
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 23 July 2018
Sec. Immunological Tolerance and Regulation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01702
This article is part of the Research Topic Key Players in Systemic Sclerosis: The Immune System and Beyond View all 22 articles
 Paoline Laurent1†
Paoline Laurent1† Vanja Sisirak1†
Vanja Sisirak1† Estibaliz Lazaro1,2
Estibaliz Lazaro1,2 Christophe Richez1,3Pierre Duffau1,2Patrick Blanco1,4
Christophe Richez1,3Pierre Duffau1,2Patrick Blanco1,4 Marie-Elise Truchetet1,3
Marie-Elise Truchetet1,3 Cécile Contin-Bordes1,4* On Behalf of the Fédération Hospitalo Universitaire ACRONIM Aquitaine’s Care and Research Organization for Inflammatory and Immune-Mediated Diseases
Cécile Contin-Bordes1,4* On Behalf of the Fédération Hospitalo Universitaire ACRONIM Aquitaine’s Care and Research Organization for Inflammatory and Immune-Mediated Diseases
Systemic sclerosis (SSc) is a heterogeneous autoimmune disease characterized by three interconnected hallmarks (i) vasculopathy, (ii) aberrant immune activation, and (iii) fibroblast dysfunction leading to extracellular matrix deposition and fibrosis. Blocking or reversing the fibrotic process associated with this devastating disease is still an unmet clinical need. Although various components of innate immunity, including macrophages and type I interferon, have long been implicated in SSc, the precise mechanisms that regulate the global innate immune contribution to SSc pathogenesis remain poorly understood. Recent studies have identified new innate immune players, such as pathogen-recognition receptors, platelet-derived danger-associated molecular patterns, innate lymphoid cells, and plasmacytoid dendritic cells in the pathophysiology of SSc, including vasculopathy and fibrosis. In this review, we describe the evidence demonstrating the importance of innate immune processes during SSc development with particular emphasis on their role in the initiation of pathology. We also discuss potential therapeutic options to modulate innate immune cells or signaling in SSc that are emerging from these recent advances.
Systemic sclerosis (SSc) is a complex autoimmune disease interconnecting vasculopathy, autoimmunity, and fibrosis features. A large body of evidence has indicated that the adaptive immune system with autoreactive T cells and autoantibodies produced by B cells plays a central role in SSc pathogenesis (1). In addition, inflammatory cytokines produced by the innate immune cells have been detected in the affected tissues of both the early and late stage of SSc, suggesting a role of innate immunity both at the onset and progression of the disease (2–6). This notion was recently reinforced by genomic and genetic approaches that have been undertaken to decipher key and conserved pathophysiological pathways within organs across disease forms (7–9). Apart from genomic approaches, the study of mechanisms governing normal tissue repair has revealed physiological pathways that may be disrupted during SSc as well. The concept of unresolved tissue repair leading to sustained fibrosis has emerged based on a persistent sterile inflammation that converts a self-limited repair response to a non-resolving pathological fibrosis (10, 11). However, the initial events leading to such sterile inflammation remain unclear. Recent data showing that an imbalance in danger-associated molecular pattern (DAMP) release and/or pathogen-recognition receptor (PRR) signaling leads to sustained inflammatory cytokine production by fibroblasts or macrophages may provide the missing link in early events of SSc pathophysiology (11). In addition, plasmacytoid dendritic cell (pDC) activation (12, 13) and type I interferon (IFNα/β, IFN-I) production has also been recently shown to contribute to SSc.
In this review, we focus on recent evidence highlighting the contribution of innate immunity during the course of SSc pathogenesis, primarily at the early stages of disease. We also discuss potential therapeutic options that may modulate innate immune cells or signaling in SSc patients.
Major technological and analytical advances in the past 10 years have allowed the extraction of critical information from transcriptomic data such as lineage-specific gene expression, networks of interactions, and functional information (14–17). This yielded a novel field of study in the integrated comprehension of SSc pathogenesis, identifying a major contribution of innate immunity.
By analyzing three independent gene expression data sets from SSc skin biopsies, the group of Whitfield proposed interconnected functional modules involved in SSc pathogenesis, two of which involve innate immunity and are dominated by IFN, IFN-inducible genes, and type 2 macrophage (M2) signatures. The three other subnetworks were linked to adaptive immunity, fibrotic processes [response to transforming growth factor beta (TGF-β) and extracellular matrix (ECM) disassembly/wound healing], cell cycle, proliferation, and apoptosis (9). The same group recently identified a common pathogenic signature related to an “innate immune-fibrotic axis” that includes IFN-I and alternatively activated macrophages commonly referred as M2 macrophages and describes new specific pathways and hubs active in the skin and lung (8). Among shared networks, the authors found that the “innate immunity-fibrotic network” is conserved between skin and lung while the internal composition and interactions of gene expression in those tissues vary.
Such large-scale genomic studies paved the way for multiple experimental approaches to determine the molecular processes involved in patients and to establish novel therapeutic options targeting specific organs or shared pathophysiological processes.
The ability of an organism to efficiently recover from injury whether traumatic, infectious, chemical, or internal is pivotal to maintain its integrity (18). During tissue repair, innate immune cell plasticity actively contributes to the development of an abnormal microenvironment, leading to a shift in the balance between the pro-inflammatory and pro-reparative sides of tissue repair, as recently reviewed (10).
Early SSc is characterized by a perivascular leukocyte infiltrate mainly composed of macrophages and T lymphocytes, reminiscent of the process induced during normal wound healing (19, 20). Whereas normal wound healing is accompanied by a remodeling or resolving stage, abnormal wound healing with chronic activation of immune cells such as macrophages or stromal cells like myofibroblasts fails to resolve fibrosis during SSc. Hence, SSc, specifically diffuse cutaneous forms of the disease, could be considered as a general form of over-repair. The initial trigger of the injury is still unknown, but several lines of recent evidence have brought new hypotheses on its nature.
Recognition of pathogen-associated molecular patterns (PAMPs) or endogenous DAMPs by innate immune cells as well as non-immune cells is the first line of response to pathogen or sterile tissue injury. DAMPs, mainly produced by epithelial cells, are heterogeneous in form encompassing early produced and highly diffusible Ca2+, H2O2, reactive oxygen species (ROS), adenosine tri-phosphate, self-nucleic acids, but also proteins like high-mobility group protein 1, heat shock protein, S100 proteins, and fragments of the ECM. The recognition of PAMPs and DAMPs relies on cell surface, endosomal, and cytosolic PRRs that include toll-like receptors (TLRs), Nod-like receptor, Rig-I-like receptors (RLRs), cyclic GMP-AMP synthase, and receptor for advanced glycation end products. Innate immune signaling triggered by DAMPs during sterile inflammation or the persistence of pathogens such as endogenous viruses might represent an important pathway responsible for converting self-limited regenerative repair into an unresolved fibrotic process during SSc. Hence, innate immune signaling via TLRs was recently proposed as a key driver of persistent fibrotic response in SSc and other fibrotic-related diseases (11).
Overexpression of TLR4 and its two co-receptors CD14 and myeloid differentiation factor 2 (MD-2) has been described in SSc-affected skin and lung. TLR4 expression was mainly associated with macrophages, fibroblasts, and myofibroblasts (21). In the skin, TLR4 expression correlated to fibrosis severity measured by modified Rodnan skin score. In vivo, chronic TLR4 activation leads to sustained nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) signaling, resulting in macrophage activation and a profibrotic profile (22). Work from the Varga lab recently demonstrated that endogenous DAMP activation of TLR4 can contribute to converting self-limited tissue repair responses into uncontrolled ECM deposition during SSc [for recent review, see Ref. (11)]. They proposed that fibronectin, containing alternatively spliced exons encoding type III repeat extra domain (EDA), and tenascin-C are constitutively produced by SSc fibroblasts leading to their accumulation in the skin but also in the blood. Together, fibronectin-EDA and tenascin-C act as strong profibrotic factors during SSc by binding to fibroblasts TLR4, leading to enhanced production of collagen and alpha-smooth muscle actin (α-SMA) expression (23, 24). Deletion of EDA or tenascin-C or disruption of TLR4 signaling resulted in reduced fibrotic response in a murine model of SSc. Furthermore, tensional forces generated within a rigid fibrotic microenvironment were reported to favor exposure of the EDA domain of fibronectin (25), suggesting that increased stiffness of the matrix in fibrotic tissue could favor the bioavailability and profibrotic activity of fibronectin-EDA.
Altered expression of multiple DAMPs/TLRs beyond TLR4 has been described during SSc. Indeed, increased expression of TLR9 was found in human SSc skin biopsies in both early and late stages of the disease and was mainly associated with α-SMA-positive myofibroblasts (26), and a TLR9 signature was detected in SSc skin. In vitro treatment of normal cutaneous fibroblasts with the TLR9 ligand unmethylated-CpG-oligodeoxynucleotides (CpG ODN) induced a profibrotic profile involving autocrine TGF-β production. Collectively, these results support the role of TLR9 signaling in SSc. Furthermore, expression of TLR2 (27) and TLR3 (28) is also increased in SSc skin fibroblasts. TLR2 was shown to respond to the endogenous ligand amyloid A, resulting in NFκB activation and increased interleukin (IL)-6 secretion causing inflammation (27). However, the role of TLR3 in SSc pathogenesis remains controversial. TLR3 activation by polyinosinic:polycytidylic acid (poly I:C) stimulates IFN-I production by fibroblasts, which in turn reduces their ability to produce ECM components (28). Conversely, such stimulation was shown to promote the expression of TGF-β by fibroblasts thus contributing to the overall fibrosis (29).
In addition to TLRs, other PRRs have been described to play a role in SSc pathogenesis. The IFN-I stimulatory property of poly I:C on SSc patient fibroblasts was shown not only to rely on TLR3 but also on intracellular RLRs (28). The inflammasome, specifically the NLRP3-inflammasome, was shown to contribute SSc pathogenesis in vivo (30) through the induction of the microRNA miR-155, which in turn favors excessive ECM production by fibroblasts, exacerbating SSc (31).
Studies on the contribution of TLR signaling to fibrosis in SSc as well as other fibrotic diseases have generated conflicting results (22, 26, 32–35), suggesting that whether TLR activation leads to pro- or anti-fibrotic effects depends on many factors. The nature of the stimulation (chronic vs acute), of the responding cells (immune or non-immune cells), as well as disease stage (inflammatory vs remodeling) might modulate the effects of TLRs in the fibrotic process. Profibrotic effects of TLR activation seem related to fibroblast and macrophage activation in the context of chronic stimulation, whereas epithelial and other immune cell activation in the context of acute stimulation might lead to anti-fibrotic effects. Although additional PRRs have recently been implicated in SSc, further studies are required to identify their endogenous ligands and mechanisms leading to disease. Nevertheless, PRRs and their signaling pathways may represent multiple novel therapeutic targets in SSc.
Macrophages and platelets have emerged as key players not only during tissue homeostasis and repair but also fibrosis, recently reviewed in Ref. (36, 37).
We and others have defined the profibrotic role of platelets in SSc. The Distler group has shown that serotonin [5-hydroxytryptamine (5-HT)] stored in platelets strongly induces ECM synthesis in interstitial fibroblasts via activation of 5-HT2B receptors (5-HT2B) in a TGF-β-dependent manner (38). Our group discovered a pathophysiological loop active in SSc that links vasculopathy and fibrosis. Indeed, we showed that platelet activation induced the production of thymic stromal lymphopoietin (TSLP) by dermal microvascular endothelial cells in an IL-1β-dependent manner. TSLP was found to be strongly expressed in SSc skin endothelial cells and correlated to the severity of skin fibrosis. In vitro, TSLP was able to induce a profibrotic profile in both normal and SSc fibroblasts (39, 40).
Infiltration of macrophages in the early skin lesions of SSc patients, particularly in perivascular areas, was first detected over 20 years ago and could lead to secondary activation of adaptative system (19, 20). Since then, numerous studies have established the involvement of macrophages in SSc pathogenesis, notably their alternatively activated counterpart called M2 macrophages as reviewed in Ref. (41). Soluble CD163, a putative marker of M2 macrophages, was shown to be elevated in SSc patients’ blood and associated with their poor clinical outcome (42). These observations have been reinforced by the recent genetic studies showing a prominent M2 macrophage signature in SSc-affected skin and lung (8). However, the activation of lung macrophages in SSc patients with pulmonary fibrosis is distinct from that observed in SSc skin. Activated lung-resident macrophages display a specific increase in the expression of genes related to lipid and cholesterol trafficking, suggesting a switch in their metabolism. Thus, while M2 macrophages are central to the fibrotic process both in skin and lung during SSc, distinct stimuli derived from the organ-specific microenvironment might differentially shape the plasticity of macrophages. In the recent FASSCINATE trial, molecular profiling of skin biopsies revealed that IL-6 receptor blockade by tocilizumab resulted in a reduced M2 macrophage signature observed in SSc skin (43). Accordingly, the blockade of cAMP-specific phosphodiesterase-4, which inhibits differentiation of M2 macrophages as well as IL-6 production, led to an amelioration of fibrosis in a murine model of SSc induced by bleomycin treatment (44). The same group demonstrated that nintedanib, a tyrosine kinase inhibitor targeting vascular endothelial-, fibroblast-, and platelet-derived growth factor receptors, effectively blocked myofibroblast differentiation and reduced pulmonary, dermal, and myocardial fibrosis in transgenic Fra2 mice. This effect was primarily mediated by preventing M2 macrophage accumulation in the affected tissues (45). However, the mechanisms leading to aberrant M2 macrophage polarization and the precise pathways through which M2 macrophages contribute to tissue fibrosis remain unclear. One elegant study by Eming et al. provided novel mechanistic insight to the role of M2 macrophages in fibrosis. Using a murine model of wound healing, IL-4Ra activation by IL-4 and IL-13 was demonstrated to induce the production of resting like molecule alpha by M2 macrophages, which in turn stimulates the production of enzyme lysyl-hydroxylase-2 (LH-2) ultimately contributing to persistent profibrotic collagen cross-linking in fibroblasts (46). This process was shown to be critical for transformation of the tissue into a persistent scar. In humans, Relm-β induces LH-2 in fibroblasts, and expression of both factors was reported to be increased in lipodermatosclerosis, a condition associated with excessive skin fibrosis. Whether this process contributes to SSc is still unknown. The fine mapping of specific macrophage subsets across tissues and during the course of disease, as well as elucidating of the molecular mechanisms underlying macrophages-induced abnormal resolution, will pave the road to the development of new drugs that prevent/limit fibrosis.
Studies in patients and animal models of SSc have demonstrated that mast cells infiltrate the fibrotic skin (47, 48). This infiltration was associated with more severe disease phenotypes (48), but the function and net contribution of mast cells to fibrosis is only beginning to be understood. Mast cells have been suggested to be an important source of TGF-β and thus contribute to the overall fibrosis (49). Furthermore, a recent report using transgenic mice that develop spontaneous skin fibrosis showed a major role for mast cells specifically in inducing inflammation of the skin and the production of ECM and α-SMA by fibroblasts (50). Together with recent observations showing that mast cell deletion ameliorates experimental SSc in vivo (47, 51), these results indicate that mast cell targeting in SSc patients may represent an effective therapeutic approach.
Finally, other innate immune players such as natural killer (NK) cells (52, 53) and neutrophils (54) were shown to display altered properties and phenotypes in the blood of SSc patients. However, further studies are required to evaluate the role of NK cells and neutrophils in the SSc pathogenesis, especially in the settings of murine experimental models.
Plasmacytoid dendritic cells are innate immune cells specialized in the production of copious amounts of IFN-I (55), and thus play a key role in the initiation of antiviral immune responses (56, 57). IFN-I production by pDCs requires recognition of viral nucleic acids by TLR7 and TLR9, respectively (56, 57). pDCs were also shown to produce IFN-I in response to self-nucleic acids and consequently contribute to the development of multiple inflammatory and autoimmune disorders (58–62). An IFN-I signature, reflected by increased expression of numerous IFN-I-stimulated genes has been reported in patients with SSc (12). Furthermore, genome-wide association studies in SSc have identified polymorphisms in genes involved in the regulation of IFN-I expression in pDCs, particularly IFN-regulatory factor (IRF)-5, IRF-7, and IRF-8 (12). Approximately half of SSc patients (~50%) display an IFN-I signature within their peripheral blood mononuclear cells (63–65) and in fibrotic skin (66). The association between IFN-I signature and SSc disease activity remains controversial as no major impact of the IFN-I signature on pathological features of SSc, including extent of skin fibrosis, autoantibody specificities, and interstitial lung disease, has been reported (63, 65). However, when the profile of IFN-induced chemokines was specifically analyzed in a large cohort of SSc patients, an association was then identified with more severe SSc (67). As pDCs are an important source of IFN-I, numerous groups have investigated their role in SSc. pDCs were indeed detected in the affected skin of SSc patients (65, 68) as well as in the fibrotic skin of mice after bleomycin treatment (13). Furthermore, mice lacking fibrillin-1 (Fbn1), which spontaneously develop a stiff skin syndrome that recapitulates the skin fibrosis observed in SSc patients, show a high infiltration of pDCs in the affected skin (69). The frequency of pDCs is reduced in the circulation of SSc patients, likely due to their preferential recruitment into the fibrotic skin (13). Anti-topoisomerase I and anti-nuclear autoantibodies in SSc patients were shown to form immune complexes with apoptotic cell-derived constituents in vitro and consequently stimulate IFN-I production by pDCs (70, 71) upon uptake via FcγRII and the stimulation of TLR7/9 (70, 71). While such “interferongenic” properties of immune complexes may contribute to the aberrant IFN-I production, an IFN-I signature was not associated with the production of specific autoantibodies detected in the sera of SSc patients (70), suggesting that additional factors may contribute to pDC activation in vivo. Furthermore, pDCs in the peripheral blood or fibrotic skin of SSc patients spontaneously secrete CXC motif ligand (CXCL)-4 and IFNα (13, 68). High levels of CXCL4 in the circulation of SSc patients were associated with disease severity including skin fibrosis and pulmonary arterial hypertension (68). CXCL4 was described to potentiate pDC ability to produce IFN-I in vitro largely in response to TLR9 stimulation. In addition, CXCL4 was shown to induce both the expression of TLR8 and the ability to produce IFN-I in response to its specific ligands in pDCs (13). Recently, the pathogenic role of TLR8 was confirmed in vivo using transgenic mice that express human TLR8 and develop exacerbated skin fibrosis after bleomycin treatment compared with control animals (13). However, whether such exacerbation of disease in TLR8 transgenic animals is dependent on pDCs remains unknown, and the association between CXCL4 levels and the IFN-I signature in SSc patients has not yet been characterized. Ah Kioon et al. showed that bleomycin-induced skin fibrosis is strongly attenuated after selective pDC depletion (13). Furthermore, this model of fibrosis was associated with an IFN-I signature and increased expression of CXCL4 in the affected skin, and pDC depletion significantly reduced the occurrence of these parameters. From a therapeutic standpoint, pDC depletion ameliorated established bleomycin-induced skin fibrosis, indicating that pDCs are critical even in the maintenance of skin fibrosis. This constitutes the first study showing the deleterious impact of pDCs on SSc development in vivo (13). Overall, pDCs play a critical role in SSc pathogenesis; however, the molecular mechanisms through which they contribute to the disease require further investigation. This recent progress nevertheless positions SSc as another autoimmune pathology that may benefit from therapeutic targeting of pDCs using depleting or inhibitory antibodies (72).
Innate lymphoid cells were recently described as novel components of the immune system that may be considered as innate counterparts of polarized T helper cells (73). Nevertheless, knowledge on the role of ILCs in SSc remains limited. Wohlfahrt and colleagues have shown elevated numbers of ILC2 in both the peripheral blood and the affected skin of patients with SSc compared with healthy individuals, and their number correlated with the extent of cutaneous fibrosis (74). However, the increased frequency of ILC2 in SSc peripheral blood was not observed in a different study, which instead reported an elevated frequency of CD4 + ILC1 and NKp44 + ILC3 (75).
Nevertheless, in animal models of lung fibrosis induced by bleomycin, IL-33, an alarmin that has been reported to be elevated in SSc patients (76), induced the expansion of ILC2s producing the profibrotic cytokine IL-13 (77). Hence, further investigations are warranted to determine the role of ILC2 in the development of SSc fibrosis.
Significant progress has recently been made in understanding the contribution of innate immunity to SSc fibrosis. Although the precise molecular mechanisms of their action must be further defined, promising new therapeutic targets for SSc have already emerged. Such strategies include blockade of TLR4/MD-2, TLR9, or downstream signaling molecules to limit chronic sterile inflammation, modulation of macrophage polarization to promote resolution and matrix remodeling, and targeting pDCs/IFN-α. This therapeutic challenge is ongoing with many attractive new therapeutic candidates, some of which are currently being tested in Phase III clinical trials (Tables 1A,B). Both the evaluation of potential side effects and identification of biomarkers of patients who would benefit from such therapies are warranted in order to maximize the efficacy of treatment.
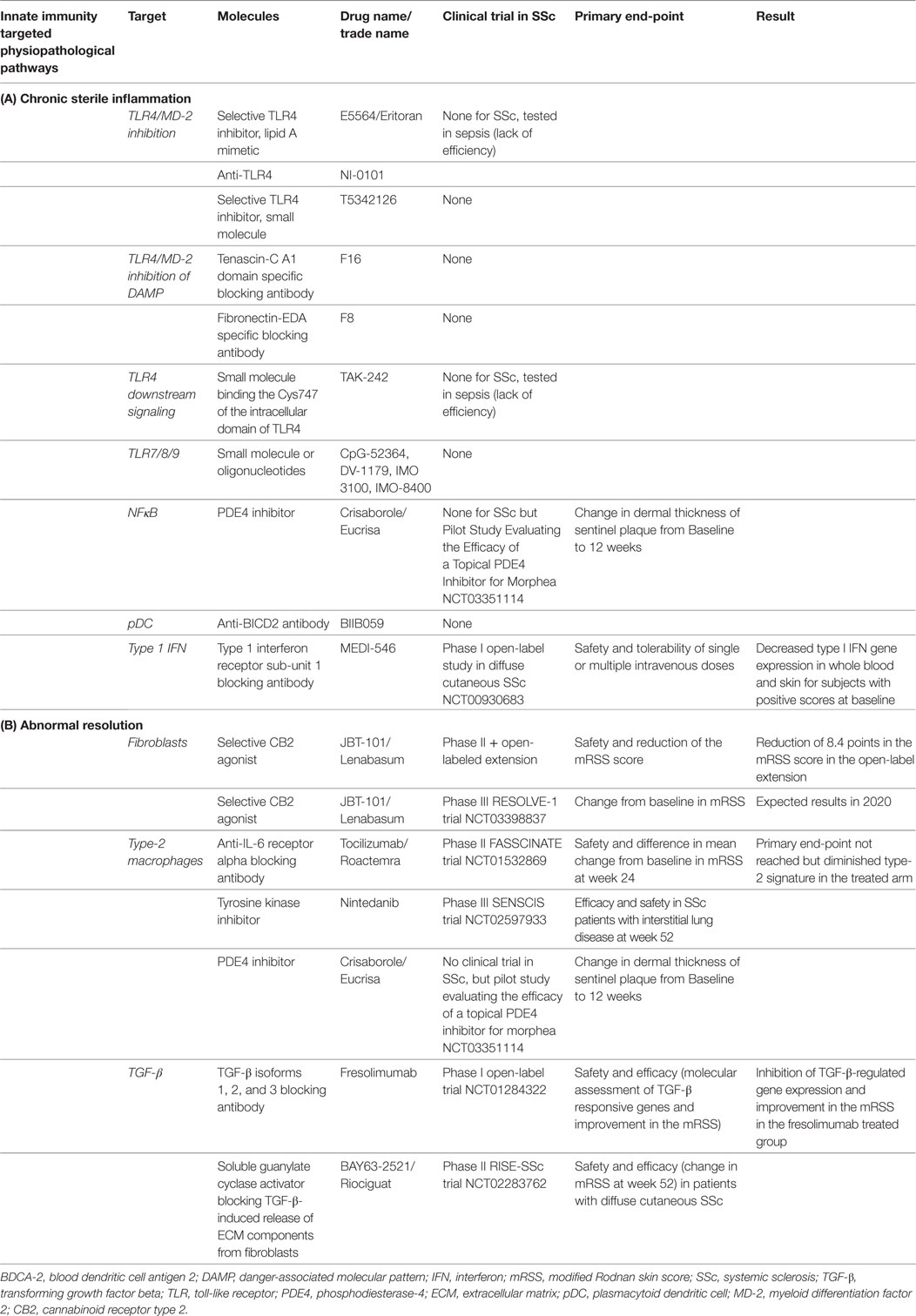
Table 1. Potential therapeutics and therapeutics in latest clinical trials specific to innate immunity and fibrosis in SSc.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that this work was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Carlo Chizzolini (University of Geneva, Switzerland) for careful reading of the manuscript and for fruitful discussions.
MET has received a grant from the Société française de rhumatologie, the SFR Rhumato-Network.
1. Chizzolini C, Brembilla NC, Montanari E, Truchetet M-E. Fibrosis and immune dysregulation in systemic sclerosis. Autoimmun Rev (2011) 10:276–81. doi:10.1016/j.autrev.2010.09.016
2. Raker V, Haub J, Stojanovic A, Cerwenka A, Schuppan D, Steinbrink K. Early inflammatory players in cutanous fibrosis. J Dermatol Sci (2017) 87:228–35. doi:10.1016/j.jdermsci.2017.06.009
3. Bhattacharyya S, Varga J. Emerging roles of innate immune signaling and toll-like receptors in fibrosis and systemic sclerosis. Curr Rheumatol Rep (2015) 17:474. doi:10.1007/s11926-014-0474-z
4. Dowson C, Simpson N, Duffy L, O’Reilly S. Innate immunity in systemic sclerosis. Curr Rheumatol Rep (2017) 19:2. doi:10.1007/s11926-017-0630-3
5. Fullard N, O’Reilly S. Role of innate immune system in systemic sclerosis. Semin Immunopathol (2015) 37:511–7. doi:10.1007/s00281-015-0503-7
6. Chia JJ, Lu TT. Update on macrophages and innate immunity in scleroderma. Curr Opin Rheumatol (2015) 27:530–6. doi:10.1097/BOR.0000000000000218
7. Johnson ME, Pioli PA, Whitfield ML. Gene expression profiling offers insights into the role of innate immune signaling in SSc. Semin Immunopathol (2015) 37:501–9. doi:10.1007/s00281-015-0512-6
8. Taroni JN, Greene CS, Martyanov V, Wood TA, Christmann RB, Farber HW, et al. A novel multi-network approach reveals tissue-specific cellular modulators of fibrosis in systemic sclerosis. Genome Med (2017) 9:27. doi:10.1186/s13073-017-0417-1
9. Mahoney JM, Taroni J, Martyanov V, Wood TA, Greene CS, Pioli PA, et al. Systems level analysis of systemic sclerosis shows a network of immune and profibrotic pathways connected with genetic polymorphisms. PLoS Comput Biol (2015) 11:e1004005. doi:10.1371/journal.pcbi.1004005
10. Laurent P, Jolivel V, Manicki P, Chiu L, Contin-Bordes C, Truchetet M-E, et al. Immune-mediated repair: a matter of plasticity. Front Immunol (2017) 8:454. doi:10.3389/fimmu.2017.00454
11. Bhattacharyya S, Varga J. Endogenous ligands of TLR4 promote unresolving tissue fibrosis: implications for systemic sclerosis and its targeted therapy. Immunol Lett (2018) 195:9–17. doi:10.1016/j.imlet.2017.09.011
12. Wu M, Assassi S. The role of type 1 interferon in systemic sclerosis. Front Immunol (2013) 4:266. doi:10.3389/fimmu.2013.00266
13. Ah Kioon MD, Tripodo C, Fernandez D, Kirou KA, Spiera RF, Crow MK, et al. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci Transl Med (2018) 10(423):eaam8458. doi:10.1126/scitranslmed.aam8458
14. Jansen R, Yu H, Greenbaum D, Kluger Y, Krogan NJ, Chung S, et al. A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science (2003) 302:449–53. doi:10.1126/science.1087361
15. Hwang S, Rhee SY, Marcotte EM, Lee I. Systematic prediction of gene function in Arabidopsis thaliana using a probabilistic functional gene network. Nat Protoc (2011) 6:1429–42. doi:10.1038/nprot.2011.372
16. Ju W, Greene CS, Eichinger F, Nair V, Hodgin JB, Bitzer M, et al. Defining cell-type specificity at the transcriptional level in human disease. Genome Res (2013) 23:1862–73. doi:10.1101/gr.155697.113
17. Chen JC, Cerise JE, Jabbari A, Clynes R, Christiano AM. Master regulators of infiltrate recruitment in autoimmune disease identified through network-based molecular deconvolution. Cell Syst (2015) 1:326–37. doi:10.1016/j.cels.2015.11.001
18. Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature (2008) 453:314–21. doi:10.1038/nature07039
19. Kräling BM, Maul GG, Jimenez SA. Mononuclear cellular infiltrates in clinically involved skin from patients with systemic sclerosis of recent onset predominantly consist of monocytes/macrophages. Pathobiology (1995) 63:48–56. doi:10.1159/000163933
20. Ishikawa O, Ishikawa H. Macrophage infiltration in the skin of patients with systemic sclerosis. J Rheumatol (1992) 19:1202–6.
21. Bhattacharyya S, Kelley K, Melichian DS, Tamaki Z, Fang F, Su Y, et al. Toll-like receptor 4 signaling augments transforming growth factor-β responses: a novel mechanism for maintaining and amplifying fibrosis in scleroderma. Am J Pathol (2013) 182:192–205. doi:10.1016/j.ajpath.2012.09.007
22. Stifano G, Affandi AJ, Mathes AL, Rice LM, Nakerakanti S, Nazari B, et al. Chronic toll-like receptor 4 stimulation in skin induces inflammation, macrophage activation, transforming growth factor beta signature gene expression, and fibrosis. Arthritis Res Ther (2014) 16:R136. doi:10.1186/ar4598
23. Bhattacharyya S, Tamaki Z, Wang W, Hinchcliff M, Hoover P, Getsios S, et al. FibronectinEDA promotes chronic cutaneous fibrosis through toll-like receptor signaling. Sci Transl Med (2014) 6:232ra50. doi:10.1126/scitranslmed.3008264
24. Bhattacharyya S, Wang W, Morales-Nebreda L, Feng G, Wu M, Zhou X, et al. Tenascin-C drives persistence of organ fibrosis. Nat Commun (2016) 7:11703. doi:10.1038/ncomms11703
25. Kelsh R, You R, Horzempa C, Zheng M, McKeown-Longo PJ. Regulation of the innate immune response by fibronectin: synergism between the III-1 and EDA domains. PLoS One (2014) 9:e102974. doi:10.1371/journal.pone.0102974
26. Fang F, Marangoni RG, Zhou X, Yang Y, Ye B, Shangguang A, et al. Toll-like receptor 9 signaling is augmented in systemic sclerosis and elicits transforming growth factor β-dependent fibroblast activation. Arthritis Rheumatol (2016) 68:1989–2002. doi:10.1002/art.39655
27. O’Reilly S, Cant R, Ciechomska M, Finnigan J, Oakley F, Hambleton S, et al. Serum amyloid A induces interleukin-6 in dermal fibroblasts via toll-like receptor 2, interleukin-1 receptor-associated kinase 4 and nuclear factor-κB. Immunology (2014) 143:331–40. doi:10.1111/imm.12260
28. Fang F, Ooka K, Sun X, Shah R, Bhattacharyya S, Wei J, et al. A synthetic TLR3 ligand mitigates profibrotic fibroblast responses by inducing autocrine IFN signaling. J Immunol (2013) 191:2956–66. doi:10.4049/jimmunol.1300376
29. Farina GA, York MR, Marzio MD, Collins CA, Meller S, Homey B, et al. Poly(I:C) drives type I IFN- and TGFβ-mediated inflammation and dermal fibrosis simulating altered gene expression in systemic sclerosis. J Invest Dermatol (2010) 130:2583–93. doi:10.1038/jid.2010.200
30. Artlett CM, Sassi-Gaha S, Rieger JL, Boesteanu AC, Feghali-Bostwick CA, Katsikis PD. The inflammasome activating caspase 1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis Rheum (2011) 63:3563–74. doi:10.1002/art.30568
31. Artlett CM, Sassi-Gaha S, Hope JL, Feghali-Bostwick CA, Katsikis PD. Mir-155 is overexpressed in systemic sclerosis fibroblasts and is required for NLRP3 inflammasome-mediated collagen synthesis during fibrosis. Arthritis Res Ther (2017) 19:144. doi:10.1186/s13075-017-1331-z
32. Takahashi T, Asano Y, Ichimura Y, Toyama T, Taniguchi T, Noda S, et al. Amelioration of tissue fibrosis by toll-like receptor 4 knockout in murine models of systemic sclerosis. Arthritis Rheumatol (2015) 67:254–65. doi:10.1002/art.38901
33. He Z, Zhu Y, Jiang H. Inhibiting toll-like receptor 4 signaling ameliorates pulmonary fibrosis during acute lung injury induced by lipopolysaccharide: an experimental study. Respir Res (2009) 10:126. doi:10.1186/1465-9921-10-126
34. Luckhardt TR, Coomes SM, Trujillo G, Stoolman JS, Vannella KM, Bhan U, et al. TLR9-induced interferon β is associated with protection from gammaherpesvirus-induced exacerbation of lung fibrosis. Fibrogenesis Tissue Repair (2011) 4:18. doi:10.1186/1755-1536-4-18
35. Abu-Tair L, Axelrod JH, Doron S, Ovadya Y, Krizhanovsky V, Galun E, et al. Natural killer cell-dependent anti-fibrotic pathway in liver injury via toll-like receptor-9. PLoS One (2013) 8:e82571. doi:10.1371/journal.pone.0082571
36. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity (2016) 44:450–62. doi:10.1016/j.immuni.2016.02.015
37. Nurden AT. The biology of the platelet with special reference to inflammation, wound healing and immunity. Front Biosci (Landmark Ed) (2018) 23:726–51. doi:10.2741/4613
38. Dees C, Akhmetshina A, Zerr P, Reich N, Palumbo K, Horn A, et al. Platelet-derived serotonin links vascular disease and tissue fibrosis. J Exp Med (2011) 208:961–72. doi:10.1084/jem.20101629
39. Truchetet M-E, Demoures B, Eduardo Guimaraes J, Bertrand A, Laurent P, Jolivel V, et al. Platelets induce thymic stromal lymphopoietin production by endothelial cells: contribution to fibrosis in human systemic sclerosis. Arthritis Rheumatol (2016) 68:2784–94. doi:10.1002/art.39817
40. Scherlinger M, Guillotin V, Truchetet M-E, Contin-Bordes C, Sisirak V, Duffau P, et al. Systemic lupus erythematosus and systemic sclerosis: all roads lead to platelets. Autoimmun Rev (2018) 17(6):625–35. doi:10.1016/j.autrev.2018.01.012
41. Stifano G, Christmann RB. Macrophage involvement in systemic sclerosis: do we need more evidence? Curr Rheumatol Rep (2016) 18:2. doi:10.1007/s11926-015-0554-8
42. Frantz C, Pezet S, Avouac J, Allanore Y. Soluble CD163 as a potential biomarker in systemic sclerosis. Dis Markers (2018) 2018:8509583. doi:10.1155/2018/8509583
43. Khanna D, Denton CP, Jahreis A, van Laar JM, Frech TM, Anderson ME, et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet (2016) 387:2630–40. doi:10.1016/S0140-6736(16)00232-4
44. Maier C, Ramming A, Bergmann C, Weinkam R, Kittan N, Schett G, et al. Inhibition of phosphodiesterase 4 (PDE4) reduces dermal fibrosis by interfering with the release of interleukin-6 from M2 macrophages. Ann Rheum Dis (2017) 76:1133–41. doi:10.1136/annrheumdis-2016-210189
45. Huang J, Maier C, Zhang Y, Soare A, Dees C, Beyer C, et al. Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis. Ann Rheum Dis (2017) 76:1941–8. doi:10.1136/annrheumdis-2016-210823
46. Knipper JA, Willenborg S, Brinckmann J, Bloch W, Maaß T, Wagener R, et al. Interleukin-4 receptor α signaling in myeloid cells controls collagen fibril assembly in skin repair. Immunity (2015) 43:803–16. doi:10.1016/j.immuni.2015.09.005
47. Yamamoto T, Takahashi Y, Takagawa S, Katayama I, Nishioka K. Animal model of sclerotic skin. II. Bleomycin induced scleroderma in genetically mast cell deficient WBB6F1-W/W(V) mice. J Rheumatol (1999) 26:2628–34.
48. Yukawa S, Yamaoka K, Sawamukai N, Shimajiri S, Kubo S, Miyagawa I, et al. Dermal mast cell density in fingers reflects severity of skin sclerosis in systemic sclerosis. Mod Rheumatol (2013) 23:1151–7. doi:10.1007/s10165-012-0813-8
49. Hügle T, Hogan V, White KE, van Laar JM. Mast cells are a source of transforming growth factor β in systemic sclerosis. Arthritis Rheum (2011) 63:795–9. doi:10.1002/art.30190
50. Pincha N, Hajam EY, Badarinath K, Batta SPR, Masudi T, Dey R, et al. PAI1 mediates fibroblast–mast cell interactions in skin fibrosis. J Clin Invest (2018) 128:1807–19. doi:10.1172/JCI99088
51. Karpec D, Rudys R, Leonaviciene L, Mackiewicz Z, Bradunaite R, Kirdaite G, et al. The safety and efficacy of light emitting diodes-based ultraviolet A1 phototherapy in bleomycin-induced scleroderma in mice. Adv Med Sci (2018) 63:152–9. doi:10.1016/j.advms.2017.09.001
52. Horikawa M, Hasegawa M, Komura K, Hayakawa I, Yanaba K, Matsushita T, et al. Abnormal natural killer cell function in systemic sclerosis: altered cytokine production and defective killing activity. J Invest Dermatol (2005) 125:731–7. doi:10.1111/j.0022-202X.2005.23767.x
53. Cossu M, van Bon L, Nierkens S, Bellocchi C, Santaniello A, Dolstra H, et al. The magnitude of cytokine production by stimulated CD56+ cells is associated with early stages of systemic sclerosis. Clin Immunol (2016) 173:76–80. doi:10.1016/j.clim.2016.09.004
54. Barnes TC, Anderson ME, Edwards SW, Moots RJ. Neutrophil-derived reactive oxygen species in SSc. Rheumatology (Oxford) (2012) 51:1166–9. doi:10.1093/rheumatology/ker520
55. Ito T, Kanzler H, Duramad O, Cao W, Liu Y-J. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood (2006) 107:2423–31. doi:10.1182/blood-2005-07-2709
56. Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol (2011) 29:163–83. doi:10.1146/annurev-immunol-031210-101345
57. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol (2015) 15:471–85. doi:10.1038/nri3865
58. Lande R, Gafa V, Serafini B, Giacomini E, Visconti A, Remoli ME, et al. Plasmacytoid dendritic cells in multiple sclerosis: intracerebral recruitment and impaired maturation in response to interferon-beta. J Neuropathol Exp Neurol (2008) 67:388–401. doi:10.1097/NEN.0b013e31816fc975
59. Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, Facchinetti V, et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med (2009) 206:1983–94. doi:10.1084/jem.20090480
60. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med (2011) 3:73ra19. doi:10.1126/scitranslmed.3001180
61. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med (2011) 3:73ra20. doi:10.1126/scitranslmed.3001201
62. Båve U, Magnusson M, Eloranta ML, Perers A, Alm GV, Rönnblom L. Fc gamma RIIa is expressed on natural IFN-alpha-producing cells (plasmacytoid dendritic cells) and is required for the IFN-alpha production induced by apoptotic cells combined with lupus IgG. J Immunol (2003) 171(6):3296–302.
63. Tan FK, Zhou X, Mayes MD, Gourh P, Guo X, Marcum C, et al. Signatures of differentially regulated interferon gene expression and vasculotrophism in the peripheral blood cells of systemic sclerosis patients. Rheumatology (Oxford) (2006) 45:694–702. doi:10.1093/rheumatology/kei244
64. York MR, Nagai T, Mangini AJ, Lemaire R, van Seventer JM, Lafyatis R. A macrophage marker, Siglec-1, is increased on circulating monocytes in patients with systemic sclerosis and induced by type I interferons and toll-like receptor agonists. Arthritis Rheum (2007) 56:1010–20. doi:10.1002/art.22382
65. Duan H, Fleming J, Pritchard DK, Amon LM, Xue J, Arnett HA, et al. Combined analysis of monocyte and lymphocyte messenger RNA expression with serum protein profiles in patients with scleroderma. Arthritis Rheum (2008) 58:1465–74. doi:10.1002/art.23451
66. Higgs BW, Liu Z, White B, Zhu W, White WI, Morehouse C, et al. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann Rheum Dis (2011) 70:2029–36. doi:10.1136/ard.2011.150326
67. Liu X, Mayes MD, Tan FK, Wu M, Reveille JD, Harper BE, et al. Correlation of interferon-inducible chemokine plasma levels with disease severity in systemic sclerosis. Arthritis Rheum (2013) 65:226–35. doi:10.1002/art.37742
68. van Bon L, Affandi AJ, Broen J, Christmann RB, Marijnissen RJ, Stawski L, et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med (2014) 370:433–43. doi:10.1056/NEJMoa1114576
69. Gerber EE, Gallo EM, Fontana SC, Davis EC, Wigley FM, Huso DL, et al. Integrin-modulating therapy prevents fibrosis and autoimmunity in mouse models of scleroderma. Nature (2013) 503:126–30. doi:10.1038/nature12614
70. Kim D, Peck A, Santer D, Patole P, Schwartz SM, Molitor JA, et al. Induction of interferon-alpha by scleroderma sera containing autoantibodies to topoisomerase I: association of higher interferon-alpha activity with lung fibrosis. Arthritis Rheum (2008) 58:2163–73. doi:10.1002/art.23486
71. Eloranta M-L, Franck-Larsson K, Lövgren T, Kalamajski S, Rönnblom A, Rubin K, et al. Type I interferon system activation and association with disease manifestations in systemic sclerosis. Ann Rheum Dis (2010) 69:1396–402. doi:10.1136/ard.2009.121400
72. Pellerin A, Otero K, Czerkowicz JM, Kerns HM, Shapiro RI, Ranger AM, et al. Anti-BDCA2 monoclonal antibody inhibits plasmacytoid dendritic cell activation through Fc-dependent and Fc-independent mechanisms. EMBO Mol Med (2015) 7:464–76. doi:10.15252/emmm.201404719
73. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells – a proposal for uniform nomenclature. Nat Rev Immunol (2013) 13:145–9. doi:10.1038/nri3365
74. Wohlfahrt T, Usherenko S, Englbrecht M, Dees C, Weber S, Beyer C, et al. Type 2 innate lymphoid cell counts are increased in patients with systemic sclerosis and correlate with the extent of fibrosis. Ann Rheum Dis (2016) 75:623–6. doi:10.1136/annrheumdis-2015-207388
75. Roan F, Stoklasek TA, Whalen E, Molitor JA, Bluestone JA, Buckner JH, et al. CD4+ group 1 innate lymphoid cells (ILC) form a functionally distinct ILC subset that is increased in systemic sclerosis. J Immunol (2016) 196:2051–62. doi:10.4049/jimmunol.1501491
76. Yanaba K, Yoshizaki A, Asano Y, Kadono T, Sato S. Serum IL-33 levels are raised in patients with systemic sclerosis: association with extent of skin sclerosis and severity of pulmonary fibrosis. Clin Rheumatol (2011) 30:825–30. doi:10.1007/s10067-011-1686-5
Keywords: innate immunity, systemic sclerosis, fibrosis, sterile inflammation, future therapeutic
Citation: Laurent P, Sisirak V, Lazaro E, Richez C, Duffau P, Blanco P, Truchetet M-E and Contin-Bordes C (2018) Innate Immunity in Systemic Sclerosis Fibrosis: Recent Advances. Front. Immunol. 9:1702. doi: 10.3389/fimmu.2018.01702
Received: 30 April 2018; Accepted: 10 July 2018;
Published: 23 July 2018
Edited by:
Jérôme Avouac, Université Paris Descartes, FranceReviewed by:
Muriel Elhai, Cochin Hospital, Paris, FranceCopyright: © 2018 Laurent, Sisirak, Lazaro, Richez, Duffau, Blanco, Truchetet and Contin-Bordes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cécile Contin-Bordes, Y2VjaWxlLmJvcmRlc0BjaHUtYm9yZGVhdXguZnI=
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.