
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 05 July 2018
Sec. Immunological Tolerance and Regulation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01551
This article is part of the Research Topic Autoantibodies View all 89 articles
 Thomas Weisenburger1Bettina von Neubeck1Andrea Schneider1Nadja Ebert1Daniel Schreyer1Andreas Acs1Thomas H. Winkler1,2*
Thomas Weisenburger1Bettina von Neubeck1Andrea Schneider1Nadja Ebert1Daniel Schreyer1Andreas Acs1Thomas H. Winkler1,2*
Autoantibodies against double-stranded DNA (anti-dsDNA) are a hallmark of systemic lupus erythematosus (SLE). It is well documented that anti-dsDNA reactive B lymphocytes are normally controlled by immune self-tolerance mechanisms operating at several levels. The evolution of high levels of IgG anti-dsDNA in SLE is dependent on somatic hypermutation and clonal selection, presumably in germinal centers from non-autoreactive B cells. Twin studies as well as genetic studies in mice indicate a very strong genetic contribution for the development of anti-dsDNA as well as SLE. Only few single gene defects with a monogenic Mendelian inheritance have been described so far that are directly responsible for the development of anti-dsDNA and SLE. Recently, among other mutations, rare null-alleles for the deoxyribonuclease 1 like 3 (DNASE1L3) and the Fc gamma receptor IIB (FCGR2B) have been described in SLE patients and genetic mouse models. Here, we demonstrate that double Dnase1l3- and FcgR2b-deficient mice in the C57BL/6 background exhibit a very early and massive IgG anti-dsDNA production. Already at 10 weeks of age, autoantibody production in double-deficient mice exceeds autoantibody levels of diseased 9-month-old NZB/W mice, a long established multigenic SLE mouse model. In single gene-deficient mice, autoantibody levels were moderately elevated at early age of the mice. Premature autoantibody production was accompanied by a spontaneous hyperactivation of germinal centers, early expansions of T follicular helper cells, and elevated plasmablasts in the spleen. Anti-dsDNA hybridomas generated from double-deficient mice show significantly elevated numbers of arginines in the CDR3 regions of the heavy-chain as well as clonal expansions and diversification of B cell clones with moderate numbers of somatic mutations. Our findings show a strong epistatic interaction of two SLE-alleles which prevent early and high-level anti-dsDNA autoantibody production. Both genes apparently synergize to keep in check excessive germinal center reactions evolving into IgG anti-dsDNA antibody producing B cells.
The formation of antibodies against DNA is considered to be the serologic hallmark of systemic lupus erythematosus (SLE). Autoantibodies against double-stranded DNA (anti-dsDNA) antibodies are the most studied and the most enigmatic autoantibodies in SLE. Their presence correlates with nephritis both in human SLE patients and in mice with a spontaneous lupus-like disease (1–3).
Systemic lupus erythematosus has a strong genetic component as demonstrated by high concordance rates for disease manifestation as well as autoantibody development in monozygotic twins (4, 5). In rare cases, a monogenic cause for the development of SLE-like disease and autoantibody production illustrates the pathways that predispose for SLE. The strongest associations were found to loss of function of genes that are involved in clearance of apoptotic cells, chromatin, and nucleic acids (6). Homozygous mutations in the C1q genes are responsible for SLE or SLE-like disease (7) and C1q-deficient mice accumulate apoptotic cells in several tissues and develop anti-DNA autoantibodies and SLE in certain genetic backgrounds but not in the C57BL/6 background (8, 9). Homozygous mutations in the TREX1 gene encoding a DNA exonuclease present in the cytoplasm are associated with the Aicardi–Goutieres syndrome-1 with increased systemic type I interferon levels and antinuclear autoantibodies (10). Likewise, Trex1-deficient mice develop autoantibodies and SLE-like disease.
Other apparently monogenic causes of SLE-like disease and anti-DNA autoantibodies were described in mutant mice. The deficiency of the IgG inhibitory Fc γ receptor IIB (FcγRIIB) is associated with the development of SLE in mice with a strong influence of background genes, however (11, 12). In the absence of FcγRIIB, autoreactive B cells are found in the germinal centers and somatic hypermutation contributed to anti-DNA reactivity (13). In mice, a promoter variant in the Fcgr2b gene results in enhanced germinal center responses and autoantibody production (14). A defunctioning single nucleotide polymorphism (SNP) in the human FCGR2B gene is associated with susceptibility to SLE (15) and the upregulation of FcγRIIB in memory B cells is decreased in SLE patients (16). In addition, FcγRIIB plays an important regulatory function on dendritic cells (DCs), macrophages, and neutrophils [reviewed in Ref. (17)].
Another single gene defect leading to anti-DNA production and SLE-like disease is demonstrated by the observation that milk fat globule-EGF factor 8 has a critical role in removing apoptotic B cells in the germinal centers and that its absence can lead to autoimmune diseases (18).
More recently, deficiency in the Deoxyribonuclease 1 like 3 gene (DNASE1L3) was associated with SLE-like disease in Arabian, Turkish, and Italian families (19–21). SLE-like symptoms were associated with a hypocomplementemic urticarial vasculitis syndrome in several clinical cases. In Dnase1l3-deficient mice, anti-dsDNA autoantibodies develop, followed by a late onset of SLE-like disease (22). Dnase1l3 is part of four homologous mammalian extracellular deoxyribonucleases of the Dnase1 family, Dnase1, Dnase1l1, Dnase1l2, and Dnase1l3 (23).
Here, we describe a new conditional mouse model for Dnase1l3 deficiency and confirm the early appearance of IgG anti-dsDNA autoantibodies at moderate levels in the serum of these mice. The development of anti-dsDNA antibodies does not require the presence of toll-like receptor 9 (TLR9) but a strong epistatic interaction with a mutation in the Fcgr2b gene leads to strikingly early and high anti-dsDNA serum levels accompanied by spontaneous hyperactivation of germinal centers.
Mice were maintained in SPF conditions at the animal facility of the Friedrich-Alexander-University, Erlangen. C57BL/6 mice were obtained from Charles River, Sulzfeld, Germany. TLR9-deficient mice (24) were a kind gift of H. Wagner, Munich. Fcgr2b-deficient mice (25) backcrossed for 12 generations to C57BL/6 were obtained from Taconic and the yaa mutation was introduced by breeding in the yaa mutation from male B6.SB-Yaa/J mice obtained from the Jackson laboratory. For the generation of Dnase1l3-deficient mice, a targeting vector was obtained from the KOMP consortium (project CSD48807, clone HTGRS01006_A_B09) and partially re-sequenced. Standard techniques were used to transfect the V6.5 embryonic stem cell line (kindly provided by R. Jaenisch, Boston) derived from a C57BL/6x129Sv F1 cross (26). Homologous recombination was screened with left arm and right arm primers by PCR using LongAmp® Taq (New England Biolabs) according to the instructions of the manufacturer. Primers are listed in the Primer List in Data Sheet S2 in Supplementary Material. Correct integration was further verified by sequencing of the PCR products from both sides of the genomic integration. ES cells were injected into C57BL/6 blastocysts and chimeric mice were crossed with C57BL/6 mice. After germline transmission, one line was kept on C57BL/6x129sv mixed background. A pure backcross to C57BL/6 background was obtained by marker-assisted speed-congenics using 74 genomic SNP markers (LGC Genomics, KASP genotyping) covering all mouse chromosomes. After five generations, heterozygous offspring mice containing all marker loci from C57BL/6 were intercrossed to obtain C57BL/6 “knockout-first” Dnase1l3-deficient mice. After crossing with flp-deleter mice (27), Dnase1l3-deficient mice with a deleted exon 2 were created. For the establishment of Dnase1l3/Fcgr2b double-deficient mice, male Fcgr2b−/− yaa mice were intercrossed with C57BL/6 “knockout-first” Dnase1l3-deficient mice. In the F2 generation, homozygous Dnase1l3/Fcgr2b double-deficient male and female mice were selected and used for further breeding. Animal care and experiments were approved by the animal ethics committee of Regierung von Mittelfranken (Animal Ethics Number: 54-2532.1-21/09 and TS-05/5).
For cell sorting of macrophage and DC cell populations, spleen, liver, lung, brain, and lamina propria tissue from 8- to 14-week-old C57BL/6 mice was mechanically dissociated with a gentle MACS™ and the respective tissue dissociation kit (Miltenyi Biotec). Bone marrow cells were flushed from femurs of C57BL/6 mice. For analysis of germinal center cells and myeloid cells, spleens were harvested, crushed through a 100 µM nylon mesh filter and resuspended in RPMI 1640 medium (PAN-Biotech) supplemented with 10% FCS. After erythrocyte lysis (5 min at room temperature in 0.15 M NH4Cl, 0.02 M HEPES, 0.1 mM EDTA), cells were washed two times and resuspended in FACS buffer (PBS, 2% FCS, 1 mM EDTA) for staining with directly conjugated antibodies. For intracellular IFN-γ staining, cells were treated with Cell Stimulation Cocktail + Protein Transport Inhibitors (eBioscience) for 2 h at 37°C. Cells were stained extracellularly for TFH markers and were fixed and permeabilized with the FIX & PERM Kit (Nordic-MUbio) according to the manufacturer’s protocol. The following antibody conjugates were obtained from BioLegend: CD45-BV421, CD19-BV421, CD38-PE, GL7-APC, CD95-PECy7, CD11b-BV510, CD11b-PECy7, CD11c-APCCy7, LY6C-PerCPCy5.5, LY6G-FITC, CXCR5-BV421, and PD1-PECy7. The following antibody conjugates were obtained from eBioscience: MHCII-PE, CD4-FITC, CD44-APC, CD8-FITC, B220-PerCPCy5.5, IgD-PE, IgM-FITC, CD21-FITC, CD23-PE, and F4/80-AF647. BV510-conjugated anti-PSGL-1 and BV421-conjugated anti-F4/80 were obtained from BD Biosciences. APC-conjugated anti-Siglec-H was obtained from Miltenyi Biotec. Flow cytometry analysis was performed on a Cytoflex instrument (Beckman Coulter) and FlowJo v10.4 analysis software (FlowJo, LLC).
Levels of anti-dsDNA were measured by ELISA. 20 µg/ml % poly-l-lysine (Sigma-Aldrich) was used to precoat MaxiSorp plates (Nunc). After 2 h at RT, plates were coated with dsDNA from calf thymus (20 µg/ml; Sigma-Aldrich) in TE (pH 7.5) buffer overnight at 4°C. Plates were washed with PBS/0.05% Tween 20 and sera was added in 1/2 serial dilutions starting at 1/100. A serum pool obtained from 9-month-old diseased NZB/W mice served as internal standard. The starting dilution of 1/200 of the NZB/W serum was arbitrarily assigned to 100 relative units. Goat-anti-mouse IgG or goat-anti-mouse IgM, Fc-specific, coupled to horseradish peroxidase was used for detection (Jackson ImmunoResearch).
For qRT-PCR, RNA from FACS sorted cells was isolated by the RNeasy kit (QIAGEN) according to the manufacturer’s instructions. cDNA was synthesized by SCRIPTUM reverse transcriptase (Bio&Sell), and qRT-PCR was performed on a quantitative PCR system (Biorad CFX96) with Absolute qPCR SYBR Green Mix 2× (Thermo Scientific) using intron-spanning primers for Dnase1l3 and HPRT (see Primer List in Data Sheet S2 in Supplementary Material). In a second set of experiments, TaqMan® Assays for Dnase1l3 and GAPDH were performed on an Applied Biosystems 7500 machine. Calculation of relative expression levels were performed according to the relative ΔCt-method (28).
Hybridomas were generated from three female double-deficient mice that previously had a rise of IgG anti-dsDNA autoantibodies in the serum. As a fusion partner, SP2/0 cells obtained originally from F. Melchers, Basel were used. Standard techniques were applied and hybridoma supernatants were tested on day 10 after fusion for IgG anti-dsDNA antibodies by ELISA. Hybridoma cells from positive wells were expanded and subcloned by single cell sorting at least once.
Total RNA was isolated from the hybridomas with RNeasy (Qiagen) and variable genes of the hybridomas were cloned with RT-PCR using universal VH- and VL-primers (see Primer List in Data Sheet S2 in Supplementary Material) and the StrataClone PCR Cloning Kit. Inserts were Sanger sequenced with M13 primers annealing to the plasmid vector (LGC Genomics). The analysis for VH and VL gene usage, CDR-assignment and for potential somatic mutations was performed by V-QUEST (29) at the IMGT website.1 Sequence alignment was created with the Geneious Software (Biomatters Ltd.).
It was described that Dnase1l3 is primarily expressed in macrophages in liver and spleen (30). To obtain a more detailed view of the expression pattern of Dnase1l3 in hematopoietic cells in different tissues, we isolated lymphocyte, monocyte, macrophage, and DC populations from blood, bone marrow, spleen, lymph node, peritoneal cavity, liver, lung, brain, and lamina propria of the colon by FACS sorting and analyzed expression of Dnase1l3 by quantitative real-time PCR. The results are summarized in Table 1. Among lymphocytes, a low-level expression of Dnase1l3 was found in B1a B cells and marginal zone B cells, whereas conventional B cells as well as T cells did show no detectable expression. We did not detect any expression of Dnase1l3 in different blood or tissue monocytes, including inflammatory monocytes from mice infected with the murine cytomegalovirus. Also, granulocytes did not exhibit detectable expression. As described before (22), conventional DCs in the spleen but also in liver and lamina propria expressed the highest levels of Dnase1l3. Among tissue resident macrophages, we detected a more complex expression pattern. Red pulp macrophages from spleen, Kupffer cells from liver, as well as macrophages from the lamina propria expressed very high levels, whereas macrophages isolated from the bone marrow, the peritoneum, the brain (microglia), and lung (alveolar macrophages) showed no detectable expression. Lower levels were detected in subcapsular sinus macrophages as well as plasmacytoid DCs. In summary, we detected Dnase1l3 expression mainly in antigen-presenting cell populations like conventional DCs and B1a and marginal zone B cells as well as in some tissue resident macrophages.
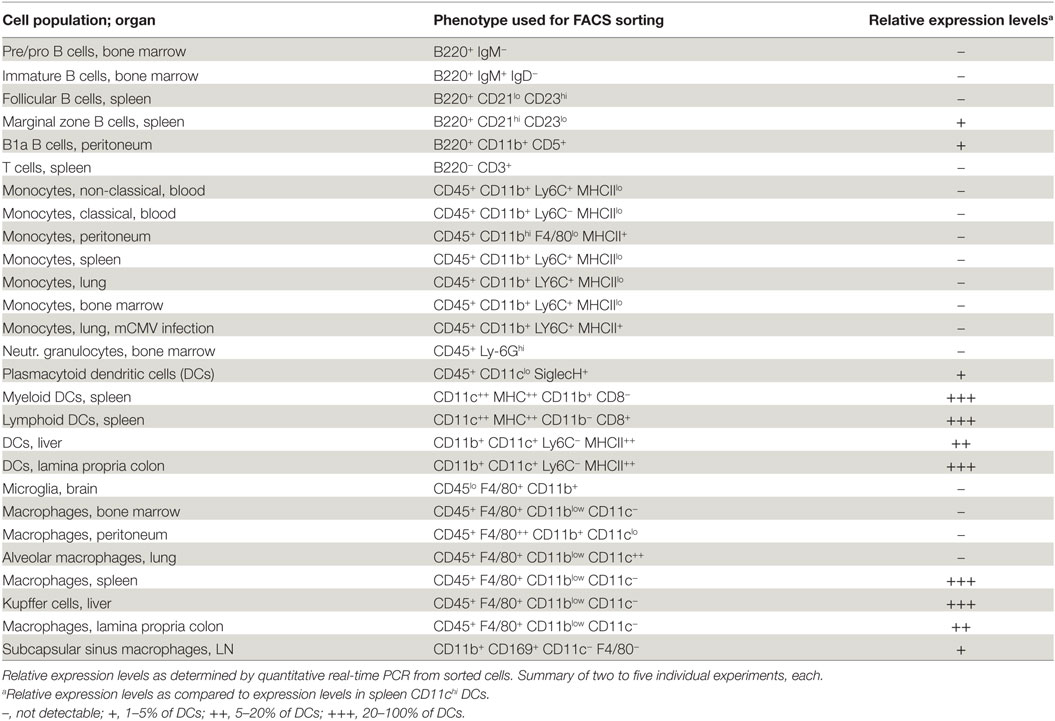
Table 1. Expression levels of Dnase1l3 in mouse hematopoietic cells.
To study the physiological function of Dnase1l3, we generated Dnase1l3-deficient mice using the “knockout-first” strategy as described by Skarnes et al. (31). The knockout-first allele is initially a non-expressive form, but can be converted to a conditional allele after recombination with Flp recombinase (Figure S1A in Supplementary Material). Transcripts are forced to be spliced from exon 1 of the Dnase1l3 gene to the splice acceptor side of a lacZ reporter and terminated by the downstream polyA sides. This should result in a knockout allele, as downstream exons are not transcribed (Figures S1A,B in Supplementary Material). To verify that downstream exons are not transcribed from the knockout-first allele [allele name termed Dnase1l3tm1a(KOMP)Wtsi according to Skarnes et al. (31), abbreviated as Dnase1l3tm1a], we used cDNA from MACS-enriched DCs from spleens of homozygous and littermate mice in an RT-PCR reaction using primers located in downstream exons 5 and 7 (Figure S1B in Supplementary Material). As shown in Figure S1C in Supplementary Material, transcripts from the mutated allele were undetectable by this sensitive PCR reaction. This shows that the knockout-first allele is a null allele, as expected from the analysis of many similar alleles generated by the KOMP program (32). We also crossed mice with the knockout-first allele with E2a-Cre deleter mice (33) to obtain an exon 2 knockout allele missing exon 2 which will be termed Dnase1l3Δex2.
We analyzed serum IgG anti-dsDNA autoantibodies from groups of mice at different ages. As shown in Figure 1A, homozygous Dnase1l3-deficient mice showed significantly elevated IgG anti-dsDNA antibody levels already at young age (3–4 months). At later time points (Figures 1B–D), these titers show only a moderate increase. Dnase1l3-deficient mice at 1-year of age still have significantly, but modestly increased IgG anti-dsDNA titers. All groups except group 4 in Figure 1B are derived from mice in a mixed 129 × C57BL/6 background. For the group of mice at the age of 18–26 weeks fully congenic C57BL/6 mice have similar anti-dsDNA antibody levels, suggesting that the mixed genetic background of the mice had little if any influence on the development of anti-dsDNA antibodies (Figure 1B). For a cohort of 16- to 28-week-old mice, the influence of the sex on the development of autoantibodies was analyzed. As shown in Figure S2 in Supplementary Material a tendency for higher anti-dsDNA autoantibodies in female versus male mice was observed, which was not statistically significant. In addition, we analyzed homozygous Dnase1l3Δex2 mice with a complete deletion of exon 2 of Dnase1l3 and found similar levels of anti-dsDNA autoantibodies as compared to mice with a homozygous Dnase1l3tm1a mutation, further supporting the notion that Dnase1l3tm1a mutation is a null mutation (Figure 2A). Thus, Dnase1l3-deficient mice spontaneously develop moderate levels of anti-DNA autoantibodies already at 3-month of age which are not increasing significantly upon aging. Neither a significant contribution of residual 129 genetic background nor any significant influence of the sex of the mice influenced anti-dsDNA autoantibodies.
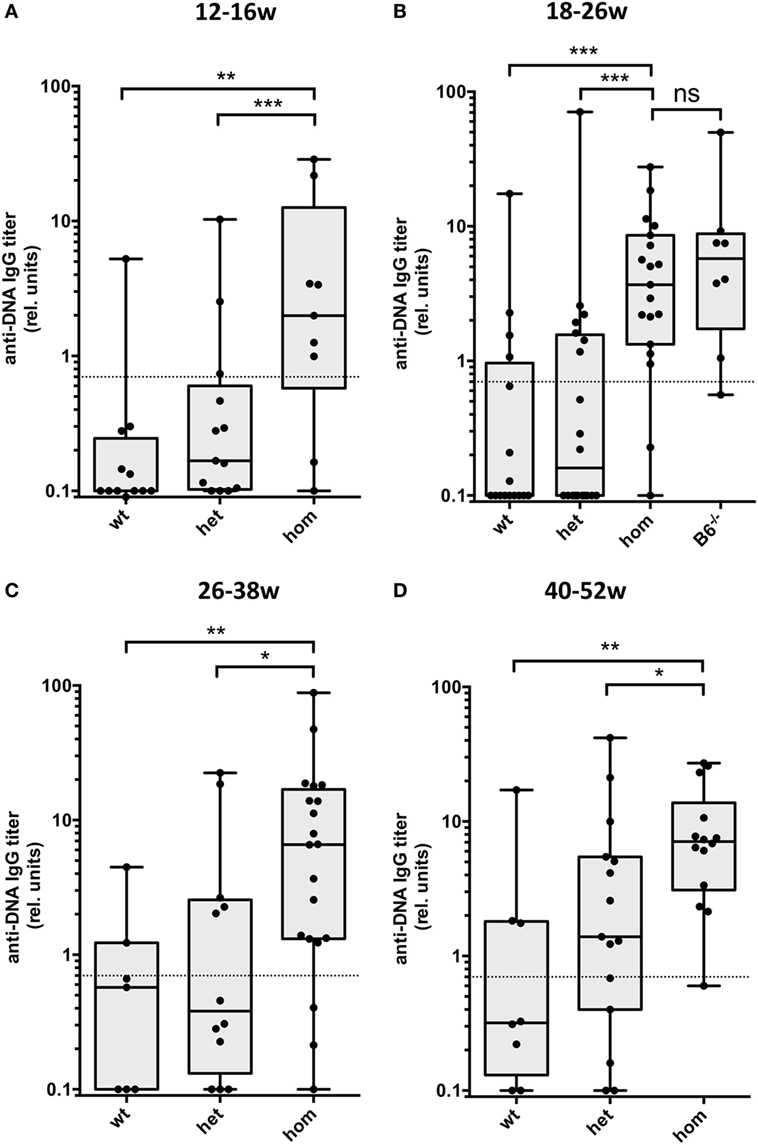
Figure 1. Development of IgG autoantibodies against double-stranded DNA (anti-dsDNA) antibodies in Dnase1l3-deficient mice. Sera from mice at the age of (A) 12–16 weeks, (B) 18–26 weeks, (C) 26–38 weeks, and (D) 40–52 weeks were tested for IgG anti-dsDNA antibodies by ELISA. The mice are grouped according to the genotype of the Dnase1l3tm1a mutation. All cohorts of mice are derived from 129 × C57BL/6 mixed background except homozygous mutant mice in (B) which are backcrossed to pure C57BL/6 background (B6−/−). Relative binding units are presented as compared to a standard serum pool from 9-month-old NZB/W mice. The dotted lines represent a cutoff derived from a panel of sera from C57BL/6 mice. Data are presented as box plot with individual data represented by filled circles (*p < 0.05; **p < 0.01; ***p < 0.001 Mann–Whitney U test).
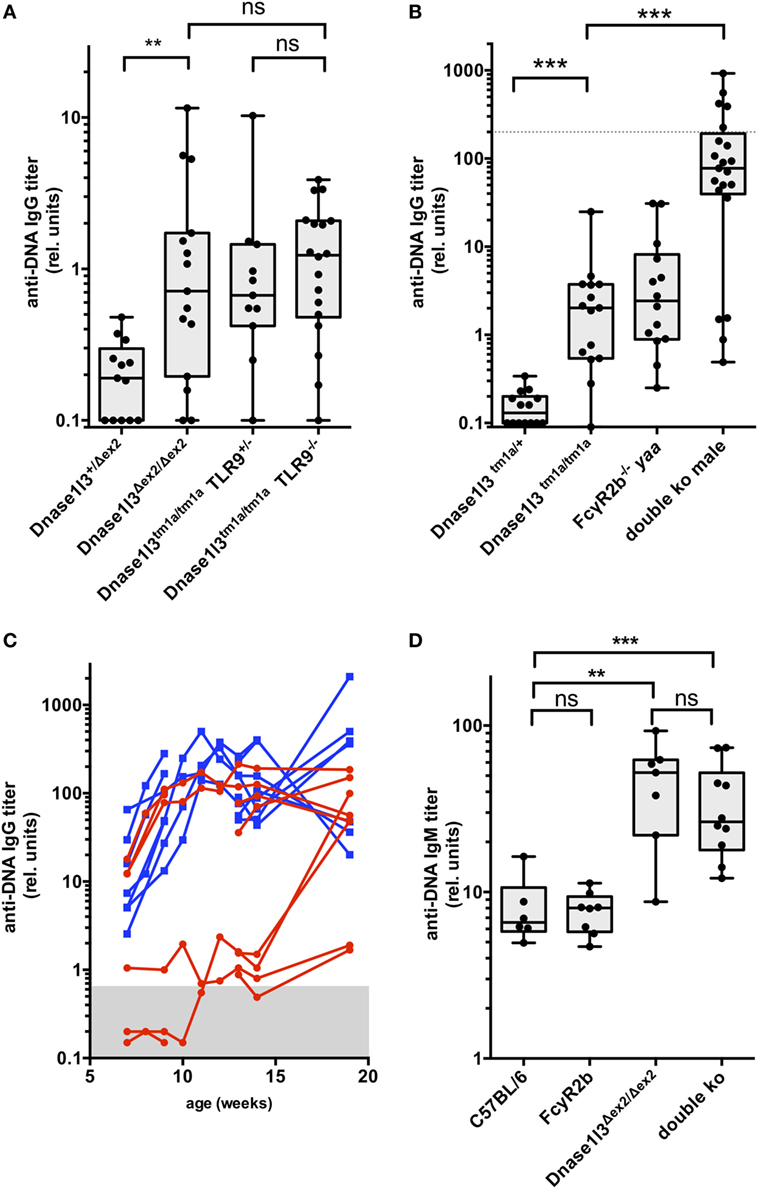
Figure 2. Development of IgG and IgM autoantibodies against double-stranded DNA (anti-dsDNA) antibodies in double-deficient mice. In (A), the results from 3- to 4-month-old toll-like receptor 9 (TLR9) × Dnase1l3 mice are displayed. The genotypes of the mice are denoted at the axis. In (B), the results from 3-month-old FcgR2b × Dnase1l3 mice are displayed. The genotypes of the mice are denoted at the axis. The dotted lines represent anti-dsDNA antibody levels of a serum pool of 9-month-old female NZB/W mice. Data are presented as box plot with individual data represented by filled circles. (C) Follow-up of IgG anti-dsDNA autoantibody levels in individual female FcgR2b × Dnase1l3 double-deficient mice (red) and male FcgR2b yaa × Dnase1l3 double-deficient mice (blue). In (D), the IgM anti-dsDNA levels from a cohort of 2- to 3-month-old FcgR2b × Dnase1l3 mice are displayed. The genotypes of the mice are denoted at the axis. Data are presented as box plot with individual data represented by filled circles (*p < 0.05; **p < 0.01; ***p < 0.001 Mann–Whitney U test).
As anti-dsDNA autoantibody levels remained moderately elevated in Dnase1l3-deficient mice up to an age of 1 year, we asked the question whether other relevant single mutations might influence anti-dsDNA autoantibody titers. First, we intercrossed Dnase1l3-deficient mice with TLR9-deficient mice to obtain double-deficient mice. As demonstrated in Figure 2A, additional TLR9-deficiency had no significant influence on anti-dsDNA titers in Dnase1l3-deficient mice. We also generated double-deficient mice for Dnase1l3 and Fcgr2b in which male mice in addition contain the yaa mutation accelerating SLE-like disease in the FcγRIIB-deficient background (12). As shown in Figure 2B, we observed extremely high anti-dsDNA titers in double-deficient male mice at the age of 3-month reaching anti-dsDNA levels similar to 9-month-old NZB/W female mice. At this age, FcγRIIB−/− yaa male mice had anti-dsDNA titers comparable to single Dnase1l3-deficient mice which were around 50-fold lower than in male double-deficient mice (Figure 2B).
Since these anti-dsDNA antibody levels in double-deficient male mice were higher than in any other SLE-prone strain of mice including MRL/lpr and NZB/W mice at the same age, we weekly analyzed anti-dsDNA antibodies in sera starting at the age of 7 weeks. We included also female mice in this analysis. As shown in Figure 2C, already at the age of 7 weeks all male mice (blue curves in Figure 2C) had clearly elevated anti-dsDNA serum levels that might have be transferred from the double-deficient mothers via placental transfer. We consider this unlikely, however, as some female littermate mice at the same age had normal levels of anti-dsDNA antibodies in their sera (Figure 2C). In all male mice of this cohort and in three out of six female mice which we analyzed at this young age, IgG anti-dsDNA antibody levels massively increased within 3–5 weeks up to 50-fold, reaching a maximum level around 10–12 weeks of age. Most of the other female mice developed these maximum levels of autoantibodies at the age of 15–20 weeks. Thus, mutations in Dnase1l3 and Fcgr2b show strong epistasis and the yaa mutation further increases the penetrance of early hyperproduction of anti-dsDNA antibodies in male mice in this model.
As IgM anti-DNA autoantibodies typically develop early in SLE models, we analyzed IgM anti-dsDNA levels in a cohort of 2- to 3-month-old animals of the different genotypes. Interestingly, Dnase1l3 single-deficient mice show highly elevated IgM anti-dsDNA autoantibodies comparable to double-deficient mice, whereas FcγRIIB-knockout mice had normal IgM anti-dsDNA levels (Figure 2D). Thus, loss of tolerance toward DNA is an early event in Dnase1l3-deficient mice.
We have proposed earlier that anti-dsDNA antibodies evolve from non-autoreactive progenitors in germinal centers (34, 35) and strong evidence has been accumulated that the FcγRIIB plays an important role in B cell tolerance in the GC (17). We therefore analyzed spontaneous germinal center development in double-deficient mice at the age of 9–14 weeks. As shown in Figure 3A, all male double-deficient mice had elevated frequencies of CD38lo, GL7+, Fas+ GC B cells in the spleen when compared to both single deficient male or female mice or wild-type C57BL/6 mice. In this cohort, one out of five female double-deficient mice showed a dramatically elevated GC B cell frequency. Therefore, the early and strong anti-dsDNA antibody response is accompanied by GC hyperactivation in double-deficient mice.
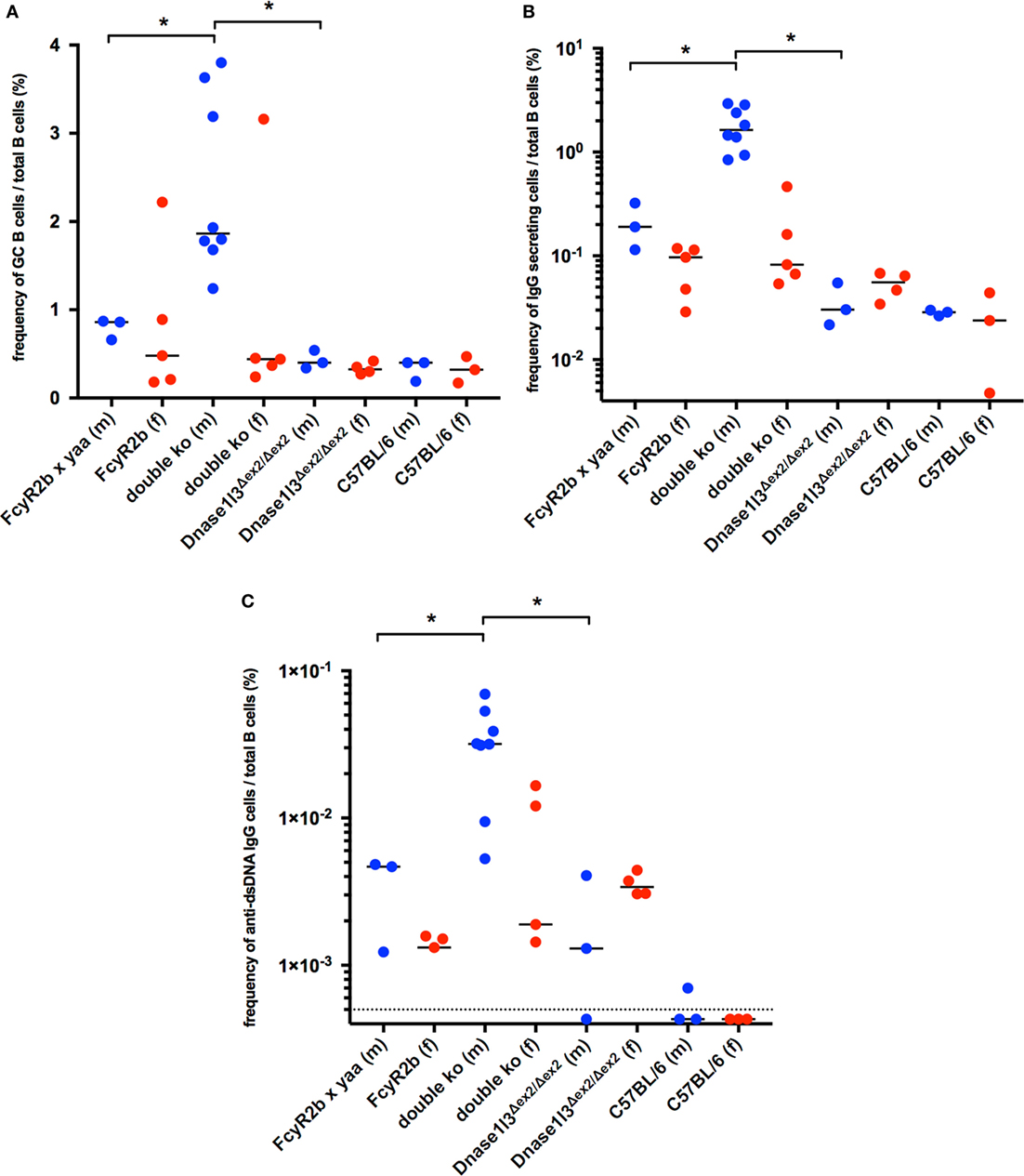
Figure 3. Spontaneous germinal center development and elevated production of autoantibodies against double-stranded DNA (anti-dsDNA) secreting cells in Dnase1l3 and FcgR2b double-deficient mice. (A) Analysis of splenic B cells from 9- to 14-week-old mice by flow cytometry. The frequency of GL7+, Fas+ germinal center B cells among all CD19+ B cells is displayed. The frequency of total IgG (B) and IgG anti-dsDNA (C) secreting B cells was determined by Elispot. Blue circles represent male mice, red circles represent female mice; dotted line in (C) represents the limit of detection (*p < 0.05; Mann–Whitney U test).
The activation of B cells in the spleens of double-deficient mice is also accompanied by high frequencies of IgG—antibody secreting plasma blasts or plasma cells (Figure 3B). Up to 3% of all B cells were found to be IgG antibody secreting cells, suggesting a considerable hyperactivation of the B cell compartment. Only a fraction of these IgG secreting B cells was anti-dsDNA specific (0.6–3.8% of all IgG secreting cells, Figures 3B,C).
To gain further insight into the mechanism of anti-dsDNA development in double-deficient mice, we prepared hybridomas from spleens of unimmunized mice and selected IgG anti-dsDNA hybridomas for the sequence analysis of VH- and in selected case also VL-genes. For this analysis, we focused on female double-deficient mice that were selected for previous rise of IgG anti-dsDNA autoantibodies before fusion. Data for the VH-genes are summarized in Table 2. Most hybridoma clones used the IgG2c isotype (25/31 clones) followed by a few IgG2b and one IgG3 anti-dsDNA hybridoma, similar to anti-DNA autoantibodies in other SLE-prone mice (36). In all three mice that were used for the generation of hybridomas we observed clonally related hybridomas clearly derived from one B cell and diversified by somatic mutations. We detected somatic mutations in the heavy chains of all hybridomas (Table 2; Figure S3 in Supplementary Material). In many cases, the numbers of mutations within the VH and VL gene region were relatively low with few replacement mutations present. For those clones that contributed with three or more individual hybridomas, we analyzed the diversification from the germline sequence as well as intraclonal diversification in more detail. Very few shared mutations were observed within the clonal relatives as shown in Figure S3 in Supplementary Material suggesting a “bush-like” diversification by somatic hypermutation from a single B cell rather than a stepwise maturation with intermediate selection.
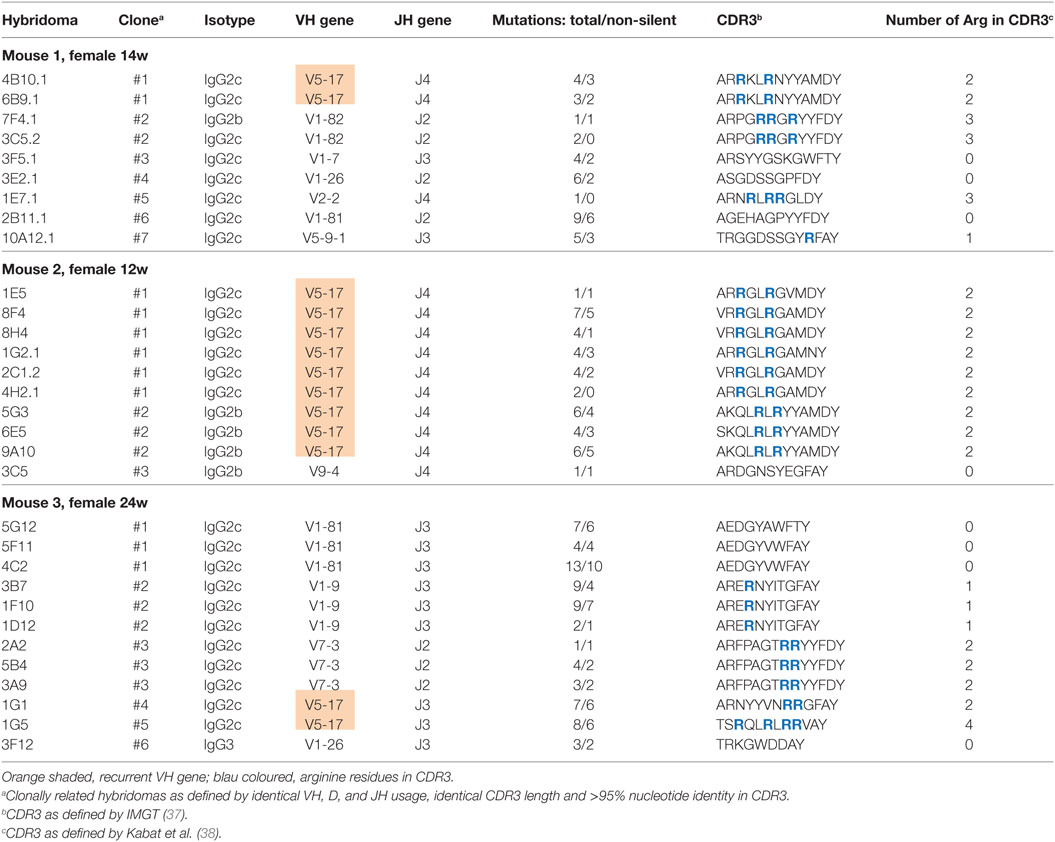
Table 2. Summary of V region gene analysis from autoantibodies against double-stranded DNA hybridomas.
We found that the CDR3 regions of the heavy chains of the hybridomas have an unusually high frequency of arginines, as it has been noted before in collections of anti-dsDNA hybridomas from SLE-prone mouse strains (39). Whereas 6,203 arginines can be found among 166,000 amino acids (3.7%) in the CDR3 region of mouse antibodies in the large abYsis antibody database,2 all anti-dsDNA hybridomas together described here have a frequency of 16.2% (47 arginines among 289 CDR3 amino acids; p < 0.001, Chi-Square). Interestingly, we found a recurrent usage of one VH gene (VH5-17) among expanded clones in all three independent mice (shaded in Table 2). In summary, sequence analysis of anti-dsDNA hybridomas showed clear evidence for clonal expansion and somatic mutation among the anti-dsDNA hybridomas from double-deficient mice.
To get some mechanistic insight into the early spontaneous formation of germinal centers, we analyzed cohorts of young (8–9 weeks of age) and older mice (14–19 weeks) of single- and double-knockout mice for myeloid cell populations as well as T helper cell subpopulations in the spleen. The analysis of the frequency of monocytes, DCs, plasmacytoid DCs, and macrophages in the spleen did not reveal significant changes among single- or double-mutant mice as compared to C57BL/6 mice of similar age (data not shown). We observed an increase of Ly6G-positive neutrophils in the spleen of older FcγRIIB- as well as double-knockout mice, most likely reflecting inflammatory activity (Figure 4A). General activation of the CD4+ T cell compartment as evidenced by high levels of CD44 expression was observed in older FcγRIIB- as well as in double-knockout mice, but interestingly not in older Dnase1l3 single knockout animals (Figure 4B). Expansions of CD44hi TH cells were not observed in any cohort of young mice. T cell hyperactivation was particularly high in male animals carrying the yaa allele as denoted by the blue symbols but not restricted to yaa-animals in the cohorts (Figure 4B). We observed an expansion of the PD1hi PSGL-1lo TFH cells in older FcγRIIB-knockout mice and particularly striking in double-knockout mice (Figure 4C). Notably, TFH cell are significantly expanded already in young double-knockout mice (Figure 4C). The frequency of IFN-γ producing CD4+ T cells was elevated in both single-knockout strains of older age and strongly elevated in older double-knockout animals (Figure 4D). Thus, early expansion of TFH cells in the spleen correlates with the enhanced spontaneous germinal center development and high-level IgG anti-dsDNA production in double-deficient mice.
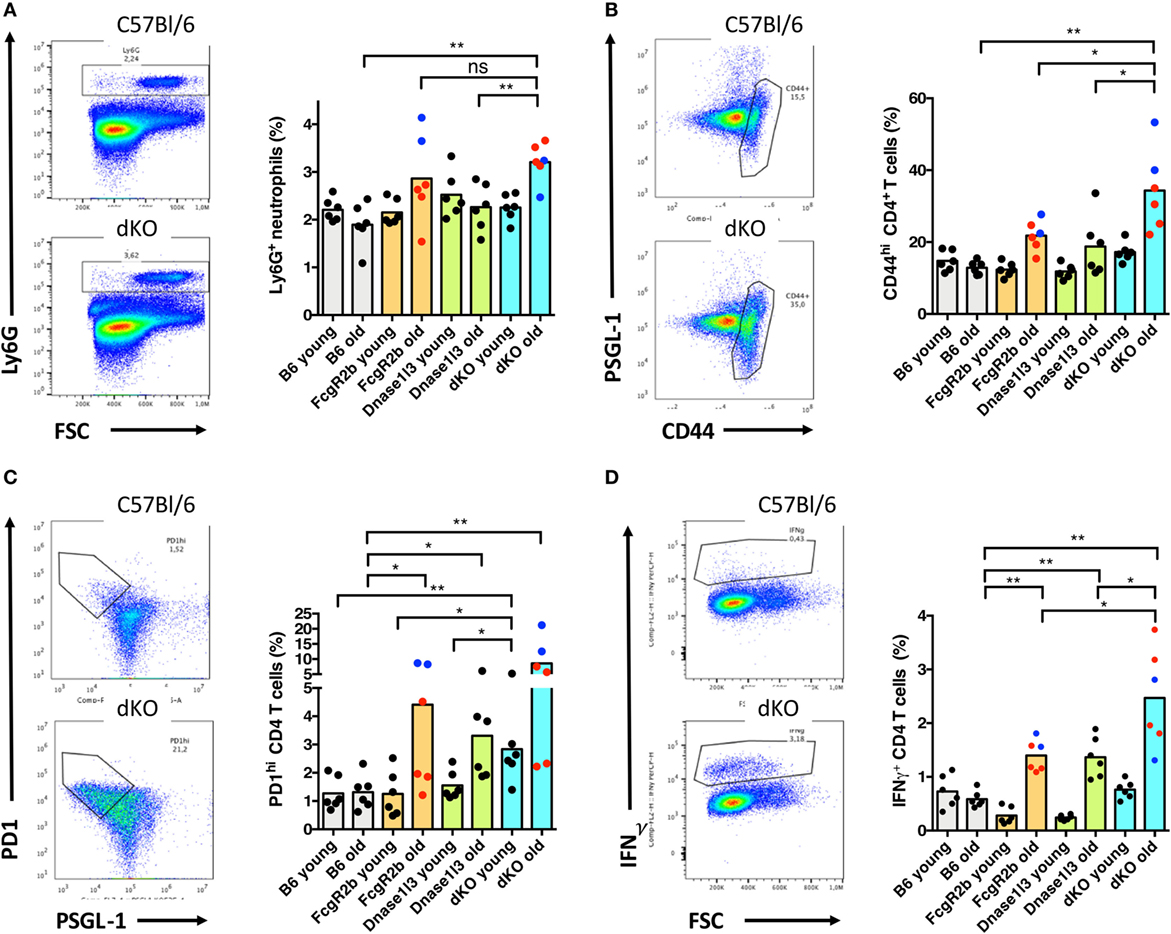
Figure 4. Early expansion of T follicular helper (TFH) cells and elevated levels of neutrophils in older Dnase1l3 and FcgR2b double-deficient mice. (A) Analysis of the expansion of Ly6G-positive neutrophils in the spleen of young (8–9 weeks old) and older (14–19 weeks old) mice. The genotypes of the mice are denoted at the axis. In (B), activation of the CD4+ T cell compartment is displayed by the frequency of CD44hi CD4 cells. (C) Analysis of the expansions of PD1hi PSGL-1lo TFH cells. In (D), the frequency of IFN-γ producing CD4+ T cells is displayed. As an example for each analysis, a density plot is shown for wild-type and double-deficient mice. Blue circles specifically highlight male mice, red circles represent female mice (*p < 0.05; **p < 0.01; Mann–Whitney U test).
The results described in this study reveal a strong genetic interaction of two individual susceptibility genes for anti-dsDNA autoantibody production in SLE that have been described in familiar lupus, as well as in knockout mouse models. Null mutations in the DNASE1L3 gene were described in familial forms of SLE in Saudi Arabia, Turkey, and Italy (19–21). Anti-dsDNA autoantibodies were observed in almost all of the cases reported. In addition, a potentially defunctioning missense SNP present in relatively high frequency in the Caucasian population (40) shows a very high association with anti-centromere antibody-positive systemic sclerosis (41). For FCGR2B a SNP with considerable variation in frequency among human populations results in threonine replacing isoleucine in the transmembrane domain of the receptor (15). This causes the variant receptor to be excluded from sphingolipid rafts, resulting in a malfunctioning receptor (15). As both SNPs have a relatively high frequency within the Caucasian and Asian population (the missense SNP in Dnase1l3 is rare among populations with African origin3), we consider it highly worthwhile to analyze the frequency of these two variations among SLE patients. Current GWAS studies can explain only a minority of the heritability of complex diseases, a phenomenon that has been termed missing heritability (42). Epistatic interactions would be difficult to detect in GWAS studies because of computational challenges and low statistical power (43). We are currently analyzing such a possible epistatic interaction of Dnase1l3 and FcγRIIB in a German/French SLE cohort from the upper Rhine.
In our analysis of cohorts of double-mutant mice, we also introduced the yaa allele that results in a TLR7 gene-duplication (44, 45). The analysis of female double-mutant mice and male double-mutant carrying the yaa mutation clearly revealed an influence of yaa on the penetrance of development of high levels of anti-dsDNA antibodies very early. Only approximately 50% of female mice developed high levels of anti-dsDNA antibodies at the age of 13 weeks. This is in accordance with the notion that yaa accelerates systemic autoimmunity as a genetic modifier in a context of a coexisting SLE background (44, 46). Also, the penetrance of early hyperactivation of germinal centers is much lower in female double-deficient mice, consistent with the described role of the yaa allele for defective germinal center selection in SLE (47). Importantly, double-deficient yaa male mice at the age of 3 months showed about 40 times higher anti-dsDNA levels when compared to FcγRIIb−/− yaa male mice of the same age.
Double-mutant mice showed a hyperactivation of spontaneously developing germinal centers which correlates with an early expansion of TFH cells in the spleen. Expansions of TFH cells were first noted in sanroque mutant mice with a lupus-like disease and autoantibody production (48) and later also in some SLE patients with severe disease (49). As TFH cells are essential for germinal center responses (50), our findings here in the context of two epistatically acting mutations in the C57BL/6 genetic background further strengthen the model that dysregulated germinal center reactions are crucially involved in the early phases of the generation of IgG anti-dsDNA autoantibodies and the development of SLE (35, 50).
In FcγRIIB-deficient mice, an increase in anti-DNA reactive GC B cells was observed and somatic mutations contributed to the generation of highly autoreactive IgG antibodies (13). Opposite to the findings in single Fcgr2b-deficient mice, however, we observed clonal expansions of anti-DNA reactive B cells as well as concomitant expansion of anti-DNA plasmablasts or plasma cells. These combined observations prompt a further development of our model of the evolution of anti-DNA autoantibodies in germinal centers (35). FcγRIIB and potentially other negative regulators for B cell signaling (51) might play a major role in the regulation of autoreactive GC B cells which otherwise would develop from non-self-reactive or low-level self-reactive precursors by somatic mutations (34). We now propose that the deficiency to eliminate nuclear material would drive such autoreactive B cell from the germinal center into clonal expansion and plasma cell differentiation, presumably outside of the germinal center. Our sequence analysis points in the direction that these B cells mutated only for limited periods of time. The “bush-like” diversification by somatic hypermutation from a single B cell rather than a stepwise maturation with intermediate selection is compatible with this model. Similar “bush-like” genealogies were also observed in SLE-prone mice (52, 53). An extraordinary high content of arginine residues in the CDR3 region of the heavy chain, which is also observed in our study here, apparently is a very common prerequisite for such an evolution of anti-dsDNA antibodies (39). Recent work attributed removal of apoptotic microparticles from the circulation as a major role of Dnase1l3 to prevent loss of tolerance to chromatin (22). We suggest an alternative explanation which is mutually not exclusive and might operate at local sites, particularly in the liver, the spleen, and lymph nodes. We found the highest levels of Dnase1l3 expression in cells with excellent antigen-presenting competence, high expression of scavenging receptors and TLRs, i.e., DCs and Kupffer cells in the liver. Local secretion of Dnase1l3 might function as a kind of shield to prevent these cells from being activated by damage-associated molecular patterns via TLRs and other receptors (54, 55).
In the same direction, local Dnase1l3 secretion might eliminate or lower the activation of DCs after uptake of DNA-containing immune complexes via Fc receptors which has the potential to trigger maturation and interferon production of DCs (17). Under non-inflammatory conditions, the inhibitory FcγRIIB would dominate activating signals from activating Fcγ receptors on DCs (17), however. This could reflect the situation in our single Dnase1l3-deficient mouse, in which anti-dsDNA production is stalled at relatively moderate levels. When FcγRIIB is blocked on human DCs, high levels of IL-12p70 can be induced when antibody coated tumor cells are delivered to the DCs (56) and type I interferon is induced after delivery of immune complexes to DCs (57). This scenario might explain the phenotype of Dnase1l3/FcγRIIB double-deficient animals described here. Under these circumstances, any DNA-containing immune complex could potentially trigger DC maturation and production of type I interferon and stimulate TH1 driven responses. A self-perpetuating feedback loop would result when anti-dsDNA autoantibodies have evolved in such a scenario (58). For the initial trigger, IgG anti-dsDNA antibodies are not an essential content in the stimulating immune complexes. Pathogen-derived DNA together with pathogen-specific antibodies would be sufficient for starting such a DC-initiated self-perpetuating feedback loop.
Our conditional Dnase1l3 allele as well as conditional deletion of FcγRIIB will allow the detailed analysis of the cell types that are essential for the protection against uncontrolled evolution of anti-dsDNA autoantibodies in germinal centers. Potentially, comprehensive understanding of this regulation might allow the development of new targeted therapies for SLE.
Animal care and experiments were approved by the animal ethics committee of Regierung von Mittelfranken (Animal Ethics Number: 54-2532.1-21/09 and TS-05/5).
TW and BN performed experiments, analyzed data, and interpreted data. AS, NE, DS, and AA performed experiments and analyzed data. THW performed experiments, conceived the experiments, analyzed data, interpreted data, and wrote the manuscript. All authors were involved in critically reading of the manuscript and approved the final version to be published.
No potential conflict of interest relevant to this publication is reported.
The reviewer, RM and the handling Editor declared their shared affiliation.
The authors wish to thank Stefanie Brey for expert technical assistance. We also thank Uwe Appelt and Markus Mroz (FACS core facility at the Nikolaus-Fiebiger-Center for Molecular Medicine, FAU, Erlangen) for cell sorting. We also thank Natalya Seredkina and Ole-Petter Rekvig (The Arctic University of Norway, Tromsø, Norway) for helpful discussions. The vector used for this research project was generated by the trans-NIH Knock-Out Mouse Project (KOMP) and obtained from the KOMP Repository (www.komp.org).
This work was funded by the Deutsche Forschungsgemeinschaft through the grant TRR130 (project P11 and C03) to THW.
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01551/full#supplementary-material.
1. Ter Borg EJ, Horst G, Hummel EJ, Limburg PC, Kallenberg CG. Measurement of increases in anti-double-stranded DNA antibody levels as a predictor of disease exacerbation in systemic lupus erythematosus. A long-term, prospective study. Arthritis Rheum (1990) 33:634–43. doi:10.1002/art.1780330505
2. Ehrenstein MR, Katz DR, Griffiths MH, Papadaki L, Winkler TH, Kalden JR, et al. Human IgG anti-DNA antibodies deposit in kidneys and induce proteinuria in SCID mice. Kidney Int (1995) 48:705–11. doi:10.1038/ki.1995.341
3. Manson JJ, Ma A, Rogers P, Mason LJ, Berden JH, Van Der Vlag J, et al. Relationship between anti-dsDNA, anti-nucleosome and anti-alpha-actinin antibodies and markers of renal disease in patients with lupus nephritis: a prospective longitudinal study. Arthritis Res Ther (2009) 11:R154. doi:10.1186/ar2831
4. Deapen D, Escalante A, Weinrib L, Horwitz D, Bachman B, Roy-Burman P, et al. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum (1992) 35:311–8. doi:10.1002/art.1780350310
5. Reichlin M, Harley JB, Lockshin MD. Serologic studies of monozygotic twins with systemic lupus erythematosus. Arthritis Rheum (1992) 35:457–64. doi:10.1002/art.1780350416
6. Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol (2010) 6:280–9. doi:10.1038/nrrheum.2010.46
7. Topaloglu R, Bakkaloglu A, Slingsby JH, Mihatsch MJ, Pascual M, Norsworthy P, et al. Molecular basis of hereditary C1q deficiency associated with SLE and IgA nephropathy in a Turkish family. Kidney Int (1996) 50:635–42. doi:10.1038/ki.1996.359
8. Botto M, Dell’agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet (1998) 19:56–9. doi:10.1038/ng0598-56
9. Mitchell DA, Pickering MC, Warren J, Fossati-Jimack L, Cortes-Hernandez J, Cook HT, et al. C1q deficiency and autoimmunity: the effects of genetic background on disease expression. J Immunol (2002) 168:2538–43. doi:10.4049/jimmunol.168.5.2538
10. Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet (2007) 39:1065–7. doi:10.1038/ng2091
11. Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity (2000) 13:277–85. doi:10.1016/S1074-7613(00)00027-3
12. Bolland S, Yim YS, Tus K, Wakeland EK, Ravetch JV. Genetic modifiers of systemic lupus erythematosus in FcgammaRIIB(-/-) mice. J Exp Med (2002) 195:1167–74. doi:10.1084/jem.20020165
13. Tiller T, Kofer J, Kreschel C, Busse CE, Riebel S, Wickert S, et al. Development of self-reactive germinal center B cells and plasma cells in autoimmune Fc gammaRIIB-deficient mice. J Exp Med (2010) 207:2767–78. doi:10.1084/jem.20100171
14. Espeli M, Clatworthy MR, Bokers S, Lawlor KE, Cutler AJ, Kontgen F, et al. Analysis of a wild mouse promoter variant reveals a novel role for FcgammaRIIb in the control of the germinal center and autoimmunity. J Exp Med (2012) 209:2307–19. doi:10.1084/jem.20121752
15. Willcocks LC, Carr EJ, Niederer HA, Rayner TF, Williams TN, Yang W, et al. A defunctioning polymorphism in FCGR2B is associated with protection against malaria but susceptibility to systemic lupus erythematosus. Proc Natl Acad Sci U S A (2010) 107:7881–5. doi:10.1073/pnas.0915133107
16. Mackay M, Stanevsky A, Wang T, Aranow C, Li M, Koenig S, et al. Selective dysregulation of the FcgammaIIB receptor on memory B cells in SLE. J Exp Med (2006) 203:2157–64. doi:10.1084/jem.20051503
17. Espeli M, Smith KG, Clatworthy MR. FcgammaRIIB and autoimmunity. Immunol Rev (2016) 269:194–211. doi:10.1111/imr.12368
18. Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science (2004) 304:1147–50. doi:10.1126/science.1094359
19. Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet (2011) 43:1186–8. doi:10.1038/ng.975
20. Ozcakar ZB, Foster J II, Diaz-Horta O, Kasapcopur O, Fan YS, Yalcinkaya F, et al. DNASE1L3 mutations in hypocomplementemic urticarial vasculitis syndrome. Arthritis Rheum (2013) 65:2183–9. doi:10.1002/art.38010
21. Carbonella A, Mancano G, Gremese E, Alkuraya FS, Patel N, Gurrieri F, et al. An autosomal recessive DNASE1L3-related autoimmune disease with unusual clinical presentation mimicking systemic lupus erythematosus. Lupus (2017) 26:768–72. doi:10.1177/0961203316676382
22. Sisirak V, Sally B, D’agati V, Martinez-Ortiz W, Ozcakar ZB, David J, et al. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell (2016) 166:88–101. doi:10.1016/j.cell.2016.05.034
23. Keyel PA. Dnases in health and disease. Dev Biol (2017) 429:1–11. doi:10.1016/j.ydbio.2017.06.028
24. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A toll-like receptor recognizes bacterial DNA. Nature (2000) 408:740–5. doi:10.1038/35047123
25. Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV. Augmented humoral and anaphylactic responses in Fc gamma RII-deficient mice. Nature (1996) 379:346–9. doi:10.1038/379346a0
26. Eggan K, Akutsu H, Loring J, Jackson-Grusby L, Klemm M, Rideout WM III, et al. Hybrid vigor, fetal overgrowth, and viability of mice derived by nuclear cloning and tetraploid embryo complementation. Proc Natl Acad Sci U S A (2001) 98:6209–14. doi:10.1073/pnas.101118898
27. Rodriguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, et al. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet (2000) 25:139–40. doi:10.1038/75973
28. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc (2008) 3:1101–8. doi:10.1038/nprot.2008.73
29. Brochet X, Lefranc MP, Giudicelli V. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res (2008) 36:W503–8. doi:10.1093/nar/gkn316
30. Liu QY, Pandey S, Singh RK, Lin W, Ribecco M, Borowy-Borowski H, et al. DNaseY: a rat DNaseI-like gene coding for a constitutively expressed chromatin-bound endonuclease. Biochemistry (1998) 37:10134–43. doi:10.1021/bi9800597
31. Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature (2011) 474:337–42. doi:10.1038/nature10163
32. White JK, Gerdin AK, Karp NA, Ryder E, Buljan M, Bussell JN, et al. Genome-wide generation and systematic phenotyping of knockout mice reveals new roles for many genes. Cell (2013) 154:452–64. doi:10.1016/j.cell.2013.06.022
33. Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, et al. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A (1996) 93:5860–5. doi:10.1073/pnas.93.12.5860
34. Wellmann U, Letz M, Herrmann M, Angermuller S, Kalden JR, Winkler TH. The evolution of human anti-double-stranded DNA autoantibodies. Proc Natl Acad Sci U S A (2005) 102:9258–63. doi:10.1073/pnas.0500132102
35. Schroeder K, Herrmann M, Winkler TH. The role of somatic hypermutation in the generation of pathogenic antibodies in SLE. Autoimmunity (2013) 46:121–7. doi:10.3109/08916934.2012.748751
36. Andrews BS, Eisenberg RA, Theofilopoulos AN, Izui S, Wilson CB, Mcconahey PJ, et al. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J Exp Med (1978) 148:1198–215. doi:10.1084/jem.148.5.1198
37. Lefranc MP, Giudicelli V, Ginestoux C, Bodmer J, Muller W, Bontrop R, et al. IMGT, the international ImMunoGeneTics database. Nucleic Acids Res (1999) 27:209–12. doi:10.1093/nar/27.1.209
38. Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of Proteins of Immunological Interest. Washington, DC: NIH Publication (1991).
39. Radic MZ, Weigert M. Genetic and structural evidence for antigen selection of anti-DNA antibodies. Annu Rev Immunol (1994) 12:487–520. doi:10.1146/annurev.iy.12.040194.002415
40. Ueki M, Kimura-Kataoka K, Takeshita H, Fujihara J, Iida R, Sano R, et al. Evaluation of all non-synonymous single nucleotide polymorphisms (SNPs) in the genes encoding human deoxyribonuclease I and I-like 3 as a functional SNP potentially implicated in autoimmunity. FEBS J (2014) 281:376–90. doi:10.1111/febs.12608
41. Mayes MD, Bossini-Castillo L, Gorlova O, Martin JE, Zhou X, Chen WV, et al. Immunochip analysis identifies multiple susceptibility loci for systemic sclerosis. Am J Hum Genet (2014) 94:47–61. doi:10.1016/j.ajhg.2013.12.002
42. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature (2009) 461:747–53. doi:10.1038/nature08494
43. Crawford L, Zeng P, Mukherjee S, Zhou X. Detecting epistasis with the marginal epistasis test in genetic mapping studies of quantitative traits. PLoS Genet (2017) 13:e1006869. doi:10.1371/journal.pgen.1006869
44. Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science (2006) 312:1669–72. doi:10.1126/science.1124978
45. Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A (2006) 103:9970–5. doi:10.1073/pnas.0603912103
46. Izui S, Merino R, Fossati L, Iwamoto M. The role of the Yaa gene in lupus syndrome. Int Rev Immunol (1994) 11:211–30. doi:10.3109/08830189409061728
47. Woods M, Zou YR, Davidson A. Defects in germinal center selection in SLE. Front Immunol (2015) 6:425. doi:10.3389/fimmu.2015.00425
48. Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature (2005) 435:452–8. doi:10.1038/nature03555
49. Simpson N, Gatenby PA, Wilson A, Malik S, Fulcher DA, Tangye SG, et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum (2010) 62:234–44. doi:10.1002/art.25032
50. Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol (2012) 8:337–47. doi:10.1038/nrrheum.2012.58
51. Jellusova J, Wellmann U, Amann K, Winkler TH, Nitschke L. CD22 x Siglec-G double-deficient mice have massively increased B1 cell numbers and develop systemic autoimmunity. J Immunol (2010) 184:3618–27. doi:10.4049/jimmunol.0902711
52. Shlomchik M, Mascelli M, Shan H, Radic MZ, Pisetsky D, Marshak-Rothstein A, et al. Anti-DNA antibodies from autoimmune mice arise by clonal expansion and somatic mutation. J Exp Med (1990) 171:265–92. doi:10.1084/jem.171.1.265
53. Guo W, Smith D, Aviszus K, Detanico T, Heiser RA, Wysocki LJ. Somatic hypermutation as a generator of antinuclear antibodies in a murine model of systemic autoimmunity. J Exp Med (2010) 207:2225–37. doi:10.1084/jem.20092712
54. Venereau E, Ceriotti C, Bianchi ME. DAMPs from cell death to new life. Front Immunol (2015) 6:422. doi:10.3389/fimmu.2015.00422
55. Mahajan A, Herrmann M, Munoz LE. Clearance deficiency and cell death pathways: a model for the pathogenesis of SLE. Front Immunol (2016) 7:35. doi:10.3389/fimmu.2016.00035
56. Dhodapkar KM, Kaufman JL, Ehlers M, Banerjee DK, Bonvini E, Koenig S, et al. Selective blockade of inhibitory Fcgamma receptor enables human dendritic cell maturation with IL-12p70 production and immunity to antibody-coated tumor cells. Proc Natl Acad Sci U S A (2005) 102:2910–5. doi:10.1073/pnas.0500014102
57. Dhodapkar KM, Banerjee D, Connolly J, Kukreja A, Matayeva E, Veri MC, et al. Selective blockade of the inhibitory Fcgamma receptor (FcgammaRIIB) in human dendritic cells and monocytes induces a type I interferon response program. J Exp Med (2007) 204:1359–69. doi:10.1084/jem.20062545
Keywords: Dnase1l3, anti-DNA autoantibodies, systemic lupus erythematosus, Fcgr2b, germinal center
Citation: Weisenburger T, von Neubeck B, Schneider A, Ebert N, Schreyer D, Acs A and Winkler TH (2018) Epistatic Interactions Between Mutations of Deoxyribonuclease 1-Like 3 and the Inhibitory Fc Gamma Receptor IIB Result in Very Early and Massive Autoantibodies Against Double-Stranded DNA. Front. Immunol. 9:1551. doi: 10.3389/fimmu.2018.01551
Received: 03 February 2018; Accepted: 22 June 2018;
Published: 05 July 2018
Edited by:
Ralf J. Ludwig, Universität zu Lübeck, GermanyReviewed by:
SunAh Kang, University of North Carolina at Chapel Hill, United StatesCopyright: © 2018 Weisenburger, von Neubeck, Schneider, Ebert, Schreyer, Acs and Winkler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas H. Winkler, thomas.winkler@fau.de
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.