 Prateek Shetty1
Prateek Shetty1 Iulian Z. Boboescu1Bernadett Pap1Roland Wirth2
Iulian Z. Boboescu1Bernadett Pap1Roland Wirth2 Kornél L. Kovács2Tibor Bíró3Zoltán Futó4
Kornél L. Kovács2Tibor Bíró3Zoltán Futó4 Richard Allen White III5,6,7,8
Richard Allen White III5,6,7,8 Gergely Maróti1*
Gergely Maróti1*- 1Biological Research Centre, Institute of Plant Biology, Hungarian Academy of Sciences, Szeged, Hungary
- 2Department of Biotechnology, University of Szeged, Szeged, Hungary
- 3Faculty of Water Sciences, National University of Public Service, Baja, Hungary
- 4Faculty of Agricultural and Economics Studies, Szent István University, Szarvas, Hungary
- 5Raw Molecular Systems LLC, Spokane, WA, United States
- 6Crop and Soil Sciences, Washington State University, Pullman, WA, United States
- 7Plant Pathology, Washington State University, Pullman, WA, United States
- 8Australian Centre for Astrobiology, University of New South Wales, Sydney, NSW, Australia
Microalgae cultivation in municipal, industrial, and agricultural wastewater is an emerging, highly effective approach for resource recovery and concomitant bioenergy generation. Wastewater effluents represent ideal sources of nutrients (especially nitrogen and phosphorous) for eukaryotic green algal species. However, the recovery performance of photosynthetic green algae is strongly dependent on the associated bacterial partners present in the effluents. Algal microbiome is a pivotal part of the algae holobiont and has a key role in modulating algal growth and functions in nature. There has been no comprehensive study on the importance of microbial communities supporting the algal hosts for the bulk of the time wastewater treatment methods have been in use. Our proposed approach applies a green microalgae-based photoheterotrophic degradation using dark fermentation effluent as substrate. The results showed that condition-dependent mutualistic relationships between the microbial and Chlorella algae populations had direct impact on the biodegradation efficiency and also on algal biohydrogen production. The genome level analysis of the novel hybrid biodegradation system provided important clues for the primary importance of the green algae partner in nitrogen and phosphorous removal. With further development and optimization this new approach can lead to a highly efficient simultaneous organic waste mitigation and renewable energy production technology.
Introduction
It is generally accepted that fundamental changes in fossil fuel policies are required in order to prevent unleashed increase in global average temperatures which could translate into amplified rates of extreme weather patterns and increasing sea levels (IPCC, 2013). In order to avoid such scenarios, the development and implementation of novel renewable energy technologies coupled with carbon capture and storage/usage systems are required (Subramanian et al., 2013). Among different proposed approaches, the bioenergy sector could play a key role in shaping the future energy vision, as it can advantageously exploit a large spectrum of organic substrates, including waste products and wastewater, while generating different types of valuable metabolites and energy carriers. The past decade has showed an expansion of interest from the established anaerobic technologies toward developing photosynthesis-based processes in order to convert organic waste into biohydrogen and other valuable end products (Xu and Lancaster, 2009; Boboescu et al., 2014; Venkateswar Reddy et al., 2014; Han et al., 2016; Wirth et al., 2018).
Biohydrogen production is a promising approach due to the renewable, low-cost, and environmentally friendly nature of this process (Cao et al., 2014). In addition, certain biohydrogen generation approaches, such as algal photoheterotrophic biodegradation, can utilize various low-priced industrial and agricultural wastes, thus coupling waste treatment with renewable energy generation (Lakatos et al., 2014; Batista et al., 2015; Chandra et al., 2015; Koók et al., 2016). During dark fermentation complex organic substrates are converted into organic acids, alcohols, carbon dioxide, and H2 by fermentative bacteria (Calusinska et al., 2015). These metabolites as well as glucose, sucrose and succinate can be further converted to H2, carbon dioxide, and a series of valuable metabolites by photo-fermentation under anaerobic conditions in the presence of light (Kim et al., 2014). Photo-fermentation was identified in a number of green algal species (Chandra and Venkata Mohan, 2011; Hwang et al., 2014; Boboescu et al., 2016).
Although most of these biohydrogen production strategies have, theoretically, low energy requirements, and could utilize various organic wastes as substrates, the practical applications are still far from being economically feasible. One way to address this economic issue could be the development of novel hybrid approaches. An overall increase in both the H2 yields and the efficiency of wastewater treatment could be achieved by using the remaining effluents of dark fermentation in a microalgae-based photoheterotrophic degradation system (Hwang et al., 2014; Turon et al., 2016). However, only a handful of studies are tackling the biohydrogen production potential of microalgae-driven photoheterotrophic degradation (Boboescu et al., 2016). The successful development of a novel hybrid dark fermentative—photoheterotrophic biohydrogen production approach requires a deep understanding of the microbial communities likely performing these metabolic processes in a tight synergy. Thus, in order to render this combined approach a viable alternative to the classic single-stage biohydrogen production methods, the metabolic waste-products generated during the dark fermentation step have to be readily available as substrates for algal biomass generation in the second step (Turon et al., 2015).
A novel hybrid biohydrogen evolving system was investigated in the present study using a two-stage biodegradation approach (microbial dark fermentation followed by photoheterotrophic treatment using Chlorella vulgaris green algae). In this study we have focused on the second photo-fermentative stage of this biodegradation process. A series of outputs ranging from nitrogen and phosphorous removal through biological oxygen demand to the composition of the biodegradation communities were carefully monitored during the photo-fermentation experiments. Special attention was paid to the correlations between the rearrangements of the involved bacterial-algal communities and the performance of the biodegradation system.
In this project we have not only used whole metagenome shotgun sequencing to characterize the microbial community during different stages of wastewater treatment, but also applied this approach to track how the major functions changed across the different stages. We have also used the contigs to bin the genomes of the most abundant species in the biodegradation system. The whole genome bins provide a better understanding of microbial functions specifically enriched, therefore being significant in the photoheterotrophic stage of our wastewater treatment pipeline.
Materials and Methods
Experimental conditions and sample specifications are provided in Table 1.
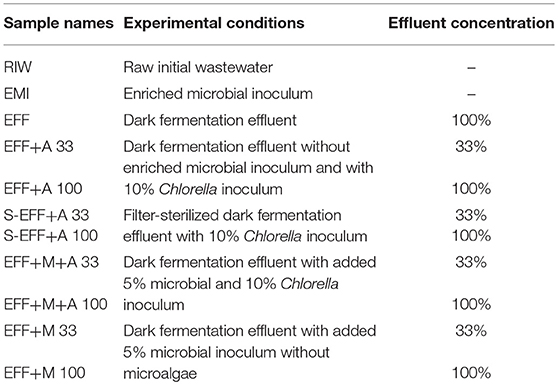
Table 1. Experimental conditions and sample specifications in the photoheterotrophic degradation phase.
Preparation of Microbial Inoculum
Samples were collected from a full-scale methane bioreactor using sludge generated by the wastewater pre-treatment process of a beer brewing factory. Once collected, the samples were incubated for 24 h at 32°C. The samples were then incubated at 70°C for 1 h in order to selectively reduce the abundance of potential hydrogen-consuming microorganisms, primarily methanogenic Archaea. This community (mostly consisting of bacteria) was applied as enriched microbial inoculum (EMI) used in a 5% volume for photo-fermentation experiments.
Microalgae Inoculum
For the photoheterotrophic degradation experiments axenic microalgae Chlorella vulgaris MACC360 was added to the dark fermentation effluents. Freshly grown algae (OD750: 0.7 equal to 2.77 * 108 algae cell mL−1) were added to the dark fermentation effluent in a 10% volume (2 mL in 20 mL). The green algae strain was obtained from the Mosonmagyaróvár Algal Culture Collection (MACC; Institute of Plant Biology, University of West-Hungary, Hungary) and grown on TAP (TRIS-Acetate-Phosphate medium) medium as described in our previous work (Lakatos et al., 2017). The algae liquid cultures and plates were continuously incubated under 50 μmol m−2 s−1 light intensity at 25°C.
Photoheterotrophic Bioreactor Operation
The microalgae-driven photoheterotrophic degradation of the dark fermentation effluent (EFF) was conducted under a number of different experimental conditions (Table 1). The dark fermentation effluent (EFF) was the result of a microbial dark fermentation of a brewery's raw initial wastewater (RIW) (Boboescu et al., 2014). The batch-mode experiments were conducted in 40 mL serum vials with 20 mL of dark fermentation effluent (either 3x diluted or non-diluted effluents), inoculated with 2 mL freshly grown Ch. vulgaris green algae (5.54 * 108 algae cell). For certain samples 1 mL (5 %) of enriched microbial inoculum (EMI) was added. Photoheterotrophic fermentation was performed at 24°C in batch mode for a period of 72 h under continuous illumination with 50 μmol m−2 s−1 light intensity, the vials were shaken at 120 rpm. Samples were taken at 72 h for the determination of substrate degradation rates and microbial community composition. All experiments were performed in triplicate.
Analytical Methods
Biomass in each individual culture was estimated by daily measurement of optical density (OD600) with a Jenway 6320D Spectrophotometer. The algae cell numbers were counted on Tris-Phosphate solid media using serial dilutions. All experiments were repeated three times.
Hydrogen was directly measured by gas chromatography using an Agilent Technologies 7890A GC system equipped with a thermal conductivity detector and argon as a carrier gas. The temperatures of the injector, detector and column were kept at 30, 200, and 230°C, respectively. An HP MolSieve column was used (Agilent). Since a concentration gradient of H2 gas can form in the vial headspace, gas samples (0.5 mL) were taken out after mixing of the headspace gas by sparging several times with a gas-tight syringe. Both daily (by sparging the headspace with nitrogen in every 24 h after the measurement) and total accumulated (without headspace sparging) hydrogen production was measured.
Wastewater degradation efficiency was monitored as a result of BOD analysis (Biological Oxygen Demand) as well as total N and total P measurements. BOD measurements were performed according to the instructions of Hach-Lange Cuvette Test LCK555 at the start of the photo-fermentation experiment and at 72 h. Total nitrogen and total phosphorous were measured according to the instructions of Hach-Lange Cuvette Tests LCK138 and LCK349, respectively, at the start of the photo-fermentation experiment and at 72 h.
Total DNA Extraction From Samples
DNA from complex samples was extracted and purified according to described methods with some modifications (Sharma et al., 2007). Samples (0.5 g) were extracted with 1.3 mL extraction buffer (100 mM Tris-Cl pH 8.0, 100 mM EDTA pH 8.0, 1.5 M NaCl, 100 mM sodium phosphate pH 8.0, 1% CTAB). After thorough mixing, 7 μL of proteinase K (20.2 mg mL−1) was added. After incubation for 45 min, 160 μL of 20% SDS was added and mixed by inversion several times with further incubation at 60°C for 1 h with intermittent shaking every 15 min. Samples were centrifuged at 13,000 rpm for 5 min and the supernatant was transferred into new Eppendorf tubes. The remaining soil pellets were treated three times with 400 μL extraction buffer and 60 μL SDS (20%) and kept at 60°C for 15 min with intermittent shaking every 5 min. Supernatants collected from all four extractions were mixed with an equal quantity of chloroform and isoamyl alcohol (25:24:1). The aqueous layer was separated and precipitated with 0.7 volume isopropanol. After centrifugation at 13,000 rpm for 15 min, the brown pellet was washed with 70% ethanol, dried at room temperature and dissolved in TE (10 mM Tris-Cl, 1 mM EDTA, pH 8.0).
Metagenomic Characterization of Microbial Communities
Total DNA from selected samples was prepared for high-throughput next generation sequencing analysis performed on the Ion Torrent PGM platform (Life Technologies). To estimate coverage in sequenced metagenomes we used Nonpareil (Rodriguez-R and Konstantinidis, 2014).
Taxonomic profiling and assessment of metabolic potential were conducted using the public MG-RAST software package, which is a modified version of RAST (Rapid Annotations based on Subsystem Technology) (Meyer et al., 2008). The sequence data were compared to M5NR using a maximum e-value of 1 × 10−5, a minimum identity of 95%, and a minimum alignment length of 15, measured in amino acids for protein and base pairs for RNA databases.
All results for taxonomy and functional assignment were downloaded from MG-RAST and visualized in R package (Version 3.5.1) using Vegan (Oksanen et al., 2016) and ggplot2 (Wickham, 2016). This also allowed us to filter assignments with read counts lower than five reads per taxonomy unit and incorrectly assigned reads.
Metagenome Assembly, Genome Binning, and Annotation
All reads were then trimmed to increase quality and remove contaminants using bbduk (http://jgi.doe.gov/data-and-tools/bb-tools/). Reads matching algal genomes were identified and separated. Multiple draft algal genomes were downloaded from NCBI and were used as reference to identify algal specific reads using bbduk. This allowed for assembly of the prokaryotic microbiome and eukaryotic algal reads individually.
The microbiome and algal reads were assembled using Megahit (Li et al., 2015). This is one of the few programs that can assemble single end metagenome reads. We then used MetaWrap to bin the bacterial contigs into species specific bins (Uritskiy et al., 2018). MetaWrap accepts bacterial assembled contigs and extracts individual draft genomes by using multiple binning software (metaBAT2, MaxBin2, and CONCOCT) and identifying those bins with the highest evidence across all software. The extracted bins were uploaded to the RAST server to annotate draft bacterial bins.
The three bacterial bins were also subsequently annotated using PROKKA (Seemann, 2014). Annotated protein sequences were then uploaded to KEGG database and BlastKoala (Kanehisa et al., 2016) was used to identify specific genes involved in biosynthesis pathways such as cobalamin, biotin, and thiamine biosynthesis.
All raw fastq files were uploaded to NCBI with SRA accession number PRJNA521112.
Results
The dark fermentation effluent (EFF) of an industrial raw wastewater (RIW) originated from a beer brewing factory was used as our basic, initial substrate for the photoheterotrophic propagation of Ch. vulgaris green microalgae. Algal growth and total biomass, biodegradation efficiency, biohydrogen production and the microbial community structure were monitored, and correlated under various experimental conditions.
Photo-Fermentative Biomass and Biohydrogen Production
An important question was whether selected microalgae strains were able to proliferate and generate considerable amount of biohydrogen when grown on dark fermentation effluents. Microalgae proliferation and survival in the effluent was followed by an algae selective cell counting assay (Figure 1). The results showed that microalgae were able to propagate in the dark fermentation effluent either the effluent was filter-sterilized or the effluent was untreated thereby harbouring its microbial community developed in the first dark fermentation stage (see Table 1 for sample specifications). Thus, Ch. vulgaris showed not only a stable survival rate in the effluent, but clear photosynthetic growth was observed regardless the addition of extra enriched microbial inoculum (Figure 1). Nevertheless, the algal growth rate was higher in the diluted dark fermentation effluent compared to the original, non-diluted effluent.
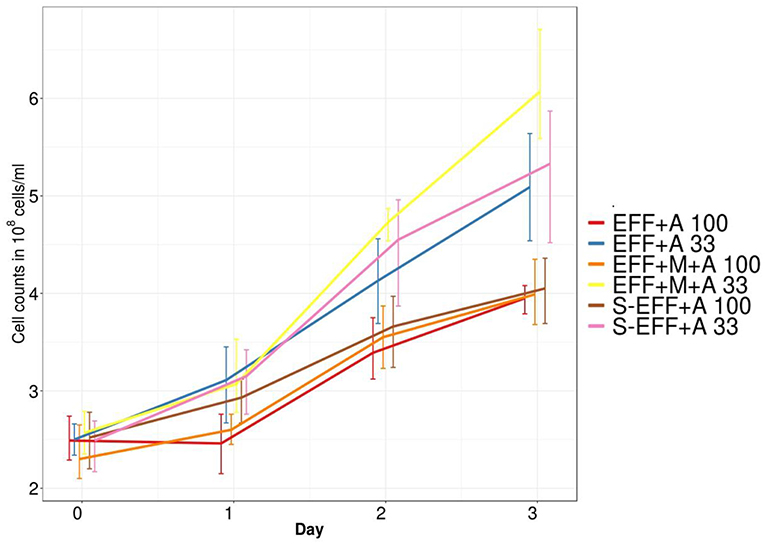
Figure 1. Variation in algal cell numbers measured by counting colony forming units. All diluted samples have higher colony forming units compared to the non-diluted samples. This variation can be attributed to the lower light penetration in the non-diluted samples.
Hydrogen production was monitored during the photoheterotrophic algae cultivation, the headspace was sampled and analyzed in every 24 h throughout the experiments (Figure 2). Significant differences in daily bio-H2 production rates were observed under the experimental conditions applied (Supplementary Table 1). The experimental conditions employing microalgae in the filter-sterilized effluent generally resulted in moderate biohydrogen yields with a maximum of 52 mL H2 L−1 d−1 when grown in the non-diluted effluent (S-EFF+A 100). No significant H2 consumption was detected in the 72 h experimental period. When growing in the diluted, filter-sterilized dark fermentation effluent (S-EFF+A 33), only minor H2 production was observed. The addition of enriched microbial inoculum together with the microalgae (EFF+M+A 100 and EFF+M+A 33) led to a considerable increase in biohydrogen production. The highest maximum yields (154 mL H2 L−1 d−1) were observed on the non-diluted dark fermentation effluent as a substrate (EFF+M+A 100). The dark fermentation effluent with enriched microbial inoculum but without Ch. vulgaris addition (EFF+M 100 and EFF+M 33) resulted in lower levels of biohydrogen with a maximum of 88 mL H2 L−1 d−1 when non-diluted effluent was used as substrate (EFF+M 100). The effluents supplemented with Ch. vulgaris but lacking additional microbial inoculum (EFF+A 33 and 100) showed comparable biohydrogen generation to that observed in the sterilized effluents with added algae (maximum of 56 mL H2 L−1 d−1 on the non-diluted effluent EFF+A 100). However, the observed strong H2 consumption in these samples (EFF+A 33 and 100) as revealed by the comparison of the daily and the accumulated hydrogen amounts made an important difference (Figure 2).
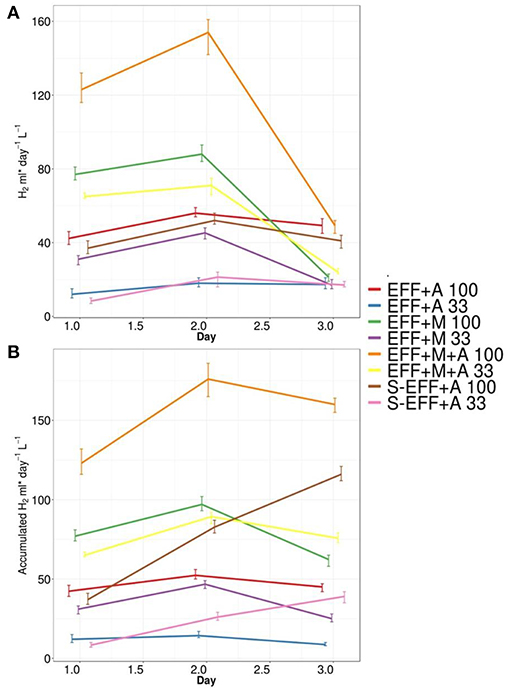
Figure 2. Daily (A) and cumulative biohydrogen production (B) in the photoheterotrophic stage. Microalgae inoculum was added to the effluent resulted from a dark-fermentative biohydrogen production process. Photoheterotrophic biohydrogen production experiments were performed using different experimental conditions according to Table 1. Each of these different experimental conditions were investigated using diluted and non-diluted dark fermentation effluent.
Substrate Utilization (Total N, P, and BOD)
In order to assess the effluent treatment potential of the added microalgae in the photoheterotrophic system, biological oxygen demand (BOD) measurements as well as total nitrogen (N) and total phosphorous (P) investigations were performed. The results showed that green algae significantly contributed to the consumption of total N, P, and BOD under all applied conditions irrespective of the presence of enriched microbial inoculum (Figure 3). Similar N and P metabolizing efficiency as well as BOD decrease were achieved by the green algae under experimental conditions using either diluted or non-diluted dark fermentation effluent as substrate (Figure 3). Significantly lower biodegradation efficiency was observed in the samples lacking green algae indicating the primary role of algae in N and P accumulation in the photoheterotrophic stage. Interestingly, the addition of enriched microbial inoculum resulted in similar biodegradation rate compared to other conditions at the photoheterotrophic stage indicating that the microbial community originated from the dark fermentation stage was not able to exert further biodegradation of the applied wastewater substrate. However, green algae proved to be highly efficient, ~75% of the remaining total N, P, and BOD could be metabolized by photosynthetic eukaryotic algae under illumination. Ch. vulgaris green algae showed survival, photosynthetic growth, and metabolic activity (N and P utilization) both in the sterilized effluent and in the effluent harbouring its natural microbial community (Figure 3).
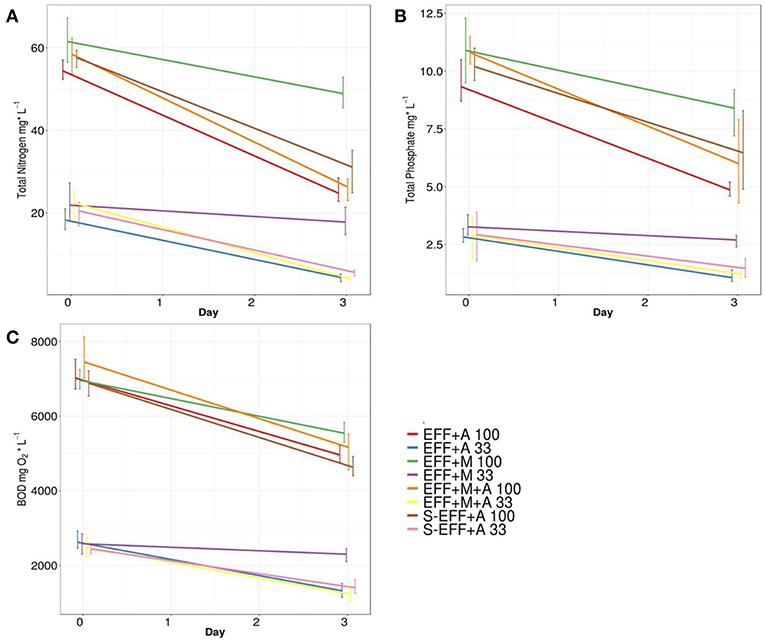
Figure 3. Substrate degradation rates as a function of total N (A), total P (B), and BOD values (C). Measurements were done at the start and at the end (72 h) of the photoheterotrophic degradation experiments. The photoheterotrophic degradation measurements were performed on diluted and non-diluted dark fermentation effluents (33 and 100% concentrations, respectively).
Metagenome Sequencing and Microbial Community Profile
An average of 232,160 sequencing reads was generated for each sample, with a mean read length of 227 nucleotides. To estimate how efficiently this sequencing approach captured the microbiome information, sample datasets were examined using Nonpareil. On an average, all samples had a coverage of 66.01%, with the exemption of the EMI (enriched microbial inoculum) which had a lower coverage of 24.02%. This is most probably due to the high diversity of microbes present in this sample (Table 2).
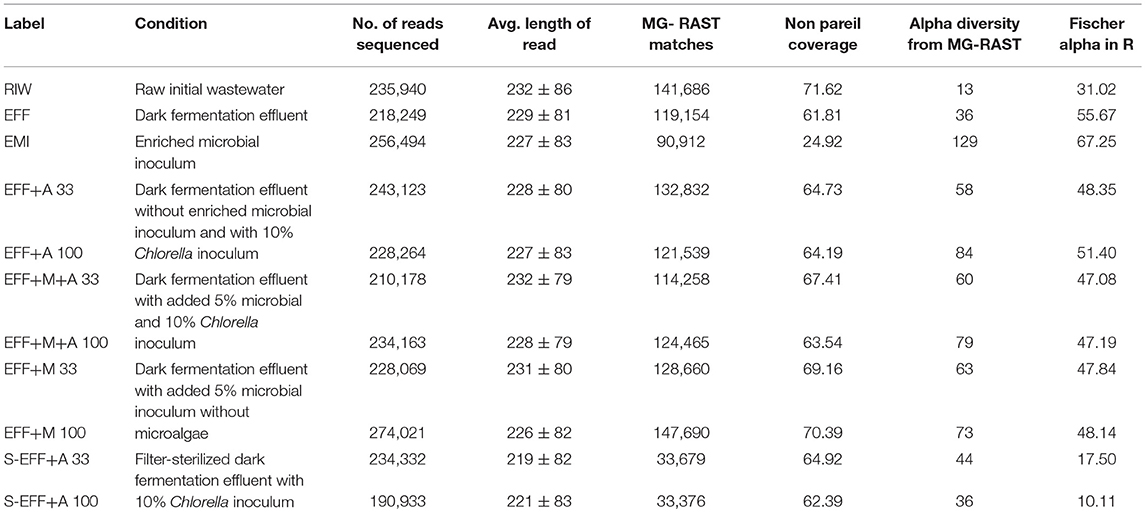
Table 2. Sequencing data and alpha diversity metrics.
Taxonomic Profiling
Reads were uploaded to MG-RAST for taxonomic and functional classification. MG-RAST classified at least 50% of the reads on average for all samples with the exception of the enriched microbial inoculum sample (35%) (Table 2). It was able to classify fewer reads for both the sterilized samples (~15%). Read counts for all samples were downloaded from MG-RAST and filtered to remove classifications that had <5 reads assigned to them. This ensured high confidence of classifications. Since an axenic Ch. vulgaris culture was used as algae inoculum, and the effluent as well as the EMI did not contain any algae (shown by their identified composition), the observed minor presence of other algae hits must be caused by the mis-assignment of the MG-RAST software. Thus, all classifications automatically assigned to genera Actinastrum, Parachlorella, Chlamydomonas, Micractinium, Volvox, Prototheca, and Helicosporidium were manually re-assigned to Chlorella sp. Alpha diversity was measured by both MG-RAST and Fisher's alpha in R (Table 2). The samples with enriched microbial inoculum (EMI) showed the highest alpha diversity, while the filter-sterilized samples had the lowest alpha diversity. The low coverage estimated by Nonpareil for the samples with added enriched microbial inoculum can be attributed to the high diversity. Further, discrepancy was observed in alpha diversity metrics between MG-RAST values and Fisher's alpha. This is due to the fact that low count assignments were filtered out. The observed high diversity of the dark fermentation effluent indicated that the raw wastewater (RIW) was dominated by only a few highly abundant strains, which reduced in abundance during the dark fermentation and instead other bacterial members originally with low relative abundance increased (Figure 4). Finally, higher diversity was found in samples with non-diluted effluents compared to the diluted effluents. This can most probably be attributed to the fact that certain rare species were lost when samples were diluted. After filtering sample counts for low abundance genera, read counts were also proportion normalized. This allowed us to calculate distance matrix downstream. The raw initial wastewater (RIW) sample had lower diversity compared to all other samples. Bacterial members of the genera Lactobacillus and Saccharomyces were present in high abundance (Figure 4). However, after dark fermentation (EFF) this profile clearly changed to a more diverse microbiome. There was an increase in the abundance of Prevotella, Clostridium, Bacterioides, Anaerolinea, Candidatus Cloacimonas, and Megasphera genera along with a concomitant decrease in Saccharomyces sp. abundance. All photoheterotrophic fermentation samples had similar profiles with increases in Prevotella, Veillonella, and Dialister genera with a concomitant sharp decrease in Lactobacillus abundance. Higher relative abundance of Acidaminococcus sp. was detected in the non-diluted wastewater effluent. However, higher abundance of Escherichia sp. was detected in the diluted wastewater effluent. Filter-sterilized samples were not completely sterile, Prevotella, and Lactobacillus genera were detected in low abundance in both samples along with a few other bacterial genera (Figure 4).
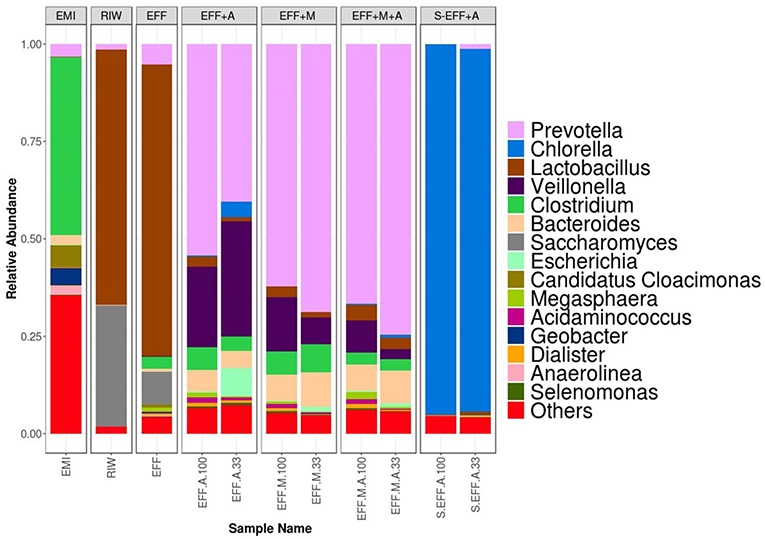
Figure 4. Taxonomic profiles for all samples at the genus level. Enriched microbial inoculum (EMI) has a different profile of microbial genera compared to both the raw initial wastewater (RIW) and dark fermented wastewater (EFF). However, during photo-fermentation all the samples have a very similar profile and abundance of genera. Higher Ch. vulgaris abundance is seen in diluted samples compared to undiluted samples. This correlates well with cell count assays. The top 15 genera are shown below.
Sample Clustering
Proportion normalized read counts were used to calculate a “bray-curtis” distance matrix. Sample clustering was carried out using this distance matrix (Figure 5). Samples primarily clustered by wastewater concentration and then by the addition of the enriched microbial inoculum. This implied that some rare microbes have driven the clustering between samples and the overall microbiome composition changed relatively little during the photoheterotrophic stage. A PCA plot was built using the same distance matrix. The PCA plot showed that the samples with the enriched microbial inoculum were highly divergent from other samples (Figure 6A). This is understandable as the microbial inoculum came from a different source. Further, when dropping the microbial inoculum sample, there is a great degree of variation between all photoheterotrophic samples and the initial raw wastewater sample (Figure 6B). All samples throughout the photoheterotrophic treatment stage clustered together, specifically driven by the effluent concentration. Adonis-test was carried out on the distance matrix of only the photoheterotrophic samples with the presence or absence of either the microbial or the algal inoculum. This allowed us to test which inoculum had a stronger role in shaping the microbial community structure. The addition of microbial inoculum changed the community structure significantly while the algal inoculum had only minor effect (Table 3).
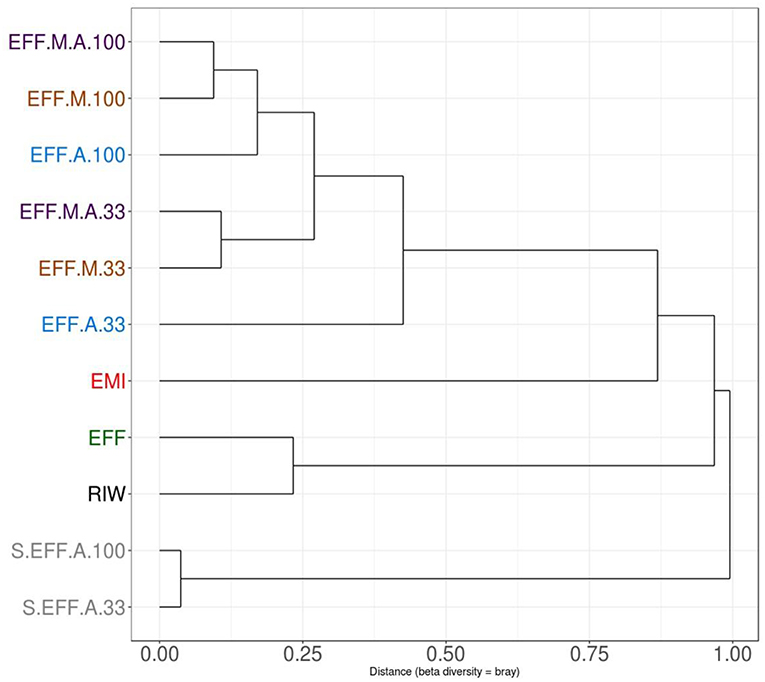
Figure 5. Clustering of samples using the Bray Curtis distance matrix. Samples clustered primarily by dilution rate. Diluted samples clustered together, away from non-diluted samples. Further clustering is seen depending on the presence or absence of enriched microbial inoculum (EMI). Samples which received additional microbial inoculum had a similar taxonomic profile compared to samples that received only algae inoculum.
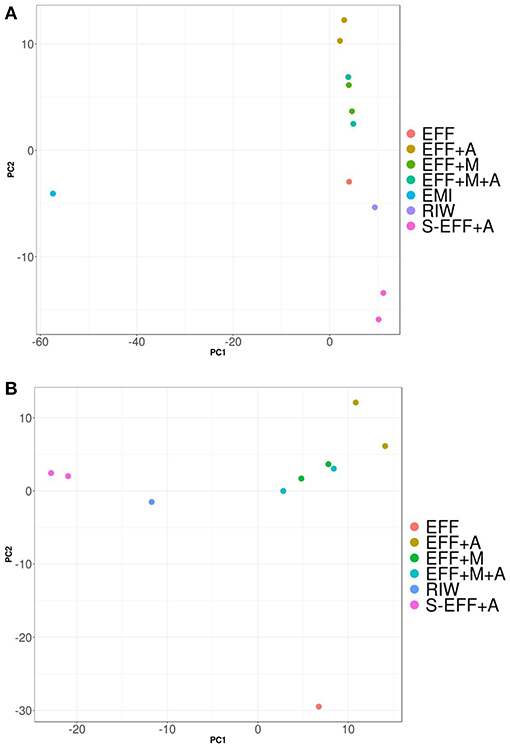
Figure 6. (A) PCA graphs when all samples are present. Sample clustering was driven by enriched microbial inoculum. Since this sample contained highly diverse microbiota, it clustered away from all other samples. All photoheterotrophic samples clustered together indicating that the photoheterotropic treatment had similar effects on all samples regardless of the presence or absence of EMI or algal inoculum. (B) PCA graphs when excluding enriched microbial inoculum sample. There was low variation in taxonomic profile before and after dark fermentation. All photoheterotrophic samples had similar microbial community profiles.
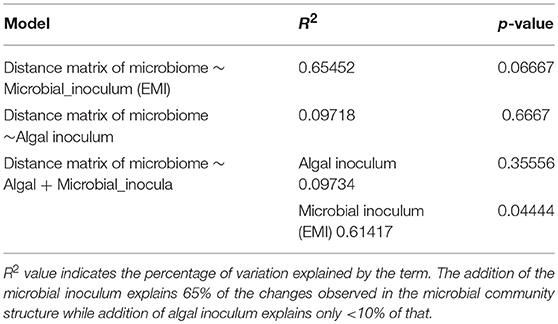
Table 3. Adonis model to test the effects of algal inoculum and microbial inoculum on overall microbial community structure.
Metagenome Assembly and Binning
Metagenome assembly was carried out by Megahit. A total of 50,165 contigs were generated. These contigs were then binned together using three different programs within MetaWrap. The generated bins were further refined and checked for contamination and completion using CheckM database (Table 4). A total of four different bins were generated, three of them showed a high degree of completeness (>70%). These three bins belonged to Lactobacillus, Prevotella, and Veillonella genera (Supplementary Image 1). These bins were uploaded to RAST to get a complete annotation. Gene and protein predictions for these bins were carried out using PROKKA. The predicted proteins were then uploaded to KEGG to identify completion of specific co-factor pathways (Table 5). The Veillonella bin genome had a complete pathway for thiamine biosynthesis and nearly complete pathways for biotin and cobalamine biosynthesis. The Prevotella bin also had a nearly complete pathway for thiamine biosynthesis. No biosynthetic pathways were found for these particular cofactors in the generated Lactobacillus bin genome.

Table 4. Basic metrics for the generated bacterial bins.

Table 5. Presence of cofactor and vitamin metabolism pathways for all bacterial genomes as identified using KEGG database and BlastKoala.
Discussion
The operation of biodegradation systems under permanently changing micro-environmental conditions is facing a series of challenges, which need to be addressed in order to develop and implement economically viable solutions for efficient biological wastewater treatment coupled with energy generation. In this work a series of microalgae-based photoheterotrophic strategies were employed. The main goal of our study was to evaluate the potential of the dark fermentation effluent as substrate for photoheterotrophic algal biomass production and concomitant biohydrogen evolution. Algal growth characteristics, biodegradation efficiency, and rearrangements of the effluent microbial community were monitored throughout the photo-fermentation phase of the hybrid (dark and photo-fermentation) biodegradation process.
Algae were able to grow in the dark fermentation effluent. However, differences in algal growth were observed, slightly higher number of algal cells, and higher algal growth rate were detected in the diluted effluent compared to the non-diluted dark fermentation effluent. This difference is most likely due to the higher accessibility of light in the diluted effluent. Importantly, the algae cells showed similar biodegradation performance as well as biohydrogen production under both conditions. The addition of enriched microbial inoculum (EMI) to the photo-fermentation had no significant effect on algae growth, the minor increase observed in algae cell number in the presence of EMI was not significant. Here, an important conclusion was the fact that EMI addition did not inhibit algal growth. However, EMI had a positive effect on the daily and also on the accumulated H2 production during photo-fermentation, higher H2 evolution was observed in the samples containing EMI. The source of H2 was shown to be cumulative, Ch. Vulgaris, and members of the EMI contributed to the total H2 production. This was supported by the H2 data of photo-fermentation samples either without EMI or without green algae (Figure 2). These data indicated a higher contribution of the EMI to the total H2 production compared to the algal contribution ~60–65% of the total H2 was produced by the EMI while 35–40% could be assigned to Ch. vulgaris. The results also indicated that the pretreatment step selectively inhibited methanogenic archaea in EMI. Since hydrogen is efficiently utilized by hydrogenotrophic archaea, restriction of methanogenesis is integral to render H2 to an end-product in the metabolic flow (Venkata Mohan, 2008). Other studies showed that pretreatment improved the availability of hydrogen producing microorganisms and resulted in higher hydrogen yield (Xiao and Liu, 2009; Kim et al., 2013; Phowan and Danvirutai, 2014). Pretreatment temperatures in different studies have ranged from 65 to 121°C applied over a time period of 10 min to 24 h (Selembo et al., 2009; Xiao and Liu, 2009; Sivagurunathan et al., 2014). Various heat treatments showed varying degree of efficiency with more intense treatments also leading to higher loss in bacterial abundance. Previous study in our lab showed that heat treatment for 1 h at 70°C was sufficient to inhibit hydrogen consuming microbes in wastewater (Boboescu et al., 2013). The algal contribution was clearly supported by the samples where algae were grown on filter-sterilized dark fermentation effluent, thereby bacterial H2 production was excluded. Similar amount of daily H2 was produced in EFF-A and S-EFF-A samples, while significantly higher amount of H2 was accumulated in the S-EFF-A samples, indicating that the effluent sterilization successfully prevented H2 consumption by certain bacteria (even if the sterilization was not complete as can be seen in Figure 4), while strong H2 uptake was observed in the dark fermentation effluent samples containing green algae (EFF-A). It is to note that increasing H2 evolution was observed in the first and the second days of the experiments in all samples, while strongly decreased daily H2 production was detected in the third day regardless the sample conditions. Thus, both green algae and hydrogen-evolving bacteria found suitable substrate conditions (presumably accessible electrons) in the first 48 h of the photo-fermentation. The photosynthetic oxygen produced by the green algae did not inhibit the hydrogenase enzymes (neither algal Fe-hydrogenases nor bacterial Fe- and NiFe-hydrogenases), the active respiration of the bacterial community (even in the S-EFF-A samples) scavenged oxygen as it was shown in earlier studies (Lakatos et al., 2014, 2017).
The changes in total nitrogen, total phosphorous and biological oxygen demand (BOD) reflected the efficiency of the biodegradation in the photo-fermentation phase and implied to the specific and important role of the green algae in the community. The presence of green algae was shown to be crucial for efficient N and P consumption as well as BOD decrease in the photo-fermentation stage. The role of the enriched microbial inoculum at this photoheterotrophic stage appeared to be marginal in further element uptake and degradation of organic molecules. EMI has around 10–20% biodegradation efficiency on the dark fermentation effluent compared to that of the green algae being around 35–50%. This result is interesting and coherent, as the composition of the dark fermentation effluent is presumably more accessible to the photosynthetic green algae than to the bacterial communities developed during anaerobic dark fermentation. Thus, the green algae metabolism represent a novel approach compared to the dark fermentation communities already performing biodegradation in the first dark fermentation phase.
Culture independent methods of microbial community analysis can be carried out by either sequencing the 16S rRNA gene or the total genome sequencing. While 16S rRNA gene sequencing is cost effective and can be used in identifying taxa, whole metagenome shotgun sequencing provides increased accuracy at the species level of characterization of a microbial community and can also be used to predict functional potential of the microbiota. When looking at the taxonomic profiles, large differences can be observed between the EMI sample and all other samples. The EMI sample was obtained from a brewery and was heat treated to remove methanogenic Archaea that readily consume H2. The heat treatment is the primary reason for the strong difference in its taxonomic profile compared to all other samples. Further, there was a sharp decrease in the abundance of Lactobacillus and Saccharomyces genera from both the RIW and EFF samples compared to all the photoheterotrophic samples. The reason for this decrease is not fully understood, if we saw this decrease only in samples with additional enriched microbial inoculum (EFF+M), it could be attributed to negative interactions between the organisms. However, this decrease in relative abundance is important in the context of biohydrogen production as Lactobacillus species are able to lower the H2 yields by diverting H2 potential to the production of lactate (Higgins et al., 2018).
Photoheterotrophic samples primarily cluster by dilution and by the presence or absence of enriched microbial inoculum. The added enriched microbial community changed the community structure over the course of 3 days. In all samples with enriched microbial inoculum an increased abundance of the Prevotella genus was observed while the relative abundance of the Veillonella genus decreased. The presence of Prevotella bacteria is common in H2 reactors (Castelló et al., 2011), the members of this genus have the potential to co-aggregate with other microbes to form granules (Li et al., 2006). The bacterial community was impacted to a greater degree by the addition of enriched microbial inoculum compared to the addition of algal inoculum. Thus, Ch. vulgaris did not have a strong effect in shaping the community, instead the various interactions taking place between different bacterial species might have made the changes. However, it could be very well-attributed to the fact that the photoheterotrophic stage was only performed for 3 days. Further research is needed to elucidate the complex metabolic interactions among the main identified bacterial groups and the photosynthetic microalgae.
The SEED based functional classification provided an overview of the functional potential of all samples (Figure 7). Interestingly, when samples were clustered by taxonomic profiles (Figure 5), RIW, and EFF clustered together, away from EMI. However, when samples were clustered by functional profiles (Figure 8), EMI, and EFF clustered together. This indicated that the relatively small taxonomic changes brought about by dark fermentation of the RIW sample enriched members to perform broad-scale functions similar to those being carried out in EMI. Further, carbohydrate metabolism was performed primarily by the bacterial members of the community as this function was found in low abundance in filter-sterilized samples and in diluted EFF+A sample. All three samples had high abundance of algal cells.
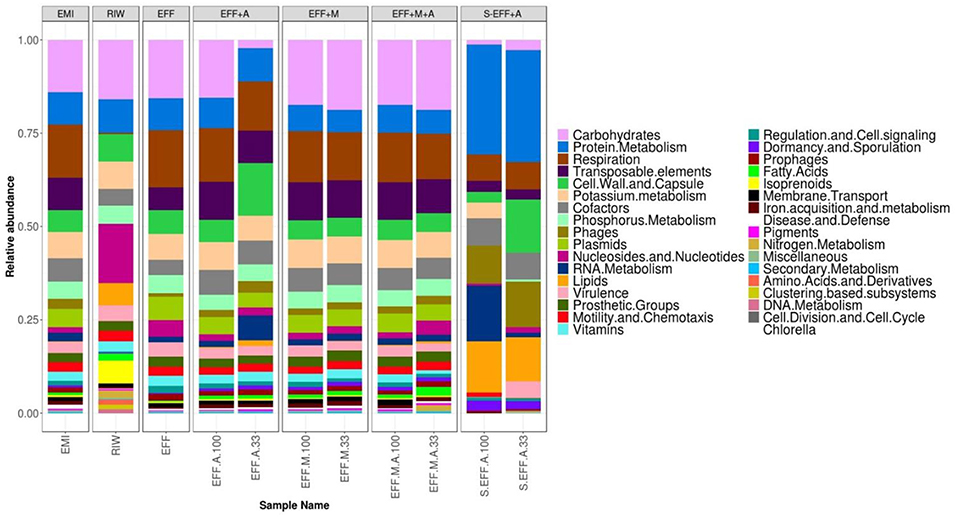
Figure 7. Distribution of functional profiles. The functions were identified using SEED classification. The enriched microbial inoculum (EMI) showed a similar functional profile compared to that of the dark fermentation effluent (EFF) despite their highly different taxonomic profiles.
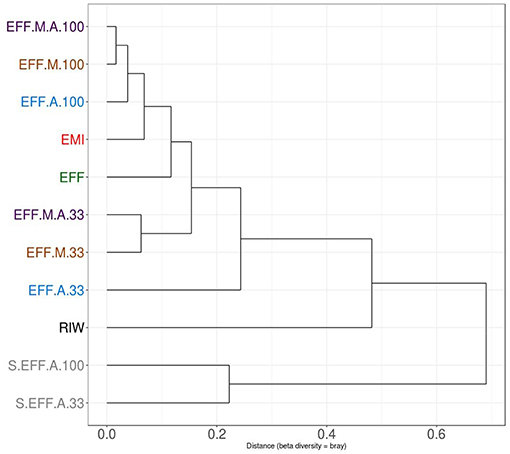
Figure 8. Clustering of samples using Bray Curtis distance matrix from functional abundance. A similar relationship is seen compared to the taxonomic profile where all diluted samples clustered together. The major difference here the EMI sample clustering with the EFF sample indicating the similarity of functional profile despite the dissimilar taxonomic profile.
We were also interested in exploring the biosynthetic potential of various important secondary metabolites. Hence, all the bacterial samples were compared against the KEGG database. Interestingly, both the Veillonella and Prevotella bins had complete pathways for thiamine biosynthesis. The Veillonella bin genome also had partially complete pathways for biotin and cobalamin biosynthesis. These cofactors are particularly important for algal growth. Previous studies involving 306 algal species showed that more than 51% required cobalamin, 22% required thiamine, and 6% required biotin (Croft et al., 2006). Thus, despite the low abundance of the Veillonella bin, it might be performing extremely integral functions for the growth of Ch. vulgaris.
Algal microbiome studies represent a nascent field, with just a handful of studies exploring the algal phycosphere (Krohn-Molt et al., 2017), and fewer still exploring the interactions between the algal host and its bacterial partner (Gonzalez and Bashan, 2000; Croft et al., 2006; Higgins et al., 2018). Understanding the nature of these interactions and the benefits and detriments caused to the host algal species will allow us to design better bioreactors and also can give us insights into improving growth and development without the use of bacterial species when necessary. Our results indicated promising perspectives for the combined approach of wastewater treatment and concomitant biohydrogen evolution using a two-stage pipeline of dark and photoheterotrophic fermentation.
Data Availability
Metagenome reads used in the study are uploaded and accessible at: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA521112/andlt/aandgt.
Author Contributions
PS and IB participated in the conception, design, experimental work, data collection, and analysis, as well as drafted the manuscript. TB and ZF participated in the microbiological sampling and pre-treatment procedures and in the critical discussion of the results. BP carried out the total DNA extraction methods as well as community composition assessments by 16S rRNA method. RW contributed to the whole shotgun metagenomic analyses. RW and KK participated in the analytical measurements and in the interpretation of the results. GM participated in the critical discussion of the results and composed the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the following international and domestic funds: HU09-0091-A1-2016 Norway Grant, NKFI-FK-123899 (GM) and NKFI-PD-121085 (RW), János Bolyai Research Scholarship of the Hungarian Academy of Sciences (GM), and by a Bolyai+ grant UNKP-18-4-SZTE-94 (GM). KK and GM participate in the GINOP-2.3.2-15-2016-00011 project and KK also received funding from GINOP-2.2.1-15-2017-00081.
Conflict of Interest Statement
RW was employed by company RAW Molecular Systems LLC.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenrg.2019.00052/full#supplementary-material
Supplementary Table 1. Results of Tukey HSD test comparing different samples for daily Hydrogen production.
Supplementary Image 1. GC distribution for the 3 bacterial bins.
References
Batista, A., Ambrosano, L., Graça, S., Sousa, C., Marques, P., Ribeiro, B., et al. (2015). Combining urban wastewater treatment with biohydrogen production – An integrated microalgae-based approach. Bioresou. Technol. 184, 230–235. doi: 10.1016/j.biortech.2014.10.064
Boboescu, I., Gherman, V., Lakatos, G., Pap, B., Bíró, T., and Maróti, G. (2016). Surpassing the current limitations of biohydrogen production systems: the case for a novel hybrid approach. Bioresour. Technol. 204, 192–201. doi: 10.1016/j.biortech.2015.12.083
Boboescu, I., Ilie, M., Gherman, V., Mirel, I., Pap, B., Negrea, A., et al. (2014). Revealing the factors influencing a fermentative biohydrogen production process using industrial wastewater as fermentation substrate. Biotechnol. Biofuels. 7:139. doi: 10.1186/s13068-014-0139-1
Boboescu, I. Z., Gherman, V. D., Mirel, I., Maroti, G., and Negrea, A. (2013). Development of a two-step fermentative biohydrogen production process using selectively enriched microbial populations as inoculum. Revista De Chim. 64, 919–924.
Calusinska, M., Hamilton, C., Monsieurs, P., Mathy, G., Leys, N., Franck, F., et al. (2015). Genome-wide transcriptional analysis suggests hydrogenase- and nitrogenase-mediated hydrogen production in Clostridium butyricum CWBI 1009. Biotechnol. Biofuels. 8:27. doi: 10.1186/s13068-015-0203-5
Cao, G., Zhao, L., Wang, A., Wang, Z., and Ren, N. (2014). Single-step bioconversion of lignocellulose to hydrogen using novel moderately thermophilic bacteria. Biotechnol. Biofuels. 7:82. doi: 10.1186/1754-6834-7-82
Castelló, E., Perna, V., Wenzel, J., Borzacconi, L., and Etchebehere, C. (2011). Microbial community composition and reactor performance during hydrogen production in a UASB reactor fed with raw cheese whey inoculated with compost. Water Sci. Technol. 64, 2265–2273. doi: 10.2166/wst.2011.706
Chandra, R., Nikhil, G., and Venkata Mohan, S. (2015). Single-stage operation of hybrid dark-photo fermentation to enhance biohydrogen production through regulation of system redox condition: evaluation with real-field wastewater. Int. J. Mol. Sci. 16, 9540–9556. doi: 10.3390/ijms16059540
Chandra, R., and Venkata Mohan, S. (2011). Microalgal community and their growth conditions influence biohydrogen production during integration of dark-fermentation and photo-fermentation processes. Int. J. Hydrogen Energy 36, 12211–12219. doi: 10.1016/j.ijhydene.2011.07.007
Croft, M., Warren, M., and Smith, A. (2006). Algae need their vitamins. Eukaryotic Cell 5:1175. doi: 10.1128/EC.00097-06
Gonzalez, L., and Bashan, Y. (2000). Increased growth of the microalga chlorella vulgaris when coimmobilized and cocultured in alginate beads with the plant-growth-promoting bacterium azospirillum brasilense. Appl. Environ. Microbiol. 66, 1527–1531. doi: 10.1128/aem.66.4.1527-1531.2000
Han, W., Huang, J., Zhao, H., and Li, Y. (2016). Continuous biohydrogen production from waste bread by anaerobic sludge. Bioresour. Technol. 212, 1–5. doi: 10.1016/j.biortech.2016.04.007
Higgins, B., Gennity, I., Fitzgerald, P., Ceballos, S., Fiehn, O., and VanderGheynst, J. (2018). Algal–bacterial synergy in treatment of winery wastewater. NPJ Clean Water 1:6. doi: 10.1038/s41545-018-0005-y
Hwang, J.-H., Kabra, A., Kim, J., and Jeon, B.-H. (2014). Photoheterotrophic microalgal hydrogen production using acetate- and butyrate-rich wastewater effluent. Energy 78, 887–894. doiL 10.1016/j.energy.2014.10.086
IPCC (2013). Climate Change 2013: “The Physical Science Basis,” in Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change (Cambridge, UK; New York, NY: Cambridge University Press). doi: 10.1017/CBO9781107415324
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kim, D., Lee, J., Kang, S., Hallenbeck, P., Kim, E., Lee, J., et al. (2014). Enhanced photo-fermentative H2production using Rhodobacter sphaeroides by ethanol addition and analysis of soluble microbial products. Biotechnol. Biofuels. 7:79. doi: 10.1186/1754-6834-7-79
Kim, M., Liu, C., Noh, J. W., Yang, Y., Oh, S., Shimizu, K., et al. (2013). Hydrogen and methane production from untreated rice straw and raw sewage sludge under thermophilic anaerobic conditions. Int. J. Hydrogen Energy 38, 8648–8656. doi: 10.1016/j.ijhydene.2013.04.079
Koók, L., Rózsenberszki, T., Nemestóthy, N., Bélafi-Bakó, K., and Bakonyi, P. (2016). Bioelectrochemical treatment of municipal waste liquor in microbial fuel cells for energy valorization. J. Cleaner Product. 112, 4406–4412. doi: 10.1016/j.jclepro.2015.06.116
Krohn-Molt, I., Alawi, M., Förstner, K., Wiegandt, A., Burkhardt, L., Indenbirken, D., et al. (2017). Insights into Microalga and bacteria interactions of selected phycosphere biofilms using metagenomic, transcriptomic, and proteomic approaches. Front. Microbiol. 8:1941. doi: 10.3389/fmicb.2017.01941
Lakatos, G., Balogh, D., Farkas, A., Ördög, V., Nagy, P., Bíró, T., et al. (2017). Factors influencing algal photobiohydrogen production in algal-bacterial co-cultures. Algal Res. 28, 161–171. doi: 10.1016/j.algal.2017.10.024
Lakatos, G., Deák, Z., Vass, I., Rétfalvi, T., Rozgonyi, S., Rákhely, G., et al. (2014). Bacterial symbionts enhance photo-fermentative hydrogen evolution of Chlamydomonas algae. Green Chem. 16, 4716–4727. doi: 10.1039/C4GC00745J
Li, C., Zhang, T., and Fang, H. (2006). Fermentative hydrogen production in packed-bed and packaging-free upflow reactors. Water Sci. Technol. 54, 95–103.
Li, D., Liu, C., Luo, R., Sadakane, K., and Lam, T. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Meyer, F., Paarmann, D., D'Souza, M., Olson, R., Glass, E. M., Kubal, M., et al. (2008). The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386. doi: 10.1186/1471-2105-9-386
Oksanen, J. F., Blanchet, G., Kindt, R., Legendre, P., Minchin, P. R., O'Hara, R., et al. (2016). vegan:Community Ecology Package. R Package Version 2.2-1. Available online at: http://CRAN.R-project.org/package=vegan. (accessed May 12, 2019)
Phowan, P., and Danvirutai, P. (2014). Hydrogen production from cassava pulp hydrolysate by mixed seed cultures: effects of initial pH, substrate and biomass concentrations. Biomass Bioenergy 64, 1–10. doi: 10.1016/j.biombioe.2014.03.057
Rodriguez-R, L., and Konstantinidis, K. (2014). Nonpareil: a redundancy-based approach to assess the level of coverage in metagenomic datasets. Bioinformatics 30, 629–635. doi: 10.1093/bioinformatics/btt584
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Selembo, P. A., Perez, J. M., Lloyd, W. A., and Logan, B. E. (2009). Enhanced hydrogen and 1, 3-propanediol production from glycerol by fermentation using mixed cultures. Biotechnol. Bioeng. 104, 1098–1106. doi: 10.1002/bit.22487
Sharma, P., Capalash, N., and Kaur, J. (2007). An improved method for single step purification of metagenomic DNA. Mol. Biotechnol. 36, 61–63. doi: 10.1007/s12033-007-0015-3
Sivagurunathan, P., Sen, B., and Lin, C. Y. (2014). Batch fermentative hydrogen production by enriched mixed culture: combinationstrategy and their microbial composition. J. Biosci. Bioeng. 117, 222–228. doi: 10.1016/j.jbiosc.2013.07.015
Subramanian, S., Barry, A., Pieris, S., and Sayre, R. (2013). Comparative energetics and kinetics of autotrophic lipid and starch metabolism in chlorophytic microalgae: implications for biomass and biofuel production. Biotechnol. Biofuels. 6:150 doi: 10.1186/1754-6834-6-150
Turon, V., Trably, E., Fayet, A., Fouilland, E., and Steyer, J.-P. (2015). Raw dark fermentation effluent to support heterotrophic microalgae growth: microalgae successfully outcompete bacteria for acetate. Algal Res. 12, 119–125. doi: 10.1016/j.algal.2015.08.011
Turon, V., Trably, E., Fouilland, E., and Steyer, J.-P. (2016). Potentialities of dark fermentation effluents as substrates for microalgae growth: a review. Process Biochem. 51, 1843–1854. doi: 10.1016/j.procbio.2016.03.018
Uritskiy, G., Diruggiero, J., and Taylor, J. (2018). MetaWRAP - A flexible pipeline for genome-resolved metagenomic data analysis 08 information and computing sciences 0803 computer software 08 information and computing sciences 0806 information systems. Microbiome. 6:158. doi: 10.1186/s40168-018-0541-1
Venkata Mohan, S. (2008). Fermentative hydrogen production with simultaneous wastewater treatment: influence of pretreatment and system operating conditions. J. Sci. Indus. Res. 67, 950–961.
Venkateswar Reddy, M., Amulya, K., Rohit, M., Sarma, P., and Venkata Mohan, S. (2014). Valorization of fatty acid waste for bioplastics production using Bacillus tequilensis: Integration with dark-fermentative hydrogen production process. Int. J. Hydrogen Energy 39, 7616–7626. doi: 10.1016/j.ijhydene.2013.09.157
Wirth, R., Lakatos, G., Bojti, T., Maroti, G., Bagi, Z., Rakhely, G., et al. (2018). Anaerobic gaseous biofuel production using microalgal biomass – a review. Anaerobe 52, 1–8. doi: 10.1016/j.anaerobe.2018.05.008
Xiao, B., and Liu, J. (2009). Biological hydrogen production from sterilized sewage sludge by anaerobic self-fermentation. J. Hazard. Mater. 168, 163–167. doi: 10.1016/j.jhazmat.2009.02.008
Keywords: photoheterotrophic degradation, green algae, metagenomics, wastewater, biohydrogen
Citation: Shetty P, Boboescu IZ, Pap B, Wirth R, Kovács KL, Bíró T, Futó Z, White RA III and Maróti G (2019) Exploitation of Algal-Bacterial Consortia in Combined Biohydrogen Generation and Wastewater Treatment. Front. Energy Res. 7:52. doi: 10.3389/fenrg.2019.00052
Received: 11 February 2019; Accepted: 16 May 2019;
Published: 04 June 2019.
Edited by:
Peter Bakonyi, University of Pannonia, HungaryReviewed by:
Dr S. Venkata Mohan, Indian Institute of Chemical Technology (CSIR), IndiaGopalakrishnan Kumar, University of Stavanger, Norway
Poonam Singh, Durban University of Technology, South Africa
Copyright © 2019 Shetty, Boboescu, Pap, Wirth, Kovács, Bíró, Futó, White and Maróti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gergely Maróti, bWFyb3RpLmdlcmdlbHkmI3gwMDA0MDticmMubXRhLmh1