Editorial
14 June 2016
Spyros Petrakis
and
Miguel A. Andrade-Navarro

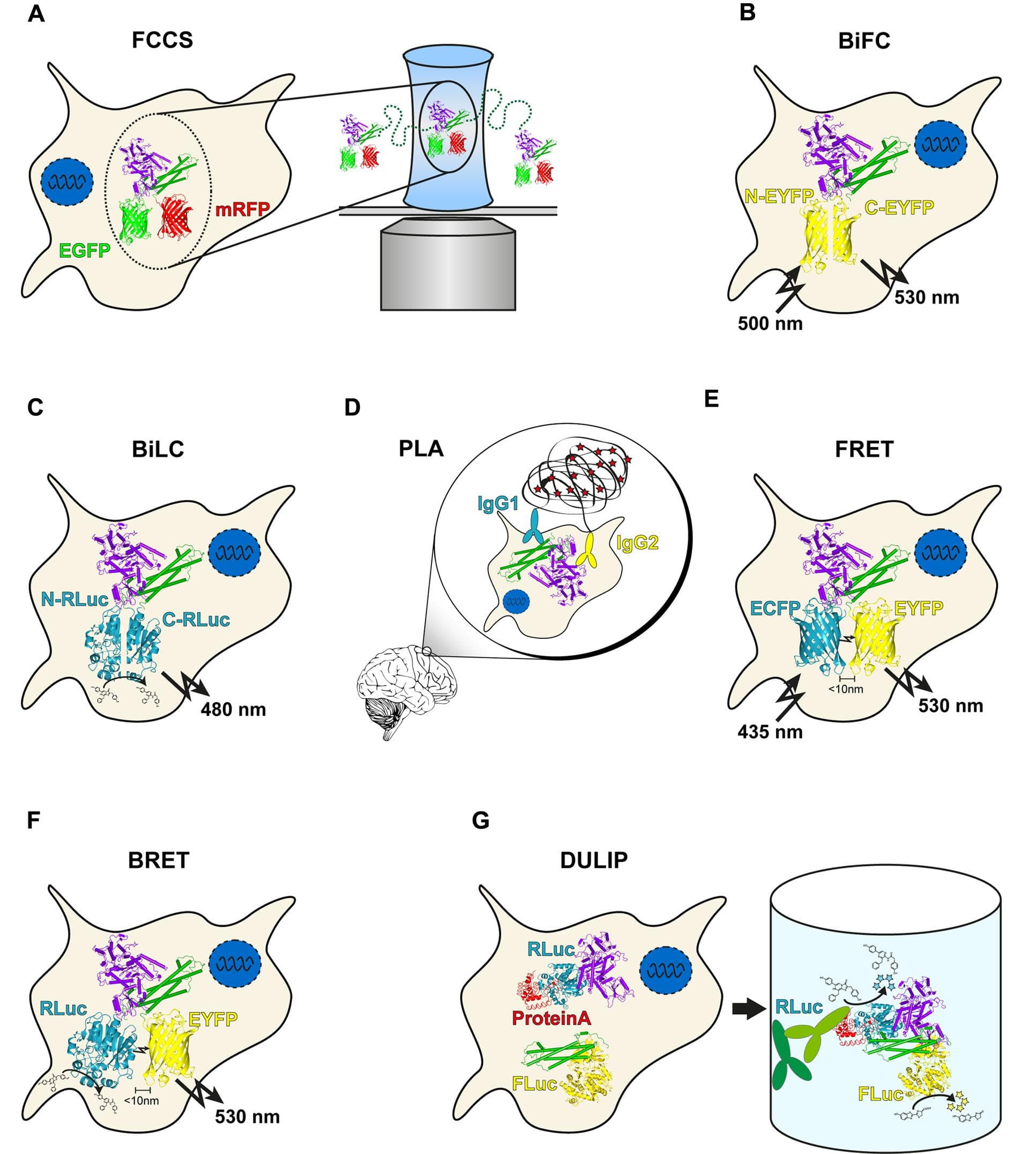
Protein–protein interactions (PPIs) play a key role in many, if not all, cellular processes. Disease is often caused by perturbation of PPIs, as recently indicated by studies of missense mutations. To understand the associations of proteins and to unravel the global picture of PPIs in the cell, different experimental detection techniques for PPIs have been established. Genetic and biochemical methods such as the yeast two-hybrid system or affinity purification-based approaches are well suited to high-throughput, proteome-wide screening and are mainly used to obtain qualitative results. However, they have been criticized for not reflecting the cellular situation or the dynamic nature of PPIs. In this review, we provide an overview of various genetic methods that go beyond qualitative detection and allow quantitative measuring of PPIs in mammalian cells, such as dual luminescence-based co-immunoprecipitation, Förster resonance energy transfer or luminescence-based mammalian interactome mapping with bait control. We discuss the strengths and weaknesses of different techniques and their potential applications in biomedical research.
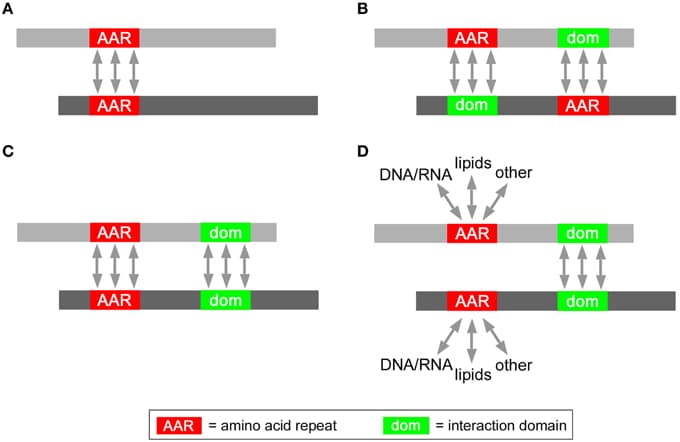
The yeast two-hybrid (Y2H) system exploits host cell genetics in order to display binary protein–protein interactions (PPIs) via defined and selectable phenotypes. Numerous improvements have been made to this method, adapting the screening principle for diverse applications, including drug discovery and the scale-up for proteome wide interaction screens in human and other organisms. Here we discuss a systematic workflow and analysis scheme for screening data generated by Y2H and related assays that includes high-throughput selection procedures, readout of comprehensive results via next-generation sequencing (NGS), and the interpretation of interaction data via quantitative statistics. The novel assays and tools will serve the broader scientific community to harness the power of NGS technology to address PPI networks in health and disease. We discuss examples of how this next-generation platform can be applied to address specific questions in diverse fields of biology and medicine.

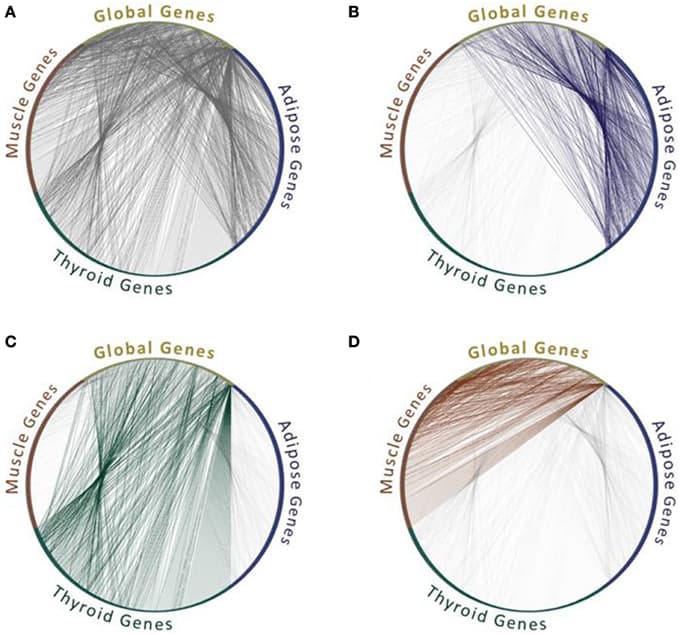
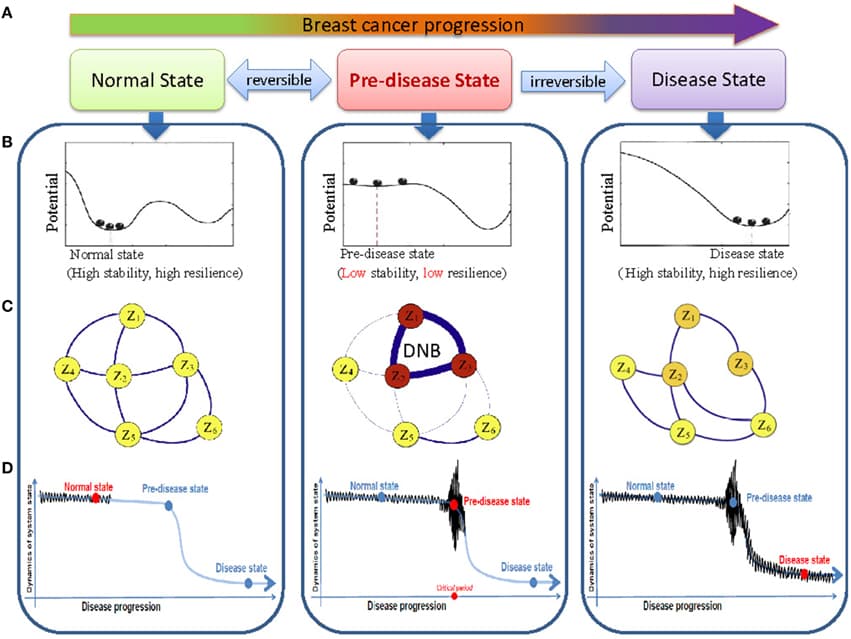
Protein interaction networks are an important framework for studying protein function, cellular processes, and genotype-to-phenotype relationships. While our view of the human interaction network is constantly expanding, less is known about networks that form in biologically important contexts such as within distinct tissues or in disease conditions. Here we review efforts to characterize these networks and to harness them to gain insights into the molecular mechanisms underlying human disease.
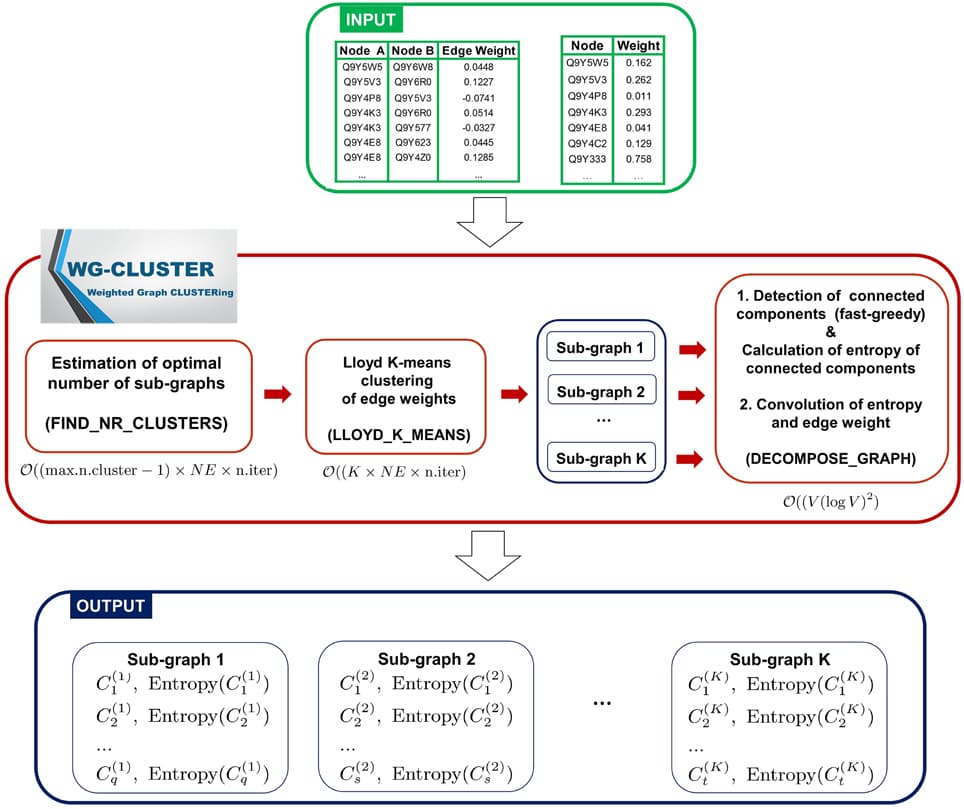
Protein–protein interaction (PPI) networks are associated with multiple types of biases partly rooted in technical limitations of the experimental techniques. Another source of bias are the different frequencies with which proteins have been studied for interaction partners. It is generally believed that proteins with a large number of interaction partners tend to be essential, evolutionarily conserved, and involved in disease. It has been repeatedly reported that proteins driving tumor formation have a higher number of PPI partners. However, it has been noticed before that the degree distribution of PPI networks is biased toward disease proteins, which tend to have been studied more often than non-disease proteins. At the same time, for many poorly characterized proteins no interactions have been reported yet. It is unclear to which extent this study bias affects the observation that cancer proteins tend to have more PPI partners. Here, we show that the degree of a protein is a function of the number of times it has been screened for interaction partners. We present a randomization-based method that controls for this bias to decide whether a group of proteins is associated with significantly more PPI partners than the proteomic background. We apply our method to cancer proteins and observe, in contrast to previous studies, no conclusive evidence for a significantly higher degree distribution associated with cancer proteins as compared to non-cancer proteins when we compare them to proteins that have been equally often studied as bait proteins. Comparing proteins from different tumor types, a more complex picture emerges in which proteins of certain cancer classes have significantly more interaction partners while others are associated with a smaller degree. For example, proteins of several hematological cancers tend to be associated with a higher number of interaction partners as expected by chance. Solid tumors, in contrast, are usually associated with a degree distribution similar to those of equally often studied random protein sets. We discuss the biological implications of these findings. Our work shows that accounting for biases in the PPI network is possible and increases the value of PPI data.
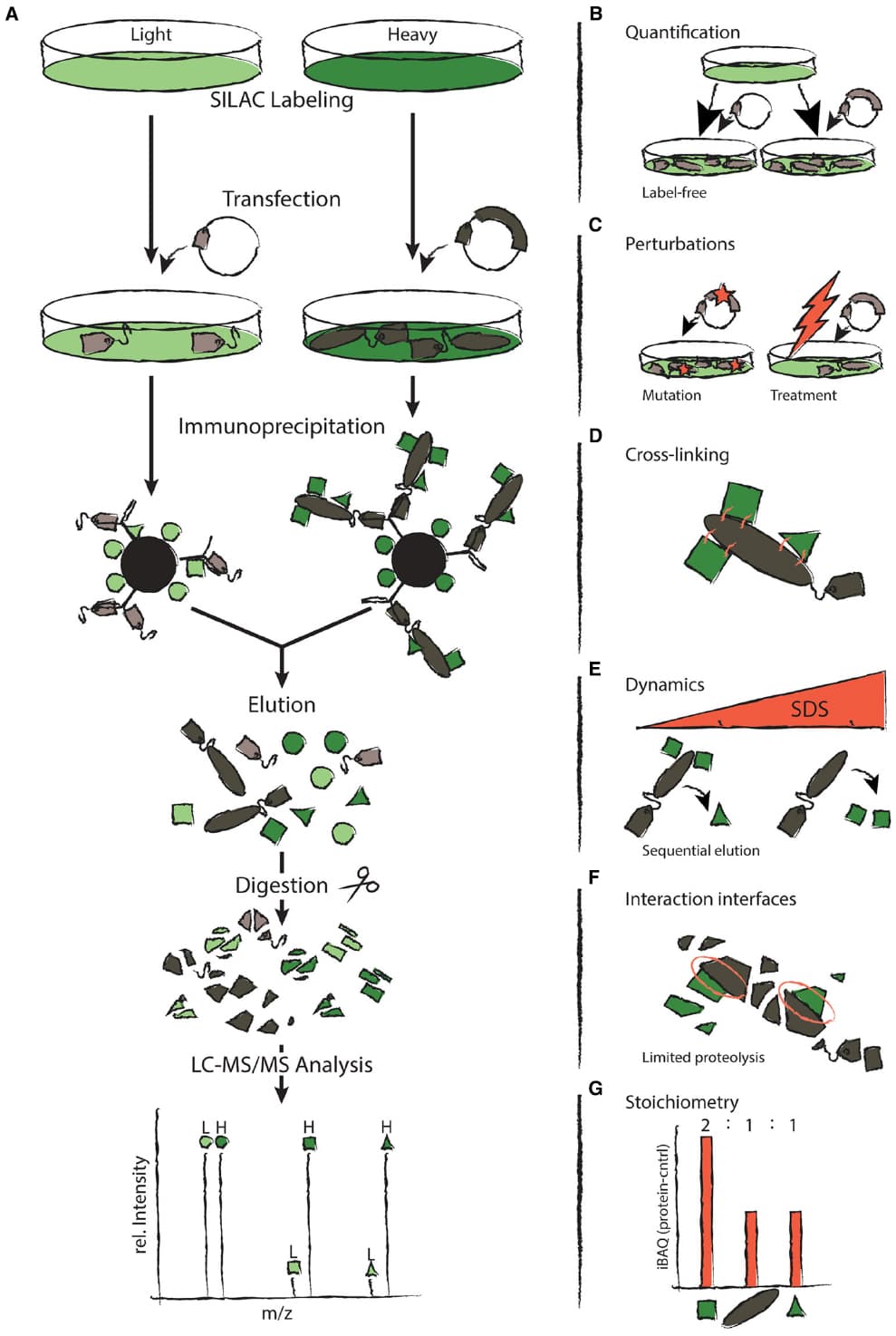
While the genomic revolution has dramatically accelerated the discovery of disease-associated genes, the functional characterization of the corresponding proteins lags behind. Most proteins fulfill their tasks in complexes with other proteins, and analysis of protein–protein interactions (PPIs) can therefore provide insights into protein function. Several methods can be used to generate large-scale protein interaction networks. However, most of these approaches are not quantitative and therefore cannot reveal how perturbations affect the network. Here, we illustrate how a clever combination of quantitative mass spectrometry with different biochemical methods provides a rich toolkit to study different aspects of PPIs including topology, subunit stoichiometry, and dynamic behavior.