 Sunil Thomas
Sunil Thomas
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Virol., 26 November 2024
Sec. Fundamental Virology
Volume 4 - 2024 | https://doi.org/10.3389/fviro.2024.1451810
In an era marked by rapid environmental transformations, the viability of numerous species hinges crucially on their ability to navigate and adjust to shifting climatic conditions through migration. Yet, amidst this natural quest for survival, a consequential and often overlooked consequence emerges: the inadvertent transportation of a diverse array of parasites and pathogens across landscapes. This phenomenon poses a substantial and tangible threat to global health, particularly in the wake of recent outbreaks and pandemics fueled by zoonotic viruses, which leap from animals to humans. These viruses, originating in animals but capable of infecting humans, serve as poignant reminders of the intricate relationship between human and wildlife health, and the potential dangers of disregarding this delicate balance. In 2015, the borealpox virus (BRPV) [formerly Alaskapox virus (AKPV)], classified as an Orthopoxvirus, was first identified in a woman living near Fairbanks, Alaska. The BRPV causes borealpox disease. The first recorded fatality attributed to BRPV infection occurred in January 2024. Current evidence suggests that BRPV primarily targets small mammals, particularly red-backed voles and shrews, as evidenced by sampling efforts in the Fairbanks North Star Borough. However, it is likely that the virus is more widespread among Alaska’s small mammal populations, potentially leading to undiagnosed human infections. Additionally, domestic pets, such as cats and dogs, may also contribute to the transmission of the virus. To date, no instances of human-to-human transmission have been documented, but the lack of diagnostics and vaccines for BRPV remains a concerning gap. This paper aims to address this issue by reporting the structure and epitopes of the major proteins of BRPV. This data could serve as a foundation for the development of novel diagnostics and vaccines to combat BRPV, thereby mitigating its impact on both human and animal populations.
The Orthopoxvirus is a genus of viruses within the Poxviridae family, notorious for causing several significant diseases in humans and animals. Among the most notable diseases caused by Orthopoxvirus include smallpox, cowpox, camelpox, and monkeypox. These diseases can indeed be debilitating, causing severe illness and potentially fatal outcomes in some cases (1). Orthopoxviruses are identified by their sizable brick-shaped viral particles, housing a double-stranded DNA genome of roughly 200,000 base pairs (2).
The diseases caused by Orthopoxvirus are debilitating not only because of their acute symptoms but also due to their potential for long-term complications, including scarring, blindness, and secondary infections (3). The eradication of smallpox through vaccination campaigns stands as one of the greatest achievements in public health, but vigilance against other Orthopoxviruses remains important to prevent potential outbreaks and mitigate their impact on human and animal populations (4).
Instances of human infection by orthopoxviruses are being increasingly reported, partly due to the discontinuation of smallpox vaccination and the subsequent decline in population-wide immunity. In 2015, the borealpox virus (BRPV) [formerly Alaskapox virus (AKPV)] (name changed on March 27, 2024) (5), classified as an Orthopoxvirus, was first identified in a woman residing near Fairbanks, Alaska (6). The first recorded fatality attributed to BRPV infection occurred in January 2024 (7).
BRPV is genetically akin to other orthopoxviruses such as smallpox, cowpox, and monkeypox (mpox). The BRPV has a genome of 210,797 bp, containing 206 predicted open reading frames (8). Presently, available evidence indicates that the BRPV primarily targets small mammals, particularly red-backed voles and shrews, as evidenced by sampling initiatives in the Fairbanks North Star Borough. Nonetheless, it is probable that the virus is more widespread among Alaska’s small mammal populations, potentially leading to undiagnosed human infections. Domestic pets, including cats and dogs, may also contribute to the transmission of the virus. To date, no instances of human-to-human transmission have been documented [Borealpox virus (9): https://health.alaska.gov/dph/Epi/id/Pages/Borealpox.aspx)].
Typical symptoms of BRPV encompass one or more skin lesions (bumps or pustules), in addition to swollen lymph nodes and joint and/or muscle pain. Immunocompromised individuals may face an elevated risk of developing severe illness if infected (https://health.alaska.gov/dph/Epi/id/Pages/Alaskapox.aspx).
The major proteins of BRPV include surface glycoprotein, hemagglutinin, major membrane protein, EEV membrane glycoprotein, trimeric virion coat protein, core protein (8). As yet the structure of the proteins of BRPV is not determined. Using bioinformatic tools we determined the structure of the major proteins of BRPV. This paper describes the structure and epitopes of the major proteins of BRPV. Our study could lead to development of diagnostics and vaccines for borealpox virus.
The sequences of the BRPV were downloaded from the NCBI (https://www.ncbi.nlm.nih.gov/protein/) protein database. The BRPV proteins, accession numbers and functions are shown in Table 1.
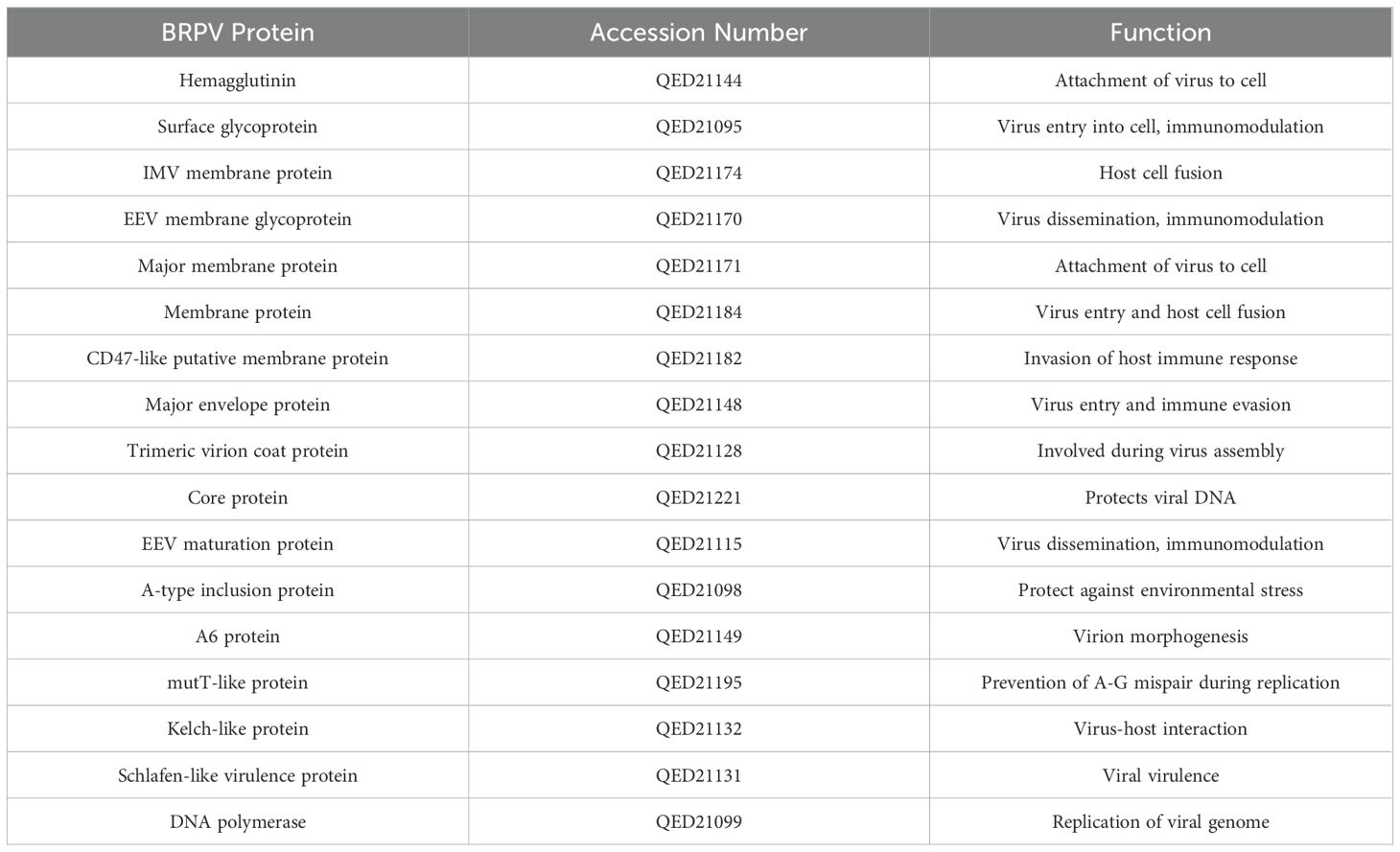
Table 1. The borealpox virus (BRPV) major proteins, accession numbers and its function.
A thorough understanding of biological systems relies on comprehending how protein complexes and networks function, which necessitates a detailed exploration of protein interactions and the overall quaternary structure. Protein plots, also known as snake diagrams, provide a 2-D representation of a protein sequence, offering insights into properties such as secondary structure (10). To generate a protein’s snake diagram model, we utilized Protter (11) ver. 1.0 (http://wlab.ethz.ch/protter), an interactive web-based application (12–15). Protter facilitates the integration and visualization of annotated and predicted protein sequence features alongside experimental proteomic evidence for peptides and post-translational modifications onto the protein’s transmembrane topology. Users can select from various annotation sources, incorporate their own proteomics data files, choose suitable peptides for targeted quantitative proteomics applications, and export high-quality illustrations (12).
For the three-dimensional homology modeling we employed the iterative threading assembly refinement [I-TASSER (16)] (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) and Phyre2 (17) (ver. 2.0) (http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index) with default settings. The protein sequences of BRPV were entered in FASTA format (13, 14).
Phobius (18) was used for the prediction of transmembrane topology and signal peptides from the amino acid sequence of proteins (https://phobius.sbc.su.se/) (15).
The B-cell epitope and T -cell epitope (MHC-II allele, HLA-DRB1) was predicted using IEDB Analysis Resource (19) (http://tools.iedb.org).
Sequence alignment is a pivotal technique utilized in bioinformatics to elucidate the relationships between different protein sequences. It involves arranging these sequences in a manner that highlights regions of similarity, thereby uncovering potential functional, structural, or evolutionary connections among them. By aligning sequences, researchers can discern conserved motifs, elucidate evolutionary pathways, and infer the functional significance of specific residues or domains within proteins. This process serves as a fundamental step in comparative genomics, aiding in the understanding of genetic variation, protein function, and evolutionary dynamics across diverse organisms (20). The BRPV proteins underwent a comparative analysis with variola proteins through the UniProt platform (21) (uniport.org/align).
The predicted solvent accessibility was determined using NetSurfP-3.0 (22) (https://services.healthtech.dtu.dk/services/NetSurfP-3.0/). NetSurfP 3.0 is an advanced tool for predicting solvent accessibility, secondary structure, structural disorder, and backbone dihedral angles for individual residues in an amino acid sequence. This latest version leverages recent advancements in pre-trained protein language models, resulting in a significant reduction in runtime—improving it by two orders of magnitude compared to its predecessor—while maintaining comparable prediction accuracy (23).
NetSurP 3.0 estimates surface accessibility, secondary structure elements, intrinsic disorder, and the phi (φ) and psi (ψ) dihedral angles. By analyzing the amino acid sequence, the server provides insights into how the protein may fold and function in a biological context. Surface accessibility predictions help identify which residues are exposed to the solvent, indicating potential interaction sites or functional areas. The secondary structure predictions categorize regions of the protein into alpha-helices, beta-strands, and coil structures, offering a clearer picture of the protein’s overall architecture (23).
Additionally, the server assesses intrinsic disorder, which is crucial for understanding regions of the protein that may lack a fixed or stable structure but still play important roles in molecular interactions and signaling pathways. The phi and psi dihedral angle predictions give detailed information about the backbone conformation, helping researchers visualize the three-dimensional arrangement of the protein (23).
The Orthopoxvirus BRPV is characterized by its substantial size, with a repertoire of 206 unique proteins. Despite its significance, the structural details and epitopes of these proteins remain elusive. In this research endeavor, we aim to elucidate the structure and epitopes of the key proteins within BRPV. By unraveling these crucial aspects, we anticipate opening new avenues for the development of innovative diagnostics and vaccines tailored specifically for BRPV. This study holds promise for advancing our understanding of BRPV pathogenesis and fostering strategies for effective disease management and control.
The BRPV structural proteins include hemagglutinin, surface glycoprotein, IMV membrane protein, major membrane protein, membrane protein, EEV membrane glycoprotein, CD47-like putative membrane protein and major envelope protein. The non-structural proteins include trimeric virion coat protein, DNA polymerase, EEV maturation protein, A-type inclusion protein, Kelch-like protein, Schlaen-like virulence protein, A6 protein, core protein and mutT-like protein.
Orthopoxviruses encode a functional hemagglutinin, a protein that facilitates the binding of viruses to host cell receptors, although it’s deemed non-essential for virus growth in cell culture (24). The hemagglutinin expressed on the surface of these viruses plays a crucial role in their interaction with host cells (25). Utilizing bioinformatics tools such as Protter and Phobius, modeling studies have revealed that the hemagglutinin of BRPV is a transmembrane protein (Figures 1A, B). The predicted protein structure of BRPV hemagglutinin (ribbon diagram) is shown in Figure 1C.
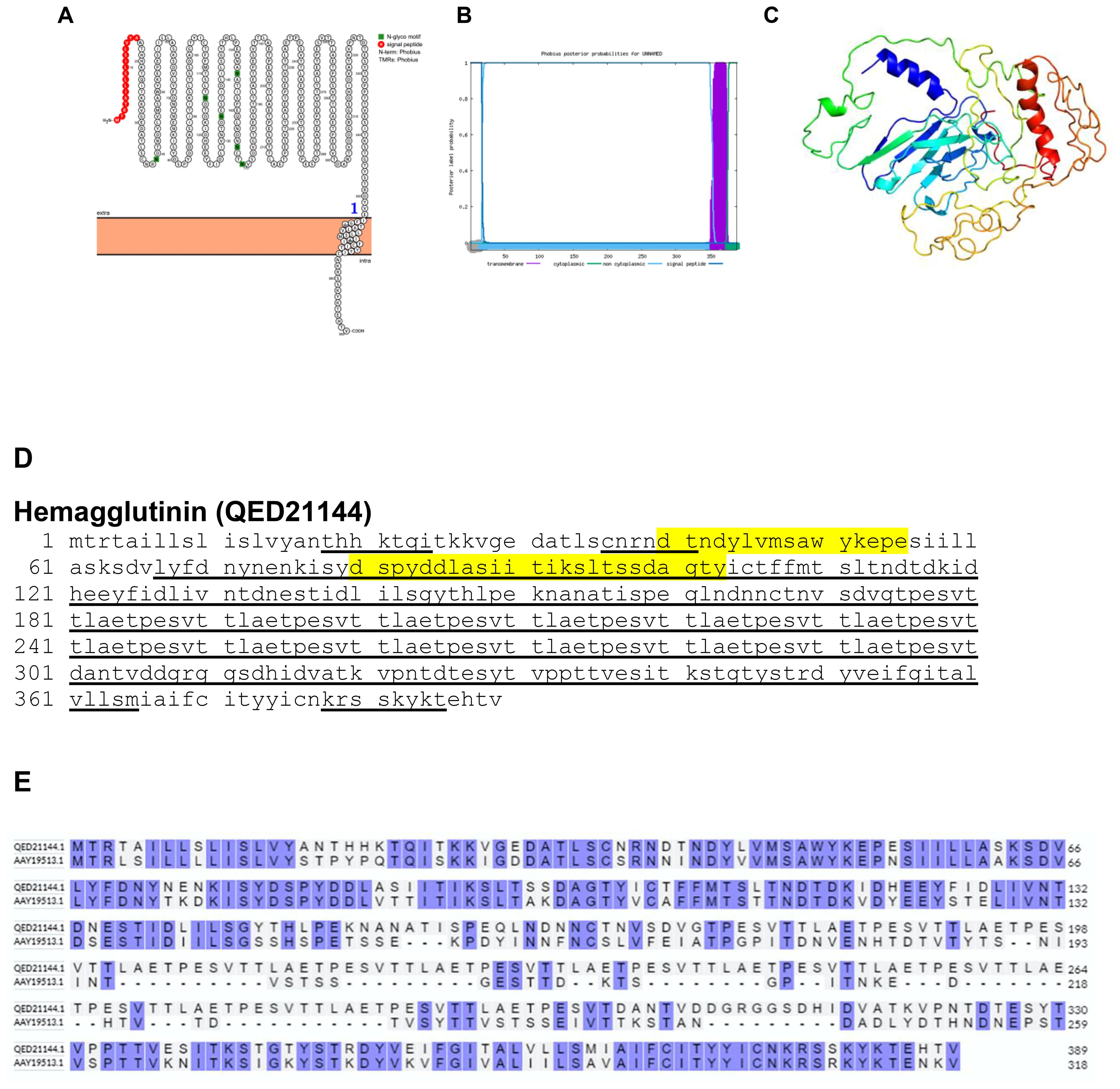
Figure 1. The topology of hemagglutinin of BRPV. (A) Protter diagram. (B) Phobius diagram. (C) Ribbon diagram determined using the software Phyre2. (D) The B-cell (underlined) and T-cell epitope (yellow shaded). (E) Alignment of BRPV hemagglutinin (QED21144) and Variola hemagglutinin (AAY19513) shows a percentage identity of 57.55%.
Furthermore, through predictive analysis using the Immune Epitope Database (IEDB), B-cell epitopes of BRPV hemagglutinin have been identified, underlined in black; the most promiscuous and high affinity binders to MHC-II allele HLA-DRB1 are shown shaded in yellow. The high affinity MHC-II binders are the most probable T-cell epitopes. Surprisingly, data unveiled that over 80% of the BRPV hemagglutinin protein comprises epitopes, implying its potential significance in immune recognition and response (Figure 1D).
A comparative analysis of the amino acid sequences between the hemagglutinins of BRPV and Variola viruses showed a sequence identity of 57.55%. This finding shed light on the evolutionary relationships and functional similarities/differences between these viral strains, providing valuable insights into their pathogenic mechanisms and potential antigenic properties (Figure 1E).
Relative solvent accessibility tends to differentiate between exposed and buried regions, often favoring the latter. This bias results in higher prediction accuracy for buried regions compared to exposed ones. Exposed regions may be more variable and subject to environmental influences, which complicates predictions. In contrast, the more stable nature of buried regions allows for more reliable assessments, leading to better accuracy in predictive models (26). The predicted solvent accessibility of BRPV hemagglutinin is shown in Supplementary Figure S1.
The surface glycoprotein is the largest structural protein of BRPV. They have a signal peptide at the N-terminal and a transmembrane domain near the C-terminal (Figures 2A, B). The predicted protein structure of BRPV surface glycoprotein (ribbon diagram) is shown in Figure 2C. The major B-cell and T-cell epitope is shown in Figure 2D. The comparative analysis of the amino acid sequences between the surface glycoprotein of BRPV and Variola viruses showed a sequence identity of 82.06% (Figure 2E). The predicted solvent accessibility of BRPV surface glycoprotein is shown in Supplementary Figure S2.

Figure 2. The topology of surface glycoprotein of BRPV. (A) Protter diagram. (B) Phobius diagram. (C) Ribbon diagram determined using I-Tasser. (D) The B-cell (underlined) and T-cell epitope (yellow shaded) of BRPV surface glycoprotein. (E) Alignment of surface glycoprotein of BRPV (QED21095) and Variola (DAA80705) shows a percentage identity of 82.06%.
The less complex of the two infectious variants of the poxvirus, known as the intracellular mature virus (IMV), demonstrates a lower efficiency in infecting cells compared to its counterpart, the extracellular enveloped virus (EEV). This discrepancy in infectivity between IMV and EEV highlights the diverse strategies employed by the virus to exploit host cells for replication and dissemination. While IMV operates predominantly within the confines of host cells, its capacity for efficient cell-to-cell spread is somewhat limited. In contrast, EEV, encased in a lipid envelope derived from host cell membranes, possesses enhanced capabilities for intercellular transmission, facilitating its dissemination throughout the host organism. This distinction underscores the intricate interplay between viral morphology and infectivity dynamics, elucidating the multifaceted nature of poxvirus pathogenesis (27). Our study demonstrated that the IMV membrane protein is a transmembrane protein as determined by Protter and Phobius (Figures 3A, B). The predicted protein structure of BRPV IMV membrane protein (ribbon diagram) is shown in Figure 3C. The epitopes of the protein were found in the extracellular domain (Figure 3D). The comparative analysis of the amino acid sequences between the IMV membrane protein of BRPV and Variola viruses showed a sequence identity of 82.51% (Figure 3E). The predicted solvent accessibility of BRPV IMV membrane protein is shown in Supplementary Figure S3.
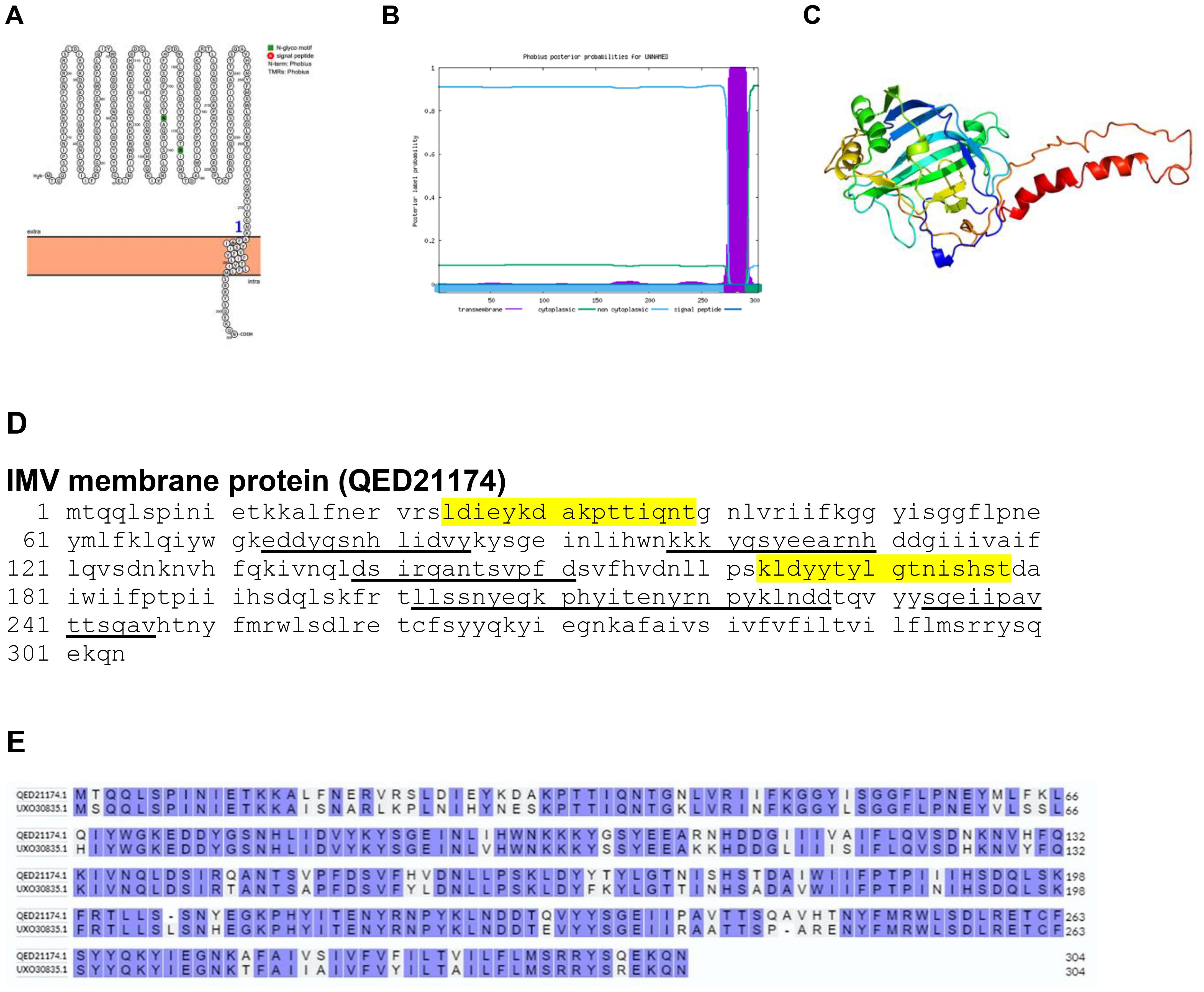
Figure 3. The topology of IMV membrane protein of BRPV. (A) Protter diagram. (B) Phobius diagram. (C) Ribbon diagram determined using Phyre2. (D) The B-cell (underlined) and T-cell epitope (yellow shaded) of BRPV IMV membrane protein. (E) Alignment of membrane protein of BRPV (QED21174) and Variola (UXO30835) IMV membrane protein shows a percentage identity of 82.51%.
Viral glycoproteins play a pivotal role as a key constituent of the outermost envelope of viruses, engaging actively in essential phases of the viral lifecycle. These glycoproteins govern the intricate relationship between viruses and their hosts, primarily through interactions with host cell receptors. Consequently, alterations such as deletion or mutation of these glycoproteins, present in the viral envelope, can significantly affect crucial processes including host cell entry, host range determination, and pathogen recognition. It is noteworthy that while extracellular enveloped viruses (EEVs) have surface proteins extensively adorned with sugar molecules (glycosylation), this feature is absent in the surface proteins of intracellular mature viruses (IMVs). This distinction underscores differing strategies employed by viruses to interact with their host environments. The absence of glycosylation in IMV surface proteins suggests potential differences in their mode of interaction with host cells compared to glycosylated counterparts in EEVs. Such distinctions may influence the efficiency and specificity of viral entry, as well as the range of host cells susceptible to infection. Moreover, variations in glycosylation patterns between EEVs and IMVs could impact how these viruses are recognized and targeted by the host immune system (28). Bioinformatics analysis demonstrated that the EEV membrane glycoprotein of BRPV is a transmembrane protein as determined by Protter and Phobius (Figures 4A, B). They have a signal peptide at the N-terminal and a transmembrane domain near the C-terminal. The predicted protein structure of BRPV EEV membrane glycoprotein (ribbon diagram) is shown in Figure 4C. The epitopes of the protein were found in the extracellular domain (Figure 4D). The comparative analysis of the amino acid sequences between the EEV membrane glycoprotein of BRPV and Variola viruses showed a sequence identity of 58.6% (Figure 4E). The predicted solvent accessibility of BRPV EEV membrane glycoprotein is shown in Supplementary Figure S4.
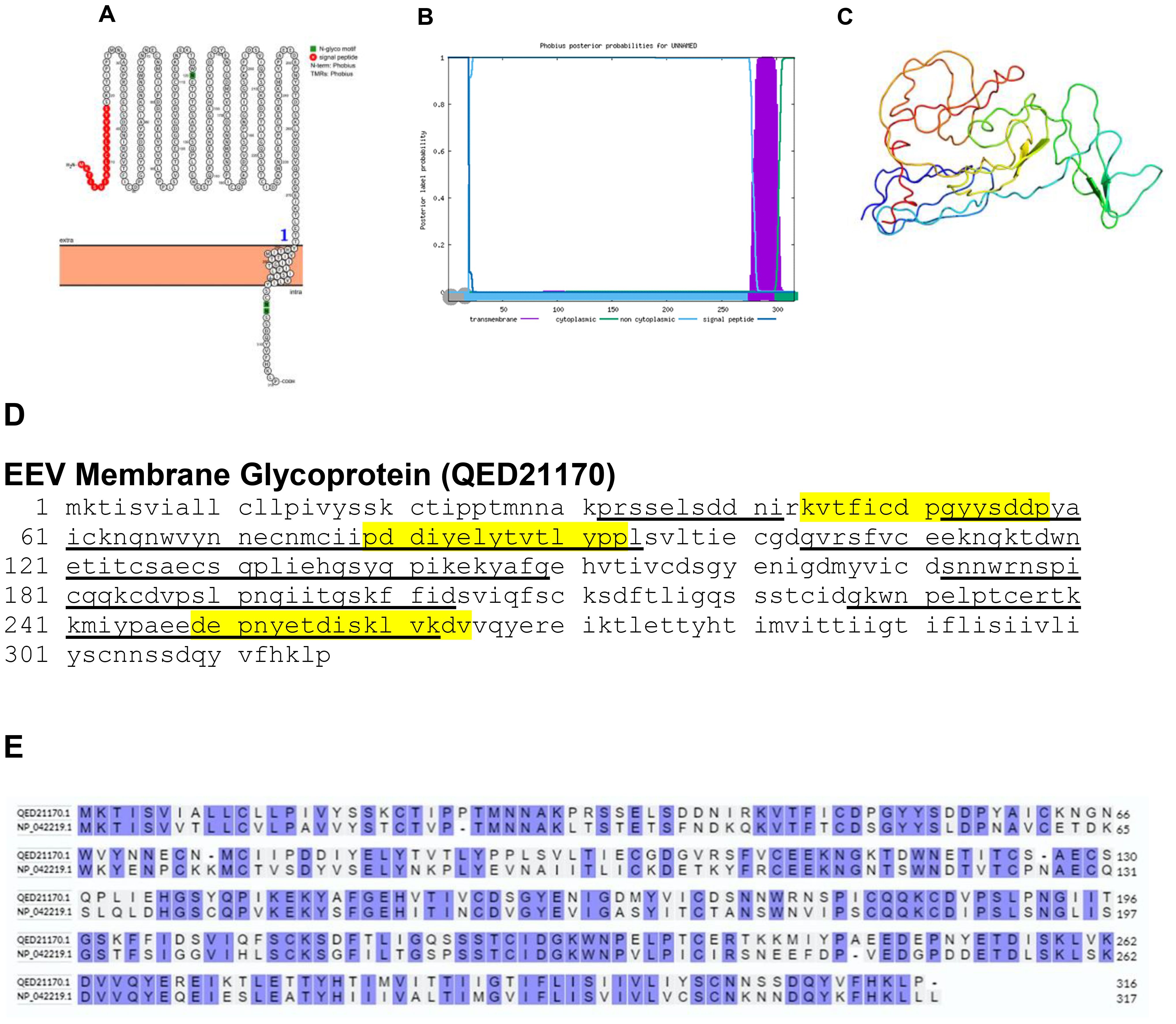
Figure 4. The topology of EEV membrane glycoprotein of BRPV. (A) Protter diagram. (B) Phobius diagram. (C) Ribbon diagram determined using Phyre2. (D) The B-cell (underlined) and T-cell epitope (yellow shaded) of BRPV EEV membrane glycoprotein. (E) Alignment of EEV membrane glycoprotein of BRPV (QED21170) and Variola (NP_042219) shows a percentage identity of 58.6%.
Bioinformatics analysis revealed that the major membrane protein of BRPV is a transmembrane protein, as confirmed by Protter and Phobius (Figures 5A, B). This protein possesses a signal peptide located at the N-terminal and a transmembrane domain in close proximity to the C-terminal region. The predicted protein structure of BRPV major membrane protein (ribbon diagram) is shown in Figure 5C. Notably, epitopes of the protein were identified within the extracellular domain (Figure 5D). Alignment of major membrane protein of BRPV and Variola shows a percentage identity of 80.13% (Figure 5E). The predicted solvent accessibility of BRPV major membrane protein is shown in Supplementary Figure S5.
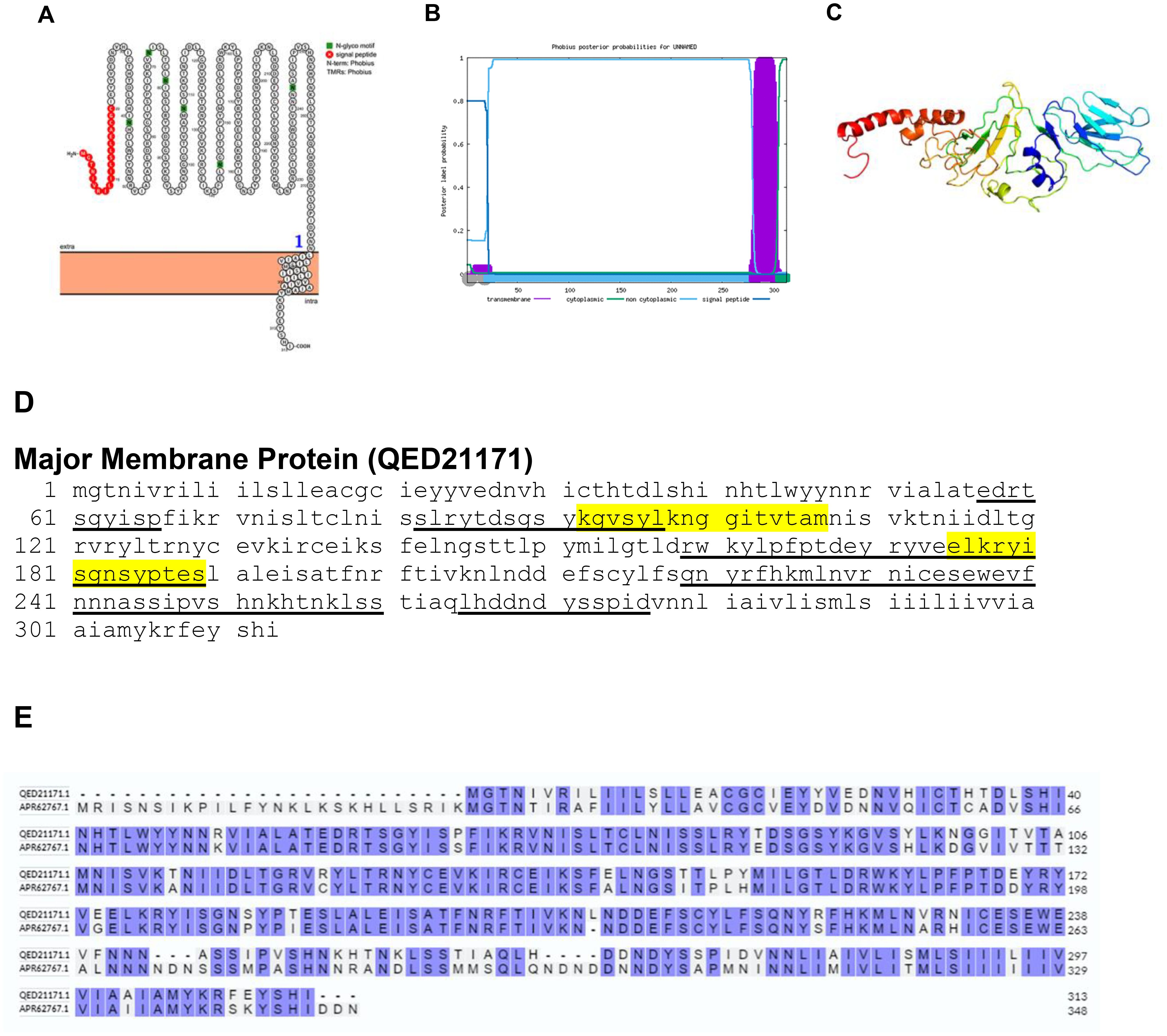
Figure 5. The topology of major membrane protein of BRPV. (A) Protter diagram. (B) Phobius diagram. (C) Ribbon diagram determined using Phyre2. (D) The B-cell (underlined) and T-cell epitope (yellow shaded) of BRPV major membrane protein. (E) Alignment of major membrane protein of BRPV (QED21171) and Variola (APR62767) shows a percentage identity of 80.13%.
Membrane proteins of the Vaccinia virus serve as promising candidates for Orthopoxvirus subunit vaccines and represent potential targets for therapeutic antibodies. Bioinformatics analysis revealed that the membrane protein of BRPV is a transmembrane protein, as confirmed by Protter and Phobius (Figures 6A, B). The predicted protein structure of BRPV membrane protein (ribbon diagram) is shown in Figure 6C. The epitopes of the membrane protein are depicted in Figure 6D. The comparative analysis of the amino acid sequences between the membrane protein of BRPV and Variola viruses showed a high sequence identity of 97.07% (Figure 6E). The predicted solvent accessibility of BRPV membrane protein is shown in Supplementary Figure S6.
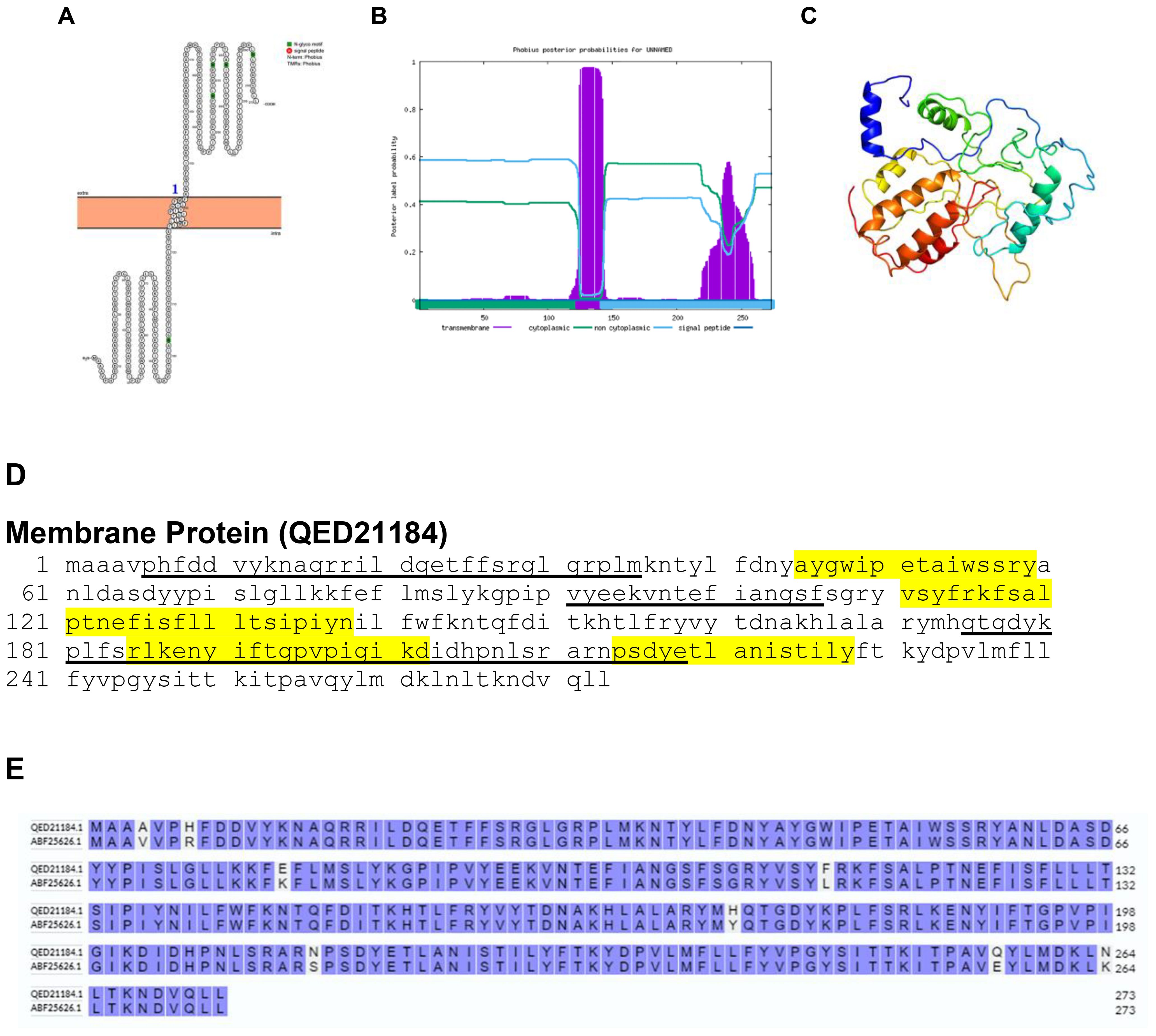
Figure 6. The topology of membrane protein of BRPV. (A) Protter diagram. (B) Phobius diagram. (C) Ribbon diagram determined using Phyre2. (D) The B-cell (underlined) and T-cell epitope (yellow shaded) of BRPV membrane protein. (E) Alignment of membrane protein of BRPV (QED21184) and Variola (ABF25626) shows a percentage identity of 97.07%.
Some of the poxviruses gene expresses a five-membrane spanning cell surface protein with significant amino acid homology to the cellular CD47 proteins. The BRPV has a five-membrane spanning cell surface CD47-like putative membrane protein (Figures 7A–C). The protein has only a single B-cell epitope (Figure 7D). The comparative analysis of the amino acid sequences between the CD47-like putative membrane protein of BRPV and Variola virus showed a sequence identity of 87.64% (Figure 7E). The predicted solvent accessibility of BRPV CD47-like putative major membrane protein is shown in Supplementary Figure S7.
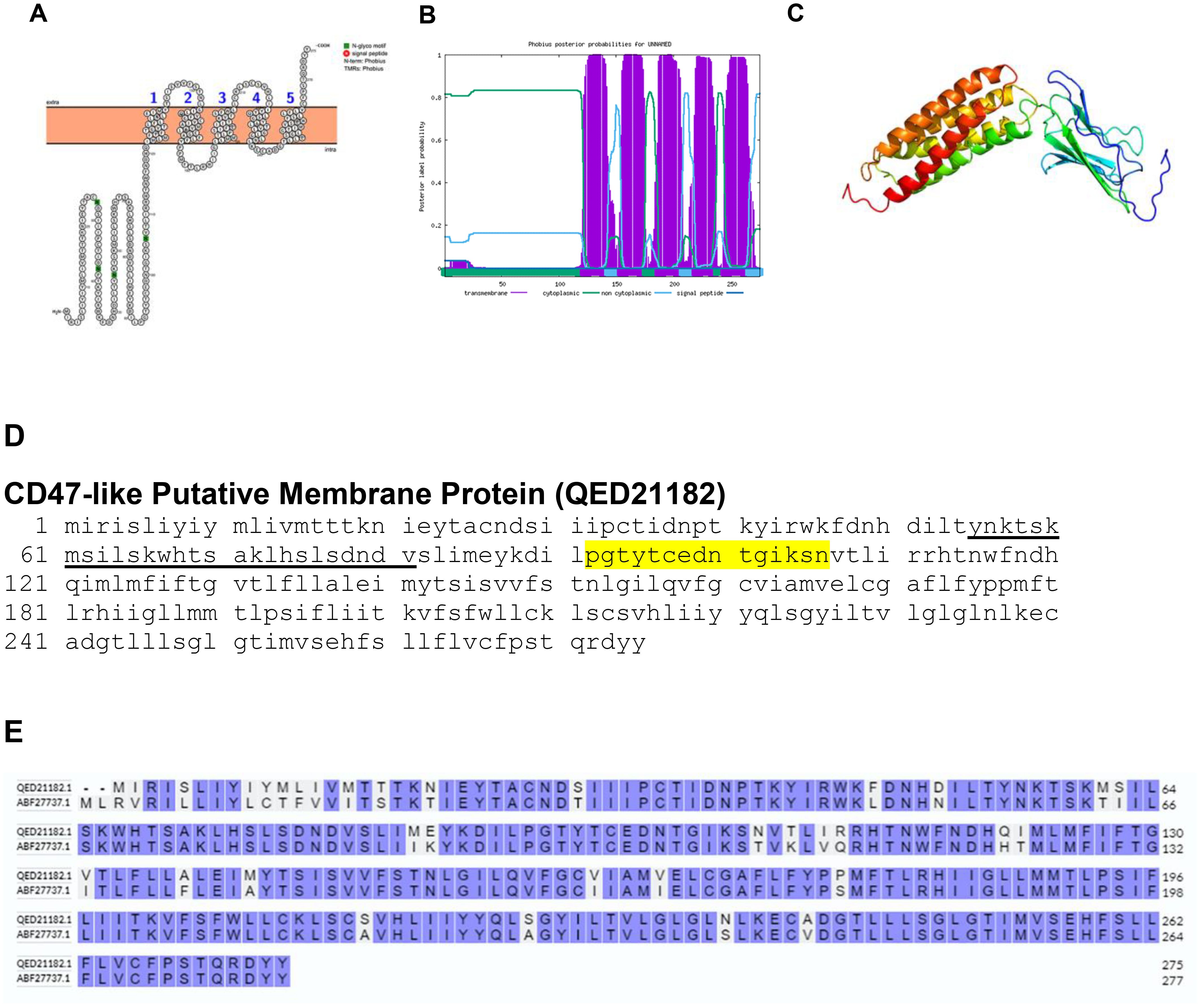
Figure 7. The topology of CD47-like putative membrane protein of BRPV. (A) Protter diagram. (B) Phobius diagram. (C) Ribbon diagram determined using Phyre2. (D) The B-cell (underlined) and T-cell epitope (yellow shaded) of BRPV CD47-like putative membrane protein. (E) Alignment of CD47-like putative membrane protein of BRPV (QED21182) and Variola (ABF27737) shows a percentage identity of 87.64%.
The major envelope protein of poxvirus is a major target of neutralizing antibodies (29). The major envelope protein of BRPV is a non-transmembrane structural protein as determined by Protter and Phobius (Figures 8A, B). The predicted protein structure of BRPV major envelope protein (ribbon diagram) is shown in Figure 8C. The B and T-cell epitopes of the major envelope protein are shown in Figure 8D. The comparative analysis of the amino acid sequences between the major envelope protein of BRPV and Variola virus showed a sequence identity of 88.98% (Figure 8E). The predicted solvent accessibility of BRPV major envelope protein is shown in Supplementary Figure S8.
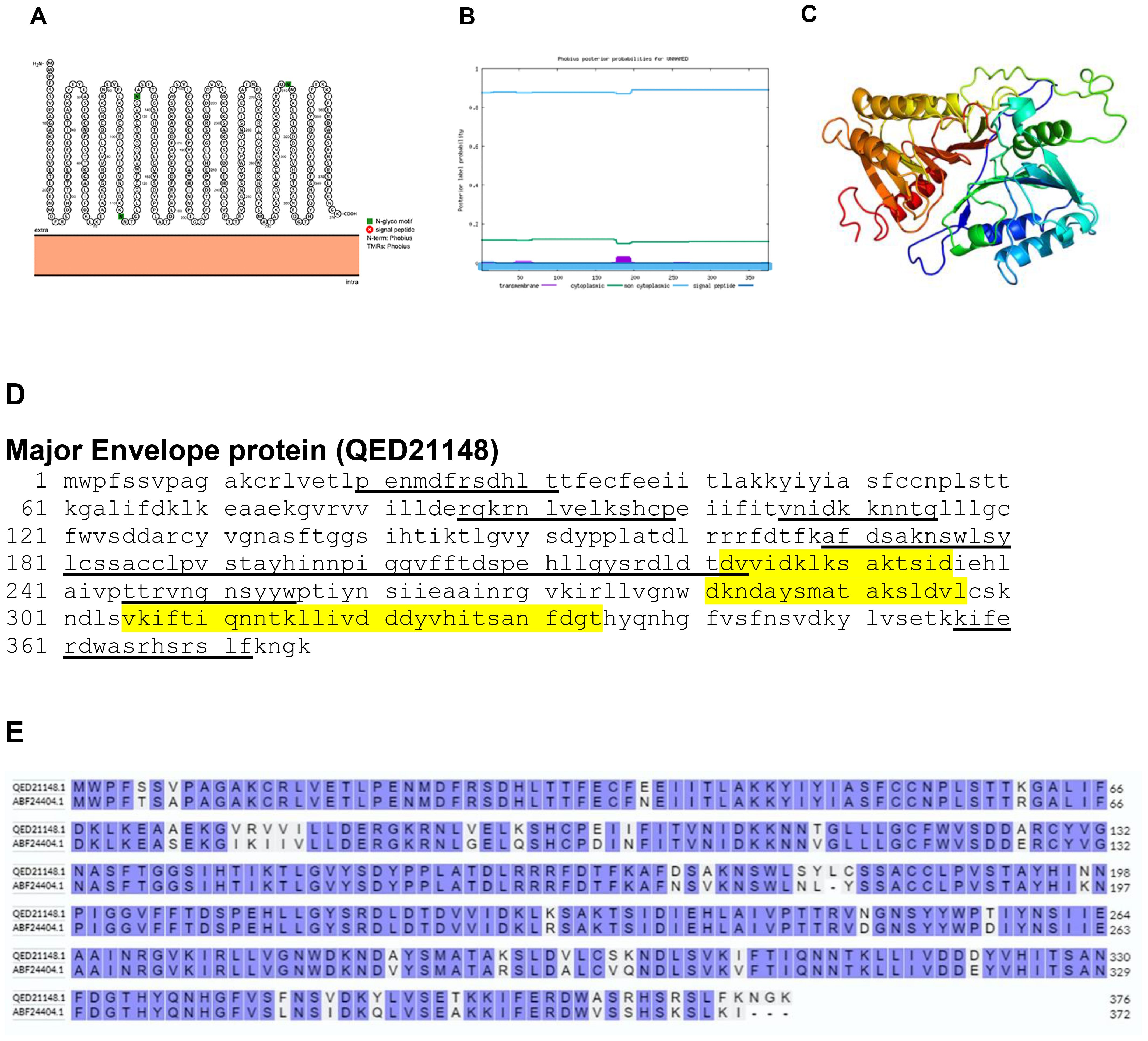
Figure 8. The topology of major envelope protein of BRPV. (A) Protter diagram. (B) Phobius diagram. (C) Ribbon diagram determined using Phyre2. (D) The B-cell (underlined) and T-cell epitope (yellow shaded) of BRPV major envelope protein. (E) Alignment of major envelope protein of BRPV (QED21148) and Variola (ABF24404) shows a percentage identity of 88.98%.
Trimeric virion coat protein is a non-structural capsid protein of BRPV. Analysis of the sequence of the trimeric virion coat protein by Protter and Phobius confirmed that the protein is a non-transmembrane protein. The comparative analysis of the amino acid sequences between the trimeric virion coat protein of BRPV and Variola virus showed a sequence identity of 94.74% (Figure 9). The predicted solvent accessibility of BRPV trimeric virion coat protein is shown in Supplementary Figure S9.
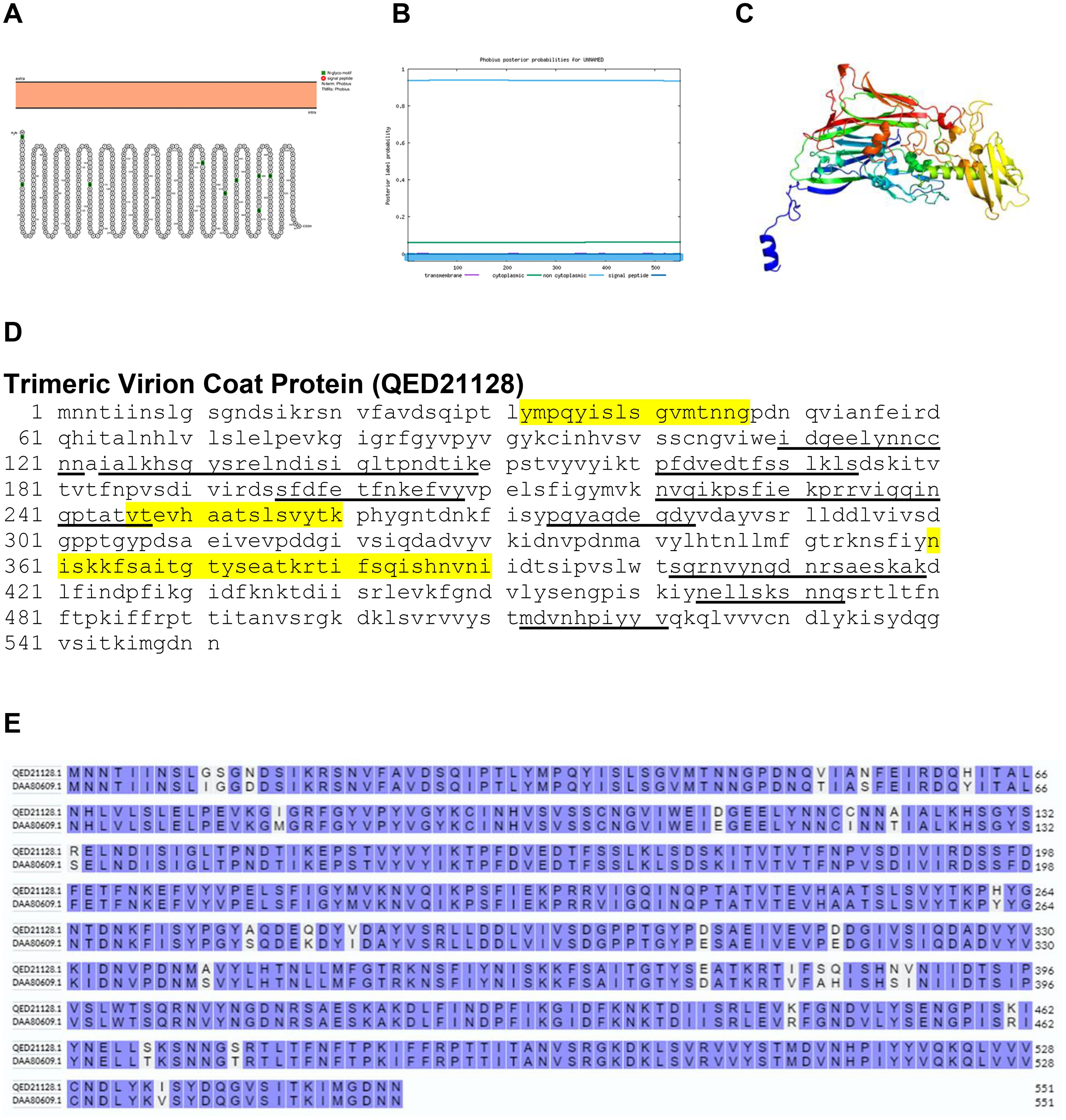
Figure 9. The topology of trimeric virion coat protein of BRPV. (A) Protter diagram. (B) Phobius diagram. (C) Ribbon diagram determined using Phyre2. (D) The B-cell (underlined) and T-cell epitope (yellow shaded) of BRPV trimeric virion coat protein. (E) Alignment of trimeric virion coat protein of BRPV (QED21128) and Variola (DAA80609) shows a percentage identity of 94.74%.
The core of the poxvirus contains a peanut shaped shell encompassing the genetic material-DNA and other proteins. Bioinformatics analysis by Protter and Phobius confirmed that the core is a non-transmembrane protein (Figures 10A, B). The predicted protein structure of BRPV core protein (ribbon diagram) is shown in Figure 10C. The B and T-cell epitopes of the core protein are shown in Figure 10D. The amino acid comparison between BRPV and Variola core protein show sequence identity of 90.48% (Figure 10E). The predicted solvent accessibility of BRPV core protein is shown in Supplementary Figure S10.
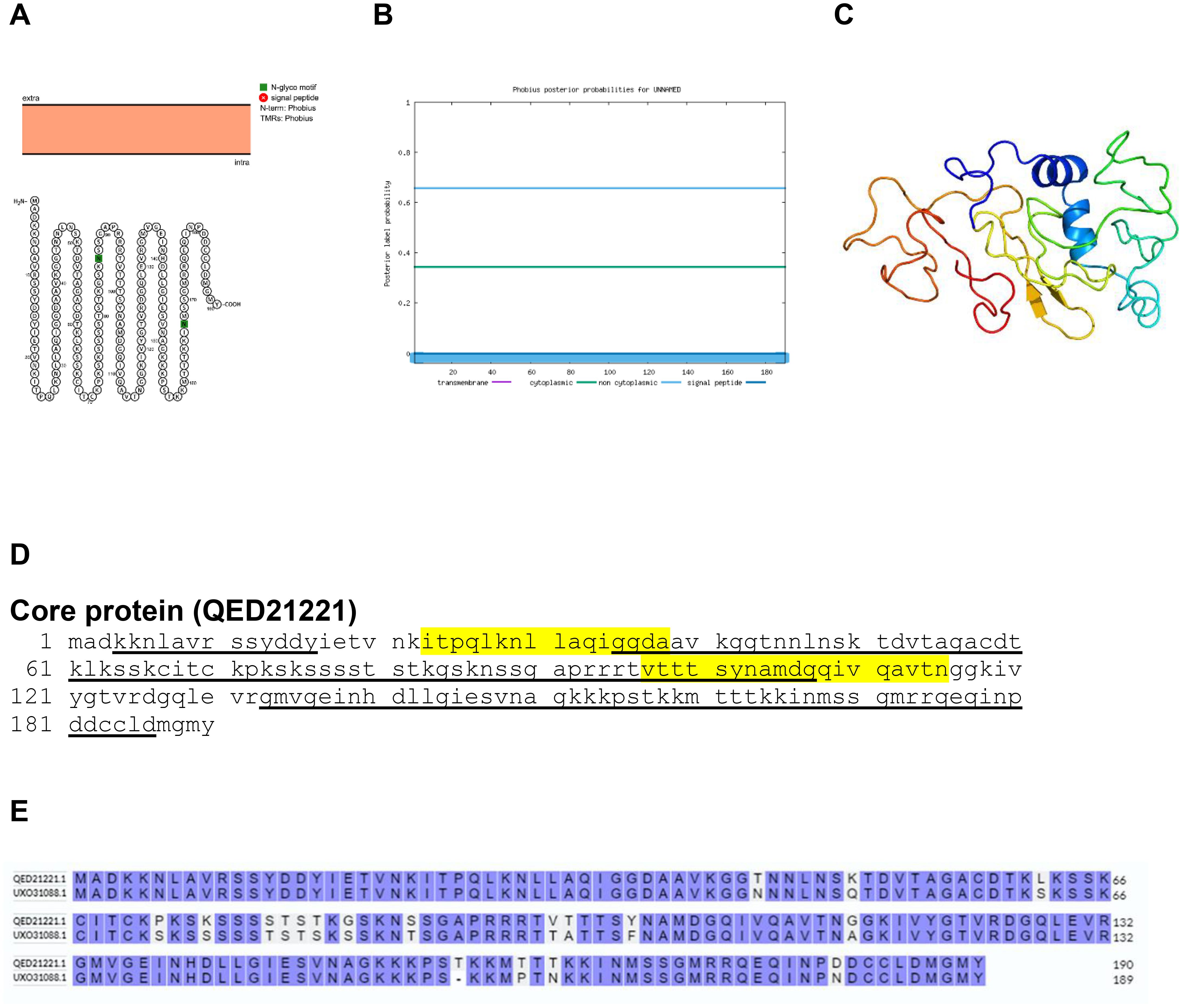
Figure 10. The topology of core protein of BRPV. (A) Protter diagram. (B) Phobius diagram. (C) Ribbon diagram determined using Phyre2. (D) The B-cell (underlined) and T-cell epitope (yellow shaded) of BRPV core protein. (E) Alignment of core protein of BRPV (QED21221) and Variola (UXO31088) shows a percentage identity of 90.48%.
The EEV maturation protein is a non-structural non-transmembrane protein as confirmed by Protter and Phobius (Figures 11A, B). The predicted protein structure of BRPV EEV maturation protein (ribbon diagram) is shown in Figure 11C. The B and T-cell epitopes of the EEV maturation protein are shown in Figure 11D. The amino acid comparison between BRPV and Variola EEV maturation protein show sequence identity of 89.73% (Figure 11E). The predicted solvent accessibility of BRPV EEV maturation protein is shown in Supplementary Figure S11.
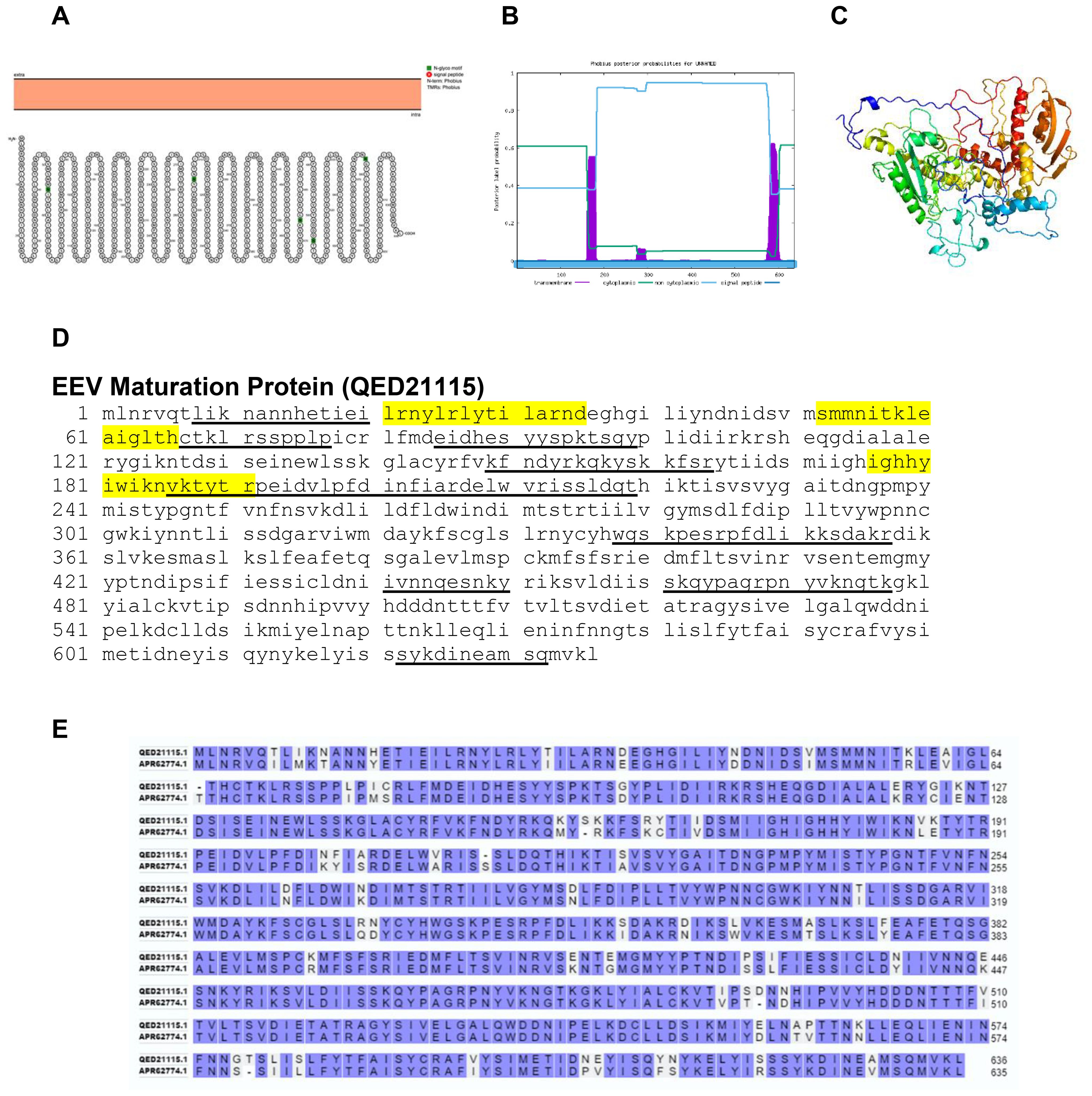
Figure 11. The topology of EEV maturation protein of BRPV. (A) Protter diagram. (B) Phobius diagram. (C) Ribbon diagram determined using Phyre2. (D) The B-cell (underlined) and T-cell epitope (yellow shaded) of BRPV EEV maturation protein. (E) Alignment of EEV maturation protein of BRPV (QED21115) and Variola (APR62774) shows a percentage identity of 89.73%.
The gene of A-type inclusion protein is used to distinguish members of orthopoxviruses by PCR (30). Protter and Phobius predicted A-type inclusion as a non-transmembrane protein (Figures 12A, B). The predicted protein structure of BRPV A-type inclusion protein (ribbon diagram) is shown in Figure 12C. The B-cell epitope covered more than 80% of the protein sequence (Figure 12D). The amino acid comparison between BRPV and Variola A-type inclusion protein show sequence identity of 73.51% (Figure 12E). The predicted solvent accessibility of BRPV A-type inclusion protein is shown in Supplementary Figure S12.
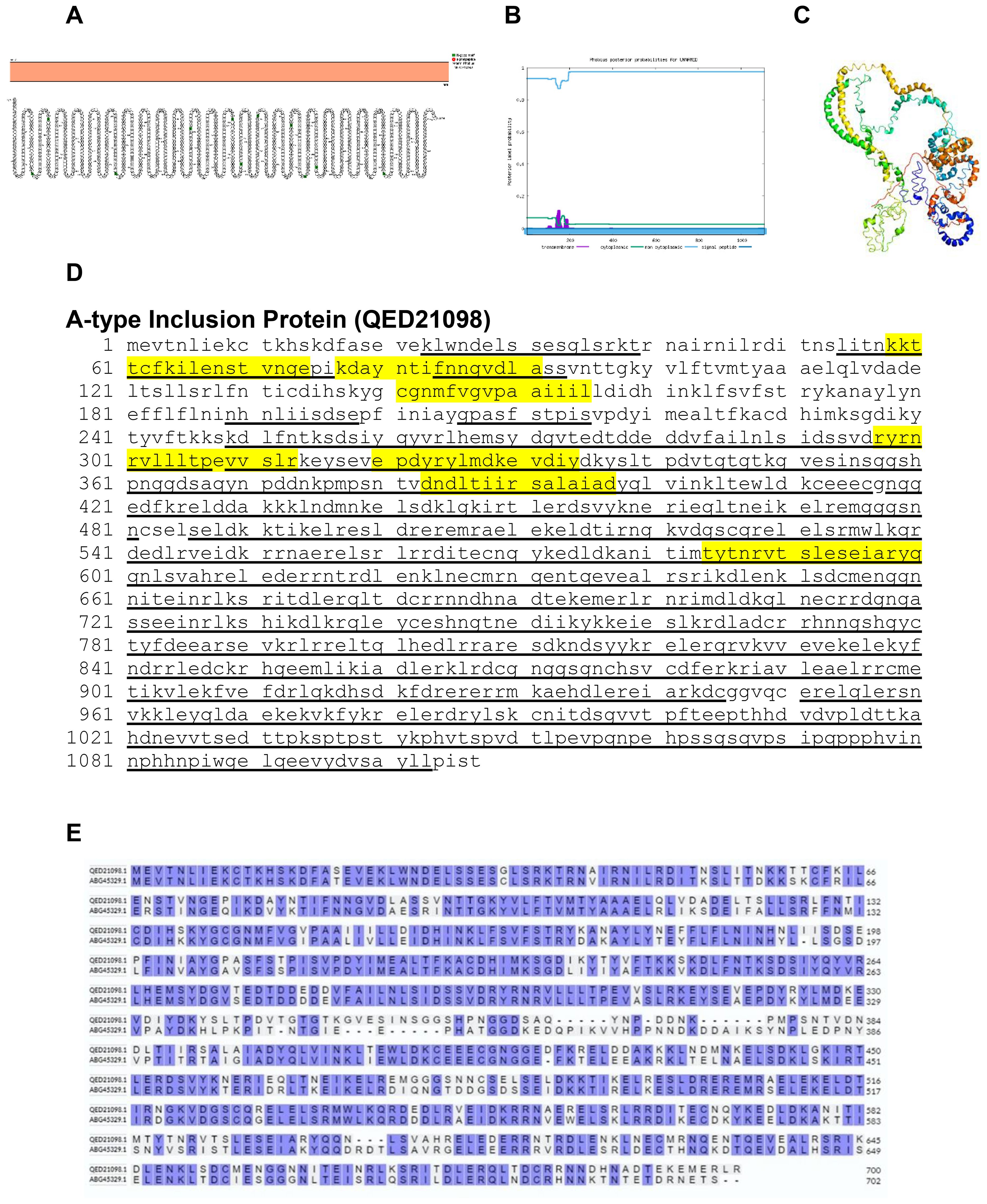
Figure 12. The topology of A-type inclusion protein of BRPV. (A) Protter diagram. (B) Phobius diagram. (C) Ribbon diagram determined using Phyre2. (D) The B-cell (underlined) and T-cell epitope (yellow shaded) of BRPV A-type inclusion protein. (E) Alignment of A-type inclusion protein of BRPV (QED21098) and Variola (ABG45329) shows a percentage identity of 73.51%.
The A6 protein of poxvirus is a key member of the viral membrane assembly proteins and is an essential factor for virion morphogenesis (31). The Protter, Phobius and Phyre2 structure of A6 protein is shown in Figures 13A, B. The predicted protein structure of BRPV A6 protein (ribbon diagram) is shown in Figure 13C. The B and T-cell epitopes of the A6 protein are shown in Figure 13D. The amino acid comparison between BRPV and Variola A6 protein show sequence identity of 93.82% (Figure 13E). The predicted solvent accessibility of BRPV A6 protein is shown in Supplementary Figure S13.
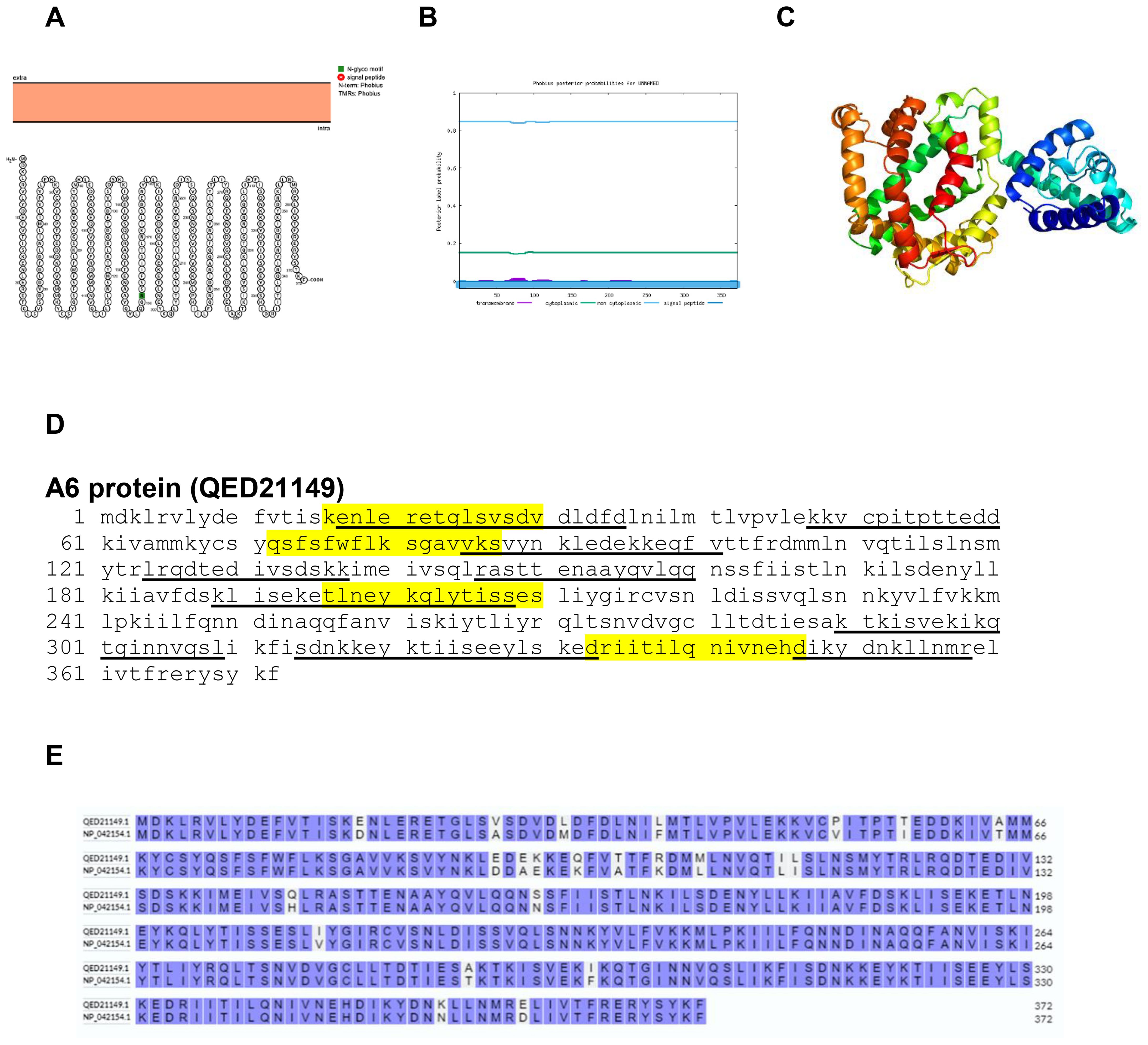
Figure 13. The topology of A6 protein of BRPV. (A) Protter diagram. (B) Phobius diagram. (C) Ribbon diagram determined using Phyre2. (D) The B-cell (underlined) and T-cell epitope (yellow shaded) of BRPV A6 protein. (E) Alignment of A6 protein of BRPV (QED21149) and Variola (NP_042154) shows a percentage identity of 93.82%.
MutT is known as a mutator gene because mutations within it specifically increase the rate of AT: GC tranversions (32). The Protter, Phobius and Phyre2 structure of mutT-like protein is shown in Figure 14. The amino acid comparison between BRPV and Variola mutt-like protein show sequence identity of 95.55% (Figure 14E). The predicted solvent accessibility of BRPV mutT-like protein is shown in Supplementary Figure S14.
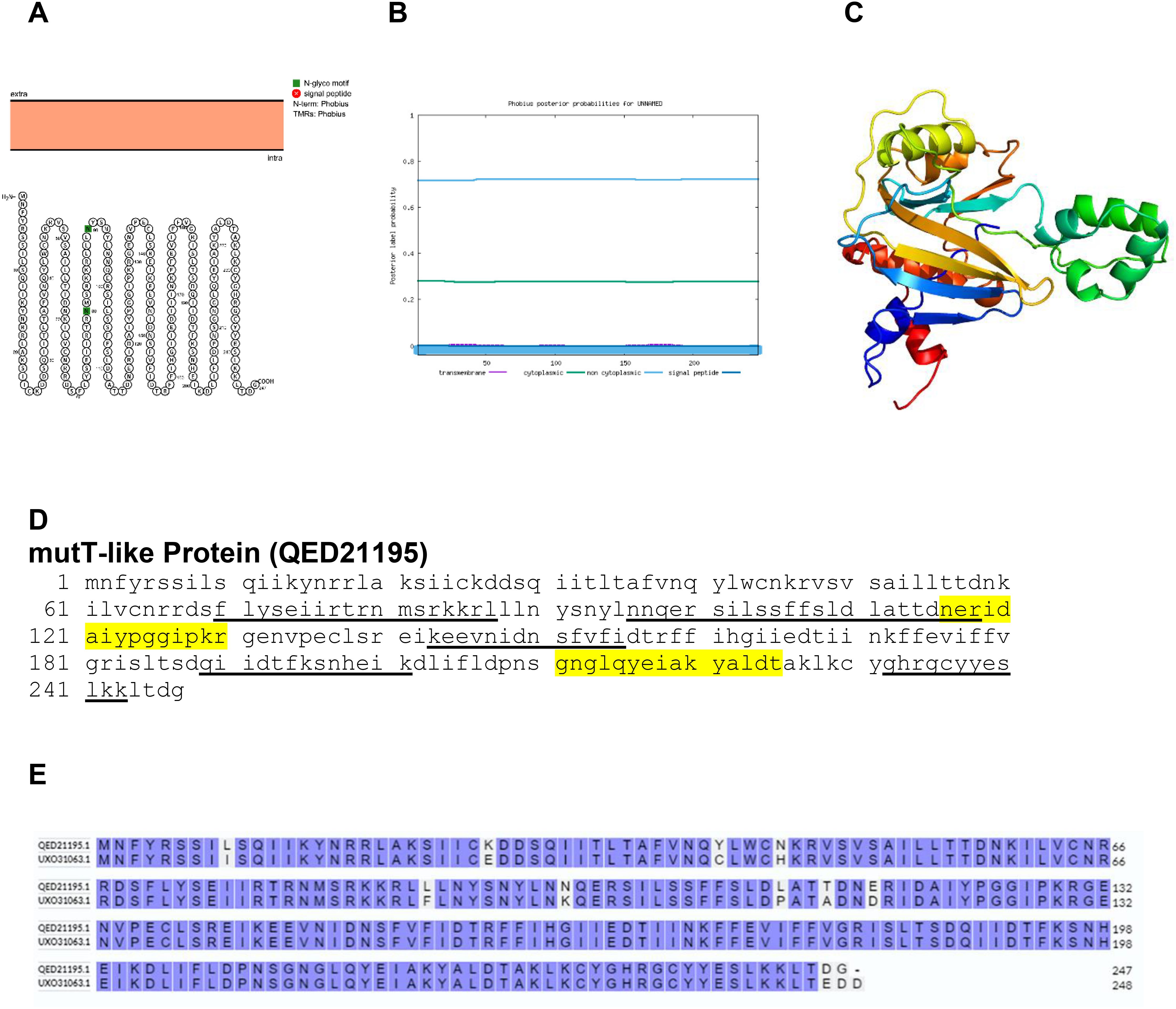
Figure 14. The topology of mutT-like protein of BRPV. (A) Protter diagram. (B) Phobius diagram. (C) Ribbon diagram determined using Phyre2. (D) The B-cell (underlined) and T-cell epitope (yellow shaded) of BRPV mutT-like protein. (E) Alignment of mutT-like protein of BRPV (QED21195) and Variola (UXO31063) shows a percentage identity of 95.55%.
Kelch-like protein genes typically exist in multiple, divergent, and occasionally fragmented forms, situated in variable and nonessential regions near genomic termini. This positioning suggests their involvement in virus-host interactions. Despite maintaining domain structure, there is generally low amino acid identity between poxviral and cellular kelch-like proteins. Studies exploring the function of poxviral kelch-like genes in virus replication and pathogenesis have primarily focused on orthopoxviruses. Deletion of single or multiple kelch-like genes in these viruses has been shown to impact factors such as lesion size, inflammatory cell infiltration, virulence in mouse models, as well as vascularization and virus yield in chorioallantoic membrane infection models (33). These findings suggest a potential role for kelch-like genes in poxvirus virulence and host range, possibly through the modulation of inflammatory responses (33). The Protter, Phobius and Phyre2 structure of kelch-like protein is shown in Figure 15. The amino acid comparison between BRPV and Variola kelch-like protein show sequence identity of only 16.92% (Figure 15E). The predicted solvent accessibility of BRPV kelch-like protein is shown in Supplementary Figure S15.
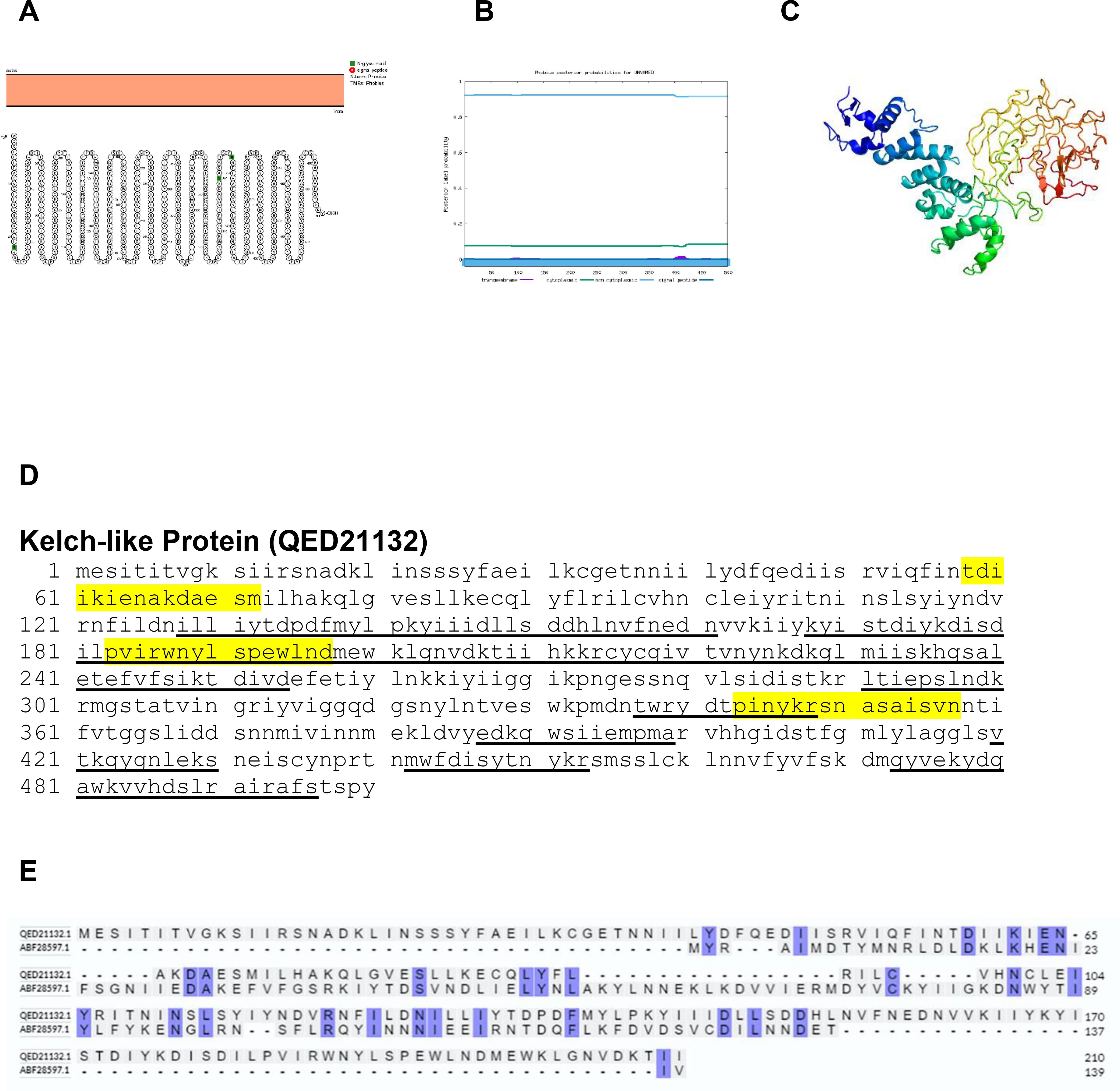
Figure 15. The topology of kelch-like protein of BRPV. (A) Protter diagram. (B) Phobius diagram. (C) Ribbon diagram determined using Phyre2. (D) The B-cell (underlined) and T-cell epitope (yellow shaded) of BRPV kelch-like protein. (E) Alignment of kelch-like protein of BRPV (QED21132) and Variola (ABF28597) shows a percentage identity of 16.92%.
Schlafen-like virulence protein of poxviruses is involved in virulence of the virus (34). The Protter, Phobius and Phyre2 structure of schlafen-like virulence protein is shown in Figures 16A, B. The predicted protein structure of BRPV Schlafen-like virulence protein (ribbon diagram) is shown in Figure 16C. The B and T-cell epitopes of the Schlafen-like virulence protein are shown in Figure 16D. Gubser et al. (34) reported that the N-terminus of the protein was more active compared to the other regions. The predicted solvent accessibility of BRPV schlafen-like virulence protein is shown in Supplementary Figure S16.
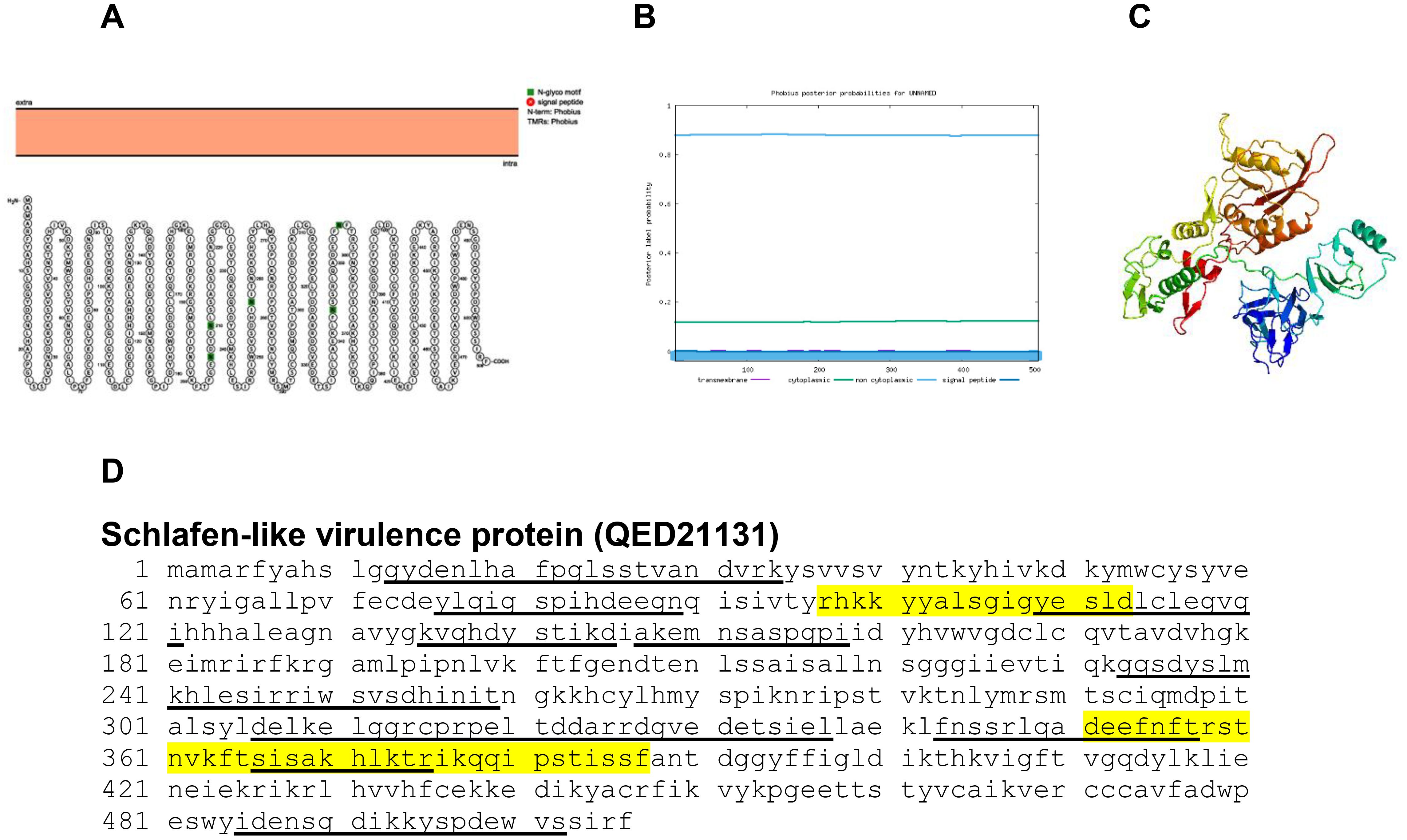
Figure 16. The topology of schlafen-like virulence protein (QED21131) of BRPV. (A) Protter diagram. (B) Phobius diagram. (C) Ribbon diagram determined using Phyre2. (D) The B-cell (underlined) and T-cell epitope (yellow shaded) of BRPV schlafen-like virulence protein.
Poxvirus DNA synthesis can usually be detected within 2 hours after infection and occurs in the cytoplasm. Proteins crucial for DNA synthesis in poxviruses comprise a 117-kDa polymerase, a helicase–primase complex, an uracil DNA glycosylase, a processivity factor, a single-stranded DNA-binding protein, a protein kinase, and a DNA ligase (35). The DNA polymerase of the poxvirus, coded by the E9L gene, falls under the B family of replicative polymerases. It possesses essential polymerase and 3′-5′ exonuclease activities crucial for sustaining virus replication (36). The Protter, Phobius and Phyre2 structure of kelch-like protein is shown in Figures 17A, B. The predicted protein structure of BRPV DNA polymerase (ribbon diagram) is shown in Figure 17C. The B and T-cell epitopes of the DNA polymerase are shown in Figure 17D. The amino acid comparison between BRPV and Variola DNA polymerase show sequence identity of 96.42% (Figure 17E). The predicted solvent accessibility of BRPV Dna polymerase is shown in Supplementary Figure S17.

Figure 17. The topology of DNA polymerase of BRPV. (A) Protter diagram. (B) Phobius diagram. (C) Ribbon diagram determined using Phyre2. (D) The B-cell (underlined) and T-cell epitope (yellow shaded) of BRPV DNA polymerase. (E) Alignment of DNA polymerase of BRPV (QED21099) and Variola (NP_042094) shows a percentage identity of 96.42%.
Climate change plays a significant role in the emergence and spread of zoonotic diseases, which are infections that can be transmitted from animals to humans. As global temperatures rise, patterns of precipitation shift, and extreme weather events become more frequent, the dynamics of infectious diseases are altered in complex ways. The complex interplay between environmental factors, human activities, and ecological dynamics creates conditions that facilitate the transmission of these diseases. Climate change-induced disruptions to ecosystems can result in changes to species composition, biodiversity, and food web dynamics. These alterations can influence the prevalence and transmission of zoonotic pathogens by affecting the abundance and behavior of reservoir hosts, such as rodents, bats, and certain bird species, as well as their interactions with humans and domestic animals (37).
Recent research indicates that the number of virus species capable of infecting humans exceeds 10,000, with the majority currently remaining dormant within wild mammal populations (38). However, as climate patterns and land utilization evolve, these conditions are increasingly fostering opportunities for viral exchange among wildlife species that were once isolated by geography. This phenomenon is significantly escalating the potential for zoonotic spillover events, establishing a direct mechanistic connection between global environmental transformations and the emergence of infectious diseases (39).
The ongoing alterations in climate and land use patterns are anticipated to break down natural barriers that historically prevented interactions between different species of wildlife. As habitats shift and species migrate in response to changing environmental conditions, formerly distinct viral populations are encountering new hosts and environments, setting the stage for cross-species transmission. This environmental reshaping not only enhances the likelihood of viruses crossing species barriers but also increases the opportunities for these viruses to adapt and potentially become infectious to humans (40, 41). The implications of these environmental changes extend beyond ecological dynamics to profound public health concerns. The interconnectedness of ecosystems underscores the urgent need for comprehensive surveillance and management strategies to monitor emerging infectious diseases. By understanding the ecological drivers of zoonotic spillover, researchers and policymakers can better anticipate and mitigate the risks posed by novel viral infections.
Poxviruses represent a diverse family of large, pleomorphic, and double-stranded DNA viruses capable of infecting a broad spectrum of hosts, spanning from vertebrates, including humans, to arthropods. Variola virus, notorious as the causative agent behind the devastating smallpox outbreaks, stands prominently within the Poxviridae family. Another prominent member is Vaccinia virus (VACV), celebrated for its pivotal role as the prototypical and extensively studied poxvirus. Remarkably, VACV also served as an attenuated vaccination strain instrumental in the successful eradication of smallpox during the late 1970s (42).
The resurgence of monkeypox virus, leading to localized outbreaks of monkeypox (mpox) globally, has reignited the urgency for a deeper comprehension of the intricate lifecycle of poxviruses. Mpox transmission rates are influenced positively by environmental factors such as temperature, relative humidity, and surface pressure. These variables play crucial roles in shaping the dynamics of the disease, affecting both its prevalence and spread (43). Temperature is a key determinant as it influences the survival and replication rates of the virus. Warmer temperatures generally create more favorable conditions for viral activity, potentially increasing transmission rates among susceptible populations. Similarly, relative humidity affects viral stability in the environment and can facilitate the persistence of infectious particles. Higher humidity levels may prolong the viability of the virus on surfaces or in the air, thereby enhancing the chances of transmission. Understanding the interplay between these environmental factors is crucial for predicting and managing outbreaks of mpox. Climate variability and changes in land use can further complicate these dynamics by altering the habitats and behaviors of reservoir hosts or vectors involved in the transmission cycle (43). The emerging threats underscore the critical necessity for comprehensive research aimed at elucidating the mechanisms underlying poxvirus pathogenesis, transmission dynamics, and host interactions. Such endeavors are pivotal in the development of effective preventive and therapeutic strategies to combat emerging poxvirus infections and mitigate potential public health crises (42).
The emergence of the Borealpox virus (BRPV) marked a significant event in 2015 when it was initially identified in Alaska. Since its discovery, the virus has continued to pose a notable threat, with multiple incidents of circulation reported within the state (6). Tragically, the gravity of its impact was underscored by the first reported death attributed to BRPV in January 2024 (7). BRPV is believed to primarily infect voles, shrews, and other rodent species indigenous to Alaska, raising concerns about its potential implications for both wildlife populations and human health. As a zoonotic pathogen, the virus has the capacity to spill over from its natural reservoir hosts to humans, posing risks of transmission and potential outbreaks.
The persistence and spread of BRPV underscore the urgent need for enhanced surveillance, research, and public health interventions to mitigate its impact. Understanding the dynamics of its transmission, host range, and potential for human infection is crucial for developing effective prevention and control strategies. Furthermore, as climate change and human activities continue to influence ecological landscapes, the risk of emerging infectious diseases like BRPV amplifies, emphasizing the importance of proactive measures to safeguard both wildlife and human populations from such threats.
Most viruses rely on one or two proteins to facilitate cell binding, membrane fusion, and entry. However, poxviruses are distinct due to their utilization of an unusually high number of proteins, which could be linked to their capacity to infect a diverse array of cells. Among poxviruses, the vaccinia virus exists in two primary infectious forms: the mature virion (MV), possessing a single membrane, and the extracellular enveloped virion (EV), characterized by an additional outer membrane that is dismantled prior to fusion (44). In this study we include proteins from different growth phases of BRPV.
Investigating the structure of proteins and pinpointing specific epitopes represents a pivotal step toward the development of effective vaccines and diagnostics (45). In our study, we conducted a comprehensive analysis to elucidate the structure and epitopes of BRPV’s key proteins. Additionally, we performed a comparative alignment of the amino acid residues of BRPV proteins with those of Variola virus to discern similarities and variations between the two. This multifaceted approach not only enhances our understanding of BRPV’s molecular architecture but also sheds light on potential targets for vaccine design and diagnostic development. By uncovering similarities and differences between BRPV and Variola, we gain valuable insights into the evolutionary relationship and antigenic profiles of these viruses, further informing the strategic design of interventions to combat BRPV and related pathogens.
Hemagglutinin and A-type inclusion protein exhibit a striking feature in their protein structure: a substantial 80% of their composition is comprised of epitopes. These epitopes play a pivotal role in various biological processes, particularly in the context of immune responses and molecular recognition. Epitopes, often referred to as antigenic determinants, are regions on the surface of proteins that are recognized by the immune system, eliciting specific antibody responses. This abundance of epitopes within the protein structure underscores their significance in mediating interactions with other molecules, such as antibodies, receptors, or other proteins, ultimately influencing the protein’s function and biological activity. This characteristic highlights the potential importance of hemagglutinin and A-type inclusion protein in immune recognition and molecular signaling pathways, warranting further investigation into their structural and functional properties. We name those proteins that has >80% of their structure composed of epitopes as octogintope protein (Latin, octoginta, eighty). This classification not only underscores the significance of epitope-rich proteins in diagnostic and vaccine design but also provides a practical framework for prioritizing targets in the development pipeline.
Our study represents a significant advancement in the understanding of BRPV biology and the development of countermeasures against this emerging pathogen. By unraveling the intricacies of protein structure, epitope identification, and comparative genomics, we pave the way for the design of more efficacious vaccines and diagnostics, ultimately bolstering our preparedness against future viral threats.
1. Limitation of author to access I-TASSER for quality score of the predicted protein structures like C-score, TM-score and RMSD.
2. Limitation in accessing data for Normalized B-Factor profile of the predicted proteins.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
ST: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The author acknowledges Abraham Thomas Foundation for providing the resources for this work.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2024.1451810/full#supplementary-material
1. Shchelkunova GA, Shchelkunov SN. Smallpox, monkeypox and other human orthopoxvirus infections. Viruses. (2022) 15:103. doi: 10.3390/v15010103
2. Massung RF, Liu LI, Qi J, Knight JC, Yuran TE, Kerlavage AR, et al. Analysis of the complete genome of smallpox variola major virus strain Bangladesh-1975. Virology. (1994) 201:215–40. doi: 10.1006/viro.1994.1288
3. Billioux BJ, Mbaya OT, Sejvar J, Nath A. Neurologic complications of smallpox and monkeypox: A review. JAMA Neurol. (2022) 79:1180–6. doi: 10.1001/jamaneurol.2022.3491
4. Strassburg MA. The global eradication of smallpox. Am J Infect Control. (1982) 10:53–9. doi: 10.1016/0196-6553(82)90003-7
5. Douglass N. Borealpox (Alaskapox) virus: will there be more emerging zoonotic orthopoxviruses? Lancet Microbe. (2024) 5(8):100883. doi: 10.1016/S2666-5247(24)00106-X
6. Springer YP, Hsu CH, Werle ZR, Olson LE, Cooper MP, Castrodale LJ, et al. Novel orthopoxvirus infection in an Alaska resident. Clin Infect Dis. (2017) 64:1737–41. doi: 10.1093/cid/cix219
7. Dyer O. Alaskapox: First human death from zoonotic virus is announced. BMJ. (2024) 384:q415. doi: 10.1136/bmj.q415
8. Gigante CM, Gao J, Tang S, McCollum AM, Wilkins K, Reynolds MG, et al. Genome of alaskapox virus, A novel orthopoxvirus isolated from Alaska. Viruses. (2019) 11:708. doi: 10.3390/v11080708
9. Borealpox virus. Formerly known as Alaskapox. Available online at: https://health.alaska.gov/dph/Epi/id/Pages/Borealpox.aspx (Accessed Sept. 27, 2024).
10. Skrabanek L, Campagne F, Weinstein H. Building protein diagrams on the web with the residue-based diagram editor RbDe. Nucleic Acids Res. (2003) 31:3856–8. doi: 10.1093/nar/gkg552
11. Protter. Interactive protein feature visualization. Available online at: https://wlab.ethz.ch/protter/start/ (Accessed March 2, 2024).
12. Omasits U, Ahrens CH, Müller S, Wollscheid B. Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics. (2014) 30:884–6. doi: 10.1093/bioinformatics/btt607
13. Thomas S. The structure of the membrane protein of SARS-CoV-2 resembles the sugar transporter SemiSWEET. Pathog Immun. (2020) 5:342–63. doi: 10.20411/pai.v5i1.377
14. Thomas S. Mapping the nonstructural transmembrane proteins of severe acute respiratory syndrome coronavirus 2. J Comput Biol. (2021) 28:909–21. doi: 10.1089/cmb.2020.0627
15. Thomas S. Mapping the accessory proteins (Orfs) of severe acute respiratory syndrome Coronavirus 2. Re: Gen Open. (2023) 3:1–10. doi: 10.20944/preprints202210.0158.v2
16. I-TASSER. Protein structure and function predictions. Available online at: https://zhanggroup.org/I-TASSER/ (Accessed March 5, 2024).
17. Phyre2. Protein fold recognition server. Available online at: http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index (Accessed March 12, 2024).
18. Phobius. A combined transmembrane topology and signal peptide predictor. Available online at: https://phobius.sbc.su.se/ (Accessed March 3, 2024).
19. IEDB Analysis Resource. Available online at: http://tools.iedb.org/main/ (Accessed March 6, 2024).
20. Jia K, Kilinc M, Jernigan RL. New alignment method for remote protein sequences by the direct use of pairwise sequence correlations and substitutions. Front Bioinform. (2023) 3:1227193. doi: 10.3389/fbinf.2023.1227193
21. UniProt. Available online at: https://www.uniprot.org/align (Accessed March 8, 2024).
22. NetSurfP-3.0. Protein secondary structure and relative solvent accessibility. Available online at: https://services.healthtech.dtu.dk/services/NetSurfP-3.0/ (Accessed October 2, 2024).
23. Høie MH, Kiehl EN, Petersen B, Nielsen M, Winther O, Nielsen H, et al. NetSurfP-3.0: accurate and fast prediction of protein structural features by protein language models and deep learning. Nucleic Acids Res. (2022) 50(W1):W510–5. doi: 10.1093/nar/gkac439
24. Turner PC, Moyer RW. An orthopoxvirus serpinlike gene controls the ability of infected cells to fuse. J Virol. (1992) 66:2076–85. doi: 10.1128/jvi.66.4.2076-2085.1992
25. Parekh P, Tang Z, Turner PC, Moyer RW, Tan W. Aptamers recognizing glycosylated hemagglutinin expressed on the surface of vaccinia virus-infected cells. Anal Chem. (2010) 82:8642–9. doi: 10.1021/ac101801j
26. Wu W, Wang Z, Cong P, Li T. Accurate prediction of protein relative solvent accessibility using a balanced model. BioData Min. (2017) 10:1. doi: 10.1186/s13040-016-0121-5
27. Locker JK, Kuehn A, Schleich S, Rutter G, Hohenberg H, Wepf R, et al. Entry of the two infectious forms of vaccinia virus at the plasma membane is signaling-dependent for the IMV but not the EEV. Mol Biol Cell. (2000) 11:2497–511. doi: 10.1091/mbc.11.7.2497
28. Monticelli SR, Bryk P, Brewer MG, Aguilar HC, Norbury CC, Ward BM. An increase in glycoprotein concentration on extracellular virions dramatically alters vaccinia virus infectivity and pathogenesis without impacting immunogenicity. PloS Pathog. (2021) 17:e1010177. doi: 10.1371/journal.ppat.1010177
29. Singh K, Gittis AG, Gitti RK, Ostazeski SA, Su HP, Garboczi DN. The vaccinia virus H3 envelope protein, a major target of neutralizing antibodies, exhibits a glycosyltransferase fold and binds UDP-glucose. J Virol. (2016) 90:5020–30. doi: 10.1128/JVI.02933-15
30. Meyer H, Ropp SL, Esposito JJ. Gene for A-type inclusion body protein is useful for a polymerase chain reaction assay to differentiate orthopoxviruses. J Virol Methods. (1997) 64:217–21. doi: 10.1016/S0166-0934(96)02155-6
31. Meng X, Embry A, Rose L, Yan B, Xu C, Xiang Y. Vaccinia virus A6 is essential for virion membrane biogenesis and localization of virion membrane proteins to sites of virion assembly. J Virol. (2012) 86:5603–13. doi: 10.1128/JVI.00330-12
32. Koonin EV. A highly conserved sequence motif defining the family of MutT-related proteins from eubacteria, eukaryotes and viruses. Nucleic Acids Res. (1993) 21:4847. doi: 10.1093/nar/21.20.4847
33. Balinsky CA, Delhon G, Afonso CL, Risatti GR, Borca MV, French RA, et al. Sheeppox virus kelch-like gene SPPV-019 affects virus virulence. J Virol. (2007) 81:11392–401. doi: 10.1128/JVI.01093-07
34. Gubser C, Goodbody R, Ecker A, Brady G, O’Neill LAJ, Jacobs N, et al. Camelpox virus encodes a schlafen-like protein that affects orthopoxvirus virulence. J Gen Virol. (2007) 88:1667–76. doi: 10.1099/vir.0.82748-0
35. Moss B. Poxvirus DNA replication. Cold Spring Harb Perspect Biol. (2013) 5:a010199. doi: 10.1101/cshperspect.a010199
36. Sèle C, Gabel F, Gutsche I, Ivanov I, Burmeister WP, Iseni F, et al. Low-resolution structure of vaccinia virus DNA replication machinery. J Virol. (2012) 87(3):1679–89. doi: 10.1128/JVI.01533-12
37. Fontúrbel FE, Nespolo RF, Amico GC, Watson DM. Climate change can disrupt ecological interactions in mysterious ways: Using ecological generalists to forecast community-wide effects. Climate Change Ecol. (2021) 2:100044. doi: 10.1016/j.ecochg.2021.100044
38. Carlson CJ, Zipfel CM, Garnier R, Bansal S. Global estimates of mammalian viral biodiversity accounting for host sharing. Nat Ecol Evol. (2019) 3:1070–5. doi: 10.1038/s41559-019-0910-6
39. Carlson CJ, Albery GF, Merow C, Trisos CH, Zipfel CM, Eskew EA, et al. Climate change increases cross-species viral transmission risk. Nature. (2022) 607:555–62. doi: 10.1038/s41586-022-04788-w
40. Casadevall A. Climate change brings the specter of new infectious diseases. J Clin Invest. (2020) 130:553–5. doi: 10.1172/JCI135003
41. Pfenning-Butterworth A, Buckley LB, Drake JM, Farner JE, Farrell MJ, Gehman AM, et al. Interconnecting global threats: climate change, biodiversity loss, and infectious diseases. Lancet Planet Health. (2024) 8:e270–83. doi: 10.1016/S2542-5196(24)00021-4
42. Datler J, Hansen JM, Thader A, Schlögl A, Bauer LW, Hodirnau VV, et al. Multi-modal cryo-EM reveals trimers of protein A10 to form the palisade layer in poxvirus cores. Nat Struct Mol Biol. (2024) 31:1114–1123. doi: 10.1101/2023.05.24.541142
43. Islam MA, Sangkham S, Tiwari A, Vadiati M, Hasan MN, Noor STA, et al. Association between global monkeypox cases and meteorological factors. Int J Environ Res Public Health. (2022) 19:15638. doi: 10.3390/ijerph192315638
44. Moss B. Poxvirus cell entry: how many proteins does it take? Viruses. (2012) 4:688–707. doi: 10.3390/v4050688
Keywords: borealpox virus, BRPV, borealpox disease, Alaskapox virus, AKPV, Alaska, Orthopoxvirus, infectious disease
Citation: Thomas S (2024) Plotting the major proteins of borealpox virus. Front. Virol. 4:1451810. doi: 10.3389/fviro.2024.1451810
Received: 19 June 2024; Accepted: 01 November 2024;
Published: 26 November 2024.
Edited by:
Madhu Khanna, University of Delhi, IndiaReviewed by:
Gaëtan Ligat, INSERM UMR1291 Institut Toulousain des Maladies Infectieuses et Inflammatoires, FranceCopyright © 2024 Thomas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sunil Thomas, dGhvbWFzcy0wMkBtbGhzLm9yZw==; c3VudG9tMkBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.