
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 03 July 2018
Sec. Systems Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.01462
This article is part of the Research TopicConnections Between Environmental and Human Microbial Communities: Causes and Consequences of ContaminationView all 4 articles
 Altina Lacerda Nascimento1†
Altina Lacerda Nascimento1† Adijailton Jose Souza1†
Adijailton Jose Souza1† Pedro Avelino Maia Andrade1
Pedro Avelino Maia Andrade1 Fernando Dini Andreote1
Fernando Dini Andreote1 Aline Renée Coscione2
Aline Renée Coscione2 Fernando Carvalho Oliveira3
Fernando Carvalho Oliveira3 Jussara Borges Regitano1*
Jussara Borges Regitano1*Sewage sludges generation and their disposal have become one of the greatest challenges of the 21st century. They have great microbial diversity that may impact wastewater treatment plant (WWTP) efficiency and soil quality whether used as fertilizers. Therefore, this research aimed to characterize microbial community diversity and structure of 19 sewage sludges from São Paulo, Brazil, as well as to draw their relations to sludge sources [domestic and mixed (domestic+industrial)], biological treatments (redox conditions and liming), and chemical attributes, using molecular biology as a tool. All sludges revealed high bacterial diversity, but their sources and redox operating conditions as well as liming did not consistently affect bacterial community structures. Proteobacteria was the dominant phylum followed by Bacteroidetes and Firmicutes; whereas Clostridium was the dominant genus followed by Treponema, Propionibacterium, Syntrophus, and Desulfobulbus. The sludge samples could be clustered into six groups (C1 to C6) according their microbial structure similarities. Very high pH (≥11.9) was the main sludge attribute segregating C6, that presented very distinct microbial structure from the others. Its most dominant genera were Propionibacterium > > Comamonas > Brevundimonas > Methylobacterium ∼Stenotrophomonas ∼Cloacibacterium. The other clusters’ dominant genera were Clostridium > > Treponema > Desulfobulbus ∼Syntrophus. Moreover, high Fe and S were important modulators of microbial structure in certain sludges undertaking anaerobic treatment and having relatively low N-Kj, B, and P contents (C5). However, high N-Kj, B, P, and low Fe and Al contents were typical of domestic, unlimed, and aerobically treated sludges (C1). In general, heavy metals had little impact on microbial community structure of the sludges. However, our sludges shared a common core of 77 bacteria, mostly Clostridium, Treponema, Syntrophus, and Comamonas. They should dictate microbial functioning within WWTPs, except by SS12 and SS13.
Urban centers fast growth and industrial activities intensification generate high volumes of effluents daily (Atashgahi et al., 2015), which are collected or discharged into the sewage network reaching wastewater treatment plants (WWTPs) (Shchegolkova et al., 2016). WWTPs comprehend efficient and low-cost processes to treat domestic and industrial effluents (Wen et al., 2015; Dezotti et al., 2017; Bassin et al., 2018). Among the treatments, the biological aims to degrade toxic organic compounds (petroleum derivatives, pharmaceutical compounds, and other xenobiotics) and reduce pathogenic organisms, mitigating effects on human health and environment (Seviour and Nielsen, 2010; Yang et al., 2011; Biswas and Turner, 2012; Xia et al., 2015). The residue (or by-product) of this activity, the sewage sludge, has great microbial diversity, which may vary depending on sewage origin, treatment condition (e.g., liming and redox conditions), industrial activity, among others.
Many factors may modulate microbial community structure within WWTPs, which may change from autotrophic to heterotrophic bacteria depending on effluent source, for example (Cydzik-Kwiatkowska and Zielińska, 2016). Proteobacteria phylum (21–65%) was predominant in municipal WWTPs (domestic sewage), mostly belonging to Betaproteobacteria that represents a class of microorganisms related to organic matter degradation and nutrient cycling. Other less dominant taxa were Bacteroidetes, Acidobacteria, and Chloroflexi (Nielsen et al., 2010; Wan et al., 2011; Wang et al., 2012). Proteobacteria was also abundant in industrial sewages that often have high concentrations of recalcitrant compounds originating from pharmaceutical industries, petroleum refineries, animal feed factories, and others (Ibarbalz et al., 2013; Ma et al., 2015). Biological treatment condition may be another important modulating factor. For example, microorganisms were most abundant in both anaerobic and anaerobic-aerobic than in aerobic system, but Proteobacteria was most abundant in aerobic whereas Bacteroidetes was most abundant in anaerobic bioreactors (Hu et al., 2012).
It is also clear that chemical attributes, such as pH and macronutrient contents (Tan et al., 2006; Ibarbalz et al., 2013; Gao et al., 2016; Meerburg et al., 2016); presence of toxic compounds, such as organic pollutants and heavy metals (Bettiol and Ghini, 2011; Balcom et al., 2016); and biological treatment (redox) conditions (Hu et al., 2012) can directly impact sludge bacterial community structure. In Brazil, sulfur oxidoreductive bacteria community were composed by 22 families, which could be clustered by sludge sources and chemical attributes, such as S, K, Zn, Mn, P, and N (Meyer et al., 2016).
Despite the relevance of the microorganisms, the literature in this field presents some shortcomings. First, there are several studies addressing sludge microbial community structure in WWTPs, but they often regard a small number of samples. Second, current knowledge was attained employing mainly laboratory bioreactors and pilot systems (Ahmed, 2012; Saia et al., 2016), but controlled operating conditions (temperature, aeration, and effluent flow) shorten microbial community diversity (Muszyński et al., 2013, 2015). Third, several studies used conventional techniques, but only 60–90% of bacteria population are cultivable. The emergence of molecular techniques allowed better characterization of microbial structure and function directly in the environment (Liaw et al., 2010; Tomazetto and Oliveira, 2013; Lee et al., 2015), as well as better description of microbial community ecological role (Dezotti et al., 2017). However, these information are still scarce under realistic conditions (Biswas and Turner, 2012; Bassin et al., 2018), and even more in tropical countries.
Therefore, this research work aimed to evaluate whether microbial community structure of several sewage sludges from São Paulo State, Brazil, is related to WWTP conditions, such as sewage source [domestic or mixed (domestic+industrial)], biological treatment (redox) conditions, liming, urbanization, and industrial activity; as well as to sludge chemical attributes. It would supply useful information about hygiene measures needed and/or potential contamination resulting from sludge application as soil amendment.
Sewage sludge samples were collected from 19 WWTPs of São Paulo State, Brazil. Five samples (SS6, SS7, SS11, SS12, and SS13) were collected from metropolitan area of São Paulo City, the most urbanized and industrialized region within São Paulo State, whereas the others were collected from other municipalities (Table 1). Sample collection was performed at the sludges dewatering points, as described by EPA SW-865.
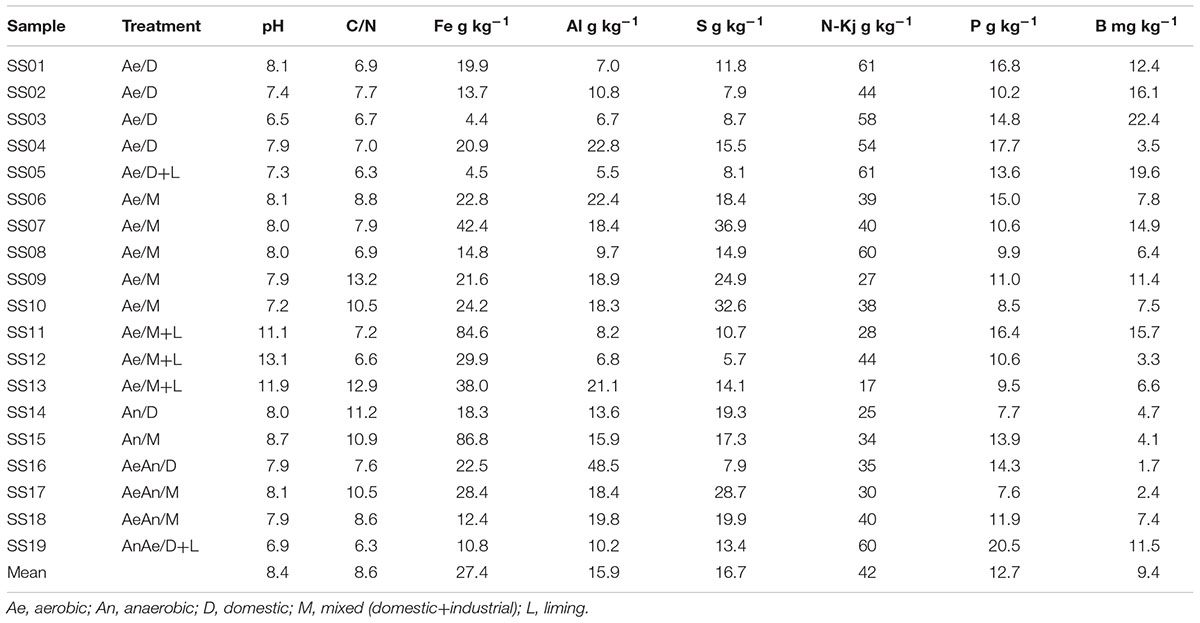
TABLE 1. Sewage sludges sources and treatments as well as main chemical attributes.
For this purpose, three samples were collected from each WWTP. Each sample was composed of five subsamples (200 g) taken in 10-min intervals, mixed, and properly homogenized. They were conditioned in glass bottles and refrigerated until analysis according CONAMA Resolution 375/2006 (BRASIL, 2006).
Moisture was determined according to EPA-SW 846. For this, sludge samples of 100 g were oven dried at 65°C, for 48 h. pH was measured using 2 g of moist sample and 20 ml of deionized water, which was stirred for 5 min at 220 rpm and rested for 30 min. For total inorganic N, 5 g of moist samples were distilled with 50 ml of 1.0 mol L-1 KCl, 0.2 g of MgO, and 0.2 g of Devarda alloy, which were taken in 5 mL of 20 g L-1 H3BO3 and titrated with 0.0025 mol L-1 H2SO4 (Bremner, 1996). Nitrite and nitrate were determined according to Mulvaney (1996). For organic N (N-Kj), 0.05 g of oven dried samples were mixed with 3 mL of concentrated H2SO4, placed in a digester block ( ± 360° C) for 3 h, distilled with 20 mL of 10 mol L-1 NaOH, which were also taken in 20 mL of 20 g L-1 H3BO3 and then titrated with 0.0025 mol L-1H2SO4 (American Public Health Association [APHA], 2005). Organic carbon (OC) was determined by the K2Cr2O7 method (Nelson and Sommers, 1996). Ca, K, P, Mg, S, Cu, Fe, Ni, Mn, Mo, Si, Zn, Al, As, Ba, Cd, Cr, Pb, Hg, and Na were extracted in microwave oven, according to EPA (2007). K and Na were quantified by flame photometry and the other elements by inductively coupled plasma atomic emission spectrometry (ICP-OES).
For total DNA, 0.4 g of each sewage sludge sample was extracted individually using MoBio Power Soil DNA Isolation Kit (MoBio, United States), according to manufacturer’s instructions. Integrity of the extracted DNA was checked by electrophoresis (1% agarose gel), which was stained with ethidium bromide and visualized under ultraviolet light.
DNA sequencing was performed by Illumina MiSeq platform and library preparation based on Nextera XT index kit (Illumina, United States), targeting the V4 region of the 16S rRNA gene. This was amplified using a mixture of 4-Forward and 4-Reverse primers with pre-adapters (Supplementary Table S1). For the PCR reaction (final volume of 25 μL), 3.0 μL of PCR Buffer, 2.5 μL of MgCl2 (50 mM), 2.0 μL of DNTPs (2.5 mM), 0.1 μL of each primer mix, 0.3 μL Taq DNA polymerase (0.05 U/μL), 16 μL mili-Q water and 1.0 μL template DNA were utilized. Amplification conditions involved initial denaturation at 95°C for 3 min, 30 cycles at 95°C for 45 s, 57°C for 1 min: 45 s; 72°C for 1 min; followed by a final extension at 72°C for 4 min (Caporaso et al., 2011). PCR products were confirmed by electrophoresis in agarose gel (1%) and resulted in amplified fragments of ∼430 bp. Amplified DNA was then purified with QiaQuick PCR kit, quantified by spectrophotometry (ND-1000), and PCR products stored (-20°C) for sequencing.
After DNA purification, another PCR reaction was performed to bind adapters (an index pair) to identify sequence origin. This consisted of 3.0 μL of PCR buffer, 2.5 μL of MgCl2 (50 mM), 2.0 μL of DNTPs (2.5 mM), 5 μL of each adapter (index), 0.3 μL of Taq DNA polymerase (0.05 U/μL), 17.2 μL of mili-Q water, and 15 μL of previous reaction product (final volume = 50 μL). Amplification conditions consisted of 95°C for 3 min, five cycles at 95°C for 45 s, 57°C for 1 min: 45 s; 72°C for 1 min; followed by a final extension at 72°C for 4 min. Sequencing was carried out at the University of São Paulo (USP/ESALQ), by the Animal Biotechnology Laboratory within the Animal Science Department.
Quantitative Insights into Microbial Ecology (QIIME) program was used for DNA sequencing analysis (Caporaso et al., 2010). Sequences quality was set at 20. Removal of poor quality sequences, primers, barcodes, and adapters were performed with CLC Genomics Workbench 6 (CLCbio). Operational taxonomic units (OTUs) were grouped in 3% distance level (97% of similarity) and classification was performed by the Ribosomal Database Project (RDP Classifier). OTUs were also used to estimate ecological parameters using Chao 1, Simpson, and Shannon diversity indexes. Clustering of the samples was performed by principal coordinate analysis (PCoA) (Ramette, 2007), and tested by similarity analysis (ANOSIM) on Past® software (v.3.2) (Hammer et al., 2001). ANOSIM was also used to verify sample similarities according sludge sources, biological (redox) treatments, and liming.
Relationship between bacterial community composition and sludges sources, treatments, and chemical attributes (pH, moisture, N-NH4+, N-NO2-/NO3-, organic N (N-Kjeldahl = N-Kj), organic carbon (OC), K, Ca, Fe, P, S, Mg, Na, Cd, Cr, Cu, Hg, Mn, Mo, Ni, Pb, Se, Zn, Al, As, and Ba) were settled by redundancy analysis (RDA) on Canoco® software (v.4.5). Graphics were plotted on Origin® software (v.10.5), but heatmap graphical scales were built in R software, using “gplots” and “RColorBrewer” packages1.
Samples identification and main chemical attributes affecting microbial community and their clustering were presented in Table 1. The other chemical attributes were summarized from a previous thesis work (Nascimento, 2015) and presented as supplementary material (Supplementary Table S2). Thirteen samples underwent aerobic (SS1 to SS13) whereas the other six (SS14-SS19) underwent either strictly anaerobic or combined aerobic-anaerobic treatments during biological digestion. Eight samples were collected from domestic (SS1-SS5, SS14, SS16, and SS19) whereas the others were collected from mixed sewers. Only five samples were limed (SS5, SS11, SS12, SS13, and SS19).
A total of 7,219,247 16S RNA gene sequences were attained. After removal of low quality sequences (cut level = 3%), OTUs matrixes showed that all sludges presented high diversity indexes (Supplementary Table S3). Although sequencing would contain inactive (dormant and dead) microorganisms, it should not impact diversity as verified by Liang et al. (2017).
RDP Classifier identified 68 phyla, 164 classes, and 665 genera of bacteria. The most abundant phyla were Proteobacteria > Bacteroidetes > Firmicutes, corresponding to >73% of the DNA sequences (Figure 1A); whereas the most abundant classes were Saprospirae > Betaproteobacteria > Bacteroidia > Clostridia > Deltaproteobacteria (Figure 1B). In addition, Betaproteobacteria was the most abundant class within the Proteobacteria phylum (∼37% of the sequences), followed by Deltaproteobacteria (∼26%), Alphaproteobacteria (∼16%), and Gammaproteobacteria (∼11%); whereas Saprospirae was the most abundant class within the Bacteroidetes (∼46%), followed by Bacteroidia (∼36) and Flavobacteria (∼3%) (Figures 1A,B). Within the Firmicutes, the most abundant classes were Clostridia (∼87%) and Bacilli (∼9%) (Figures 1A,B). Finally, the most abundant genera were Clostridium > Treponema > Propionibacterium > Syntrophus > Desulfobulbus > Brevundimonas > Paludibacter > Cloaci-bacterium > Methylobacterium (Figure 1C). Despite distinctions in sewage sources and treatments, their bacterial community presented a common core of 77 genera, being Clostridium, Treponema, Syntrophus, and Comamonas the most abundant ones (Supplementary Figure S1).
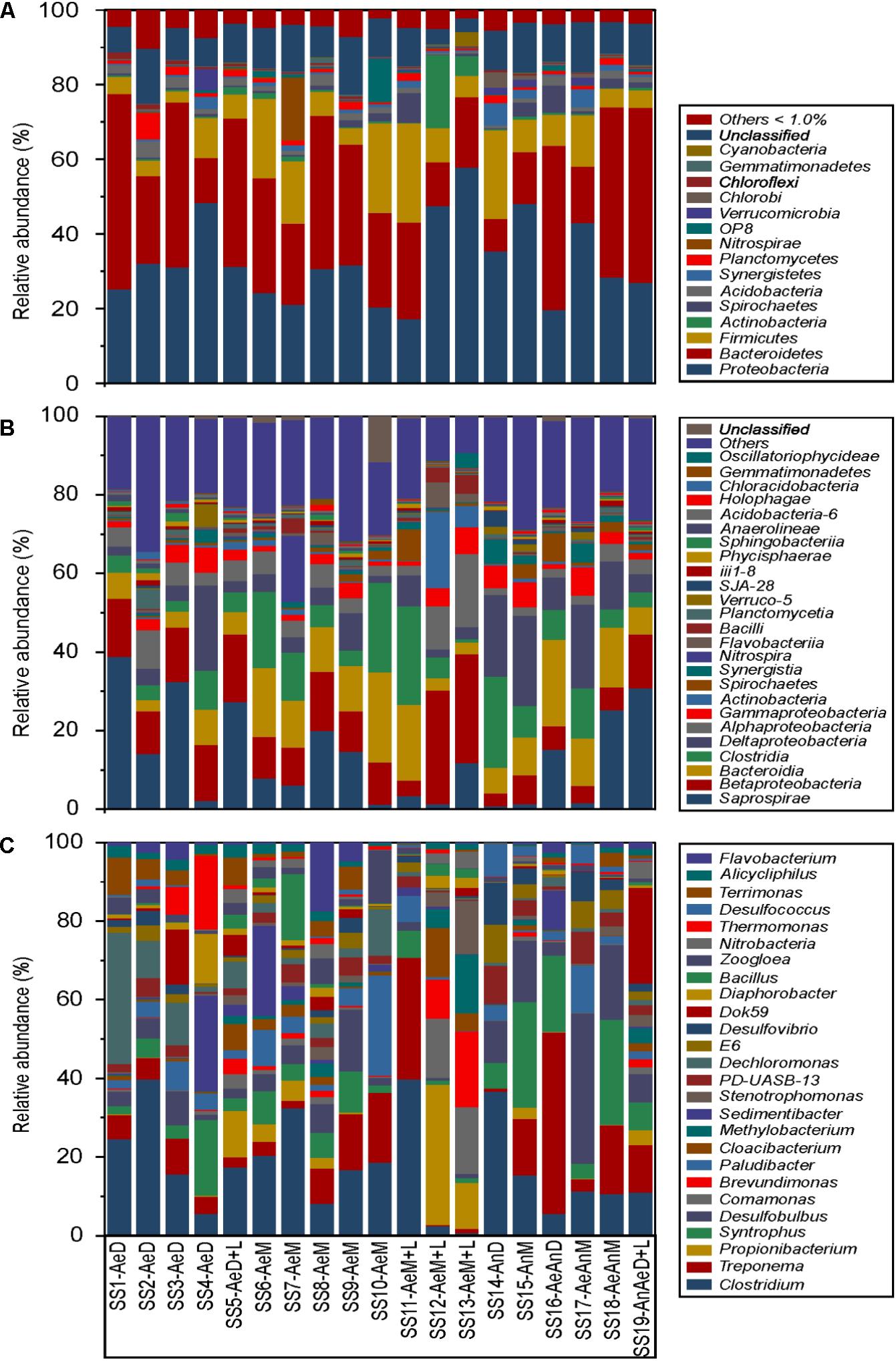
FIGURE 1. Relative microbial community abundance of 19 sewage sludges from São Paulo State, Brazil. (A, phyla; B, classes; C, genera; Others, members with relative abundance < 1%).
The sludge samples could be grouped in six clusters according to PCoA: C1 (SS1, SS2, and SS3), C2 (SS9 and SS16), C3 (SS11 and SS18), C4 (SS4, SS5, SS6, SS7, SS8, and SS19), C5 (SS10, SS14, SS15, and SS17), C6 (SS12 and SS13). Its main two coordinates explained 36.7% of sludges’ bacterial community structures (Figure 2). This result was also validated by similarity analysis of their bacterial communities (Table 2).
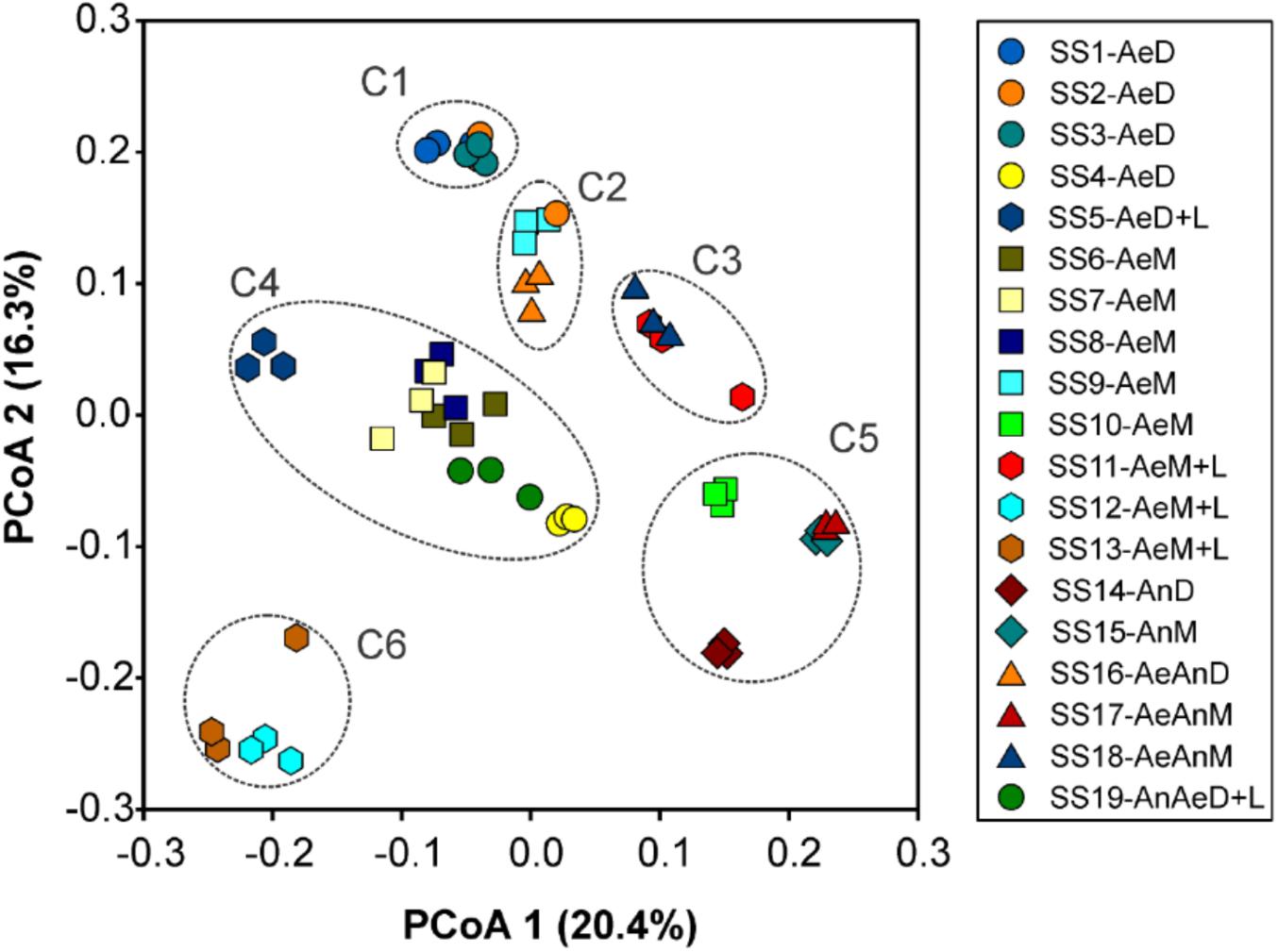
FIGURE 2. Principal coordinate analysis (PCoA) for bacterial community structure of 19 sewage sludges from São Paulo State, Brazil (n = 3). Axis values indicated percentage of variance.
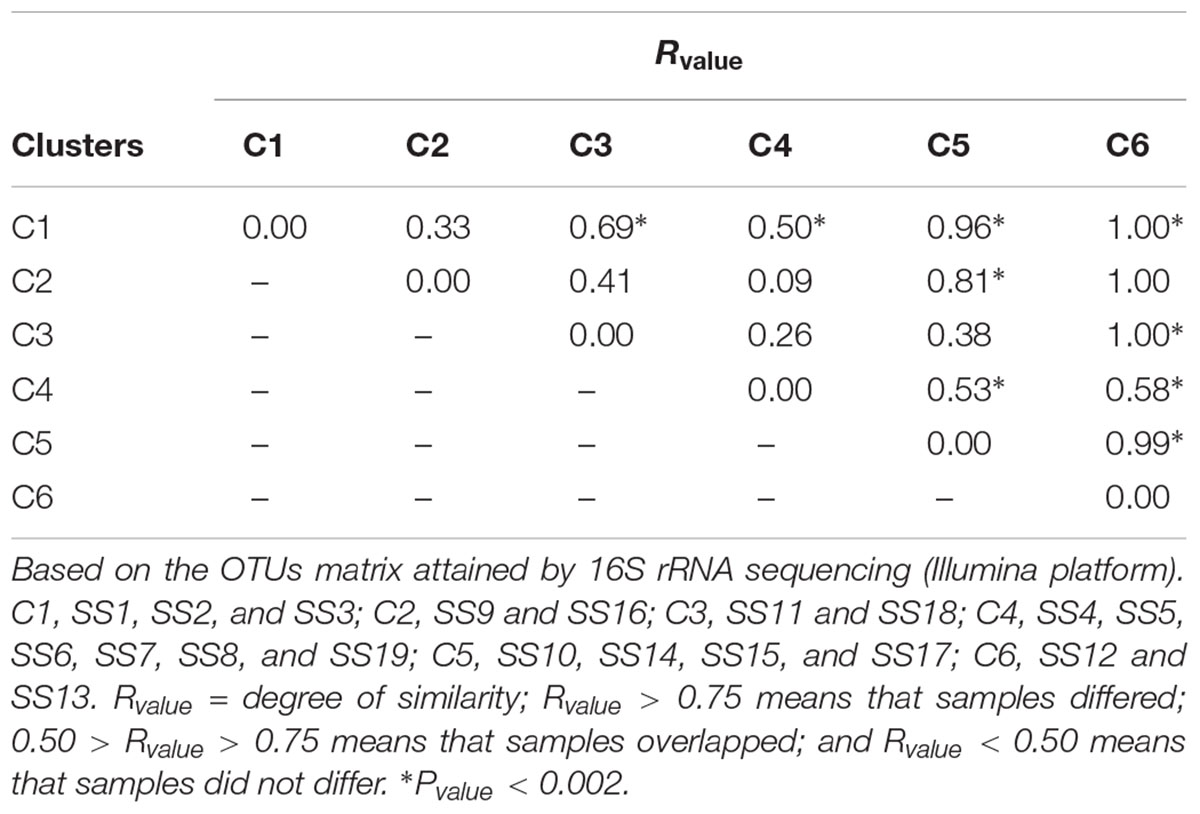
TABLE 2. Analysis of similarity (ANOSIM) for microbial community structure and clusters formation for 19 sewage sludges from São Paulo State, Brazil.
C6 showed bacterial community very distinct from the others, with relative dominance of Propionibacterium, Comamonas, Brevundimonas, Methylobacterium, Stenotrophomonas, and Cloacibacterium (Figure 3). The other clusters (C1-C5) generally presented great abundance of Clostridium, Treponema, Syntrophus, and Desulfobulbus, except that C1 showed low abundance of Syntrophus and high abundance of Dechloromonas; C2 showed relative high abundance of Sedimentibacter; C3 showed relative high abundance of Paludibacter; C4 showed relative high abundance of Sedimentibacter and also of Dok59 and Bacillus; and C5 showed relative high abundance of Paludibacter, PD-UASB-13, Desulfovibrio, and E6 (Figure 3).
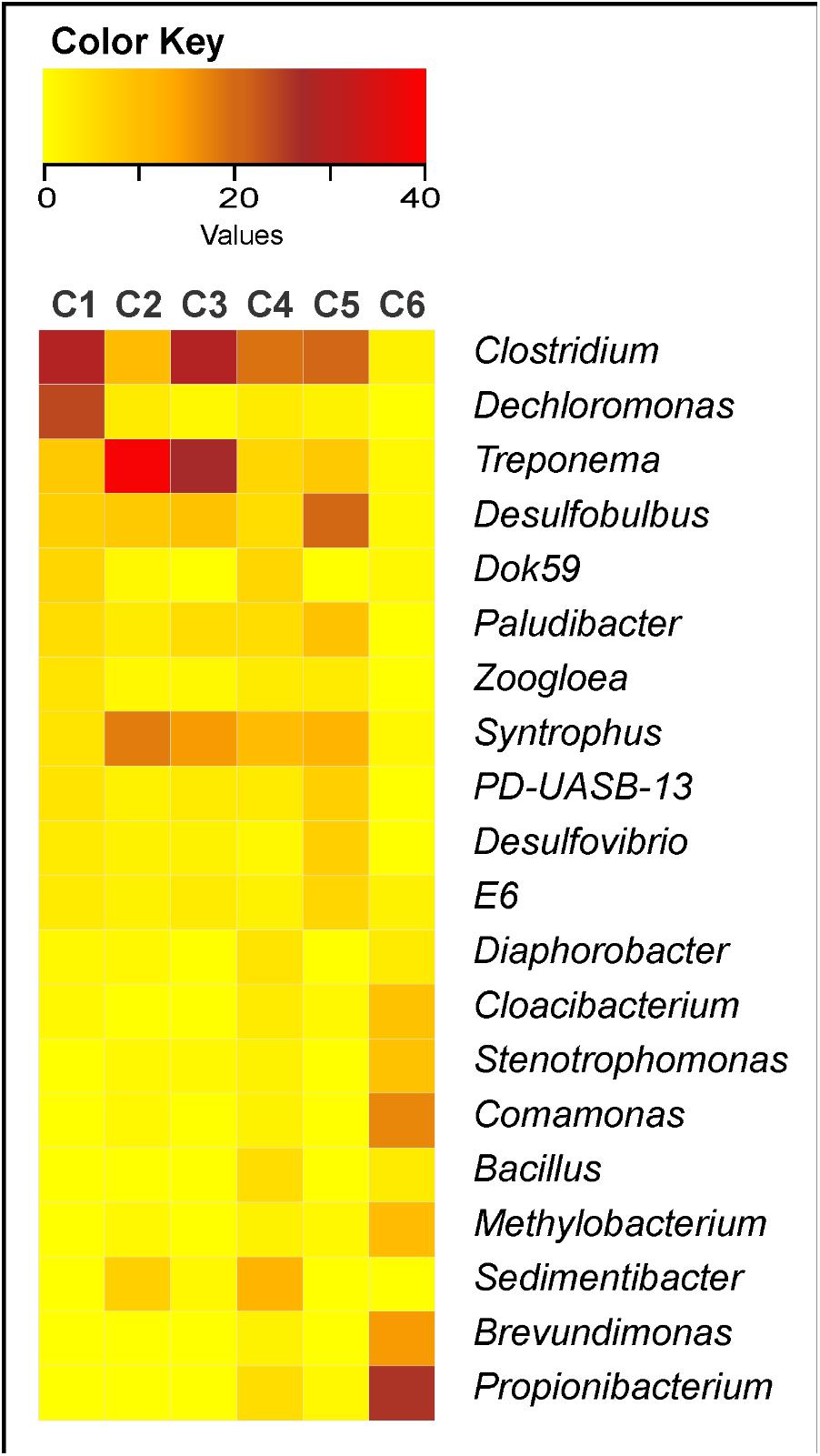
FIGURE 3. Heatmap of the 20 most abundant bacterial genera found in 19 sewage sludges from São Paulo State, Brazil. C1, cluster 1 (n = 3 samples); C2, cluster 2 (n = 2 samples); C3, cluster 3 (n = 2 samples); C4, cluster 4 (n = 6 samples); C5, cluster 5 (n = 4 samples); and C6, cluster 6 (n = 2 samples). Red color, most abundant genus; Yellow, least abundant genus.
Sewage sources (domestic or mixed) and biological treatments (redox conditions) did not affect consistently the bacterial community structuring (Table 3), suggesting that clusters were formed due to other factors, likely related with sludges chemical attributes as suggested by RDA (Figure 4). In fact, pH (λ = 0.11, P-value < 0.002), Fe (λ = 0.07, P-value < 0.002); B and Mg (λ = 0.06, P-value < 0.002); Na (λ = 0.05, P-value < 0.002); and P, Ba, organic N (N-Kj), and Ca (λ = 0.04, P-value < 0.002) contents were the sludge attributes most related to microbial community structuring and clustering; whereas organic carbon (OC), inorganic N (in the different forms), Hg, Se, and As contents, C/N ratio, and moisture were not correlated with sludges bacterial community structures (λ < 0.01 and P-value > 0.05) (Supplementary Table S2).
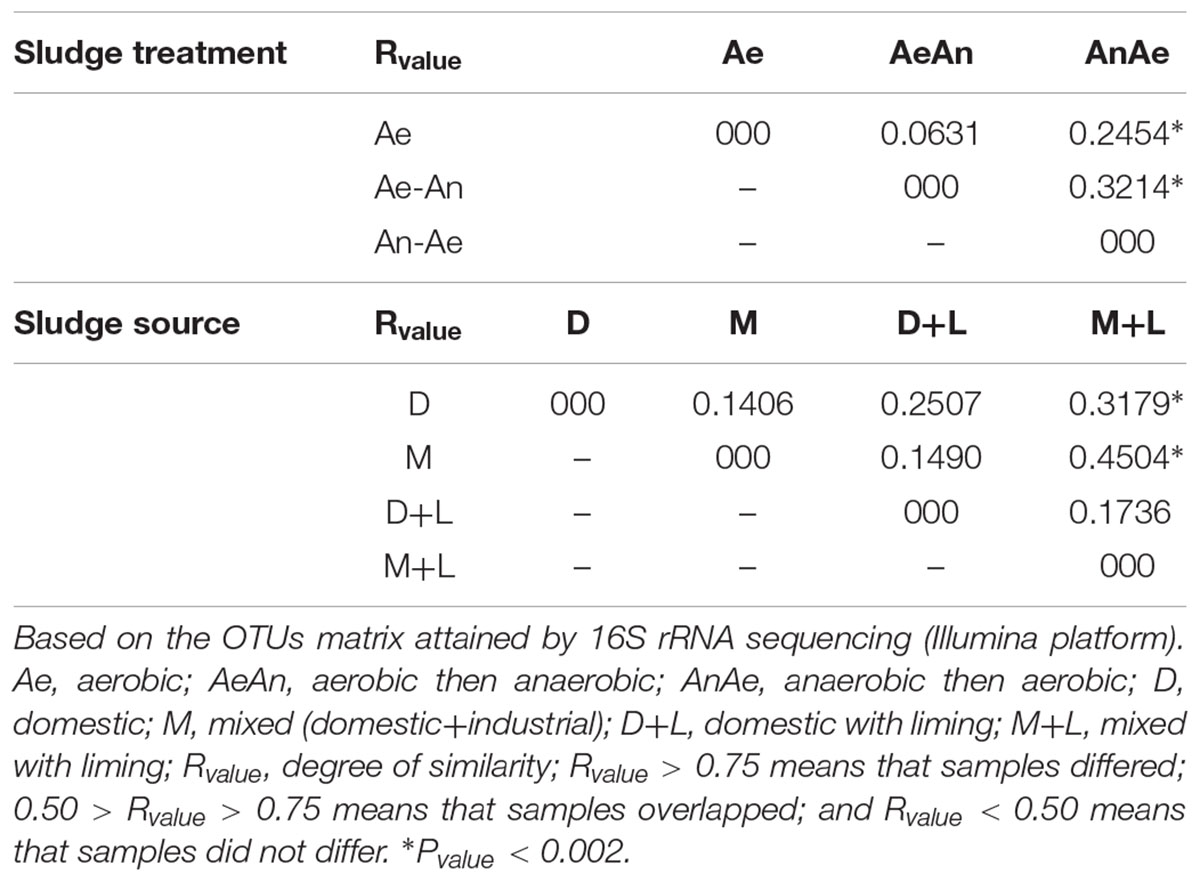
TABLE 3. Analysis of similarity (ANOSIM) for microbial community structure as affected by sources and treatments of 19 sewage sludges from São Paulo State, Brazil.
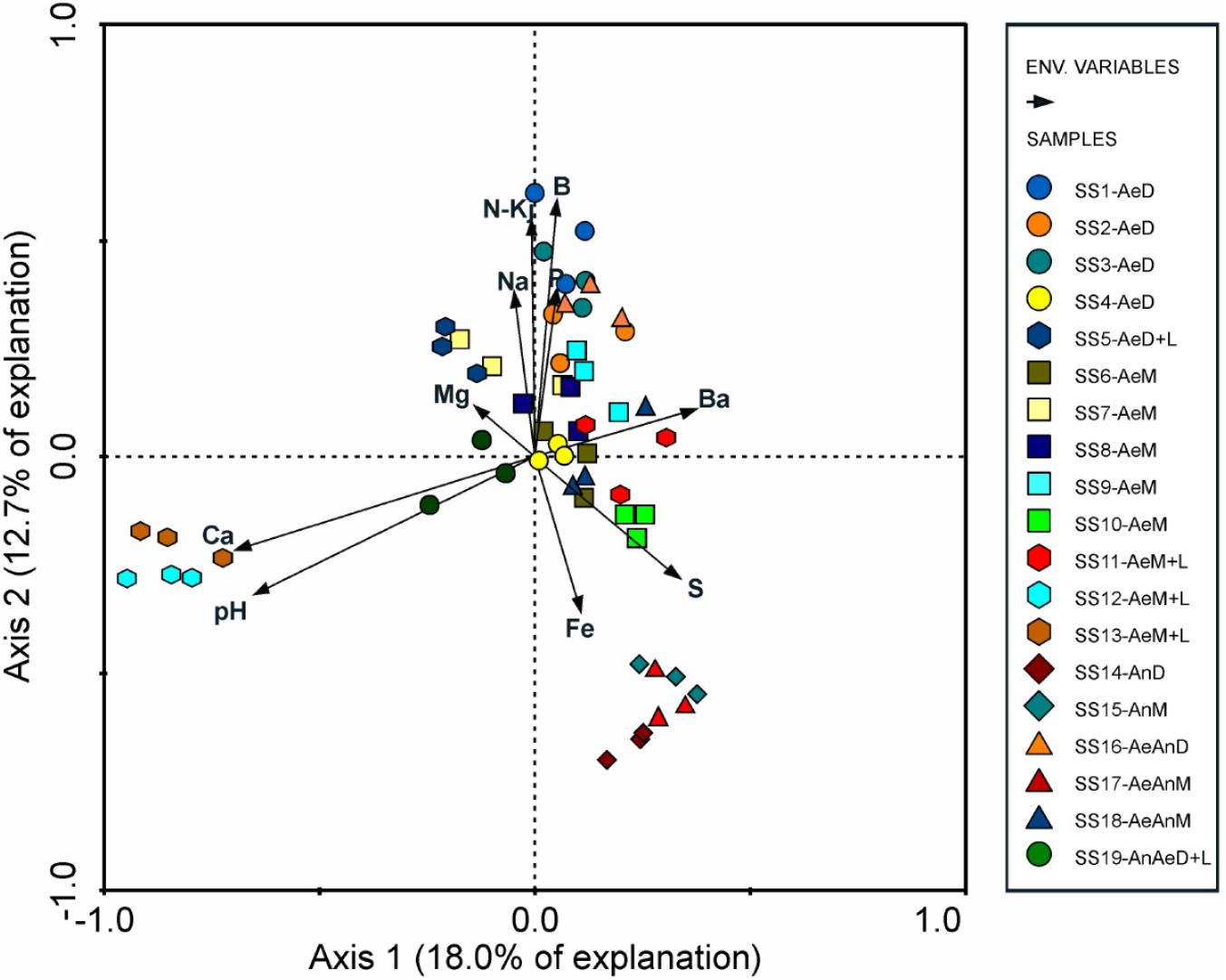
FIGURE 4. Redundancy analysis (RDA) between chemical attributes and bacterial community of 19 sewage sludges from São Paulo State, Brazil (n = 3 replicates).
WWTPs bacterial community exhibited low variation at higher taxonomical levels (e.g., phylum) even for distinct geographic regions and sludge treatments (Philippot et al., 2010; Ibarbalz et al., 2013; Hatamoto et al., 2017) (Figure 1A). In all samples, independently of sewer operating condition, the most abundant phyla were Proteobacteria > Bacteroidetes > Firmicutes (Figure 1A). Similar results were reported for sludges from China (Zhang et al., 2012; Shu et al., 2015b; Gao et al., 2016; Liang et al., 2017). However, the literature shows some contrasting results. Meerbergen et al. (2017) found predominantly Proteobacteria, Bacteroidetes, and Actinobacteria for domestic sludges, but Planctomycetes, Chloroflexi, Acidobacteria, and Chlorobi for industrial sludges. Proteobacteria usually predominated in domestic sewage sludges, corresponding from 30 to 65% of the total sequences (Liang et al., 2017; Meerbergen et al., 2017), as well as in various other environments, such as soil (Roesch et al., 2007; Spain et al., 2009; Sun et al., 2015) and rhizosphere (Jiang et al., 2016). Proteobacteria usually presented wide diversity and metabolic capacity, acting in important environmental functions such as the cycles of C, N, S, and P (Friedrich et al., 2005; Meyer et al., 2016). Bacteroidetes were often reported as proteolytic bacteria, involved in degrading protein to volatile phenolic acids and ammonia (NH3) (Yi et al., 2014). Their abundance was correlated with total solid contents when submitted to anaerobiosis (Liu et al., 2016). Firmicutes were often widely distributed in anaerobic sludge treatment systems (Yang et al., 2014) and were versatile in degrading a vast array of environmental substrates (Liu et al., 2016). They may act on metabolic pathways responsible for producing volatile fatty acids, which can be used by other microbial groups.
The most abundant classes were Saprospirae > Betaproteo-bacteria > Bacteroidia > Clostridia > Deltaproteobacteria > Alphaproteobacteria > Gammaproteobacteria > Actinobacteria > Spirochaetes (Figure 1B). Liang et al. (2017) and Shu et al. (2015a) reported relative high abundance of Betaproteobacteria, which is often associated with organic matter degradation and S cycle (Friedrich et al., 2005; Takai et al., 2005). In Denmark, however, several studies reported low occurrence of Saprospirae in full scale WWTPs (Nielsen et al., 2012; Kong et al., 2007; Muszyński et al., 2015). The members of this class were predominant in marine environments, but could also be found in fresh water and sewage sludges degrading complex organic compounds (Nielsen and McMahon, 2014).
Lower taxonomic levels (e.g., genus) showed higher bacterial community differentiation among WWTPs (Figure 1C), corroborating with the literature (Philippot et al., 2010; Ibarbalz et al., 2013). The most abundant genera were Clostridium > Treponema > Propionibacterium > Propionibacterium > Syntrophus > Desulfobulbus > Comamonas > Brevundimonas > Paludibacter > Cloacibacterium > Methylobacterium > Sedimentibacter > Stenotrophomonas (Figure 1C). A great diversity of bacterial genera were also described in the literature (Lee et al., 2015), which several times differed from ours (Ibarbalz et al., 2013; Stiborova et al., 2015; Gao et al., 2016). It could be explained by the fact that WWTPs comprise open and very dynamic systems allowing rapid succession among microbial community members during spatial and temporal scales (Shu et al., 2015a). Nevertheless, our samples showed a common nucleus of 77 bacteria, represented mostly by Clostridium, Treponema, Syntrophus, and Comamonas (Supplementary Figure S1). Gao et al. (2016) identified a common nucleus of 177 genera for sewage sludges from China. This shared core of bacteria is usually responsible for the main functions in the environment (Shu et al., 2015b). Several pathogenic bacteria, such as Clostridium, Treponema, Stenotrophomonas, Bacillus, Mycobacterium, and Acinetobacter were also identified, in accordance to Stiborova et al. (2015).
Sewage sources (domestic or mixed) and biological treatments (redox conditions) did not affect microbial community structure (Table 3) and diversity (Chao1, Simpson and Shannon) (Supplementary Table S3), at least not consistently, similarly to Hai et al. (2014). However, Gao et al. (2016) found that biological treatment (redox conditions) influenced microbial community, which was more diverse in aerobic tanks; whereas Meerburg et al. (2016) reported structural differences in the bacterial community of domestic and industrial sludges. Gao et al. (2016) found a common core of 177 bacteria genera to their samples and we found a common core of 77 bacteria that corresponded to 85% of the identified sequences. They considered only seven samples from strictly domestic sewers that would explain their greater similarity. Normally, bacterial communities of domestic sewers are more diverse due to its larger fraction of readily degradable organic material (Meerbergen et al., 2017). Industrial sewers receive recurrent discharges of more recalcitrant and toxic pollutants (Gao et al., 2016), such as heavy metals and antimicrobial agents (Bettiol and Ghini, 2011; Balcom et al., 2016), thus limiting microbial diversity. Hu et al. (2012) reported high similarity between bacterial communities of five sludges from China, whereas Zhao et al. (2014) reported substantial disparity, mainly due to their spatial variation and biological composition. Meyer et al. (2016) also reported significant variation in the structure of S oxidoreductive bacteria from south Brazil.
On the other side, certain chemical attributes showed direct connections to sludge microbial community structures (Figure 4 and Supplementary Table S2), favoring samples segregation in clusters (Figure 2 and Table 2). High pH values (≥11.9) resulted from liming were responsible for segregating C6 (SS12 and SS13) and enhancing Ca contents (Figure 4). Its most abundant phyla were Actinobacteria, Proteobacteria, and Bacteroidetes; whereas the most abundant genera were Propionibacterium, Comamonas, Brevundimonas, Methylobacterium, Stenotrophomonas, and Cloacibacterium (Figure 3). Other limed samples (SS5, SS11, and SS19) presented lower pH (Table 1) and; therefore, very distinct microbial structure from C6. Despite having similar operating conditions as SS12 and SS13, SS11 also showed slightly lower pH as well as lower Cu and Zn and higher Fe and Pb contents (Table 1). It has been demonstrated that 1 pH-unit may considerably affect bacterial community structure and composition (Fierer and Jackson, 2006). Gao et al. (2016) also observed distinct phylogeny (Proteobacteria, Bacteroidetes, Acidobacteria, Chloroflexi, and Firmicutes) at lower pH values (∼8.0). Liming to high pH values usually decreases microbial community diversity (Blaszczyk and Krzysko-Lupicka, 2013; Farzadkia and Bazrafshan, 2014), being an important tool promoting sludge hygienization (i.e., pathogens control). In our case, high pH did not affect microbial diversity (Supplementary Table S3) but affected its structure inclusive favoring extremotolerant bacterial groups, such as Actinobacteria (Figure 1A). Several studies showed that pH affected microbial community diversity and composition in soils (Rousk et al., 2009; Cho et al., 2016; Wu et al., 2016) and sewage sludges (Maspolim et al., 2015). Lauber et al. (2009) found that bacterial phyla (Acidobacteria, Actinobacteria, Bacteroidetes, and α, β, and γ-Proteobacteria) relative abundance did not depend on sludge location, but on pH instead. Therefore, pH may modulate microbial community by controlling nutrients availability and enzymatic processes that are essential to microbial metabolism (Fierer and Jackson, 2006; Madigan et al., 2016).
C5 (SS10, SS14, SS15, and SS17) segregation was related mostly to Fe but also to S, B, P, and N-kj, and contents (Figure 4). These samples underwent anaerobic treatment, except for SS10 (Table 1). Under anaerobiosis, both Fe and S have important roles in redox reactions (Ma et al., 2014), acting as final electron acceptors (Moreira and Siqueira, 2006; Shrestha et al., 2009; Alexandre et al., 2012). Fe is reduced to its most soluble form (Fe3+ → Fe2+) (Shrestha et al., 2009) and reducing bacteria are important mediators of C and N transformations (Tan et al., 2006; Wang et al., 2009; Ding et al., 2014). Several bacteria associated with Fe reduction were identified, such as Acidithiobacillus, Ferrimicrobium, and Nitrospira (Figure 1B). In parallel, S reduction generates energy in anaerobic environments (Aida et al., 2014); and it is crucial in structuring microbial community. It could be ratified by relative high abundance of Desulfobulbus and presence of Desulfovibrio as well as Desulfococcus, Desulforhabdus, and Desulfovirga in the samples (Figure 1C). Desulfovibrio, Desulforhabdus, and Smithella were very efficient in removing S from anaerobically treated sludges (Aida et al., 2015). SS10 was the only sample having aerobic treatment and showed the highest S concentration (Table 1). C5 samples had lower N-Kj likely due to denitrification (Yamashita and Yamamoto-Ikemoto, 2014), resulting sludges with slightly higher C/N ratios (10.8 versus 8.0) (Table 1). Systems operated initially under anaerobic followed by aerobic conditions usually contribute most to N loss since they warranty anoxic denitrification and aerobic nitrification, thus converting ammonia (NH4+) to gaseous N (N2, NO2, and N2O) (Ruiz et al., 2006; Kassab et al., 2010; Yao et al., 2013a,b; Zhang et al., 2014). These samples also showed low P and B contents (Table 1).
All C1 samples (SS1, SS2, and SS3) derived from domestic sewers, aerobically treated and without liming (Table 1). They presented higher B, P, and N-Kj as would be expected from their higher organic matter pool, thus generating sludges with lower C/N ratios (Table 1). They also presented high Na and low Fe and Al contents (Table 1), which would be expected by their source nature (domestic). The most abundant genera were Clostridium > Dechloromonas >> Treponema > Desulfobulbus > Dok59 (Figure 3). Likewise, Clostridium, Treponema, and Desulfobulbus were also abundant in C2 and C3 (Figure 3). High abundance of Clostridium in domestic sludges was expected since it represents 10–40 % of human intestinal microbiota (Manson et al., 2008; Lopetuso et al., 2013). Clostridium was usually the most abundant genera in activated sludges, whereas Desulfobulbus and Dechloromonas were often associated with nutrients (such as N and S) removal from WWTPs (Aida et al., 2015).
Toxic inorganic elements, such as heavy metals (excluding the micronutrients), did not impact microbial community structure (Supplementary Table S2 and Figure 4). These element contents were below those set by the Brazilian legislation for sludge use in agriculture (CONAMA 375/2006). Only one sample (SS7) exceeded threshold concentration for Ni and three (SS1, SS12, and SS13) for Zn, but both are plant micronutrients. On the other side, Cd, Cr, and Ag inhibited important microorganisms for biological treatment, thus impacting sludge bacterial community (Wells et al., 2011).
All sewage sludges presented high bacterial diversity. Their sources and biological treatment (redox) conditions did not consistently affect bacterial community structures. Overall, Proteobacteria was the dominant phylum, followed by Bacteroidetes and Firmicutes. Their predominant classes were Betaproteobacteria (∼37%), Saprospirae (∼46%), and Clostridia (∼87%), respectively. Clostridium was the dominant genera, followed by Treponema, Propionibacterium, Syntrophus, and Desulfobulbus. Moreover, the samples were clustered into six groups according similarity of microbial community structures, which were related to their chemical attributes. High pH values (≥11.9) resulted from liming impacted mostly bacterial community structures and segregated C6, in which predominated Propionibacterium, Comamonas, Brevundimonas, Methylobacterium, and Cloacibacterium that are extremotolerant organisms. However, Clostridium, Treponema, Desulfobulbus, and Syntrophus were usually the most abundant ones in the other clusters, except that C1 presented relatively high abundance of Dechloromonas; C2 and C4 presented relatively high abundance of Sedimentibacter, and C3 and C5 presented relatively high abundance of Paludibacter. High Fe and S contents were important modulators of microbial structure for certain sludges undertaking anaerobic treatments and having relatively low N-kj, B, and P contents (C5); whereas high N-Kj, B, and P contents were important modulator for domestic, aerobically treated, and unlimed sludges having low Fe and Al contents (C1). Toxic inorganic elements, such as heavy metals (excluding micronutrients), had little impact on microbial community structure of the sludges. Nevertheless, the sludges shared a common core of 77 bacteria, being Clostridium, Treponema, Syntrophus, and Comamonas the most abundant ones.
AN contributed to sample collection and processing as well as chemical and microbiological analyses. AS contributed to overall data analyses and manuscript writing. PA performed bioinformatic analyses. FA contributed to research idealization and manuscript revision. AC performed chemical attributes analyses. FO contributed to sample collection and research idealization. JR idealized, wrote, and revised the manuscript and coordinated the research.
São Paulo State Research Foundation (FAPESP) provided the financial support and scholarship for the author AN (PROCESS 2013/00897-0).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01462/full#supplementary-material
Ahmed, Z. (2012). Microbial communities in nutrient-removing membrane bioreactors: a review. J. Environ. Sci. Technol. 5, 16–28. doi: 10.3923/jest.2012.16.28
Aida, A. A., Hatamoto, M., Yamamoto, M., Ono, S., Nakamura, A., Takahashi, M., et al. (2014). Molecular characterization of anaerobic sulfur-oxidizing microbial communities in up-flow anaerobic sludge blanket reactor treating municipal sewage. J. Biosci. Bioeng. 118, 540–545. doi: 10.1016/j.jbiosc.2014.04.011
Aida, A. A., Kuroda, K., Yamamoto, M., Nakamura, A., Hatamoto, M., and Yamaguchi, T. (2015). Diversity profile of microbes associated with anaerobic sulfur oxidation in an upflow anaerobic sludge blanket reactor treating municipal sewage. Microbes Environ. 30, 157–63. doi: 10.1264/jsme2.ME14105
Alexandre, J. R., Oliveira, M. L. F., Santos, T. C., Canton, G. C., Conceição, J. M., Eutrópio, F. J., et al. (2012). Zinco e ferro: de micronutrientes a contaminantes do solo. Nat. On Line 10, 23–28.
American Public Health Association [APHA] (2005). Standard Methods for the Examination of Water and Wastewater, 21st Edn. Washington, DC: American Public Health Association.
Atashgahi, S., Aydin, R., Dimitrov, M. R., Sipkema, D., Hamots, K., Lahti, L., et al. (2015). Impact of a wastewater treatment plant on microbial community composition and function in a hyporheic zone of a eutrophic river. Sci. Rep. 5:17284. doi: 10.1038/srep17284
Balcom, I. N., Driscoll, H., Vincent, J., and Leduc, M. (2016). Metagenomic analysis of an ecological wastewater treatment plant’s microbial communities and their potential to metabolize pharmaceuticals. F1000Research 5:1881. doi: 10.12688/f1000research.9157.1
Bassin, J. P., Dezotti, M., and Rosado, A. (2018). “Molecular biology techniques applied to the study of microbial diversity of wastewater treatment systems,” in Advanced Biological Processes for Wastewater Treatment: Emerging, Consolidated Technologies and Introduction to Molecular Techniques, eds M. Dezotti, G. Lippel, and J. P. Bassin (Cham: Springer International Publishing), 205–299. doi: 10.1007/978-3-319-58835-3_6
Bettiol, W., and Ghini, R. (2011). Impacts of sewage sludge in tropical soil: a case study in Brazil. Appl. Environ. Soil Sci. 2011:212807. doi: 10.1155/2011/212807
Biswas, K., and Turner, S. J. (2012). Microbial community composition and dynamics of moving bed biofilm reactor systems treating municipal sewage. Appl. Environ. Microbiol. 78, 855–864. doi: 10.1128/AEM.06570-11
Blaszczyk, K., and Krzysko-Lupicka, T. (2013). Microbial diversity of sewage sludge. Proc. ECOpole 7, 451–466. doi: 10.2429/proc.2013.7(2)059
Bremner, M. (1996). “Chapter 37: Nitrogen-total,” in Methods of Soil Analysis. Part 3: Chemical Methods-SSSA Book Series 5, eds D. L. Sparks, A. L. Page, P. A. Helmke, R. H. Loeppert, P. N. Soltanpour, M. A. Tabatabai, et al. (Madison, WI: Segoe Rd), 1085–1121. doi: 10.2136/sssabookser5.3.c37.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108, 4516–4522. doi: 10.1073/pnas.1000080107
Cho, S.-J., Kim, M.-H., and Lee, Y.-O. (2016). Effect of pH on soil bacterial diversity. J. Ecol. Environ. 40:10. doi: 10.1186/s41610-016-0004-1
Cydzik-Kwiatkowska, A., and Zielińska, M. (2016). Bacterial communities in full-scale wastewater treatment systems. World J. Microbiol. Biotechnol. 32:66. doi: 10.1007/s11274-016-2012-9
Dezotti, M., Lippel, G., and Bassin, J. P. (2017). Advanced biological processes for wastewater treatment: emerging, consolidated technologies and introduction to molecular techniques. Cham: Springer International Publishing.
Ding, G.-C., Radl, V., Schloter-Hai, B., Jechalke, S., Heuer, H., Smalla, K., et al. (2014). Dynamics of soil bacterial communities in response to repeated application of manure containing sulfadiazine. PLoS One 9:e92958. doi: 10.1371/journal.pone.0092958
EPA (2007). SW-846 Test Method 3051A: Microwave Assisted Acid Digestion of Sediments, Sludges, Soils, and Oils. Available at: https://www.epa.gov/hw-sw846/sw-846-test-method-3051a-microwave-assisted-acid-digestion-sediments-sludges-soils-and-oils [accessed March 5, 2018].
Farzadkia, M., and Bazrafshan, E. (2014). Lime stabilization of waste activated sludge. Health Scope 3:e16035. doi: 10.17795/jhealthscope-16035
Fierer, N., and Jackson, R. B. (2006). The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. U.S.A. 103, 626–631. doi: 10.1073/pnas.0507535103
Friedrich, C. G., Bardischewsky, F., Rother, D., Quentmeier, A., and Fischer, J. (2005). Prokaryotic sulfur oxidation. Curr. Opin. Microbiol. 8, 253–259. doi: 10.1016/j.mib.2005.04.005
Gao, P., Xu, W., Sontag, P., Li, X., Xue, G., Liu, T., et al. (2016). Correlating microbial community compositions with environmental factors in activated sludge from four full-scale municipal wastewater treatment plants in Shanghai, China. Appl. Microbiol. Biotechnol. 100, 4663–4673. doi: 10.1007/s00253-016-7307-0
Hai, R., Wang, Y., Wang, X., Li, Y., and Du, Z. (2014). Bacterial community dynamics and taxa-time relationships within two activated sludge bioreactors. PLoS One 9:e90175. doi: 10.1371/journal.pone.0090175
Hammer,Ø., Harper, D. A. T., and Ryan, P. D. (2001). Past: paleontological statistics software package for education and data analysis. Palaeontol. Electron. 4:9.
Hatamoto, M., Kaneko, T., Takimoto, Y., Ito, T., Miyazato, N., Maki, S., et al. (2017). Microbial community structure and enumeration of Bacillus species in activated sludge. J. Water Environ. Technol. 15, 233–240. doi: 10.2965/jwet.17-037
Hu, M., Wang, X., Wen, X., and Xia, Y. (2012). Microbial community structures in different wastewater treatment plants as revealed by 454-pyrosequencing analysis. Bioresour. Technol. 117, 72–79. doi: 10.1016/j.biortech.2012.04.061
Ibarbalz, F. M., Figuerola, E. L., and Erijman, L. (2013). Industrial activated sludge exhibit unique bacterial community composition at high taxonomic ranks. Water Res. 47, 3854–3864. doi: 10.1016/j.watres.2013.04.010
Jiang, L., Song, M., Yang, L., Zhang, D., Sun, Y., Shen, Z., et al. (2016). Exploring the influence of environmental factors on bacterial communities within the Rhizosphere of the Cu-tolerant plant, Elsholtzia splendens. Sci. Rep. 6:36302. doi: 10.1038/srep36302
Kassab, G., Halalsheh, M., Klapwijk, A., Fayyad, M., and Van Lier, J. B. (2010). Sequential anaerobic–aerobic treatment for domestic wastewater – a review. Bioresour. Technol. 101, 3299–3310. doi: 10.1016/j.biortech.2009.12.039
Kong, Y., Xia, Y., Nielsen, J. L., and Nielsen, P. H. (2007). Structure and function of the microbial community in a full-scale enhanced biological phosphorus removal plant. Microbiology 153, 4061–4073. doi: 10.1099/mic.0.2007/007245-0
Lauber, C. L., Hamady, M., Knight, R., and Fierer, N. (2009). Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 75, 5111–5120. doi: 10.1128/AEM.00335-09
Lee, S. H., Kang, H. J., and Park, H. D. (2015). Influence of influent wastewater communities on temporal variation of activated sludge communities. Water Res. 73, 132–144. doi: 10.1016/j.watres.2015.01.014
Liang, H., Ye, D., and Luo, L. (2017). Unravelling diversity and metabolic potential of microbial consortia at each stage of leather sewage treatment. RSC Adv. 7, 41727–41737. doi: 10.1039/C7RA07470K
Liaw, R.-B., Cheng, M.-P., Wu, M.-C., and Lee, C.-Y. (2010). Use of metagenomic approaches to isolate lipolytic genes from activated sludge. Bioresour. Technol. 101, 8323–8329. doi: 10.1016/j.biortech.2010.05.091
Liu, C., Li, H., Zhang, Y., Si, D., and Chen, Q. (2016). Evolution of microbial community along with increasing solid concentration during high-solids anaerobic digestion of sewage sludge. Bioresour. Technol. 216, 87–94. doi: 10.1016/J.BIORTECH.2016.05.048
Lopetuso, L. R., Scaldaferri, F., Petito, V., and Gasbarrini, A. (2013). Commensal clostridia: leading players in the maintenance of gut homeostasis. Gut Pathog. 5, 23. doi: 10.1186/1757-4749-5-23
Ma, Q., Qu, Y., Shen, W., Zhang, Z., Wang, J., Liu, Z., et al. (2015). Bacterial community compositions of coking wastewater treatment plants in steel industry revealed by Illumina high-throughput sequencing. Bioresour. Technol. 179, 436–443. doi: 10.1016/j.biortech.2014.12.041
Ma, X., Zhou, W., Fu, Z., Cheng, Y., Min, M., Liu, Y., et al. (2014). Effect of wastewater-borne bacteria on algal growth and nutrients removal in wastewater-based algae cultivation system. Bioresour. Technol. 167, 8–13. doi: 10.1016/J.BIORTECH.2014.05.087
Madigan, M. T., Martinko, J., Bender, K., Buckley, D., and Stahl, D. (2016). “Microrganismos e microbiologia,” in Microbiologia de Brock, eds M. T. Madigan, J. Martinko, K. Bender, D. Buckley, D. A-V. Stahl Alice Freitas, A. A. S. Barbosa, et al. (Porto Alegre: Artmed).
Manson, J. M., Rauch, M., and Gilmore, M. S. (2008). “The commensal microbiology of the gastrointestinal tract,” in GI Microbiota and Regulation of the Immune System, eds G. B. Huffnagle and M. C. Noverr (New York, NY: Springer New York), 15–28. doi: 10.1007/978-0-387-09550-9_2
Maspolim, Y., Zhou, Y., Guo, C., Xiao, K., and Ng, W. J. (2015). The effect of pH on solubilization of organic matter and microbial community structures in sludge fermentation. Bioresour. Technol. 190, 289–298. doi: 10.1016/j.biortech.2015.04.087
Meerbergen, K., Van Geel, M., Waud, M., Willems, K. A., Dewil, R., Van Impe, J., et al. (2017). Assessing the composition of microbial communities in textile wastewater treatment plants in comparison with municipal wastewater treatment plants. Microbiologyopen 6:e00413. doi: 10.1002/mbo3.413
Meerburg, F. A., Vlaeminck, S. E., Roume, H., Seuntjens, D., Pieper, D. H., Jauregui, R., et al. (2016). High-rate activated sludge communities have a distinctly different structure compared to low-rate sludge communities, and are less sensitive towards environmental and operational variables. Water Res. 100, 137–145. doi: 10.1016/j.watres.2016.04.076
Meyer, D. D., de Andrade, P. A., Durrer, A., Andreote, F. D., Corção, G., and Brandelli, A. (2016). Bacterial communities involved in sulfur transformations in wastewater treatment plants. Appl. Microbiol. Biotechnol. 100, 10125–10135. doi: 10.1007/s00253-016-7839-3
Moreira, F. M. S., and Siqueira, J. O. (2006). Microbiologia e Bioquímica do Solo, 2 Edn. Lavras: Editora UFLA.
Mulvaney, R. L. (1996). “Nitrogen - inorganic forms,” in Methods of Soil Analysis. Part 3. Chemical Methods, eds D. L. Sparks, A. L. Page, P. A. Helmke, R. H. Loeppert, P. N. Soltanpour, M. A. Tabatabai, et al. (Madison, WI: Segoe Rd), 1123–1184.
Muszyński, A., Łebkowska, M., Tabernacka, A., and Miłobêdzka, A. (2013). From macro to lab-scale: changes in bacterial community led to deterioration of EBPR in lab reactor. Cent. Eur. J. Biol. 8, 130–142. doi: 10.2478/s11535-013-0116-2
Muszyński, A., Tabernacka, A., and Miłobedzka, A. (2015). Long-term dynamics of the microbial community in a full-scale wastewater treatment plant. Int. Biodeterior. Biodegradation 100, 44–51. doi: 10.1016/j.ibiod.2015.02.008
Nascimento, A. L. (2015). Caracterização Microbiológica, Química e Presença de Poluentes Orgânicos em Amostras de Lodo de Esgoto de São Paulo. Tese de Doutorado, Escola Superior de Agricultura Luiz de Queiroz, University of São Paulo, Piracicaba.
Nelson, D. W., and Sommers, L. E. (1996). “Total carbon, organic carbon, and organic matter,” in Methods of Soil Analysis. Part 3-Chemical Methods, eds D. L. Sparks, A. L. Page, P. A. Helmke, R. H. Loeppert, P. N. Soltanpour, M. A. Tabatabai, et al. (Madison, WI: Segoe Rd), 961–1010. doi: 10.2136/sssabookser5.3.c34
Nielsen, P. H., and McMahon, K. D. (2014). Microbiology and Microbial Ecology of the Activated Sludge Process. London: IWA Publishing.
Nielsen, P. H., Mielczarek, A. T., Kragelund, C., Nielsen, J. L., Saunders, A. M., Kong, Y., et al. (2010). A conceptual ecosystem model of microbial communities in enhanced biological phosphorus removal plants. Water Res. 44, 5070–5088. doi: 10.1016/j.watres.2010.07.036
Nielsen, P. H., Saunders, A. M., Hansen, A. A., Larsen, P., and Nielsen, J. L. (2012). Microbial communities involved in enhanced biological phosphorus removal from wastewater—a model system in environmental biotechnology. Curr. Opin. Biotechnol. 23, 452–459. doi: 10.1016/j.copbio.2011.11.027
Philippot, L., Andersson, S. G., Battin, T. J., Prosser, J. I., Schimel, J. P., Whitman, W. B., et al. (2010). The ecological coherence of high bacterial taxonomic ranks. Nat. Rev. Microbiol. 8, 523–529. doi: 10.1038/nrmicro2367
Ramette, A. (2007). Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 62, 142–160. doi: 10.1111/j.1574-6941.2007.00375.x
Roesch, L. F. W., Fulthorpe, R. R., Riva, A., Casella, G., Hadwin, A. K. M., Kent, A. D., et al. (2007). Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 1, 283–290. doi: 10.1038/ismej.2007.53
Rousk, J., Brookes, P. C., and Bååth, E. (2009). Contrasting soil pH effects on fungal and bacterial growth suggest functional redundancy in carbon mineralization. Appl. Environ. Microbiol. 75, 1589–1596. doi: 10.1128/AEM.02775-08
Ruiz, G., Jeison, D., Rubilar, O., Ciudad, G., and Chamy, R. (2006). Nitrification–denitrification via nitrite accumulation for nitrogen removal from wastewaters. Bioresour. Technol. 97, 330–335. doi: 10.1016/j.biortech.2005.02.018
Saia, F. T., Souza, T. S. O., Duarte, R. T. D., Pozzi, E., Fonseca, D., and Foresti, E. (2016). Microbial community in a pilot-scale bioreactor promoting anaerobic digestion and sulfur-driven denitrification for domestic sewage treatment. Bioprocess Biosyst. Eng. 39, 341–352. doi: 10.1007/s00449-015-1520-6
Seviour, R., and Nielsen, P. H. (2010). Microbial Ecology of Activated Sludge. London: IWA Publishing Company.
Shchegolkova, N. M., Krasnov, G. S., Belova, A. A., Dmitriev, A. A., Kharitonov, S. L., Klimina, K. M., et al. (2016). Microbial community structure of activated sludge in treatment plants with different wastewater compositions. Front. Microbiol. 7:90. doi: 10.3389/fmicb.2016.00090
Shrestha, J., Rich, J. J., Ehrenfeld, J. G., and Jaffe, P. R. (2009). Oxidation of ammonium to nitrite under iron-reducing conditions in Wetland soils. Soil Sci. 174, 156–164. doi: 10.1097/SS.0b013e3181988fbf
Shu, D., He, Y., Yue, H., and Wang, Q. (2015a). Bioresource technology microbial structures and community functions of anaerobic sludge in six full-scale wastewater treatment plants as revealed by 454 high-throughput pyrosequencing. Bioresour. Technol. 186, 163–172. doi: 10.1016/j.biortech.2015.03.072
Shu, D., He, Y., Yue, H., Zhu, L., and Wang, Q. (2015b). Metagenomic insights into the effects of volatile fatty acids on microbial community structures and functional genes in organotrophic anammox process. Bioresour. Technol. 196, 621–633. doi: 10.1016/j.biortech.2015.07.107
Spain, A. M., Krumholz, L. R., and Elshahed, M. S. (2009). Abundance, composition, diversity and novelty of soil Proteobacteria. ISME J. 3, 992–1000. doi: 10.1038/ismej.2009.43
Stiborova, H., Wolfram, J., Demnerova, K., Macek, T., and Uhlik, O. (2015). Bacterial community structure in treated sewage sludge with mesophilic and thermophilic anaerobic digestion. Folia Microbiol. 60, 531–539. doi: 10.1007/s12223-015-0396-9
Sun, M., Xiao, T., Ning, Z., Xiao, E., and Sun, W. (2015). Microbial community analysis in rice paddy soils irrigated by acid mine drainage contaminated water. Appl. Microbiol. Biotechnol. 99, 2911–2922. doi: 10.1007/s00253-014-6194-5
Takai, K., Campbell, B. J., Cary, S. C., Suzuki, M., Oida, H., Nunoura, T., et al. (2005). Enzymatic and genetic characterization of carbon and energy metabolisms by deep-sea hydrothermal chemolithoautotrophic isolates of Epsilonproteobacteria. Appl. Environ. Microbiol. 71, 7310–7320. doi: 10.1128/AEM.71.11.7310-7320.2005
Tan, W.-F., Liu, F., Li, Y.-H., Hu, H.-Q., and Huang, Q.-Y. (2006). Elemental composition and geochemical characteristics of iron-manganese nodules in main soils of China. Pedosphere 16, 72–81. doi: 10.1016/S1002-0160(06)60028-3
Tomazetto, G., and Oliveira, V. M. (2013). Investigation of the FeFe-hydrogenase gene diversity combined with phylogenetic microbial community analysis of an anaerobic domestic sewage sludge. World J. Microbiol. Biotechnol. 29, 2003–2014. doi: 10.1007/s11274-013-1363-8
Wan, C. Y., De Wever, H., Diels, L., Thoeye, C., Liang, J. B., and Huang, L. N. (2011). Biodiversity and population dynamics of microorganisms in a full-scale membrane bioreactor for municipal wastewater treatment. Water Res. 45, 1129–1138. doi: 10.1016/j.watres.2010.11.008
Wang, X., Hu, M., Xia, Y., Wen, X., and Ding, K. (2012). Pyrosequencing analysis of bacterial diversity in 14 wastewater treatment systems in china. Appl. Environ. Microbiol. 78, 7042–7047. doi: 10.1128/AEM.01617-12
Wang, X.-J., Yang, J., Chen, X.-P., Sun, G.-X., and Zhu, Y.-G. (2009). Phylogenetic diversity of dissimilatory ferric iron reducers in paddy soil of Hunan, South China. J. Soils Sediments 9, 568–577. doi: 10.1007/s11368-009-0113-x
Wells, G. F., Park, H. D., Eggleston, B., Francis, C. A., and Criddle, C. S. (2011). Fine-scale bacterial community dynamics and the taxa-time relationship within a full-scale activated sludge bioreactor. Water Res. 45, 5476–5488. doi: 10.1016/j.watres.2011.08.006
Wen, Y., Jin, Y., Wang, J., and Cai, L. (2015). MiSeq sequencing analysis of bacterial community structures in wastewater treatment plants. Pol. J. Environ. Stud. 24, 1809–1815. doi: 10.15244/pjoes/38456
Wu, S., Li, Y., Wang, P., Zhong, L., Qiu, L., and Chen, J. (2016). Shifts of microbial community structure in soils of a photovoltaic plant observed using tag-encoded pyrosequencing of 16S rRNA. Appl. Microbiol. Biotechnol. 100, 3735–3745. doi: 10.1007/s00253-015-7219-4
Xia, Z., Xiao-chun, W., Zhong-lin, C., Hao, X., and Qing-fang, Z. (2015). Microbial community structure and pharmaceuticals and personal care productsremoval in a membrane bioreactor seeded with aerobic granular sludge. Appl. Microbiol. Biotechnol. 99, 425–433. doi: 10.1007/s00253-014-5984-0
Yamashita, T., and Yamamoto-Ikemoto, R. (2014). Nitrogen and phosphorus removal from wastewater treatment plant effluent via bacterial sulfate reduction in an anoxic bioreactor packed with wood and iron. Int. J. Environ. Res. Public Health 11, 9835–53. doi: 10.3390/ijerph110909835
Yang, C., Zhang, W., Liu, R., Li, Q., Li, B., Wang, S., et al. (2011). Phylogenetic diversity and metabolic potential of activated sludge microbial communities in full-scale wastewater treatment plants. Environ. Sci. Technol. 45, 7408–7415. doi: 10.1021/es2010545
Yang, Y., Yu, K., Xia, Y., Lau, F. T. K., Tang, D. T. W., Fung, W. C., et al. (2014). Metagenomic analysis of sludge from full-scale anaerobic digesters operated in municipal wastewater treatment plants. Appl. Microbiol. Biotechnol. 98, 5709–5718. doi: 10.1007/s00253-014-5648-0
Yao, S., Ni, J., Chen, Q., and Borthwick, A. G. (2013a). Enrichment and characterization of a bacteria consortium capable of heterotrophic nitrification and aerobic denitrification at low temperature. Bioresour. Technol. 127, 151–157. doi: 10.1016/j.biortech.2012.09.098
Yao, S., Ni, J., Ma, T., and Li, C. (2013b). Heterotrophic nitrification and aerobic denitrification at low temperature by a newly isolated bacterium, Acinetobacter sp. HA2. Bioresour. Technol. 139, 80–86. doi: 10.1016/j.biortech.2013.03.189
Yi, J., Dong, B., Jin, J., and Dai, X. (2014). Effect of increasing total solids contents on anaerobic digestion of food waste under mesophilic conditions: performance and microbial characteristics analysis. PLoS One 9:e102548. doi: 10.1371/journal.pone.0102548
Zhang, T., Shao, M. F., and Ye, L. (2012). 454 Pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J. 6, 1137–1147. doi: 10.1038/ismej.2011.188
Zhang, X., Li, X., Zhang, Q., Peng, Q., Zhang, W., and Gao, F. (2014). New insight into the biological treatment by activated sludge: the role of adsorption process. Bioresour. Technol. 153, 160–164. doi: 10.1016/j.biortech.2013.11.084
Keywords: bacteria, wastewater, biological treatment, liming, molecular biology
Citation: Nascimento AL, Souza AJ, Andrade PAM, Andreote FD, Coscione AR, Oliveira FC and Regitano JB (2018) Sewage Sludge Microbial Structures and Relations to Their Sources, Treatments, and Chemical Attributes. Front. Microbiol. 9:1462. doi: 10.3389/fmicb.2018.01462
Received: 08 March 2018; Accepted: 12 June 2018;
Published: 03 July 2018.
Edited by:
Dimitrios Georgios Karpouzas, University of Thessaly, GreeceReviewed by:
Seung Gu Shin, Pohang University of Science and Technology, South KoreaCopyright © 2018 Nascimento, Souza, Andrade, Andreote, Coscione, Oliveira and Regitano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jussara Borges Regitano, cmVnaXRhbm9AdXNwLmJy
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.