 Nan Song
Nan Song Yuehua Geng
Yuehua Geng Xinghao Li
Xinghao Li- College of Plant Protection, Henan Agricultural University, Zhengzhou, China
A number of species in Bipolaris are important plant pathogens. Due to a limited number of synapomorphic characters, it is difficult to perform species identification and to estimate phylogeny of Bipolaris based solely on morphology. In this study, we sequenced the complete mitochondrial genome of Bipolaris sorokiniana, and presented the detailed annotation of the genome. The B. sorokiniana mitochondrial genome is 137,775 bp long, and contains two ribosomal RNA genes, 12 core protein-coding genes, 38 tRNA genes. In addition, two ribosomal protein genes (rps3 gene and rps5 gene) and the fungal mitochondrial RNase P gene (rnpB) are identified. The large genome size is mostly determined by the presence of numerous intronic and intergenic regions. A total of 28 introns are inserted in eight core protein-coding genes. Together with the published mitochondrial genome sequences, we conducted a preliminary phylogenetic inference of Dothideomycetes under various datasets and substitution models. The monophyly of Capnodiales, Botryosphaeriales and Pleosporales are consistently supported in all analyses. The Venturiaceae forms an independent lineage, with a distant phylogenetic relationship to Pleosporales. At the family level, the Mycosphaerellaceae, Botryosphaeriaceae. Phaeosphaeriaceae, and Pleosporaceae are recognized in the majority of trees.
Introduction
The genus Bipolaris belongs to the family Pleosporaceae (Ascomycota, Dothideomycetes, Pleosporales). Shoemaker (1959) originally established the generic Bipolaris. The species of Bipolaris were once classified in Helminthosporium (Link, 1824). Subramanian (1971) provided a key to separate the species of Bipolaris from members of the Helminthosporium. Further study divided the Helminthosporium into several genera including Bipolaris, Curvularia, Drechslera, and Exserohilum (Sivanesan, 1987). Modern descriptions and illustrations for the species in the genus Bipolaris were given in the studies of Manamgoda et al. (2012, 2014). Many morphological similarities are shared by Bipolaris and Curvularia, both of which have sexual morphs in the genus Cochliobolus (Alcorn, 1983; Manamgoda et al., 2014). The genera Cochliobolus, Bipolaris and Curvularia form a complex, and the taxonomy of this complex are uncertain with the morphological characters (Manamgoda et al., 2012). A combined analysis of ITS, GPDH, EF1-α and LSU gene sequences resulted in two main groups corresponding to the complex (Manamgoda et al., 2012). One clade included the majority of Bipolaris and Cochliobolus, and another contained the Curvularia and the partial members of Bipolaris and Cochliobolus (Manamgoda et al., 2012). Manamgoda et al. (2014) recovered Bipolaris as a sister group to Curvularia based on a combined analysis of ITS, GPDH and EF1-α gene sequences.
Bipolaris sorokiniana (Sacc.) Shoemaker [teleomorph: Cochliobolus sativus (Ito and Kuribayashi) Drechs. ex Dastur] is a seed and soil borne pathogen, which causes spot blotch, root rot, leaf spot, seedling blight, head blight, and black point disease in cereal crops (Wiese, 1998; Kumar et al., 2002). Among these diseases, the leaf spot blotch is recognized as the major biotic stress hampering commercial production of wheat, because it results in significant yield losses (Chowdhury et al., 2013; Mina et al., 2016). The type strain was isolated from Russia with number of MBT197973 in 1890, which was originally named as Helminthosporium sorokinianum. Several synonyms of B. sorokiniana were used as following: H. sorokinianum, H. sativum, Drechslera sorokiniana (Maraite et al., 1998). The detailed description of B. sorokiniana can be seen from Commonwealth Mycological Institute’s Sivanesan and Holliday (1981). In addition to B. sorokiniana, the genus Bipolaris contains many other important plant pathogens with worldwide distribution. The distinction of species in Bipolaris and the classification of Bipolaris with other fungus species appear difficult when only morphological characters are considered. Because some species have overlapping morphological features. Molecular phylogenetic studies based on gene sequences have shown promise in resolving the fungi classification problems (Swann and Taylor, 1995; Berbee et al., 1999; Fitzpatrick et al., 2006; James et al., 2006a, b; Robbertse et al., 2006; Mangold et al., 2008; Wang et al., 2009; Manamgoda et al., 2012, 2014). Ohm et al. (2012) determined 14 dothideomycete whole-genomes, including that from the sexual morph of B. sorokiniana (i.e., Cochliobolus sativus). Condon et al. (2013) reported three additional whole-genome sequences of Bipolaris, namely the B. victoriae (as C. victoriae), B. zeicola (as C. carbonum), and B. maydis (as C. heterostrophus).
Mitochondrial (mt) DNA has been widely used in evolutionary biology and systematics of Fungi (Paquin et al., 1997; Hausner, 2003; Kouvelis et al., 2004; Formighieri et al., 2008; Pantou et al., 2008; Sethuraman et al., 2009; Duo et al., 2012; Aguileta et al., 2014; Lin et al., 2015; Utomo et al., 2019). As a class of molecular markers, mtDNA sequences have some preferable characteristics in resolving systematic issues, such as faster rate of evolution, the virtual absence of recombination, and conserved gene content (Simon et al., 1994, 2006; Caterino et al., 2000; Lin and Danforth, 2004). Recently, development of high-throughput sequencing technologies and related bioinformatics tools have allowed considerably greater numbers of nucleotides to be characterized and the genome-scale data to be more easily assembled. As a case in point, next-generation sequencing (NGS) technologies have been successfully used to reconstruct the fungal mitochondrial genomes (Losada et al., 2014; Mardanov et al., 2014; Salavirta et al., 2014; Torriani et al., 2014; Lin et al., 2015; Kanzi et al., 2016; Nowrousian, 2016; Kang et al., 2017; Deng et al., 2018). Compared to the whole-genome data, mitochondrial genomes are more readily sequenced with broader taxon sampling and a reasonable cost. The fungal mitochondrial genome size varied greatly between species. For example, the mitochondrial genome of Hanseniaspora uvarum has a length of 18,844 bp (Pramateftaki et al., 2006), while the Sclerotinia borealis has a length of 203,051 bp (Mardanov et al., 2014). Large variation of mitochondrial genome size in fungi is mainly attributed to different lengths of intronic sequences and intergenic regions (Belcour et al., 1997; Hausner, 2003; Formighieri et al., 2008). Despite with the continuously increasing number of fungal mitochondrial genome studies (Paquin et al., 1997; Hausner, 2003; Formighieri et al., 2008; Pantou et al., 2008; Duo et al., 2012; Torriani et al., 2014; Kang et al., 2017; Zaccaron and Bluhm, 2017; Deng et al., 2018; Utomo et al., 2019), the complete mtDNA sequences available for some important lineages are still limited. There are only 16 complete mitochondrial genomes published from the class Dothideomycetes in GenBank as of December 2019. The Bipolaris cookei was the first species having complete mtDNA sequence (Zaccaron and Bluhm, 2017) in the genus Bipolaris. It provided a reference for further investigation of the Bipolaris mitochondrial genomes.
In the present study, we use an NGS based approach to determine a complete mitochondrial genome of B. sorokiniana. The description of the mitochondrial genome organization is presented. One of our primary objectives is to explore the utility of mtDNA sequence data in inferring phylogeny of Dothideomycetes.
Materials and Methods
Fungal Isolate
The strains of B. sorokiniana were obtained from wheat root and leaves in Luyi County (38.48°N, 115.08°E), Henan province, China, with the single spore isolation (Matusinsky et al., 2010). The living strains were harvested in 10% aqueous glycerol in a 2 ml-Eppendorf tube, and then were stored at −80°C. The fungus was cultured on PDA culture medium at 25°C, with ampicillin resistant to bacterial growth. Species identification was based on morphological features and confirmed using ITS sequence. The dried cultures and living cultures of specimen have been deposited in the Herbarium of Henan Agricultural University: Fungi (HHAUF).
DNA Extraction and Genome Sequencing
Total genomic DNA was extracted by using the Cetyl Trimethyl Ammonium Bromide (CTAB) method (Rogers and Bendich, 1985), with minor modification. DNA concentration (avg. 14.20 ng/μL) was determined using a Qubit Flurometer (Invitrogen, United States) and a NanoDrop Spectrophotometer (Thermo Scientific, United States).
Genomic DNA were sent to Shanghai Personal Biotechnology Co., Ltd. for library preparation and high-throughput sequencing. Library was constructed by using the Illumina TruSeqTM DNA Sample Prep Kit (Illumina, San Diego, CA, United States), with the insert size of 400 bp. The genome sequencing was conducted on an Illumina NovaSeq 6000 platform, with a strategy of 150 paired-end sequencing.
Genome Assembly and Annotation
Raw data set was filtered through AdapterRemoval (Lindgreen, 2012) and SOAPec_v2.01 (Luo et al., 2012). The high-quality reads (Q20 = 97.71%, and Q30 = 93.43%) were used to assemble the mitochondrial contig, with the software MITObim v1.9 (Hahn et al., 2013). The mitochondrial genome of B. cookei (Zaccaron and Bluhm, 2017) was used as the reference genome. We also used IDBA-tran v. 1.1.1 (Peng et al., 2013) to conduct de novo assembly of B. sorokiniana, with the following parameters: the minimum size of contig of 200, an initial k-mer size of 41, an iteration size of 10, and a maximum k-mer size of 91. The assembled result produced by IDBA-tran were used to build a Blast database, using the program makeblastdb implemented in the BLAST package v 2.9.0+ (Camacho et al., 2009). The mitochondrial gene sequences of B. cookei were used as queries in Blastn searches against the database. BLAST hit contigs were retrieved through the program blastdbcmd in BLAST package v 2.9.0+ (Camacho et al., 2009).
To check the quality of assembly, we used BWA v. 0.7.5 (Li and Durbin, 2009) to align the sequenced reads to the mtDNA sequence assembled. SAMtools v. 0.1.19 (Li et al., 2009) was used to convert SAM output to a sorted BAM file. Qualimap v. 2.2.1 (Okonechnikov et al., 2016) was applied to calculate correctly mapped reads to the mitochondrial genome.
The mitochondrial genome annotation was conducted in MITOS web (Bernt et al., 2013). The following settings were implemented: Reference, “RefSeq 63 Fungi”; Genetic Code, “4 Mold.” We also used the automated organelle genome annotation tool MFannot (Valach et al., 2014) to perform annotation of the mitochondrial genome of B. sorokiniana, under the genetic code of “4 Mold, Protozoan, and Coelenterate Mitochondrial; Mycoplasma/Spiroplasma.” The gene boundaries were further refined by sequence alignment to the B. cookei mitochondrial genome (Zaccaron and Bluhm, 2017). The secondary structures of tRNA genes were predicted by MITOS (Bernt et al., 2013), and redrawn in Adobe Illustrator CS6. The genome structure image (Figure 1) was generated using OGDRAW (Greiner et al., 2019). The newly determined mitochondrial genome sequence of B. sorokiniana has been submitted to GenBank under the accession number MN978926. Besides the B. sorokiniana mitochondrial genome sequenced in this study, all other 19 complete mtDNA sequences (sixteen representing the Dothideomycetes and three representing the Sordariomycetes, see details in Table 1) downloaded from GenBank were re-annotated in MITOS web (Bernt et al., 2013) following the settings described above for uniform annotations.
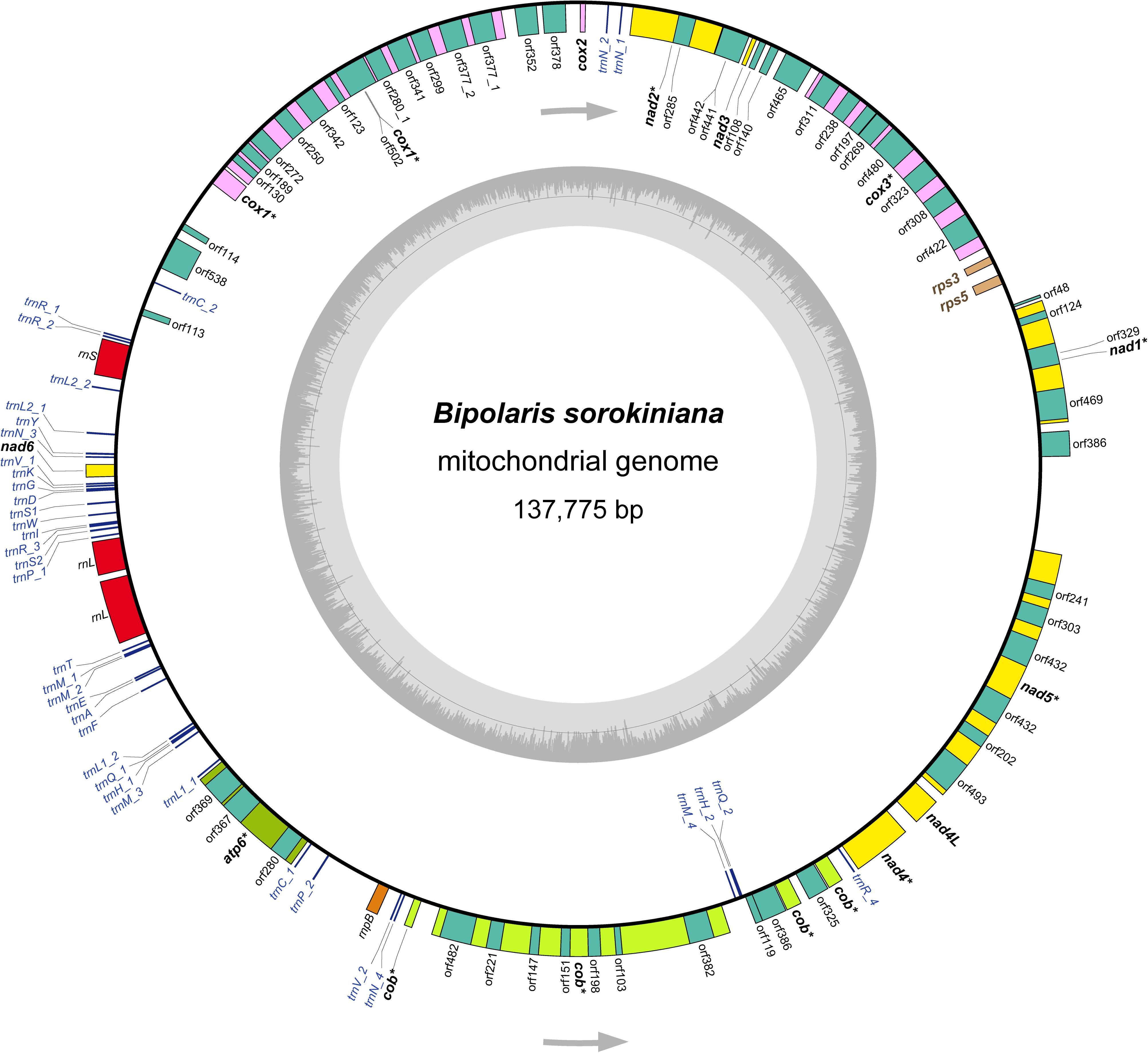
Figure 1. The structure of the mitochondrial genome of Bipolaris sorokiniana. Arrows indicate the direction of gene transcription. The inner circles show the GC content. All genes identified are indicated in italics. The core 12 protein-coding genes are highlighted in bold. Of which, eight protein-coding genes including introns are denoted by asterisk. The detailed annotation of the mitochondrial genome of B. sorokiniana are shown in Supplementary Table S2.
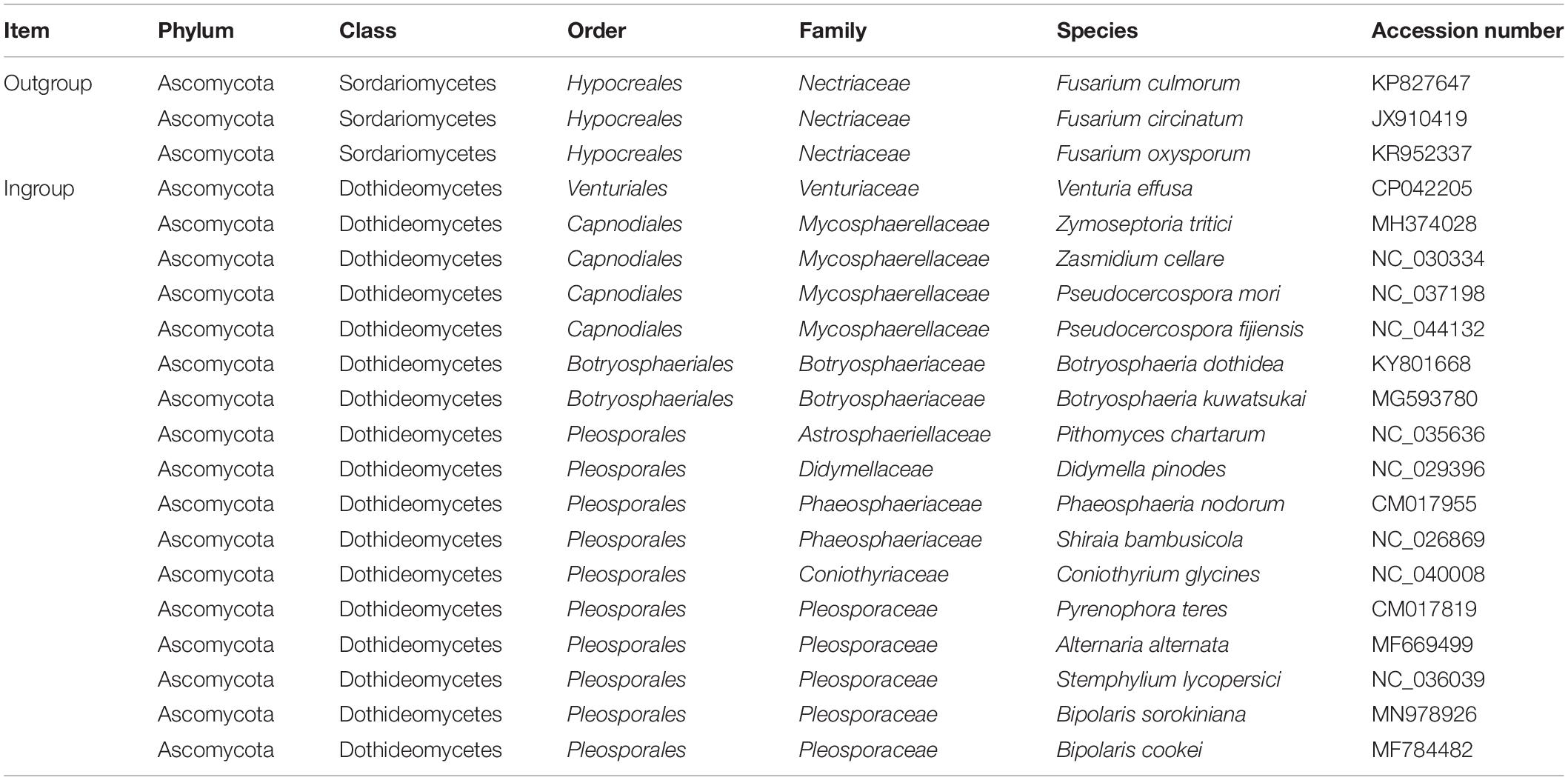
Table 1. Species included in the phylogenetic analyses.
Composition of Mitochondrial Genome
The nucleotide composition of the mtDNA sequence of B. sorokiniana and other 16 dothideomycete fungi downloaded from GenBank were computed with MEGA 7 (Kumar et al., 2016). The AT and GC skews were calculated according to the formula AT skew = (A - T)/(A + T) and GC skew = (G - C)/(G + C) (Perna and Kocher, 1995), in order to measure the strand-specific bias of nucleotide composition. Moreover, codon usage in the core mitochondrial protein-encoding genes were analyzed in MEGA 7 (Kumar et al., 2016).
Sequence Alignments
Prior to aligning each mitochondrial gene, we merged the FASTA files generated by MITOS for all species into a single file. Using the custom Perl script (selectSeqs.pl, commands used are provided in Supplementary File S1), the annotated genes collected from each species were extracted to compile the matrices. For the core protein-coding gene matrix, we firstly used TranslatorX (Abascal et al., 2010) to construct a preliminary multiple sequence alignment. The following parameters were set: Genetic code = “Mold mitochondrial,” and Protein alignment = “MAFFT.” Because many protein-coding genes were split into fragments by introns, the initial alignment contained many long gap sequences. In order to remove these gap sequences, we divided an alignment into several shorter ones based on the sequence homology checked by eye. After removal of long gap sequences, the alignments were concatenated into a single matrix, with the Perl script FASconCAT_v1.0 (Kuck and Meusemann, 2010). Ambiguous positions in the genes’ alignment were trimmed with Gblocks (Talavera and Castresana, 2007), under less stringent options. Stop codons in each alignment were checked in MEGA 7 (Kumar et al., 2016) and deleted. Individual gene alignments were concatenated into an alignment comprising all 13 core protein-coding genes but atp8. Because this gene is missing in nine out of 20 fungal mitochondrial genomes analyzed. The mitochondrial rRNA and tRNA genes were individually aligned using MAFFT online server with E-INS-i strategy (Katoh and Standley, 2013). For the tRNA gene with more than one copy, only the one most similar to other species was used in the sequence alignment. Poorly aligned sections were eliminated by Gblocks (Talavera and Castresana, 2007). The individual alignments were concatenated to create the data matrices of rRNA gene (rrn) and tRNA gene (trn), respectively.
Characteristics of Sequence Alignments
Sequence potential saturation was assessed in DAMBE software (Xia, 2013). Sequence divergence heterogeneity was evaluated using AliGROOVE (Kuck et al., 2014), with the default sliding window size. Indels in nucleotide data set were treated as ambiguity and a BLOSUM62 matrix was used as default amino acid substitution matrix. The number of synonymous substitutions per synonymous site (Ks) and the number of non-synonymous substitutions per non-synonymous site (Ka) for protein-coding genes were estimated with DnaSP v5 (Librado and Rozas, 2009), under the genetic code of mtDNA Mold-Protozoan. Congruence between different gene type alignments (protein-coding genes, rRNA genes, and tRNA genes) was assessed by using the Incongruence Length Difference (ILD) tests (Farris et al., 1994) implemented in PAUP∗4.0b10 (Swofford, 2002), under the parsimony optimality criterion and using 1,000 additional replicates.
Phylogenetic Reconstructions
In the phylogenetic analyses, our taxon sample included 17 fungus species representing eight families of Dothideomycetes, namely Venturiaceae, Mycosphaerellaceae, Botryosphaeriaceae, Astrosphaeriellaceae, Didymellaceae, Phaeosphaeriaceae, Coniothyriaceae, and Pleosporaceae (Table 1). In addition, three mitochondrial genome sequences from the class Sordariomycetes were selected as outgroups (Fourie et al., 2013; Kulik et al., 2016).
A total of five concatenated datasets were compiled as following: (1) PCG_nt (10,561 nucleotide sites), nucleotide alignment including 12 protein-coding genes; (2) PCG_aa (3,364 amino acid sites), amino acid alignment including 12 protein-coding genes; (3) PCG-rrn (14,612 nucleotide sites), nucleotide alignment including 12 protein-coding genes and two rRNA genes; (4) rrn (4,051 nucleotide sites), nucleotide alignment including rrnL and rrnS genes; and (5) trn (1,637 nucleotide sites), nucleotide alignment including 22 tRNA genes. The sequence alignments supporting the phylogenetic results of this article are presented in Supplementary File S2.
Phylogenetic trees were built using maximum likelihood (ML) method and Bayesian inference (BI) method. For the combined datasets of PCG_nt, PCG_aa, and PCG-rrn, data partition schemes and best-fitting substitution models (Supplementary Table S1) were estimated using PartitionFinder 2 (Lanfear et al., 2012). Data blocks were predefined by genes. The PartitionFinder 2 analyses were run using a greedy search scheme (Lanfear et al., 2012), with all models considered under the Akaike information criteria.
For ML inferences, we conducted separate analyses of each protein-coding gene (i.e., atp6, cob, cox1-3, and nad1-6), and then performed combined analysis based on the dataset of PCG_nt. In addition, other four concatenated datasets (i.e., PCG_aa, PCG-rrn, rrn, and trn) were also used in the ML analyses. ML tree searches were performed in IQ-TREE 1.6.10 (Nguyen et al., 2015) implemented in the Cipres Science Gateway (Miller et al., 2010). Partitioned analyses were conducted for datasets of PCG_nt, PCG_aa, and PCG-rrn, with the data partitions and the best-fitting models selected by PartitionFinder 2 (Lanfear et al., 2012). Allowing partitions to have different speeds (-spp) was selected. Nodal support values (BP) were evaluated through an ultrafast bootstrap approach (Minh et al., 2013), with 10,000 replicates.
To circumvent the long computational time required, only the five concatenated datasets (i.e., PCG_nt, PCG_aa, PCG-rrn, rrn, and trn) were used in Bayesian inferences. Bayesian tree searches were conducted in MrBayes 3.2.6 (Ronquist et al., 2012) implemented in the CIPRES Science Gateway (Miller et al., 2010). We applied the MrBayes blocks for partition definitions generated from PartitionFinder 2 (Lanfear et al., 2012). All model parameters were set as unlinked across partitions. Each analysis involved two independent runs and started from random topology. Each run implemented four Markov chain Monte Carlo chains in parallel for 10 million generations, and sampled every 1,000 generations. The program Tracer 1.7 (Rambaut et al., 2018) was used to analyze the trace files from two Bayesian MCMC runs for monitoring convergence. The first 25% of sampled trees were discarded as burn-in, and the remaining trees were used to calculate a majority-rule consensus tree. Branch support was assessed by clade posterior probabilities (PP).
We also used PhyloBayes (Lartillot et al., 2009) to conduct Bayesian inferences. This software implements the site-heterogeneous CAT-GTR or CAT model accounting for the heterogeneity present in the data. Each analysis involved two independent runs, and started from random topology, respectively. Each run implemented two Markov chain Monte Carlo chains in parallel for at least 20,000 iterations. The CAT-GTR model was used for nucleotide dataset analyses, while the CAT-MTZOA model for amino acid dataset. The “bpcomp” program contained in the package of PhyloBayes was used to calculate the largest (maxdiff) and mean (meandiff) discrepancy observed across all bipartitions. The program “tracecomp” was also used to summarize the discrepancies and the effective sizes estimated for each column of the trace file. When the maxdiff was <0.1 and minimum effective size was >100, the Bayesian runs were recognized to be reached good convergence. The first 1,000 trees of each MCMC were treated as the burn-in, and the majority-rule consensus tree was calculated from the saved trees.
Hypothesis Testing
We tested the statistical robustness of critical nodes defining deep level relationships in Dothideomycetes, by means of the four-cluster likelihood-mapping (FcLM) approach implemented in IQ-TREE 1.6.10 (Nguyen et al., 2015). The datasets of PCG_nt, PCG_aa, PCG-rrn, rrn, and trn were used for this test. The partition schemes and the corresponding best-fitting models were applied as those in the ML tree searches.
Results
Genome Assembly
The total number of raw reads are 22,819,080. After filtering, 22,111,146 high-quality PE reads are produced. A 137,775 bp mitochondrial contig is reconstructed by the assembler MITObim. Statistical analysis from Qualimap show that 465,656 single-mate reads are mapped on this mitochondrial contig. This accounts for 4.2% of the total number of reads. The mean coverage of this mitochondrial contig reach 499.92-fold. Blastn searches against the IDBA-tran assembly identify 19 possible mitochondrial contigs. Twelve contigs with a length larger than 1,000 bp match the 137,775 bp mitochondrial contig assembled by MITObim. Of which, five largest contigs have the length of 42,635, 20,394, 16,519, 7,563, and 5,602 bp, respectively. The IDBA-tran assembly confirms the result from MITObim to some extent. Considering the completeness of the mitochondrial genome assembled, the following sections will focus on the result from the assembler MITObim.
General Features of B. sorokiniana Mitochondrial Genome
The B. sorokiniana mtDNA sequence shows similar values concerning AT content, AT skew, and GC skew to most other dothideomycete fungi (mean AT content of 70.5%, mean AT skew of 0.0043, and mean GC skew of 0.0203). The mean AT content of the complete mitochondrial genome calculated for B. sorokiniana is 69.5%. The AT skew is slightly negative (-0.0043), while the GC skew is positive (0.0263). These results suggest that there is no significant strand-specific bias of nucleotide composition in the mitochondrial genome of B. sorokiniana. The gene content of B. sorokiniana mitochondrial genome is characteristic of fungal mitochondria (Figure 1 and Supplementary Table S2), including the usual set of genes interrupted by numerous introns (up to 60,277 bp in length) and separated by long intergenic spacers (up to 53,712 bp in length). A total of 12 core mitochondrial protein-coding genes are identified. In addition to the core protein-coding genes, 52 free-standing open reading frames of unknown function (uORFs) are annotated. The remaining conserved genes identified include two ORFs coding for the putative ribosomal protein S3 (rps3) gene and the putative ribosomal protein S5 (rps5) gene, the fungal mitochondrial RNase P (rnpB) gene, 38 tRNA genes and two mitochondrial ribosomal RNA genes (rrnL and rrnS).
Protein-Coding Genes and Codon Usage
The 12 core protein-coding genes (atp6, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, and nad6) are typical to fungal mtDNA, which are related to the mitochondrial oxidative phosphorylation pathway. The atp8 gene is missing, which resembles that in the mitochondrial genome of B. cookei. Five protein-coding genes (cox1-3 and nad2-3) are coded at the light strand, while the remaining seven ones are coded at the heavy strand (Figure 1). Eight out of 12 protein-coding genes are interrupted by introns, namely atp6, cob, cox1, cox3, nad1, nad2, nad4, and nad5. The highest number of intron sequences (8) are present in the cox1 gene.
In the core protein-coding genes of B. sorokiniana, Ile (I), Leu2 (L2), and Phe (F) are among the most frequently found amino acids with the frequency of AUU (14.8%), AUA (5.48%), and AUC (1.40%) for Ile, UUA (10.17%) and UUG (0.82%) for Leu2, UUU (5.63%) and UUC (2.65%) for Phe, respectively. In B. sorokiniana mitochondrial genome, 82.33% codons terminate with A or T. The codon usage pattern can be due to the relatively high AT content found in the whole mitochondrial genome. The average AT content of the core protein-coding genes is 70.5%, and the third codon positions have the highest AT content (82.3%).
Ribosomal Protein Genes and Mitochondrial RNase P Gene
The rps3 gene and the rps5 gene are found between cox3 and nad1, with the lengths of 399 and 555 bp, respectively. Both genes have the same positions and the similar sequence lengths as those in the mitochondrial genome of B. cookei. The genes of rps3 and rps5 are coded at the light strand. The AT content of rps3 gene and rps5 gene are 78.9 and 73.6%, which are higher than that of the core protein-coding genes. In addition, the fungal mitochondrial RNase P gene (rnpB) is identified between trnP_2 and trnV_2, with a length of 563 bp. The AT content of rnpB gene is 73.4%.
Transfer RNA and Ribosomal RNA Genes
A total of 38 tRNA genes are identified in the mitochondrial genome of B. sorokiniana coding for 22 amino acids (Supplementary Figure S1), with length ranging from 68 bp (trnC_1) to 85 bp (trnY). Seven tRNA genes (trnC, trnH, trnL1, trnL2, trnP, trnQ, and trnV) are duplicated. Four trnM genes are present in the mitochondrial genome, as the genes of trnN and trnR. The majority of tRNA genes are located between rrnS and nad4, and coded at the heavy strand in an anti-clockwise direction. No introns have been found in tRNA genes. All tRNA genes can be folded into the cloverleaf secondary structure. However, the trnC_1 displays an unusual acceptor stem due to nucleotide missing. In addition, the anticodon loop in trnP_1 is replaced with a large loop structure due to redundant nucleotides.
Two mitochondrial rRNA genes are identified, namely rrnL gene and rrnS gene. The rrnL gene is 4,440 bp in length, which is placed between trnP_1 and trnT. The rrnL gene is separated into two fragments by an intronic spacer with length of 273 bp. The rrnS gene is 1,659 bp, and positioned between trnR_2 and trnL2_2. The rrnS is a complete gene sequence, without intronic spacer identified in this gene. The AT content of rrnL and rrnS gene are 65.1 and 63.3%, respectively.
Phylogenetic Inference
Partition homogeneity tests showed the dataset of PCG_nt to be congruent with the dataset of rrn (P = 0.13). Therefore, the alignments of PCG_nt and rrn can be combined. The trn alignment is significantly incongruent when evaluated against PCG_nt (P = 0.01) or rrn (P = 0.01).
The separate ML analyses for individual protein-coding genes (Supplementary Table S3) recover three order groups, namely, Capnodiales, Botryosphaeriales and Pleosporales. The Venturiales is often a deeply-diverging clade, while the Pleosporales is placed in a relatively derived position. Despite with these results, the relationships among orders vary greatly across separate analyses. The discordance indicates the conflicting signals in the individual protein-coding gene data.
Combined protein-coding gene datasets (i.e., PCG_nt and PCG_aa) and the PCG-rrn dataset also recovered the three order-level lineages outlined above (Figures 2, 3 and Supplementary Figures S2–S4). Compared to separate ML analyses on individual protein-coding genes, combined analyses yield higher support values for main nodes in Dothideomycetes. Based on the PCG-rrn data, ML analysis and MrBayes analysis result in an identical tree topology (Figure 2). In which, the Venturiales is supported as the sister group to all other Dothideomycetes. The Botryosphaeriales is placed as sister group to Pleosporales (BP = 62, PP = 0.95). This arrangement receive 46.3% quartet support from the FcLM analysis on PCG-rrn data (Figure 4). The datasets of PCG_nt and PCG_aa show weaker signal for this relationship (30% of quartets from PCG_nt, and 27.5% of quartets from PCG_aa). Within Pleosporales, the family Astrosphaeriellaceae is the first clade to diverge, followed by the families Phaeosphaeriaceae and Coniothyriaceae in a sequential order. The Pleosporaceae forms a sister group to Didymellaceae. Within Pleosporaceae, our analyses maximally support B. sorokiniana as a sister group to B. cookei. This sister group relationship is also recovered in the analyses of PCG_nt and PCG_aa (BP = 100, PP = 1), irrespective of method of phylogenetic inference.
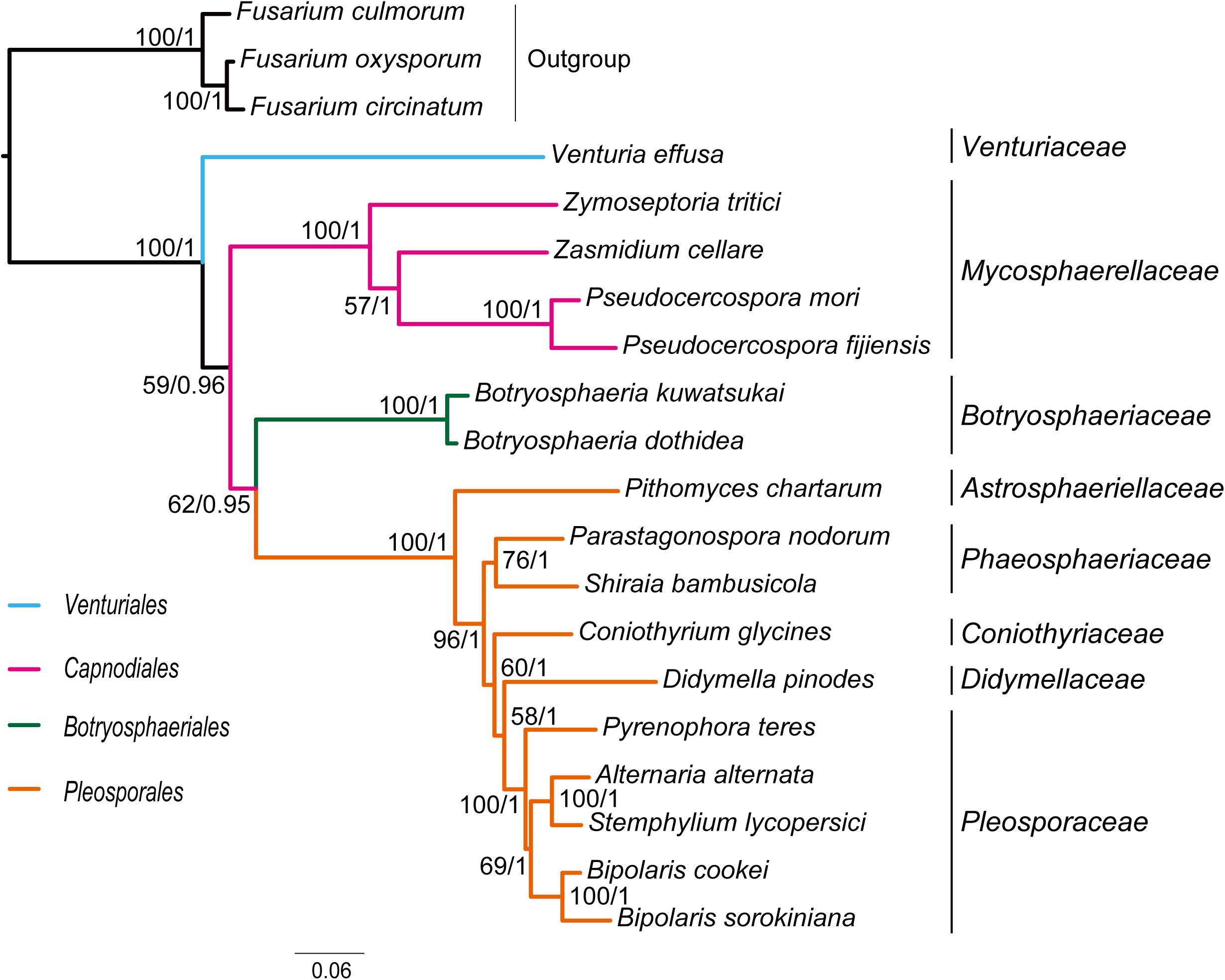
Figure 2. Maximum likelihood tree inferred from the dataset of PCG-rrn using IQ-TREE, under the partition schemes and best-fitting models selected by PartitionFinder 2. Node numbers show bootstrap support values (left) and poster probability values from MrBayes (right).
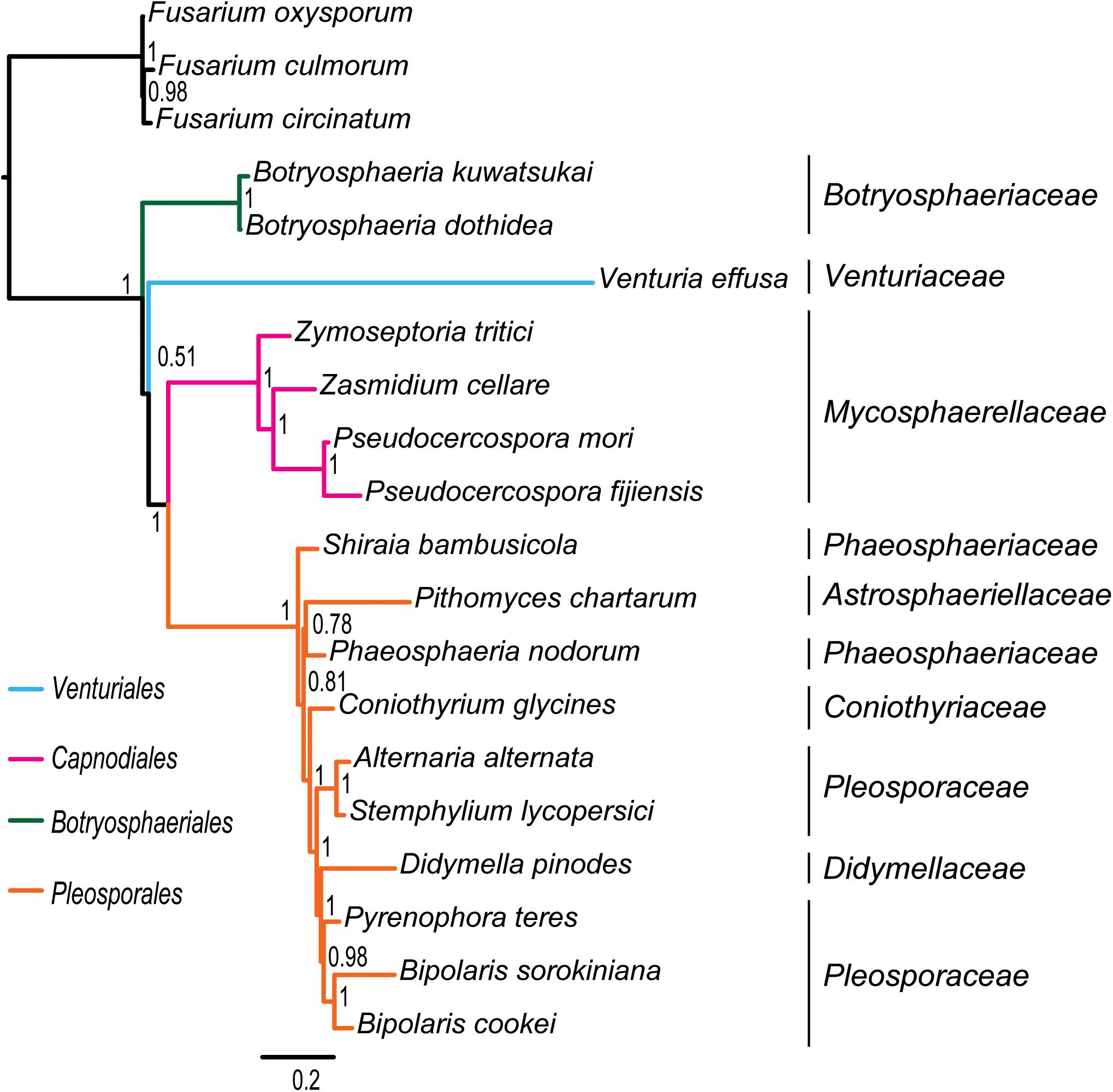
Figure 3. Bayesian tree inferred from the dataset of PCG_aa using PhyloBayes, under the site-heterogeneous CAT-MTZOA model. Node numbers show the poster probability values.
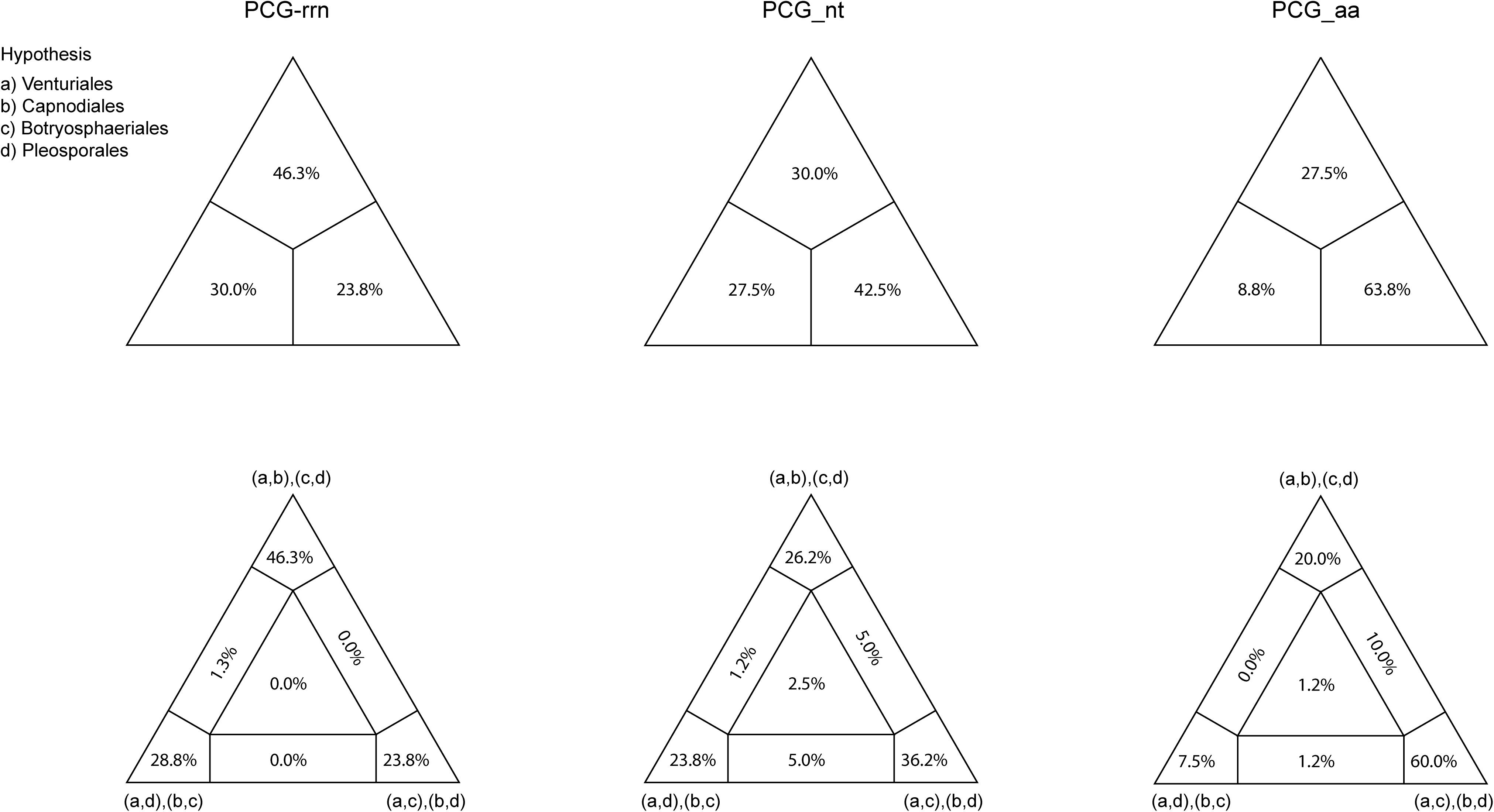
Figure 4. Results obtained from the four-cluster likelihood-mapping analyses on the datasets of PCG-rrn, PCG_nt, and PCG_aa. The triangles above are the three posterior probabilities for the three possible hypotheses; and the triangles down are the two-dimensional simplex graphs supporting different evolutionary information.
Under the site-homogeneous models, trees obtained from the analyses of PCG_nt data (Supplementary Figures S2A,B) are identical in the major lineages to those from the PCG-rrn analyses, but varied in the relationships among orders and families. The similar patterns are revealed in the analyses of PCG_aa data (Supplementary Figures S3A,B). The ML tree and MrBayes tree inferred from the amino acid data of PCG_aa differ from the nucleotide data of PCG-rrn in that the Capnodiales is retrieved as the sister group to Pleosporales. This sister group relationship is also recovered in the ML analysis from the nucleotide data of PCG_nt (Supplementary Figure S2A). The FcLM results show weak to moderate signal for the placement of Capnodiales as sister group to Pleosporales (23.8, 42.5, and 63.8% of quartets, PCG-rrn, PCG_nt, and PCG_aa, respectively).
Under the site-heterogeneous models, phylogenetic trees resulting from PhyloBayes analyses based on datasets of PCG_aa, PCG_nt and PCG-rrn consistently place Botryosphaeriales as the first diverging clade in Dothideomycetes (Figure 3 and Supplementary Figures S4A,B). In the PhyloBayes trees of PCG_nt and PCG-rrn, Venturiales and Capnodiales are recovered as sister group. But this relationship lacks significant support from both datasets (PP ≤ 0.62). By contrast, Venturiales is sister to a clade comprising Capnodiales and Pleosporales in the PhyloBayes trees from PCG_aa (Figure 3). Moreover, the Capnodiales is supported as a sister-group of Pleosporales (PP = 1). Within Pleosporales, the Phaeosphaeriaceae is non-monophyletic. Both B. sorokiniana and B. cookei are consistently grouped together with strong support (PP = 1).
The inference methods do not change the tree topology greatly in the phylogenetic analyses based on the datasets of rrn and trn (Figures 5, 6). Under the ML and MrBayes inferences, the rrn data recovers a very similar inter-order relationship as the PCG-rrn data. The Venturiales is the deepest lineage in Dothideomycete, while Botryosphaeriales forms a sister-group of Pleosporales (BP = 86 and PP = 1 in Figure 5). The trn data also supports a sister-group relationship between Botryosphaeriales and Pleosporales (BP = 98 and PP = 1 in Figure 6). But it recovers Venturiales and Capnodiales as sister group, which is not favored in analyses from rrn data. The PhyloBayes trees from the rrn and trn datasets show very similar topological structures as those in the ML and MrBayes trees, but with lower support values for several nodes. The results of the FcLM analyses on the rrn and trn datasets strongly support the (Botryosphaeriales, Pleosporales) clade (Supplementary Figure S5).
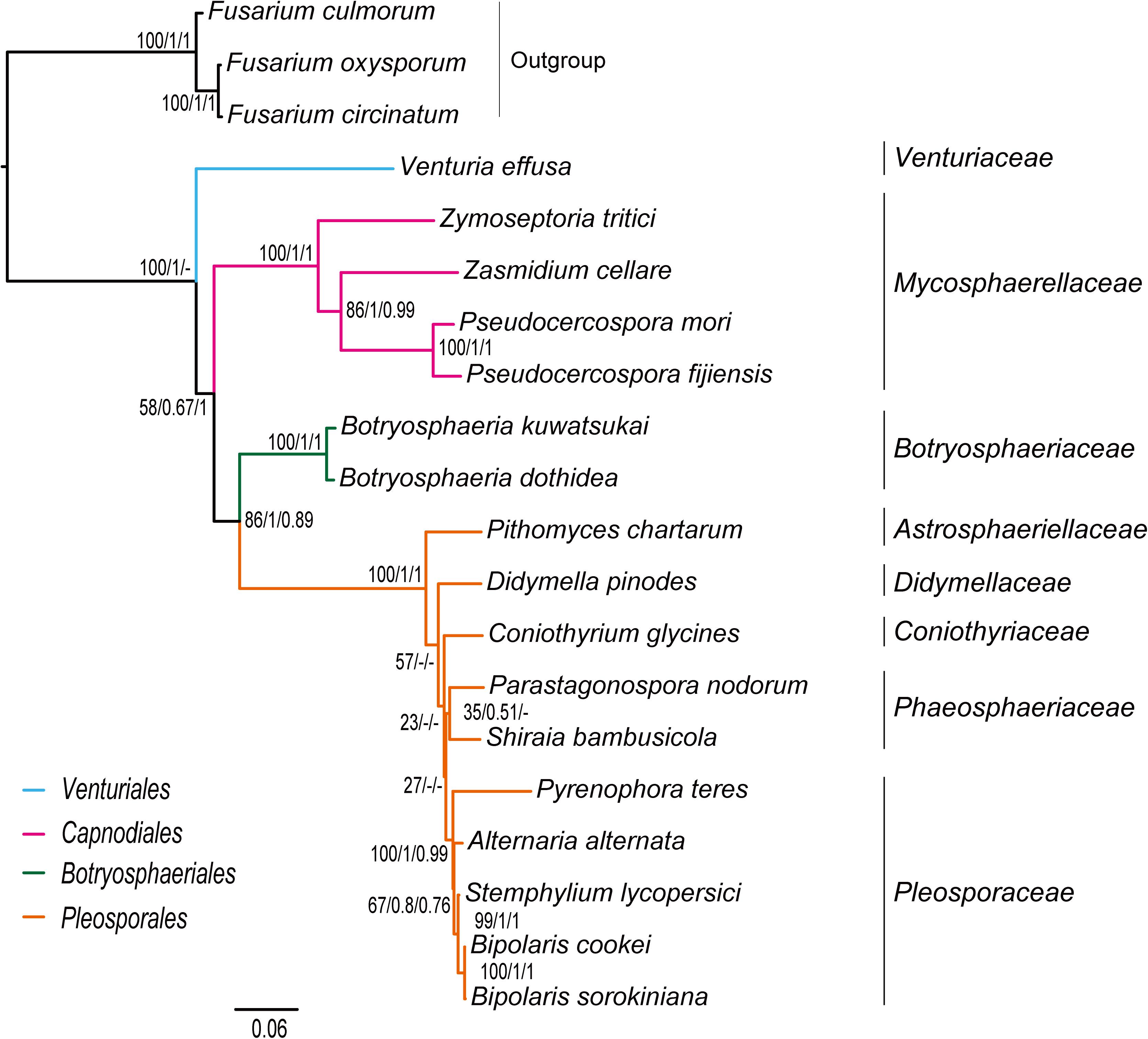
Figure 5. Maximum likelihood tree inferred from the dataset of rrn using IQ-TREE. Node numbers show bootstrap support values (left), poster probability values from MrBayes (middle), and poster probability values from PhyloBayes (right).
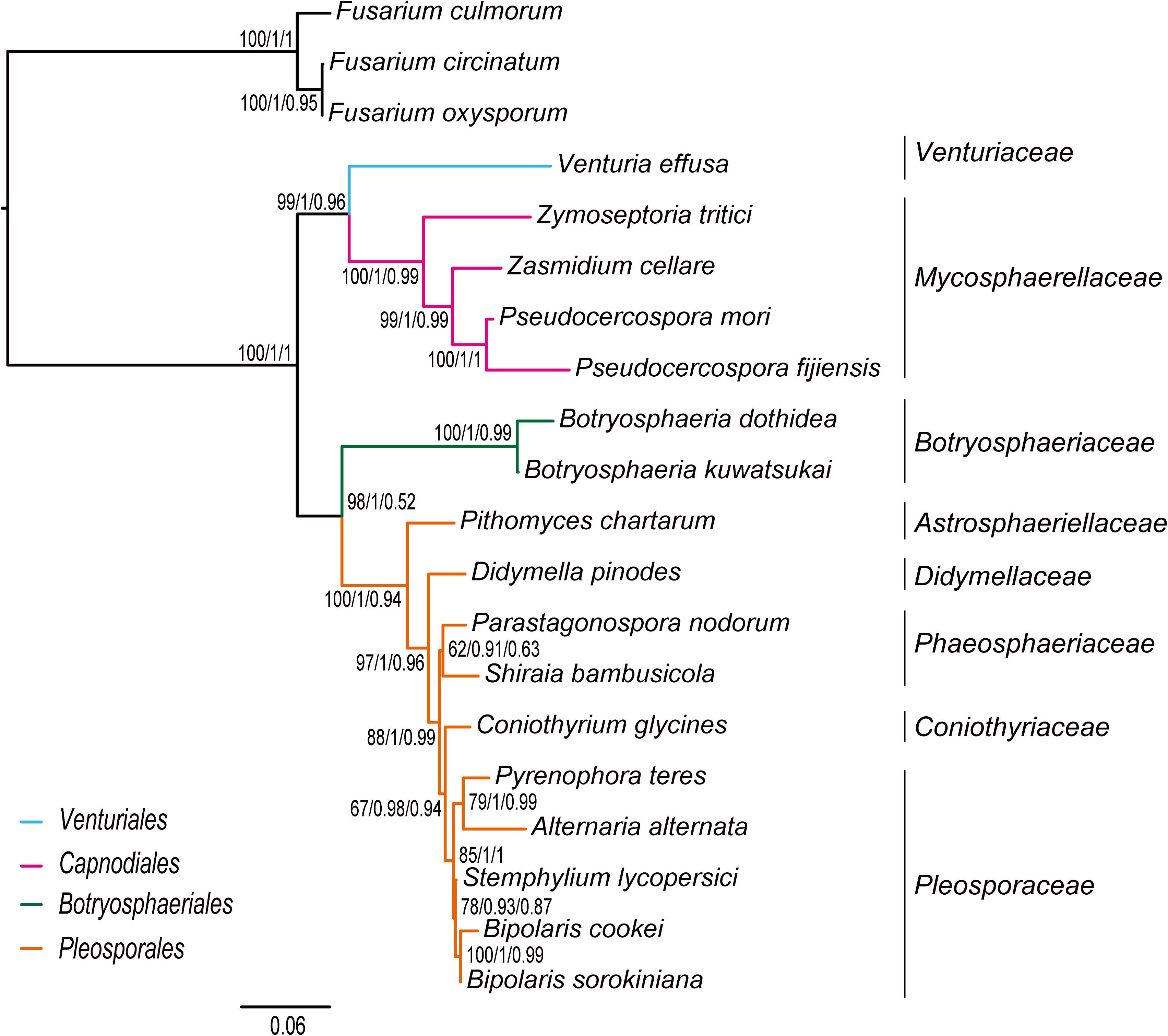
Figure 6. Maximum likelihood tree inferred from the dataset of trn using IQ-TREE. Node numbers show bootstrap support values (left), poster probability values from MrBayes (middle), and poster probability values from PhyloBayes (right).
Discussion
Thus far, the complete mtDNA sequences available in Dothideomycete are scarce. Compared to animal mitochondrial genomes, fungal mitochondrial genomes have some characteristic features. In particular, presence of numerous introns within the fungal mitochondrial protein-coding genes makes it difficult to align sequences. This study presents an effective procedure to deal with this problem. Most of the nodes in our phylogenies have maximal statistical support, demonstrating the utility of our approach for generating mitochondrial genome data matrix useful in resolving the relationships in Dothideomycetes.
In addition to the enormous variations in size of mitochondrial genomes, the gene content and gene order may be different among fungal species. The gene content and gene order of the B. sorokiniana mitochondrial genome are very similar to those of B. cookei (Zaccaron and Bluhm, 2017). The major difference is the occurrence of a variable number of tRNA genes. There are only 28 tRNA genes present in the mitochondrial genome of B. cookei, in contrast with 38 tRNA genes in B. sorokiniana. Besides tRNA genes, the numbers of unique ORFs, introns inserted in the protein-coding genes and intergenic spacers are different between two Bipolaris species. Compared with animal mitochondrial genomes, the atp9 is an additional gene occurring in many fungi (Paquin et al., 1997; Losada et al., 2014; Salavirta et al., 2014; Torriani et al., 2014; Lin et al., 2015; Ellenberger et al., 2016; Kanzi et al., 2016; Kang et al., 2017; Deng et al., 2018). However, the atp9 gene is missing in B. sorokiniana and in B. cookei. Although plasmids and plasmid-like elements have been reported from mitochondria and mitochondrial genomes in some fungal species (Griffiths, 1995; Sakurai et al., 2004; Formighieri et al., 2008; Monteiro-Vitorello et al., 2009), our sequence analysis detected no apparent linear or circular plasmids in B. sorokiniana.
Mitochondrial phylogenomic analyses have been extensively used in the studies of animals (Boore, 1999; Garesse and Vallejo, 2001; Xu et al., 2006; Mulcahy et al., 2012; Cameron, 2014; Li et al., 2017; Du et al., 2019; Song F. et al., 2019; Song N. et al., 2019; Tang et al., 2019; Klucnika and Ma, 2020), but few attempts to produce fungal phylogenies based on mitochondrial genome sequences. Dothideomycetes is the largest class of Ascomycota (Hyde et al., 2013), which includes many economically significant plant pathogens. Phylogeny of the Dothideomycetes has attracted systematists’ attention in recent years (Schoch et al., 2006, 2009a,b; Shearer et al., 2009; Suetrong et al., 2009; Nelsen et al., 2011a, b; Zhang et al., 2011; Liu et al., 2017). Some authors have initiated studies using single-gene and multi-gene phylogenetic analyses to investigate the relationships of Dothideomycetes (Schoch et al., 2006, 2009a; Shearer et al., 2009; Suetrong et al., 2009; Hyde et al., 2013). These molecular studies more frequently applied nuclear gene sequence data (e.g., nuc SSU rDNA, nuc LSU rDNA, EF1-α and ITS) to establish the phylogenetic framework (Schoch et al., 2006, 2009a,b; Shearer et al., 2009; Hyde et al., 2013; Manamgoda et al., 2012, 2014). Nuclear genes have different mechanisms of inheritance in comparison with mitochondrial genes. In general, nuclear genes show slower rates of evolution than mitochondrial genes (Brown et al., 1979; Moriyama and Powell, 1997; Johnson and Clayton, 2000; San Mauro et al., 2004). Comparisons of phylogenies based on nuclear and mitochondrial gene sequences are essential to our understanding of the phylogenetic affinities of Dothideomycetes.
Traditionally, members of Venturiaceae were classified in the order Pleosporales (Hyde et al., 2013). Schoch et al. (2009a, b) recovered Venturiaceae as a separated clade from the core members of Pleosporales, with nuclear gene fragments data. Based on combined analysis of molecular, morphological and ecological evidence, Zhang et al. (2011) established the order Venturiales comprising Venturiaceae and Sympoventuriaceae. Our phylogenetic estimates on the mitochondrial genome sequence data consistently support the Venturiaceae as an independent clade. The Botryosphaeriales is another recently proposed order (Schoch et al., 2006). Based on the PCG-rrn data, ML and MrBayes analyses returned a sister-group relationship between Botryosphaeriales and Pleosporales. The earlier molecular studies based on the nuclear gene sequences also suggested a relatively close affiliation of Botryosphaeriales to Pleosporales (Schoch et al., 2006; Hyde et al., 2013). Concerning the deep relationships among four orders surveyed, the branching pattern of {Venturiales, [Capnodiales, (Botryosphaeriales, Pleosporales)]} inferred from the PCG-rrn data and the rrn data is congruent with the multi-locus phylogenies from nuclear genes (Schoch et al., 2009a; Hyde et al., 2013).
Our analyses using a variety of datasets and different inference methods largely recover the same substantial clades in Dothideomycetes, albeit with discordant relationships between them. No significant saturation was found in any data type alignments (Iss < Iss.cSym and Iss < Iss.cAsym in Supplementary Table S4). The presence of heterogeneous sequences and lineage-specific substitution rates may provide some explanation for the incongruence between gene trees. The instability of the phylogenetic placement of Venturiales among analyses deserves special attention. This clade displays the obviously long branch length when compared with other dothideomycete lineages. The sequence heterogeneity analyses revealed that V. effusa has the notably higher heterogeneity than other dothideomycete species in the matrix of rrnL gene sequences (Figure 7). The analyses of synonymous substitution and non-synonymous substitution showed that the Venturiales has the elevated frequencies of synonymous substitutions and non-synonymous substitutions (Table 2). Both factors may lead to random similarity associated with long-branch effect. In addition to V. effusa, four species from Capnodiales (Zymoseptoria tritici, Zasmidium cellare, Pseudocercospora mori, and Pseudocercospora fijiensis) also have higher synonymous and non-synonymous substitution rates (Table 2). Therefore, the grouping of Venturiales and Capnodiales might be the result of long-branch attraction.
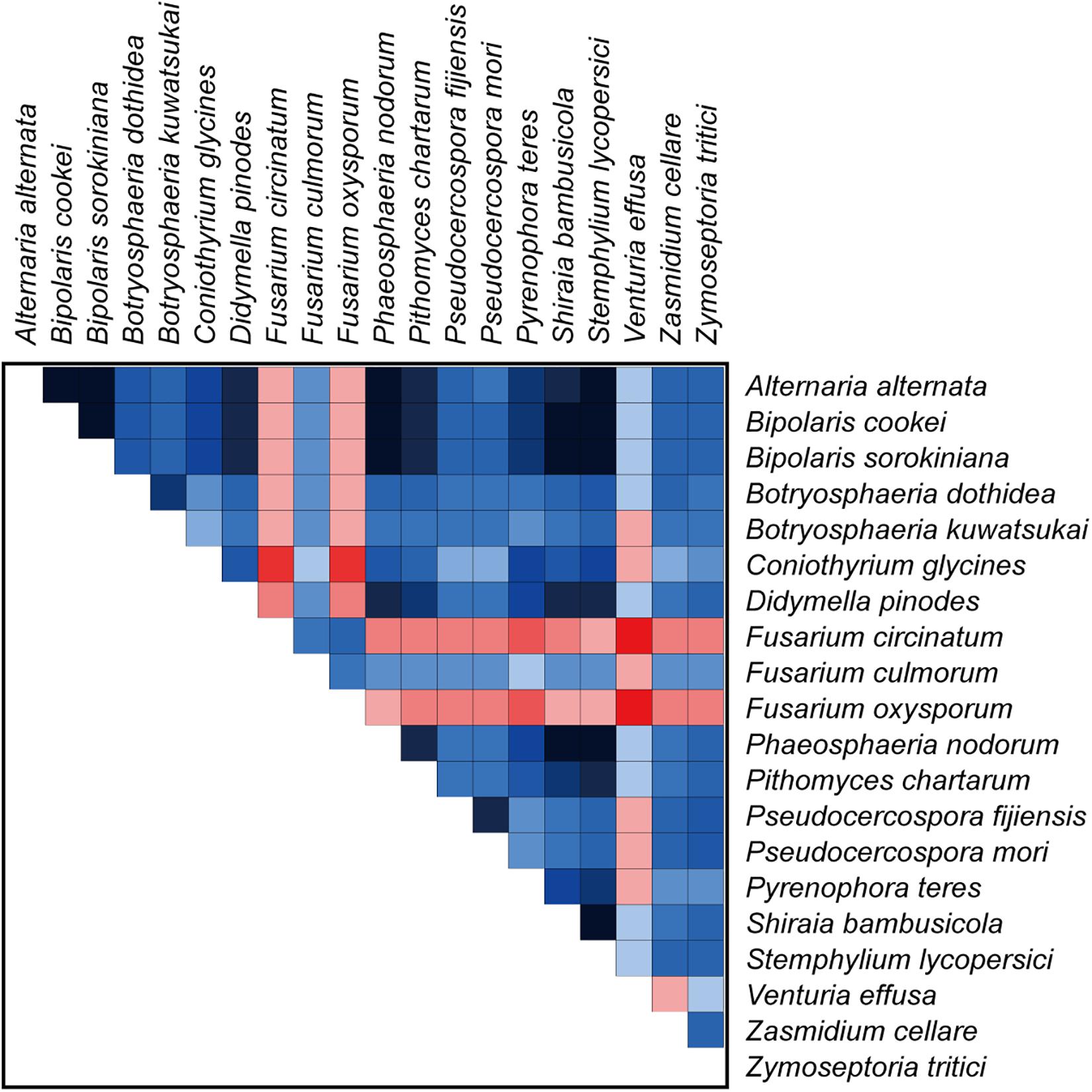
Figure 7. The AliGROOVE graph shows the mean similarity scores between sequences, based on the analysis of the rrnL gene alignment. AliGROOVE scores range from −1 (indicating great difference in rates from the remainder of the data set, i.e., red coloring implies the significant heterogeneity) to +1 (indicating rates match all other comparisons). The results from AliGROOVE analyses based on other datasets are shown in Supplementary Figure S6.
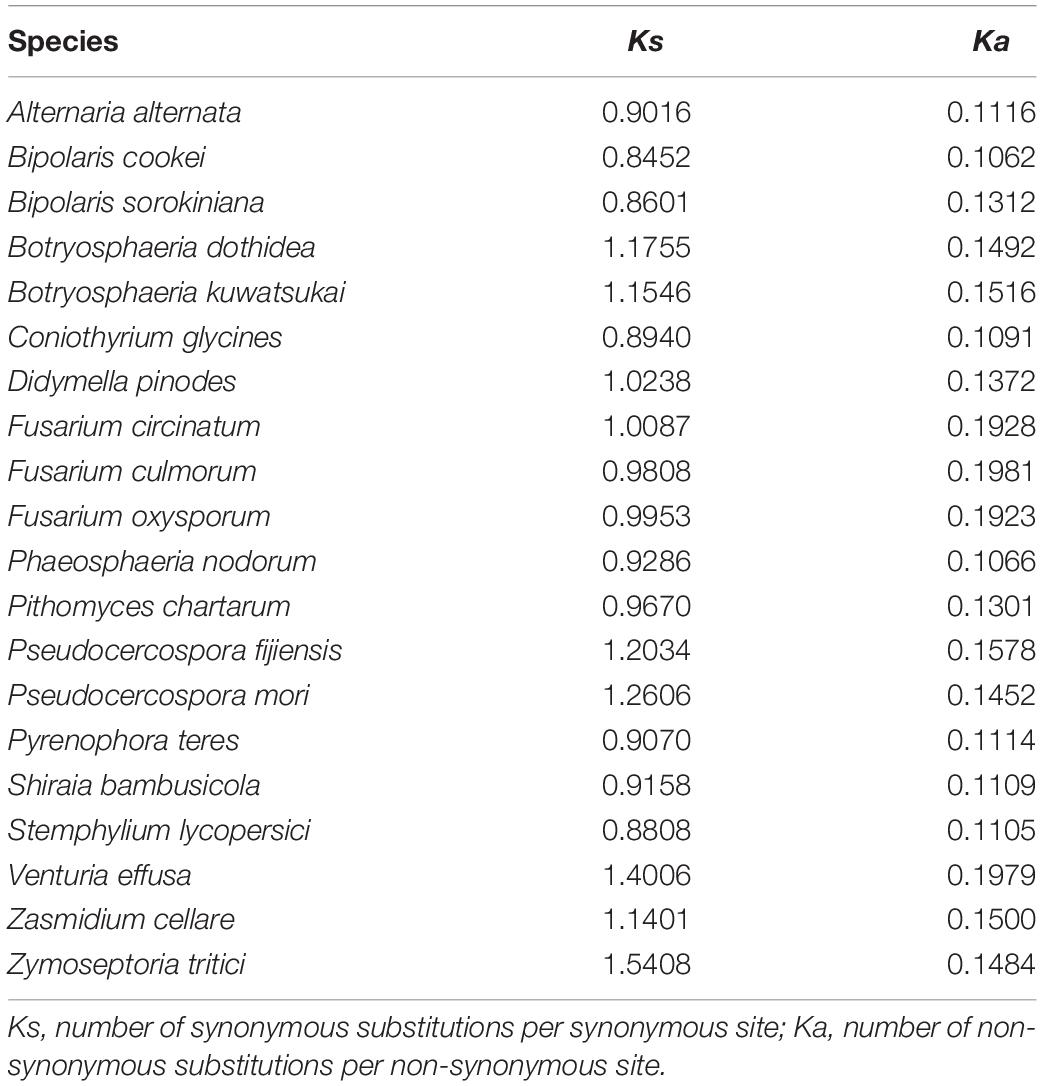
Table 2. The synonymous and non-synonymous nucleotide substitutions calculated for each species.
Due to rapid evolution of mtDNA (Brown et al., 1979; Simon et al., 2006), long branches occur frequently in the phylogenetic analyses based on mitochondrial genome sequences (Li et al., 2015; Song et al., 2016, Song N. et al., 2019; Timmermans et al., 2016; Liu et al., 2018; Tang et al., 2019). Long branches may have a negative effect on the accuracy of estimation of phylogenetic relationships. Previous studies (Lartillot et al., 2007; Li et al., 2015; Liu et al., 2018; Song F. et al., 2019; Song N. et al., 2019) showed that the site-heterogeneous model implemented in PhyloBayes could reduce the effects of compositional and mutational bias, and further suppress the long-branch attraction artifacts in the animal phylogeny. However, analyzing the current fungal mitochondrial genome data under different models did not resolve the observed incongruence. The PhyloBayes analysis on amino acid data under site-heterogeneous model breaks the grouping of Venturiales and Capnodiales, and recovers the Pleosporales as a sister group to Capnodiales (Figure 3). The PhyloBayes analyses on nucleotide datasets (PCG_nt and PCG-rrn) still retrieve a sister group of Venturiales and Capnodiales (Supplementary Figures S4A,B).
In contrast to varied results from the datasets compiled by protein-coding genes, consistency between phylogenetic reconstructions at deep level relationships within Dothideomycetes was remarkably improved by the datasets of rRNA gene and tRNA gene. The variable rates of molecular evolution in different gene sequences introduce a significant source of conflict. Slow evolving genes are more suitable for resolving deep level phylogenies (Donoghue and Sanderson, 1992; Giribet, 2002; Nosenko et al., 2013). Rates of evolutionary substitution in rRNA or tRNA genes are unusually lower than those for protein-coding genes (Ochman and Wilson, 1987; Yamamoto and Harayama, 1998; Nosenko et al., 2013). Thus, rRNA or tRNA data partitions are likely to give reliable estimates of the phylogenetic relationships among dothideomycete orders.
The lack of resolution in phylogenetics may be attributed to low levels of phylogenetic signal or highly conflicting phylogenetic signal (Whitfield and Kjer, 2008; Suh, 2016). Our FcLM analyses showed substantial levels of phylogenetic conflict for the interrelationships of four dothideomycete orders. In addition, deficient taxon sampling can also lead to poor resolution of phylogenetic relationships (Nabhan and Sarkar, 2012). We acknowledge the sensitivity of taxon sampling to phylogenetic reconstructions of a highly diverse fungal lineage. Yet, the effectively analytical approaches presented in this study are expected to inspire more mitochondrial phylogenomic analyses of Dothideomycetes. Increasing the available mitogenomic data will inevitably contribute to addressing the persisting phylogenetic uncertainties in this important fungal group.
Conclusion
We sequenced and annotated the complete mitochondrial genome of B. sorokiniana, which is only the second mitochondrial genome sequence published in the genus Bipolaris. The gene content and organization are similar to those of the B. cookei. The large number and size of introns and intergenic spacers result in the large genome size. Several order- and family-level taxa are robustly supported by the current mtDNA sequence data. The Venturiaceae is consistently recovered to be an independent clade and phylogenetically distant from Pleosporales. This result confirms the view of prior studies (Schoch et al., 2009a, b; Zhang et al., 2011). Incongruence between phylogenies constructed using various data types may stem from conflicting signals and unequal evolutionary rates. Tremendous advances of new DNA sequencing technologies have opened a window into the field of phylogenomic research, which will provide more alternative methods to tackle the problems. An understanding of mitochondrial genome evolution and phylogenetic relationships within Dothideomycetes would require the sequencing of mitochondrial genomes from more members of this group.
Data Availability Statement
The datasets generated for this study can be found in the GenBank under the accession number MN978926.
Author Contributions
NS and YG conceived this study. NS analyzed the data, prepared the figures, and drafted the manuscript. YG and XL participated in early analysis of preliminary data and manuscript writing and revision. YG provided suggestion for the research, contributed to the data interpretation, writing, and revising the manuscript critically. All authors have read and approved the final version of the manuscript.
Funding
This research was funded by Science and Technology Innovation Fund of Henan Agricultural University (KJCX2019A10) and Key Scientific Research Projects of Henan Province (18B210006).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00863/full#supplementary-material
References
Abascal, F., Zardoya, R., and Telford, M. J. (2010). TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 38, W7–W13. doi: 10.1093/nar/gkq291
Aguileta, G., De Vienne, D. M., Ross, O. N., Hood, M. E., Giraud, T., Petit, E., et al. (2014). High variability of mitochondrial gene order among fungi. Genome Biol. Evol. 6, 451–465. doi: 10.1093/gbe/evu028
Belcour, L., Rossignol, M., Koll, F., Sellem, C. H., and Oldani, C. (1997). Plasticity of the mitochondrial genome in Podospora. Polymorphism for 15 optional sequences: group-I, group-II introns, intronic ORFs and an intergenic region. Curr. Genet. 31, 308–317. doi: 10.1007/s002940050210
Berbee, M., Pirseyedi, M., and Hubbard, S. (1999). Cochliobolus phylogenetics and the origin of known, highly virulent pathogens, inferred from ITS and glyceraldehyde-3-phosphate dehydrogenase gene sequences. Mycologia 91, 964–977.
Bernt, M., Donath, A., Juhling, F., Externbrink, F., Florentz, C., Fritzsch, G., et al. (2013). MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319. doi: 10.1016/j.ympev.2012.08.023
Brown, W. M., George, M. Jr., and Wilson, A. C. (1979). Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. U.S.A. 76, 1967–1971. doi: 10.1098/rspb.2007.0169
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Cameron, S. L. (2014). Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117. doi: 10.1146/annurev-ento-011613-162007
Caterino, M. S., Cho, S., and Sperling, F. A. (2000). The current state of insect molecular systematics: a thriving Tower of Babel. Annu. Rev. Entomol. 45, 1–54. doi: 10.1146/annurev.ento.45.1.1
Chowdhury, A. K., Singh, G., Tyagi, B. S., Ojha, A., Dhar, T., and Bhattacharya, P. M. (2013). Spot blotch disease of wheat–a new thrust area for sustaining productivity. J. Wheat Res. 5, 1–11.
Condon, B. J., Leng, Y. Q., Wu, D. L., Bushley, K. E., Ohm, R. A., Otillar, R., et al. (2013). Comparative genome structure, secondary metabolite, and effector coding capacity across Cochliobolus pathogens. PLoS Genet. 9:e1003233. doi: 10.1371/journal.pgen.1003233
Deng, Y., Hsiang, T., Li, S., Lin, L., Wang, Q., Chen, Q., et al. (2018). Comparison of the mitochondrial genome sequences of six Annulohypoxylon stygium Isolates suggests short fragment insertions as a potential factor leading to larger genomic size. Front. Microbiol. 9:2079. doi: 10.3389/fmicb.2018.02079
Donoghue, M., and Sanderson, M. (1992). “The suitability of molecular and morphological evidence in reconstructing plant phylogeny,” in Molecular Systematics in Plants, eds P. Soltis, D. Soltis, and J. Doyle (New York: Chapman and Hall), 340–368.
Du, Z., Hasegawa, H., Cooley, J. R., Simon, C., Yoshimura, J., Cai, W., et al. (2019). Mitochondrial genomics reveals shared phylogeographic patterns and demographic history among three periodical cicada species groups. Mol. Biol. Evol. 36, 1187–1200. doi: 10.1093/molbev/msz051
Duo, A., Bruggmann, R., Zoller, S., Bernt, M., and Grunig, C. R. (2012). Mitochondrial genome evolution in species belonging to the Phialocephala fortinii s.l. - Acephala applanata species complex. BMC Genomics 13:166. doi: 10.1186/1471-2164-13-166
Ellenberger, S., Burmester, A., and Wöstemeyer, J. (2016). Complete mitochondrial DNA sequence of the mucoralean fungus Absidia glauca, a model for studying host-parasite interactions. Genome Announc. 4:e00153-16. doi: 10.1128/genomeA.00153-16
Farris, T. S., Källersjö, M., Kluge, A. G., and Bult, C. (1994). Testing significance of congruence. Cladistics 10, 315–319. doi: 10.1111/j.1096-0031.1994.tb00181.x
Fitzpatrick, D. A., Logue, M. E., Stajich, J. E., and Butler, G. (2006). A fungal phylogeny based on 42 complete genomes derived from supertree and combined gene analysis. BMC Evol. Biol. 6:99. doi: 10.1186/1471-2148-6-99
Formighieri, E. F., Tiburcio, R. A., Armas, E. D., Medrano, F. J., Shimo, H., Carels, N., et al. (2008). The mitochondrial genome of the phytopathogenic basidiomycete Moniliophthora perniciosa is 109 kb in size and contains a stable integrated plasmid. Mycol. Res. 112, 1136–1152. doi: 10.1016/j.mycres.2008.04.014
Fourie, G., Van Der Merwe, N. A., Wingfield, B. D., Bogale, M., Tudzynski, B., Wingfield, M. J., et al. (2013). Evidence for inter-specific recombination among the mitochondrial genomes of Fusarium species in the Gibberella fujikuroi complex. BMC Genomics 14:605. doi: 10.1186/1471-2164-14-605
Garesse, R., and Vallejo, C. G. (2001). Animal mitochondrial biogenesis and function: a regulatory cross-talk between two genomes. Gene 263, 1–16. doi: 10.1016/s0378-1119(00)00582-5
Giribet, G. (2002). Current advances in the phylogenetic reconstruction of metazoen evolution. a new paradigm for the cambrian explosion? Mol. Phylogenet. Evol. 24, 345–357. doi: 10.1016/s1055-7903(02)00206-3
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3. 1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Hahn, C., Bachmann, L., and Chevreux, B. (2013). Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-a baiting and iterative mapping approach. Nucleic Acids Res. 41, 1–9. doi: 10.1093/nar/gkt371
Hausner, G. (2003). Fungal mitochondrial genomes, plasmids and introns. Appl. Mycol. Biotech. 3, 101–131.
Hyde, K. D., Jones, E. B. G., Liu, J. K., Ariyawansa, H., Boehm, E., Boonmee, S., et al. (2013). Families of dothideomycetes. Fungal Divers. 63, 1–313.
James, T. Y., Kauff, F., Schoch, C. L., Matheny, P. B., Hofstetter, V., Cox, C. J., et al. (2006a). Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature 443, 818–822. doi: 10.1038/nature05110
James, T. Y., Letcher, P. M., Longcore, J. E., Mozley-Standridge, S. E., Porter, D., Powell, M. J., et al. (2006b). A molecular phylogeny of the flagellated fungi (Chytridiomycota) and description of a new phylum (Blastocladiomycota). Mycologia 98, 860–871. doi: 10.3852/mycologia.98.6.860
Johnson, K. P., and Clayton, D. H. (2000). Nuclear and mitochondrial genes contain similar phylogenetic signal for pigeons and doves (Aves: Columbiformes). Mol. Phylogenet. Evol. 14, 141–151. doi: 10.1006/mpev.1999.0682
Kang, X., Hu, L., Shen, P., Li, R., and Liu, D. (2017). SMRT Sequencing revealed mitogenome characteristics and mitogenome-wide DNA modification pattern in Ophiocordyceps sinensis. Front. Microbiol. 8:1422. doi: 10.3389/fmicb.2017.01422
Kanzi, A. M., Wingfield, B. D., Steenkamp, E. T., Naidoo, S., and Van Der Merwe, N. A. (2016). Intron derived size polymorphism in the mitochondrial genomes of closely related chrysoporthe species. PLoS One 11:e0156104. doi: 10.1371/journal.pone.0156104
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Klucnika, A., and Ma, H. (2020). Mapping and editing animal mitochondrial genomes: can we overcome the challenges? Philos. Trans. R. Soc. Lond. B. Biol. Sci. 375:20190187. doi: 10.1098/rstb.2019.0187
Kouvelis, V. N., Ghikas, D. V., and Typas, M. A. (2004). The analysis of the complete mitochondrial genome of Lecanicillium muscarium (synonym Verticillium lecanii) suggests a minimum common gene organization in mtDNAs of Sordariomycetes: phylogenetic implications. Fungal Genet. Biol. 41, 930–940. doi: 10.1016/j.fgb.2004.07.003
Kuck, P., Meid, S. A., Gross, C., Wagele, J. W., and Misof, B. (2014). AliGROOVE - visualization of heterogeneous sequence divergence within multiple sequence alignments and detection of inflated branch support. BMC Bioinformatics 15:294. doi: 10.1186/1471-2105-15-294
Kuck, P., and Meusemann, K. (2010). FASconCAT: convenient handling of data matrices. Mol. Phylogenet. Evol. 56, 1115–1118. doi: 10.1016/j.ympev.2010.04.024
Kulik, T., Brankovics, B., Sawicki, J., and Van Diepeningen, A. (2016). The complete mitogenome of Fusarium culmorum. Mitochondrial DNA A 27, 2425–2426. doi: 10.3109/19401736.2015.1030626
Kumar, J., Schafer, P., Huckelhoven, R., Langen, G., Baltruschat, H., Stein, E., et al. (2002). Bipolaris sorokiniana, a cereal pathogen of global concern: cytological and molecular approaches towards better control. Mol. Plant Pathol. 3, 185–195. doi: 10.1046/j.1364-3703.2002.00120.x
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lanfear, R., Calcott, B., Ho, S. Y. W., and Guindon, S. (2012). PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 29, 1695–1701. doi: 10.1093/molbev/mss020
Lartillot, N., Brinkmann, H., and Philippe, H. (2007). Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol. Biol. 7:S4. doi: 10.1186/1471-2148-7-S1-S4
Lartillot, N., Lepage, T., and Blanquart, S. (2009). PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 25, 2286–2288. doi: 10.1093/bioinformatics/btp368
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, H., Leavengood, J. M. Jr., Chapman, E. G., Burkhardt, D., Song, F., et al. (2017). Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc. Biol. Sci. 284:20171223. doi: 10.1098/rspb.2017.1223
Li, H., Shao, R. F., Song, N., Song, F., Jiang, P., Li, Z. H., et al. (2015). Higher-level phylogeny of paraneopteran insects inferred from mitochondrial genome sequences. Sci. Rep. 5:8527. doi: 10.1038/srep08527
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lin, C. P., and Danforth, B. N. (2004). How do insect nuclear and mitochondrial gene substitution patterns differ? Insights from Bayesian analyses of combined datasets. Mol. Phylogenet. Evol. 30, 686–702. doi: 10.1016/S1055-7903(03)00241-0
Lin, R., Liu, C., Shen, B., Bai, M., Ling, J., Chen, G., et al. (2015). Analysis of the complete mitochondrial genome of Pochonia chlamydosporia suggests a close relationship to the invertebrate-pathogenic fungi in Hypocreales. BMC Microbiol. 15:5. doi: 10.1186/s12866-015-0341-8
Lindgreen, S. (2012). AdapterRemoval: easy cleaning of next-generation sequencing reads. BMC Res. Notes 5:337. doi: 10.1186/1756-0500-5-337
Link, J. H. F. (1824). “Species hyphomycetum et gymnomycetum,” in Species Plantarum, ed. C. Linne (Germany: G.C. Nauk), 1–33.
Liu, J. K., Hyde, K. D., Jeewon, R., Phillips, A. J. L., Maharachchikumbura, S. S. N., Ryberg, M., et al. (2017). Ranking higher taxa using divergence times: a case study in Dothideomycetes. Fungal Divers. 84, 75–99.
Liu, Y., Song, F., Jiang, P., Wilson, J. J., Cai, W., and Li, H. (2018). Compositional heterogeneity in true bug mitochondrial phylogenomics. Mol. Phylogenet. Evol. 118, 135–144. doi: 10.1016/j.ympev.2017.09.025
Losada, L., Pakala, S. B., Fedorova, N. D., Joardar, V., Shabalina, S. A., Hostetler, J., et al. (2014). Mobile elements and mitochondrial genome expansion in the soil fungus and potato pathogen Rhizoctonia solani AG-3. FEMS Microbiol. Lett. 352, 165–173. doi: 10.1111/1574-6968.12387
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1:18. doi: 10.1186/s13742-015-0069-2
Manamgoda, D. S., Cai, L., Mckenzie, E. H. C., Crous, P. W., Madrid, H., Chukeatirote, E., et al. (2012). A phylogenetic and taxonomic re-evaluation of the Bipolaris - Cochliobolus - curvularia complex. Fungal Divers. 56, 131–144.
Manamgoda, D. S., Rossman, A. Y., Castlebury, L. A., Crous, P. W., Madrid, H., Chukeatirote, E., et al. (2014). The genus Bipolaris. Stud. Mycol. 79, 221–288. doi: 10.1016/j.simyco.2014.10.002
Mangold, A., Martin, M. P., Lucking, R., and Lumbsch, T. (2008). Molecular phylogeny suggests synonymy of Thelotremataceae within Graphidaceae (Ascomycota: Ostropales). Taxon 57, 476–486. doi: 10.1016/j.ympev.2011.04.025
Maraite, H., Di Zinno, T., Longrée, H., Daumerie, V., and Duveiller, E. (1998). “Fungi associated with foliar blight of wheat in warmer areas,” in Proceedings of the International Workshop on Helminthosporium Diseases of Wheat: Spot Blotch and Tan Spot, eds E. Duveiller, H. J. Dubin, J. Reeves, and A. McNab (Mexico: CIMMYT, El Batán), 293–300.
Mardanov, A. V., Beletsky, A. V., Kadnikov, V. V., Ignatov, A. N., and Ravin, N. V. (2014). The 203 kbp mitochondrial genome of the phytopathogenic fungus Sclerotinia borealis reveals multiple invasions of introns and genomic duplications. PLoS One 9:e107536. doi: 10.1371/journal.pone.0107536
Matusinsky, P., Frei, P., Mikolasova, R., Svacinova, I., Tvaruzek, L., and Spitzer, T. (2010). Species-specific detection of Bipolaris sorokiniana from wheat and barley tissues. Crop Prot. 29, 1325–1330.
Miller, M., Pfeiffer, W., and Schwartz, T. (2010). Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Gateway Comput. Environ. Workshop 14, 1–8.
Mina, U., Fuloria, A., and Aggarwal, R. (2016). Effect of ozone and antioxidants on wheat and its pathogen - Bipolaris sorokininana. Cereal Res. Commun. 44, 594–604.
Minh, B. Q., Nguyen, M. A. T., and Von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi: 10.1093/molbev/mst024
Monteiro-Vitorello, C. B., Hausner, G., Searles, D. B., Gibb, E. A., Fulbright, D. W., and Bertrand, H. (2009). The Cryphonectria parasitica mitochondrial rns gene: plasmid-like elements, introns and homing endonucleases. Fungal Genet. Biol. 46, 837–848. doi: 10.1016/j.fgb.2009.07.005
Moriyama, E. N., and Powell, J. R. (1997). Synonymous substitution rates in Drosophila: mitochondrial versus nuclear genes. J. Mol. Evol. 45, 378–391. doi: 10.1007/pl00006243
Mulcahy, D. G., Noonan, B. P., Moss, T., Townsend, T. M., Reeder, T. W., and Sites, J. W. Jr. (2012). Estimating divergence dates and evaluating dating methods using phylogenomic and mitochondrial data in squamate reptiles. Mol. Phylogenet. Evol. 65, 974–991. doi: 10.1016/j.ympev.2012.08.018
Nabhan, A. R., and Sarkar, I. N. (2012). The impact of taxon sampling on phylogenetic inference: a review of two decades of controversy. Brief. Bioinform. 13, 122–134. doi: 10.1093/bib/bbr014
Nelsen, M. P., Lucking, R., Mbatchou, J. S., Andrew, C. J., Spielmann, A. A., and Lumbsch, H. T. (2011a). New insights into relationships of lichen-forming Dothideomycetes. Fungal Divers. 51, 155–162.
Nelsen, M. P., Plata, E. R., Andrew, C. J., Lucking, R., and Lumbsch, H. T. (2011b). Phylogenetic diversity of Trentepohlialean Algae associated with lichen-forming fungi. J. Phycol. 47, 282–290. doi: 10.1111/j.1529-8817.2011.00962.x
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nosenko, T., Schreiber, F., Adamska, M., Adamski, M., Eitel, M., Hammel, J., et al. (2013). Deep metazoan phylogeny: when different genes tell different stories. Mol. Phylogenet. Evol. 67, 223–233. doi: 10.1016/j.ympev.2013.01.010
Nowrousian, M. (2016). Complete mitochondrial genome sequence of the Pezizomycete Pyronema confluens. Genome Announc. 4:e00355-16. doi: 10.1128/genomeA.00355-16
Ochman, H., and Wilson, A. C. (1987). Evolution in bacteria: evidence for a universal substitution rate in cellular genomes. J. Mol. Evol. 26, 74–86. doi: 10.1007/bf02111283
Ohm, R. A., Feau, N., Henrissat, B., Schoch, C. L., Horwitz, B. A., Barry, K. W., et al. (2012). Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog. 8:e1003037. doi: 10.1371/journal.ppat.1003037
Okonechnikov, K., Conesa, A., and García-Alcalde, F. (2016). Qualimap 2: advanced multisample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294. doi: 10.1093/bioinformatics/btv566
Pantou, M. P., Kouvelis, V. N., and Typas, M. A. (2008). The complete mitochondrial genome of Fusarium oxysporum: insights into fungal mitochondrial evolution. Gene 419, 7–15. doi: 10.1016/j.gene.2008.04.009
Paquin, B., Laforest, M. J., Forget, L., Roewer, I., Wang, Z., Longcore, J., et al. (1997). The fungal mitochondrial genome project: evolution of fungal mitochondrial genomes and their gene expression. Curr. Genet. 31, 380–395. doi: 10.1007/s002940050220
Peng, Y., Leung, H. C., Yiu, S. M., Lv, M. J., Zhu, X. G., and Chin, F. Y. (2013). IDBA-tran: a more robust de novo de Bruijn graph assembler for transcriptomes with uneven expression levels. Bioinformatics 29, i326–i334. doi: 10.1093/bioinformatics/btt219
Perna, N. T., and Kocher, T. D. (1995). Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358. doi: 10.1007/bf00186547
Pramateftaki, P. V., Kouvelis, V. N., Lanaridis, P., and Typas, M. A. (2006). The mitochondrial genome of the wine yeast Hanseniaspora uvarum: a unique genome organization among yeast/fungal counterparts. FEMS Yeast Res. 6, 77–90. doi: 10.1111/j.1567-1364.2005.00018.x
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Robbertse, B., Reeves, J. B., Schoch, C. L., and Spatafora, J. W. (2006). A phylogenomic analysis of the Ascomycota. Fungal Genet. Biol. 43, 715–725.
Rogers, S. O., and Bendich, A. J. (1985). Extraction of DNA from milligram amounts of fresh, herbarium, and mummified plant tissues. Plant Mol. Biol. 5, 69–76. doi: 10.1007/BF00020088
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Sakurai, T., Nomura, H., Moriyam, Y., and Kawano, S. (2004). The mitochondrial plasmid of the true slime mold Physarum polycephalum bypasses uniparental inheritance by promoting mitochondrial fusion. Curr. Genet. 46, 103–114. doi: 10.1007/s00294-004-0512-x
Salavirta, H., Oksanen, I., Kuuskeri, J., Makela, M., Laine, P., Paulin, L., et al. (2014). Mitochondrial genome of Phlebia radiata is the second largest (156 kbp) among Fungi and features signs of genome flexibility and recent recombination events. PLoS One 9:e97141. doi: 10.1371/journal.pone.0097141
San Mauro, D., Gower, D. J., Oommen, O. V., Wilkinson, M., and Zardoya, R. (2004). Phylogeny of Caecilian amphibians (Gymnophiona) based on complete mitochondrial genomes and nuclear RAG1. Mol. Phylogenet. Evol. 33, 413–427. doi: 10.1016/j.ympev.2004.05.014
Schoch, C. L., Crous, P. W., Groenewald, J. Z., Boehm, E. W., Burgess, T. I., De Gruyter, J., et al. (2009a). A class-wide phylogenetic assessment of Dothideomycetes. Stud. Mycol. 64, 1–15.
Schoch, C. L., Shoemaker, R. A., Seifert, K. A., Hambleton, S., Spatafora, J. W., and Crous, P. W. (2006). A multigene phylogeny of the Dothideomycetes using four nuclear loci. Mycologia 98, 1041–1052. doi: 10.3852/mycologia.98.6.1041
Schoch, C. L., Sung, G. H., Lopez-Giraldez, F., Townsend, J. P., Miadlikowska, J., Hofstetter, V., et al. (2009b). The Ascomycota tree of life: a phylum-wide phylogeny clarifies the origin and evolution of fundamental reproductive and ecological traits. Syst. Biol. 58, 224–239. doi: 10.1093/sysbio/syp020
Sethuraman, J., Majer, A., Iranpour, M., and Hausner, G. (2009). Molecular evolution of the mtDNA encoded rps3 gene among filamentous ascomycetes fungi with an emphasis on the ophiostomatoid fungi. J. Mol. Evol. 69, 372–385. doi: 10.1007/s00239-009-9291-9
Shearer, C. A., Raja, H. A., Miller, A. N., Nelson, P., Tanaka, K., Hirayama, K., et al. (2009). The molecular phylogeny of freshwater Dothideomycetes. Stud. Mycol. 64, 145–153.
Shoemaker, R. A. (1959). Nomenclature of Drechslera and Bipolaris, grass parasites segregated from Helminthosporium. Can. J. Bot. 37, 879–887.
Simon, C., Buckley, T. R., Frati, F., Stewart, J. B., and Beckenbach, A. T. (2006). Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 37, 545–579.
Simon, C., Frati, F., Beckenbach, A., Crespi, B., Liu, H., and Flook, P. (1994). Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soci. Am. 87, 651–701.
Sivanesan, A. (1987). Graminicolous species of Bipolaris, Curvularia, Drechslera, Exserohilum and their teleomorphs. Mycol. Pap. 158, 1–261. doi: 10.1094/PDIS.2001.85.11.1206B
Sivanesan, A., and Holliday, P. (1981). CMI Descriptions of Pathogenic Fungi and Bacteria Sheet-No. 701. London: CAB—International Mycological Institute.
Song, F., Li, H., Jiang, P., Zhou, X. G., Liu, J. P., Sun, C. H., et al. (2016). Capturing the phylogeny of Holometabola with mitochondrial genome data and Bayesian site-heterogeneous mixture models. Genome Biol. Evol. 8, 1411–1426. doi: 10.1093/gbe/evw086
Song, F., Li, H., Liu, G. H., Wang, W., James, P., Colwell, D. D., et al. (2019). Mitochondrial genome fragmentation unites the parasitic lice of Eutherian mammals. Syst. Biol. 68, 430–440. doi: 10.1093/sysbio/syy062
Song, N., Zhang, H., and Zhao, T. (2019). Insights into the phylogeny of Hemiptera from increased mitogenomic taxon sampling. Mol. Phylogenet. Evol. 137, 236–249. doi: 10.1016/j.ympev.2019.05.009
Subramanian, C. V. (1971). “Bipolaris shoemaker,” in Hyphomycetes: An Account of Indian Species, Except Cercosporae, ed. C. V. Subramanian (New Delhi: Indian Council of Agricultural Research).
Suetrong, S., Schoch, C. L., Spatafora, J. W., Kohlmeyer, J., Volkmann-Kohlmeyer, B., Sakayaroj, J., et al. (2009). Molecular systematics of the marine Dothideomycetes. Stud. Mycol. 64, 155–173.
Suh, A. (2016). The phylogenomic forest of bird trees contains a hard polytomy at the root of Neoaves. Zool. Scr. 45, 50–62.
Swann, E. C., and Taylor, J. W. (1995). Phylogenetic perspectives on Basidiomycete systematics - evidence from the 18s ribosomal-rna gene. Can. J. Bot. 73, S862–S868.
Swofford, D. L. (2002). PAUP∗, Phylogenetic Analysis Using Parsimony (∗and other methods). Version. 4. Sunderland, MA: Sinauer Associates.
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Tang, P., Zhu, J. C., Zheng, B. Y., Wei, S. J., Sharkey, M., Chen, X. X., et al. (2019). Mitochondrial phylogenomics of the Hymenoptera. Mol. Phylogenet. Evol. 131, 8–18.
Timmermans, M., Barton, C., Haran, J., Ahrens, D., Culverwell, C. L., Ollikainen, A., et al. (2016). Family-level sampling of mitochondrial genomes in Coleoptera: compositional heterogeneity and phylogenetics. Genome Biol. Evol. 8, 161–175. doi: 10.1093/gbe/evv241
Torriani, S. F., Penselin, D., Knogge, W., Felder, M., Taudien, S., Platzer, M., et al. (2014). Comparative analysis of mitochondrial genomes from closely related Rhynchosporium species reveals extensive intron invasion. Fungal Genet. Biol. 62, 34–42. doi: 10.1016/j.fgb.2013.11.001
Utomo, C., Tanjung, Z. A., Aditama, R., Buana, R. F. N., Pratomo, A. D. M., Tryono, R., et al. (2019). Complete mitochondrial genome dequence of the phytopathogenic Basidiomycete Ganoderma boninense Strain G3. Microbiol. Resour. Announc. 8:e00968-18. doi: 10.1128/MRA.00968-18
Valach, M., Burger, G., Gray, M. W., and Lang, B. F. (2014). Widespread occurrence of organelle genome-encoded 5S rRNAs including permuted molecules. Nucleic Acids Res. 42, 13764–13777. doi: 10.1093/nar/gku1266
Wang, H., Xu, Z., Gao, L., and Hao, B. L. (2009). A fungal phylogeny based on 82 complete genomes using the composition vector method. BMC Evol. Biol. 9: 195. doi: 10.1186/1471-2148-9-195
Whitfield, J. B., and Kjer, K. M. (2008). Ancient rapid radiations of insects: challenges for phylogenetic analysis. Annu. Rev. Entomol. 53, 449–472. doi: 10.1146/annurev.ento.53.103106.093304
Xia, X. H. (2013). DAMBE5: a comprehensive software package for data analysis in Mol. Biol. Evol. 30, 1720–1728. doi: 10.1007/s12539-016-0158-7
Xu, W., Jameson, D., Tang, B., and Higgs, P. G. (2006). The relationship between the rate of molecular evolution and the rate of genome rearrangement in animal mitochondrial genomes. J. Mol. Evol. 63, 375–392. doi: 10.1007/s00239-005-0246-5
Yamamoto, S., and Harayama, S. (1998). Phylogenetic relationships of Pseudomonas putida strains deduced from the nucleotide sequences of gyrB, rpoD and 16S rRNA genes. Int. J. Syst. Bacteriol. 48, 813–819. doi: 10.1099/00207713-48-3-813
Zaccaron, A. Z., and Bluhm, B. H. (2017). The genome sequence of Bipolaris cookei reveals mechanisms of pathogenesis underlying target leaf spot of sorghum. Sci. Rep. 7:17217. doi: 10.1038/s41598-017-17476-x
Keywords: fungi, Ascomycota, Pleosporales, next-generation sequencing, mitogenome, phylogenetic
Citation: Song N, Geng Y and Li X (2020) The Mitochondrial Genome of the Phytopathogenic Fungus Bipolaris sorokiniana and the Utility of Mitochondrial Genome to Infer Phylogeny of Dothideomycetes. Front. Microbiol. 11:863. doi: 10.3389/fmicb.2020.00863
Received: 27 January 2020; Accepted: 09 April 2020;
Published: 08 May 2020.
Edited by:
Tomasz Kulik, University of Warmia and Mazury in Olsztyn, PolandReviewed by:
Johannes Wöstemeyer, Friedrich Schiller University Jena, GermanyYongjie Zhang, Shanxi University, China
Bruno Andrade, Universidade Estadual do Sudoeste da Bahia, Brazil
Copyright © 2020 Song, Geng and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nan Song, songnan@henau.edu.cn; Yuehua Geng, gyh@henau.edu.cn