Corrigendum: Alterations of Gut Microbiome in the Patients With Severe Fever With Thrombocytopenia Syndrome
 Honghai Xu1,2,3†
Honghai Xu1,2,3† Yuanyuan Wei
Yuanyuan Wei Lifen Hu
Lifen Hu- 1Department of Infectious Diseases, The First Affiliated Hospital of Anhui Medical University, Hefei, China
- 2Department of Pathology, The First Affiliated Hospital of Anhui Medical University, Hefei, China
- 3Anhui Center for Surveillance of Bacterial Resistance, Hefei, China
- 4Department of Hospital Infection Control, The First Affiliated Hospital of Anhui Medical University, Hefei, China
Severe fever with thrombocytopenia syndrome (SFTS) is an emerging tick-borne infectious disease caused by SFTS virus, and the number of SFTS cases increased year by year in China. Previous studies had indicated that gut microbiome closely associated with human health and diseases, including infection diseases, liver diseases, gastrointestinal diseases and metabolic diseases. The aim of this study is to investigate the alterations and involvements of gut microbial in SFTS patients. We compared the gut microbiome of 26 SFTS patients between 20 health controls using the Illumina MiSeq sequencing platform. Reduced gut microbiota diversity and dramatic shifts of fecal microbial composition in SFTS patients were observed compared with health controls. In the intestinal microbial of SFTS patients, the Lachnospiraceae and Ruminococcaceae which could produce short-chain fatty acids were clearly dropped compared with health people, meanwhile, Sutterella which have anti-inflammation properties were reduced too. On the contrary, some common opportunistic pathogens like Enterococcus and Streptococcus and endotoxin-producing bacteria Escherichia which could rise the risk of infections were increased in SFTS patients than healthy people, in addition lactate-producing bacteria Lactobacillaceae also significantly increased in SFTS patients. In addition, research findings on the correlation between gut microbiota and biochemical data found that the changes of gut microbiota of SFTS patients were closely associated with clinical symptoms, key serum enzymes, infection and mortality. These alterations of gut microbiome in SFTS patients suggest the potential contributions of gut microbial to the pathogenesis of SFTS.
Introduction
Sever fever with thrombocytopenia syndrome (SFTS) is a recent confirmed tick-borne disease caused by the severe fever with thrombocytopenia syndrome virus (SFTSV), the virus which is classified in the family Phenuiviridae, genus Phlebovirus (Yu et al., 2011). In 2009, the SFTSV was first identified in the rural area in central China, and after then although SFTS cases had been reported in other East Asia, including Japan (Kurihara et al., 2016) and Korean (Park et al., 2016), the most SFTS cases were reported in China. From 2011 to 2016, the annual case numbers increased year by year in China (Sun et al., 2017), SFTS has already posed as a severe threat to public health. The clinical symptoms of SFTS include fever, lymphadenopathy, vomiting, diarrhea, anorexia, headache, fatigue, chill, myalgia, abdominal pain, gingival hemorrhage and so on (Sun J. et al., 2014). The severe cases could present with hemorrhagic, neurologic, multiple organs dysfunction, and even developing fatal outcome (Sun Y. et al., 2014). Although recently studies had reported that dysfunction of innate immune response (Song et al., 2017), cytokine storm and endothelial activation/dysfunction (Li et al., 2017) were associated with SFTS, the pathogenesis of SFTS has not been fully defined.
Gut microbiota, act as a real organ, the interactions between gut microbial and multiple diseases had received considerable attention. Previous literature had confirmed that the dysfunction of gut microbiome was closely related to the liver diseases, including liver cirrhosis (Chen et al., 2011; Bajaj et al., 2014), liver cancer (Yoshimoto et al., 2013), non-alcoholic steatohepatitis (Zhu et al., 2013). And the alterations of intestinal microbial in human diseases, such as obese (Turnbaugh et al., 2009; Henao-Mejia et al., 2012), inflammatory bowel disease (Palm et al., 2014), colorectal cancer (Gagnière et al., 2016), diabetes mellitus (Qin J. et al., 2014) are becoming increasing clear through numerous studies.
It is noteworthy that no study had focused on the potential role of gut microbiome played in the pathogenesis of SFTS. The alterations and involvements of gut microbiome in SFTS are still not clear, and elucidating the characteristics of the gut micobiome in SFTS patients will help unravel the detailed pathogenic role of gut dysbiosis in the pathogenesis of SFTS. Thus, the aim of this study was to investigate the changes in gut microbial of SFTS patients. Here, to fulfill the research purpose, we used the Illumina MiSeq high-throughput sequencing platform targeting the 16S rDNA gene, to compare the fecal microbial communities in SFTS patients with health people. And a comprehensive study was conducted on the correlation between clinical manifestations, key serum enzymes, infection and gut microbiota in SFTS patients.
Materials and Methods
Patients Information
The study was approved by the Ethical Review Board of the first affiliated hospital of Anhui Medical University and conformed to the ethical guidelines of the 1975 Declaration of Helsinki. All subjects obtained informed consent and all experiments were performed in accordance with relevant guidelines and regulations. The 26 SFTS patients who were included in were had been treated in the first affiliated hospital of Anhui Medical University during 2016–2017, and the 20 health volunteer subjects were randomly recruited during their annual health survey in the same hospital. The health controls have comparable age and gender with SFTS patients. And all subjects from the same mountain areas, the Ta-pieh Mountains (Supplementary Table S1), which located in southwest of Anhui Province. For all subjects, the exclusion criteria included the following: gastrointestinal disease, a history of cancer, diabetes mellitus, liver diseases, inflammation, others infectious diseases, intake history of any medicine, antibiotics or probiotics within the preceding 3 months. The SFTS patients was confirmed by real-time reverse transcription-polymerase chain reaction (RT-PCR) tests or serological tests as guided by the Ministry of Health, China (Ministry of Health China, 2011). The baseline characteristics of subjects are shown in Table 1.
TABLE 1
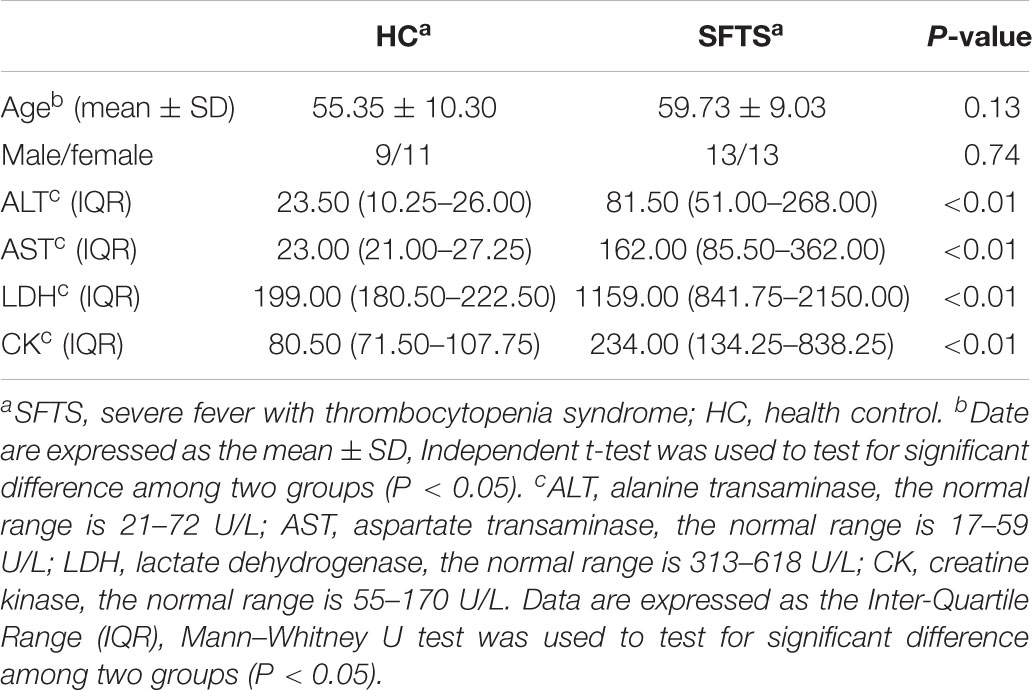
TABLE 1. Baseline characteristics of subjects.
Sample Collection
Each SFTS patient and healthy volunteer provided a fresh stool sample that was delivered immediately from our hospital to the laboratory, and it was divided into three aliquots of 200 mg and immediately stored at -80°C. For SFTS patients, an additional blood sample were required. 5 mL of blood was collected immediately in gel-separator vacuum test tubes (Hubei Jingxing Tech Co. Ltd., Hubei, China) from SFTS patients who was admitted to the hospital. All samples were kept at room temperature for 30 min until clotted. All blood samples were centrifuged to separate the serum at 1200 g for 10 min within 2 h after collection, and serum was analyzed within 2 h after separation.
DNA Extraction
DNA was extracted from 200 mg of each stool sample using a QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. The amount of DNA was measured by NanoDrop (Thermo Scientific) and its molecular size was estimated by agarose gel electrophoresis. All DNA was stored at -20°C until further analysis.
16S rDNA V4 PCR Amplification and MiSeq Sequencing
PCR amplification of the V4 hypervariable region of the16S rDNA gene was performed using the universal bacterial primers 515F(5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′). All crucial steps during sample processing, DNA isolation and the entire PCR set up were performed in a laminar air flow bench, illuminated with a UV lamp prior to use in order to avoid possible contaminants. The amplicons were normalized, pooled and sequenced on the Illumina MiSeq platform according to the manufacturer’s instructions.
Accession Codes
The 16S rDNA V4 amplicon sequencing data have been deposited in the NCBI Short Read Archive (SRA) database with accession (the details will be uploaded later).
Biochemical Data of SFTS Patients
Serum samples were measured for alanine transaminase (ALT), aspartate transaminase (AST) on an Architect C-8000 (Abbott Laboratories, United States) automated chemistry analyzer. All reagents except quality control were provided by Abbott Laboratories. ALT and AST data normal range are 21–72 U/L and 17–59 U/L, respectively. Creatine kinase (CK) and lactate dehydrogenase (LDH) were, respectively, measured with N-acetyl-L-cysteine method and the oxidation of lactate on pyruvate method. The normal range was 55–170 U/L and 313–618 U/L, respectively.
Bioinformatics Analysis and Statistical Analysis
Overlapping paired-end reads form original DNA fragments were been filtered to obtain clean reads by using QIIME (version 1.17) as following (Fadrosh et al., 2014): (1) truncation of sequence reads not having an average quality of 20 over a 30 bp sliding window based on the phred algorithm, and trimmed reads having less than 75% of their original length, as well as its paired read, will be removed; (2) removal of reads contaminated by adapter (default parameter: 15 bases overlapped by reads and adapter with maximal 3 bases mismatch allowed); (3) removal of reads with ambiguous basa (N base), and its paired reads; (4) removal of reads with low complexity (default: reads with 10 consecutive same base). Then, to get tags, clean reads were paired using FLASH (version 1.2.11) as follows (Magoč and Salzberg, 2011): (1) minimal overlapping length: 15 bp; (2) mismatching ratio of overlapped region: ≤0.1; (3) removal of paired end reads without overlaps.
The tags were clustered to Operational Taxonomic Unit (OTUs) by utilizing the software USEARCH (v7.0.1090), detailed as follows: (1) the tags were clustered into OTU with a 97% threshold by using UPARSE (Edgar, 2013); (2) chimeras were filtered out by using UCHIME (v4.2.40) (Edgar et al., 2011). Next, the OTU representative sequences were taxonomically classified using Ribosomal Database Project (RDP) Classifier v.2.2, using 0.8 confidence values as cutoff.
To describe the alpha diversity, Chao 1 index, the Observed species value (the number of OTUs) and Shannon diversity index were calculated. Chao1 index and Observed species which indicated the species richness of community were calculated using R package “fossil,” and Shannon diversity index which indicated the species diversity of the community were calculated using R package “diversity.” Principal component analysis (PCA) was used to display the differences of OTU composition in different samples using R program (v3.1.1).
Continuous variables were reported as means ± standard deviations (SD), and comparisons were made with the Independent t-test. For variables that were not normally distributed were reported as Inter-Quartile Range (IQR), comparisons were made with the Mann–Whitney U test. In addition, Spearman test was applied to analyze correlation between the clinical variables and gut microbiome. P-value of <0.05 was considered statistically significant. All data were analyzed by SPSS (version 20.0; SPSS Inc., Chicago, IL, United States).
Results
Cohort Description and Sequencing Data
Altogether, 46 subjects (SFTS = 26, HC = 20) were enrolled in this study, Table 1 displays the baseline characteristics of the participants in each cohort. We obtained 3,056,284 overlapping paired-end sequences from the 46 samples, after filtering and removing the chimeric sequences, we obtained 2,894,352 clean reads for further analysis. And then, the clean reads were combined to tags based on overlaps. 1,442,505 tags were obtained in total with a mean of 3,1358 tags per sample, and the mean read length is 252 bp. A total of 648 Operational Taxonomic Units (OTUs) were obtained by clustering the 1,442,505 tags at 97% similarity. The number of OTUs among the SFTS and HC groups were 572 and 472, respectively, and 60.8% of the OTUs (394 OTUs) were overlapped in group SFTS and healthy controls. PCA were used to compare the difference of microbial composition among the SFTS and HC groups, despite the inter-individual variability, Figure 1 showing the significant separation between the SFTS group and health control groups. The Observed species value, Chao1 index and Shannon index were significantly lower in the SFTS compared to in HC (Table 2), indicating that richness and diversity estimators in SFTS are much lower than in HC.
FIGURE 1
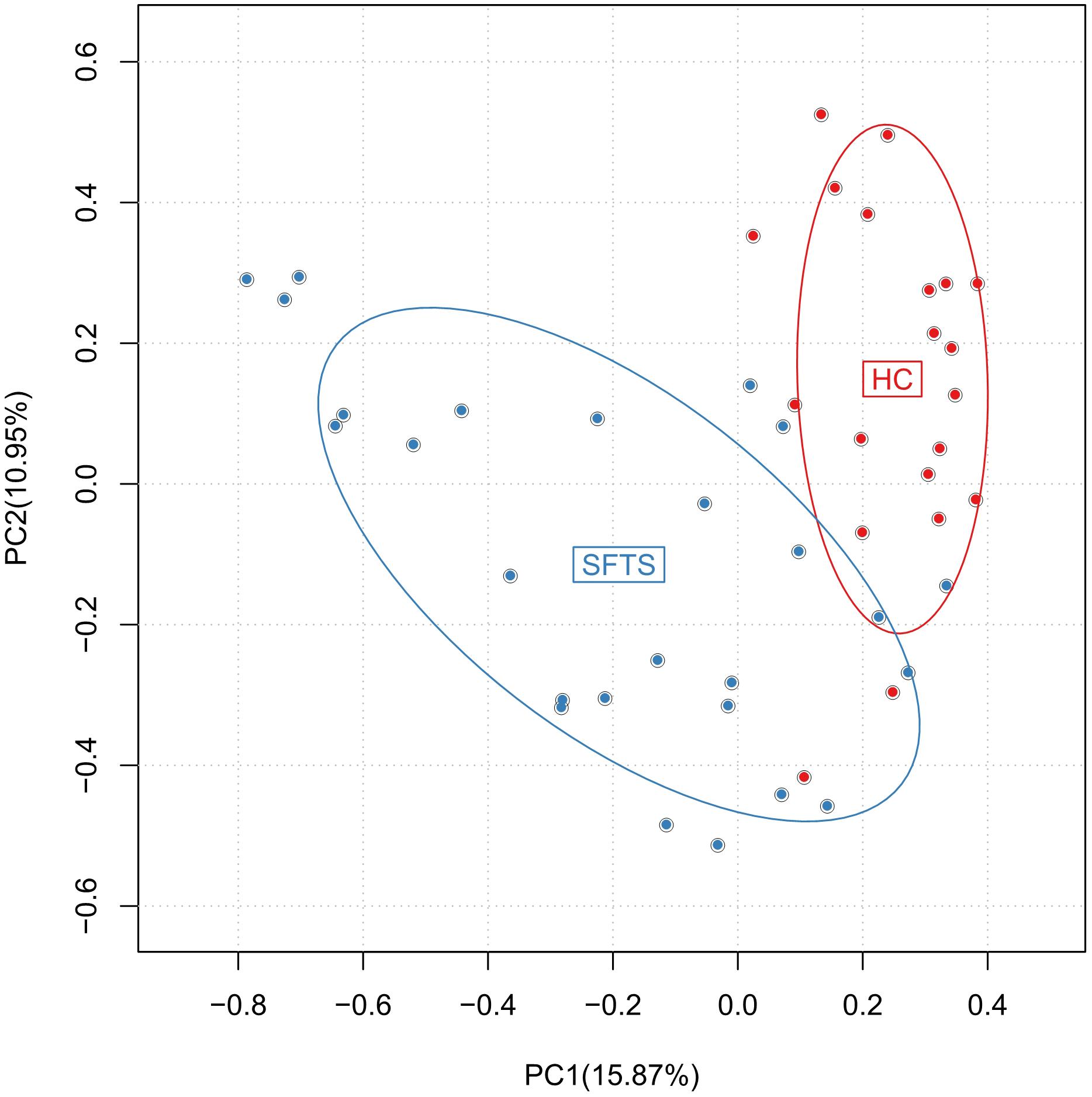
FIGURE 1. Differences in gut microbiome composition across SFTS and health control groups were assessed by PCA.
Variations in Microbial Compositions
The gut microbiome from 46 fecal samples belonging to 118 different genera in 14 different phyla. Further analysis of microbiome data showed that SFTS was associated with changes in the fecal microbiota at the phylum, class, family and genus level. At the phylum level, Bacteroidetes, Firmicutes, and Proteobacteria dominated the fecal microbial communities of both groups (percentage standard deviation of the three major phyla Bacteroidetes, Firmicutes, and Proteobacteria, were 48.16%, 27.27%, 10.16% for the SFTS, respectively, and 58.57%, 33.20%, 3.95% for health controls). Compared with healthy controls, SFTS patients had fewer Bacteroidetes and Firmicutes, but higher level of Proteobacteria compared with health controls (Figure 2A). Additionally, there are more significant difference between SFTS and health controls were discovered at microbial genera level by using univariate analyses.
Bacteroidetes
Whatever in the SFTS group or the health controls, the predominant phylum was Bacteroidetes, and numerically lower percentage of Bacteroidetes were found in SFTS group compared to health controls. And Bacteroidia (order Bacteroidales) in SFTS group were also numerically lower than in health controls. Further analysis found that although in the same class, different families had different situations. As the dominant family of Bacteroidetes, the differences of Bacteroidaceae family between two groups were not obvious (Figure 2C). However, SFTS patients had much lower level of Prevotellaceae family than healthy people, which was attributed to the decrease of Prevotella (Figure 2D).
FIGURE 2
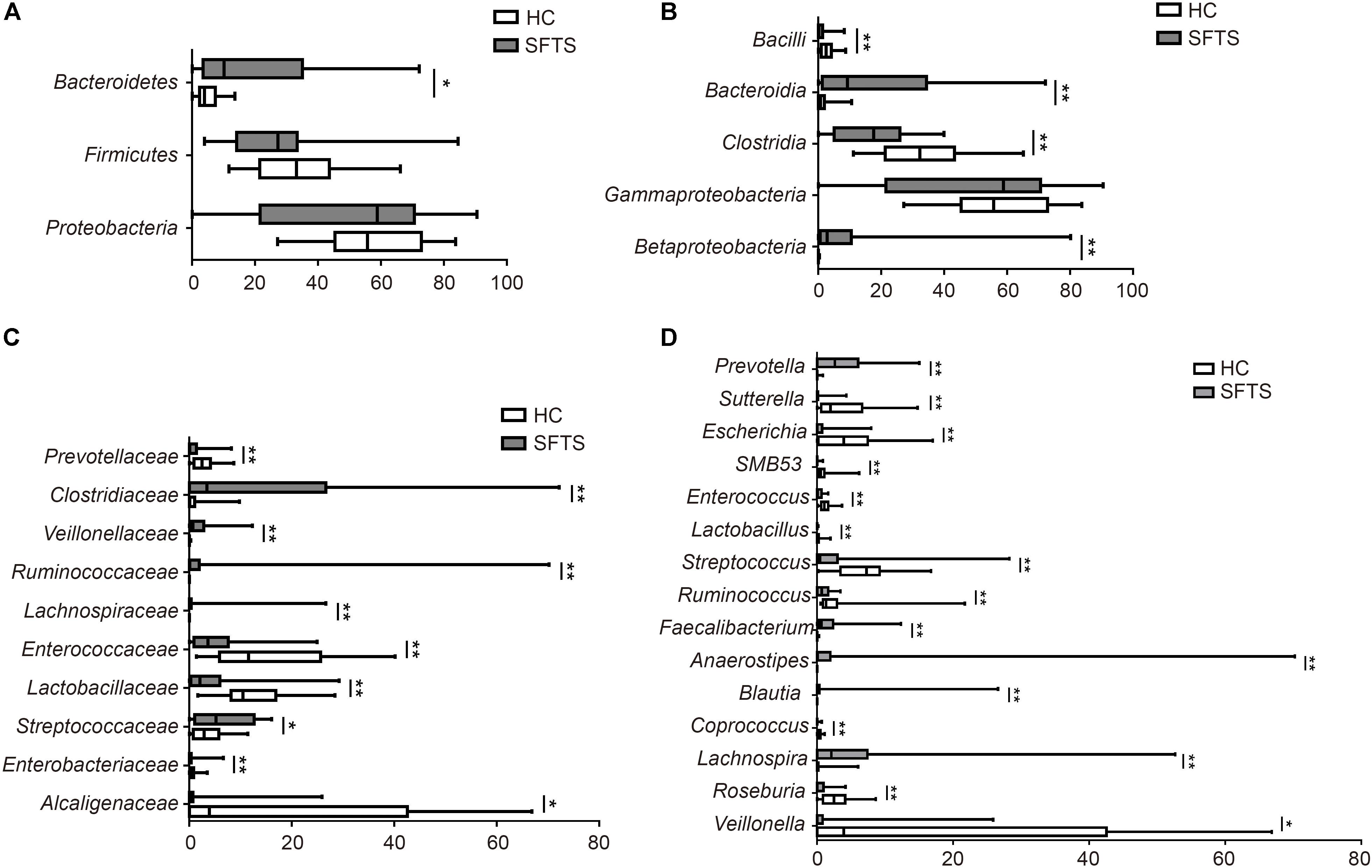
FIGURE 2. Taxonomic differences of gut microbiota between SFTS, health control groups. Comparison of relative abundance at the bacterial phylum (A), class (B), family (C), genus (D) between the two groups. ∗P < 0.05, ∗∗P < 0.01.
Firmicutes
Numerically lower percentages of Firmicutes were observed in SFTS patients than in healthy controls. At the class level, Clostridia (order Clostridiales) were significantly reduced in SFTS patients than in health controls, but Bacilli (order Lactobacillales) were obviously increased in SFTS patients. The predominant bacterial families in the class Clostridia were Lachnospiraceae, Ruminococcaceae, Veillonellaceae, and Clostridiaceae. The Lachnospiraceae and Ruminococcaceae families which are the representative types of putative butyrate-producing anaerobic bacteria were dramatically decreased in the SFTS group. Compared to health controls, SFTS patients had less Lachnospiraceae. And these results were partly due to significant differences in Lachnospira, Roseburia, Blautia, Coprococcus, and Anaerostipes. Ruminococcaceae were also discovered to be significantly underrepresented in SFTS patients compared with healthy controls (Figure 2D). Further analysis of this family showed that two genera, Faecalibacterium and Ruminococcus, led to the differences. On contrast, obviously higher percentages of Veillonellaceae family were observed in SFTS patients than in healthy controls (Figure 2C), which caused by the increase Veillonella. Although the proportion of Clostridiaceae family were both low in the two groups, the SFTS patients had less Clostridiaceae than health controls, especially SMB53. In addition, the increase of Bacilli class (order Lactobacillales) in SFTS patients was attributed to the presence of lactic acid-producing bacteria families such as Streptococcaneae family, Enterococcaceae and Lactobacillaceae, especially Streptococcus, Lactobacillus, and Enterococcus (Figure 2D).
Proteobacteria
The abundance of Proteobacteria was significantly higher in SFTS patients than in healthy controls. For the SFTS patients, the predominant class of Proteobacteria phylum was Gammaproteobacteria, whereas the dominant phylum in the health subjects was Betaproteobacteria. At the Betaproteobacteria class, the Alcaligenaceae family was higher in health people than in SFTS patients (Figure 2B), which was induced by the increase of Sutterella. On the other way, the higher level of Gammaproteobacteria in SFTS patients was caused by the increase of Enterobacteriaceae family, especially Escherichia, which are usually considered as potential opportunistic pathogens (Figure 2D).
TABLE 2

TABLE 2. The richness and diversity estimators in HC and SFTS groups.
Correlation Between Clinical Characteristics and Gut Microbiota
In this study, a total of 26 patients with SFTS were included, 4 (15.38%) patients died. SFTSV-infected patients had acute issue damage as indicated by multiple elevated key serum enzymes, as shown in Table 1, ALT, AST, LDH, and CK were all highly elevated in SFTS patients. Our analysis further demonstrated the correlation between gut microbiota and serum enzymes (Table 3). Notably, Lachnospira showed strong positive association with serum ALT and serum AST, respectively. Interestingly, Prevotella and Lactobacillus exhibited only moderate positive association with serum ALT. On the contrary, Ruminococcus manifested moderate negative association with serum CK. And gut microbiomes showed no relationship between serum LDH.
TABLE 3
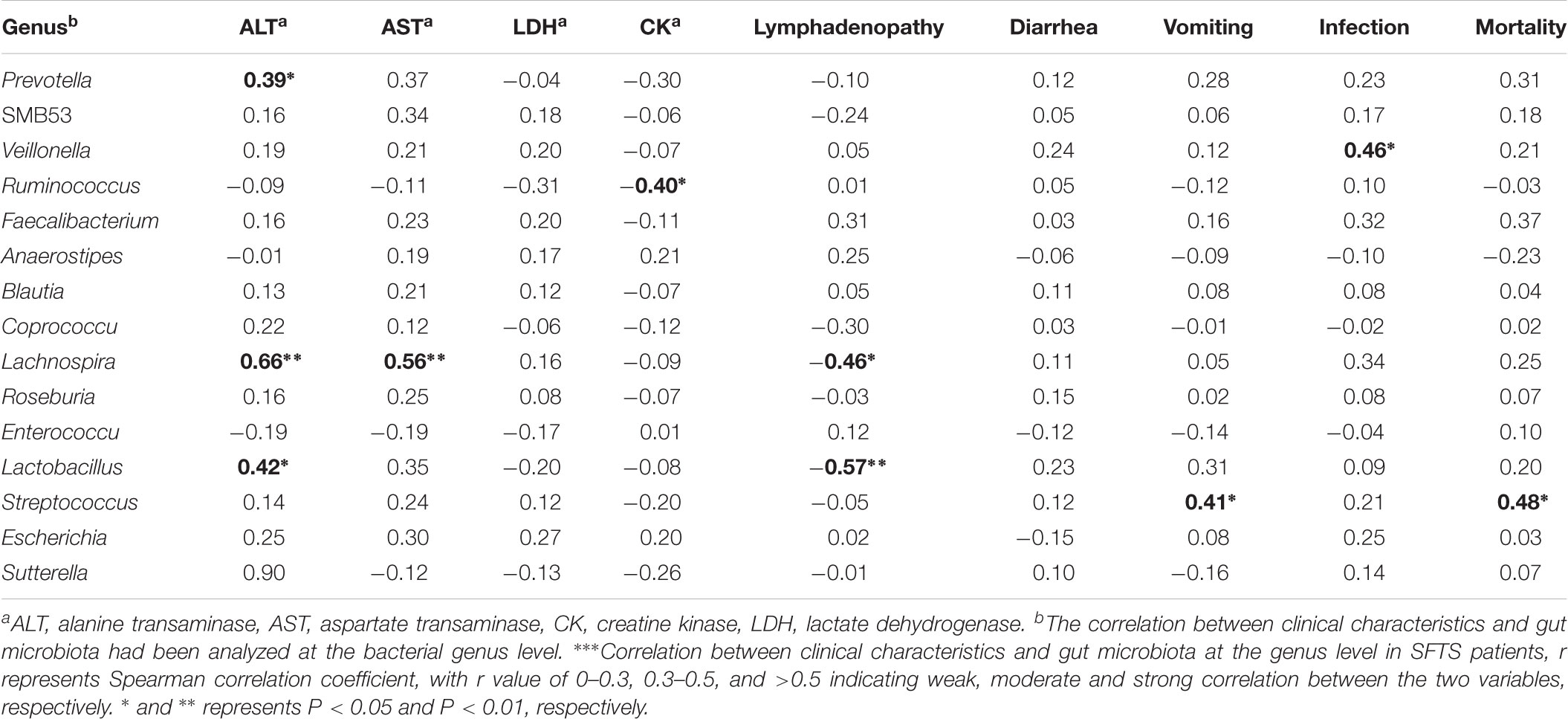
TABLE 3. Correlation between clinical characteristics and gut microbiota.
For SFTS patients, as its name described, patients suffered high fever and thrombocytopenia. Besides, the clinical characteristics include lymphadenopathy, diarrhea, vomiting, infections, and so on. As shown in Table 3, Lactobacillus and Lachnospira showed strong and moderate negative correlation between lymphadenopathy in SFTS patients, respectively. Since Streptococcus are considered as a common opportunistic pathogen, we found that Streptococcus exhibited positive association between vomiting and death, respectively. In addition, Veillonella showed positive correlation with bacterial infections in SFTS patients. In conclusion, the data suggest that the clinical characteristics were related with changes of gut microbiota in SFTS patients.
Discussion
Severe fever with thrombocytopenia syndrome is an emerging infectious disease characterized with high fever, thrombocytopenia, leukocytopenia, gastrointestinal symptoms, hemorrhage, and multiple organ failure, with high fatalities ranging from 12 to 30% (Li et al., 2014; Song et al., 2017). In recent years, the number of SFTS cases have increased considerably in China from 511 in 2011 to nearly 1,500 cases in 2012 (Silvas and Aguilar, 2017). SFTS imposes great challenge to the public health because its increased prevalence and limited clinical interventions (Zhang et al., 2012). In this study, the mean age of SFTS patients were 59 years. And a previous research had reported that the majority of SFTS (86%) are detected in subjects 50 years of age or older (Liu et al., 2015), which is same with our finding. Previous studies had pay attention to the ecology and transmission cycle (Wang et al., 2015; Li et al., 2016), pathogenesis (Wu et al., 2014; Ning et al., 2015; Silvas et al., 2015) and treatment (Kim et al., 2016; Yoo et al., 2017) of SFTS, but no one has pointed to the alteration of gut microbiota for SFTS patients, this study aim to find some differences of gut microbiome between SFTS patients and health people.
Severe fever with thrombocytopenia syndrome appears to decrease intestinal microbiome richness as those in SFTS patients had less number of OTUs and lower Chao 1 index compared to those in the health controls. And the Shannon index for SFTS patients was lower than it for health controls indicated that SFTS appears to decrease the diversity of the gut microbiota. The results were similar at both the phylum and genus level. Cotillard et al. (2013) had illustrated that obesity not only influenced the by the numbers and richness of gut microbiota and the diversity of gut microbiome. According to this, we speculate that the richness and diversity of gut microbiome both have some influence on pathogenesis of SFTS, and the specific mechanism needs further study.
In the human intestinal microbiome, Bacteroidetes and Firmicutes are the dominant bacteria, which accounting for approximately 99% of the whole bacteria (Li et al., 2008). However, in our study, at the phylum level, the relative abundance of Bacteroidetes and Firmicutes were numerically lower in SFTS patients, but the proportion of Proteobacteria was significantly higher in SFTS than healthy people. Different from previous studies, which had found that fat people had lower Firmicutes and higher Bacteroidetes (Zhu et al., 2013) and liver cirrhosis patients had less Bacteroidetes and more Proteobacteria compared with health people (Qin N. et al., 2014). The novelty of our study is that we provide the first report of a significant alteration of intestinal microbiota of SFTS patients compared with healthy subjects. In our research, the higher Proteobacteria and lower Bacteroidetes and Firmicutes showing a severe ratio of “bad vs. good” taxa abundance associated with SFTS. Previous study had suggested that Bacteroidetes and Firmicutes were increased after successful fecal microbial transplantation (FMT), and Proteobacteria were less abundant than before (Hamilton et al., 2013).
At the family level, the decrease of the phylum Firmicutess in SFTS patients was mostly associated with the families Lachnospiraceae and Ruminococcaceae, which were the predominant members of Clostridia class, usually considered as ‘healthy’ gene-rich microbiota as it has anti-inflammatory properties. These bacteria, as typical representatives of short-chain fatty acids (SCFAs)-producing bacteria, play a key role in producing nutrients for the host and supplying energy for the colonic epithelium, regulating host gene expression, inflammation and differentiation (Canani et al., 2011). A decrease of SCFAs would result in deteriorated intestinal integrity and increased intestinal permeability, which may cause worse gastrointestinal symptoms in SFTS. The SMB53 which belong to Clostridiaceae family in Clostridia class were also reduced in SFTS patients. Besides the reduction of Lachnospiraceae and Ruminococcaceae, there were also some common opportunistic pathogens significantly increased in SFTS patients, including Enterococcaceae and Streptococcaceae, especially Enterococcus and Streptococcus. However, the increase of Enterococcaceae and Streptococcaceae could be a result, or a cause of progression in SFTS, and further studies are necessary to assess the causal relationship. Meanwhile, Lactobacillaceae family and its most abundant genus Lactobacillus were clearly increased in SFTS patients compared with health people. Antharam et al. (2013) had also revealed an increase of Lactobacillus in antibiotic-associated diarrhea compared with healthy people. Actually, Lactobacillaceae family, and the bacteria family Enterococcaceae and Streptococcaceae, belong to the same order Bacilli, and all of these are good at producing lactic acid. We speculate that besides the increasing of Enterococcaceae and Streptococcaceae may lead more risk of opportunistic infections of SFTS patients, the function of producing lactic acid by these bacteria especially Lactobacillus may also paly indeed role in the progression of SFTS. It is worth noting that a recent study had reported that the reduction of Lactobacillus spp. caused by high salt diet could contribute to hypertension, which by increasing T helper 17 cells (Wilck et al., 2017). Interestingly, Ritze et al. (2014) demonstrated that Lactobacillus could attenuate liver pathologies through anti-inflammatory actions and stabilization of the intestinal barrier. The inevitable discrepancies among different studies may due to the different diseases and host factors influencing the intestinal microbiome. In addition, we firstly found that Veillonellaceae were much higher in SFTS patients compared with healthy controls. The finding was in accordance with a previous study which reported that liver cirrhosis had more Veillonellaceae which came from oral (Qin N. et al., 2014). We suspect the origin of Veillonellaceae in the gut microbiota of SFTS patients, but it needs further validation.
Our present study found that the higher proportion of Proteobacteria was contributed by the increase of lipopolysaccharide-producing G-bacteria, such as Enterobacteriaceae family, especially Escherichia. The higher abundance of these opportunistic pathogens would may result in endotoxemia in SFST patients. Differ from the increase of Enterobacteriaceae, we found that the Sutterella in Alcaligenaceae family was reduced in SFTS patients. Morgan et al. (2015) had reported that host inflammation processes were inversely correlated with Sutterella and positively correlated with Escherichia in inflammatory bowel disease. We guess that these bacteria may have same influence on SFST which need to confirm. A previous study had found that high level of Prevotella was associated with the development of hypertension and hyperlipidemia (Liu F. et al., 2017), which contrary in our study, the Prevotella was reduced in the SFTS patients. Furthermore, a former research had revealed that the content of Prevotella was same in patients with non-alcoholic fatty liver disease compared with healthy people (Wang et al., 2016). It demonstrated that the alteration of gut microbiota in multiple diseases have its own characteristics.
There have been a limited number of studies in detail on the correlation between gut microbiota and clinical characteristics after SFTSV infection. In accordance with our study Streptococcaceae found to be significantly associated with vomiting and mortality of SFTS patients. The higher abundance of Streptococcaceae which are opportunistic pathogens, may be a consequence of imbalance in microbiota resulting in the fetal outcome of SFTS patients. In fact, in SFTS patients, Veillonellaceae were related with bacterial infections. Indeed, previous study had reported that SFTS patients had a higher risk of secondary bacterial infection, which resulted in an increased morality (Liu et al., 2014). The link between Veillonellaceae and bacterial infections indicate that the Veillonellaceae may contribute to the aggravation of SFTS, but it needs further validation. In this study, Lactobacillus had showed strong negative correlation between lymphadenopathy and moderate positive association with serum ALT. As we all know that the function of Lactobacillus was producing lactic acid, but the role of Lactobacillus in the pathogenesis of SFTS needs further analysis. Prevotella manifested positive association with serum ALT, and the specific mechanism needs further study. Meanwhile, Lachnospiraceae and Ruminococcaceae, which were the predominant members of SCFAs-producing bacteria, exhibited different relationship with clinical characteristics in SFTS patients. For Ruminococcaceae, it showed significant negative correlation with serum CK. One of the characteristic laboratory findings in the severity SFTS was higher serum CK (Liu J. et al., 2017). We think that decreased Ruminococcaceae may play an important role in the pathogenesis of SFTS, and increased the level of Ruminococcaceae may be a new idea for the therapy for SFTS patients. On the contrary, we observed the phenomenon that Lachnospiraceae were strong positive related with ALT and AST and negative associated with lymphadenopathy. We feel confused about the causes of this phenomenon and will continue to explore it in subsequent research.
Conclusion
Our present study firstly demonstrated that the gut microbiota of SFTS patients were significant different with health people. Not only the richness and diversity of intestinal microbiome were reduced in SFTS, but also the composition of it had significant differences. The good bacteria such as Lachnospiraceae and Ruminococcaceae which could produce short-chain fatty acids were clearly dropped in SFTS. Meanwhile, Sutterella which have anti-inflammation properties were reduced too. On the contrary, some common opportunistic pathogens like Enterococcus and Streptococcus which could rise the risk of infections were increased in SFTS patients than healthy people, and high level of lactate-producing bacteria Lactobacillaceae could take a heavy on the development of SFTS. Moreover, the increase of endotoxin-producing bacteria Escherichia also adversely affected the SFTS patients. Besides, the analysis of the correlations between clinical index and gut microbiota indicated that gut microbiota was strong associated with key serum enzymes level, lymphadenopathy, vomiting, infection and mortality in SFTS. So far, the evidence supporting the role of intestinal dysbiosis in SFTS is limited, and the impact of some changes of bacteria in SFTS are still confused, further studies underlying the regulatory mechanisms and functions of gut microbiome are essential.
Author Contributions
HX and YW performed the laboratory work. YW performed the data analysis. HX wrote the manuscript. JL and LH provided ideas and checked the manuscript. HM, YL, and YZ provided suggestions.
Funding
This study was supported by National Natural Science Foundation of China (81172737).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02315/full#supplementary-material
References
Antharam, V. C., Li, E. C., Ishmael, A., Sharma, A., Mai, V., Rand, K. H., et al. (2013). Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J. Clin. Microbiol. 51, 2884–2892. doi: 10.1128/JCM.00845-13
Bajaj, J. S., Heuman, D. M., Hylemon, P. B., Sanyal, A. J., White, M. B., Monteith, P., et al. (2014). Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 60, 940–947. doi: 10.1016/j.jhep.2013.12.019
Canani, R. B., Costanzo, M. D., Leone, L., Pedata, M., Meli, R., and Calignano, A. (2011). Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J. Gastroenterol. 17, 1519–1528. doi: 10.3748/wjg.v17.i12
Chen, Y., Yang, F., Lu, H., Wang, B., Chen, Y., Lei, D., et al. (2011). Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 54, 562–572. doi: 10.1002/hep.24423
Cotillard, A., Kennedy, S. P., Kong, L. C., Prifti, E., Pons, N., Le Chatelier, E., et al. (2013). Dietary intervention impact on gut microbial gene richness. Nature 500, 585–588. doi: 10.1038/nature12480
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fadrosh, D. W., Ma, B., Gajer, P., Sengamalay, N., Ott, S., Brotman, R. M., et al. (2014). An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2:6. doi: 10.1186/2049-2618-2-6
Gagnière, J., Raisch, J., Veziant, J., Barnich, N., Bonnet, R., Buc, E., et al. (2016). Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 22, 501–518. doi: 10.3748/wjg.v22.i2.501
Hamilton, M. J., Weingarden, A. R., Unno, T., Khoruts, A., and Sadowsky, M. J. (2013). High-throughput DNA sequence analysis reveals stable engraftment of gut microbiota following transplantation of previously frozen fecal bacteria. Gut Microbes 4, 125–135. doi: 10.4161/gmic.23571
Henao-Mejia, J., Elinav, E., Jin, C., Hao, L., Mehal, W. Z., Strowig, T., et al. (2012). Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185. doi: 10.1038/nature10809
Kim, U. J., Kim, D. M., Ahn, J. H., Kang, S. J., Jang, H. C., Park, K. H., et al. (2016). Successful treatment of rapidly progressing severe fever with thrombocytopenia syndrome with neurological complications using intravenous immunoglobulin and corticosteroid. Antivir. Ther. 21, 637–640. doi: 10.3851/IMP3036
Kurihara, S., Satoh, A., Yu, F., Hayasaka, D., Shimojima, M., Tashiro, M., et al. (2016). The world first two cases of severe fever with thrombocytopenia syndrome: an epidemiological study in Nagasaki. Japan. J. Infect. Chemother. 22, 461–465. doi: 10.1016/j.jiac.2016.04.001
Li, J., Han, Y., Xing, Y., Li, S., Kong, L., Huang, Z., et al. (2014). Concurrent measurement of dynamic changes in viral load, serum enzymes, T cell subsets, and cytokines in patients with severe fever with thrombocytopenia syndrome. PLoS One 9:e91679. doi: 10.1371/journal.pone.0091679
Li, M., Wang, B., Zhang, M., Rantalainen, M., Chen, Y., Zhou, H., et al. (2008). Symbiotic gut microbes modulate human metabolic phenotypes. Proc. Natl. Acad. Sci. U.S.A. 105, 2117–2122. doi: 10.1073/pnas.0712038105
Li, X. K., Yang, Z. D., Du, J., Xing, B., Cui, N., Zhang, P. H., et al. (2017). Endothelial activation and dysfunction in severe fever with thrombocytopenia syndrome. PLoS Negl. Trop. Dis. 11:e0005746. doi: 10.1371/journal.pntd.0005746
Li, Z., Bao, C., Hu, J., Liu, W., Wang, X., Zhang, L., et al. (2016). Ecology of the tick- borne Phlebovirus causing severe fever with thrombocytopenia syndrome in an endemic area of China. PLoS Negl. Trop. Dis. 10:e0004574. doi: 10.1371/journal.pntd.0004574
Liu, F., Ling, Z., Xiao, Y., Yang, Q., Wang, B., Zheng, L., et al. (2017). Alterations of urinary microbiota in type 2 diabetes mellitus with hypertension and/or hyperlipidemia. Front. Physiol. 8:126. doi: 10.3389/fphys.2017.00126
Liu, J., Wang, L., Feng, Z., Geng, D., Sun, Y., and Yuan, G. (2017). Dynamic changes of laboratory parameters and peripheral blood lymphocyte subsets in severe fever with thrombocytopenia syndrome patients. Int. J. Infect. Dis. 58, 45–51. doi: 10.1016/j.ijid.2017.02.017
Liu, K., Zhou, H., Sun, R. X., Yao, H. W., Li, Y., Wang, L. P., et al. (2015). A national assessment of the epidemiology of severe fever with thrombocytopenia syndrome. China. Sci. Rep. 5:9679. doi: 10.1038/srep09679
Liu, Q., He, B., Huang, S. Y., Wei, F., and Zhu, X. Q. (2014). Severe fever with thrombocytopenia syndrome, an emerging tick-borne zoonosis. Lancet Infect. Dis. 14, 763–772. doi: 10.1016/S1473-3099(14)70718-2
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Ministry of Health China (2011). Guideline for prevention and treatment of severe fever with thrombocytopenia syndrome (2010 version). Chin. J. Clin. Infect. Dis. 4, 193–194.
Morgan, X. C., Kabakchiev, B., Waldron, L., Tyler, A. D., Tickle, T. L., Milgrom, R., et al. (2015). Associations between host gene expression, the mucosal microbiome, and clinical outcome in the pelvic pouch of patients with inflammatory bowel disease. Genome Biol. 16:67. doi: 10.1186/s13059-015-0637-x
Ning, Y. J., Feng, K., Min, Y. Q., Cao, W. C., Wang, M., Deng, F., et al. (2015). Disruption of type I interferon signaling by the nonstructural protein of severe fever with thrombocytopenia syndrome virus via the hijacking of STAT2 and STAT1 into inclusion bodies. J. Virol. 89, 4227–4236. doi: 10.1128/JVI.00154-15
Palm, N. W., de Zoete, M. R., Cullen, T. W., Barry, N. A., Stefanowski, J., Hao, L., et al. (2014). Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 158, 1000–1010. doi: 10.1016/j.cell
Park, S. W., Ryou, J., Choi, W. Y., Han, M. G., and Lee, W. J. (2016). Epidemiological and clinical features of severe fever with thrombocytopenia syndrome during an outbreak in South Korea, 2013-2015. Am. J. Trop. Med. Hyg. 95, 1358–1361. doi: 10.4269/ajtmh.16-0251
Qin, J., Li, Y., Cai, Z., Li, S., Zhu, J., Zhang, F., et al. (2014). A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60. doi: 10.1038/nature11450
Qin, N., Yang, F., Li, A., Prifti, E., Chen, Y., Li, L., et al. (2014). Alterations of the human gut microbiome in liver cirrhosis. Nature 513, 59–64. doi: 10.1038/nature13568
Ritze, Y., Bárdos, G., Claus, A., Ehrmann, V., Bergheim, I., Schwiertz, A., et al. (2014). Lactobacillus rhamnosus GG protects against non-alcoholic fatty liver disease in mice. PLoS One 9:e80169. doi: 10.1371/journal.pone.0080169
Silvas, J. A., and Aguilar, P. V. (2017). The Emergence of Severe Fever with Thrombocytopenia Syndrome Virus. Am. J. Trop. Med. Hyg. 97, 992–996. doi: 10.4269/ajtmh.16-0967
Silvas, J. A., Popov, V. L., Paulucci-Holthauzen, A., and Aguilar, P. V. (2015). Extracellular vesicles mediate receptor-independent transmission of novel tick-borne bunyavirus. J. Virol. 90, 873–886. doi: 10.1128/JVI.02490-15
Song, P., Zheng, N., Zhang, L., Liu, Y., Wu, Z., Bao, C., et al. (2017). Downregulation of interferon-β and inhibition of TLR3 expression are associated with fatal outcome of severe fever with Thrombocytopenia Syndrome. Sci. Rep. 7:6532. doi: 10.1038/s41598-017-06921-6
Sun, J., Chai, C., Lv, H., Lin, J., Wang, C., Chen, E., et al. (2014). Epidemiological characteristics of severe fever with thrombocytopenia syndrome in Zhejiang Province. China. Int. J. Infect. Dis. 25, 180–185. doi: 10.1016/j.ijid.2014.02.022
Sun, J., Lu, L., Wu, H., Yang, J., Ren, J., and Liu, Q. (2017). The changing epidemiological characteristics of severe fever with thrombocytopenia syndrome in China, 2011-2016. Sci. Rep. 7:9236. doi: 10.1038/s41598-017-08042-6
Sun, Y., Qi, Y., Liu, C., Gao, W., Chen, P., Fu, L., et al. (2014). Nonmuscle myosin heavy chain IIA is a critical factor contributing to the efficiency of early infection of severe fever with thrombocytopenia syndrome virus. J. Virol. 88, 237–248. doi: 10.1128/JVI.02141-13
Turnbaugh, P. J., Hamady, M., Yatsunenko, T., Cantarel, B. L., Duncan, A., Ley, R. E., et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. doi: 10.1038/nature07540
Wang, B., Jiang, X., Cao, M., Ge, J., Bao, Q., Tang, L., et al. (2016). Altered fecal microbiota correlates with liver biochemistry in nonobese patients with non-alcoholic fatty Liver Disease. Sci. Rep. 6:32002. doi: 10.1038/srep32002
Wang, S., Li, J., Niu, G., Wang, X., Ding, S., Jiang, X., et al. (2015). SFTS virus in ticks in an endemic area of China. Am. J. Trop. Med. Hyg. 92, 684–689. doi: 10.4269/ajtmh.14-0008
Wilck, N., Matus, M. G., Kearney, S. M., Olesen, S. W., Forslund, K., Bartolomaeus, H., et al. (2017). Salt-responsive gut commensal modulates TH17 axis and disease. Nature 551, 585–589. doi: 10.1038/nature24628
Wu, X., Qi, X., Liang, M., Li, C., Cardona, C. J., Li, D., et al. (2014). Roles of viroplasm-like structures formed by nonstructural protein NSs in infection with severe fever with thrombocytopenia syndrome virus. FASEB J. 28, 2504–2516. doi: 10.1096/fj.13-243857
Yoo, J. R., Kim, S. H., Kim, Y. R., Lee, K. H., Oh, W. S., and Heo, S. T. (2017). Application of therapeutic plasma exchange in patients having severe fever with thrombocytopenia syndrome. Korean J. Intern. Med. doi: 10.3904/kjim.2016.194 [Epub ahead of print].
Yoshimoto, S., Loo, T. M., Atarashi, K., Kanda, H., Sato, S., Oyadomari, S., et al. (2013). Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101. doi: 10.1038/nature12347
Yu, X. J., Liang, M. F., Zhang, S. Y., Liu, Y., Li, D. X., Sun, Y. L., et al. (2011). Fever with thrombocytopenia associated with a novel bunyavirus in China. N. Engl. J. Med. 364, 1523–1532. doi: 10.1056/NEJMoa1010095
Zhang, Y. Z., He, Y. W., Dai, Y. A., Xiong, Y., Xu, J., Zheng, H., et al. (2012). Hemorrhagic fever caused by a novel Bunyavirus in China: pathogenesis and correlates of fatal outcome. Clin. Infect. Dis. 54, 527–533. doi: 10.1093/cid/cir804
Keywords: sever fever with thrombocytopenia syndrome (SFTS), gut microbiome, 16S rDNA sequencing, Bacteroidetes, Firmicutes, Proteobacteria, clinical symptoms, key serum enzymes
Citation: Xu H, Wei Y, Ma H, Liu Y, Zhang Y, Hu L and Li J (2018) Alterations of Gut Microbiome in the Patients With Severe Fever With Thrombocytopenia Syndrome. Front. Microbiol. 9:2315. doi: 10.3389/fmicb.2018.02315
Received: 23 June 2018; Accepted: 11 September 2018;
Published: 01 October 2018.
Edited by:
George Grant, University of Aberdeen, United KingdomReviewed by:
Keun Hwa Lee, Jeju National University, South KoreaSang Taek Heo, Jeju National University, South Korea
Keita Matsuno, Hokkaido University, Japan
Copyright © 2018 Xu, Wei, Ma, Liu, Zhang, Hu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lifen Hu, lifen.hu@163.com Jiabin Li, lijiabin@ahmu.com.cn
†Co-first authors