrov, D. V., Boore, J. L., and Brown, W. M. (2002). Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: duplication and nonrandom loss. Mol. Biol. Evol. 19, 163–169. doi: 10.1093/oxfordjournals.molbev.a004068
Li, H., Shao, R., Song, N., Song, F., Jiang, P., Li, Z., et al. (2015). Higher-level phylogeny of paraneopteran insects inferred from mitochondrial genome sequences. Sci. Rep. 5:8527.
Li, H., Wu, S., Ma, X., Chen, W., Zhang, J., Duan, S., et al. (2018). The genome sequences of 90 mushrooms. Sci. Rep. 8:9982.
Li, Q., Chen, C., Xiong, C., Jin, X., Chen, Z., and Huang, W. (2018a). Comparative mitogenomics reveals large-scale gene rearrangements in the mitochondrial genome of two Pleurotus species. Appl. Microbiol. Biotechnol. 102, 6143–6153. doi: 10.1007/s00253-018-9082-6
Li, Q., Liao, M., Yang, M., Xiong, C., Jin, X., Chen, Z., et al. (2018b). Characterization of the mitochondrial genomes of three species in the ectomycorrhizal genus Cantharellus and phylogeny of Agaricomycetes. Int. J. Biol. Macromol. 118(Pt A), 756–769. doi: 10.1016/j.ijbiomac.2018.06.129
Li, Q., Wang, Q., Chen, C., Jin, X., Chen, Z., Xiong, C., et al. (2018c). Characterization and comparative mitogenomic analysis of six newly sequenced mitochondrial genomes from ectomycorrhizal fungi (Russula) and phylogenetic analysis of the Agaricomycetes. Int. J. Biol. Macromol. 119, 792–802. doi: 10.1016/j.ijbiomac.2018.07.197
Li, Q., Wang, Q., Jin, X., Chen, Z., Xiong, C., Li, P., et al. (2019a). Characterization and comparative analysis of six complete mitochondrial genomes from ectomycorrhizal fungi of the Lactarius genus and phylogenetic analysis of the Agaricomycetes. Int. J. Biol. Macromol. 121, 249–260. doi: 10.1016/j.ijbiomac.2018.10.029
Li, Q., Wang, Q., Jin, X., Chen, Z., Xiong, C., Li, P., et al. (2019b). Characterization and comparison of the mitochondrial genomes from two Lyophyllum fungal species and insights into phylogeny of Agaricomycetes. Int. J. Biol. Macromol. 121, 364–372. doi: 10.1016/j.ijbiomac.2018.10.037
Li, Q., Yang, L., Xiang, D., Wan, Y., Wu, Q., Huang, W., et al. (2020). The complete mitochondrial genomes of two model ectomycorrhizal fungi (Laccaria): features, intron dynamics and phylogenetic implications. Int. J. Biol. Macromol. 145, 974–984. doi: 10.1016/j.ijbiomac.2019.09.188
Li, Q., Yang, M., Chen, C., Xiong, C., Jin, X., Pu, Z., et al. (2018d). Characterization and phylogenetic analysis of the complete mitochondrial genome of the medicinal fungus Laetiporus sulphureus. Sci. Rep. 8:9104.
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). OrganellarGenomeDRAW–a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581.
Lowe, T. M., and Chan, P. P. (2016). tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57.
Poliseno, A., Feregrino, C., Sartoretto, S., Aurelle, D., Worheide, G., McFadden, C. S., et al. (2017). Comparative mitogenomics, phylogeny and evolutionary history of Leptogorgia (Gorgoniidae). Mol. Phylogenet. Evol. 115, 181–189. doi: 10.1016/j.ympev.2017.08.001
Pulman, J. A., Childs, K. L., Sgambelluri, R. M., and Walton, J. D. (2016). Expansion and diversification of the MSDIN family of cyclic peptide genes in the poisonous agarics Amanita phalloides and A-bisporigera. BMC Genomics 17:1038. doi: 10.1186/s12864-016-3378-7
Qian, L., Wang, H., Yan, J., Pan, T., Jiang, S., Rao, D., et al. (2018). Multiple independent structural dynamic events in the evolution of snake mitochondrial genomes. BMC Genomics 19:354. doi: 10.1186/s12864-018-4717-7
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Saarma, U., Jogisalu, I., Moks, E., Varcasia, A., Lavikainen, A., Oksanen, A., et al. (2009). A novel phylogeny for the genus Echinococcus, based on nuclear data, challenges relationships based on mitochondrial evidence. Parasitology 136, 317–328. doi: 10.1017/s0031182008005453
Sanchez-Ramirez, S., Tulloss, R. E., Amalfi, M., and Moncalvo, J. M. (2015). Palaeotropical origins, boreotropical distribution and increased rates of diversification in a clade of edible ectomycorrhizal mushrooms (Amanita section Caesareae). J. Biogeogr. 42, 351–363. doi: 10.1111/jbi.12402
Sandor, S., Zhang, Y., and Xu, J. (2018). Fungal mitochondrial genomes and genetic polymorphisms. Appl. Microbiol. Biotechnol. 102, 9433–9448. doi: 10.1007/s00253-018-9350-5
Sankoff, D., Leduc, G., Antoine, N., Paquin, B., Lang, B. F., and Cedergren, R. (1992). Gene order comparisons for phylogenetic inference: evolution of the mitochondrial genome. Proc. Natl. Acad. Sci. U.S.A. 89, 6575–6579. doi: 10.1073/pnas.89.14.6575
Schubert, M., Lindgreen, S., and Orlando, L. (2016). AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 9:88. doi: 10.1186/s13104-016-1900-2
Stajich, J. E., Wilke, S. K., Ahren, D., Au, C. H., Birren, B. W., Borodovsky, M., et al. (2010). Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus). Proc. Natl. Acad. Sci. U.S.A. 107, 11889–11894.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Sun, J., Li, H. J., Zhang, H. S., Zhang, Y. Z., Xie, J. W., Ma, P. B., et al. (2018). Investigating and analyzing three cohorts of mushroom poisoning caused by Amanita exitialis in Yunnan, China. Hum. Exp. Toxicol. 37, 665–678. doi: 10.1177/0960327117721960
Thongbai, B., Miller, S. L., Stadler, M., Wittstein, K., Hyde, K. D., Lumyong, S., et al. (2017). Study of three interesting Amanita species from Thailand: morphology, multiple-gene phylogeny and toxin analysis. PLoS One 12:e0182131. doi: 10.1371/journal.pone.0182131
Thongbai, B., Tulloss, R. E., Miller, S. L., Hyde, K. D., Chen, J., Zhao, R. L., et al. (2016). A new species and four new records of Amanita (Amanitaceae, Basidiomycota) from Northern Thailand. Phytotaxa 286, 211–231.
Vaidya, G., Lohman, D. L., and Meier, R. (2011). SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27, 171–180. doi: 10.1111/j.1096-0031.2010.00329.x
Valach, M., Burger, G., Gray, M. W., and Lang, B. F. (2014). Widespread occurrence of organelle genome-encoded 5S rRNAs including permuted molecules. Nucleic Acids Res. 42, 13764–13777. doi: 10.1093/nar/gku1266
Wang, J., Zhang, L., Zhang, Q. L., Zhou, M. Q., Wang, X. T., Yang, X. Z., et al. (2017). Comparative mitogenomic analysis of mirid bugs (Hemiptera: Miridae) and evaluation of potential DNA barcoding markers. PeerJ 5:e3661. doi: 10.7717/peerj.3661
Wolfe, B. E., Kuo, M., and Pringle, A. (2012a). Amanita thiersii is a saprotrophic fungus expanding its range in the United States. Mycologia 104, 22–33. doi: 10.3852/11-056
Wolfe, B. E., Tulloss, R. E., and Pringle, A. (2012b). The irreversible loss of a decomposition pathway marks the single origin of an ectomycorrhizal symbiosis. PLoS One 7:e39597. doi: 10.1371/journal.pone.0039597
Ye, Y. Z., and Liu, Z. N. (2018). Management of Amanita phalloides poisoning: a literature review and update. J. Crit. Care 46, 17–22. doi: 10.1016/j.jcrc.2018.03.028
Keywords: Amanita, mitochondrial genome, ectomycorrhiza, ecological adaptation, evolution
Citation: Li Q, He X, Ren Y, Xiong C, Jin X, Peng L and Huang W (2020) Comparative Mitogenome Analysis Reveals Mitochondrial Genome Differentiation in Ectomycorrhizal and Asymbiotic Amanita Species. Front. Microbiol. 11:1382. doi: 10.3389/fmicb.2020.01382
Received: 19 December 2019; Accepted: 28 May 2020;
Published: 19 June 2020.
Edited by:
Nuria Ferrol, Zaidín Experimental Station (EEZ), SpainCopyright © 2020 Li, He, Ren, Xiong, Jin, Peng and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lianxin Peng, cGVuZ2xpYW54aW5AY2R1LmVkdS5jbg==; Wenli Huang, d2VubGloMTFAMTI2LmNvbQ==
†Present address: Lianxin Peng, Key Laboratory of Coarse Cereal Processing, Ministry of Agriculture and Rural Affairs, Chengdu, China
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.