 Roberta Listro1†
Roberta Listro1† Silvia Stotani1,2†
Silvia Stotani1,2† Giacomo Rossino1†
Giacomo Rossino1† Marta Rui1†
Marta Rui1† Alessio Malacrida3†
Alessio Malacrida3† Guido Cavaletti3†
Guido Cavaletti3† Michela Cortesi4†
Michela Cortesi4† Chiara Arienti4†
Chiara Arienti4† Anna Tesei4†
Anna Tesei4† Daniela Rossi1†
Daniela Rossi1† Marcello Di Giacomo1†
Marcello Di Giacomo1† Mariarosaria Miloso3†
Mariarosaria Miloso3† Simona Collina1*†
Simona Collina1*†- 1Medicinal Chemistry and Pharmaceutical Technology Section, Department of Drug Sciences, University of Pavia, Pavia, Italy
- 2Medicinal Chemistry, Taros Chemicals GmbH and Co. KG, Dortmund, Germany
- 3Experimental Neurology Unit, School of Medicine and Surgery & Milan Center for Neuroscience, University of Milan Bicocca, Monza, Italy
- 4Biosciences Laboratory, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (IRCCS), Meldola, Italy
Despite the fact that significant advances in treatment of common cancers have been achieved over the years, orphan tumors still represent an important unmet medical need. Due to their complex multifactorial origin and limited number of cases, such pathologies often have very limited treatment options and poor prognosis. In the search for new anticancer agents, our group recently identified RC-106, a Sigma receptor modulator endowed with proteasome inhibition activity. This compound showed antiproliferative activity toward different cancer cell lines, among them glioblastoma (GB) and multiple myeloma (MM), two currently unmet medical conditions. In this work, we directed our efforts toward the exploration of chemical space around RC-106 to identify new active compounds potentially useful in cancer treatment. Thanks to a combinatorial approach, we prepared 41 derivatives of the compound and evaluated their cytotoxic potential against MM and GB. Three novel potential anticancer agents have been identified.
Introduction
Cancer represents one of the leading causes of death worldwide (9.6 million deaths in 2018) (“Cancer” n.d.1). Despite the relevant progresses accomplished in the diagnosis and treatment of common cancers, rare tumors are still considered a global issue, in virtue of their negative prognosis (Pillai and Jayasree, 2017). Among the numerous rare cancers listed by the competent organizations, in this work, we focused the attention on glioblastoma (GB) and multiple myeloma (MM), for which effective treatment options are still needed (Shergails et al., 2018; Willenbacher et al., 2018). Glioblastoma is a malignant brain tumor that develops from astrocytes; it is often aggressive and grows into surrounding normal brain tissues (Hambardzumyan and Bergers, 2015). Signs and symptoms of GB are strictly related to the size and location of the tumor (Esmaeili et al., 2018). The availability of only palliative treatments, as well as the risk of relapses, makes GB the most malignant and lethal form of primary brain tumors (Hanif et al., 2017). Multiple myeloma led to abnormal and uncontrolled growth of plasma cells in the bone marrow (Fairfield et al., 2016). Patients in early stages of the pathology have no concerning signs or symptoms, and therefore, the diagnosis is confirmed too late (Rajkumar, 2009). MM is mostly associated with anemia, which exacerbates secondarily to the suppression of erythropoiesis by cytokine networks. Although novel target therapies prolonged from 2.5 to over 10 years the life expectancy, nowadays, a concrete cure is still missing (Banaszkiewicz et al., 2019).
Despite the fact that the molecular basis underlying their pathogenesis has yet to be fully clarified, both cancers share a complex multifactorial origin, where genetic and environmental factors concur in promoting the pathological manifestations (Kanu et al., 2009; Kyrtsonis et al., 2010). Among others, proteasome machinery plays a role in the major degradation of misfolded proteins involved in cancer etiology (Cvek and Dvorak, 2008; Chen Y. et al., 2017). Proteasome inhibitors like Bortezomib (commercially known as Velcade), carfilzomib, and ixazomib (Okazuka and Ishida, 2018) are already used in therapy for the treatment of MM (Chen et al., 2011; Gelman et al., 2013; Ao et al., 2017; Lee et al., 2018). Several other proteasome inhibitors are undergoing clinical trials and testing, including disulfiram (Lövborg et al., 2006), epigallocatechin-3-gallate (Mereles and Hunstein, 2011), Salinosporamide A (Groll et al., 2018), ONX 0912 (Chauhan et al., 2010), CEP-18770 (Sanchez et al., 2010), and MLN9708 (Lee et al., 2011). Recently it has also been demonstrated that Bortezomib is cytotoxic against patient-derived GB cells (Wang et al., 2018) in vitro and that it is able to enhance the effect of natural killer significantly reducing tumor volumes in GB-bearing mice (Gras Navarro et al., 2019). Moreover, increasing evidence suggests the involvement of Sigma receptors (SRs) in proteasomal dysfunction and in molecular cascades of cell proliferation and survival (Tesei et al., 2018). Several molecules able to modulate SR-mediated pathways are able to inhibit growth, migration, and invasion of cancer cells (Aydar et al., 2006; Collina et al., 2017). A high density of SRs has been found in numerous cancer cell lines, including Roswell Park Memorial Institute (RPMI) 8226, a human MM cell line, and U87MG, a glioblastoma cell line (Brune et al., 2012). Moreover, GB malignancy and aggressiveness are strictly related to the expression level of SRs (Kranz et al., 2018; Liu et al., 2019).
Our research team has been active in this field, studying potential anticancer compounds acting through proteasome complex inhibition and SR modulation (Collina et al., 2013; Rui et al., 2016a; Rossi et al., 2017; Malacrida et al., 2019). Our medicinal chemistry campaign recently led to the identification of (E)-4-benzyl-1-[3-(naphthalen-2-yl)but-2-en-1-yl]piperidine (henceforth RC-106) (Figure 1). This compound is able to activate terminal unfolded protein response (UPR) and to inhibit proteasome complex activity, through the induction of endoplasmic reticulum stress (ER) (Tesei et al., 2019). Moreover, RC-106 possesses a pan-SRs profile—ability to bind both S1R and S2R—and shows a cytotoxic effect against a wide panel of cancer cell lines, all expressing SRs, acting as a proapoptotic drug, which induces a fast triggering of cell death program (Rui et al., 2016b). Despite the promising antitumor profile of RC-106, this compound still suffers from some drawbacks, namely, solubility issues, which might interfere significantly with further investigations and development. Accordingly, we planned to develop a series of analogs in the search for molecules with comparable or enhanced in vitro efficacy and improved pharmacokinetic properties.
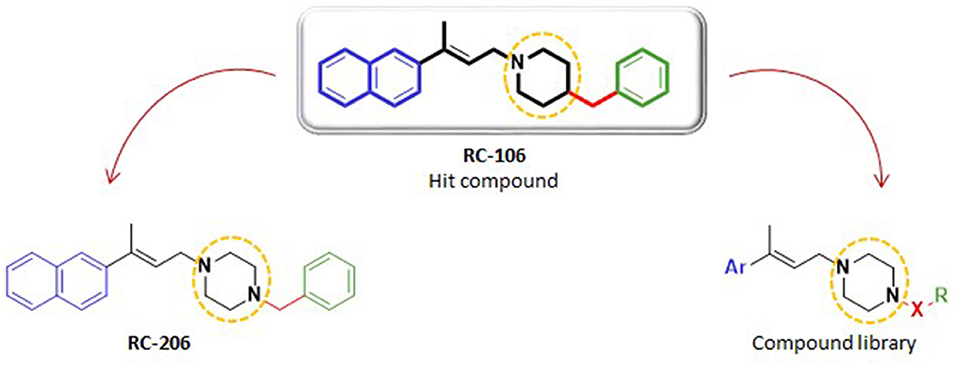
Figure 1. Structures of hit compound RC-106, its analog RC-206, and common scaffold of the new compound library.
Herein, we report the results of a study broadening the structural diversity of the hit RC-106. We replaced the piperidine ring with a piperazine moiety, speculating that this structural elaboration might not affect the anticancer profile. To validate our hypothesis, we first synthesized the piperazine analog named RC-206, and then we built and synthesized via a combinatorial approach a compound library of 40 molecules. The designed members were in silico studied to evaluate their drug-likeness (employing the swissADME free web tool Daina et al., 2017), and their synthetic feasibility was evaluated. Lastly, their cytotoxic profile against U87 and RPMI 8226 cell lines, representative of GB and MM, respectively, was evaluated.
Materials and Methods
Chemistry
General Remarks
Chemicals and solvents were obtained from commercial suppliers and were used without further purification. All dry reactions were performed under nitrogen atmosphere using commercial dry solvents. For Fourier-transform infrared spectroscopy (FTIR) analysis, a Spectrum One Perkin Elmer spectrophotometer equipped with a MIRacle™ ATR device was used. The IR spectra were scanned over a wavenumber range of 4,000–650 cm−1 with a resolution of 4 cm−1. Analytical thin-layer chromatography (TLC) was carried out on silica-gel-precoated glassbacked plates (Fluka Kieselgel 60 F254, Merck), visualized by ultraviolet (UV) radiation, acidic ammonium molybdate (IV), or potassium permanganate. Flash chromatography (FC) was performed with Silica Gel 60 (particle size, 230–400 mesh, purchased from Sigma Aldrich) or Grace Reveleris X2 flash chromatography system using silica-gel-packed Macherey Nagel Chromabond Flash BT cartridges (60 Å, 45 μm) and Grace Reveleris flash Cartridges (60 Å, 40 μm). Proton nuclear magnetic resonance (NMR) spectra were recorded on Bruker Avance 400 and 300 spectrometers operating at 400 and 300 MHz, respectively. Proton chemical shifts (δ) are reported in parts per million with the solvent reference relative to tetramethylsilane (TMS) internal standard (CDCl3, δ = 7.26 ppm). The following abbreviations are used to describe spin multiplicity: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad signal; dd, doublet–doublet; td, triplet–doublet. The coupling constant values are reported in Hertz. 13C NMR spectra were recorded on 400 and 300 MHz spectrometers operating at 100 MHz, with complete proton decoupling. Carbon chemical shifts (δ) are reported in parts per million relative to TMS with the respective solvent resonance as the internal standard (CDCl3, δ = 77.23 ppm). Ultraperformance liquid chromatography-UV-electron spray ionization/mass spectrometry (UPLC-UV-ESI/MS) analyses were carried out on an Acuity UPLC Waters LCQ FLEET system using an ESI source operating in positive ion mode, controlled by Acquidity PDA and 4 MICRO (Waters). Analyses were run on a Acquity BEH Phenyl (ABP) (50 × 2.1, 1.7 mm) or Acquity BEH Shield (ABS) (100 × 2.1, 1.7 mm) columns, at room temperature, with gradient elution (solvent A: water containing 0.1% of formic acid; solvent B: methanol containing 0.1% of formic acid; gradient: 10% B in A to 100% B in 3 min, followed by isocratic elution 100% B for 1.5 min, return to the initial conditions in 0.2 min) at a flowrate of 0.5 ml min−1. All the final compounds had 95% or greater purity.
Analytical, preparative high-performance liquid chromatography (HPLC) and ESI condition mass spectra were performed on an Agilent UHPLC (1290 Infinity) and an Agilent Prep-HPLC (1260 Infinity) both equipped with a diode array detector and a quadrupole MS Dusing mixture gradient of formic acid/water/acetonitrile as system solvent. High-resolution ESI Fourier transform mass spectrometry (ESI-FTMS) mass spectra were recorded on a Thermo LTQ Orbitrap (high-resolution mass spectrometer from Thermo Electron) coupled to an “Accela” HPLC system supplied with a “Hypersil GOLD” column (Thermo Electron).
General Procedure for the Synthesis of Allylic Esters 1[1–4]
In a two-neck round-bottom flask, preserving the anhydrous conditions, solid reagents are added quickly. After AcONa (2 equiv.) addition, Palladium EnCat® (loading, 0.4 mmol/g, 1%, 0.01 equiv.) is added with TEAC (2 equiv.). Afterwards, the liquid reagent (E)-ethyl but-2-enoate (1.5 equiv.; d = 0.918 g/ml) and the dry solvent dimethylformamide (DMF) are added, too. Separately, in a round-bottom flask, a solution of aryl bromide (Ar-Br, 1 equiv.) in dry DMF is prepared. This solution is then transferred to a dropping funnel, preventively added to the second neck of the reaction flask. The total volume of dry DMF used within these two steps has to provide a final concentration in the reaction ambient close to 0.35 M. This reaction mixture, kept in inert medium and under magnetic stirring, is heated under reflux with an oil bath. At the beginning, the temperature is settled at 50°C. During the addition of the aryl bromide solution, that should last for 1 h, the temperature is gradually increased until it reaches 75°C. Once the additions are finished, the heating is increased again until reaching a temperature of 105°C. After almost 2 h from the beginning of the aryl bromide addition, the reaction mixture starts getting darker, and after one more hour, it gets totally black. This color change means that the reaction has gone to completion, as confirmed by TLC with MP Hex/AcOEt 95:5 where the aryl bromide spot disappears. The raw product is then filtered on a paper filter and simultaneously transferred in a separating funnel. The reaction mixture is diluted and extracted three times with Et2O. The collected organic phases are washed with water, dried with Na2SO4 anhydrous, filtrated, and evaporated to dryness at reduced pressure. The dark-brown raw oil obtained after solvent evaporation is purified by chromatography on silica gel (see below for more detail). The purified intermediate products were characterized by 1H and 13C NMR.
(E)-Ethyl 3-phenylbut-2-enoate 1[1]: by following the general procedure, starting from bromobenzene (4.9 g), the desired product was obtained as clear oil (2.8 g). Yield: 46.7%. Rf: 0.5 (TLC Hex/AcOEt, 95:5 v/v). Purification: flash chromatography, gradient elution, MP Hex/AcOEt 99:1, 97:3, and 96:4 v/v, 5 cm diameter column. IR (cm−1): 601.682; 628.68; 692.32; 724.139; 734.746; 764.637; 870.703; 1,041.37; 1,094.4; 1,155.15; 1,270.86; 1,343.18; 1,365.35; 1,445.39; 1,493.6; 1,539.88; 1,576.52; 1,626.66; 1,708.62; 2,116.49; 2,301.63; 2,359.48; 2,959.23; 3,024.8. 1H NMR (400 MHz, CDCl3): δ 7.50 (d, J = 5.1 Hz, 2H), 7.44–7.33 (m, 3H), 6.16 (s, 1H), 4.24 (q, J = 7.1 Hz, 2H), 2.60 (s, 3H), 1.34 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 166.77, 155.41, 142.11, 128.86, 128.37, 126.19, 117.06, 59.73, 17.83, 14.23.
(E)-Ethyl 3-(4-methoxyphenyl)but-2-enoate 1[2]: by following the general procedure, starting from 1-bromo-4-methoxybenzene (4.5 g), the desired product was obtained as a clear oil (2.6 g). Yield: 49%. Rf: 0.35 (TLC Hex/AcOEt, 95:5 v/v). Purification: flash chromatography, gradient elution, MP Hex/AcOEt 98:2, 97:3, and 96:4 v/v, 5 cm diameter column. IR (cm−1): 627.716; 697.141; 735.71; 828.277; 870.703; 1,030.77; 1,152.26; 1,249.65; 1,273.75; 1,343.18; 1,365.35; 1,440.56; 1,511.92; 1,573.63; 1,601.59; 1,624.73; 1.705.73; 2,143.49; 2,338.27; 2,836.77; 2,901.38; 2,988.16. 1H NMR (400 MHz, CDCl3): δ 7.47 (d, J = 8.8 Hz, 2H), 6.91 (d, J = 8.8 Hz, 2H), 6.13 (d, J = 0.7 Hz, 1H), 4.22 (q, J = 7.1 Hz, 2H), 3.84 (s, 3H), 2.58 (d, J = 0.5 Hz, 3H), 1.33 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 166.98, 160.28, 154.78, 134.15, 127.54, 115.15, 113.67, 59.61, 55.20, 17.52, 14.25.
(E)-Ethyl 3-(naphthalen-2-yl)but-2-enoate 1[3]: by following the general procedure, starting from 2-bromonaphtalene (4.2 g), the desired product was obtained as a white solid (2.4 g). Yield: 49.3%. Rf: 0.44 (TLC Hex/AcOEt, 95:5 v/v). mp: 54°C. Purification: flash chromatography, gradient elution, MP Hex/AcOEt 97:3 and 96:4 v/v, 5 cm diameter column. IR (cm−1): 601.682; 627.716; 665.321; 707.747; 724.139; 735.71; 816.706; 1,129.12; 1,413.57; 1,540.85; 1,704.76; 2,143.49; 2,286.2; 2,337.3; 2,391.3; 2,847.38; 2,911.99; 2,953.45; 2,997.8. 1H NMR (400 MHz, CDCl3): δ 7.98 (s, 1H), 7.92–7.81 (m, 3H), 7.63 (dd, J = 8.6, 1.7 Hz, 1H), 7.57–7.49 (m, 2H), 6.33 (d, J = 0.9 Hz, 1H), 4.28 (q, J = 7.1 Hz, 2H), 2.73 (d, J = 0.9 Hz, 3H), 1.37 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 167.39, 155.70, 139.53, 133.93, 133.30, 128.71, 128.45, 127.77, 126.92, 126.80, 126.24, 124.39, 118.01, 60.20, 17.95, 14.69.
(E)-Ethyl 3-(6-methoxynaphthalen-2-yl)but-2-enoate 1[4]: by following the general procedure, starting from 2-bromo-6-methoxynaphthalene (6.6 g), the desired product was obtained as a white solid (2.9 g). Yield: 50%. Rf: 0.3 (TLC Hex/AcOEt, 95:5 v/v). mp: 78°C. Purification: flash chromatography, MP Hex/AcOEt 9:1 v/v, 5 cm diameter column. IR (cm−1): 601.682; 618.074; 627.716; 665.321; 706.783; 735.71; 815.742; 850.454; 1,157.08; 1,424.17; 1,540.85; 1,565.92, 1,647.88, 1,747.19, 2,217.74; 2,995.87; 3,613.95. 1H NMR (400 MHz, CDCl3): δ 7.91 (s, 1H), 7.76 (dd, J = 15.0, 8.8 Hz, 2H), 7.61 (d, J = 8.6 Hz, 1H), 7.22–7.16 (m, 1H), 7.15 (d, J = 1.3 Hz, 1H), 6.30 (s, 1H), 4.27 (q, J = 7.1 Hz, 2H), 3.95 (s, 3H), 2.71 (s, 3H), 1.36 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 166.89, 158.26, 155.14, 136.92, 134.72, 129.92, 128.43, 126.81, 125.67, 124.32, 119.22, 116.53, 105.46, 59.69, 55.20, 17.64, 14.26.
General Procedure for the Reduction to Allylic Alcohols 2[1–4]
In a one-neck round-bottom flask, preserving the anhydrous conditions, the allylic ester (1 equiv.) from the previous synthetic step is solubilized in dry Et2O under magnetic stirring. Once the solution is homogeneous, the reaction flask is put in a 0°C ice bath. Afterwards, LiAlH4 [1 equiv., 1 M solution in dry tetrahydrofuran (THF)] is added, slowly and dropwise, to the reaction mixture. After almost 30 min from the end of the reducing agent addition, the reaction is monitored by TLC with MP Hex/AcOEt 7:3 and results completed. The reaction is then quenched by adding a few drops of AcOEt and, afterwards, some of NH4Cl saturated aqueous solution. The workup procedure follows three extractions with Et2O and washing of the collected organic phases with brine. After that, the organic phase is dried with anhydrous Na2SO4, filtrated, and evaporated to dryness at reduced pressure. Depending on the purity of the raw product, as reported below, this is either purified by chromatography on silica gel or used directly in the subsequent reaction. The products were characterized by 1H and 13C NMR.
(E)-3-Phenylbut-2-en-1-ol 2[1]: by following the general procedure, starting from compound 1[1] (0.8 g), the desired product was obtained as a clear oil (0.617 g). Yield: 95.4%. Rf: 0.375 (TLC Hex/AcOEt, 6:4 v/v). Purification: flash chromatography, gradient elution, MP Hex/AcOEt 95:5, 7:3, and 6:4 v/v, 5 cm diameter column. IR (cm−1): 601.682; 609.396; 627.716; 698.105; 735.71; 758.852; 871.667; 1,026.91; 1,061.62; 1,149.37; 1,375.96; 1,445.39; 1,475.28; 1,493.6; 1,540.85; 1,648.84; 1,703.8; 1,722.12; 2,062.5; 2,337.3; 2,888.84; 2,985.27; 3,310.21. 1H NMR (400 MHz, CDCl3): δ 7.44 (d, J = 6.8 Hz, 2H), 7.36 (t, J = 6.8 Hz, 2H), 7.29 (m, 1H), 6.00 (t, J = 6.4 Hz, 1H), 4.39 (d, J = 6.4 Hz, 2H), 2.11 (s, 3H), 1.76 (s, 1H). 13C NMR (101 MHz, CDCl3): δ 142.73, 137.71, 128.18, 127.18, 126.38, 125.67, 59.83, 15.92.
(E)-3-(4-Methoxyphenyl)but-2-en-1-ol 2[2]: by following the general procedure, starting from compound 1[2] (1.17 g), the desired product was obtained as a white solid (0.95 g). Yield: ≥99.9%. Rf: 0.3 (TLC Hex/AcOEt, 6:4 v/v). mp: 96°C. Purification: none; after the NMR analysis, the crude product obtained from the reaction workup displayed a suitable purity for the following step. IR (cm−1): 601.682; 627.716; 665.321; 706.783; 735.71; 798.385; 1,025.94; 1,180.22; 1,247.72; 1,285.32; 1,440.56; 1,509.99; 1,605.45; 2,338.27; 2,871.49; 2,952.48; 2,999.73; 3,244.65. 1H NMR (400 MHz, CDCl3): δ 7.38 (d, J = 8.7 Hz, 2H), 6.89 (d, J = 8.7 Hz, 2H), 5.94 (t, J = 6.7 Hz, 1H), 4.37 (d, J = 6.7 Hz, 2H), 3.83 (s, 3H), 2.08 (s, 3H), 1.50 (s, 1H). 13C NMR (101 MHz, CDCl3): δ 158.88, 137.27, 135.18, 126.71, 124.70, 113.49, 59.83, 55.17, 15.91, 15.79.
(E)-3-(Naphthalen-2-yl)but-2-en-1-ol 2[3]: by following the general procedure, starting from compound 1[3] (2.00 g), the desired product was obtained as a white solid (1.35 g). Yield: 81.8%. Rf: 0.36 (TLC Hex/AcOEt, 6:4 v/v). mp: 63°C. Purification: flash chromatography, gradient elution, MP Hex/AcOEt 7:3 and 4:6 v/v, 5 cm diameter column. IR (cm−1): 609.396; 627.716; 665.321; 724.139; 738.603; 814.777; 858.168; 893.844; 1,008.59; 1,099.23; 1,375; 1,506.13; 1,540.85; 1,596.77; 1,670.05; 1,705.73; 1,747.19, 2,217.74, 2,372.01; 2,985.27; 3,339.14. 1H NMR (400 MHz, CDCl3): δ 7.94–7.78 (m, 4H), 7.70–7.58 (m, 1H), 7.55–7.43 (m, 2H), 6.17 (t, J = 6.6 Hz, 1H), 4.46 (d, J = 6.6 Hz, 2H), 2.22 (s, 3H), 1.60 (s, 1H). 13C NMR (101 MHz, CDCl3): δ 139.83, 137.51, 133.24, 132.58, 128.02, 127.68, 127.40, 126.91, 126.00, 125.71, 124.39, 124.03, 59.95, 15.93.
(E)-3-(6-Methoxynaphthalen-2-yl)but-2-en-1-ol 2[4]: by following the general procedure, starting from compound 1[4] (1.11 g), the desired product was obtained as a white solid (0.8 g). Yield: 84.7%. Rf: 0.28 (TLC Hex/AcOEt, 6:4 v/v). mp: 105°C. Purification: none; after the NMR analysis, the raw product obtained from the reaction workup results adequately pure to move forward with the following step. IR (cm−1): 602.646; 627.716; 665.321; 735.71; 809.956; 1,028.84; 1,164.79; 1,540.85; 1,646.91; 1,705.73; 1,747.19; 2,217.74; 2,996.84; 3,208. 1H NMR (400 MHz, CDCl3): δ 7.75 (m, 3H), 7.60 (d, J = 8.8 Hz, 1H), 7.15 (m, 1H), 6.14 (t, J = 6.5 Hz, 1H), 4.44 (d, J = 6.5 Hz, 2H), 3.95 (s, 3H), 2.20 (s, 3H), 1.55 (s, 1H). 13C NMR (101 MHz, CDCl3): δ 197.91, 157.56, 137.64, 137.60, 133.72, 129.54, 128.64, 126.53, 124.49, 124.24, 118.85, 105.42, 59.90, 55.20, 15.89.
General Procedure for the Synthesis of Allylic Amines 3[1–4]
In a one-neck round-bottom flask, a mixture of allylic alcohol (1 equiv.) and PPh3 (1.5 equiv.) is solubilized in dry THF (2/3 of the total volume calculated in order to have a final concentration of ~0.5 M) under magnetic stirring and preserving the anhydrous conditions. Separately, a Dewar containing ice, NaCl, and MeOH is set up to reach the temperature of −18°C. Once the solution is homogeneously stirred, the flask is cooled in the ice bath. Afterwards, the reaction mixture is treated with N-bromosuccinimide (NBS) (1.4 equiv.): this operation should be done carefully, adding the NBS portion-wise in six equal fractions each 5–10 min. During this step, it is extremely important to pay attention to the NBS solubilization: these white crystals tend to precipitate as a yellow solid or form a yellow oil phase above the reaction mixture. Therefore, for each two additions, it is useful to bring out the flask from the ice bath and let it heat to room temperature, improving the NBS solubilization. Nevertheless, every addition must be done at −18°C. Once the entire amount of NBS is added, the reaction flask is allowed to warm to room temperature and stirred for 20–30 min. A color variation from a clear solution (in some case lightly yellow) to a brownish suspension (more or less dark, depending on the substrate) is observed. Monitoring the reaction by TLC with MP Hex/AcOEt 7:3, the alcohol spot slowly disappears in favor of the one representing the hypothetical alcohol-PPh3-NBS abduct. Meanwhile, in another anhydrous round-bottom flask, the amines 1-Boc-piperazine (1.2 equiv.) and Et3N (2 equiv.) are solubilized in the residual part of dry THF (1/3 of the total volume calculated in order to have a final concentration of almost 0.5 M). This solution is later added to the reaction flask, previously cooled again to −18°C. After the addition of these last reagents, the reaction flask is brought out from the ice bath one more time and allowed to react overnight at room temperature under magnetic stirring. The day after, the reaction is monitored by TLC with MP AcOEt/Hex 8:2, and then, the TLC plate is developed with the stain reagent ninhydrin to confirm the presence of the amine and the absence of the alcohol-PPh3-NBS abduct previously observed. The raw product is worked up by dilution with Et2O, filtration directly into the separating funnel, and three times washing with a Na2CO3 saturated aqueous solution. The washed organic phase is then dried with Na2SO4 anhydrous, filtrated, and evaporated to dryness at reduce pressure. This way, a raw dark oil to be purified by chromatography on silica gel is obtained. The purified key intermediates were characterized by 1H and 13C NMR.
(E)-Tert-butyl 4-(3-phenylbut-2-en-1-yl)piperazine-1-carboxylate 3[1]: by following the general procedure, starting from compound 2[1] (0.60 g), the desired product was obtained as a red-brown oil (1.0 g). Yield: 78.9%. Overall yield: 35.2%. Rf: 0.41 (TLC AcOEt/Hex, 7:3 v/v). Purification: flash chromatography, MP Hex/AcOEt 3:7 v/v, 5 cm diameter column. IR (cm−1): 601.682; 627.716; 696.177; 755.959; 865.882; 914.093; 1,001.84; 1,122.37; 1,169.62; 1,244.83; 1,287.25; 1,364.39; 1,417.42; 1,693.19; 2,217.74; 2,764.46; 2,843.52; 2,981.41. 1H NMR (400 MHz, CDCl3): δ 7.41 (d, J = 7.6 Hz, 2H), 7.33 (t, J = 7.6 Hz, 2H), 7.26 (m, 1H), 5.89 (t, J = 6.8 Hz, 1H), 3.51–3.45 (m, 4H), 3.21 (d, J = 6.8 Hz, 2H), 2.55–2.42 (m, 4H), 2.08 (s, 3H), 1.47 (s, 9H). 13C NMR (101 MHz, CDCl3): δ 154.60, 143.05, 138.02, 128.11, 126.94, 125.55, 123.85, 79.49, 56.55, 52.89, 28.30, 16.09.
(E)-Tert-butyl 4-(3-(4-methoxyphenyl)but-2-en-1-yl) piperazine-1-carboxylate 3[2]: by following the general procedure, starting from compound 2[2] (0.90 g), the desired product was obtained as a yellow-orange oil (1.0 g). Yield: 57.2%. Overall yield: 28%. Rf: 0.34 (TLC AcOEt/Hex, 7:3 v/v). Purification: flash chromatography, MP Hex/AcOEt 3:7 v/v, 5 cm diameter column. IR (cm−1): 627.716; 670.142; 735.71; 826.348; 864.917; 916.986; 999.91; 1,032.69; 1,119.48; 1,170.58; 1,243.86; 1,287.25; 1,364.39; 1,417.42; 1,511.92; 1,607.38; 1,692.23; 2,340.19; 2,361.41; 2,388.41; 2,814.6; 2,871.49; 2,983.34. 1H NMR (400 MHz, CDCl3): δ 7.36 (d, J = 8.7 Hz, 2H), 6.87 (d, J = 8.7 Hz, 2H), 5.82 (t, J = 6.8 Hz, 1H), 3.82 (s, 3H), 3.48 (brs, 4H), 3.19 (d, J = 6.8 Hz, 2H), 2.48 (brs, 4H), 2.05 (s, 3H), 1.47 (s, 9H). 13C NMR (101 MHz, CDCl3): δ 177.45, 158.66, 154.64, 137.32, 135.51, 128.84, 126.59, 122.19, 113.44, 79.54, 56.57, 55.14, 52.87, 29.51, 28.31, 16.09.
(E)-Tert-butyl 4-(3-(naphthalen-2-yl)but-2-en-1-yl) piperazine-1-carboxylate 3[3]: by following the general procedure, starting from compound 2[3] (0.81 g), the desired product was obtained as a white-yellow solid (1.16 g). Yield: 77.7%. Overall yield: 31.6%. Rf: 0.37 (TLC AcOEt/Hex, 7:3 v/v). mp: 93°C. Purification: flash chromatography, MP Hex/AcOEt 4:6 v/v, 5 cm diameter column. IR (cm−1): 601.682; 627.716; 665.321; 735.71; 816.706; 1,094.4; 1,424.17; 1,540.85; 1,647.88; 1,705.73; 1,747.19, 2,217.74, 2,372.01; 2,912.95; 2,991.05. 1H NMR (400 MHz, CDCl3): δ 7.83 (m, 4H), 7.61 (d, J = 8.6 Hz, 1H), 7.54–7.40 (m, 2H), 6.07 (t, J = 6.7 Hz, 1H), 3.52 (brs, 4H), 3.29 (d, J = 6.7 Hz, 2H), 2.54 (brs, 4H), 2.20 (s, 3H), 1.49 (s, 9H). 13C NMR (101 MHz, CDCl3): δ 154.60, 140.13, 133.27, 132.50, 127.97, 127.62, 127.37, 126.01, 125.59, 124.25, 124.16, 124.06, 79.57, 56.65, 52.92, 29.57, 28.22, 16.12.
(E)-Tert-butyl 4-(3-(6-methoxynaphthalen-2-yl)but-2-en-1-yl)piperazine-1-carboxylate 3[4]: by following the general procedure, starting from compound 2[4] (0.80 g), the desired product was obtained as a white-yellow solid (1.1 g). Yield: 79.2%. Overall yield: 38.1%. mp: 133°C. Rf: 0.34 (TLC AcOEt/Hex, 7:3 v/v). Purification: flash chromatography, MP Hex/AcOEt 3:7 v/v, 5 cm diameter column. IR (cm−1): 601.682; 617.109; 627.716; 665.321; 706.783; 724.139; 7735.71; 815.742; 1,114.65; 1,397.17; 1,565.92; 1,646.91; 1,745.26; 2,284.27; 2,389.37; 2,996.84. 1H NMR (400 MHz, CDCl3): δ 7.84–7.66 (m, 3H), 7.58 (d, J = 8.6 Hz, 1H), 7.21–7.08 (m, 2H), 6.03 (t, J = 6.7 Hz, 1H), 3.93 (s, 3H), 3.51 (brs, 4H), 3.26 (d, J = 6.7 Hz, 2H), 2.52 (brs, 4H), 2.17 (s, 3H), 1.49 (s, 9H). 13C NMR (101 MHz, CDCl3): δ 157.49, 154.63, 138.03, 137.70, 133.60, 129.47, 128.70, 126.47, 124.53, 123.97, 123.73, 118.78, 105.45, 79.50, 56.70, 55.17, 52.97, 28.35, 16.07.
General Procedure for the de-boc Reactions
Into a two-necked round bottomed flask of the appropriate volume, the Boc-protected intermediate was dissolved in 10 ml of 1,4-dioxane. The mixture was cooled to 0°C using an ice bath and 10 ml of 4 N HCl in dioxane were added dropwise. The mixture was allowed to reach room temperature and stirred at such temperature overnight. Solvent was evaporated, and the resulting de-Boc products (as HCl salts) were used without further purification.
1-[(2E)-3-Phenylbut-2-en-1-yl]piperazine 4[1], orange oil, 88%, Rf = 0.55 (CHCl3/MeOH 5:1), UHPLC-ESI-MS: Rt = 1.34, m/z = 217.3 [M + H]+. 1H NMR (300 MHz, CDCl3), δ 7.42–7.38 (m, 2H), 7.34–7.28 (m, 3H), 5.90 (dt, J = 1.3 Hz, J = 6.8 Hz, 1H), 3.18 (dd, J = 0.7 Hz, J = 6.8 Hz, 2H), 2.94 (t, J = 4.9 Hz, 4H), 2.52 (s, 4H), 2.07 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3), δ 143.3, 137.7, 128.2, 127.0, 125.7, 124.4, 57.3, 54.4, 46.0, 16.2 ppm.
1-[(2E)-3-(4-Methoxyphenyl) but-2-en-1-yl]piperazine 4[2], orange oil, 87%, Rf = 0.62 (CHCl3/MeOH 5:1), UHPLC-ESI-MS: Rt = 1.71, m/z = 247.3 [M + H]+. 1H NMR (300 MHz, CDCl3), δ 7.35 (d, J = 8.7 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 5.82 (dt, J = 1.0 Hz, J = 6.9 Hz, 1H), 3.81 (s, 3H), 3.17 (d, J = 6.5 Hz, 2H), 2.95–2.92 (m, 4H), 2.51 (s, 4H), 2.04 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3), δ 158.7, 137.1, 135.8, 126.7, 122.7, 113.6, 57.3, 55.3, 54.4, 46.0, 16.2 ppm.
1-[(2E)-3-(Naphthalen-2-yl)but-2-en-1-yl]piperazine 4[3], orange solid, 93%, Rf = 0.65 (CHCl3/MeOH 5:1), UHPLC-ESI-MS: Rt = 1.91, m/z = 267.2 [M + H]+. 1H NMR (300 MHz, CDCl3), δ 7.81–7.77 (m, 4H), 7.60 (d, J = 8.3 Hz, 1H), 7.48–7.41 (m, 2H), 6.05 (t, J = 7.1 Hz, 1H), 3.25 (d, J = 6.7 Hz, 2H), 3.00 (s, 4H), 2.59 (s, 4H), 2.18 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3), δ 140.3, 137.8, 133.4, 132.6, 128.1, 127.7, 127.5, 126.1, 125.7, 124.8, 124.2, 124.2, 57.2, 53.7, 45.6, 16.2 ppm.
1-[(2E)-3-(6-Methoxynaphthalen-2-yl)but-2-en-1-yl]piperazine 4[4], white solid, 80%, Rf = 0.68 (CHCl3/MeOH 5:1), UHPLC-ESI-MS: Rt = 1.99, m/z = 297.2 [M + H] +. 1H NMR (300 MHz, CDCl3), δ 7.75–7.66 (m, 4H), 7.57 (dd, J = 1.9 Hz, J = 8.6 Hz, 1H), 7.15–7.11 (m, 2H), 6.02 (dt, J = 1.4 Hz, J = 6.9 Hz 1H), 3.92 (s, 3H), 3.25 (d, J = 6.6 Hz, 2H), 2.98 (s, 4H), 2.58 (s, 4H), 2.16 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3), δ 159.0, 142.5, 139.8, 137.9, 132.2, 128.8, 127.4, 126.6, 124.4, 121.8, 119.1, 105.6, 54.9, 54.2, 50.7, 43.1, 16.2 ppm.
General Procedure for Sulfonylation
Reactions were performed in parallel in 15-ml reaction tubes in a 24-position Mettler-Toledo Miniblock® equipped with a heat transfer block and inert gas manifold. Each reaction tube was loaded with a previously prepared solution of 30 mg of the corresponding amine (1.0 equiv.) in 2 ml of DCM and TEA (5.0 equiv.). The corresponding sulfonyl chlorides (1.5 equiv.) were added. The reaction mixtures were stirred at room temperature overnight. Reaction conversion was confirmed through UHPLC check of some representative samples. The mixtures were evaporated until dryness. The crudes were redissolved in 1.0 ml of acetonitrile (ACN), filtered and purified with preparative HPLC (gradient acetonitrile/water with 0.1% formic acid, 2–98%). Fractions containing pure product were combined and evaporated to dryness in Mettler vials.
(E)-1-(Cyclopentylsulfonyl)-4-(3-phenylbut-2-en-1-yl) piperazine SU[1,1], yellow oil, 75%, UHPLC-ESI-MS: Rt = 2.21, m/z = 349.2 [M + H]+. Purity (UHPLC) = 97%
(E)-1-(Cyclohexylsulfonyl)-4-(3-phenylbut-2-en-1-yl) piperazine SU[1,2], white solid, 52%, UHPLC-ESI-MS: Rt = 2.40, m/z = 363.2 [M + H]+. Purity (UHPLC) = 96%
1-[(2E)-3-Phenylbut-2-en-1-yl]-4-[4-(trifluoromethyl) benzenesulfonyl]piperazine SU[1,3], white solid, 37%, Rf = 0.90 (DCM/MeOH 19:1), UHPLC-ESI-MS: Rt = 2.46, m/z = 425.2 [M + H]+. Purity (UHPLC) = 99%. 1H NMR (300 MHz, CDCl3), δ 7.88 (d, J = 8.4 Hz, 1H), 7.83–7.80 (m, 2H), 7.57 (d, J = 8.2 Hz, 1H), 7.34–7.28 (m, 5H), 5.80 (dt, J = 0.9 Hz, J = 7.2 Hz, 1H), 3.61 (s, 3H), 3.39 (s, 4H), 3.06 (s, 4H), 2.09 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3), δ 142.0, 139.2, 135.2, 134.7, 128.4, 128.2, 127.8, 126.4, 126.3, 125.7, 125.4, 55.9, 51.7, 44.8, 16.4 ppm.
1-(4-Methylbenzenesulfonyl)-4-[(2E)-3-phenylbut-2-en-1-yl] piperazine SU[1,4], yellow oil, 31%, Rf = 0.66 (DCM/MeOH 19:1), UHPLC-ESI-MS: Rt = 2.30, m/z = 371.0 [M + H]+. Purity (UHPLC) = 89%. 1H NMR (300 MHz, CDCl3), δ 7.63–7.59 (m, 3H), 7.34–7.28 (m, 5H), 7.10 (d, J = 8.0 Hz, 1H), 5.80 (dt, J = 1.0 Hz, J = 7.3 Hz, 1H), 3.57 (s, 2H), 3.27 (s, 4H), 2.98 (s, 4H), 2.43 (s, 3H), 2.07 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3), δ 144.3, 141.8, 140.1, 132.2, 129.9, 128.8, 128.3, 127.7, 125.8, 125.7, 55.7, 51.4, 44.5, 21.5, 16.4 ppm.
1-(2,6-Difluorobenzenesulfonyl)-4-[(2E)-3-phenylbut-2-en-1-yl] piperazine SU[1,5], yellow oil, 37%, UHPLC-ESI-MS: Rt = 2.24, m/z = 393.0 [M + H]+. Purity (UHPLC) = 99%.
1-(Cyclopentanesulfonyl)-4-[(2E)-3-(4-methoxyphenyl)but-2-en-1-yl] piperazine SU[2,1], white solid, 9%, Rf = 0.36 (DCM/MeOH 19:1), UHPLC-ESI-MS: Rt = 2.30, m/z = 379.2 [M + H]+. Purity (UHPLC) = 97%. 1H NMR (300 MHz, CDCl3), δ 7.38 (dd, J = 2.1 Hz, J = 6.9 Hz, 2H), 6.87 (dd, J = 2.1 Hz, J = 6.9 Hz, 2H), 5.93 (t, J = 7.7 Hz, 1H), 3.82 (s, 3H), 3.80–3.77 (m, 4H), 3.50–3.40 (m, 1H), 3.11–2.94 (m, 4H), 2.10 (s, 3H), 2.03–1.91 (m, 8H) ppm; 13C NMR (100 MHz, CDCl3), δ 160.0, 146.1, 133.3, 127.3, 113.9, 111.4, 61.4, 55.7, 55.3, 51.5, 43.1, 28.0, 25.5, 16.6 ppm.
1-(Cyclohexanesulfonyl)-4-[(2E)-3-(4-methoxyphenyl) but-2-en-1-yl]piperazine SU[2,2], yellow solid, 17%, UHPLC-ESI-MS: Rt = 2.39, m/z = 393.2 [M + H]+. Purity (UHPLC) = 98%.
1-[(2E)-3-(4-Methoxyphenyl)but-2-en-1-yl]-4-[4-(trifluoromethyl)benzenesulfonyl]piperazine SU[2,3], white solid, 6%, UHPLC-ESI-MS: Rt = 2.62, m/z = 455.2 [M + H]+. Purity (UHPLC) = 85%.
1-[(2E)-3-(4-Methoxyphenyl)but-2-en-1-yl]-4-(4-methyl benzenesulfonyl)piperazine SU[2,4], yellow solid, 12%, UHPLC-ESI-MS: Rt = 2.39, m/z = 401.2 [M + H]+. Purity (UHPLC) = 85%.
1-(Cyclopentanesulfonyl)-4-[(2E)-3-(naphthalen-2-yl)but-2 -en-1-yl]piperazine SU[3,1], white solid, 22%, Rf = 0.44 (DCM/MeOH 19:1), UHPLC-ESI-MS: Rt = 2.60, m/z = 399.2 [M + H]+. Purity (UHPLC) = 97%. 1H NMR (300 MHz, CDCl3), δ 7.85–7.78 (m, 4H), 7.58 (dd, J = 1.8 Hz, J = 8.6 Hz, 1H), 7.49–7.46 (m, 2H), 6.13 (t, J = 7.3 Hz, 1H), 3.74 (s, 6H), 3.47–3.41 (m, 1H), 2.21 (s, 3H), 2.05–1.96 (m, 7H), 1.79–1.75 (m, 3H), 1.63–1.59 (m, 2H) ppm; 13C NMR (100 MHz, CDCl3), δ 141.3, 138.5, 135.8, 133.1, 131.4, 128.3, 128.1, 127.6, 126.5, 126.4, 125.2, 123.9, 61.2, 55.7, 51.8, 30.9, 27.9, 25.5, 16.6 ppm.
1-(Cyclohexanesulfonyl)-4-[(2E)-3-(naphthalen-2-yl)but-2-en-1-yl]piperazine SU[3,2], white solid, 20%, UHPLC-ESI-MS: Rt = 2.72, m/z = 413.2 [M + H]+. Purity (UHPLC) = 97%.
1-[(2E)-3-(Naphthalen-2-yl)but-2-en-1-yl]-4-[4-(trifluoromethyl) benzenesulfonyl]piperazine SU[3,3], yellow solid, 22%, UHPLC-ESI-MS: Rt = 2.85, m/z = 477.0 [M + H]+. Purity (UHPLC) = 93%.
1-(4-Methylbenzenesulfonyl)-4-[(2E)-3-(naphthalen-2-yl)but-2-en-1-yl]piperazine SU[3,4], white solid, 5%, UHPLC-ESI-MS: Rt = 2.79, m/z = 421.0 [M + H]+. Purity (UHPLC) = 96%.
General Procedure for Reductive Amination
Reactions were performed in parallel in 15-ml reaction tubes in a 24-position Mettler-Toledo Miniblock® equipped with a heat transfer block and inert gas manifold. Each reaction tube was loaded with a previously prepared solution of 30 mg of the corresponding amine (1.0 equiv.) in 2 mL of DCE and acetic acid (2.0 equiv). The corresponding aldehydes were added, and the mixtures were stirred at room temperature for 20 min. Afterwards, NaBH(OAc)3 (2.5 equiv.) was added. The reactions were stirred at room temperature overnight. Reaction conversion was confirmed through UHPLC check of some representative samples. The reaction mixtures were washed with 1 ml of water, and the organic layers were evaporated to dryness. The crudes were redissolved in 1.0 ml of ACN, filtered and purified with preparative HPLC (gradient acetonitrile/water with 0.1% formic acid, 2–98%). Fractions containing pure product were combined and evaporated to dryness in Mettler vials.
(E)-1-Benzyl-4-(3-(naphthalen-2-yl)but-2-en-1-yl)piperazine RC-206, brown solid, 28%, m.p. = 98°C, Rf = 0.37 (DCM/MeOH 95:5), 1H-NMR [400 MHz, (CD3)2CO], δ (ppm) 7.93–7.91 (m, 2H), 7.89–7.86 (m, 2H), 7.69–7.67 (m, 1H), 7.50–7.46 (m, 3H), 7.35–7.33 (m, 5H), 6.08 (t,1H), 3.52 (s, 2H), 3.25 (d, 2H), 2.62–2.39 (brs, 8H), 2.20 (s,3H) ppm; 13C-NMR [100 MHz (CD3)2CO], δ (ppm) 128.86,128.08,127.65,127.39,126.85,126.07,125.64, 124.19, 124.08, 62.49, 56.24, 53.08, 52.88, 15.55.
1-(Cyclopentylmethyl)-4-[(2E)-3-phenylbut-2-en-1-yl]piperazine RA[1,1], orange oil, 99%, UHPLC-ESI-MS: Rt = 1.82, m/z = 299.2 [M + H]+. Purity (UHPLC) = 99%.
1-(Cyclohexylmethyl)-4-[(2E)-3-phenylbut-2-en-1-yl]piperazine RA[1,2], orange solid, 52%, UHPLC-ESI-MS: Rt = 1.98, m/z = 313.2 [M + H]+. Purity (UHPLC) = 99%.
1-[(2E)-3-Phenylbut-2-en-1-yl]-4-{[4-(trifluoromethyl)phenyl]methyl}piperazine RA[1,3], brown solid, 60%, UHPLC-ESI-MS: Rt = 2.33, m/z = 375.2 [M + H]+. Purity (UHPLC) = 99%.
1-[(4-Methylphenyl)methyl]-4-[(2E)-3-phenylbut-2-en-1-yl]piperazine RA[1,4], orange oil, 55%, Rf = 0.41 (DCM/MeOH 19:1), UHPLC-ESI-MS: Rt = 2.07, m/z = 321.2 [M + H]+. Purity (UHPLC) = 99%. 1H NMR (300 MHz, CDCl3), δ 7.41–7.37 (m, 2H), 7.34–7.31 (m, 1H), 7.32–7.25 (m, 2H), 7.20 (d, J = 8.0 Hz, 2H), 7.13 (d, J = 7.9 Hz, 2H), 5.87 (dt, J = 1.3 Hz, J = 7.2 Hz, 1H), 3.58 (s, 2H), 3.39 (d, J = 7.2 Hz, 2H), 2.79 (s, 4H), 2.67 (s, 4H), 2.33 (s, 3H), 2.06 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3), δ 142.7, 140.1, 137.2, 133.2, 129.4, 129.0, 128.2, 127.3, 125.7, 121.1, 61.9, 55.5, 51.7, 51.4, 21.1, 16.2 ppm.
1-[(2,6-Difluorophenyl)methyl]-4-[(2E)-3-phenylbut-2-en-1-yl]piperazine RA[1,5], orange oil, 42%, UHPLC-ESI-MS: Rt = 2.09, m/z = 343.2 [M + H]+. Purity (UHPLC) = 93%.
1-(Cyclopentylmethyl)-4-[(2E)-3-(4-methoxyphenyl)but-2-en-1-yl]piperazine RA[2,1], white solid, 37%, UHPLC-ESI-MS: Rt = 1.85, m/z = 329.2 [M + H]+. Purity (UHPLC) = 99%.
1-(Cyclohexylmethyl)-4-[(2E)-3-(4-methoxyphenyl)but-2-en-1-yl]piperazine RA[2,2], yellow oil, 45%, Rf = 0.22 (DCM/MeOH 19:1), UHPLC-ESI-MS: Rt = 1.99, m/z = 343.2 [M + H]+. Purity (UHPLC) = 99%. 1H NMR (300 MHz, CDCl3), δ 7.34 (d, J = 8.9 Hz, 2H), 6.85 (d, J = 8.9 Hz, 2H), 5.81 (dt, J = 1.2 Hz, J = 7.2 Hz, 1H), 3.80 (s, 3H), 3.36 (d, J = 7.2 Hz, 2H), 2.77 (s, 4H), 2.64 (s, 4H), 2.25 (d, J = 7.1 Hz, 2H), 2.04 (s, 3H), 1.77–1.68 (m, 5H), 1.52–1.46 (m, 1H), 1.27–1.11 (m, 3H) ppm; 13C NMR (100 MHz, CDCl3), δ 159.0, 139.3, 135.2, 126.8, 119.7, 113.6, 64.8, 55.6, 55.3, 52.2, 51.7, 34.6, 31.7, 26.5, 26.0, 16.2 ppm.
1-[(2E)-3-(4-Methoxyphenyl)but-2-en-1-yl]-4-{[4-(trifluoromethyl)phenyl]methyl}piperazine RA[2,3], orange oil, 21%, Rf = 0.30 (DCM/MeOH 19:1), UHPLC-ESI-MS: Rt = 2.36, m/z = 405.2 [M + H]+. Purity (UHPLC) = 99%. 1H NMR (300 MHz, CDCl3), δ 7.57 (d, J = 8.1 Hz, 2H), 7.44 (d, J = 8.0 Hz, 2H), 7.34 (d, J = 8.9 Hz, 2H), 6.86 (d, J = 8.9 Hz, 2H), 5.82 (dt, J = 1.3 Hz, J = 7.2 Hz, 1H), 3.81 (s, 3H), 3.59 (s, 2H), 3.40 (d, J = 7.3 Hz, 2H), 2.79 (s, 4H), 2.61 (s, 4H), 2.05 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 159.1, 141.9, 139.6, 135.1, 129.7, 129.2, 128.8, 125.3 (q, J = 3.8Hz), 119.3, 118.6, 62.0, 55.6, 55.3, 52.1, 51.8, 16.2 ppm.
1-[(2E)-3-(4-Methoxyphenyl)but-2-en-1-yl]-4-[(4-methylphenyl)methyl]piperazine RA[2,4], yellowish solid, 15%, UHPLC-ESI-MS: Rt = 2.12, m/z = 351.2 [M + H]+. Purity (UHPLC) = 99%.
1-[(2,6-Difluorophenyl)methyl]-4-[(2E)-3-(4-methoxyphenyl) but-2-en-1-yl]piperazine RA[2,5], yellow oil, 43%, UHPLC-ESI-MS: Rt = 2.13, m/z = 373.2 [M + H]+. Purity (UHPLC) = 95%.
1-(Cyclopentylmethyl)-4-[(2E)-3-(naphthalen-2-yl)but-2-en-1-yl]piperazine RA[3,1], colorless oil, 28%, Rf = 0.33 (DCM/MeOH 19:1), UHPLC-ESI-MS: Rt = 2.17, m/z = 349.2 [M + H]+. Purity (UHPLC) = 99%. 1H NMR (300 MHz, CDCl3), δ 7.85–7.78 (m, 4H), 7.57 (dd, J = 1.9 Hz, J = 8.6 Hz, 1H), 7.49–7.45 (m, 2H), 5.97 (dt, J = 1.3 Hz, J = 7.3 Hz, 1H), 3.53–3.43 (m, 2H), 3.00 (s, 8H), 2.70 (d, J = 7.3 Hz, 2H), 2.21 (s, 3H), 1.89–1.80 (m, 2H), 1.68–1.55 (m, 5H), 1.29–1.20 (m, 2H) ppm; 13C NMR (100 MHz, CDCl3), δ 141.4, 139.5, 133.2, 132.8, 128.2, 127.9, 127.5, 126.3, 126.0, 124.6, 124.0, 120.4, 63.0, 55.4, 51.7, 50.3, 35.8, 31.5, 25.0, 16.5 ppm.
1-(Cyclohexylmethyl)-4-[(2E)-3-(naphthalen-2-yl)but-2-en-1-yl]piperazine RA[3,2], white solid, 53%, UHPLC-ESI-MS: Rt = 2.30, m/z = 363.2 [M + H]+. Purity (UHPLC) = 95%.
1-[(2E)-3-(Naphthalen-2-yl)but-2-en-1-yl]-4-{[4-(trifluoromethyl)phenyl]methyl}piperazine RA[3,3], yellow oil, 42%, UHPLC-ESI-MS: Rt = 2.56 m/z = 425.2 [M + H]+. Purity (UHPLC) = 98%.
1-[(4-Methylphenyl)methyl]-4-[(2E)-3-(naphthalen-2-yl) but-2-en-1-yl]piperazine RA[3,4], yellow oil, 16%, UHPLC-ESI-MS: Rt = 2.38 m/z = 371.2 [M + H]+. Purity (UHPLC) = 88%.
1-[(2,6-Difluorophenyl)methyl]-4-[(2E)-3-(naphthalen-2-yl)but-2-en-1-yl]piperazine RA[3,5], yellow oil, 26%, UHPLC-ESI-MS: Rt = 2.39 m/z = 393.2 [M + H]+. Purity (UHPLC) = 88%.
1-(Cyclopentylmethyl)-4-[(2E)-3-(6-methoxynaphthalen-2-yl)but-2-en-1-yl]piperazine RA[4,1], colorless oil, 5%, UHPLC-ESI-MS: Rt = 2.18 m/z = 379.2 [M + H]+. Purity (UHPLC) = 99%.
1-[(2E)-3-(6-Methoxynaphthalen-2-yl)but-2-en-1-yl]-4-{[4-(trifluoromethyl)phenyl]methyl}piperazine RA[4,3], colorless oil, 5%, UHPLC-ESI-MS: Rt = 2.57 m/z = 455.2 [M + H]+. Purity (UHPLC) = 88%.
1-[(2,6-Difluorophenyl)methyl]-4-[(2E)-3-(6-methoxynaphthalen-2-yl)but-2-en-1-yl]piperazine RA[4,5], white solid, 21%, Rf = 0.50 (DCM/MeOH 19:1), UHPLC-ESI-MS: Rt = 2.38, m/z = 423.0 [M + H]+. Purity (UHPLC) = 95%. Purity (UHPLC) = 97%. 1H NMR (300 MHz, CDCl3), δ 7.75–7.68 (m, 3H), 7.53 (dd, J = 1.9 Hz, J = 8.6 Hz, 1H), 7.32–7.27 (m, 1H), 7.16–7.11 (m, 2H), 6.93–6.87 (m, 2H), 5.99 (dd, J = 6.6 Hz, J = 7.9 Hz, 1H), 3.92 (s, 3H), 3.76 (s, 2H), 3.61 (d, J = 7.5 Hz, 2H), 2.99 (s, 4H), 2.80 (s, 4H), 2.18 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3), δ 160.3 (d, J = 8.3 Hz), 157.9, 142.9, 136.8, 134.2, 129.9, 129.8, 128.7, 128.6, 126.9, 124.7, 124.4, 119.2, 111.5, 111.1, 105.5, 55.3, 55.1, 51.7, 50.0, 48.1, 16.4 ppm.
General Procedure for the Amide Formation
Reactions were performed in parallel in 15-ml reaction tubes in a 24-position Mettler-Toledo Miniblock® equipped with a heat transfer block and inert gas manifold. Each reaction tube was loaded with a previously prepared solution of 30 mg of the corresponding amine (1.0 equiv.) in 2 ml of DMF, DIPEA (5.0 equiv.), HOBt (2.0 equiv.), EDC*HCl (2.5 equiv.). The corresponding acids were added (2.0 equiv.). The reaction mixtures were stirred at room temperature overnight. Reaction conversion was confirmed through UHPLC check of some representative samples. The mixtures were evaporated until dryness. The crudes were redissolved in 1.0 ml of ACN, filtered and purified with preparative HPLC (gradient acetonitrile/water with 0.1% formic acid, 2–98%). Fractions containing pure product were combined and evaporated to dryness in Mettler vials.
1-Cyclopentanecarbonyl-4-[(2E)-3-phenylbut-2-en-1-yl]piperazine AM[1,1], brown oil, 72%, UHPLC-ESI-MS: Rt = 2.13 m/z = 313.2 [M + H]+. Purity (UHPLC) = 96%.
1-Cyclohexanecarbonyl-4-[(2E)-3-phenylbut-2-en-1-yl]piperazine AM[1,2], orange oil, 70%, UHPLC-ESI-MS: Rt = 2.20 m/z = 327.2 [M + H]+. Purity (UHPLC) = 99%.
1-[(2E)-3-Phenylbut-2-en-1-yl]-4-[4-(trifluoromethyl)benzoyl]piperazine AM[1,3], orange oil, 54%, UHPLC-ESI-MS: Rt = 2.39 m/z = 389.2 [M + H]+. Purity (UHPLC) = 99%.
1-(4-Methylbenzoyl)-4-[(2E)-3-phenylbut-2-en-1-yl]piperazine AM[1,4], orange oil, 78%, UHPLC-ESI-MS: Rt = 2.21 m/z = 335.2 [M + H] +. Purity (UHPLC) = 99%.
1-(2,6-Difluorobenzoyl)-4-[(2E)-3-phenylbut-2-en-1-yl]piperazine AM[1,5], orange oil, 31%, Rf = 0.50 (DCM/MeOH 19:1), UHPLC-ESI-MS: Rt = 2.13, m/z = 357.2 [M + H]+. Purity (UHPLC) = 95%. 1H NMR (300 MHz, CDCl3), δ 7.41–7.28 (m, 6H), 6.95 (dd, J = 7.3 Hz, J = 8.4 Hz, 2H), 5.87 (dt, J = 1.3 Hz, J = 7.0 Hz, 1H), 3.94–3.91 (m, 2H), 3.44–3.41 (m, 2H), 3.32 (d, J = 7.1 Hz, 2H), 2.73–2.70 (m, 2H), 2.63–2.59 (m, 2H), 2.08 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3), δ 160.0, 157.2 (d, J = 7.9 Hz), 142.9, 139.5, 131.1, 128.3, 127.3, 125.7, 122.3, 113.5, 111.8 (d, J = 24.9 Hz), 56.1, 52.9, 52.3, 46.3, 41.4, 16.3 ppm.
1-Cyclopentanecarbonyl-4-[(2E)-3-(4-methoxyphenyl)but-2-en-1-yl]piperazine AM[2,1], yellow oil, 37%, UHPLC-ESI-MS: Rt = 2.11m/z = 343.2 [M + H]+. Purity (UHPLC) = 99%.
1-Cyclohexanecarbonyl-4-[(2E)-3-(4-methoxyphenyl)but-2-en-1-yl]piperazine AM[2,2], yellow oil, 32%, UHPLC-ESI-MS: Rt = 2.21 m/z = 357.2 [M + H]+. Purity (UHPLC) = 99%.
1-[(2E)-3-(4-Methoxyphenyl)but-2-en-1-yl]-4-[4-(trifluoromethyl)benzoyl]piperazine AM[2,3], yellow oil, 36%, UHPLC-ESI-MS: Rt = 2.35 m/z = 419.0 [M + H]+. Purity (UHPLC) = 99%.
1-[(2E)-3-(4-Methoxyphenyl)but-2-en-1-yl]-4-(4-methylbenzoyl)piperazine AM[2,4], yellow oil, 34%, Rf = 0.35 (DCM/MeOH 19:1), UHPLC-ESI-MS: Rt = 2.23, m/z = 365.2 [M + H]+. Purity (UHPLC) = 98%. 1H NMR (300 MHz, CDCl3), δ 7.35–7.29 (m, 4H), 7.21 (d, J = 7.8 Hz, 2H), 6.86 (dd, J = 2.1 Hz, J = 9.0 Hz, 2H), 5.79 (dt, J = 1.2 Hz, J = 7.2 Hz, 1H), 3.96–3.87 (m, 1H), 3.81 (s, 3H), 3.71–3.59 (m, 2H), 3.40 (d, J = 7.3 Hz, 2H), 2.80–2.61 (m, 5H), 2.37 (s, 3H), 2.05 (d, J = 1.0 Hz, 3H), ppm; 13C NMR (100 MHz, CDCl3), δ 170.6, 159.1, 140.3, 140.1, 135.0, 132.1, 129.1, 127.2, 126.8, 118.7, 113.6, 55.7, 55.3, 52.2, 41.0, 21.4, 16.3 ppm.
1-(2,6-Difluorobenzoyl)-4-[(2E)-3-(4-methoxyphenyl)but-2-en-1-yl]piperazine AM[2,5], yellow oil, 15%, UHPLC-ESI-MS: Rt = 2.16 m/z = 387.2 [M + H]+. Purity (UHPLC) = 95%.
1-Cyclopentanecarbonyl-4-[(2E)-3-(naphthalen-2-yl)but-2-en-1-yl]piperazine AM[3,1], yellow oil, 35%, Rf = 0.37 (DCM/MeOH 19:1), UHPLC-ESI-MS: Rt = 2.42, m/z = 363.2 [M + H]+. Purity (UHPLC) = 98%. 1H NMR (300 MHz, CDCl3), δ 9.75 (s br, 1H), 7.85–7.78 (m, 4H), 7.57 (dd, J = 1.8 Hz, J = 8.6 Hz, 1H), 7.48–7.44 (m, 2H), 6.03 (dt, J = 1.2 Hz, J = 7.1 Hz, 1H), 3.79–3.76 (m, 2H), 3.69–3.65 (m, 2H), 3.43 (d, J = 7.1 Hz, 2H), 2.90–2.85 (m, 1H), 2.76–2.72 (m, 4H), 2.18 (s, 3H), 1.83–1.67 (m, 5H), 1.60–1.55 (m, 3H) ppm; 13C NMR (100 MHz, CDCl3), δ 174.6, 140.1, 139.8, 133.3, 132.7, 128.1, 127.8, 127.5, 126.2, 125.9, 124.5, 124.1, 121.6, 55.9, 52.6, 44.5, 40.9, 30.1, 26.0, 16.3 ppm.
1-Cyclohexanecarbonyl-4-[(2E)-3-(naphthalen-2-yl)but-2-en-1-yl]piperazine AM[3,2], yellow oil, 99%, UHPLC-ESI-MS: Rt = 2.52 m/z = 377.2 [M + H]+. Purity (UHPLC) = 97%.
1-(2,6-Difluorobenzoyl)-4-[(2E)-3-(naphthalen-2-yl)but-2-en-1-yl]piperazine AM[3,5], colorless oil, 6%, Rf = 0.42 (DCM/MeOH 19:1), UHPLC-ESI-MS: Rt = 2.53, m/z = 407.0 [M + H]+. Purity (UHPLC) = 85%. 1H NMR (300 MHz, CDCl3), δ 7.81 (d, J = 4.4 Hz, 3H), 7.70 (d, J = 8.3 Hz, 1H), 7.57 (dd, J = 1.8 Hz, J = 8.6 Hz, 1H), 7.48–7.44 (m, 2H), 7.38–7.35 (m, 1H), 6.99–6.94 (m, 2H), 6.07 (dt, J = 1.0 Hz, J = 7.2 Hz, 1H), 4.04 (s, 2H), 3.56–3.49 (m, 4H), 2.93–2.89 (m, 2H), 2.83–2.80 (m, 2H), 2.21 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3), δ 168.5, 160.0, 139.7, 133.3, 132.8, 131.3, 130.3, 128.1, 127.9, 127.5, 126.2, 126.0, 125.4 (d, J = 3.7 Hz), 124.6, 124.1, 112.1 (d, J = 3.0 Hz), 111.8 (d, J = 3.4 Hz), 55.9, 52.5, 52.0, 45.8, 40.8, 16.4 ppm.
In silico Studies
Prediction of some basic absorption, distribution, metabolism, and excretion (ADME) parameters, most importantly solubility, was performed with the SwissADME web tool. This open-access and user-friendly tool is accessible at http://www.swissadme.ch. SMILES strings corresponding to all the designed compounds, both as free bases and protonated species, were submitted to the software.
Biological Assays
Cell Culture
Human multiple myeloma RPMI 8226 cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1% L-glutamine, and 1% penicillin and streptomycin (Euroclone, Italy). Human glioblastoma U87-MG cells were cultured in Dulbecco's modified Eagle's medium (DMEM) low glucose supplemented with 10% fetal bovine serum, 1% L-glutamine, and 1% penicillin and streptomycin (Euroclone, Italy). Cells were incubated in a humidified incubator at 37°C and 5% CO2. A stock solution of all compounds in dimethyl sulfoxide (DMSO) (50 mM) has been prepared and then directly diluted in culture medium.
MTT Assay
Cells were seeded in 96-well plates at a density of 1 × 104 cells/well and were treated after 24 h with RA, AM, or SU molecules. After 24 h, a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma Aldrich, United States) solution was added to each well to reach a final concentration of 0.5 mg/ml. After 2 h of incubation (4 h for RPMI 8226 cells), formazan salt was solubilized in ethanol, and absorbance was measured at 570 nm in a microplate reader (BMG-Labtech, Germany).
Trypan Blue Cell Viability Assay
Cells were seeded in six-well plates at a density of 2.5 × 105 cells/well and were treated after 24 h with different concentrations of RA molecules. After 24 h of treatment, cells were collected and stained with Trypan blue vital dye (Sigma Aldrich, United States). Viable and dead cells were then counted in a hemocytometer under a light microscope.
Proteasome Activity Assay
Cells not used in Trypan blue assay were lysed to assess proteasome activity. Briefly, cells were resuspended in a lysis buffer [50 mM Hepes pH 7.5, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1.5 mM MgCl2, 5 mM ethylene glycol tetraacetic acid (EGTA)] and were mechanically lysed with a vortex. Obtained protein lysates were centrifuged at 13,500 rpm for 15 min and were quantified using the Bradford method. Forty micrograms of proteins was then loaded in black 96-well plate to perform the proteasome activity assay. In each well, 7.6 mg/ml proteasome substrate (N-succinyl-Leu-Leu-Val-Tyr-7-Amido-4-methylcoumarin, Sigma Aldrich, United States) and proteasome buffer [250 mM Hepes pH 7.5, 5 mM ethylenediaminetetraacetic acid (EDTA) pH 8.0, 0.5% NP-40, 0.01% sodium dodecyl sulfate (SDS)] were added to proteins. Fluorescence was measured after 2 h of incubation in a microplate reader (excitation, 380 nm; emission, 460 nm; BMG-Labtech, Germany).
Statistical Analysis
Data showed the mean ± standard deviation (SD) from at least three independent experiments. Statistical analysis was performed using GraphPad Prism 3 software. The differences between control and treated cells were evaluated using one-way ANOVA analysis of variance followed by Dunnet's multiple comparison test. Statistical significance was set at p < 0.05 and p < 0.01.
Results
Compound Library Design and in silico Evaluation
As a first step of this work, we synthesized compound RC-206 (Figure 1), characterized by the presence of a piperazine as a versatile moiety suitable for different types of derivatization. Once we verified that this structural change does not significantly affect the cytotoxic activity, three different series of compounds, named RA, SU, and AM, have been designed, thus obtaining the library of 60 members (Table 1). Compounds are identified by the series name (which refers to the final reaction exploited) and matrix, indicating the variable portions of the final molecules. For each compound, water solubility and lipophilicity were predicted with the SwissADME web tool (Daina et al., 2017), which allows to compare the outputs of different computational methods. In detail, five freely available predictive models are employed in SwissADME to estimate log PO/W (i.e., XLOGP3 (Cheng et al., 2007), WLOGP (Wildman and Crippen, 1999), MLOGP (Souza et al., 2011), SILICOS-IT (“Silicos-It | Filter-It™” n.d.2), and iLOGP Daina et al., 2014), and the consensus log PO/W is calculated as the arithmetic mean of the values derived from these methods. On the other hand, three different models used to predict water solubility: the ESOL model (Delaney, 2004), an adaptation of the one developed by Ali et al. (2012) and the one by SILICOS-IT (“Silicos-It | Filter-It™” n.d.). The solubility predicted for the majority of the compound library, including RC-206, is enhanced with only a few compounds having log S and log P-values close to that of RC-106. Moreover, most of the designed compounds are predicted to cross both the gastrointestinal (GI) tract and the blood–brain barrier (BBB). Lastly, no pan-assay interference compounds (PAINS) (Baell and Walters, 2014; Dahlin et al., 2015) have been identified among our molecules. Data retrieved from the software are presented in Tables S1, S2. Overall, results thus obtained suggested a promising drug-like profile for the designed compound library, with improved solubility respect to hit compound and the desired BBB permeability and absorption requirements. Hence, all these molecules have been selected for the synthesis and experimental investigation.

Table 1. Compound library of RC-106 analogs.
Chemistry
For the synthesis of compound RC-206 (Figure 1) and the library of derivatives (Table 1) the procedure already set up for RC-106 (Rui et al., 2016b) has been optimized, and a final diversification step, suitable for combinatorial synthesis, was added as reported in Scheme 1. In detail, Heck reaction was exploited to prepare the α,β-unsaturated esters 1[1–4]. Particularly, the reaction was optimized using Pd EnCat® 40 (palladium acetate microencapsulated in polyurea matrix) instead of ordinary palladium acetate, to both simplify the workup and reduce its exposure to air while limiting palladium contamination in the resulting products. The isolated (E) α,β-unsaturated esters were then reduced with LiAlH4 to prepare allylic alcohols 2[1–4]. The latter were reacted with N-Boc-piperazine according to the procedure described by Frøyen (Frøyen and Juvvik, 1995). This reaction consists in a nucleophilic substitution via alkoxyphosphonium salt, generated by the addition of triphenylphosphine (PPh3) and N-bromosuccinimide (NBS) to the alcohol. The protocol reported in the literature was slightly modified in order to find the optimal conditions (i.e., reagent equivalents, temperature, and timing of reagent additions) to access compounds 3[1–4]. Upon Boc- deprotection with TFA, key intermediates 4[1–4] were isolated in quantitative yield. At this point, to prepare RC-206, a bench-scale reductive amination was performed on intermediate 4[3]. Conversely, for the preparation of the library, a combinatorial approach was exploited, and the piperazinic nitrogen was derivatized using three different reactions: sulfonylation (Scheme 1, SU[1,1]–SU[4,5]), reductive amination (Scheme 1, RA[1,1]–RA[4,5]), and amide coupling (Scheme 1, AM[1,1]–AM[4,5]). The preparation of the derivatives was performed in a parallel fashion using a 24-position Mettler-Toledo block equipped with 15-ml reaction tubes (Scheme 1).
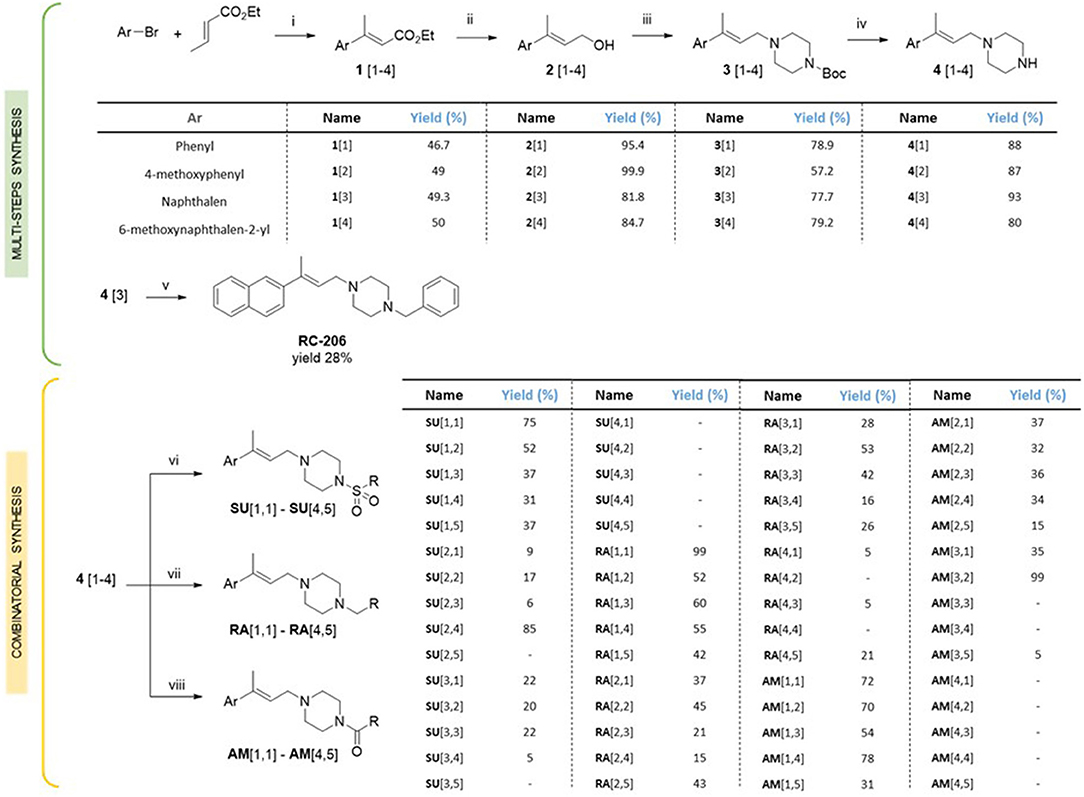
Scheme 1. Synthesis of the compound library. Reagents and reaction conditions: (i) Pd EnCat (0.01 equiv.), TEAC (2 equiv.), AcONa (2 equiv.), dry DMF, 105 °C; (ii) LiAlH4 (1M in THF, 1 equiv.), dry Et2O, 0°C; (iii) PPh3 (1.5 equiv.), NBS (1.4 equiv.), 1-Boc-piperazine (1.2 equiv.), Et3N (2 equiv.), THF, −18°C; (iv) HCl 4 N, 1,4-dioxane, 0°C to RT; (v) benzaldehyde (1 equiv.), AcOH (2 equiv.), DCE (2 equiv.), RT, 20 min, then NaBH(OAc)3 (2.5 equiv.), RT, o.n.; (vi) RSO2Cl (1.5 equiv.), TEA (5 equiv.), DCM, RT, o.n.; (vii) RCHO (1 equiv.), AcOH (2 equiv.), DCE (2 equiv.), RT, 20 min, then NaBH(OAc)3 (2.5 equiv.), RT, o.n.; (viii) RCO2H (2 equiv.), DIPEA (5 equiv.), HOBt (2 equiv.), EDC·HCl (2.5 equiv.), DMF, RT, o.n.
The compounds were characterized by NMR, UPLC-MS, and IR analyses. Overall, 44 compounds were obtained from the combinatorial synthesis in suitable amount and purity for subsequent biological investigations.
Biological Investigation
The cytotoxic activities of all synthesized compounds were evaluated in vitro via MTT assay against the two human cancer cell lines U87MG and RPMI 8226, representative of human glioblastoma multiforme (GBM) and multiple myeloma, respectively.
In particular, the cell viability of U87-MG cells was assessed by MTT assay after 24 h of continuous treatment with all synthesized compounds, at the concentration of 60 μM, corresponding to the RC-106 IC50 value observed on the same cell lines after a similar time exposure. In this preliminary screening, RC-206 showed a moderate activity against U87-MG cells after 24 h of treatment, reducing the cell viability to 34% (Figure 2A). Conversely, all compounds belonging to RA, SU, and AM series (Figures 2B–D) did not show any cell viability reduction. Nevertheless, compound AM[3,1] induced an alteration of cell morphology. Actually, treated U87-MG cells showed an epithelial-like morphology, characterized by a flat cell body, and by the presence of tight junctions between cells, whereas U87-MG control cells had an elongated cell body, and no or few tight junctions between near cells (Figure 3A). Therefore, the cytotoxicity of AM[3,1] compound at long-term exposure times was evaluated, determining the IC50 values at 48 and 72 h (Figure 3B). A time-dependent effect in the micromolar range on cell viability was observed (IC50, 39.05 and 11.21 μM, at 48 and 72 h, respectively).
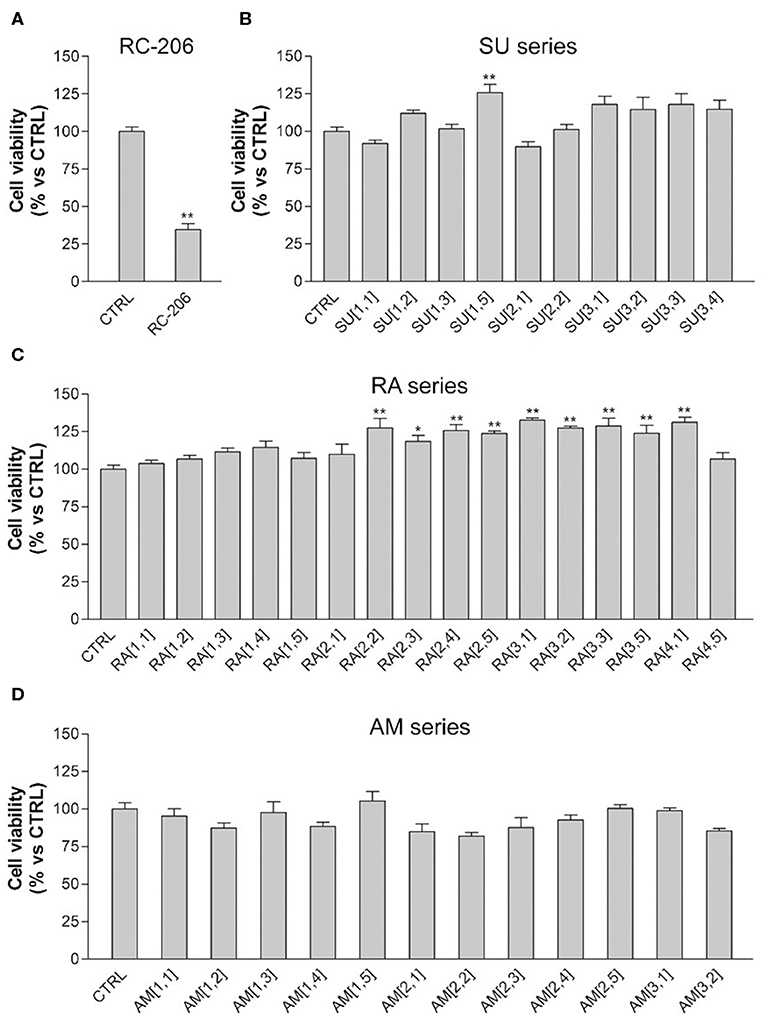
Figure 2. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, U87-MG cells treated with test compounds for 24 h at 40 μM concentration. Results are expressed as cell viability (%). (A) RC-206. (B) SU molecules. (C) RA molecules. (D) AM molecules. All graphs are represented as the mean percentage ± SD of three independent experiments and are compared to untreated controls (CTRL) arbitrarily set to 100%. *p < 0.05 vs. CTRL; **p < 0.01 vs. CTRL.
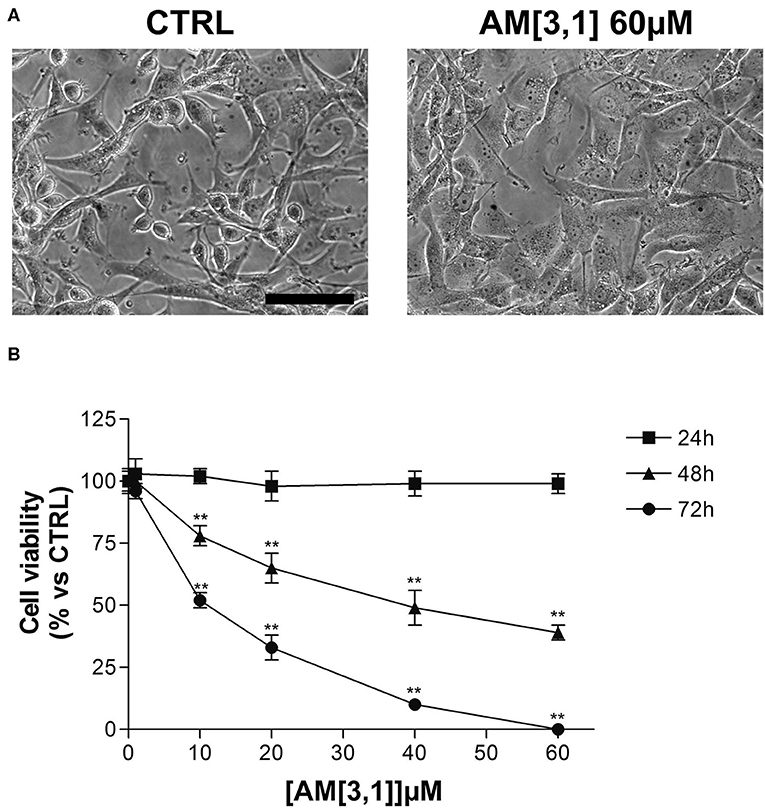
Figure 3. U87-MG cell morphological alterations and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay after AM[3,1] treatment. (A) Morphology of U87-MG cells, untreated (CTRL), or treated with AM[3,1] 60 μM. Scale bar, 80 μm. (B) MTT assay of U87-MG cells treated with increasing concentrations of AM[3,1] (1–60 μM) for 24, 48, and 72 h. Graph represents the mean percentage ± SD of viable cells compared to untreated control cells arbitrarily set to 100%. **p < 0.01 vs. CTRL.
We also observed in this preliminary screening that 3 out of 20 compound of RA series (RA[2,2], RA[3,1], RA[4,1]) and only 1 out of 20 molecules belonging to SU series (SU[1,5]) induced a slight, but significant, increase in cell viability, which is ~25% higher than in untreated controls (Figures 2A–C).
Regarding the effect of the novel compounds against RPMI 8226 cells, the cell viability was preliminarily assessed by MTT assay after 24 h of treatment, at the concentration of 40 μM, corresponding to the IC50 24 h of RC-106. Results were compared to RPMI 8226 untreated control cells (Figure 4). RC-206 resulted to be effective, thus confirming that the replacement of the piperidine moiety by the piperazine one does not greatly affect the antiproliferative activity (Figure 4A). Within the compound library, the most interesting compounds belong to RA series, followed by those of SU and AM series. Indeed, 9 out 19 of RA compounds, i.e., RA[1,3], RA[1,4], RA[2,2], RA[2,3], RA[2,4], RA[3,1], RA[3,2], RA[3,3], and RA[4,1], impaired cell viability in a more effective way than RC-106 (Figure 4A). Only three compounds of the SU series significantly impaired cell viability of RPMI 8226 cells, i.e., SU[1,3], SU[3,1], and SU[3,2]. Of particular interest is the compound SU[3,2], which is able to induce a greater reduction in cell viability compared to RC-106 effect (Figure 4C). Only four molecules of AM series (AM[1,3], AM[2,3], AM[2,4], and AM[3,1]) impaired cell viability of RPMI 8226 cells, but they are less effective than RC-106. Among all compounds tested, only AM[1,1], characterized by the presence of a cycloalkyl as R substituent, was found to induce a slight, but significant, increase in cell viability (Figure 4B), and for this reason, it must be discarded. Of note, none of the assayed compounds caused relevant changes in RPMI 8226 cell morphology. The IC50 of the 10 most effective compounds against RPMI 8226 cell viability were then calculated (Table 2).
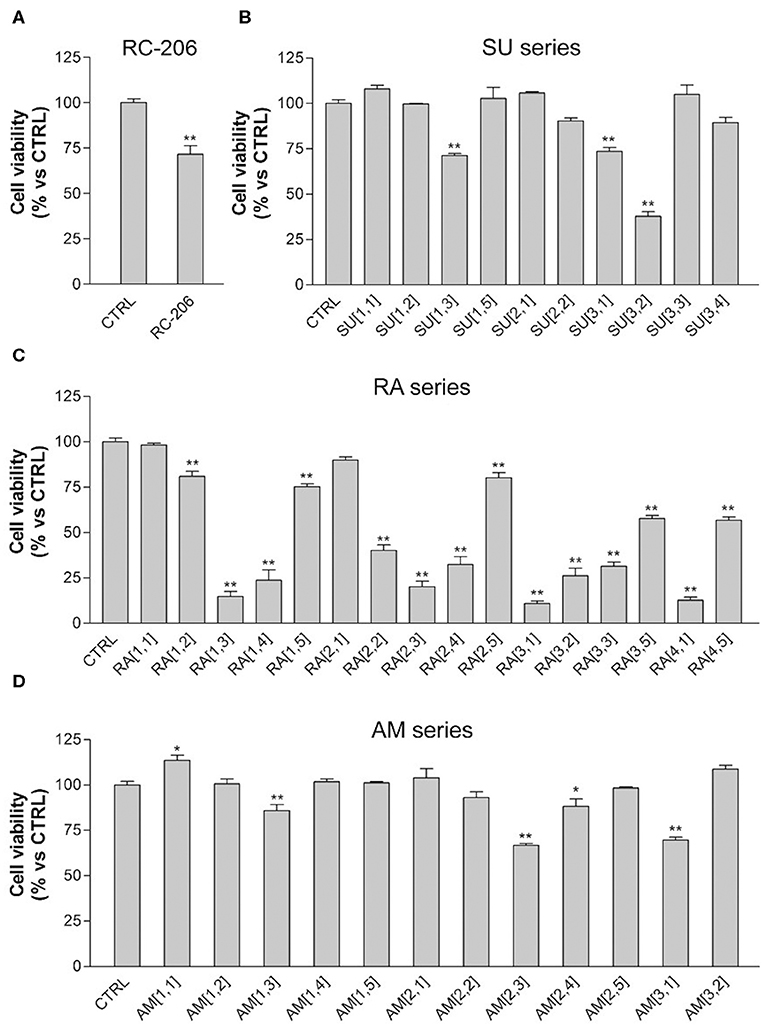
Figure 4. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, Roswell Park Memorial Institute (RPMI) 8226 cells treated with test compounds for 24 h at 40 μM concentration. Results are expressed as cell viability (%). (A) RC-206. (B) SU molecules. (C) RA molecules. (D) AM molecules. All graphs are represented as the mean percentage ± SD of three independent experiments and are compared to untreated controls (CTRL) arbitrarily set to 100%. *p < 0.05 vs. CTRL; **p < 0.01 vs. CTRL.
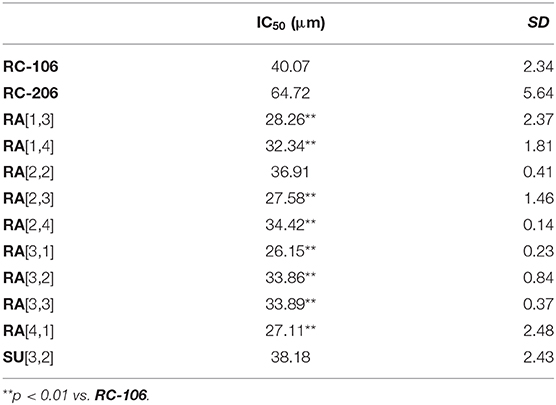
Table 2. IC50 of Roswell Park Memorial Institute (RPMI) 8226 cell viability after treatment with the 10 most effective compounds.
Results described so far clearly suggest that, against MM, the change in piperidine ring into piperazine is an allowed modification and that the “X” portion of the general structure reported in Figure 1 plays a key role in the activity: indeed, the optimal results were obtained for compounds belonging to RA series, i.e., when the linker between piperazine and the “R” substituent consists of a simple methylene. Finally, a small aryl group (i.e., phenyl, 4-methoxy-phenyl) is preferred when combined with cyclohexyl and p-substituted aromatics attached to the piperazine ring, whereas the bulkier naphthalene group is allowed when “R” consists of the small cyclopentyl and cyclohexyl ring (RA[3,1], RA[3,2], RA[4,1]). An exception is represented by RA[3,3].
Cell viability of the four most effective compound (RA[1,3], RA[2,3], RA[3,1], and RA[4,1]) was evaluated at different time points (24, 48, and 72 h) and different concentrations (1–40 μM) by MTT assay. All compounds were able to significantly reduce in a dose- and time-dependent manner the cell viability of RPMI 8226 cells (Figure 5).
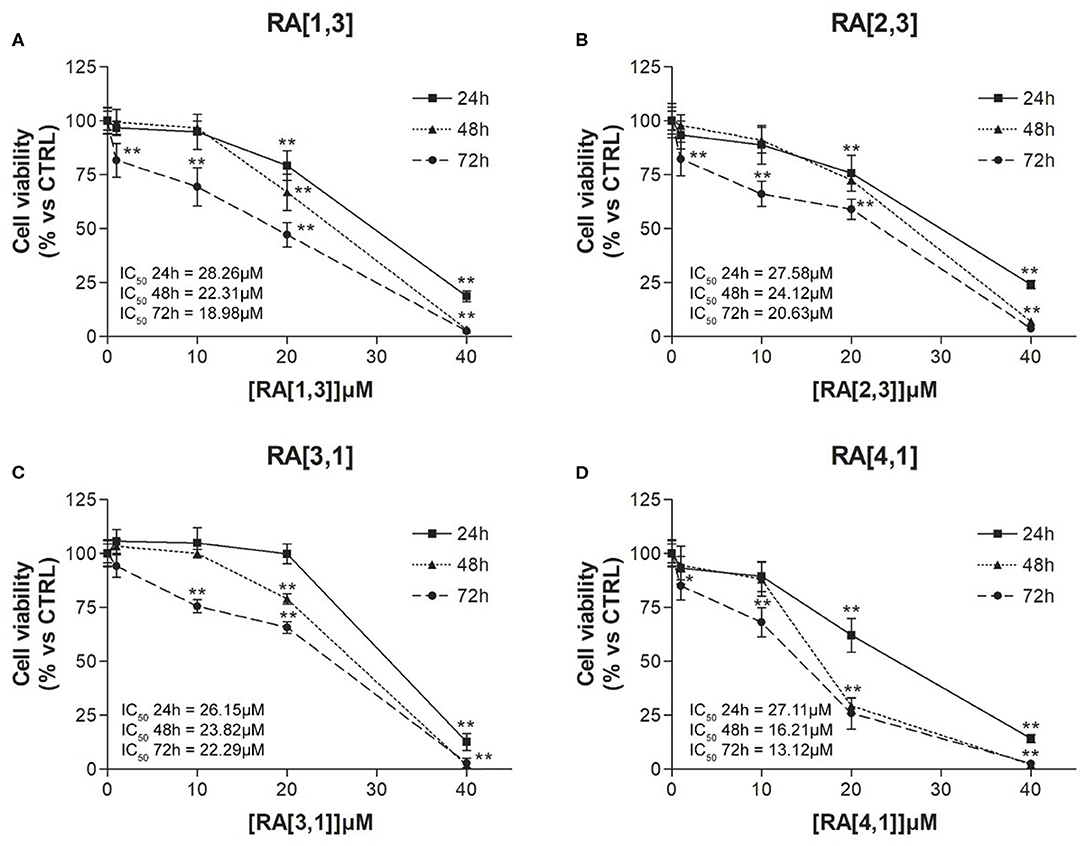
Figure 5. Cell viability (%) curves of Roswell Park Memorial Institute (RPMI) 8226 cells treated with RA[1,3], RA[2,3], RA[3,1], and RA[4,1] molecules. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay of RPMI 8226 cells treated with different concentration (1–40 μM) of (A) RA[1,3], (B) RA[2,3], (C) RA[3,1], and (D) RA[4,1] for 24, 48, and 72 h. All graphs are represented as the mean percentage ± SD of three independent experiments and are compared to untreated controls (CTRL) arbitrarily set to 100%. **p < 0.01 vs. CTRL.
Prompted by the positive results obtained, the proteasome activity of the compounds belonging to the RA series was also evaluated. After 24 h treatment of RPMI 8226 cells, RA[1,3] and RA[2,3] showed higher proteasome inhibition activity than RC-106. Both compounds significantly inhibited proteasome in a dose-dependent manner and showed an IC50 value, for the inhibition of proteasome, ~22 μM (versus an RC-106 IC50 value of 35 μM) (Figure 6A). Moreover, both RA[1,3] and RA[2,3] reduced in a dose-dependent manner the number of viable cells counted by trypan blue vital count (Figure 7). However, the induction of cell death was quite different. After RA[1,3] 20 μM treatment, the percentage of RPMI 8226 dead cells were comparable to untreated controls, then at 40 μM concentration, a significant increase in cell mortality was observable. Instead, after RA[2,3] treatment, the number of RPMI 8226 dead cells increased in a dose-dependent manner. This could suggest a cytostatic effect of RA[1,3] and a cytotoxic effect of RA[2,3].
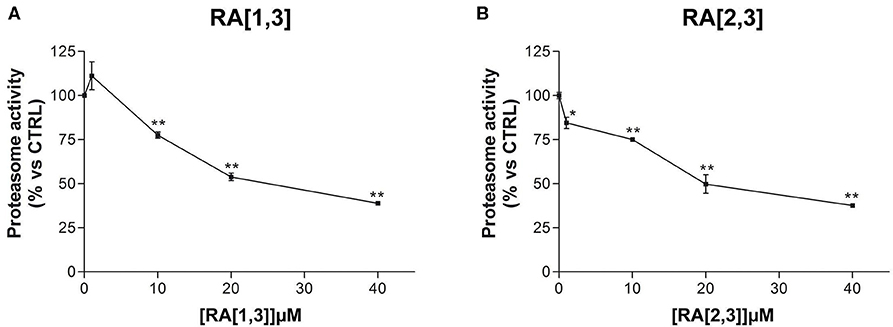
Figure 6. Proteasome activity of Roswell Park Memorial Institute (RPMI) 8226 cells after treatment with RA[1,3] and RA[2,3]. (A) Proteasome activity of RPMI 8226 cells after treatment with increasing concentrations of RA[1,3] (1–40 μM) for 24 h. (B) Proteasome activity of RPMI 8226 cells after treatment with increasing concentrations of RA[2,3] (1–40 μM) for 24 h. All graphs are represented as the mean percentage ± SD of three independent experiments, and results are compared to untreated control cells (CTRL) arbitrarily set to 100%. *p < 0.05 vs. CTRL; **p < 0.01 vs. CTRL.
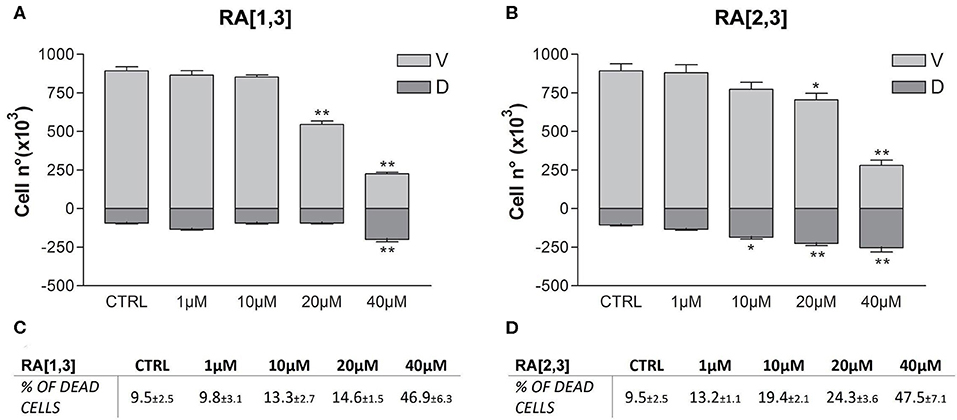
Figure 7. Trypan blue vital count of Roswell Park Memorial Institute (RPMI) 8226 cells treated with RA[1,3] and RA[2,3]. (A) Vital count of RPMI 8226 cells after treatment with increasing concentrations of RA[1,3] (1–40 μM) for 24 h. (B) Vital count of RPMI 8226 cells after treatment with increasing concentrations of RA[2,3] (1–40 μM) for 24 h. In graphs (A,B), both viable (V) and dead (D) cells number are represented. (C) Table represents the percentage of dead cells compared to all counted cells after treatment with increasing concentrations of RA[1,3] (1–40 μM). (D) Table represents the percentage of dead cells compared to all counted cells after treatment with increasing concentrations of RA8 (1–40 μM). *p < 0.05 vs. CTRL; **p < 0.01 vs. CTRL.
Discussion
To identify novel chemical entities active against aggressive tumors with poor prognosis, i.e., GB and MM for which effective drugs or therapeutic strategies are still needed, we investigated the chemical space around compound RC-106 (Rui et al., 2016b), characterized by a promising cytotoxic activity against glioblastoma (U87MG) and multiple myeloma (RPMI-8226) cell lines. To determine which positions are amenable to the lead optimization extension strategy and to find effective developable compounds, we designed a small compounds library, replacing the piperidine moiety of RC-106 with the piperazine ring, thus gaining a third point of derivatization. Moreover, the derivatization of piperazine allows the attachment of a wide range of alkyl or aryl substituents through a combinatorial approach. At first, we synthesized compound RC-206, replacing the piperidine moiety with piperazine nucleus, and evaluated its antiproliferative properties against GB and MM. In vitro results showed that such a minor structural change is allowed. Therefore, we built a compound library on the new scaffold, taking into account the synthetic feasibility and commercial availability of building blocks. In detail, we choose phenyl, 4-methoxy-phenyl, naphthyl, and 6-methoxy-naphthyl scaffolds for the exploration of stereoelectronic features of the primary aryl group, and we envisaged to adopt three different approaches for piperidine derivatization: (i) sulfonylation (SU), (ii) reductive amination and (RA), and (iii) amidation (AM). As a result, a compound library of 60 members was designed. Since it is well known that pharmacokinetic studies performed in the early stages of drug discovery may reduce attrition rate (Kola and Landis, 2004; Merlot, 2010), we evaluated the in silico ADME profile and drug-likeness of the compounds. In particular, the prediction of log P and log S-values indicates that the majority of designed compounds is endowed with an improved water-solubility with respect to hit compound RC-106. Only a few compounds have a predicted solubility profile comparable to that of RC-106. In addition, good GI and BBB permeability were predicted, and no PAINS structures were identified.
Only a few compounds have a predicted solubility profile comparable to that of RC-106. In addition, good GI and BBB permeability were predicted, and no PAINS structures were identified. Considering the results obtained for the whole library, in terms of solubility and barriers permeability, we moved forward to synthesis and experimental evaluation of the designed compounds. The synthetic strategy previously reported for RC-106 (Rui et al., 2016b) was successfully optimized and adapted to obtain a library of structurally diverse analogs. All the designed compounds, with few exceptions, were obtained in suitable amounts and purity for biological investigation. Moreover, the adopted approach could be exploited for the synthesis of other RC-106 analogs to further expand the exploration of chemical space around our hit compound. All compounds have been evaluated in two different human cancer cell lines, U87-MG glioblastoma cells and RPMI 8226 multiple myeloma cells, and screened their ability in inhibiting or hampering the tumor growth. In general, new compounds showed poor or no effect against glioblastoma U87-MG cell line, whereas nine molecules of RA series and one of SU series were significantly more effective in reducing RPMI 8226 cells viability than RC-106. In particular, compounds RA[1,3], RA [2,3], RA [3,1], and RA [4,1] deserved to be mentioned: they showed a good dose–response curve by MTT assay (IC50 values <30 μM) and therefore the capability to significantly slow the metabolic activity of tumor cells after only 24 h exposure. Such effect was mirrored by the impairment of proteasome activity (Figure 6), an enzyme involved in the protein homeostasis. This activity was also documented in our previous work focused on the antitumor activity of the hit RC-106 and where we showed its capability to trigger the UPR response machinery (Tesei et al., 2019). Accordingly, the compounds also hamper tumor cell growth starting from the lowest concentrations tested, as evidenced by the proliferation test (Figure 7) showing also a significant induction of cell death (~50%) at the highest concentration tested (Figure 5).
For U87-MG, none of the new molecules of RA, AM, and SU series was able to reduce cell viability of U87-MG after 24 h of treatment. Only RC-206 showed a cytotoxic activity higher than the hit compound, thus representing the most promising molecules against U87-MG cells. However, of particular interest is molecule AM[3,1] that even if it was not able to reduce U87-MG cell viability after 24 h of treatment, it caused evident alteration of cell morphology. U87-MG cells lost the elongated cell body with long processes and acquired an epithelial-like phenotype with a flat cell body without processes and presence of tight junctions between cells. These results suggest that the process inducing a reduction in U87-MG cell viability triggered by AM[3,1], evident only after 48 and 72 h of treatment, starts already from 24 h preceded by specific morphological changes caused by AM[3,1]. Such morphology alteration strongly suggests a transition of U87-MG cell to an epithelial cell morphology (MET). Cellular morphological changes are evident in epithelial–mesenchymal transition (EMT) and in the reverse process mesenchymal–epithelial transition (MET) and are reported in previous works on U87-MG cells (Guan et al., 2017; Yang et al., 2020, p. 3). EMT and MET play an important role in development, reprogramming, and tumorigenesis (Chen T. et al., 2017; Pei et al., 2019). Different compounds are able to exert antitumoral effects reversing ETM and promoting MET (Peng et al., 2016; Cheng et al., 2018; Li et al., 2019; Yuan et al., 2019). In addition, a cocktail of reprogramming transcription factors is able to promote MET attenuating the malignancy of cancer cells (Takaishi et al., 2016). It is worth noting that glioblastoma is the most aggressive (WHO grade IV) and common of the human gliomas. The invasive characteristic of glioblastoma, at least in part, is due to their high migratory potential to invade the surrounding tissue. EMT has been pointed as one of the mechanisms that confer to GBM cells this invasive property (Iser et al., 2017). Therefore, transition to polarized epithelial cells (MET) increases efficacy of antineoplastic agents and makes tumors less aggressive and with a better patient prognosis (Takaishi et al., 2016). Further experiments will be necessary to demonstrate the property of AM[3,1] to induce the MET transition of U87-MG cells and confirm our hypothesis.
Conclusions
To conclude, exploration of chemical space around our previously reported hit RC-106 led us to identify compounds endowed with interesting antitumor properties. In particular, molecules derived from RA-106 have different effects on RPMI 8226 and U87-MG cell lines, resulting in RPMI 8226 myeloma cells more sensitive than glioblastoma U87-MG cell line. These differences could be due to different mechanisms of action of the molecules in the cell lines used for biological testing. It is not surprising, since the compound may have different effects depending on the tumoral cell line examined (Bittkau et al., 2019; Chen et al., 2019) and on the concentrations evaluated (Jiménez-Orozco et al., 2011; Navarro-Villarán et al., 2016).
Regarding compounds effective against RPMI 8226 cells, of particular interest are RA[1,3] and RA[2,3] (Figure 8), able to significantly inhibit proteasome in a dose-dependent manner. Indeed, proteasome inhibitors, like bortezomib (BTZ), ixazomib, and carfilzomib, are currently used for the treatment of multiple myeloma (Cvek and Dvorak, 2008; Chen et al., 2011; Chen T. et al., 2017; Okazuka and Ishida, 2018; Kim et al., 2019). Conversely, AM[3,1] (Figure 8) emerged for its activity on U87-MG cell lines since it is able to induce a significant cellular morphology alteration and a time-dependent effect on cell viability.
Figure 8. Structures of the three compounds emerged as the most interesting for their promising anticancer profile.
In view of the results discussed so far, the biological profile of RA[1,3] and RA[2,3] and AM[3,1] will be deepened. Studies of mechanism of action and of the potential in improving the efficacy of cancer treatment and reducing the side effects are ongoing and will be presented in due course.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The authors gratefully acknowledge MIUR for the doctoral fellowship to RL and GR and the University of Pavia for the postdoctoral fellowship to MR.
Conflict of Interest
SS performed part of her Ph.D. project in the company Taros Chemicals GmbH and Co. KG, working on a different topic.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thankfully acknowledge Paolo Dognini for the contribution to the synthesis of the compound library.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00495/full#supplementary-material
Footnotes
1. ^Cancer. n.d. Available online at: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed February 7, 2020).
2. ^Silicos-It Filter-ItTM. n.d. Available online at: http://silicos-it.be.s3-website-eu-west-1.amazonaws.com/software/filter-it/1.0.2/filter-it.html (accessed March 16, 2020).
References
Ali, J., Camilleri, P., Brown, M. B., Hutt, A. J., and Kirton, S. B. (2012). Revisiting the general solubility equation: in silico prediction of aqueous solubility incorporating the effect of topographical polar surface area. J. Chem. Inform. Model. 52, 420–428. doi: 10.1021/ci200387c
Ao, N., Chen, Q., and Liu, G. (2017). The small molecules targeting ubiquitin-proteasome system for cancer therapy. Comb. Chem. High Throughput Screen. 20, 403–413. doi: 10.2174/1386207320666170710124746
Aydar, E., Onganer, P., Perrett, R., Djamgoz, M. B., and Palmer, C. P. (2006). The expression and functional characterization of sigma (sigma) 1 receptors in breast cancer cell lines. Cancer Lett. 242, 245–257. doi: 10.1016/j.canlet.2005.11.011
Baell, B. J., and Walters, M. A. (2014). Chemistry: chemical con artists foil drug discovery. Nat. News 513:481. doi: 10.1038/513481a
Banaszkiewicz, M., Małyszko, J., Vesole, D. H., Woziwodzka, K., Jurczyszyn, A., Zórawski, M., et al. (2019). New biomarkers of ferric management in multiple myeloma and kidney disease-associated anemia. J. Clin. Med. 8:1828. doi: 10.3390/jcm8111828
Bittkau, K. S., Dörschmann, P., Blümel, M., Tasdemir, D., Roider, J., Klettner, A., et al. (2019). Comparison of the effects of fucoidans on the cell viability of tumor and non-tumor cell lines. Mar. Drugs 17:441. doi: 10.3390/md17080441
Brune, S., Schepmann, D., Lehmkuhl, K., Frehland, B., and Wünsch, B. (2012). Characterization of ligand binding to the σ(1) receptor in a human tumor cell line (RPMI 8226) and establishment of a competitive receptor binding assay. Assay Drug Dev. Technol. 10, 365–374. doi: 10.1089/adt.2011.0376
Chauhan, D., Singh, A. V., Aujay, M., Kirk, C. J., Bandi, M., Ciccarelli, B., et al. (2010). A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood 116, 4906–4915. doi: 10.1182/blood-2010-04-276626
Chen, D., Frezza, M., Schmitt, S., Kanwar, J., and Dou, Q. P. (2011). Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr. Cancer Drug Targets 11, 239–253. doi: 10.2174/156800911794519752
Chen, T., You, Y., Jiang, H., and Wang, Z. Z. (2017). Epithelial-mesenchymal transition (EMT): a biological process in the development, stem cell differentiation, and tumorigenesis. J. Cell. Physiol. 232, 3261–3272. doi: 10.1002/jcp.25797
Chen, X., Huang, Y., Wang, Y., Wu, Q., Hong, S., and Huang, Z. (2019). THBS4 predicts poor outcomes and promotes proliferation and metastasis in gastric cancer. J. Physiol. Biochem. 75, 117–123. doi: 10.1007/s13105-019-00665-9
Chen, Y., Zhang, Y., and Guo, X. (2017). Proteasome dysregulation in human cancer: implications for clinical therapies. Cancer Metast. Rev. 36, 703–716. doi: 10.1007/s10555-017-9704-y
Cheng, C., Ji, Z., Sheng, Y., Wang, J., Sun, Y., Zhao, H., et al. (2018). Aphthous ulcer drug inhibits prostate tumor metastasis by targeting IKKε/TBK1/NF-KB signaling. Theranostics 8, 4633–4648. doi: 10.7150/thno.26687
Cheng, T., Zhao, Y., Li, X., Lin, F., Xu, Y., Zhang, X., et al. (2007). Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J. Chem. Inform. Model. 47, 2140–2148. doi: 10.1021/ci700257y
Collina, S., Bignardi, E., Rui, M., Rossi, D., Gaggeri, R., Zamagni, A., et al. (2017). Are sigma modulators an effective opportunity for cancer treatment? a patent overview (1996-2016). Exp. Opin. Ther. Patents 27, 565–78. doi: 10.1080/13543776.2017.1276569
Collina, S., Gaggeri, R., Marra, A., Bassi, A., Negrinotti, S., Negri, F., and Rossi, D. (2013). Sigma receptor modulators: a patent review. Exp. Opin. Ther. Patents 23, 597–613. doi: 10.1517/13543776.2013.769522
Cvek, B., and Dvorak, Z. (2008). The value of proteasome inhibition in cancer. can the old drug, disulfiram, have a bright new future as a novel proteasome inhibitor? Drug Discov. Today 13, 716–22. doi: 10.1016/j.drudis.2008.05.003
Dahlin, J. L., Nissink, J. W., Strasser, J. M., Francis, S., Higgins, L., Zhou, H., et al. (2015). PAINS in the assay: chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J. Med. Chem. 58, 2091–2113. doi: 10.1021/jm5019093
Daina, A., Michielin, O., and Zoete, V. (2014). ILOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J. Chem. Inform. Model. 54, 3284–3301. doi: 10.1021/ci500467k
Daina, A., Michielin, O., and Zoete, V. (2017). SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7:42717. doi: 10.1038/srep42717
Delaney, J. S. (2004). ESOL: estimating aqueous solubility directly from molecular structure. J. Chem. Inform. Comput. Sci. 44, 1000–1005. doi: 10.1021/ci034243x
Esmaeili, M., Stensjøen, A. L., Berntsen, E. M., Solheim, O., and Reinertsen, I. (2018). The direction of tumour growth in glioblastoma patients. Sci. Rep. 8:1199. doi: 10.1038/s41598-018-19420-z
Fairfield, H., Falank, C., Avery, L., and Reagan, M. R. (2016). Multiple myeloma in the marrow: pathogenesis and treatments. Ann. N. Y. Acad. Sci. 1364, 32–51. doi: 10.1111/nyas.13038
Frøyen, P., and Juvvik, P. (1995). One-pot synthesis of secondary or tertiary amines from alcohols and amines via alkoxyphosphonium salts. Tetrahedron Lett. 36, 9555–9558. doi: 10.1016/0040-4039(95)02046-2
Gelman, J. S., Sironi, J., Berezniuk, I., Dasgupta, S., Castro, L. M., Gozzo, F. C., et al. (2013). Alterations of the intracellular peptidome in response to the proteasome inhibitor bortezomib. PLoS ONE 8:e53263. doi: 10.1371/journal.pone.0053263
Gras Navarro, A., Espedal, H., Joseph, J. V., Trachsel-Moncho, L., Bahador, M., Gjertsen, B. T., et al. (2019). Pretreatment of glioblastoma with bortezomib potentiates natural killer cell cytotoxicity through TRAIL/DR5 mediated apoptosis and prolongs animal survival. Cancers 11:996. doi: 10.3390/cancers11070996
Groll, M., Nguyen, H., Vellalath, S., and Romo, D. (2018). Homosalinosporamide A and its mode of proteasome inhibition: an X-Ray crystallographic study. Mar. Drugs 16:240. doi: 10.3390/md16070240
Guan, Y., Cheng, W., Zou, C., Wang, T., and Cao, Z. (2017). Gremlin1 promotes carcinogenesis of glioma in vitro. Clin. Exp. Pharmacol. Physiol. 44, 244–256. doi: 10.1111/1440-1681.12697
Hambardzumyan, D., and Bergers, G. (2015). Glioblastoma: defining tumor niches. Trends Cancer 1, 252–265. doi: 10.1016/j.trecan.2015.10.009
Hanif, F., Muzaffar, K., Perveen, K., Malhi, S. M., and Simjee, S. U. (2017). Glioblastoma multiforme: a review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac. J. Cancer Prev. APJCP 18, 3–9. doi: 10.22034/APJCP.2017.18.1.3
Iser, I. C., Pereira, M. B., Lenz, G., and Wink, M. R. (2017). The epithelial-to-mesenchymal transition-like process in glioblastoma: an updated systematic review and in silico investigation. Med. Res. Rev. 37, 271–313. doi: 10.1002/med.21408
Jiménez-Orozco, F. A., Rosales, A. A., Vega-López, A., Domínguez-López, M. L., García-Mondragón, M. J., Maldonado-Espinoza, A., et al. (2011). Differential effects of esculetin and daphnetin on in vitro cell proliferation and in vivo estrogenicity. Eur. J. Pharmacol. 668, 35–41. doi: 10.1016/j.ejphar.2011.06.024
Kanu, O. O., Hughes, B., Di, C., Lin, N., Fu, J., Bigner, D. D., et al. (2009). Glioblastoma multiforme oncogenomics and signaling pathways. Clin. Med. Oncol. 3, 39–52. doi: 10.4137/CMO.S1008
Kim, K. H., Cheong, H. J., Lee, M. Y., Lee, N., Lee, K. T., Park, S. K., et al. (2019). Bortezomib is more effective to side population of RPMI8226 myeloma cells than classical anti-myeloma agents. Anticancer Res. 39, 127–133. doi: 10.21873/anticanres.13088
Kola, I., and Landis, J. (2004). Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 3, 711–715. doi: 10.1038/nrd1470
Kranz, M., Bergmann, R., Kniess, T., Belter, B., Neuber, C., Cai, Z., et al. (2018). Bridging from brain to tumor imaging: (S)-(–)- and (R)-(+)-[18F]Fluspidine for Investigation of Sigma-1 receptors in tumor-bearing mice. Molecules 23:702. doi: 10.3390/molecules23030702
Kyrtsonis, M. C., Bartzis, V., Papanikolaou, X., Koulieris, E., Georgiou, G., Dimou, M., et al. (2010). Genetic and molecular mechanisms in multiple myeloma: a route to better understand disease pathogenesis and heterogeneity. Appl. Clin. Genet. 3, 41–51. doi: 10.2147/TACG.S7456
Lee, E. C., Fitzgerald, M., Bannerman, B., Donelan, J., Bano, K., Terkelsen, J., et al. (2011). Antitumor activity of the investigational proteasome inhibitor MLN9708 in mouse models of B-cell and plasma cell malignancies. Clin. Cancer Res. 17, 7313–7323. doi: 10.1158/1078-0432.CCR-11-0636
Lee, J. H., Jung, K. H., Quach, C. H. T., Park, J. W., Moon, S. H., Cho, Y. S., et al. (2018). Reporter PET images bortezomib treatment-mediated suppression of cancer cell proteasome activity. Sci. Rep. 8:12290. doi: 10.1038/s41598-018-29642-w
Li, T., Timmins, H. C., King, T., Kiernan, M. C., Goldstein, D., and Park, S. B. (2019). Characteristics and risk factors of bortezomib induced peripheral neuropathy: a systematic review of phase III trials. Hematol. Oncol. 1–15. doi: 10.1002/hon.2706
Liu, C. C., Yu, C. F., Wang, S. C., Li, H. Y., Lin, C. M., Wang, H. H., et al. (2019). Sigma-2 receptor/TMEM97 agonist PB221 as an alternative drug for brain tumor. BMC Cancer 19:473. doi: 10.1186/s12885-019-5700-7
Lövborg, H., Oberg, F., Rickardson, L., Gullbo, J., Nygren, P., and Larsson, R. (2006). Inhibition of proteasome activity, nuclear factor-KB translocation and cell survival by the antialcoholism drug disulfiram. Int. J. Cancer 118, 1577–1580. doi: 10.1002/ijc.21534
Malacrida, A., Cavalloro, V., Martino, E., Cassetti, A., Nicolini, G., Rigolio, R., et al. (2019). Anti-multiple myeloma potential of secondary metabolites from hibiscus sabdariffa. Molecules 24:2500. doi: 10.3390/molecules24132500
Mereles, D., and Hunstein, W. (2011). Epigallocatechin-3-gallate (EGCG) for clinical trials: more pitfalls than promises? Int. J. Mol. Sci. 12, 5592–5603. doi: 10.3390/ijms12095592
Merlot, C. (2010). Computational toxicology–a tool for early safety evaluation. Drug Discov. Today 15, 16–22. doi: 10.1016/j.drudis.2009.09.010
Navarro-Villarán, E., Tinoco, J., Jiménez, G., Pereira, S., Wang, J., Aliseda, S., et al. (2016). Differential antitumoral properties and renal-associated tissue damage induced by tacrolimus and mammalian target of rapamycin inhibitors in hepatocarcinoma: in vitro and in vivo studies. PLoS ONE 11:e0160979. doi: 10.1371/journal.pone.0160979
Okazuka, K., and Ishida, T. (2018). Proteasome inhibitors for multiple myeloma. Jpn. J. Clin. Oncol. 48, 785–793. doi: 10.1093/jjco/hyy108
Pei, D., Shu, X., Gassama-Diagne, A., and Thiery, J. P. (2019). Mesenchymal-epithelial transition in development and reprogramming. Nat. Cell. Biol. 21, 44–53. doi: 10.1038/s41556-018-0195-z
Peng, C., Li, Z., Niu, Z., Niu, W., Xu, Z., Gao, H., et al. (2016). Norcantharidin suppresses colon cancer cell epithelial-mesenchymal transition by inhibiting the vβ6-ERK-Ets1 signaling pathway. Sci. Rep. 6:20500. doi: 10.1038/srep20500
Pillai, R. K., and Jayasree, K. (2017). Rare cancers: challenges & issues. Indian J. Med. Res. 145, 17–27. doi: 10.4103/ijmr.IJMR_915_14
Rajkumar, S. V. (2009). Multiple myeloma. Curr. Probl. Cancer 33, 7–64. doi: 10.1016/j.currproblcancer.2009.01.001
Rossi, D., Rui, M., Di Giacomo, M., Schepmann, D., Wünsch, B., Monteleone, S., et al. (2017). Gaining in pan-affinity towards sigma 1 and sigma 2 receptors. SAR studies on arylalkylamines. Bioorg. Med. Chem. 25, 11–19. doi: 10.1016/j.bmc.2016.10.005
Rui, M., Marra, A., Pace, V., Juza, M., Rossi, D., and Collina, S. (2016a). Novel enantiopure sigma receptor modulators: quick (Semi-)preparative chiral resolution via HPLC and absolute configuration assignment. Molecules 21:1210. doi: 10.3390/molecules21091210
Rui, M., Rossi, D., Marra, A., Paolillo, M., Schinelli, S., Curti, D., et al. (2016b). Synthesis and biological evaluation of new aryl-alkyl(alkenyl)-4-benzylpiperidines, novel sigma receptor (SR) modulators, as potential anticancer-agents. Eur. J. Med. Chem. 124, 649–665. doi: 10.1016/j.ejmech.2016.08.067
Sanchez, E., Li, M., Steinberg, J. A., Wang, C., Shen, J., Bonavida, B., et al. (2010). The proteasome inhibitor CEP-18770 enhances the anti-myeloma activity of bortezomib and melphalan. Br. J. Haematol. 148, 569–581. doi: 10.1111/j.1365-2141.2009.08008.x
Shergails, A., Bankhead, A. 3rd., Luesakul, U., Muangsin, N., and Neamati, N. (2018). Current challenges and opportunities in treating glioblastoma. Pharmacol. Rev. 70, 412–445. doi: 10.1124/pr.117.014944
Souza, E. S., Zaramello, L., Kuhnen, C. A., Junkes Bda, S., Yunes, R. A., and Heinzen, V. E. (2011). Estimating the octanol/water partition coefficient for aliphatic organic compounds using semi-empirical electrotopological index. Int. J. Mol. Sci. 12, 7250–7264. doi: 10.3390/ijms12107250
Takaishi, M., Tarutani, M., Takeda, J., and Sano, S. (2016). Mesenchymal to epithelial transition induced by reprogramming factors attenuates the malignancy of cancer cells. PLoS ONE 11:e0156904. doi: 10.1371/journal.pone.0156904
Tesei, A., Cortesi, M., Pignatta, S., Arienti, C., Dondio, G. M., Bigogno, C., et al. (2019). Anti-tumor efficacy assessment of the sigma receptor pan modulator RC-106. A promising therapeutic tool for pancreatic cancer. Front. Pharmacol. 10:490. doi: 10.3389/fphar.2019.00490
Tesei, A., Cortesi, M., Zamagni, A., Arienti, C., Pignatta, S., Zanoni, M., et al. (2018). Sigma receptors as endoplasmic reticulum stress ‘gatekeepers' and their modulators as emerging new weapons in the fight against cancer. Front. Pharmacol. 9:711. doi: 10.3389/fphar.2018.00711
Wang, W., Cho, H. Y., Rosenstein-Sisson, R., Marín Ramos, N. I., Price, R., Hurth, K., et al. (2018). Intratumoral delivery of bortezomib: impact on survival in an intracranial glioma tumor model. J. Neurosurg. 128, 695–700. doi: 10.3171/2016.11.JNS161212
Wildman, S. A., and Crippen, G. M. (1999). Prediction of physicochemical parameters by atomic contributions. J. Chem. Inform. Comput. Sci. 39, 868–873. doi: 10.1021/ci990307l
Willenbacher, W., Seeber, A., Steiner, N., Willenbacher, E., Gatalica, Z., Swensen, J., et al. (2018). Towards molecular profiling in multiple myeloma: a literature review and early indications of its efficacy for informing treatment strategies. Int. J. Mol. Sci. 19:2087. doi: 10.3390/ijms19072087
Yang, Z., Bian, E., Xu, Y., Ji, X., Tang, F., Ma, C., et al. (2020). Meg3 induces EMT and invasion of glioma cells via autophagy. Onco Targets Ther. 13, 989–1000. doi: 10.2147/OTT.S239648
Yuan, L., Zhou, M., Huang, D., Wasan, H. S., Zhang, K., Sun, L. , et al. (2019). Resveratrol inhibits the invasion and metastasis of colon cancer through reversal of epithelial- mesenchymal transition via the AKT/GSK-3β/snail signaling pathway. Mol. Med. Rep. 20, 2783–2795. doi: 10.3892/mmr.2019.10528
Keywords: cancer, multiple myeloma, glioblastoma, drug discovery, compound library
Citation: Listro R, Stotani S, Rossino G, Rui M, Malacrida A, Cavaletti G, Cortesi M, Arienti C, Tesei A, Rossi D, Giacomo MD, Miloso M and Collina S (2020) Exploring the RC-106 Chemical Space: Design and Synthesis of Novel (E)-1-(3-Arylbut-2-en-1-yl)-4-(Substituted) Piperazine Derivatives as Potential Anticancer Agents. Front. Chem. 8:495. doi: 10.3389/fchem.2020.00495
Received: 19 March 2020; Accepted: 14 May 2020;
Published: 30 June 2020.
Edited by:
Clemens Zwergel, Sapienza University of Rome, ItalyReviewed by:
Antonello Mai, Sapienza University of Rome, ItalySonja M. Kessler, Martin Luther University of Halle-Wittenberg, Germany
Copyright © 2020 Listro, Stotani, Rossino, Rui, Malacrida, Cavaletti, Cortesi, Arienti, Tesei, Rossi, Giacomo, Miloso and Collina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simona Collina, c2ltb25hLmNvbGxpbmEmI3gwMDA0MDt1bmlwdi5pdA==
†These authors have contributed equally to this work