
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med., 30 July 2019
Sec. Atherosclerosis and Vascular Medicine
Volume 6 - 2019 | https://doi.org/10.3389/fcvm.2019.00099
This article is part of the Research TopicEstablished and Novel Roles of Platelets in Health and DiseaseView all 20 articles
 Magdolna Nagy1
Magdolna Nagy1 Johanna P. van Geffen1
Johanna P. van Geffen1 David Stegner2David J. Adams3Attila Braun2Susanne M. de Witt1
David Stegner2David J. Adams3Attila Braun2Susanne M. de Witt1 Margitta Elvers4
Margitta Elvers4 Mitchell J. Geer5
Mitchell J. Geer5 Marijke J. E. Kuijpers1Karl Kunzelmann6
Marijke J. E. Kuijpers1Karl Kunzelmann6 Jun Mori5
Jun Mori5 Cécile Oury7
Cécile Oury7 Joachim Pircher8Irina Pleines2
Joachim Pircher8Irina Pleines2 Alastair W. Poole9Yotis A. Senis5Remco Verdoold1
Alastair W. Poole9Yotis A. Senis5Remco Verdoold1 Christian Weber1,10Bernhard Nieswandt2Johan W. M. Heemskerk1*
Christian Weber1,10Bernhard Nieswandt2Johan W. M. Heemskerk1* Constance C. F. M. J. Baaten1,11*
Constance C. F. M. J. Baaten1,11*Genetically modified mice are indispensable for establishing the roles of platelets in arterial thrombosis and hemostasis. Microfluidics assays using anticoagulated whole blood are commonly used as integrative proxy tests for platelet function in mice. In the present study, we quantified the changes in collagen-dependent thrombus formation for 38 different strains of (genetically) modified mice, all measured with the same microfluidics chamber. The mice included were deficient in platelet receptors, protein kinases or phosphatases, small GTPases or other signaling or scaffold proteins. By standardized re-analysis of high-resolution microscopic images, detailed information was obtained on altered platelet adhesion, aggregation and/or activation. For a subset of 11 mouse strains, these platelet functions were further evaluated in rhodocytin- and laminin-dependent thrombus formation, thus allowing a comparison of glycoprotein VI (GPVI), C-type lectin-like receptor 2 (CLEC2) and integrin α6β1 pathways. High homogeneity was found between wild-type mice datasets concerning adhesion and aggregation parameters. Quantitative comparison for the 38 modified mouse strains resulted in a matrix visualizing the impact of the respective (genetic) deficiency on thrombus formation with detailed insight into the type and extent of altered thrombus signatures. Network analysis revealed strong clusters of genes involved in GPVI signaling and Ca2+ homeostasis. The majority of mice demonstrating an antithrombotic phenotype in vivo displayed with a larger or smaller reduction in multi-parameter analysis of collagen-dependent thrombus formation in vitro. Remarkably, in only approximately half of the mouse strains that displayed reduced arterial thrombosis in vivo, this was accompanied by impaired hemostasis. This was also reflected by comparing in vitro thrombus formation (by microfluidics) with alterations in in vivo bleeding time. In conclusion, the presently developed multi-parameter analysis of thrombus formation using microfluidics can be used to: (i) determine the severity of platelet abnormalities; (ii) distinguish between altered platelet adhesion, aggregation and activation; and (iii) elucidate both collagen and non-collagen dependent alterations of thrombus formation. This approach may thereby aid in the better understanding and better assessment of genetic variation that affect in vivo arterial thrombosis and hemostasis.
Current concepts of platelet activation pathways in thrombosis and hemostasis rely to a large extent on the summation of single observations. Frequently, the role of a particular protein or signaling pathway is deduced from the consequences of a genetic knockout in mouse on platelet responses, such as in comparison to changes in experimental arterial thrombosis and tail bleeding. A large set of such studies has resulted in the concept of collagen-induced arterial thrombus formation (1–4). Herein, it is stipulated that the exposure of subendothelial collagen to flowing blood is a key trigger to start shear-dependent thrombus formation. Collagen causes platelet adhesion and furthermore binds von Willebrand factor (VWF), which can decelerate flowing platelets at high shear rate. Firm VWF/collagen-mediated adhesion and subsequent platelet activation requires synergy between the VWF receptor, glycoprotein (GP)Ib-V-IX, and the collagen receptors, GPVI and integrin α2β1 (5–8).
Collagen is known to induce a number of stimulating pathways via GPVI, in particular: (i) activation of Src-family and Syk tyrosine kinases, resulting in phospholipase C (PLC)γ2 activation and intracellular Ca2+ mobilization (2, 9); (ii) additional Ca2+ influx via ORAI1 channels which couple to the calcium sensor STIM1 in the reticular membrane (10); (iii) activation of several isoforms of protein kinase C (PKC) (11), phosphoinositide 3-kinases (PI3K) (12), and small GTPases, the latter including CDC42, RAC1 and RHOA (13). Additional, modifying signaling pathways include: (iv) activation of phospholipase D, augmenting flow-dependent platelet activation (14); and (v) activation of multiple (tyrosine) phosphatases, a part of which act downstream of immunoreceptor tyrosine-based inhibition motif (ITIM)-containing receptors. Such phosphatases can have a direct or indirect platelet-stimulating (e.g., CD148, DUSP) or a platelet-inhibiting effect (PECAM1, G6b-B) in response to collagen (15). Together, these signaling routes co-operate to control the activation state of the platelet fibrinogen receptor, integrin αIIbβ3, the secretion of granular contents, and the release of thromboxane-A2. Locally secreted or generated soluble agonists, acting via G-protein coupled receptors (GPCR) ensure the capture and incorporation of passing platelets into an aggregate or thrombus (2–4). In a subset of platelets in the thrombus, procoagulant activity is generated by Ca2+-dependent activation of the anoctamin-6 (TMEM16F) channel (16), regulating phosphatidylserine (PS) exposure and platelet ballooning (17–19).
Although all these signaling components are known to play a certain role in collagen-dependent thrombus formation, there is still limited insight into the relative contribution of individual proteins. In addition, it is unclear to which extent C-type lectin-like receptor-2 (CLEC2), another receptor that signals via tyrosine kinases (20, 21), is capable to regulate the process of thrombus formation. The same holds for integrin α6β1, an adhesive receptor, which mediates flow-dependent adhesion of platelets to the matrix protein laminin (22).
In a recent synthesis approach, a quantitative evaluation was made of the contribution of 431 mouse genes to experimental arterial thrombosis and hemostasis in vivo, thereby revealing several genes with a role in thrombosis without affecting bleeding (23). For the total cohort of studies and mouse genes, it appeared that microfluidics assays where thrombus formation is measured in vitro—by whole blood perfusion over a collagen surface—predict the consequences of a gene knockout on thrombosis models in vivo (23). However, the standard microfluidic tests only report on changes in platelet adhesion (surface area coverage, SAC%), which is a limitation given that the recorded microscopic images also contain information on platelet aggregate formation (24, 25). In comparison, for human blood samples from a large cohort of healthy subjects, it could be shown that a multi-parameter image analysis can provide detailed information on the sub-processes of platelet adhesion, aggregation and activation at the same time (26).
To better understand the alterations in thrombus phenotypes using microfluidics, we applied a similar multi-parameter approach to quantitatively compare the effects of deficiency of 37 signaling proteins on collagen-induced pathways. We therefore re-analyzed earlier recorded microscopic images, in all cases from thrombi generated using the same microfluidic flow chamber setup.
Mice were included from 38 strains, in each case with a monogenetic or antibody-induced deficiency, as well as 22 sets of corresponding wild-type or control mice, as described in the original publications (see Table 1). As a selection criterion for inclusion, microscopic images needed to be available from n ≥ 3 animals per modified group and n ≥ 4 for wild-types. Scaled effects on in vivo arterial thrombosis and tail bleeding are for the majority of strains described previously (23).
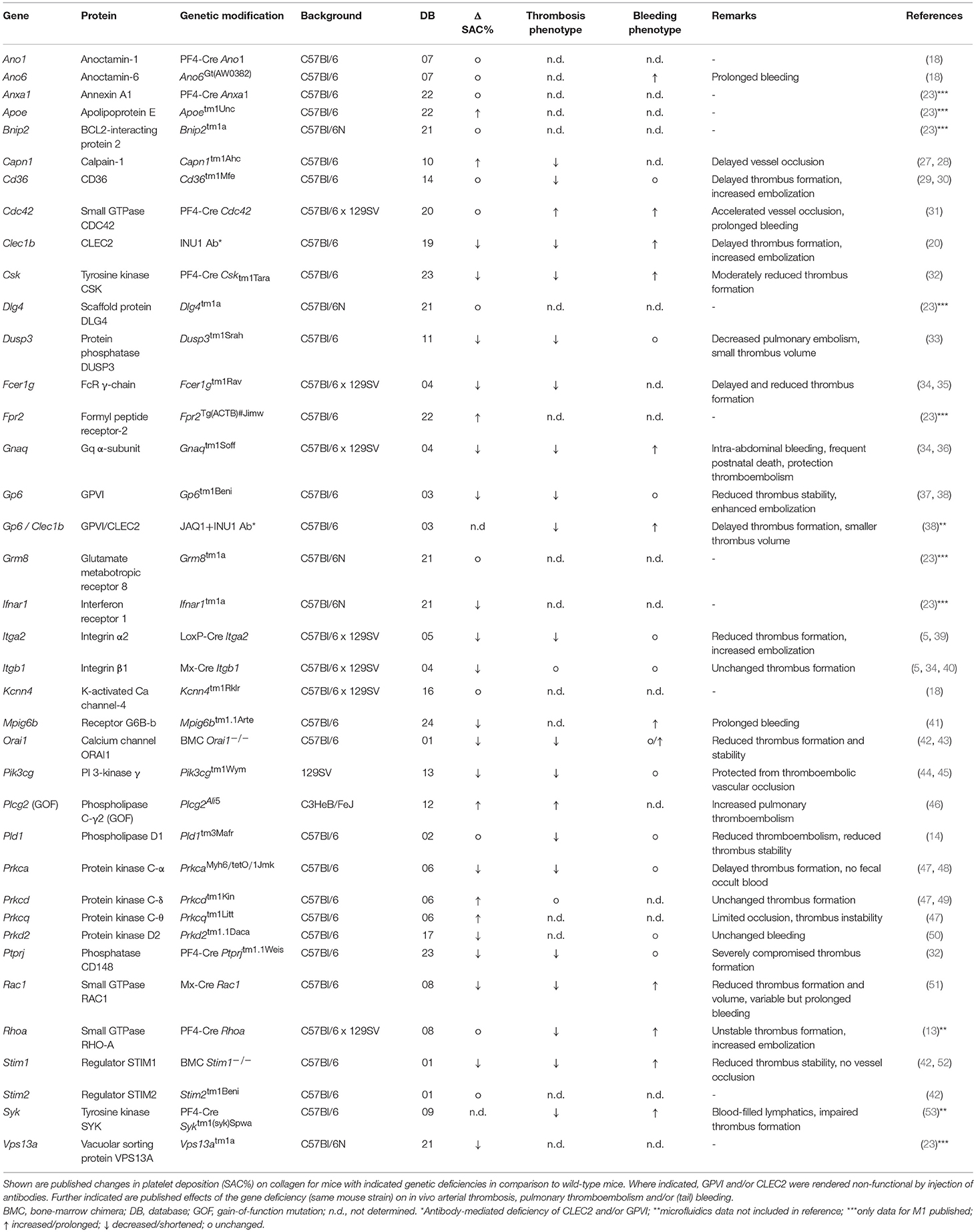
Table 1. Overview of re-analyzed data sets on thrombus formation using the Maastricht flow chamber.
In all cases, mouse blood was collected into anticoagulant medium, consisting of PPACK (40 μM), unfractionated heparin (5 U/ml) and low molecular weight heparin (fragmin, 50 U/ml). Samples were immediately processed. For original data, experiments were approved by the district government of Lower Franconia (Bezirksregierung Unterfranken) and by the Animal Experimental Committee in Hinxton/Cambridge. For previously published data, experiments were approved by the local Animal Experimental Committees as indicated in the original publications (see references in Table 1).
For all data sets, thrombus formation in whole blood under flow was assessed with the Maastricht flow chamber (depth 50 μm, width 3 mm, length 30 mm) (24). In short, PPACK/heparin anticoagulated blood (400–500 μl) was perfused for 3.5–4.0 min at room temperature at a wall shear rate of 1000 s−1 (where indicated 1700 s−1) over (i) Horm-type collagen (100 μg/ml, Nycomed Pharma, Munich, Germany), allowing platelet interaction via GPIb, GPVI and integrin α2β1.
Where specified, two additional surfaces were used, similarly as described before (24): (ii) VWF-binding peptide (VWF-BP, 12.5 μg/ml, obtained from Prof. Dr. R. Farndale, Cambridge University, UK) + laminin (50 μg/ml, from human plasma, Sigma-Aldrich, St. Louis MO, USA; binding integrin α6β1) (22), and (iii) VWF-BP + laminin + rhodocytin (250 μg/ml, activating CLEC2) (54). Rhodocytin purified from Calloselasma rhodostoma (55), was a kind gift of Prof. Dr. K. Clemetson (Bern University, Switzerland).
After blood perfusion, platelet thrombi were rinsed with modified Tyrode's Hepes buffer (pH 7.45, 5 mM Hepes, 136 mM NaCl, 2.7 mM KCl, 0.42 mM NaH2PO4, 1 mg/ml glucose, 1 mg/ml bovine serum albumin, 2 mM CaCl2, 2 mM MgCl2 and 1 U/ml heparin), and then stained during 1.5 min flow with one to three fluorescently labeled platelet activation markers. These were: Alexa Fluor (AF)647 (or FITC) conjugated annexin A5 (AF647: 1:200, Invitrogen Life Technologies, Carlsbad, CA, USA/FITC: 1:1000, PharmaTarget, Maastricht, The Netherlands); FITC anti-mouse CD62P mAb (1:40, rat-anti-mouse, Emfret Analytics, Würzburg, Germany); and phycoerythrin (PE)-labeled JON/A mAb (1:20, Emfret Analytics); all diluted in modified Tyrode's Hepes buffer. Residual labels were removed by another perfusion for 2 min with label-free Tyrode's Hepes buffer. Multiple brightfield and (if applicable) fluorescence microscopic images were captured per surface, of which three representative images were re-analyzed in a systematic way (26).
For image recording, different fluorescence microscopic systems were used (see references in Table 1), but always containing a 60/63x oil objective and a sensitive CCD camera for capturing enhanced-contrast, brightfield images.
For all mouse strains, the recorded 16-bit or 8-bit brightfield and fluorescence images were re-analyzed using the same newly developed scripts (one per image type), written in Fiji (56). Scripts always opened a series of images one-by-one with a loop. In each loop run, background illumination was corrected using a fast Fourier transform bandpass filter, followed by manual setting of a threshold and measurement of the surface area coverage. For brightfield images, a series of Gray morphology conversions was applied to reduce striping and to improve the detection quality. Image conversion steps were as follows: a diamond large sized close, followed by a medium sized circle close and a small circle shaped dilate. Note that the first step increased the pixels that were stronger in regions with many neighboring pixels, the second step then rounded the shapes and additionally reduced straight lines, while the final step (allowing alteration by user-interface) served to obtain the best match with the original image. The Fourier transform filter served to flatten the background areas sufficiently for good analysis, with a minimal impact on the structures. For brightfield images, large structures were filtered down to 60 pixels, for the fluorescence images of annexin A5, integrin and P-selectin images large structures were filtered down to 65 pixels. Small structures were not filtered down, as these contained structures of interest within the adhered platelets (Supplementary Figure 1).
Using these scripts and by visually scoring the unprocessed brightfield images, the following parameters of thrombus formation were obtained (Table 2): surface area coverage of adhered platelets (P1, %SAC); platelet aggregate coverage (P2, %SAC); thrombus morphology score (P3, scale 0-5); thrombus multilayer score (P4, scale 0-3); and thrombus contraction score (P5, scale 0-3). Scoring was performed in comparison to a set of pre-defined standard images (Supplementary Figure 1). For the assessment of platelet activation, fluorescence images were analyzed for PS exposure (P6, %SAC), P-selectin expression (P7, %SAC) and integrin αIIbβ3 activation (P8, %SAC) (24).
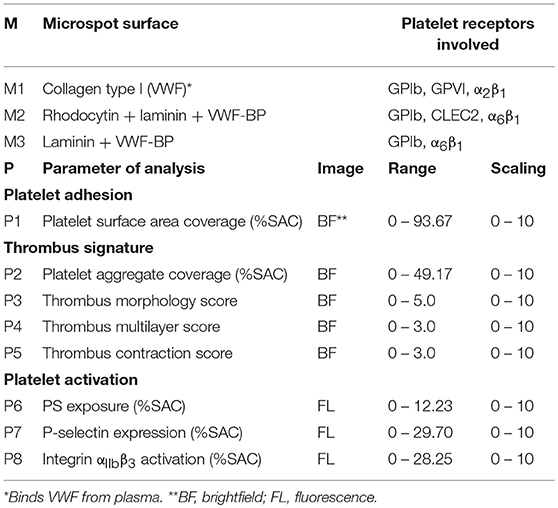
Table 2. Overview of microspotted surfaces (M) and parameters of image analysis (P) for the assessment of thrombus formation.
Regardless of the image type, parameter values from three images per experiment were averaged, thus resulting in a single value per parameter and experiment. The image analysis and scoring parameters were verified by different observers, who were blinded to the experimental condition. Parameters of experiments from the same mouse strain were combined as proxy measures of platelet adhesion (P1), thrombus signature (P2-5), and platelet activation (P6-8), as described before for human platelets (26).
A network of protein-protein interactions was built based on 37 investigated genes, using the STRING database (57), taking into consideration the following settings: 1st shell interactors: <20 interactors, 2nd shell interactors: <60 interactors, confidence level: medium to high. Networks were visualized in Cytoscape version 3.7.0 (58).
Network clustering analysis was performed with the Cytoscape app, MCODE to identify highly interacting nodes using the following settings: degree cutoff: 2, node score cutoff: 0.2, K-core: 2 and maximum depth: 100 (59, 60).
For each genetically modified strain and corresponding wild-types, image data were averaged to obtain one parameter per surface, of which mean and SD values were calculated. For heatmap representation, mean values were univariate scaled from 0 to 10 per parameter (24). Gene effect heatmaps were constructed by subtracting scaled average values of the wild-type (control) strain from those of the modified strain. For statistical evaluation, a filter was applied, considering changes outside the range of composite mean ± SD as a relevant difference between modification and wild-type (26). Heatmap data were visualized by (unsupervised hierarchical) cluster analysis using the program R (61). For comparison of raw parameter values, a Kendall's tau-b correlation analysis was performed using SPSS (IBM SPSS version 24, Armonk, NY, USA).
Microscopic brightfield and (annexin A5) fluorescence images were collected from earlier performed whole blood perfusion experiments with blood from 38 different strains of modified mice and 22 corresponding wild-types (Table 1). Included were experiments with strains (in majority published), that were judged to be of sufficiently high quality and power to allow re-analysis by newly developed image analysis scripts (Supplementary Figure 1). From each experiment, five parameters of thrombus formation were obtained (Table 2). Platelet adhesion was quantified by the conventional analysis of platelet SAC% (P1). Thrombus signature (26) was composed of four parameters related to the thrombus buildup (ΣP2-5), i.e., platelet aggregate SAC% (P2), thrombus morphology score (P3), thrombus multilayer score (P4), and thrombus contraction score (P5). As far as available, platelet activation was assessed from fluorescence images of PS exposure (P6) (see Supplementary Figure 1).
To establish the overall consistency of the combined databases of the whole blood experiments, we compared the mean values plus variation for each of the image analysis parameters P1-5 for the 22 wild-type control datasets. The calculated coefficients of variation of means across the wild-type datasets ranged from <12% (P3-5) to 23-25% (P1,2) (Supplementary Table 1). For heatmap presentation, mean values for all wild-types (and corresponding transgenic animals) were univariate scaled from 0-10 (Figure 1A). The obtained heatmap illustrated an overall high cohesion of the data, yet also suggesting that wild-type strains with mixed C57Bl/6 x 129SV background (databases 04, 05, 08, 16, 20) had a tendency for smaller P3-5 values. Overall, these findings point to a high degree of comparability between the various wild-type datasets.
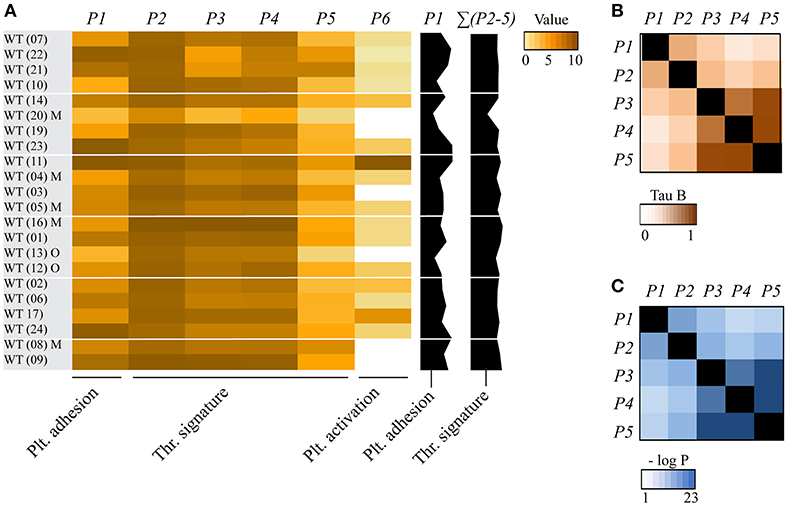
Figure 1. Consistency of collagen-dependent thrombus formation between multiple wild-type mouse datasets. Blood from wild-type (WT) mice (databases as indicated in brackets) was perfused over collagen at a shear rate of 1000 s−1 (in two cases 1700 s−1), and parameters of thrombus formation were obtained by re-analysis of random brightfield images. Investigated wild-type mice (n = 22) had a C57Bl6 genetic background or, where indicated, a mixed C57Bl6 x 129SV background (M) or other background (O). For full details, see Table 1. (A) Heatmap of mean parameters, univariate scaled (0–10) across all mouse strains. Parameter clustering was as follows: platelet adhesion: P1 (platelet SAC%); thrombus signature: P2 (platelet aggregate SAC%), P3 (thrombus morphology score), P4 (thrombus multilayer score), P5 (thrombus contraction score); and platelet activation: P6 (PS exposure). Also indicated (black bars on the right) are the overall scaled values of platelet adhesion (P1) and thrombus signature (ΣP2-5). The wild-type datasets were arranged based on the alphabetical order of the (genetically) modified mice. (B,C) Correlations between parameters of thrombus formation for all cohorts of mice strains. Shown are Kendall's tau-b correlation coefficients (B) and corresponding p-values (C).
To determine how the different parameters of collagen-mediated thrombus formation correlated within our dataset of wild-type and genetically modified mice, a correlation matrix was constructed based on the Kendall's tau-b correlation analysis (Figure 1). All parameters showed a significant moderate to strong correlation to each other (Kendall's tau-b: 0.424–0.829). Platelet adhesion (P1) correlated strongly to platelet aggregation (P2), while it correlated moderately to thrombus morphology (P3), multilayer (P4) and contraction (P5) (Kendall's tau-b: 0.424-0.572). Strongest associations were observed between platelet aggregation (P2) and the thrombus scores (P3-5) (Kendall's tau-b 0.55–0.65, p < 0.001). Furthermore, even stronger correlations were seen between P3-5, i.e., the thrombus morphology, multilayer and contraction scores (Kendall's tau-b 0.76–0.83, p < 10−10).
As detailed in Table 1, the included 38 modified mouse strains concern animals with defects of single genes, encoding for proteins implicated in GPVI- and/or GPCR-related platelet activation pathways, i.e., Csk, Fcer1g (FcR γ-chain), Gnaq (Gαq), Orai1, Pik3cg (PI3Kγ), Pld1 (PLD1), Prkca (PKCα), Prkcd (PKCδ), Prkcq (PKCθ), Prkd2 (PKD2), Stim1, Stim2 or Syk. In addition, the list contains mice with genetic deficiencies of the small GTPases, Cdc42, Rhoa or Rac1; deficiencies of protein (tyrosine) phosphatases Dusp3, Mpig6b (G6b-B), or Ptprj (CD148); deficiencies of other adhesive receptors, such as Itga2 (integrin α2), Itgb1 (integrin β1) or Cd36 (GPIV); defects linked to altered PS exposure, i.e., Ano1 (TMEM16A), Ano6 (TMEM16F), Capn1 (calpain-1) or Kcnn4. Other mice with single gene deficiencies came from an earlier undertaking to find novel proteins implicated in thrombosis and hemostasis (23), namely Anxa1 (annexin 1), Apoe (plasma lipoprotein component), Bnip2 (CBL2-interacting protein), Dlg4 (scaffold protein), Fpr2 (formyl peptide receptor), Grm8 (glutamate receptor), Ifnar1 (interferon receptor) or Vps13a (vacuolar sorting protein). In addition, a mutated mouse strain with a Plcg2 gain-of-function (GOF, constitutive active PLCγ2) was incorporated (46). Given the ability of antibodies JAQ1 and INU1 to specifically cause platelet depletion from GPVI or CLEC2, respectively, after in vivo injection into mice (5) we also included strains with such antibody-induced GPVI and/or CLEC2 deficiencies.
For assessment of the effects of (genetic) deficiency, scaled values per parameter were calculated for each of the 38 modified mouse strains and compared to those of the corresponding wild-types (Supplementary Figure 2). A subtraction heatmap was generated to pinpoint the effects of genetic modification, in which differences outside the range of (composite) means ± SD were considered as being relevant. The heatmap data could be ranked based on alterations in thrombus signature (Figure 2A) or on differences in platelet adhesion (Figure 2B). With either way of ranking, profound quantitative differences were observed in the majority of the thrombus formation parameters, when comparing the 38 modified mouse strains. Thrombus signatures were suppressed, in a decreasing order (Figure 2A), by deficiencies in Syk, GPVI/CLEC2, GPVI, Fcer1g, Gnaq, Rac1, Prkd2, Prkca, Stim1, Itga2, Orai1, CLEC2, Mpig6b, Pik3cg, Csk, and Ptprj. In contrast, this thrombus marker was elevated, in an increasing order, by deficiencies in Prkcq, Capn1, Plcg2 (gain-of-function mutation), Apoe, Prkcd, and Fpr2.
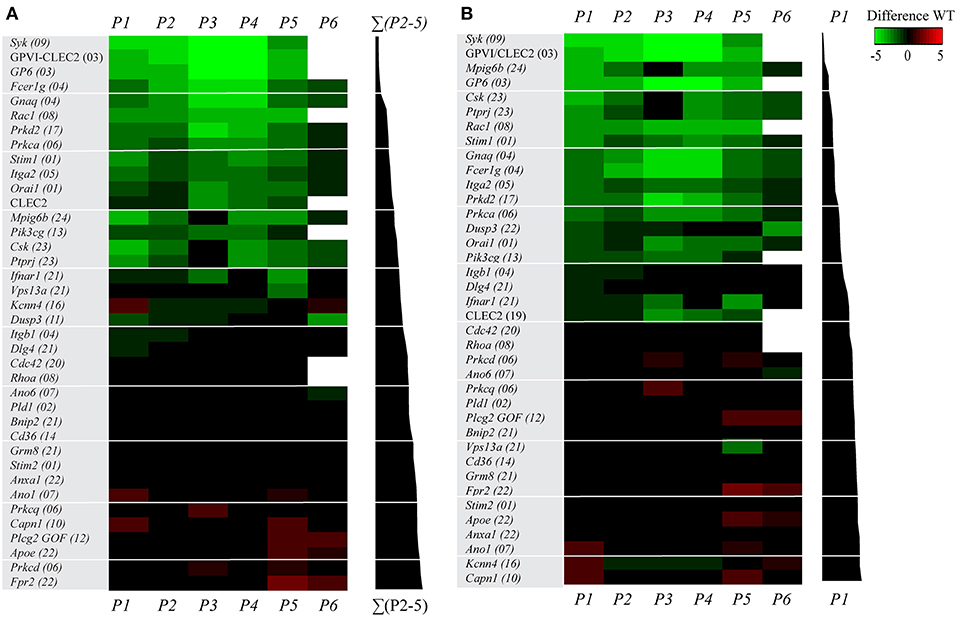
Figure 2. Multi-parameter comparison of collagen-dependent thrombus formation for 38 (genetically) deficient mice. Whole blood from mice with indicated genetic or antibody-induced defects as well as from corresponding wild-type mice was perfused over collagen-I. Parameters of thrombus formation were as indicated in Figure 1. For detailed information on mouse strains, see Table 1. Mean parameters per strain were scaled (0–10) across all wild-types (n = 22) and (genetically) modified mice (n = 38), as for Figure 1. Subtraction heatmaps showing differences between indicated genetic (or antibody-mediated) deficiency in comparison to wild-type, after filtering for differences outside the range of (composite) mean ± SD, to select relevant changes. (A) Ranking of genes based on effect on thrombus signature (ΣP2-5), as shown in black bars on the right. Colors indicate unchanged (black), decreased (green) or increased (red) parameters. (B) Ranking of genes based on effect on platelet adhesion (P1), as indicated in black bars. Colors represent unchanged (black), decreased (green) or increased (red) parameters. Non-subtracted heatmap values are provided in Supplementary Figure 2.
The ranking based on altered platelet adhesion revealed several similarities, but also marked differences. Defects in Syk, GPVI, Rac1, Stim1, and Itga2 resulted in a strong suppression of both platelet adhesion and thrombus signature. Relative larger effects on thrombus signature—in comparison to platelet adhesion—were apparent for mice with defects in Fcer1g, Prkd2, and Prkca (reduced thrombus formation), as well as for mice with defective members of the PKC family, Prkcd and Prkcq (increased thrombus formation). Relative increases in platelet adhesion were only seen for mice with deficiencies in Kcnn4, Ano1, and Capn1. In general, these heatmaps demonstrated that the ranking based on changes in thrombus signature (Figure 2A) can provide additional insight into the 'thrombogenic” consequences of a gene defect, in comparison to a ranking based on altered platelet adhesion (Figure 2B).
For a subset of 11 genetically modified mice strains, the same microfluidic device was used to assess whole blood thrombus formation on collagen-I (M1) plus two additional microspots, i.e., rhodocytin/laminin (M2) and laminin (M3), both co-coated with VWF-BP to induce shear-dependent platelet adhesion (Table 2). As clarified before, immobilized rhodocytin triggers CLEC2-induced platelet activation (26, 54), whereas laminin surfaces allow platelet adhesion via integrin α6β1 (22). For the same subset of mice, the formed thrombi were post-stained in three colors to quantify PS exposure (P6), P-selectin expression (P7) and integrin αIIbβ3 activation (P8), using procedures previously established for human platelet thrombi (26). Representative images from each of the microspots using wild-type blood are depicted in Figure 3. In comparison to collagen-I (M1), rhodocytin/laminin (M2) was less thrombogenic, with only moderately activated platelets that formed small aggregates. The laminin microspot (M3) only triggered adhesion of a monolayer of spreading platelets.
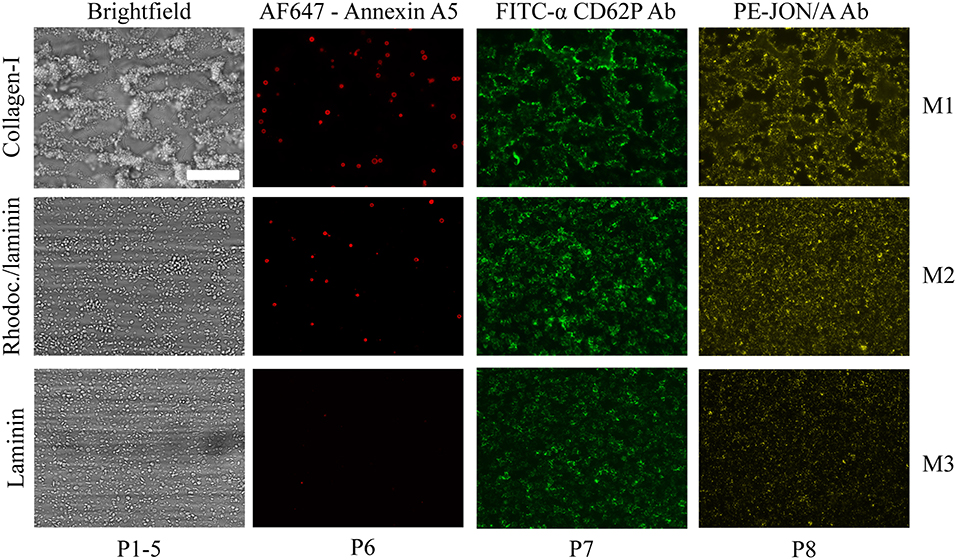
Figure 3. Microscopic imaging of whole blood thrombus formation on adjacent microspots. Whole blood from wild-type mice (DB 24) was flowed for 3.5 min at 1000 s−1 over three consecutive microspots of collagen-I (M1, upper row), rhodocytin/laminin/VWF-BP (M2, middle row), and laminin/VWF-BP (M3, lower row). Shown are representative brightfield and fluorescence microscopic images of the thrombi formed. Triple staining was performed with AF647-annexin A5, FITC-anti-CD62P Ab, and PE-JON/A Ab. Also indicated are the types of parameters (P1-8) taken from the image sets. Bar, 50 μm.
For these 11 genetically modified mouse strains plus corresponding wild-type mice, we again listed the scaled parameters (P1-8) for each microspot (M1-3) (Supplementary Figure 3). The ensuing subtraction heatmap revealed major reductions in platelet adhesion and thrombus signature at surface M1 for the animals with deficiencies in Csk, Ptprj, Mpig6b (tyrosine protein kinase, phosphatases and ITIM receptor, respectively); and, to a clearly lesser extent, for deficiencies in Ifnar1, Vps13a and Anxa1 (Figure 4). For the kinase and phosphatase knockouts, this extended to a reduction in platelet activation parameters (P6-8) at M1, and furthermore to lower P1-2 values at the other microspots M2-3.
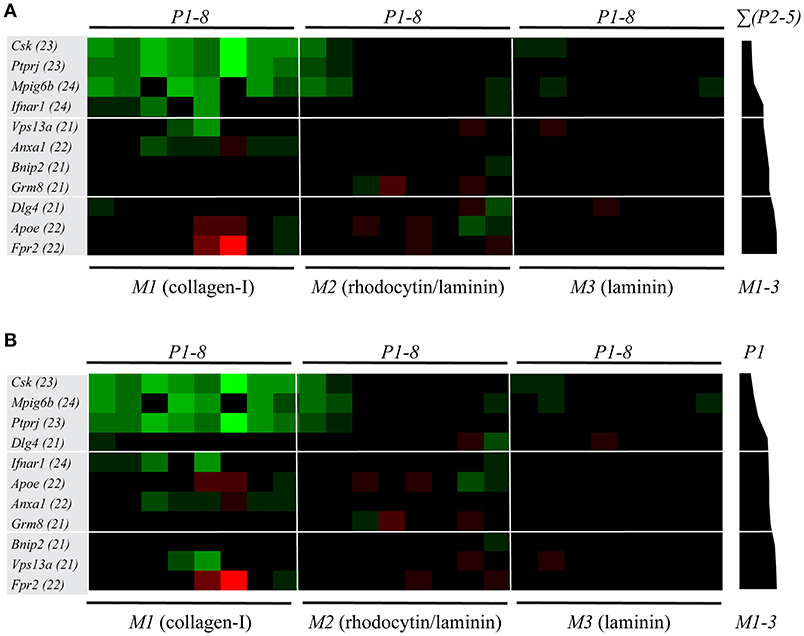
Figure 4. Multi-parameter comparison of thrombus formation on three microspots for 11 mouse strains with genetic deficiencies. Whole blood from mice with indicated genetic defects or corresponding wild-types was perfused over three microspots (M1-3), as for Figure 3. Brightfield images (P1-5) and fluorescence images (P6, AF647-annexin A5; P7, FITC anti-CD62P Ab; P8, PE-JON/A Ab) were analyzed for each microspot. Per microspot, parameter values were univariate scaled (0-10) across all mouse strains. (A) Subtraction heatmap of differences between deficient and wild-type strains, filtered for changes outside the range of (composite) mean ± SD, to select relevant changes. Genes were ranked based on effects on thrombus signature (ΣP2-5) across microspots M1-3, as shown in black bars. Colors represent unchanged (black), decreased (green) or increased (red) parameters. (B) Subtraction heatmap with ranking based on gene effects on platelet adhesion (P1), as indicated in right black bars.
Markedly, for several of these mice, also gain-of-platelet-functions could be detected. In this case, increased parameters P4-6 were distributed over M1 (deficiencies in Apoe and Fpr2) and M2 (deficiency in Grm8). Another remarkable finding was that, for the majority of mice, parameters at the laminin surface (M3) were unchanged. This suggested that laminin-platelet interactions are relatively insensitive to these genetic modifications. Next to the ranking based on changes in platelet adhesion (Figure 4B), the ranking of genes according to changes in overall thrombus signature (Figure 4A) appeared to be a valuable addition in the description of the alterations in platelet properties.
Recently, we described a systematic procedure to compare the consequences of genetic knockout in mice for experimentally induced (collagen-dependent) arterial thrombosis and hemostasis (23). For the 38 deficiencies, it was thus of interest to compare the results of the current extended microfluidic assay (M1, P1-5) with the previously reported changes in arterial thrombosis tendency and tail bleeding in vivo. Hence, per modified mouse strain, the recorded changes (Table 1) were listed as being a reduced/unchanged/increased thrombosis phenotype and as a prolonged/unchanged/shortened bleeding time. Figure 5 gives the comparison of these in vivo effects with altered parameters of in vitro thrombus formation using microfluidics.
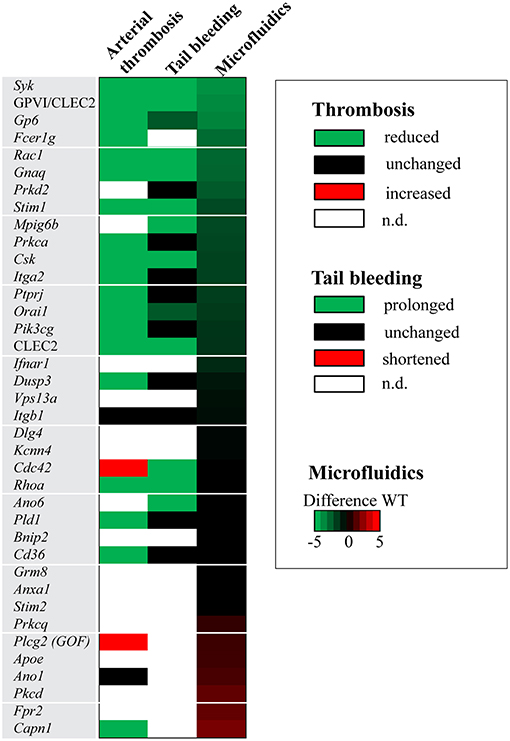
Figure 5. Effects of genetic modification on arterial thrombosis and tail bleeding in comparison to collagen-dependent thrombus formation using microfluidics. Effects of genetic or antibody-induced defects of concerning mouse strains on arterial thrombosis and tail bleeding was obtained from the literature (see Table 1). Effects were classified as being unchanged (black), antithrombotic/prolonged bleeding (green) or prothrombotic/shortened bleeding (red), according to procedures described before (23). White color indicates that information is lacking. Heat mapped data were ranked based on summative effects on all thrombus formation parameters (ΣP1-5) from surface M1.
Strikingly, the majority of mice demonstrating an antithrombotic phenotype in vivo manifested with a larger or smaller reduction in collagen-dependent thrombus formation in vitro (Figure 5). We noted only a few exceptions: (i) mice deficient in Cdc42 with an apparently prothrombotic phenotype, but for unknown reasons no effect in vitro; (ii) mice deficient in Pld1 (where in vitro thrombus formation was only impaired at a higher shear rate of 1700 s−1) (14); and (iii) Cd36-deficient mice (requiring a thrombospondin surface for altered in vitro thrombus formation) (29). Also, the impaired arterial thrombosis reported for Capn1−/− mice (27) did not match with a measured higher platelet adhesion under flow, although it should be noted that the latter mice showed a complex pattern of increased and decreased platelet activation parameters in vitro (28).
Markedly, in the mouse strains with a reduced arterial thrombosis tendency in vivo, only approximately half of these were accompanied with a hemostatic defect (Figure 5). This was also reflected by comparing in vitro thrombus formation (by microfluidics) with alterations in bleeding time. Mice with reduced in vitro thrombus formation, but unchanged or slightly prolonged bleeding times, included animals with deficiencies in the collagen receptors Gp6 and Itga2; the protein kinases Prkd2 and Prkca; and the protein phosphatases Ptprj and Dusp3. This may suggest that the collagen receptors (and hence collagen itself) and the (de)phosphorylating proteins within the downstream signaling pathways are not essential for hemostasis. This may suggest that the collagen receptors (and hence collagen itself) and the (de)phosphorylating proteins are not uniquely—perhaps redundantly—required for platelet functions at lower shear rates such as during hemostasis. The same mouse genes were also previously characterized as having a distinct role in arterial thrombosis and hemostasis (23).
Based on the 37 different mouse genes/proteins that were analyzed for effects on collagen-dependent whole blood thrombus formation, we constructed a network using the biological STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) database, in order to be capable to depict additional protein-protein interactions. Accordingly, a network was established containing in total 117 nodes (37 core and 80 novel nodes) and 1142 edges (interaction score: medium to highest confidence: 0.40–0.99). Reactome pathways that were highly represented in the network were: platelet activation, signaling and aggregation (count in gene set: 37 of 242; false discovery rate: 3.25e−39), hemostasis (count in gene set: 44 of 489; false discovery rate: 1.5e−38), signal transduction (count in gene set: 68 of 2430; false discovery rate: 2.14e−32), GPVI-mediated activation cascade (count in gene set: 18 of 34; false discovery rate: 2.07e−26) and G alpha (12/13) signaling events (count in gene set: 18 of 68; false discovery rate: 2.64e−22). The 37 core nodes were color- and size-coded, based on the established gene effects on thrombus signature M1PΣ(2-5), and then indicated three typical clusters of genes/proteins with large size effects (Figure 6): (i) Gp6 with associated receptors Fcer1g and kinase Syk (cluster score: 7; #nodes: 19; #edges: 63); (ii) the low-molecular weight GTPases Rac1 and Cdc42 together with Itga2 and Itgb1 (cluster score: 5.24; #nodes: 22; #edges: 55); and (iii) Ca2+-regulating signaling components, Stim1, Stim2 and Orai1 (cluster score: 4; #nodes: 4; #edges: 6). Out of the 80 novel nodes, 12 genes have been previously shown to modify in vivo arterial thrombosis and/or bleeding (Cblb, Cttn, Gnai2, Gria1, Itga6, Lat, Lcp2, Ldlr, Pik3cb, Pik3r1, Rock2, Vav1) (23). Color-coding of the same network nodes for gene effects on platelet adhesion revealed as most notable changes the above-mentioned gain-of-function effects of Ano1, Kcnn4 and Capn1 (Supplementary Figure 4).
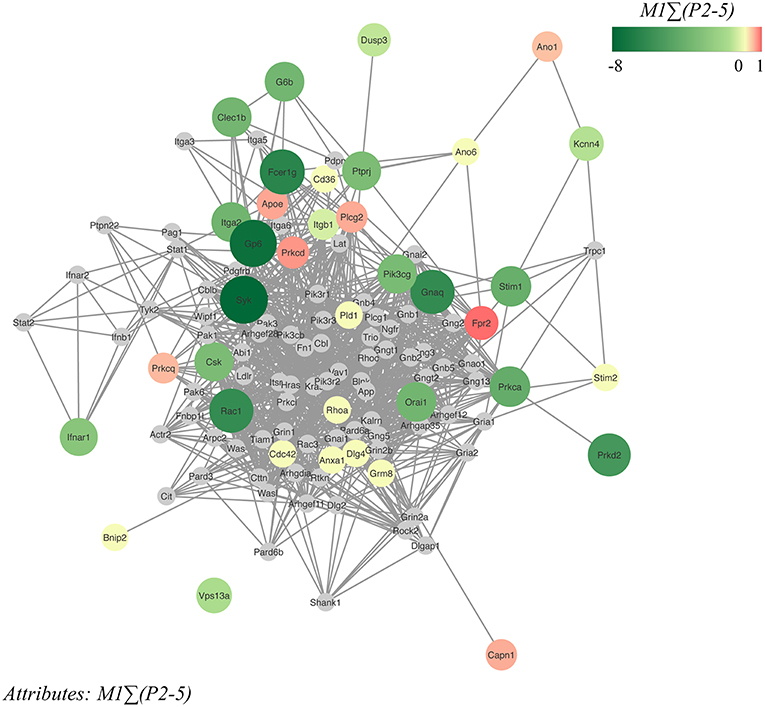
Figure 6. Network of protein-protein interactions in collagen-dependent thrombus formation. Network, constructed from the STRING database and visualized in Cytoscape, of murine protein-protein interactions with as seed the 37 investigated core genes. The network contained 117 nodes (37 core nodes, 80 novel nodes) and 1142 edges. Core nodes of the network were color- and size-coded based on altered thrombus signature (ΣP2-5) at surface M1. Green color intensity (larger size) of nodes points to a stronger reducing effect, and red color intensity (large size) to a stronger stimulating effect in comparison to wild-type. Novel nodes are indicated in gray.
In this paper, we applied standardized analysis procedures in order to allow detailed and quantitative comparison of the changes in platelet functions in multiple genetically modified mice, using microfluidic methods of collagen-dependent thrombus formation under shear conditions. For this purpose, we re-analyzed sets of microscopic brightfield and fluorescence images, using defined semi-automated scripts, resulting in quantitative parameters of platelet adhesion, platelet aggregation (defined as thrombus signature) (26), and platelet activation. The underlying rationale was that earlier captured images provide more relevant information than only a platelet surface area coverage, and thus can provide additional insight into the complex process of thrombus formation. For a subset of mice, it was also possible to extend this complex phenotyping of thrombus formation to other, non-collagen surfaces with additional parameters of platelet activation.
Recent work has shown that multiple platelet function assessment by whole blood microfluidic assays provides novel insights, for instance into the changes in human platelets linked to normal genetic variation (26, 62) and in mouse platelets due to co-activation by interacting chemokines (63). The present comparative quantitative analysis of changes in thrombus formation in multiple genetically or antibody-induced modified mice now also allows to evaluate the assay outcomes for changes in specific platelet functions. Here, we could identify a partial distinction between genes/protein affecting platelet adhesion and those altering platelet aggregation properties (collectively termed as thrombus signature). For instance, it seems that the murine Csk, Mpig6b and Ptprj have a relatively large role in flow-dependent platelet adhesion. Markedly, for Csk, Mpig6b, and Ptprj this effect is extended to also a reduced adhesion at the non-collagen surface M2. On the other hand, the processes of platelet adhesion and aggregate formation are also related, as shown by an overall consistent correlation between adhesion and aggregation parameters (Kendall's tau-b = 0.42-0.68). A similar conclusion was drawn earlier from the analysis of thrombus formation in blood samples from 94 healthy subjects, also pointing to the existence of a subject-dependent thrombus signature (26).
In comparison to the collagen surface (M1), for the 11 mouse strains analyzed, we observed in general smaller gene effects at the two other surfaces (M2-3), which mediate platelet adhesion via GPIb-V-IX and α6β1 with or without CLEC2 (rhodocytin). Accordingly, it seems that, at least in part, distinct sets of genes/proteins are implicated in the platelet adhesion to non-collagen surfaces than to the collagen surface. Clearly in vivo thrombus formation is not purely mediated by collagen, but rather is the result of platelet interactions with a mixture of different extracellular matrix proteins.
The present data set also includes new findings with unpublished blood samples from mice deficient in Rhoa and Syk. The Rhoa defect did not appear to influence platelet adhesion nor thrombus signature parameters at the shear rate of 1000 s−1, which is in support of the earlier conclusion that platelet RHO-A becomes relevant for thrombus formation at high (pathological) shear rates (13). In contrast, genetic deficiency in Syk resulted in a strong reduction of all platelet and thrombus parameters on collagen. This highlights the importance of SYK signaling in collagen-dependent thrombus formation.
Given the earlier established correlation between the outcome of microfluidic tests (in terms of platelet surface area coverage) and experimental arterial thrombosis in mice (23), it was of interest to evaluate for the current mouse strains how a more detailed analysis (considering more thrombus parameters) contributes to this relationship. Such a comparison clearly has limitations, such as the wide variety of methods and test outcomes of the in vivo thrombosis measurements, making a clear differentiation between moderate and strong phenotypes difficult (23); and furthermore, the absence of coagulation and vessel wall components other than collagen in the in vitro approach. Nevertheless, a ranking of the investigated mouse strains according to overall changes in thrombus parameters (including platelet adhesion and aggregation) showed a good reflection with published changes in arterial thrombosis in vivo. On the other hand, for a considerable set of genes, a changed thrombus formation in vitro (and mostly in vivo) was not associated with an altered bleeding time. In several cases, a discrepancy is well explainable. For instance, defects in platelet Clec1b (as a non-collagen receptor) (64) or in Ano6 (an isolated defect of platelet-dependent coagulation) (18), will not be picked up by flow assays over collagen with anticoagulated blood. A striking example is Gp6. Whereas, GPVI deficiency overall impairs thrombus formation in vitro as well as arterial thrombosis in vivo, mouse tail bleeding times are only moderately prolonged (37, 38). In line with that, mild bleeding symptoms have been reported in patients with a defect in the GP6 gene (65, 66). Hence, this gene does likely have a restricted role, which is in line with the constructed network indicating multiple GPVI-linked proteins that contribute to both thrombosis and hemostasis.
Translation of the current evaluation of genes in murine thrombus formation to human pathophysiology can—taking into account the above—provide better insight into the genetic background of human platelet function abnormalities and how these can relate to thrombotic and bleeding disorders. Indeed, for homologs of several of the genes analyzed in this paper, e.g., human ORAI1 and STIM1, mutations have been identified that link to defective collagen-dependent thrombus formation in vitro (67). By applying an extended image analysis, a detailed description of the formed thrombi can be generated. This can aid in the identification of the defective part (platelet adhesion, aggregation or procoagulant response) of the process of thrombus formation in patients with an unexplained increased risk for either thrombosis or bleeding. Extended analysis of the available mouse data and summation in the form of a network can also contribute to a more targeted approach to select novel candidate genes and proteins, possibly affecting platelet functions in a positive or negative way.
Moreover, by applying new knowledge and techniques, in the present paper, new insights were gained from previously performed mouse experiments. In such a manner, our study may contribute to the 3R approaches by reducing, refining and replacing animal experiments.
Taken together, we conclude that the presently developed multi-parameter analysis of thrombus formation on microspots using microfluidics can be used to: (i) determine the severity of platelet abnormalities; (ii) distinguish between altered platelet adhesion, aggregation and activation; and (iii) elucidate both collagen and non-collagen dependent platelet changes. This approach may thereby aid in the better understanding and better assessment of the changes in platelets that affect arterial thrombosis and hemostasis.
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
For original data, experiments were approved by the district government of Lower Franconia (Bezirksregierung Unterfranken) and by the Animal Experimental Committee in Hinxton/Cambridge. For previously published data, experiments were approved by the local Animal Experimental Committees as indicated in the original publications (see references in Table 1).
MN and JvG analyzed and interpreted the data and wrote the manuscript. SdW and MK analyzed the data. RV created scripts used for image analysis. DA, AB, ME, KK, CO, JP, IP, AP, JM, MG, YS, and CW provided the mice, other tools, and revised the manuscript. DS and BN provided the mice, original published and unpublished images, and revised the manuscript. JH and CB provided expert supervision, analyzed and interpreted data, and wrote the manuscript.
MN, MK, and JH have received funding from the Cardiovascular Centre (HVC) MUMC+ Maastricht, the Interreg V Euregio Meuse-Rhine program (Poly-Valve). DS, AB, IP, and BN were supported by the Deutsche Forschungsgemeinschaft (project-number 374031971 – TRR240). CO is Senior Research Associate at the Belgian Fund for Scientific Research (F.R.S.-FNRS). YS received founding from a BHF Fellowship (FS/13/1/29894 and a Programme Grant RG/15/13/31673. CB is supported by the Alexander von Humboldt Foundation.
JH is a co-founder and shareholder of FlowChamber.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2019.00099/full#supplementary-material
1 Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circ Res. (2007) 100:1673–85. doi: 10.1161/01.RES.0000267878.97021.ab
2 Stegner D, Nieswandt B. Platelet receptor signaling in thrombus formation. J Mol Med. (2011) 89:109–21. doi: 10.1007/s00109-010-0691-5
3 Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev. (2013) 93:327–58. doi: 10.1152/physrev.00016.2011
4 Van der Meijden PE, Heemskerk JW. Platelet biology and functions: new concepts and future clinical perspectives Nat. Rev Cardiol. (2019) 16:166–79. doi: 10.1182/blood-2008-07-171066
5 Nieswandt B, Brakebusch C, Bergmeier W, Schulte V, Bouvard D, Mokhtari-Nejad R, et al. Glycoprotein VI but not α2β1 integrin is essential for platelet interaction with collagen. EMBO J. (2001) 20:2120–30. doi: 10.1093/emboj/20.9.2120
6 Siljander PR, Munnix IC, Smethurst PA, Deckmyn H, Lindhout T, Ouwehand WH, et al. Platelet receptor interplay regulates collagen-induced thrombus formation in flowing human blood. Blood. (2004) 103:1333–41. doi: 10.1182/blood-2003-03-0889
7 Auger JM, Kuijpers MJ, Senis YA, Watson SP, Heemskerk JW. Adhesion of human and mouse platelets to collagen under shear: a unifying model. FASEB J. (2005) 19:825–7. doi: 10.1096/fj.04-1940fje
8 Varga-Szabo D, Pleines I, Nieswandt B. Cell adhesion mechanisms in platelets. Arterioscler Thromb Vasc Biol. (2008) 28:403–12. doi: 10.1161/ATVBAHA.107.150474
9 Senis YA, Mazharian A, Mori J. Src family kinases: at the forefront of platelet activation. Blood. (2014) 124:2013–24. doi: 10.1182/blood-2014-01-453134
10 Mammadova-Bach E, Nagy M, Heemskerk JW, Nieswandt N, Braun A. Store-operated calcium entry in blood cells in thrombo-inflammation. Cell Calcium. (2019) 77:39–48. doi: 10.1016/j.ceca.2018.11.005
11 Harper MT, Poole AW. Diverse functions of protein kinase C isoforms in platelet activation and thrombus formation. J Thromb Haemost. (2010) 8:454–62. doi: 10.1111/j.1538-7836.2009.03722.x
12 Guidetti GF, Canobbio I, Torti M. PI3K/Akt in platelet integrin signaling and implications in thrombosis. Adv Biol Regul. (2015) 59:36–52. doi: 10.1016/j.jbior.2015.06.001
13 Pleines I, Hagedorn I, Gupta S, May F, Chakarova L, van Hengel J, et al. Megakaryocyte-specific RhoA deficiency causes macrothrombocytopenia and defective platelet activation in hemostasis and thrombosis. Blood. (2012) 119:1054–63. doi: 10.1182/blood-2011-08-372193
14 Elvers M, Stegner D, Hagedorn I, Kleinschnitz C, Braun A, Kuijpers ME, et al. Impaired αIIbβ3 integrin activation and shear-dependent thrombus formation in mice lacking phospholipase D1. Sci. Signal. (2010) 3:ra1. doi: 10.1126/scisignal.2000551
15 Coxon CH, Geer MJ, Senis YA. ITIM receptors: more than just inhibitors of platelet activation. Blood. (2017) 129:3407–18. doi: 10.1182/blood-2016-12-720185
16 Kunzelmann K, Nilius B, Owsianik G, Schreiber R, Ousingsawat J, Sirianant L, et al. Molecular functions of anoctamin 6 (TMEM16F): a chloride channel, cation channel, or phospholipid scramblase? Pflugers Arch. (2014) 466:407–14. doi: 10.1007/s00424-013-1305-1
17 De Witt SM, Verdoold R, Cosemans JM, Heemskerk JW. Insights into platelet-based control of coagulation. Thromb Res. (2014) 133:S139–S148. doi: 10.1016/s0049-384850024-2
18 Mattheij NJ, Braun A, van Kruchten R, Castoldi E, Pircher J, Baaten CC, et al. Survival protein anoctamin-6 controls multiple platelet responses including phospholipid scrambling, swelling, and protein cleavage. FASEB J. (2016) 30:727–37. doi: 10.1096/fj.15-280446
19 Agbani EO, Poole AW. Procoagulant platelets: generation, function, and therapeutic targeting in thrombosis. Blood. (2017) 130:2171–9. doi: 10.1182/blood-2017-05-787259
20 May F, Hagedorn I, Pleines I, Bender M, Vögtle T, Eble J, et al. CLEC-2 is an essential platelet-activating receptor in hemostasis and thrombosis. Blood. (2009) 114:3464–72. doi: 10.1182/blood-2009-05-222273
21 Watson SP, Herbert JM, Pollitt AY. GPVI and CLEC-2 in hemostasis and vascular integrity. J Thromb Haemost. (2010) 8:1457–67. doi: 10.1111/j.1538-7836.2010.03875.x
22 Schaff M, Tang C, Maurer E, Bourdon C, Receveur N, Eckly A, et al. Integrin α6β1 is the main receptor for vascular laminins and plays a role in platelet adhesion, activation, and arterial thrombosis. Circulation. (2013) 128:541–52. doi: 10.1161/circulationaha.112.000799
23 Baaten CC, Meacham S, de Witt SM, Feijge MA, Adams DJ, Akkerman JW, et al. A synthesis approach of mouse studies to identify genes and proteins in arterial thrombosis and bleeding. Blood. (2018) 132:e35–46. doi: 10.1182/blood-2018-02-831982
24 De Witt SM, Swieringa F, Cavill R, Lamers MM, van Kruchten R, Mastenbroek T, et al. Identification of platelet function defects by multi-parameter assessment of thrombus formation. Nat Commun. (2014) 5:4257. doi: 10.1038/ncomms5257
25 Brouns S, van Geffen JP, Heemskerk JW. High throughput measurement of platelet aggregation under flow. Platelets. (2018) 29:662–9. doi: 10.1080/09537104.2018.1447660
26 Van Geffen JP, Brouns S, Batista J, McKinney H, Kempster CK, Sivapalaratnam S, et al. High-throughput elucidation of thrombus formation reveals sources of platelet function variability. Haematologica. (2019) 104:1256–67. doi: 10.3324/haematol.2018.198853
27 Kuchay SM, Kim N, Grunz EA, Fay WP, Chishti AH. Double knockouts reveal that protein tyrosine phosphatase 1β is a physiological target of calpain-1 in platelets. Mol Cell Biol. (2007) 27:6038–52. doi: 10.1128/MCB.00522-07
28 Mattheij NJ, Gilio K, van Kruchten R, Jobe SM, Wieschhaus AJ, Chishti AH, et al. Dual mechanism of integrin αIIbβ3 closure in procoagulant platelets. J Biol Chem. (2013) 288:13325–11336. doi: 10.1074/jbc.M112.428359
29 Kuijpers MJ, de Witt S, Nergiz-Unal R, van Kruchten R, Korporaal SJ, Verhamme P, et al. Supporting roles of platelet thrombospondin-1 and CD36 in thrombus formation on collagen. Arterioscler Thromb Vasc Biol. (2014) 34:1187–92. doi: 10.1161/atvbaha.113.302917
30 Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. (2007) 13:1086–95. doi: 10.1038/nm1626
31 Pleines I, Eckly A, Elvers M, Hagedorn I, Eliautou S, Bender M, et al. Multiple alterations of platelet functions dominated by increased secretion in mice lacking Cdc42 in platelets. Blood. (2010) 115:3364–73. doi: 10.1182/blood-2009-09-242271
32 Mori J, Nagy Z, Di Nunzio G, Smith CW, Geer MJ, Al Ghaithi R, et al. Maintenance of murine platelet homeostasis by the kinase Csk and phosphatase CD148. Blood. (2018) 131:1122–44. doi: 10.1182/blood-2017-02-768077
33 Musumeci L, Kuijpers MJ, Gilio K, Hego A, Theatre E, Maurissen L, et al. Dual-specificity phosphatase 3 deficiency or inhibition limits platelet activation and arterial thrombosis. Circulation. (2015) 131:656–68. doi: 10.1161/circulationaha.114.010186
34 Kuijpers MJ, Schulte V, Bergmeier W, Lindhout T, Brakebusch C, Offermanns S, et al. Complementary roles of glycoprotein VI and α2β1 integrin in collagen-induced thrombus formation in flowing whole blood ex vivo. Faseb J. (2003) 17:685–7. doi: 10.1096/fj.02-0381fje
35 Munnix IC, Strehl A, Kuijpers MJ, Auger JM, van der Meijden PE, van Zandvoort MA, et al. The glycoprotein VI-phospholipase Cγ2 signaling pathway controls thrombus formation induced by collagen and tissue factor in vitro and in vivo. Arterioscler Thromb Vasc Biol. (2005) 25:2673–8. doi: 10.1161/01.ATV.0000193568.71980.4a
36 Offermanns S, Toombs CF, Hu YH, Simon MI. Defective platelet-activation in Gaq-deficient mice. Nature. (1997) 389:183–6. doi: 10.1038/38284
37 Nieswandt B, Schulte V, Bergmeier W, Mokhtari-Nejad R, Rackebrandt K, Cazenave JP, et al. Long-term antithrombotic protection by in vivo depletion of platelet glycoprotein VI in mice. J Exp Med. (2001) 193:459–69. doi: 10.1084/jem.193.4.459
38 Bender M, May F, Lorenz V, Thielmann I, Hagedorn I, Finney BA, et al. Combined in vivo depletion of glycoprotein VI and C-type lectin-like receptor 2 severely compromises hemostasis and abrogates arterial thrombosis in mice. Arterioscler Thromb Vasc Biol. (2013) 33:926–34. doi: 10.1161/ATVBAHA.112.300672
39 Kuijpers MJ, Pozgajova M, Cosemans JM, Munnix IC, Eckes B, Nieswandt B, et al. Role of murine integrin α2β1 in thrombus stabilization and embolization: contribution of thromboxane A2. Thromb Haemost. (2007) 98:1072–80. doi: 10.1160/TH07–02–0089
40 Grüner S, Prostredna M, Schulte V, Krieg T, Eckes B, Brakebusch C, et al. Multipe integrin-ligand interaction synergize in shear-resistant platelet adhesion at sites of arterial injury in vivo. Blood. (2003) 102:4021–7. doi: 10.1182/blood-2003-05-1391
41 Hofmann I, Geer MJ, Vögtle T, Crispin A, Campagna DR, Barr A, et al. Congenital macrothrombocytopenia with focal myelofibrosis due to mutations in human G6b-B is rescued in humanized mice. Blood. (2018) 132:1399–412. doi: 10.1182/blood-2017-08-802769
42 Gilio K, van Kruchten R, Braun A, Berna-Erro A, Feijge MA, Stegner D, et al. Roles of platelet STIM1 and Orai1 in glycoprotein VI- and thrombin-dependent procoagulant activity and thrombus formation. J Biol Chem. (2010) 285:23629–38. doi: 10.1074/jbc.M110.108696
43 Braun A, Varga-Szabo D, Kleinschnitz C, Pleines I, Bernder M, Austinat M, et al. Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation. Blood. (2009) 113:2056–63. doi: 10.1182/blood-2008-07-171611
44 Cosemans JM, Munnix IC, Wetzker R, Heller R, Jackson SP, Heemskerk JW. Continuous signaling via PI3K isoforms beta and gamma is required for platelet ADP receptor function in dynamic thrombus stabilization. Blood. (2006) 108:3045–52. doi: 10.1182/blood-2006-03-006338
45 Hirsch E, Bosco O, Tropel P, Laffargue M, Calvez R, Altruda F, et al. Resistance to thromboembolism in PI3Kγ-deficient mice. FASEB J. (2001) 15:2019–21. doi: 10.1096/fj.00-0810fje
46 Elvers M, Pozgai R, Varga-Szabo D, May F, Pleines I, Kuijpers MJ, et al. Platelet hyperactivity and a prothrombotic phenotype in mice with a gain-of-function mutation in phospholipase Cγ2. J Thromb Haemost. (2010) 8:1353–63. doi: 10.1111/j.1538-7836.2010.03838.x
47 Gilio K, Harper MT, Cosemans JM, Konopatskaya O, Munnix IC, Prinzen L, et al. Functional divergence of platelet protein kinase C (PKC) isoforms in thrombus formation on collagen. J Biol Chem. (2010) 285:23410–9. doi: 10.1074/jbc.M110.136176
48 Konopatskaya O, Gilio K, Harper MT, Zhao Y, Cosemans JM, Karim AZ, et al. PKCα regulates platelet granule secretion and thrombus formation in mice. J Clin Invest. (2009) 119:399–407. doi: 10.1172/JCI34665
49 Chari R, Getz T, Nagy BJr, Bhavaraju K, Mao Y, Bynagari YS, et al. Protein kinase Cδ differentially regulates platelet functional responses. Arterioscler Thromb Vasc Biol. (2009) 29:699–705. doi: 10.1161/atvbaha.109.184010
50 Konopatskaya O, Matthews SA, Harper MT, Gilio K, Cosemans JM, Williams CM, et al. Protein kinase C mediates platelet secretion and thrombus formation through protein kinase δ2. Blood. (2011) 118:416–24. doi: 10.1182/blood-2010-10-312199
51 Pleines I, Elvers M, Strehl A, Pozgajova M, Varga-Szabo D, May F, et al. Rac1 is essential for phospholipase C-γ2 activation in platelets. Pflugers Arch. (2009) 457:1173–85. doi: 10.1007/s00424-008-0573-7
52 Varga-Szabo D, Braun A, Kleinschnitz C, Bender M, Pleines I, Pham M, et al. The calcium sensor STIM1 is an essential mediator of arterial thrombosis and ischemic brain infarction. J Exp Med. (2008) 205:1583–91. doi: 10.1084/jem.20080302
53 Hughes CE, Finney BA, Koentgen F, Lowe KL, Watson SP. The N-terminal SH2 domain of Syk is required for (hem)ITAM, but not integrin, signaling in mouse platelets. Blood. (2015) 125:144–54. doi: 10.1182/blood-2014-05-579375
54 Suzuki-Inoue K, Kato Y, Kaneko MK, Mishima K, Yatomi Y, Yamazaki Y, et al. Involvement of the snake toxin receptor CLEC-2, in podoplanin-mediated platelet activation, by cancer cells. J Biol Chem. (2007) 282:25993–6001. doi: 10.1074/jbc.M702327200
55 Hooley E, Papagrigoriou E, Navdaev A, Pandey AV, Clemetson JM, Clemetson KJ, et al. The crystal structure of the platelet activator aggretin reveals a novel (alphabeta)2 dimeric structure. Biochemistry. (2008) 47:7831–7. doi: 10.1021/bi800528t
56 Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. (2012) 9:676–82. doi: 10.1038/nmeth.2019
57 Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acid Res. (2015) 43:D447–D452. doi: 10.1093/nar/gku1003
58 Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction network. Genome Res. (2003) 13:2498–504. doi: 10.1101/gr.1239303
59 Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformat. (2003) 4:2. doi: 10.1186/1471-2105-4-2
60 Miryala SK, Anbarasu A, Ramaiah S. Discerning molecular interactions: a comprehensive review on biomolecular interaction databases and network analysis tools. Gene. (2018) 642:84–94. doi: 10.1016/j.gene.2017.11.028
61 Team RC. R: A Language and Environment for Statistical Computing. Vienna: Foundation for statistical computing. Available online at: https://www.R-project.org (accessed 2019 2018)
62 Petersen R, Lambourne JJ, Javierre BM, Grassi L, Kreuzhuber R, Ruklisa D, et al. Platelet function is modified by common sequence variation in megakaryocyte super enhancers. Nat Commun. (2017) 8:16058. doi: 10.1038/ncomms16058
63 Von Hundelshausen P, Agten SM, Eckardt V, Blanchet X, Schmitt MM, Ippel H, et al. Chemokine interactome mapping enables tailored intervention in acute and chronic inflammation. Sci. Transl. Med. (2017) 9:eaah6650. doi: 10.1126/scitranslmed.aah6650
64 Suzuki-Inoue K, Inoue O, Ozaki Y. Novel platelet activation receptor CLEC-2: from discovery to prospects. J. Thromb. Haemost. (2011) 9 :44–55. doi: 10.1111/j.1538-7836.2011.04335.x
65 Matus V, Valenzuela G, Sáez CG, Hidalgo P, Lagos M, Aranda E, et al. An adenine insertion in exon 6 of human GP6 generates a truncated protein associated with a bleeding disorder in four Chilean families. J Thromb Haemost. (2013) 11:1751–9. doi: 10.1111/jth.12334
66 Dumont B, Lasne D, Rothschild C, Bouabdelli M, Ollivier V, Oudin C, et al. Absence of collagen-induced platelet activation caused by compound heterozygous GPVI mutations. Blood. (2009) 114:1900–3. doi: 10.1182/blood-2009-03-213504
Keywords: arterial thrombus formation, bleeding, collagen, glycoprotein VI, platelets, microfluidics
Citation: Nagy M, van Geffen JP, Stegner D, Adams DJ, Braun A, de Witt SM, Elvers M, Geer MJ, Kuijpers MJE, Kunzelmann K, Mori J, Oury C, Pircher J, Pleines I, Poole AW, Senis YA, Verdoold R, Weber C, Nieswandt B, Heemskerk JWM and Baaten CCFMJ (2019) Comparative Analysis of Microfluidics Thrombus Formation in Multiple Genetically Modified Mice: Link to Thrombosis and Hemostasis. Front. Cardiovasc. Med. 6:99. doi: 10.3389/fcvm.2019.00099
Received: 06 May 2019; Accepted: 03 July 2019;
Published: 30 July 2019.
Edited by:
Paul Jurasz, University of Alberta, CanadaReviewed by:
Plinio Cirillo, University of Naples Federico II, ItalyCopyright © 2019 Nagy, van Geffen, Stegner, Adams, Braun, de Witt, Elvers, Geer, Kuijpers, Kunzelmann, Mori, Oury, Pircher, Pleines, Poole, Senis, Verdoold, Weber, Nieswandt, Heemskerk and Baaten. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johan W. M. Heemskerk, andtLmhlZW1za2Vya0BtYWFzdHJpY2h0dW5pdmVyc2l0eS5ubA==; Constance C. F. M. J. Baaten, Y2JhYXRlbkB1a2FhY2hlbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.