 Alexandre Couture1
Alexandre Couture1 Anthony Garnier2
Anthony Garnier2 Fabian Docagne2
Fabian Docagne2 Olivier Boyer3
Olivier Boyer3 Denis Vivien2,4
Denis Vivien2,4 Brigitte Le-Mauff2,5
Brigitte Le-Mauff2,5 Jean-Baptiste Latouche1,6†
Jean-Baptiste Latouche1,6† Olivier Toutirais2,5,7*
Olivier Toutirais2,5,7*- 1UNIROUEN, Inserm U1245, Institute for Research and Innovation in Biomedicine, Normandie University, Rouen, France
- 2Inserm U1237, Physiopathology and Imaging of Neurological Disorders, Caen University Hospital, Caen, France
- 3Department of Immunology and Biotherapy, Inserm U1234, Institute for Research and Innovation in Biomedicine, UNIROUEN, Rouen University Hospital, Normandie University, Rouen, France
- 4Department of Clinical Research, Caen University Hospital, Caen, France
- 5Department of Immunology and Immunopathology, Caen University Hospital, Caen, France
- 6Department of Genetics, Rouen University Hospital, Rouen, France
- 7French Blood Service (Etablissement Français du Sang), Caen, France
CD4+ T cells differentiate into various T helper subsets characterized by distinct cytokine secreting profiles that confer them effector functions adapted to a variety of infectious or endogenous threats. Regulatory CD4+ T cells are another specialized subset that plays a fundamental role in the maintenance of immune tolerance to self-antigens. Manipulating effector or regulatory CD4+ T cells responses is a promising immunotherapy strategy for, respectively, chronical viral infections and cancer, or severe autoimmune diseases and transplantation. Adoptive cell therapy (ACT) is an emerging approach that necessitates defining robust and efficient methods for the in vitro expansion of antigen-specific T cells then infused into patients. To address this challenge, artificial antigen presenting cells (AAPCs) have been developed. They constitute a reliable and easily usable platform to stimulate and amplify antigen-specific CD4+ T cells. Here, we review the recent advances in understanding the functions of CD4+ T cells in immunity and in immune tolerance, and their use for ACT. We also describe the characteristics of different AAPC models and the way to improve their stimulating functions. Finally, we discuss the potential interest of these AAPCs, both as fundamental tools to decipher CD4+ T cell responses and as reagents to generate clinical grade antigen-specific CD4+ T cells for immunotherapy.
Introduction
Functionally distinct CD4+ T cell subsets orchestrate immune responses against pathogenic microorganisms or transformed cells (1, 2). Each type of CD4+ T helper (Th) cells is endowed with a specific cytokine profile that regulates adaptive and innate immunity. Since a long time, it has been known that CD4+ T cells have a crucial role to support CD8 and B cell responses. CD4+ T cells also exert direct anti-tumor and anti-viral roles based on their cytolytic activity and effector cytokine secretion. Besides CD4+ effector cells, CD4+ regulatory T cells (Tregs), consisting of thymus-derived or induced cells, maintain peripheral tolerance to self-antigens by regulating other types of immune cells (3). In many autoimmune diseases, a defect in peripheral blood Treg number or immunosuppressive function has been described (4). Due to their multiple functions in immunity and immune tolerance, targeting CD4+ T cells has important clinical applications to treat cancer and chronic viral diseases, or to induce tolerance in autoimmune diseases and allograft. In vivo and/or in vitro approaches could be harnessed to develop CD4+ T cell-based immunotherapy. Several types of artificial antigen presenting cells (AAPCs) have been engineered through gene transfer allowing expression of presentation and costimulatory molecules required to stimulate antigen-specific CD4+ T cells. In this review, we describe our current understanding of CD4+ T cell functions in immunity and immune tolerance and discuss their contribution in adoptive cell therapy (ACT). We then focus on AAPCs as potent tools to induce specific CD4+ T cells in vitro. Finally, we examine the different ways to optimize AAPC models with the goals of reinforcing our basic knowledge of human CD4 responses and to propose efficient specific CD4+ T cell expansion protocols for ACT.
Antigen Presentation By Mhc Class Ii Molecules and Cd4+ T Cell Activation
The function of the major histocompatibility complex class II (MHC-II) molecules is to present antigens to CD4+ T cells. Their expression is preferentially restricted to professional antigen-presenting cells (APCs), including dendritic cells (DCs), monocytes/macrophages and B lymphocytes. In humans, HLA-DR, HLA-DQ, and HLA-DP molecules are the three classical and highly polymorphic MHC-II molecules. Both glycoproteins α and β chains of the MHC-II molecules are synthetized in the endoplasmic reticulum (ER) and are associated with the invariant chain (Ii), a chaperone molecule which stabilizes the conformation of the MHC dimer, avoiding the binding of endogenous peptides in the groove of MHC-II molecules. The heterotrimer is then transported out of the ER through the Golgi apparatus to a specialized endosome, the MHC-II compartment (MIIC), in which proteases, such as cathepsins degrade Ii. Only a part of Ii, the class II invariant chain peptide (CLIP), is maintained in the MHC-II groove (5). The non-classical MHC-II molecule HLA-DM interacts with MHC-II molecules and catalyses the exchange of CLIP with a high-affinity peptide. Action of HLA-DM is inhibited by another non-classical MHC-II molecule, HLA-DO (6). Peptides not covalently binding the MHC-II molecules are mainly derived from exogenous self-and non-self-proteins degraded by the endocytic pathway. Of note, a substantial fraction of MHC-II-associated peptides comes from the cytosolic proteins through the autophagic pathway (7). Then, peptide-MHC-II (pMHC-II) complexes are transferred and displayed on the plasma membrane of APCs for recognition by CD4+ T cells (Figure 1).
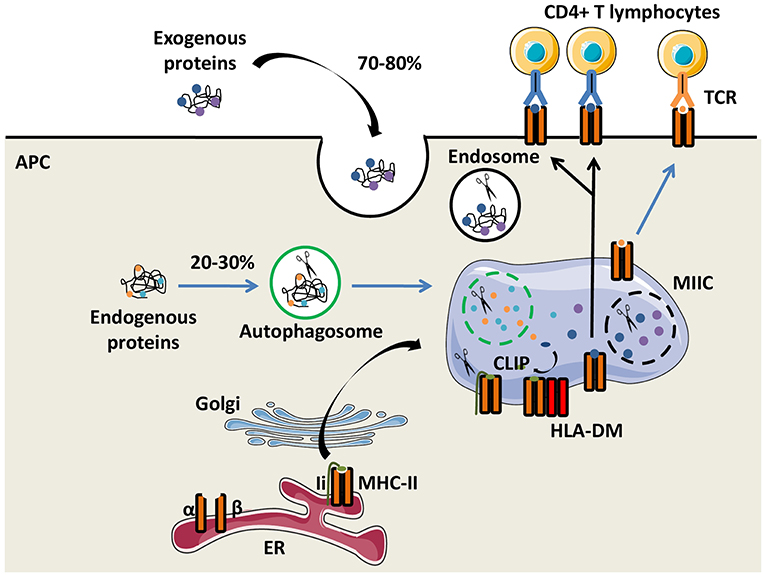
Figure 1. The MHC-II antigen presentation pathways. Major histocompatibility complex class II (MHC-II) α and β chains, expressed by antigen presenting cells (APCs), are synthetized in the endoplasmic reticulum (ER) where they form a heterotrimer with the invariant chain (Ii). After maturation in the Golgi apparatus, the heterotrimer (α/β/Ii) is delivered to the MHC class II compartment (MIIC) in which endocytosed and exogenous proteins but also Ii are degraded by proteases for generating peptides. Ii is progressively degraded into the Class II Invariant chain Peptide (CLIP) which binds to the MHC-II groove. The chaperone protein HLA-DM induces CLIP replacement by an antigenic peptide. Then, the peptide/MHC-II complexes move to the plasma membrane and are presented to T-cell receptors (TCRs) of CD4+ T lymphocytes.
The activation of naïve CD4+ T cells is initiated by the interaction of T Cell Receptors (TCRs) with specific pMHC-II complexes presented by professional APCs. Close contact between T cell and APC leads to the formation of a specialized structure named the immunological synapse (8). To fully prime CD4+ T cells, antigenic signal is reinforced by costimulatory molecule interactions with APCs and by cytokines secreted in the local environment (Figure 2). The main costimulatory molecule expressed by T cells is CD28, which interacts with CD80 and CD86 on APCs. CD40 molecule on APCs that binds CD40L on activated T cells is also critical for CD4+ T cell responses, at least, in part, by amplifying APC activation.
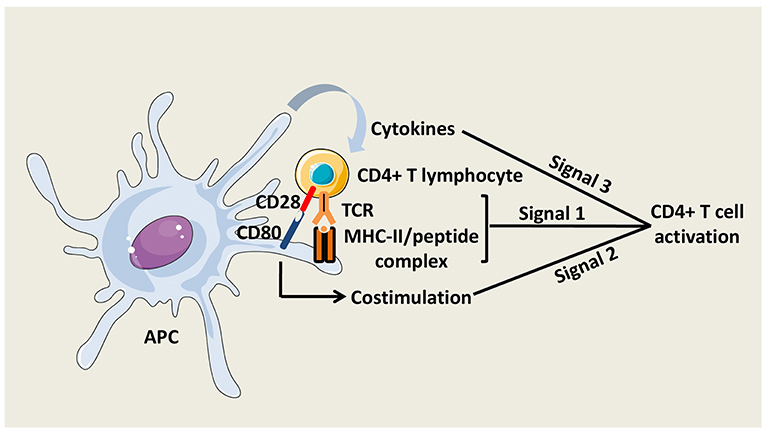
Figure 2. CD4+ T cell activation. Three signals are necessary to fully activate CD4+ T cells. The first signal is mediated by the interaction between TCRs and peptide/MHC-II complexes at the APC surface. The second signal is mediated by costimulatory molecules, such as CD80 on APCs, which interacts with CD28 on CD4+ T cells. The third signal is delivered by soluble factors including cytokines, which are notably secreted by the APCs.
Effector Cd4+ T Subsets
CD4+ T cells differentiate into diverse effector T helper (Th) subsets producing characteristic cytokines adapted to their functions and providing to the host the capacity to develop optimal immune responses against different types of pathogens (Table 1) (2, 9–13). Polarization of Th cells is governed by distinct transcription factors induced by cytokine environments. The stability of polarized CD4+ T cell subsets is still debated since many extracellular and cytosolic signals may contribute to their complex plasticity (14).
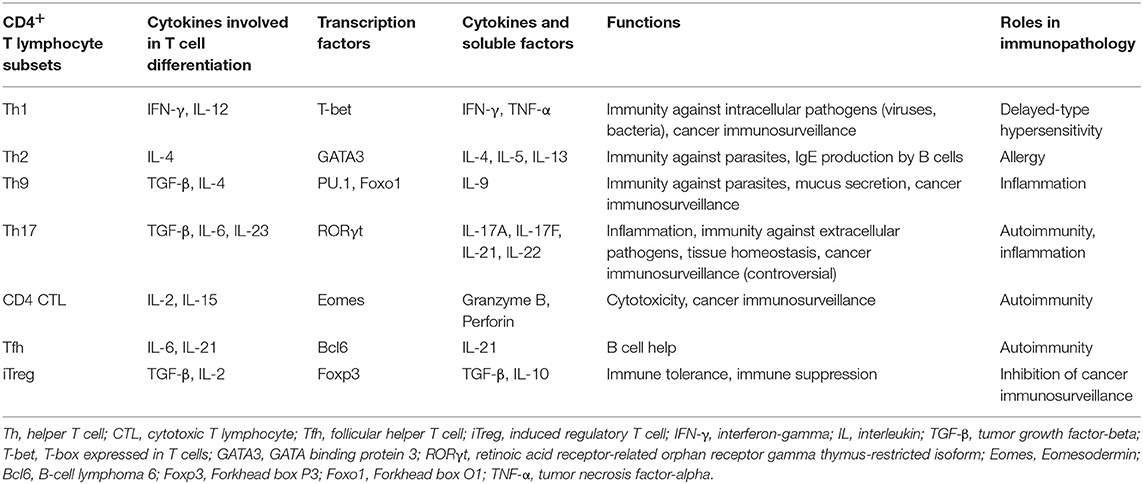
Table 1. Main features of CD4+ TL subsets arising from naïve CD4+ T cells.
Th1 cells that produce large amounts of IFN-γ and express T-bet are involved in the defense against intracellular pathogens. They activate macrophages and help the generation of effector and memory CD8+ T cells (2). The response against extracellular parasites is controlled by IL-4-producing Th2 cells that stimulate IgE production by B cells and activate eosinophils. Th2 cells have a crucial role in the pathogenesis of many allergic diseases (11).
Th17 cells secrete IL-17, IL-22, and GM-CSF, and have a high degree of plasticity (9, 10). They contribute to immunity against fungi and extracellular bacteria, mainly by inducing the release of anti-microbial peptides and attracting myeloid cells to infected mucosal surfaces, especially gut and skin. These cells are also involved in microbiota maintenance and epithelium integrity and regeneration, and promote humoral response in the gut (15). However, data from animal models and patients indicate a pathogenic role of Th17 cells in chronic inflammatory and autoimmune diseases, such as multiple sclerosis, psoriasis or systemic lupus erythematosus (16–18). Conversion of self-reactive effector Th17 cells into Th1-like Th17 cells producing IFN-γ and/or GM-CSF is a predominant mechanism of their pathogenicity identified in experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis (19, 20).
T follicular helper (Tfh) cells are involved in B cell help and humoral response against extracellular antigens, supporting germinal center formation and antibody production. Th9 cells that are mainly characterized by the secretion of IL-9, have protective roles against parasite infections and tumor development (21). Th9 promote worm clearance by inducing mucus secretion by epithelial cells. Th9 have a skin tropism but their functions in cutaneous immunity and skin disorders are currently under investigation (22–24). PU.1 is one of the major transcription factors associated with Th9 cells but recently another transcription factor, Foxo1, has been shown to be essential in their development (25). Detrimental functions of Th9 are also described in inflammatory and autoimmune diseases (26). For instance, IL-9 impairs intestinal tissue repair and exacerbates the disease in a mouse colitis model (27).
Another population recently described is the CD4+ T cells that harbor cytolytic activity against MHC-II+ target cells. Cell killing mechanisms involve perforin/granzyme and Fas-FasL pathways. In vivo, it was shown in the EAE model that Eomesodermin (Eomes) transcription factor drives the development of CD4+ cytotoxic T lymphocytes (CTLs) (28).
Regulatory Cd4+ T Cell Subsets
CD4+ regulatory T cells (Tregs) are critical for maintaining peripheral tolerance and preventing autoimmunity. Depending on the expression of the transcription factor Forkhead box protein 3 (Foxp3), Tregs may be divided into two subsets: the classical Foxp3+ Tregs and the Foxp3-negative type 1 regulatory T (Tr1) cells. Foxp3+ Tregs can be generated in the thymus (thymus-derived Treg cells, tTregs) or also be induced from naïve T cells in the periphery (peripherally-induced Treg cells, pTregs) (29). A third subtype of Foxp3+ Tregs, termed iTregs, represents the in vitro-induced Tregs. Tr1 cells induced in the periphery, secrete IL-10 and/or transforming growth factor beta (TGF-β). Distinct intracellular and surface markers help distinguishing Foxp3+ Tregs and Tr1 cells. In addition to the expression of their specific transcription factor, Foxp3+ Tregs are identified by their constitutively high expression of cell surface IL-2Rα chain (CD25) in the absence of the IL-7Rα chain (CD127). Recently, coexpression of the lymphocyte-activation gene 3 (LAG-3) molecule and the integrin alpha-2 subunit (CD49b) was shown to characterize Tr1 cells (30). Tregs have a potent suppressive capacity toward a very broad range of immune cells. Foxp3+ Tregs and Tr1 cells share common mechanisms of immunosuppression involving inhibitory cytokine production, metabolic disruption via the expression of ectoenzymes, cytotoxic activity and inhibition of APCs (31). The potential of Treg-based immunotherapies in preventing autoimmune diseases or controlling graft vs. host disease (GVHD) and allograft rejection is attested by several studies in preclinical models (32–34). In these contexts, Treg-based therapeutic strategies rely on the in vitro or in vivo activation of natural or induced Tregs. They include adoptive transfer of Tregs and vaccination with autoantigen-derived peptides or other pharmalogical approaches (see below) (35, 36).
Role of Cd4+ T Cells in Anti-Tumor and Anti-Viral Adaptive responses
Growing evidences in the literature indicate that CD4+ T cells have direct roles in anti-tumor and anti-viral responses without contribution of CD8 or B cells. Several effector mechanisms have been described depending on the experimental models and the investigated Th subsets. Quezada et al. have demonstrated that transfer of tumor-specific CD4+ cells in lymphopenic mice resulted in rejection of melanoma tumors (37). In this study, CD4+ T cells had a Th1-like phenotype, produced granzyme B and displayed a MHC class II-dependent cytotoxic activity. In another mouse adoptive transfer model, Th17-polarized T cells were also capable of rejecting melanoma tumors via an IFN-γ dependent mechanism (38). Nevertheless, Th17 cells can also have a protumor effect by inducing angiogenic factors (39).
More recently, several studies highlighted anti-tumor properties of IL-9 producing CD4+ T cells (40). Purwar et al. have found in the B16 melanoma mouse model that tumor growth was accelerated in IL-9 receptor-deficient mice while injection of recombinant IL-9 prevented tumor progression in wild-type mice (41). Other studies reported that anti-cancer effects of Th9 cells were mediated by production of IL-21 and their cytolytic activity (42).
CD8+ T cells are considered as the main effector cells of cancer and virus immunosurveillance, capable of killing tumors or infected cells and secreting immunostimulatory cytokines. Nevertheless, CD4+ T cell help is critical for maintaining CD8+ T cell functions during anti-tumor response and chronic infection (2, 43, 44). Indeed, CD4+ T cells are required to fully activate and “license” DCs which can effectively prime CD8+ T cells. CD40L-CD40 interactions between activated CD4+ T cells and DCs, respectively, are crucial to increase DC antigen-presentation and costimulation capacities (45). However, primary CD8+ T cell responses could be induced in a T cell help independent manner by microbial pathogen infections that provide potent inflammatory stimuli. Additionally, cognate interactions between activated CD4+ T cells and DCs lead to the production of chemokines that facilitate the recruitment of naïve CD8+ T cells toward antigen-bearing APCs in the secondary lymphoid organs (46). Although there is a consensus on the requirement of T cell help for the generation of long-lived memory CD8+ T cells, it is still discussed whether CD4+ T cells deliver a differentiation program during the priming phase or subsequently at later stages during the CD8+ T cell memory maintenance (47–49). Production of IL-2 by Th cells during the priming phase is crucial for an effective secondary CD8+ response (50). However, it has been shown that “licensed” DCs may provide signals that enable autocrine secretion of IL-2 by memory CD8+ T cells (51). CD4+ helper T cells also stimulate IL-15 production by DCs that favors induction of long-lived CD8+ T cells by increasing expression of anti-apoptotic molecule Bcl-xL (52). In the context of viral chronic infection, IL-21 is an essential component of CD4+ T cell help by avoiding clonal deletion and maintaining CD8+ T cell function (53). Regulation of activation-induced cell death (AICD) by CD4+ T cells is a putative mechanism for the maintenance of CD8+ T cell response. It has been reported that CD8+ T cells primed in the absence of CD4+ T cells could undergo AICD-mediated by TNF-related apoptosis-inducing ligand (TRAIL) signaling (54). However, other studies using TRAIL-deficient mice, do not confirm this mechanism (55). Furthermore, recently, CD4+ T cells have been shown to upregulate the expression of CD70 on DCs, a costimulatory molecule that triggers CD27 receptor on CD8+ T cells. CD70-CD27 interactions result in the delivery of a help program that amplifies CTL functions and downregulates inhibitory receptors, such as PD-1 (56).
Neutralizing antibodies are a central component of adaptive immune responses against microbial pathogens. In the previous decade, Tfh cells characterized by the expression of CXCR5 have been identified as the main providers of B cell help. CD40L on Tfh cells is the most prominent costimulatory molecule to promote survival and proliferation of CD40-expressing B cells. Cytokines IL-4 and IL-21 are necessary for the formation of germinal centers and differentiation of B cells into long-lived plasma cells (57). Recently the role of Tfh in anti-tumor immunity was underlined by several reports. In colorectal cancer, gene expression and tissue microarray analyses of tumor biopsies showed that tumor infiltrating Tfh and B cells were correlated with patient disease-free survival (58). The authors also found that chemokine CXCL13 and IL-21 were key factors of adaptive immunity in tumor environment. Infiltration of CXCL13-producing CD4+ Tfh cells was also associated with a better disease-free survival or preoperative response to chemotherapy (59).
CD4+ T Cell-Based Immunotherapy
ACT clinical trials mainly focus on CD8+ effector cells and very few studies have investigated the therapeutic potential of CD4+ T cells. One of the first CD4+ T cell-based immunotherapy approaches was based on the infusion of an autologous CD4+ T-cell clone. Hunder et al. had expanded in vitro, a New York Esophageal Squamous Cell Carcinoma 1 (NY-ESO-1)-specific CD4+ T-cell clone from a refractory metastatic melanoma patient. After one infusion of a few billion cells, the patient presented a durable clinical remission (60). Another clinical trial has been conducted with autologous CD4+ T cells from patients transduced with a melanoma-associated antigen 3 (MAGE-3)-specific MHC-II-restricted TCR transgene (61). In a cohort of 17 patients with metastatic solid cancer and treated with 107-1011 cells, one patient had a complete response and a partial response was observed in three patients.
With the breakthrough of next generation sequencing (NGS) technologies, it is now possible to characterize mutations within patient's tumor and finally identify potential immunogenic neoantigens in many cancers, such as melanoma or colon cancer (62–64). These findings could be translated into clinical applications. Tran et al. have identified Th1 cells specific for a neoantigen derived from the erbb2 interacting protein (ERBB2IP) in a metastatic cholangiosarcoma patient's tumor (65). Two infusions of several billion neoantigen-specific CD4+ tumor infiltrating lymphocytes led to a disease stabilization for more than 1 year. Ott et al. have identified neoantigens in six advanced melanoma patients and vaccinated them with a pool of synthetic long-peptides derived from the predicted neoantigens (66). No recurrence 25 months after treatment was observed in four patients.
A major problem associated with hematopoietic stem cell transplantation (HSCT), is Epstein-Barr Virus (EBV) or cytomegalovirus (CMV) reactivation, as well as opportunistic virus, such as adenovirus infections which are fatal in 20% of cases (67, 68). To avoid these complications, in vitro protocols have been developed to rapidly generate CD4+ and CD8+ multivirus-specific T cells by a single stimulation of donor peripheral blood mononuclear cells (PBMCs) with a pool of synthetic peptides covering the target viral antigens (69–71). In a recent clinical trial including 37 patients with viral infection after allogeneic HSCT, 92% had a complete or partial response after one infusion of 107 virus-specific T cells (72).
Regulatory T Cell-Based Immunotherapy
Compelling experimental data from mouse models indicate that adoptive immunotherapy harnessing immunosuppressive properties of CD4+ regulatory T cells is a promising therapeutic strategy against autoimmune diseases and GVHD or allograft rejection. This type of strategy has the advantages of allowing the accurate definition of the phenotype and functions of the infused cells and of avoiding the toxicity of general immunosuppressive drugs. Human peripheral blood CD4+ CD25+ Tregs, potentially including tTregs and pTregs, are the main source for such a cell therapy but they constitute only 1–2% of human CD4+ T cells. Due to the low number of circulating Tregs, translating Treg transfer approaches from mice studies to humans requires efficient protocols to generate large numbers of highly purified Tregs. Usually, cell therapy protocols consist in a first step of CD4+ CD25+ polyclonal Treg isolation using either magnetic bead-or flow cytometry-based systems. Next, isolated Tregs are either directly infused into patients or stimulated in vitro with anti-CD3/anti-CD28-coated beads (73).
So far, the therapeutic potential of polyclonal Tregs has been investigated in the context of GVHD and type 1 diabetes, with some evidences of safety and clinical benefits (74, 75). A possible drawback of using polyclonal Tregs in immunotherapy is the risk of inducing an overall immune suppression compromising beneficial immune responses. Autoantigen-specific Tregs may represent a better alternative for the treatment of autoimmune diseases, conferring a more localized and targeted immunosuppression at the site of inflammation. Noteworthy, several reports highlighted that antigen-specific Tregs were significantly more efficient than polyclonal Tregs in regulating autoimmune or allogenic responses in animal models (35, 76). To generate antigen-specific Tregs, polyclonal CD4+ CD25+ T cells could be stimulated with antigen-loaded APCs or transduced with a viral vector to express a TCR that recognizes a specific peptide. More recently, chimeric antigen receptors (CARs) designed to redirect human Tregs toward donor-MHC class I HLA-A2 molecule have been successfully used to prevent rejection in a skin xenograft transplant model (77, 78).
Additional immunoregulatory strategies that have already proven clinical efficacy could be combined with adoptive Treg immunotherapy to maximize a tolerogenic response in patients. IL-2 signaling is indispensable for Treg development and functions (79). Data from murine models and clinical trials in autoimmune diseases and HSCT have clearly shown that low-dose IL-2 administration led to increased numbers of Tregs in vivo and to beneficial therapeutic effects (80–83). Moreover, several immunosuppressive drugs, such as rapamycin, or immunomodulatory treatments including intravenous immunoglobulins could efficiently boost Treg response in vivo (84–86).
Mesenchymal stromal cells (MSCs) represent another major immune regulatory population currently assessed in cell therapy. MSCs inhibit T cell proliferation and DC maturation and also promote the expansion of Tregs (87–89). Therapeutic effects of MSCs have been reported in various autoimmune diseases, such as multiple sclerosis and diabetes, as well as in GVHD (90–92).
Hla Class Ii-Aapcs: an Attractive Tool to Activate and Expand Antigen-Specific Cd4+ T Cells In vitro
Beads coated with anti-CD3 and anti-CD28 antibodies are commonly used to amplify polyclonal human effector T cells or Tregs in vitro (93, 94). The development of protocols to efficiently expand human antigen-specific CD4+ T cells is utterly needed and requires the availability of potent APCs. As mentioned above, an interesting approach to generate specific CD4+ T cells consist in priming autologous PBMCs loaded with peptides. This approach that is mostly appropriated for highly immunogenic antigens, usually viral antigens, may involve a direct selection step, such as the one based on magnetic bead-enrichment of IFN-γ-secreting T cells (69–71, 95). In vivo, DCs are the most efficient professional APCs to trigger functional adaptive T cell immunity. Indeed, it is well-established that DCs, mainly arising from monocytes, can elicit robust specific CD4+ T cell responses in vitro (96, 97). Nevertheless, the use of DCs has limitations. Their production in vitro is difficult to standardize in terms of cell number, phenotype and functionality (98). In addition, it requires supplementary blood from healthy donors or patients. In this context, AAPCs are an interesting alternative because they constitute cell lines especially engineered for more easily and reproducibly amplifying functional antigen-specific T cells. AAPCs have the advantages of being quickly available and suitable for any healthy donor or patient in a given HLA context.
Butler and Hirano have developed an AAPC model derived from the human erythroleukemic cell line K562 which constitutively expresses the adhesion molecules CD54 (intercellular adhesion molecule-1, ICAM-1) and CD58 (leukocyte function-associated antigen-3, LFA-3) but not HLA class I and II molecules (Figure 3A) (99). To mount human CD4+ T cell responses, K562 cells were genetically modified to express costimulatory molecules CD80 and CD83, and a single HLA-DR allele. In addition, K562 cells were equipped with Ii and HLA-DM molecules to foster antigen processing and presentation. After multiple stimulations, these AAPCs exogenously loaded with peptides were able to expand Th1 central memory (CD45RO+, CD62L+, CCR7+) antigen-specific CD4+ T cells against infectious antigens without growth of Treg cells. Unfortunately, no comparison data are available regarding specific T cell activation capacities of these AAPCs vs. autologous APCs. Spontaneously, K562 cells have a low ability to take up exogenous proteins. Transduction with CD64, a high affinity Immunoglobulin G (IgG) Fragment crystallizable (Fc) receptor (Fc gamma RI), improved uptake and presentation of CMV pp65, a whole protein antigen, under immune complex forms. K562-derived AAPCs have also been used to stimulate CD4+ T cells against Survivin, a tumor associated antigen (TAA) (100). However, TAAs are inherently poorly immunogenic and CD4+ T cells needed to be initially sensitized with antigen-pulsed autologous monocytes before restimulation with K562-derived AAPCs. Interestingly, CD4+ T cells were long-lived effector cells able to secrete IFN-γ (Th1) or IL-4 (Th2) after challenge with MHC-II-positive tumor cells.
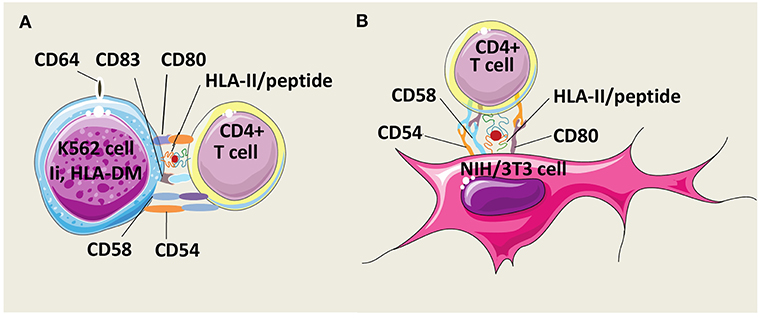
Figure 3. Models of HLA-II-AAPCs for the expansion of human antigen-specific CD4+ T cells. (A) Model of artificial antigen presenting cells (AAPCs) derived from erythroleukemic K562 cell line. K562-AAPCs express the invariant chain (Ii), HLA-DM, CD54, CD58, CD64, CD80, CD83, and one HLA class II (HLA-II) molecule. (B) Model of AAPCs derived from the mouse fibroblast NIH/3T3 cell line. NIH/3T3-AAPCs express CD54, CD58, CD80, and one HLA-II molecule.
Our group has designed another AAPC system using the mouse fibroblast cell line NIH/3T3 that has been retrovirally transduced to express a single HLA class II molecule, the costimulatory molecule CD80, and two adhesion molecules, CD54 and CD58 (Figure 3B). Previously, NIH/3T3 cells have been used to generate CTL responses after gene transfer with vectors encoding a single HLA class I molecule and an antigen in the form of a peptide or of a whole protein (101–103). These AAPCs consistently elicit strong stimulation and expansion of anti-tumor or anti-viral CD8+ CTLs. Thereafter, using the same murine fibroblast backbone, novel AAPCs expressing a single HLA class II molecule have been constructed to attempt to mount CD4+ T cell responses (104). We have demonstrated that these peptide-loaded AAPCs strongly activated specific CD4+ T-cell clones. In addition, they were able to efficiently and spontaneously take up, process extracellular whole proteins and present the correct epitope to specific T cells. In primary in vitro stimulation, AAPCs enabled expansion of antigen-specific CD4+ T cells, but with a lower stimulating ability than autologous APCs. Importantly, a major advantage of these AAPCs is their higher ability than autologous APCs to reactivate and amplify a high number of specific memory CD4+ T cells, at a scale compatible with therapeutic applications. Expanded T cells harbored transitional memory (CD45RO+, CD62L+, CCR7−) and effector memory (CD45RO+, CD62L−, CCR7−) phenotypes, and were highly functional Th1 cells that secreted IFN-γ upon antigen-specific stimulation.
Optimization Of Aapc Systems and their Use Perspectives
We and others have shown that AAPCs constitute reliable systems to expand human CD4+ T cells (104, 105). Since AAPC models are easily customizable, they could be used as a tool for basic and/or applied research on CD4+ T cell responses. As a key step, antigen presentation could be optimized by genetic approaches to address antigens into the various cell compartments (cytoplasm, ER, endosome) involved in the HLA class II presentation pathway. Indeed, endogenous antigen expression in the form of peptide or whole protein might foster their presentation as it was observed in HLA class I-AAPCs (101, 103). In addition, these antigen-targeting strategies could also be exploited to dissect the alternative MHC-II pathway which remains poorly understood (106, 107).
Manipulating expression of different costimulatory molecules on AAPCs is another opportunity to increase T cell functions in vitro. The tumor necrosis factor receptor superfamily (TNFRSF) molecules CD27 and 4-1BB (CD137) are among the costimulatory molecules that appear to be important to enhance CD4+ T cell response. CD27 is expressed on naïve and memory T cells and interactions with its ligand CD70 on APCs promote both Th1 differentiation via IL-12Rβ2 up-regulation and survival of CD4+ T cells (108). Similarly, 4-1BB ligand on APCs enhance proliferation and effector functions of activated CD4+ T cells that express 4-1BB (109, 110). Co-signaling receptors also modulate differentiation of Tregs. Interestingly, programmed-death-ligand 1 (PD-L1) binding to programmed death 1 (PD-1), expressed by T cells, has been shown to induce the conversion of naïve CD4+ CD25− T cells into iTregs in mouse models (111, 112). PD-L1 expressing AAPCs could be a useful tool to study the role of this co-inhibitory molecule in a human context.
Cytokines are critical parameters to promote the differentiation of specific CD4+ T cell subsets and to sustain their expansion in vitro. To date, culture protocols with AAPCs include IL-2 as a T cell growth factor and enable expansion of T cells with Th1 or Th2 phenotype (100, 104). Several studies have documented the cytokine requirement for the differentiation of Th subsets upon polyclonal stimulation. For instance, the cytokine mixture TGF-β1, IL-1β, IL-6, and IL-23 is known to promote Th17 differentiation whereas TGF-β1 and IL-4 enable generation of Th9 cells (113, 114). Whether AAPCs with appropriate cytokine cocktails could induce antigen-specific CD4+ T cell subsets remains to be examined. An important challenge for ACT is to generate T cells that persist in vivo and maintain long-term proliferative and functional abilities. The complex role of cytokines in CD4+ T cell responses is not fully understood. Among them, IL-7, IL-15 as well as IL-6 and IL-21 are involved in CD4+ T cell proliferation and survival, and AAPC systems should allow to better define their functions (115–120).
In clinical immunology and for research purposes, AAPCs represent a convenient platform to monitor CD4+ T cell responses in patients against allergens, autoantigens, or infectious antigens, as it was shown with the antigen ADAMTS13 involved in acquired thrombotic thrombocytopenic purpura (121). In addition, since NIH/3T3-derived AAPCs are able to internalize and process whole protein antigens, and to present derived T cell epitopes, they could be useful to characterize new epitopes of therapeutic interest by mass spectrometry after HLA molecule immunoprecipitation and peptide elution.
Conclusion and Perspectives
CD4+ T cells orchestrate a broad range of immune responses. These last years, our understanding of the CD4+ T cell biology has largely progressed but remains incomplete. Indeed, diversity of CD4+ cell subsets is more complex than expected, as attested by the discovery of rare functional subsets and reports showing the influence of environmental elements in the plasticity of Th cells and Tregs (3, 14, 122). Understanding the mechanisms underlying functional heterogeneity of CD4+ T cells will provide the opportunity to better exploit their properties in protective immunity or in immune tolerance with the ultimate goal of developing potent immunotherapy strategies. Recent studies revealing the roles of Tfh and Th9 subsets in anti-infectious and anti-tumor immunity raise the question of which subtype of Th cells is more adapted to mediate a protective response against specific diseases. In the Treg field, future works are required to characterize the best source of Tregs for immunotherapy by determining relevant markers to distinguish tTregs from iTregs, and carrying out clinical trials to compare clinical benefits of autoantigen-specific Tregs vs. polyclonal Tregs in ACT.
The development of several AAPC systems has led to reproducible and robust expansion protocols for generating high numbers of human antigen-specific CD4+ T cells. In addition, some AAPCs could be used under Good Manufacturing Practice conditions and their safety and harmlessness have already been demonstrated in several clinical trials (123–125). AAPCs also have the advantage of being easily customizable by gene transfer to express any costimulatory or co-inhibitory molecule, and are therefore ideal for analyzing all kinds of CD4+ T cell responses and providing key knowledges to maximize the success of T cell expansion protocols for ACT.
Author Contributions
AC, AG, FD, OB, DV, BL-M, J-BL, and OT substantially contributed to the conception of this work, drafted and revised the manuscript, before finally approving its submission. AC, AG, FD, OB, DV, BL-M, J-BL, and OT agreed to be accountable for all aspects of the work.
Funding
This work was supported by grants from la Ligue contre le cancer de Normandie, l'Etablissement Français du Sang, la Fondation ARC pour la recherche sur le cancer, the European Regional Development Fund (ERDF) from European Union and Normandie Regional Council.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kim H-J, Cantor H. CD4 T-cell subsets and tumor immunity: the helpful and the not-so-helpful. Cancer Immunol Res. (2014) 2:91–8. doi: 10.1158/2326-6066.CIR-13-0216
2. Swain SL, McKinstry KK, Strutt TM. Expanding roles for CD4+ T cells in immunity to viruses. Nat Rev Immunol. (2012) 12:136–48. doi: 10.1038/nri3152
3. Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol. (2018) 19:665–73. doi: 10.1038/s41590-018-0120-4
4. Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol. (2010) 10:849–59. doi: 10.1038/nri2889
5. ten Broeke T, Wubbolts R, Stoorvogel W. MHC class II antigen presentation by dendritic cells regulated through endosomal sorting. Cold Spring Harb Perspect Biol. (2013) 5:a016873. doi: 10.1101/cshperspect.a016873
6. Poluektov YO, Kim A, Sadegh-Nasseri S. HLA-DO and its role in MHC class II antigen presentation. Front Immunol. (2013) 4:260. doi: 10.3389/fimmu.2013.00260
7. Adamopoulou E, Tenzer S, Hillen N, Klug P, Rota IA, Tietz S, et al. Exploring the MHC-peptide matrix of central tolerance in the human thymus. Nat Commun. (2013) 4:2039. doi: 10.1038/ncomms3039
8. Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. (1999) 285:221–227.
9. Stockinger B, Omenetti S. The dichotomous nature of T helper 17 cells. Nat Rev Immunol. (2017) 17:535–44. doi: 10.1038/nri.2017.50
10. Bystrom J, Clanchy FIL, Taher TE, Al-Bogami M, Ong VH, Abraham DJ, et al. Functional and phenotypic heterogeneity of Th17 cells in health and disease. Eur J Clin Invest. (2019) 49:e13032. doi: 10.1111/eci.13032
11. Nakayama T, Hirahara K, Onodera A, Endo Y, Hosokawa H, Shinoda K, et al. Th2 cells in health and disease. Annu Rev Immunol. (2017) 35:53–84. doi: 10.1146/annurev-immunol-051116-052350
12. Li J, Chen S, Xiao X, Zhao Y, Ding W, Li XC. IL-9 and Th9 cells in health and diseases-from tolerance to immunopathology. Cytokine Growth Factor Rev. (2017) 37:47–55. doi: 10.1016/j.cytogfr.2017.07.004
13. Vinuesa CG, Linterman MA, Yu D, MacLennan ICM. Follicular helper T cells. Annu Rev Immunol. (2016) 34:335–68. doi: 10.1146/annurev-immunol-041015-055605
14. DuPage M, Bluestone JA. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat Rev Immunol. (2016) 16:149–63. doi: 10.1038/nri.2015.18
15. Hirota K, Turner J-E, Villa M, Duarte JH, Demengeot J, Steinmetz OM, et al. Plasticity of Th17 cells in Peyer's patches is responsible for the induction of T cell-dependent IgA responses. Nat Immunol. (2013) 14:372–9. doi: 10.1038/ni.2552
16. van Langelaar J, van der Vuurst de Vries RM, Janssen M, Wierenga-Wolf AF, Spilt IM, Siepman TA, et al. T helper 17.1 cells associate with multiple sclerosis disease activity: perspectives for early intervention. Brain J Neurol. (2018) 141:1334–49. doi: 10.1093/brain/awy069
17. Cheuk S, Wikén M, Blomqvist L, Nylén S, Talme T, Ståhle M, et al. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol. (2014) 192:3111–20. doi: 10.4049/jimmunol.1302313
18. López P, de Paz B, Rodríguez-Carrio J, Hevia A, Sánchez B, Margolles A, et al. Th17 responses and natural IgM antibodies are related to gut microbiota composition in systemic lupus erythematosus patients. Sci Rep. (2016) 6:24072. doi: 10.1038/srep24072
19. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. (2011) 12:568–75. doi: 10.1038/ni.2031
20. Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol. (2011) 12:255–63. doi: 10.1038/ni.1993
21. Licona-Limón P, Henao-Mejia J, Temann AU, Gagliani N, Licona-Limón I, Ishigame H, et al. Th9 cells drive host immunity against gastrointestinal worm infection. Immunity. (2013) 39:744–57. doi: 10.1016/j.immuni.2013.07.020
22. Hong C-H, Chang K-L, Wang H-J, Yu H-S, Lee C-H. IL-9 induces IL-8 production via STIM1 activation and ERK phosphorylation in epidermal keratinocytes: a plausible mechanism of IL-9R in atopic dermatitis. J Dermatol Sci. (2015) 78:206–14. doi: 10.1016/j.jdermsci.2015.03.004
23. Clark RA, Schlapbach C. TH9 cells in skin disorders. Semin Immunopathol. (2017) 39:47–54. doi: 10.1007/s00281-016-0607-8
24. Schlapbach C, Gehad A, Yang C, Watanabe R, Guenova E, Teague JE, et al. Human TH9 cells are skin-tropic and have autocrine and paracrine proinflammatory capacity. Sci Transl Med. (2014) 6:219ra8. doi: 10.1126/scitranslmed.3007828
25. Malik S, Sadhu S, Elesela S, Pandey RP, Chawla AS, Sharma D, et al. Transcription factor Foxo1 is essential for IL-9 induction in T helper cells. Nat Commun. (2017) 8:815. doi: 10.1038/s41467-017-00674-6
26. Zhou Y, Sonobe Y, Akahori T, Jin S, Kawanokuchi J, Noda M, et al. IL-9 promotes Th17 cell migration into the central nervous system via CC chemokine ligand-20 produced by astrocytes. J Immunol Baltim Md 1950. (2011) 186:4415–21. doi: 10.4049/jimmunol.1003307
27. Gerlach K, Hwang Y, Nikolaev A, Atreya R, Dornhoff H, Steiner S, et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat Immunol. (2014) 15:676–86. doi: 10.1038/ni.2920
28. Raveney BJE, Oki S, Hohjoh H, Nakamura M, Sato W, Murata M, et al. Eomesodermin-expressing T-helper cells are essential for chronic neuroinflammation. Nat Commun. (2015) 6:8437. doi: 10.1038/ncomms9437
29. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol. (2013) 14:307–8. doi: 10.1038/ni.2554
30. Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med. (2013) 19:739–46. doi: 10.1038/nm.3179
31. Zeng H, Zhang R, Jin B, Chen L. Type 1 regulatory T cells: a new mechanism of peripheral immune tolerance. Cell Mol Immunol. (2015) 12:566–71. doi: 10.1038/cmi.2015.44
32. Hoffmann P, Ermann J, Edinger M, Fathman CG, Strober S. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J Exp Med. (2002) 196:389–99. doi: 10.1084/jem.20020399
33. Szanya V, Ermann J, Taylor C, Holness C, Fathman CG. The subpopulation of CD4+CD25+ splenocytes that delays adoptive transfer of diabetes expresses L-selectin and high levels of CCR7. J Immunol. (2002) 169:2461–5. doi: 10.4049/jimmunol.169.5.2461
34. McGeachy MJ, Stephens LA, Anderton SM. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J Immunol. (2005) 175:3025–32. doi: 10.4049/jimmunol.175.5.3025
35. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. (2004) 199:1455–65. doi: 10.1084/jem.20040139
36. Serr I, Fürst RW, Achenbach P, Scherm MG, Gökmen F, Haupt F, et al. Type 1 diabetes vaccine candidates promote human Foxp3(+)Treg induction in humanized mice. Nat Commun. (2016) 7:10991. doi: 10.1038/ncomms10991
37. Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. (2010) 207:637–50. doi: 10.1084/jem.20091918
38. Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. (2008) 112:362–73. doi: 10.1182/blood-2007-11-120998
39. Numasaki M, Watanabe M, Suzuki T, Takahashi H, Nakamura A, McAllister F, et al. IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J Immunol. (2005) 175:6177–89. doi: 10.4049/jimmunol.175.9.6177
40. Lu Y, Wang Q, Xue G, Bi E, Ma X, Wang A, et al. Th9 cells represent a unique subset of CD4+ T cells endowed with the ability to eradicate advanced tumors. Cancer Cell. (2018) 33:1048–60.e7. doi: 10.1016/j.ccell.2018.05.004
41. Purwar R, Schlapbach C, Xiao S, Kang HS, Elyaman W, Jiang X, et al. Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells. Nat Med. (2012) 18:1248–53. doi: 10.1038/nm.2856
42. Végran F, Berger H, Boidot R, Mignot G, Bruchard M, Dosset M, et al. The transcription factor IRF1 dictates the IL-21-dependent anticancer functions of TH9 cells. Nat Immunol. (2014) 15:758–66. doi: 10.1038/ni.2925
43. Borst J, Ahrends T, Babała N, Melief CJM, Kastenmüller W. CD4+ T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. (2018) 18:635–47. doi: 10.1038/s41577-018-0044-0
44. Dobrzanski MJ. Expanding roles for CD4 T cells and their subpopulations in tumor immunity and therapy. Front Oncol. (2013) 3:63. doi: 10.3389/fonc.2013.00063
45. Laidlaw BJ, Craft JE, Kaech SM. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat Rev Immunol. (2016) 16:102–11. doi: 10.1038/nri.2015.10
46. Castellino F, Huang AY, Altan-Bonnet G, Stoll S, Scheinecker C, Germain RN. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature. (2006) 440:890–5. doi: 10.1038/nature04651
47. Bourgeois C, Rocha B, Tanchot C. A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science. (2002) 297:2060–3. doi: 10.1126/science.1072615
48. Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. (2003) 421:852–6. doi: 10.1038/nature01441
49. Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol. (2004) 5:927–33. doi: 10.1038/ni1105
50. Williams MA, Tyznik AJ, Bevan MJ. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature. (2006) 441:890–3. doi: 10.1038/nature04790
51. Feau S, Arens R, Togher S, Schoenberger SP. Autocrine IL-2 is required for secondary population expansion of CD8(+) memory T cells. Nat Immunol. (2011) 12:908–13. doi: 10.1038/ni.2079
52. Oh S, Perera LP, Terabe M, Ni L, Waldmann TA, Berzofsky JA. IL-15 as a mediator of CD4+ help for CD8+ T cell longevity and avoidance of TRAIL-mediated apoptosis. Proc Natl Acad Sci USA. (2008) 105:5201–6. doi: 10.1073/pnas.0801003105
53. Elsaesser H, Sauer K, Brooks DG. IL-21 is required to control chronic viral infection. Science. (2009) 324:1569–72. doi: 10.1126/science.1174182
54. Janssen EM, Droin NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD, et al. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature. (2005) 434:88–93. doi: 10.1038/nature03337
55. Sacks JA, Bevan MJ. TRAIL deficiency does not rescue impaired CD8+ T cell memory generated in the absence of CD4+ T cell help. J Immunol. (2008) 180:4570–6. doi: 10.4049/jimmunol.180.7.4570
56. Ahrends T, Spanjaard A, Pilzecker B, Babała N, Bovens A, Xiao Y, et al. CD4+ T cell help confers a cytotoxic T cell effector program including coinhibitory receptor downregulation and increased tissue invasiveness. Immunity. (2017) 47:848–61.e5. doi: 10.1016/j.immuni.2017.10.009
57. Ozaki K, Spolski R, Feng CG, Qi C-F, Cheng J, Sher A, et al. A critical role for IL-21 in regulating immunoglobulin production. Science. (2002) 298:1630–4. doi: 10.1126/science.1077002
58. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. (2013) 39:782–95. doi: 10.1016/j.immuni.2013.10.003
59. Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, et al. CD4+ follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest. (2013) 123:2873–92. doi: 10.1172/JCI67428
60. Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. (2008) 358:2698–703. doi: 10.1056/NEJMoa0800251
61. Lu Y-C, Parker LL, Lu T, Zheng Z, Toomey MA, White DE, et al. Treatment of patients with metastatic cancer using a major histocompatibility complex class II-restricted T-cell receptor targeting the cancer germline antigen MAGE-A3. J Clin Oncol. (2017) 35:3322–9. doi: 10.1200/JCO.2017.74.5463
62. Linnemann C, van Buuren MM, Bies L, Verdegaal EME, Schotte R, Calis JJA, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. (2015) 21:81–5. doi: 10.1038/nm.3773
63. Mennonna D, Maccalli C, Romano MC, Garavaglia C, Capocefalo F, Bordoni R, et al. T cell neoepitope discovery in colorectal cancer by high throughput profiling of somatic mutations in expressed genes. Gut. (2017) 66:454–63. doi: 10.1136/gutjnl-2015-309453
64. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. (2015) 348:69–74. doi: 10.1126/science.aaa4971
65. Tran E, Turcotte S, Gros A, Robbins PF, Lu Y-C, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. (2014) 344:641–5. doi: 10.1126/science.1251102
66. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. (2017) 547:217–21. doi: 10.1038/nature22991
67. Drylewicz J, Schellens IMM, Gaiser R, Nanlohy NM, Quakkelaar ED, Otten H, et al. Rapid reconstitution of CD4 T cells and NK cells protects against CMV-reactivation after allogeneic stem cell transplantation. J Transl Med. (2016) 14:230. doi: 10.1186/s12967-016-0988-4
68. Leen AM, Heslop HE, Brenner MK. Antiviral T-cell therapy. Immunol Rev. (2014) 258:12–29. doi: 10.1111/imr.12138
69. Bollard CM, Heslop HE. T cells for viral infections after allogeneic hematopoietic stem cell transplant. Blood. (2016) 127:3331–40. doi: 10.1182/blood-2016-01-628982
70. Lin R, Liu Q. Diagnosis and treatment of viral diseases in recipients of allogeneic hematopoietic stem cell transplantation. J Hematol OncolJ Hematol Oncol. (2013) 6:94. doi: 10.1186/1756-8722-6-94
71. Gerdemann U, Keirnan JM, Katari UL, Yanagisawa R, Christin AS, Huye LE, et al. Rapidly generated multivirus-specific cytotoxic T lymphocytes for the prophylaxis and treatment of viral infections. Mol Ther J Am Soc Gene Ther. (2012) 20:1622–32. doi: 10.1038/mt.2012.130
72. Tzannou I, Papadopoulou A, Naik S, Leung K, Martinez CA, Ramos CA, et al. Off-the-shelf virus-specific T cells to treat BK virus, human herpesvirus 6, cytomegalovirus, Epstein-Barr virus, and adenovirus infections after allogeneic hematopoietic stem-cell transplantation. J Clin Oncol. (2017) 35:3547–57. doi: 10.1200/JCO.2017.73.0655
73. Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood. (2016) 127:1044–51. doi: 10.1182/blood-2015-06-653667
74. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. (2015) 7:315ra189. doi: 10.1126/scitranslmed.aad4134
75. Trzonkowski P, Bieniaszewska M, Juścinska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127− T regulatory cells. Clin Immunol. (2009) 133:22–6. doi: 10.1016/j.clim.2009.06.001
76. Zhang H, Podojil JR, Chang J, Luo X, Miller SD. TGF-beta-induced myelin peptide-specific regulatory T cells mediate antigen-specific suppression of induction of experimental autoimmune encephalomyelitis. J Immunol. (2010) 184:6629–36. doi: 10.4049/jimmunol.0904044
77. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MA, Hannen RF, Cooper D, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant. (2017) 17:931–43. doi: 10.1111/ajt.14185
78. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. (2016) 126:1413–24. doi: 10.1172/JCI82771
79. Almeida ARM, Legrand N, Papiernik M, Freitas AA. Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J Immunol. (2002) 169:4850–60. doi: 10.4049/jimmunol.169.9.4850
80. Sykes M, Romick ML, Sachs DH. Interleukin 2 prevents graft-versus-host disease while preserving the graft-versus-leukemia effect of allogeneic T cells. Proc Natl Acad Sci USA. (1990) 87:5633–7.
81. Grinberg-Bleyer Y, Baeyens A, You S, Elhage R, Fourcade G, Gregoire S, et al. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med. (2010) 207:1871–8. doi: 10.1084/jem.20100209
82. Koreth J, Kim HT, Jones KT, Lange PB, Reynolds CG, Chammas MJ, et al. Efficacy, durability, and response predictors of low-dose interleukin-2 therapy for chronic graft-versus-host disease. Blood. (2016) 128:130–7. doi: 10.1182/blood-2016-02-702852
83. Rosenzwajg M, Lorenzon R, Cacoub P, Pham HP, Pitoiset F, El Soufi K, et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Ann Rheum Dis. (2019) 78:209–17. doi: 10.1136/annrheumdis-2018-214229
84. Ephrem A, Chamat S, Miquel C, Fisson S, Mouthon L, Caligiuri G, et al. Expansion of CD4+CD25+ regulatory T cells by intravenous immunoglobulin: a critical factor in controlling experimental autoimmune encephalomyelitis. Blood. (2008) 111:715–22. doi: 10.1182/blood-2007-03-079947
85. Trinath J, Hegde P, Sharma M, Maddur MS, Rabin M, Vallat J-M, et al. Intravenous immunoglobulin expands regulatory T cells via induction of cyclooxygenase-2-dependent prostaglandin E2 in human dendritic cells. Blood. (2013) 122:1419–27. doi: 10.1182/blood-2012-11-468264
86. Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo M-G. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol. (2006) 177:8338–47. doi: 10.4049/jimmunol.177.12.8338
87. Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. (2002) 99:3838–43. doi: 10.1182/blood.V99.10.3838
88. Casiraghi F, Azzollini N, Cassis P, Imberti B, Morigi M, Cugini D, et al. Pretransplant infusion of mesenchymal stem cells prolongs the survival of a semiallogeneic heart transplant through the generation of regulatory T cells. J Immunol. (2008) 181:3933–46. doi: 10.4049/jimmunol.181.6.3933
89. English K, Ryan JM, Tobin L, Murphy MJ, Barry FP, Mahon BP. Cell contact, prostaglandin E(2) and transforming growth factor beta 1 play non-redundant roles in human mesenchymal stem cell induction of CD4+CD25(High) forkhead box P3+ regulatory T cells. Clin Exp Immunol. (2009) 156:149–60. doi: 10.1111/j.1365-2249.2009.03874.x
90. Cohen JA. Mesenchymal stem cell transplantation in multiple sclerosis. J Neurol Sci. (2013) 333:43–9. doi: 10.1016/j.jns.2012.12.009
91. Bassi ÊJ, Moraes-Vieira PMM, Moreira-Sá CSR, Almeida DC, Vieira LM, Cunha CS, et al. Immune regulatory properties of allogeneic adipose-derived mesenchymal stem cells in the treatment of experimental autoimmune diabetes. Diabetes. (2012) 61:2534–45. doi: 10.2337/db11-0844
92. Ringdén O, Uzunel M, Rasmusson I, Remberger M, Sundberg B, Lönnies H, et al. Mesenchymal stem cells for treatment of therapy-resistant graft-versus-host disease. Transplantation. (2006) 81:1390–7. doi: 10.1097/01.tp.0000214462.63943.14
93. Li Y, Kurlander RJ. Comparison of anti-CD3 and anti-CD28-coated beads with soluble anti-CD3 for expanding human T cells: differing impact on CD8 T cell phenotype and responsiveness to restimulation. J Transl Med. (2010) 8:104. doi: 10.1186/1479-5876-8-104
94. Godfrey WR, Ge YG, Spoden DJ, Levine BL, June CH, Blazar BR, et al. In vitro-expanded human CD4(+)CD25(+) T-regulatory cells can markedly inhibit allogeneic dendritic cell-stimulated MLR cultures. Blood. (2004) 104:453–61. doi: 10.1182/blood-2004-01-0151
95. Peggs KS, Thomson K, Samuel E, Dyer G, Armoogum J, Chakraverty R, et al. Directly selected cytomegalovirus-reactive donor T cells confer rapid and safe systemic reconstitution of virus-specific immunity following stem cell transplantation. Clin Infect Dis. (2011) 52:49–57. doi: 10.1093/cid/ciq042
96. Rubio MT, Means TK, Chakraverty R, Shaffer J, Fudaba Y, Chittenden M, et al. Maturation of human monocyte-derived dendritic cells (MoDCs) in the presence of prostaglandin E2 optimizes CD4 and CD8 T cell-mediated responses to protein antigens: role of PGE2 in chemokine and cytokine expression by MoDCs. Int Immunol. (2005) 17:1561–72. doi: 10.1093/intimm/dxh335
97. Peggs KS, Verfuerth S, Pizzey A, Khan N, Guiver M, Moss PA, et al. Adoptive cellular therapy for early cytomegalovirus infection after allogeneic stem-cell transplantation with virus-specific T-cell lines. Lancet Lond Engl. (2003) 362:1375–7. doi: 10.1016/S0140-6736(03)14634-X
98. da Silva Simoneti G, Saad STO, Gilli SCO. An efficient protocol for the generation of monocyte derived dendritic cells using serum-free media for clinical applications in post remission AML patients. Ann Clin Lab Sci. (2014) 44:180–188.
99. Butler MO, Ansén S, Tanaka M, Imataki O, Berezovskaya A, Mooney MM, et al. A panel of human cell-based artificial APC enables the expansion of long-lived antigen-specific CD4+ T cells restricted by prevalent HLA-DR alleles. Int Immunol. (2010) 22:863–73. doi: 10.1093/intimm/dxq440
100. Tanaka M, Butler MO, Ansén S, Imataki O, Berezovskaya A, Nadler LM, et al. Induction of HLA-DP4-restricted anti-Survivin Th1 and Th2 responses using an artificial antigen-presenting cell. Clin Cancer Res. (2011) 17:5392. doi: 10.1158/1078-0432.CCR-10-3083
101. Latouche JB, Sadelain M. Induction of human cytotoxic T lymphocytes by artificial antigen-presenting cells. Nat Biotechnol. (2000) 18:405–9. doi: 10.1038/74455
102. Maby P, Tougeron D, Hamieh M, Mlecnik B, Kora H, Bindea G, et al. Correlation between density of CD8+ T-cell infiltrate in microsatellite unstable colorectal cancers and frameshift mutations: a rationale for personalized immunotherapy. Cancer Res. (2015) 75:3446–55. doi: 10.1158/0008-5472.CAN-14-3051
103. Papanicolaou GA, Latouche J-B, Tan C, Dupont J, Stiles J, Pamer EG, et al. Rapid expansion of cytomegalovirus-specific cytotoxic T lymphocytes by artificial antigen-presenting cells expressing a single HLA allele. Blood. (2003) 102:2498–505. doi: 10.1182/blood-2003-02-0345
104. Garnier A, Hamieh M, Drouet A, Leprince J, Vivien D, Frébourg T, et al. Artificial antigen presenting cells expressing HLA class II molecules as an effective tool for amplifying human specific memory CD4+ T cells. Immunol Cell Biol. (2016) 94:662–72. doi: 10.1038/icb.2016.25
105. Butler MO, Hirano N. Human cell-based artificial antigen-presenting cells for cancer immunotherapy. Immunol Rev. (2014) 257:191–209. doi: 10.1111/imr.12129
106. Miller MA, Ganesan APV, Eisenlohr LC. Toward a network model of MHC class II-restricted antigen processing. Front Immunol. (2013) 4:464. doi: 10.3389/fimmu.2013.00464
107. Miller MA, Ganesan APV, Luckashenak N, Mendonca M, Eisenlohr LC. Endogenous antigen processing drives the primary CD4+ T cell response to influenza. Nat Med. (2015) 21:1216–22. doi: 10.1038/nm.3958
108. van Oosterwijk MF, Juwana H, Arens R, Tesselaar K, van Oers MHJ, Eldering E, et al. CD27-CD70 interactions sensitise naive CD4+ T cells for IL-12-induced Th1 cell development. Int Immunol. (2007) 19:713–8. doi: 10.1093/intimm/dxm033
109. Cannons JL, Lau P, Ghumman B, DeBenedette MA, Yagita H, Okumura K, et al. 4-1BB ligand induces cell division, sustains survival, and enhances effector function of CD4 and CD8 T cells with similar efficacy. J Immunol. (2001) 167:1313–24. doi: 10.4049/jimmunol.167.3.1313
110. Wen T, Bukczynski J, Watts TH. 4-1BB ligand-mediated costimulation of human T cells induces CD4 and CD8 T cell expansion, cytokine production, and the development of cytolytic effector function. J Immunol. (2002) 168:4897–906. doi: 10.4049/jimmunol.168.10.4897
111. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. (2009) 206:3015–29. doi: 10.1084/jem.20090847
112. Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, Noelle RJ. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc Natl Acad Sci USA. (2008) 105:9331–6. doi: 10.1073/pnas.0710441105
113. Yu RY, Gallagher G. A naturally occurring, soluble antagonist of human IL-23 inhibits the development and in vitro function of human Th17 cells. J Immunol. (2010) 185:7302–8. doi: 10.4049/jimmunol.1002410
114. Putheti P. Polarizing cytokines for human Th9 cell differentiation. Methods Mol Biol. (2017) 1585:73–82. doi: 10.1007/978-1-4939-6877-0_6
115. Chetoui N, Boisvert M, Gendron S, Aoudjit F. Interleukin-7 promotes the survival of human CD4+ effector/memory T cells by up-regulating Bcl-2 proteins and activating the JAK/STAT signalling pathway. Immunology. (2010) 130:418–26. doi: 10.1111/j.1365-2567.2009.03244.x
116. Van Belle TL, Dooms H, Boonefaes T, Wei X-Q, Leclercq G, Grooten J. IL-15 augments TCR-induced CD4+ T cell expansion in vitro by inhibiting the suppressive function of CD25high CD4+ T cells. PLoS ONE. (2012) 7:e45299. doi: 10.1371/journal.pone.0045299
117. Silva SL, Albuquerque AS, Matoso P, Charmeteau-de-Muylder B, Cheynier R, Ligeiro D, et al. IL-7-induced proliferation of human naive CD4 T-cells relies on continued thymic activity. Front Immunol. (2017) 8:20. doi: 10.3389/fimmu.2017.00020
118. Geginat J, Sallusto F, Lanzavecchia A. Cytokine-driven proliferation and differentiation of human naive, central memory, and effector memory CD4(+) T cells. J Exp Med. (2001) 194:1711–9. doi: 10.1084/jem.194.12.1711
119. Onoda T, Rahman M, Nara H, Araki A, Makabe K, Tsumoto K, et al. Human CD4+ central and effector memory T cells produce IL-21: effect on cytokine-driven proliferation of CD4+ T cell subsets. Int Immunol. (2007) 19:1191–9. doi: 10.1093/intimm/dxm090
120. Twohig JP, Cardus Figueras A, Andrews R, Wiede F, Cossins BC, Derrac Soria A, et al. Activation of naïve CD4+ T cells re-tunes STAT1 signaling to deliver unique cytokine responses in memory CD4+ T cells. Nat Immunol. (2019) 20:458–70. doi: 10.1038/s41590-019-0350-0
121. Gilardin L, Delignat S, Peyron I, Ing M, Lone Y-C, Gangadharan B, et al. The ADAMTS131239-1253 peptide is a dominant HLA-DR1-restricted CD4+ T-cell epitope. Haematologica. (2017) 102:1833–41. doi: 10.3324/haematol.2015.136671
122. Sallusto F. Heterogeneity of human CD4(+) T cells against microbes. Annu Rev Immunol. (2016) 34:317–34. doi: 10.1146/annurev-immunol-032414-112056
123. Mavilio F, Pellegrini G, Ferrari S, Di Nunzio F, Di Iorio E, Recchia A, et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat Med. (2006) 12:1397–402. doi: 10.1038/nm1504
124. Nakamura T, Inatomi T, Sotozono C, Amemiya T, Kanamura N, Kinoshita S. Transplantation of cultivated autologous oral mucosal epithelial cells in patients with severe ocular surface disorders. Br J Ophthalmol. (2004) 88:1280–4. doi: 10.1136/bjo.2003.038497
Keywords: CD4+ T lymphocytes, adoptive cell therapy, cancer, autoimmunity, artificial antigen presenting cells, HLA class II molecules
Citation: Couture A, Garnier A, Docagne F, Boyer O, Vivien D, Le-Mauff B, Latouche J-B and Toutirais O (2019) HLA-Class II Artificial Antigen Presenting Cells in CD4+ T Cell-Based Immunotherapy. Front. Immunol. 10:1081. doi: 10.3389/fimmu.2019.01081
Received: 10 January 2019; Accepted: 29 April 2019;
Published: 17 May 2019.
Edited by:
Nadia Caccamo, University of Palermo, ItalyReviewed by:
Giuliana Guggino, University of Palermo, ItalyValerie Dardalhon, UMR5535 Institut de Génétique Moléculaire de Montpellier (IGMM), France
Copyright © 2019 Couture, Garnier, Docagne, Boyer, Vivien, Le-Mauff, Latouche and Toutirais. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olivier Toutirais, dG91dGlyYWlzLW9AY2h1LWNhZW4uZnI=
†Present Address: Jean-Baptiste Latouche, UNIROUEN, Inserm U1234, IRIB, Department of Immunology and Biotherapy, Rouen University Hospital, Normandie University, Rouen, France