Editorial
29 June 2022
Leonardo L. G. Ferreira
and
Adriano D. Andricopulo

Advances in computer hardware and the availability of high-performance supercomputing platforms and parallel computing, along with artificial intelligence methods are successfully complementing traditional approaches in medicinal chemistry. In particular, machine learning is gaining importance with the growth of the available data collections. One of the critical areas where this methodology can be successfully applied is in the development of new antibacterial agents. The latter is essential because of the high attrition rates in new drug discovery, both in industry and in academic research programs. Scientific involvement in this area is even more urgent as antibacterial drug resistance becomes a public health concern worldwide and pushes us increasingly into the post-antibiotic era. In this review, we focus on the latest machine learning approaches used in the discovery of new antibacterial agents and targets, covering both small molecules and antibacterial peptides. For the benefit of the reader, we summarize all applied machine learning approaches and available databases useful for the design of new antibacterial agents and address the current shortcomings.



Computational modeling is an essential component of modern drug discovery. One of its most important applications is to select promising drug candidates for pharmacologically relevant target proteins. Because of continuing advances in structural biology, putative binding sites for small organic molecules are being discovered in numerous proteins linked to various diseases. These valuable data offer new opportunities to build efficient computational models predicting binding molecules for target sites through the application of data mining and machine learning. In particular, deep neural networks are powerful techniques capable of learning from complex data in order to make informed drug binding predictions. In this communication, we describe Pocket2Drug, a deep graph neural network model to predict binding molecules for a given a ligand binding site. This approach first learns the conditional probability distribution of small molecules from a large dataset of pocket structures with supervised training, followed by the sampling of drug candidates from the trained model. Comprehensive benchmarking simulations show that using Pocket2Drug significantly improves the chances of finding molecules binding to target pockets compared to traditional drug selection procedures. Specifically, known binders are generated for as many as 80.5% of targets present in the testing set consisting of dissimilar data from that used to train the deep graph neural network model. Overall, Pocket2Drug is a promising computational approach to inform the discovery of novel biopharmaceuticals.


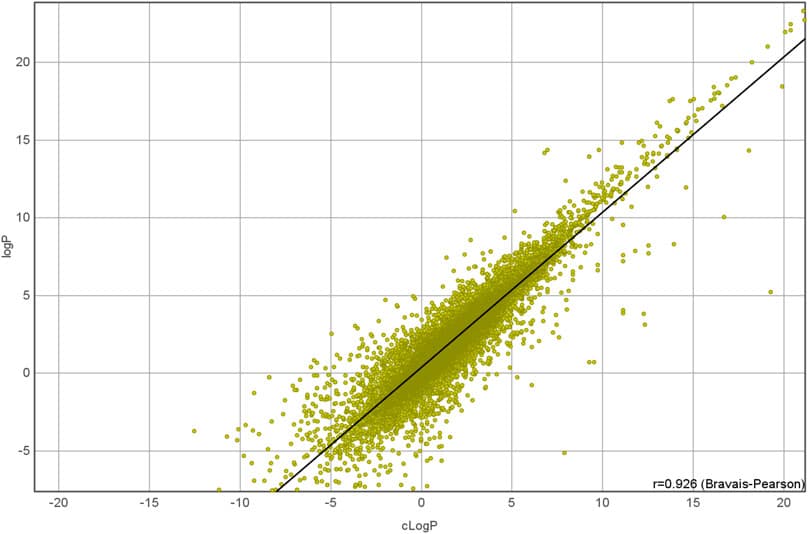

The BioChemical Library (BCL) cheminformatics toolkit is an application-based academic open-source software package designed to integrate traditional small molecule cheminformatics tools with machine learning-based quantitative structure-activity/property relationship (QSAR/QSPR) modeling. In this pedagogical article we provide a detailed introduction to core BCL cheminformatics functionality, showing how traditional tasks (e.g., computing chemical properties, estimating druglikeness) can be readily combined with machine learning. In addition, we have included multiple examples covering areas of advanced use, such as reaction-based library design. We anticipate that this manuscript will be a valuable resource for researchers in computer-aided drug discovery looking to integrate modular cheminformatics and machine learning tools into their pipelines.










