 Ali Khanifar
Ali Khanifar Azin Najafi2,3
Azin Najafi2,3 Seyed Ali Hosseini
Seyed Ali Hosseini Mohammad Taheri
Mohammad Taheri
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Virol. , 16 July 2024
Sec. Viral Disease Investigation
Volume 4 - 2024 | https://doi.org/10.3389/fviro.2024.1405680
The COVID-19 virus is a single-stranded virus from the Coronaviridae family and has a genome of about 29881 bp, which causes acute respiratory disease. One way of transmission of the COVID-19 virus is respiratory, which is the reason for the significant transmission of the disease. The COVID-19 virus causes mutagenesis in different parts of the virus genome with its mechanisms, including -1 ribosomal frameshifting. Various parts that have undergone the most severe mutations include the spike protein, which leads to the emergence of new variants. Most of the mutations observed in the COVID-19 virus are located in the S protein and a region known as the RBD (Receptor-Binding Domain), which binds to the ACE2 (Angiotensin-converting enzyme 2) receptor in human cells. The variations in the RBD region will determine how it binds to the ACE2 receptor. Essentially, the changes created because of the mutation determine the affinity of the RBD to ACE2. On the other hand, the COVID-19 virus, because of its NSP13, NSP14, and NSP16 proteins, helps the mutation of the virus by consuming magnesium ions (Mg2+). Since the ribosome is stable with Mg2+, the COVID-19 virus, by consuming Mg2+, causes the ribosome to convert from the polysome to the monosome state, which causes a break in translation and finally leads to the formation of -1 ribosomal frameshifting.
The COVID-19 virus has emerged as a significant pathogen in recent years, causing a global pandemic of unprecedented proportions. The disease first appeared on December 8th, 2019, in Wuhan, China. This report pertained to patients afflicted with severe pneumonia symptoms (1). Researchers identified the virus in China on January 7th, 2020 (2). The World Health Organization subsequently classified it as nCoV-2019. Ultimately, they named this disease COVID-19 (3). Coronaviruses, such as COVID-19 (SARS-CoV-2), pertain to the category of ssRNA viruses that possess an envelope and display a size variation between 80 and 120 nanometers. The distinguishing feature of the virus is its positive-sense ssRNA (4), which means the virus genome is being directly translated within the cellular environment of the host (Figure 1). The fundamental nature of the virus’s genetic material is such that it comprises a single-stranded ribonucleotide chain. This chain encodes four distinct structural proteins, including N, M, E, and S. Alongside these, encoding sixteen non-structural proteins that are identified as NSP 1–16 (5–7). Researchers acknowledge that coronaviruses, on the whole, are pathogenic microorganisms capable of infecting both human and animal hosts. The classification of these viruses is based on the order Nidovirales, which include Coronaviridae, Roniviridae, and Arteriviridae families (8).
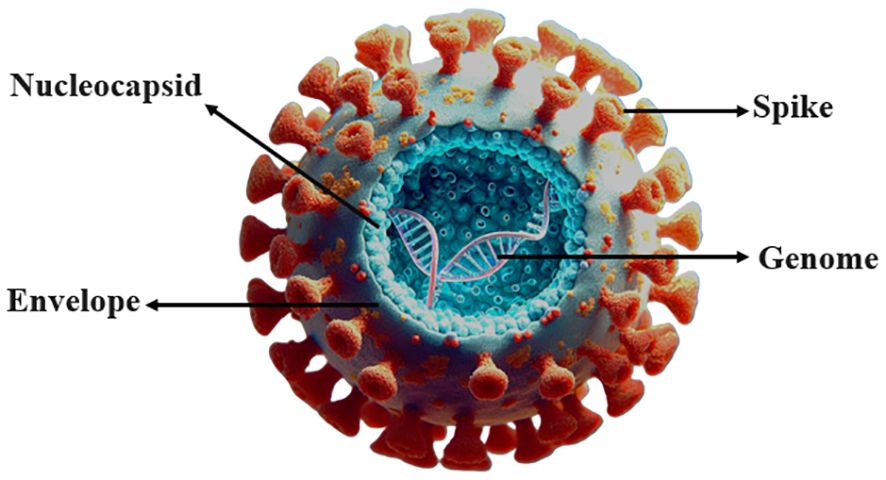
Figure 1 COVID-19 virus.
The Coronaviridae family has two subfamilies, Coronavirinae and Torovirinae. The Coronavirinae subfamily causes respiratory infections, while the Torovirinae subfamily in animals leads to intestinal infections (9). The Coronavirinae family includes four genera: Alpha, Beta, Gamma, and Delta (10). In terms of pathogenicity in humans, the two genera of Alpha and Beta play a crucial role. The Alpha genus includes 229E and NL-63, and the Beta genus includes HKU-1, MERS, SARS, and OC43. Scientists have placed the SARS-CoV-2 virus in the Beta genus after identifying its genome sequence. In this way, the number of human coronaviruses reached seven, of which two are in the Alpha genus and five are in the Beta genus (11). The coronavirus genome has a cap at the 5’ end and a poly(A) tail at the 3’ end. After the COVID-19 virus enters the cell and releases its genome, the replication process begins by translating ORF1a and ORF1b (12). The Long Replicase Gene (ORF1ab Gene) is over 21 Kb in size and encodes sixteen unstructured proteins (NSP 1 -> 16), which translates as Pp1ab polyprotein. In addition, next to this gene (late genes) are four structural proteins S, E, M, N, and 6 non-structural proteins that are also encoded by subgenomic mRNAs (NS 3a -> 14). ORF1a encodes the immediate early genes, while ORF1b can encode the early genes. However, ORF1a must repress the native immune response of the host, whereas ORF1b has a duty to replication and synthesis of RNA (13, 14). Virus NSPs use the target cell membrane to collect transcription and replication complexes which take part in negative-strand RNA synthesis (15). The NSP1 protein binds to the 40S subunit of the ribosome in the cell, inhibiting translation in the host cell. This complex induces endonucleolytic cleavage of the 5’UTR region in host mRNAs, eventually degrading them. The presence of an end-conductor sequence in the 5’ region protects viral mRNAs from endonucleolytic cleavage. By suppressing gene expression in the host cell, the NSP1 protein facilitates viral gene expression and evades the host immune response (16). The NSP2 protein interacts with host PHB1 and PHB2 molecules to regulate the cell survival transmission pathway. These two proteins play a crucial role in stabilizing mitochondrial function and protecting cells from stress (17). The NSP3 protein is involved in the N-terminal cleavage of polyproteins. Additionally, this protein, PL-PRO, exhibits deubiquitinating or DeISGylating activity that inhibits the immune response and targets lys63 and 48Lys polyubiquitin chains on cellular substrates (18). Alongside the NSP4 protein, it participates in forming membrane vesicles essential for virus replication. Moreover, it inhibits induction of type I interferon innate immunity by blocking phosphorylation, dimerization, and translocation between cell nuclei while also suppressing NF-kappa-B signaling (19, 20). In its coding gene region resides a SUD domain sequence unique to SARS-type coronaviruses which binds to G4 mRNA to inhibit apoptotic signaling and promote cell survival (21). NSP5 (Proteinase 3 CL) is responsible for cleaving the C-terminal end sequence of replicase polyprotein at eleven different regions. Known substrates for this enzyme contain [ILMVF]-Q-|-[SGACN] sequences. It also interacts with ADRP (22). The NSP6 protein initiates host endoplasmic reticulum autophagosome formation but limits their expansion while facilitating transmission of virus components to lysosomes (23). The NSP7 protein forms a hexadecamer with NSP8 acting as a primase during virus replication (24). As an ssRNA binding protein, the NSP9 protein is also involved in virus replication (25). Finally, the NSP10 protein plays a crucial role in the viral translation system by synchronizing the 3’-5’ exoribonuclease activities of the NSP14 protein and the 2’-O-methyltransferase activity of the NSP16 protein. It is also involved in the cap methylation of viral mRNAs sequences (26). The NSP12 comprises 932 amino acids. The NSP12 form consists of a NiRAN domain and an RNA-dependent RNA polymerase (RDRP) domain, which are connected by a connector domain (27). NSP12 functions as an RDRP. NSP12 proteins, together with NSP7 and NSP8 cofactors, participate in the replication and translation of the RNA virus genome (28). NSP13 (helicase or Hel) is a multifunctional protein with a binding domain located at the N-terminal region. This protein is responsible for unwinding the double helix structure of RNA and DNA from the 5’ to 3’ direction. The helicase activity of this protein relies on magnesium ions (Mg2+) (29, 30). The E (envelope) protein plays a central role in virus morphogenesis and formation. Acting as a viroporin, this protein facilitates ion passage through protein-lipid pentameric pores. Additionally, it contributes to apoptosis induction. Comparative alignment studies have identified genomic sequences of this coronavirus protein in bats and scaly anteaters. Structural modifications observed in the E protein include substitutions at position 69 (R69N/D/E), deletion at position 70 (70G/C), and substitutions at positions 55 and 56 (T, V -> S, F). These alterations highlight the significant role played by this small but crucial protein during virus infection and replication stages (31). The glycoprotein M (Membrane) helps orchestrate virus morphogenesis by facilitating proper juxtapositioning of different viral components while also playing an important role in virus germination.
HE is involved in a component of envelope proteins in some (but not all) coronaviruses. Some coronaviruses, such as human coronavirus OC43, contain the glycoprotein HE, which has hemagglutination and acetyl esterase activity. HE has a similar function to both the influenza virus receptor-binding protein (HA) and receptor-destructive enzyme (NA). The virus binds to the mucous membranes of the respiratory tract by HE. Type C influenza also contains this enzyme.
The protein on the surface of the virion is called HE. Scientists do not consider this protein an essential structural component of the virus because not all Coronaviruses express HE. However, HE may play an important role in binding coronaviruses to the target cell and facilitating the release of the virus from the cell surface.
The S region of the virus genome encodes the glycoprotein spike, which is located on the surface of the virus and plays an important role in binding to host cells and determining pathogenicity. The S protein is responsible for infection and transmission in patients (32, 33). Spike stimulates the immune system and stands as one of the crucial immunogenic antigens for this virus. By triggering antibody production, it effectively neutralizes viral activity (34). The total length of glycoprotein S is 1273 amino acids and includes a guide peptide (amino acids 1–13) in the part of the N-terminus. This part has two sub-groups, one S1 (14–685) and the other S2 (686–1273). S1 is responsible for receptor binding, and S2 is responsible for the fusion of the virus membrane with the host cell membrane. On the other hand, Spike is a homotrimeric glycoprotein that binds to the type I integral membrane in ACE2. The glycoprotein S at the furin cleavage site contains several base amino acids (PRRA) at the junction of the S1 and S2 subunits, which increase the infectivity of the virus. The S1 protein has a receptor-binding domain (RBD) that is involved in binding to the ACE2 receptor and virus entry into the host cell and induces structural changes in glycoprotein S (35), while the S2 domain mainly contains the HR (Heptad Repeat). However, HR1 and HR2, which are closely related to them, have virus membrane fusion (34, 36) (Figure 2). This protein causes the virus to enter human lung cells using TMPRSS2. The acidic pH of lysosomes and endosomes activates the enzymes cathepsin B and L. The virus also requires an acidic environment with a pH of about 3 to enter the cell cytosol. Proteolysis of glycoprotein S by CatB/L cathepsin activates the S2 fusion peptide and activates virus membrane integration into endosomes. Also, host cells express the spike protein at their surface, causing membrane attachment of adjacent cells to each other and forming syncytium. Syncytium formation, besides disrupting the function of the organ involved, also allows the virus to spread further and escape the immune system. By creating double-layered vesicles, coronaviruses block the expression of the Pattern Recognition Receptor, and as a result, the innate immune system does not recognize them and continues to multiply within the vesicle. They disrupt the production of type I interferons as one of the most important anti-viral factors. Coronaviruses disrupt the process of presenting antigens by the immune cells. Investigations into changes in the RBD sequence have indicated that mutation N501 remains stable and enhances the interaction with Y41 through hydrogen bonding, resulting in reinforcing the connection between RBD and the ACE2 receptor. This particular region exhibits a complex role because of its involvement in numerous binding mechanisms. In addition, researchers have observed that mutations in residues N479, T487, L455A, F456A, and Q493A weaken the binding to the ACE2 receptor (34, 37–40). The NSP3a protein is involved in viroporin formation and possibly virus release. This protein causes the upstream expression of fibrinogen subunits FGA, FGB, and FGG in host lung epithelial cells. It also induces apoptosis in cell culture. NSP3a induces serine phosphorylation in interferon alpha receptor subunit 1 (IFNAR1), causing low expression of type 1 interferon receptor and increased ubiquitination in IFNAR1 (41–44).
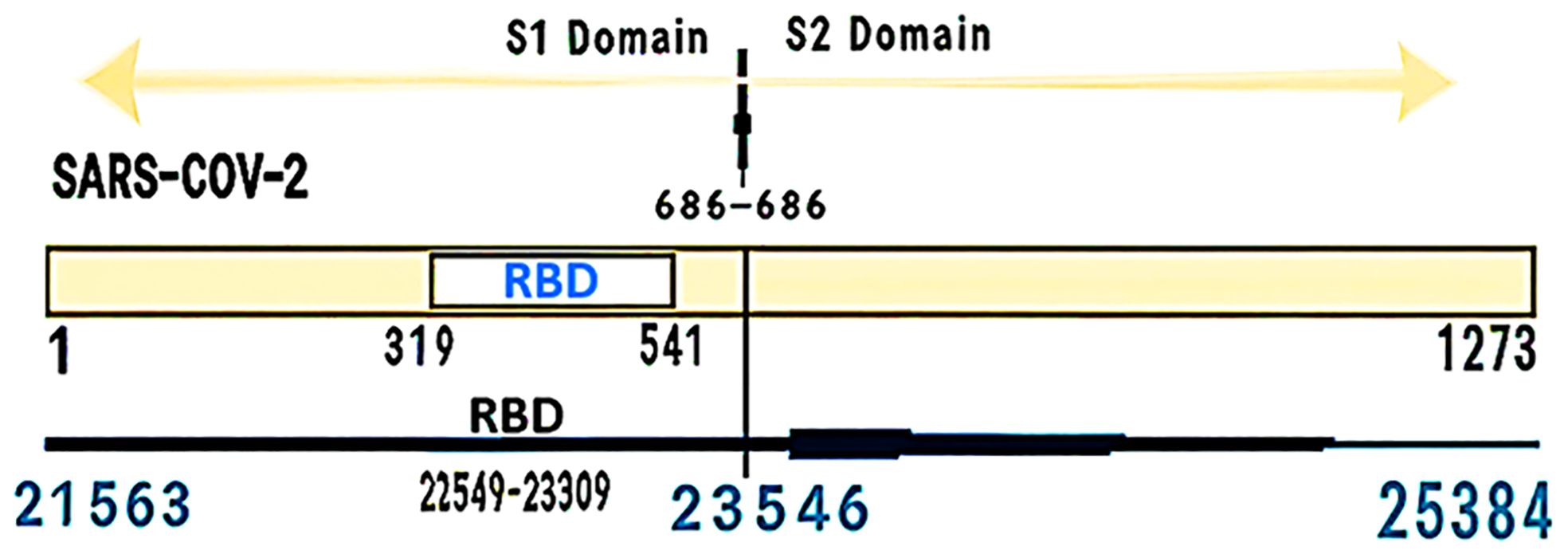
Figure 2 Positions of the S gene and RBD in terms of the number of amino acids and nucleotides.
Researchers believe that a mutation in the S1 protein may cause a malfunction in the function of the virus. Researchers have found that some mutations change the intensity of transmission and contagion of the virus. This can affect the effectiveness of the vaccine and the neutralization of the virus. Although the stem portion of the S2 appendages has experienced fewer mutational changes. However, the breaking of the S appendage into two parts, S1 and S2, is very important in virus attachment and infection.
The study findings suggest that the COVID-19 virus uses ACE2 as a receptor to enter the target cell (45). Besides, the research findings have determined that the S protein of the COVID-19 virus underwent mutations and became glycosylated, resulting in an increased virulence of the virus. However, the study of protein S mutations at a biological level has revealed the existence of a region known as the RBD within the virus’s glycoprotein S. This region enables the virus to attach to its receptor (46). The RBD in protein S comprises five beta strands, specifically β1, β2, β3, β4, and β7, organized in an antiparallel manner and linked by brief spirals and loops in the central protein region (core). The protein’s center contains β4 and β7 strands, as well as short segments of β5 and β6 strands, which attach to α4 and α5 helices and loops. This section contains the RBM and includes amino acid residues that attach to the ACE2 enzyme in the virus. Among these 4 pairs, 3 bonds (Cys336-Cys361, Cys379-Cys432, Cys391-Cys525) in the center help stabilize the structure of β-sheets. While the other (Cys480-Cys488) binds the loops at the distal end of RBM (47).
Protein modeling experiments on glycoprotein S show that the SARS-CoV-2 virus has a sufficient affinity to attach to the ACE2 enzyme in human cells. This enzyme, on the outer surface of the membrane of cells of tissues such as lungs, arteries, heart, kidney, and intestine, is also present. This virus uses this enzyme as a mediator to enter the cell (48). Several research groups have shown this enzyme as the cell surface receptor of SARS-CoV-2 (47, 49–53). Researchers have even observed that HeLa cells expressing ACE2 are susceptible to infection by this virus, while cells lacking ACE2 show no response to this virus (54). Therefore, binding to the ACE2 receptor is an important initial step for SARS-CoV-2 to enter target cells (38, 48). In vitro binding measurements show that the RBD in SARS-CoV-2 binds to ACE2 with an affinity in the nanomolar range. This indicates that the RBD is a key component in the S1 subunit responsible for the binding of SARS-CoV-2 to ACE2 (34, 37). Two repeating regions of heptad peptides 1 and 2, named Membrane-distal Heptad Repeat 1 (HR1) and Membrane-proximal Heptad Repeat 2 (HR2) on S protein, are important for binding to the receptor. After binding of the RBD in the S1 subunit of the S protein to the ACE2 receptor on the target cell, the heptad repeat domains 1 and 2 in the S2 subunit of the S protein interact with each other and form a six-helix bundle. This bundle has a length of 115 Å (angstroms) and a diameter of 25 Å. The three helices in HR1 form a central entangled twist, and the three strands of HR2 join it in an antiparallel shape. This sextuplet complex is not very stable and is likely to form after the initial fusion of the HR1 segment in the receptor membrane (55, 56).
In early January 2020, scientists sequenced the entire genome of the new coronavirus for the first time, which they had isolated from thousands of infected patients worldwide, and they reported the resulting genomic changes. This caused a turning point in the disease control process. Researchers then showed that the rapid spread was because of a slight change in the amino acid or spike proteins on the virus’s surface (Figure 3).
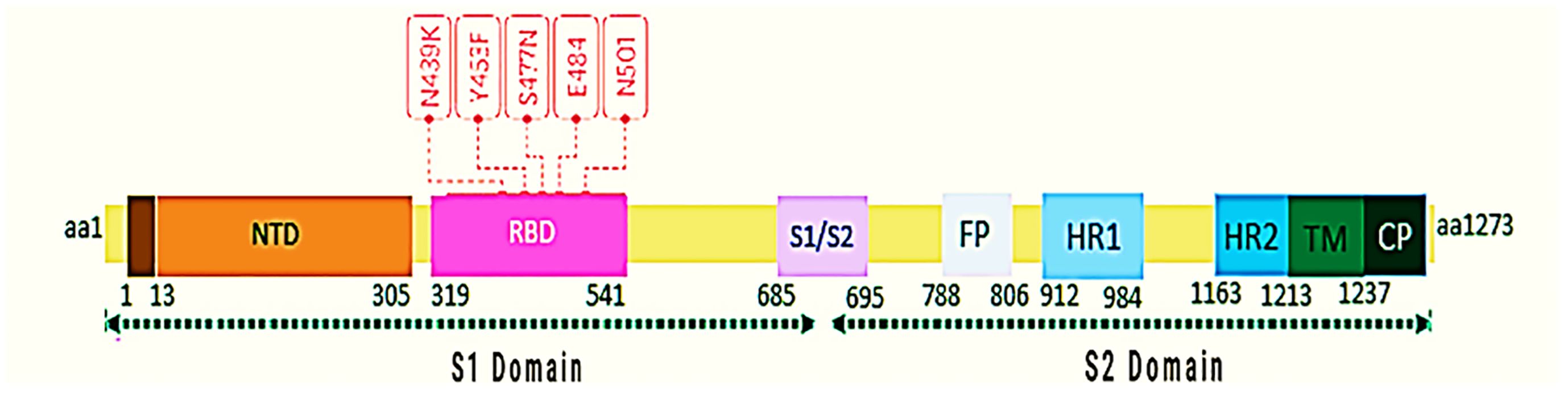
Figure 3 Key mutations in RBD are shown. Mutation data from January 1, 2020, to December 26, 2020, were obtained from the GISAID database, and the structure of protein S was designed and presented with bioinformatics software. The numbers below the protein S structure indicate the location and complete amino acid sequence.
The virus strengthens its ability to attach to its receptor and performs this step faster and with a stronger connection. In this way, by making these changes, it escapes from the patient’s immune system and remains safe; therefore, there is always the possibility that a more effective virus will emerge as a locally or regionally dominant variant. However, the quick transmission doesn’t always indicate a connection to more severe COVID-19 variants or increased mortality rates. This mutation does not always affect the course of the disease, and it can affect the speed of the disease.
The mutated type of COVID-19 occurs because of the replacement of the amino acid phenylalanine (F) with tyrosine (Y). According to the study, the Y453F mutant glycoprotein has a weaker spike binding property to human ACE2 compared to the COVID-19 glycoprotein, which is attributed to the substitution of tyrosine at position 453 with phenylalanine (57). Y453F has occurred in the Netherlands and Denmark in associated with the transmission of disease through mink and humans and in RBD. The existing report shows that Y453F, named mutation B.1.1.298, has infected about 214 humans until November 5, 2020 (58). The N439K mutation increases the affinity of the RBD for ACE2 (59). There are reports in England and later in South Africa of N501Y mutations (60). The N501Y mutation in treated subjects led to alterations in amino acids, resulting in heightened binding affinity to ACE2 (61, 62). The Alpha variant (B.1.1.7) is one of the most dangerous mutations in the UK, the N501Y mutation, and it is very worrying; because it contains one of the six key amino acids that cause the RBD to bind tightly to the ACE2 receptor (Table 1). Modeling analysis suggests that the mutation N501Y has enabled the potential for an aromatic ring connection via an extra hydrogen bond between RBD and ACE2 (63). Therefore, the mutation N501Y has increased the binding affinity of protein S for the ACE2 receptor (64). The Beta variant (B.1.351), an important mutation of concern in the Republic of South Africa, proved that besides N501Y, two mutations K417N and E484K strengthen the binding of RBD in the S region to ACE2 receptor, causing escape from the immune system and increasing speed of virus transmission (65). The Gamma variant (P.1/B.1.1.28) is another worry that was introduced to Japan from Brazil, particularly because of the E484K mutation, which boosts the evasion capability of the immune system, underscoring its significance (66). The Delta variant (B.1.617.2) contains ten mutations, including T19R, del156, del157, R158G, L452R, T478K, D614G, P681R, and D950N, which occurred in the spike protein coding region. Plus Delta variant (B.1.617.2.1), which is the latest mutated variant of corona and the difference is K417N compared to the Delta variant. However, mutations of Alpha (B.1.1.7), Beta (B.1.351); Mutations Gamma (P.1/B.1.1.28), and Delta (B.1.617.2) occur in the RBD and NTD (N-terminal domain) regions. The N501Y mutation found in the RBD is present in all variants except Delta, leading to heightened spike affinity. Together with NTD, the RBD acts as the primary target for neutralization and facilitates an antibody production response to antiserum or vaccine. The N501Y single mutation alone increases affinity between RBD and ACE2 tenfold higher than the ancestral strain (N501- RBD) has increased (67). According to the announcement of the World Health Organization on June 14, 2021, the Lambda variant (B.1.1.1/C.37) was introduced as a significant variant of interest (VOI). Some analysts considered it worrying, and they even predicted that it would probably replace the Delta variant. The spike protein of this mutant strain has point mutations, such as G75V, T76I, del247/253, L452Q, F490S, D614G, and T859N. These point mutations may affect the ability of the mutant strain to transmit and escape the immune system. Lambda and Delta randomly undergo mutations at position L452, and these mutations in this position may have caused the rapid spread of these two virus strains (68, 69). Another variant that the World Health Organization has introduced in the category of worrying cases because of its resistance to the vaccine is the Mu variant (B.1.1.1/C.37), and because it has the same behavior as the beta mutant variant (escape from the immune system), therefore, they are among the cases being monitored, and the likelihood of its widespread occurrence is not far from anticipated. This mutation in P681H is associated with increased transmissibility, while E484K and K417N mutations might aid the virus in evading the immune system. The most recent report is of the C.1.2 variant mutation, which was first identified in South Africa in May 2021 and contains multiple mutations in all four VOCs (Alpha, Beta, Delta, and Gamma) and three VOIs (Kappa, Eta, and Lambda). Additionally, other mutations in NTD (C136F) and RBD (Y449H) have also been discovered. The functional impact of these mutations probably includes reduced antibody effectiveness and evasion of the immune system (70). The latest variant classified by the World Health Organization as a VOC is called the Omicron variant (B.1.1.529), and the first sample was confirmed on November 9, 2021, from South Africa, the first sequence indicating 30 changes in Spike protein that are similar to the Alpha and Delta variants, which are associated with increased infection and the ability to escape infection-inhibiting antibodies (71).
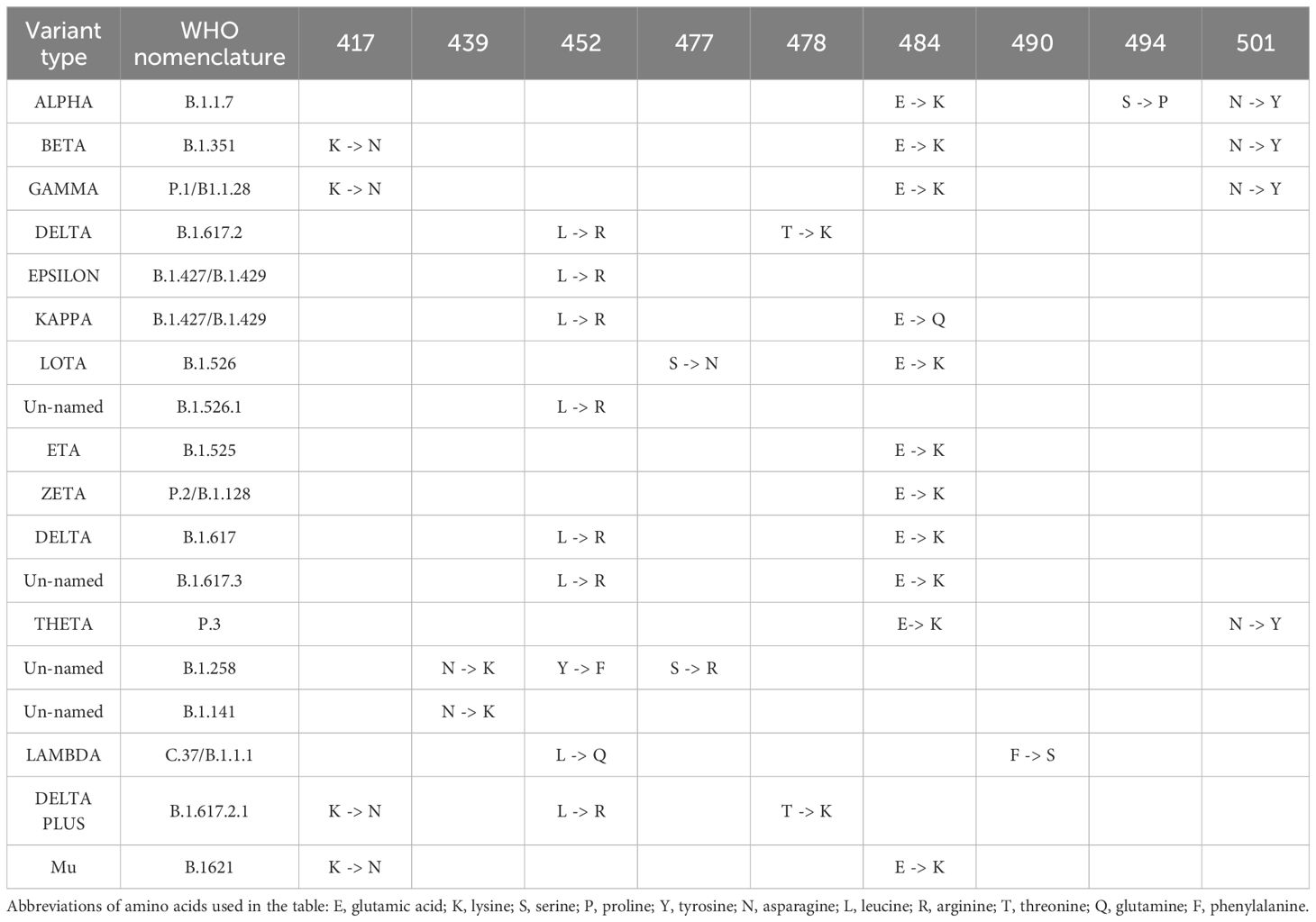
Table 1 The most important mutations occurring in the S gene of the RBD region of the COVID-19 virus are presented.
The role played by RNA helicase NSP13 in viral replication is critically significant, as it showcases the activity of 5′-RNA triphosphatase, which triggers the elimination of the 5′-γ-phosphate from the developing ppp-RNA molecule (72). Afterwards, NSP12 transfers a guanosine monophosphate to the 5′-end of the mRNA (73, 74), followed by methylation of the guanosine residue by the NSP14-NSP10 heterodimer, leading to the formation of N7-methylated Cap-0-RNA. The ultimate step in the formation of Cap-1-RNA involves the transfer of a methyl group from S-adenosylmethionine to the 2′-OH of the first adenosine ribonucleotide. However, 2′-O-methyltransferase, a heterodimeric complex that includes NSP16 and the activator NSP10, plays a facilitating role in reaction to coronaviruses (72, 75, 76). The enzymatic processes play a pivotal role in the replication of coronaviruses, as they promote the formation of Cap-1-RNA. The creation of Cap-1-RNA is a crucial stage in the existence of coronaviruses since it permits the virus to avoid the host’s immune monitoring by assisting the processing of viral mRNA in the cytoplasm of infected cells, resulting in enabling it to replicate efficiently (77, 78) (Figure 4).
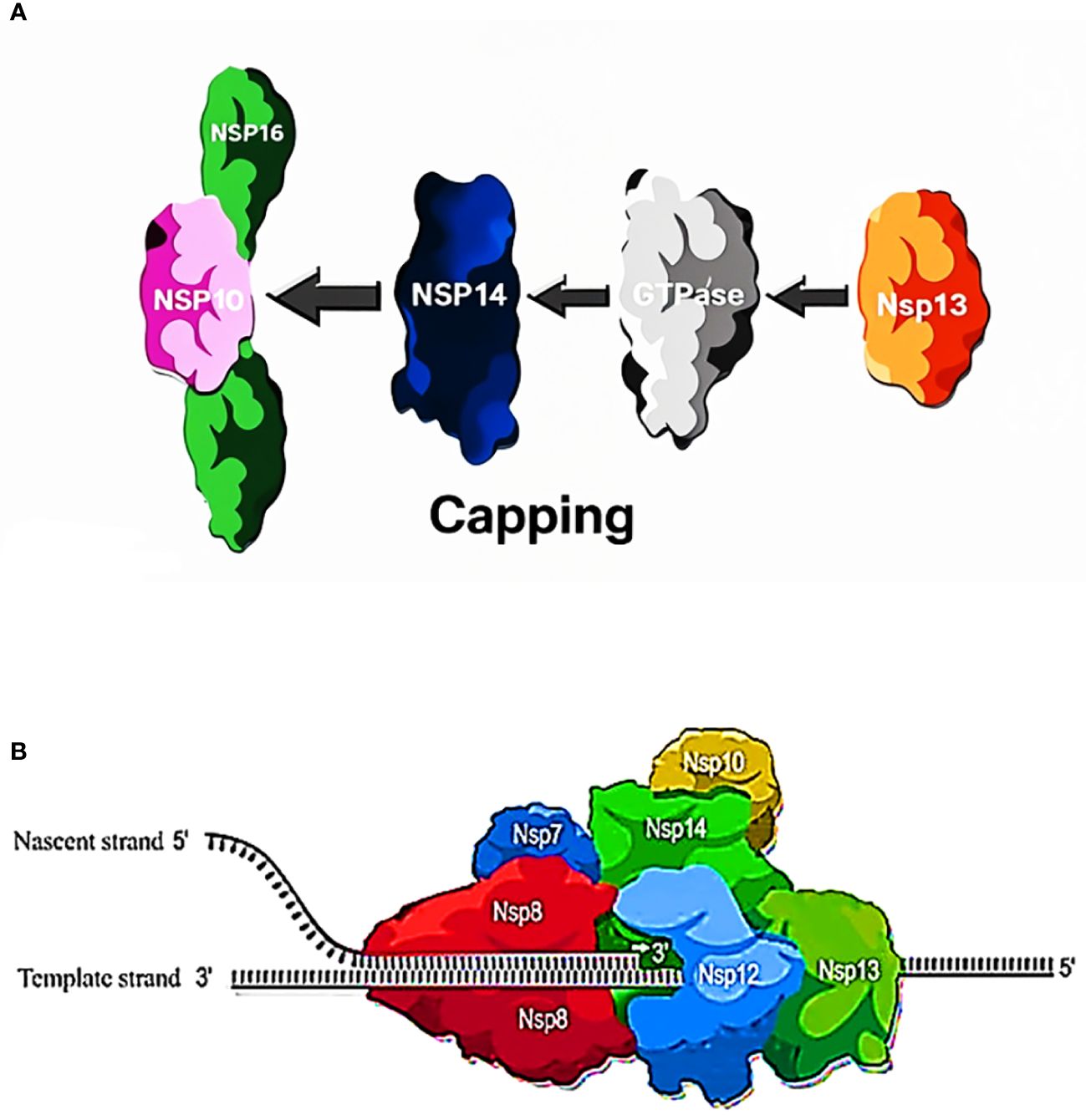
Figure 4 (A) Process of capping: The RNA capping process in SARS-CoV-2 involves three key non-structural proteins: NSP13, NSP14, and NSP16 play unique roles in kick-starting and adjusting RNA caps crucial to translation and evading the immune system. (B) Function of NSP13, NSP14, NSP16 in capping. NSP13 (RNA Triphosphatase): involved in early RNA capping stages, it hydrolyzes the nascent RNA (resulting in a pp-RNA product). NSP14 (Guanylyl-Transferase and Exoribonuclease): 1) capping process: guanylyl-transferase (GTase) hydrolyzes GTP, transferring GMP to pp-RNA, creating Gppp-RNA. 2) proofreading process: functions as an exoribonuclease, maintaining RNA integrity during replication. NSP16 (2’-O-Methyltransferase): critical for RNA capping, requires cofactor NSP10, catalyzes the transfer of a methyl group from S-adenosyl methionine (SAM) to the 2’-OH group of the first RNA nucleotide (N1), converts RNA cap from Cap-0 to Cap-1 (7-methylguanosine).
The precise function of NSP13 in the COVID-19 virus remains undefined. However, reports have suggested that NSP13 plays a crucial role in the replication of the COVID-19 virus genome (79, 80). Based on conserved sequence motifs, scientists have established the classification of helicases, leading to the identification of six distinct superfamilies labeled as SF1 to SF6 (79, 81, 82). NSP13 of the COVID-19 virus belongs to the SF1 superfamily (79, 81, 83). NSP13 weighs 67 kDa and uses the energy released from the breakdown of nucleotide triphosphates to aid in the unwinding of double-stranded DNA. This biophysical process relies on a specific orientation, occurring from 5’ to 3’, essential for its successful execution (29). A virus’s successful replication heavily depends on the complex interplay of the replication-transcription mechanism, wherein the NSP13 protein assumes a vital role within this intricate web of interactions. The collaboration of NSP13 with the non-structural proteins 7,8 and 12 proteins manifests a synergistic effect that culminates in the creation of the replication-transcription mechanism. The interaction between these non-structural proteins is critical for the virus’s life cycle, and understanding their mechanism can help in developing targeted antiviral strategies. NSP13 plays a crucial role in starting the replication process by unwinding the RNA helix. The synergistic actions of NSP13 and other proteins, which ultimately culminate in the generation of viral particles, guaranteed the efficient operation of the replication-transcription mechanism (30, 80, 83–85). The NSP13 helicase protein of SARS-CoV-2 demonstrates an exceptionally high sequence similarity index of 99.8% compared to the NSP13 helicase protein of SARS-CoV. However, a distinctive change in the amino acid sequence found at the I570V position is noticeable. Therefore, it is likely that the complete range of functions remains intact in the helicase component of SARS-CoV-2. The efficacy of helicase targeting in impeding viral replication has been demonstrated by several viral families, such as Picornaviruses, hepatitis C, Flaviviruses, and dengue viruses (86–88). Hence, this underscores the prospective usefulness of this approach.
The activities of NSP13 encompass helicase (29, 79, 83, 89), ATPase, and RNA 5′-triphosphatase (79, 90). A class of enzymes known as helicases are widespread types of enzymes that play a role in the unwinding of nucleic acids. Biological motors use the energy derived from the breakdown of nucleotide triphosphate to produce mechanical energy, which is then transmitted through nucleic acids, leading to the unwinding of the helical structure. As a result, scientists use the term ‘heli case’ to describe the necessity of utilizing ssDNA or ssRNA as a template for optimal genome replication, recombination, and repair. This is because of their minimal presence of secondary structures, which gives them a significant edge in these processes (91). The NSP13 protein comprises five unique domains, which include the N-terminal ZBD, the helical stalk domain, the 1B domain that contains a beta-barrel structure, and two subdomains namely 1 A and 2 A that exhibit RecA-like helicase architecture (92, 93) (Figure 5).
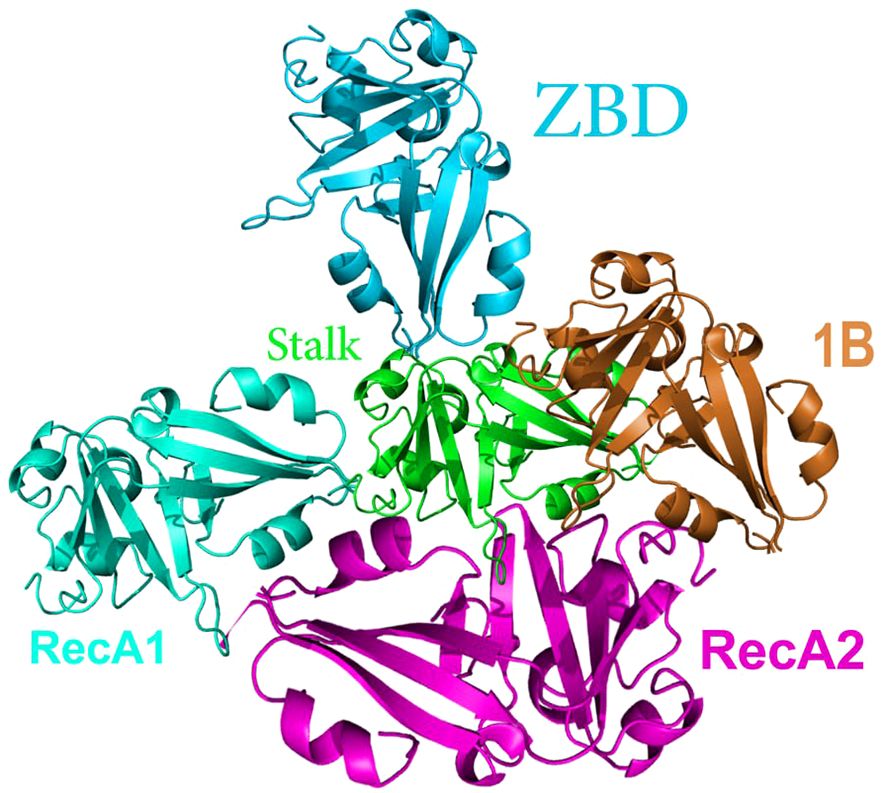
Figure 5 General structure of NSP13. NSP13 forms a triangular structure with the 1A and 2A domains as the base. RecA-Like Domains 1A and 2A contain residues responsible for nucleotide binding and hydrolysis. Beta-Barrel (1B) Domain is a part of the helicase structure. Stalk Domain links the zinc-binding domain to the rest of the helicase and serves as an essential signal transduction mechanism.
Other helicases that pertain to the Nidovirus family, namely EVA and PRRS, have, to a certain extent, preserved their fundamental 5-domain arrangement (93, 94). However, recent research has indicated that NSP13 comprises two copies. The presence of two copies of the helicase remains elusive in terms of its underlying reason; however, the mutagenesis performed on specific residues implicated in discrete domain contacts reveals the significance of both domains in the complex’s augmented helicase activity (84). On the other hand, prior investigations in SARS-CoV-1 have revealed the effect of adenosine triphosphate (ATP) on the operation of the helicase of NSP13 (95). The vital importance of the ZBD domain within the NSP13 of COVID-19 is associated with its essential function in aiding the helicase activity of the protein (80). NSP13 interacts with NSP8 through its N-terminal ZBD (74, 83, 84). Furthermore, a research study has revealed that the unwinding of dsRNA by NSP13 necessitates the presence of 1–2 mM of Mg2+ and 2 mM of ATP (96).
The NSP13 protein of SARS-CoV-2, also known as the helicase, is crucial for the viral replication process. It unwinds double-stranded RNA, a necessary step for the replication and transcription of the viral genome. NSP13 belongs to the helicase super-family 1B and uses the energy from nucleotide triphosphate hydrolysis to unwind double-stranded DNA or RNA. Mg2+ ions are essential cofactors for the ATPase activity of NSP13 (97). They are required for the binding and hydrolysis of ATP, which provides the energy needed for the helicase to unwind RNA. The structure of NSP13 reveals that magnesium ions are bound at specific sites that are crucial for enzymatic activity. These highly conserved sites play an essential role in viral replication, making them a prime target for antiviral drug development (97). Upon binding of ATP and Mg2+ ions, NSP13 undergoes conformational changes that are necessary for its helicase function. These changes allow NSP13 to interact with the RNA substrate and perform the unwinding process. In summary, NSP13 consumes Mg2+ ions as they play an integral role in the ATPase activity of the protein, allowing it to perform its function in the viral replication cycle by unwinding RNA strands necessary for the synthesis of new viral components (97).
NSP14 is a 60 kDa protein that has undergone the least mutations compared to other proteins. NSP14’s participation in the replication process necessitates its cooperation with other proteins of SARS-CoV-2 (98). However, NSP14 plays a role in the replication of the virus and escapes from the host immune system (99). NSP14 is a protein with two enzyme functions: Exonuclease (ExoN) and N7-methyltransferase (N7‐MTase). NSP14 has Exonuclease activity in its N-terminal domain and N7-methyltransferase activity in its C-terminal domain (100, 101). NSP14 increases its ExoN activity as much as 35-fold, by forming a complex with NSP10 (102). ExoN plays the role of proofreading the genome of a virus in a 3’ to 5’ direction by removing mismatched nucleotides (26, 102, 103). ExoN uses divalent cations (Mg2+, Zn2+) and one reactive water molecule to remove nucleoside monophosphates from the genome (104–106) (Figure 6).
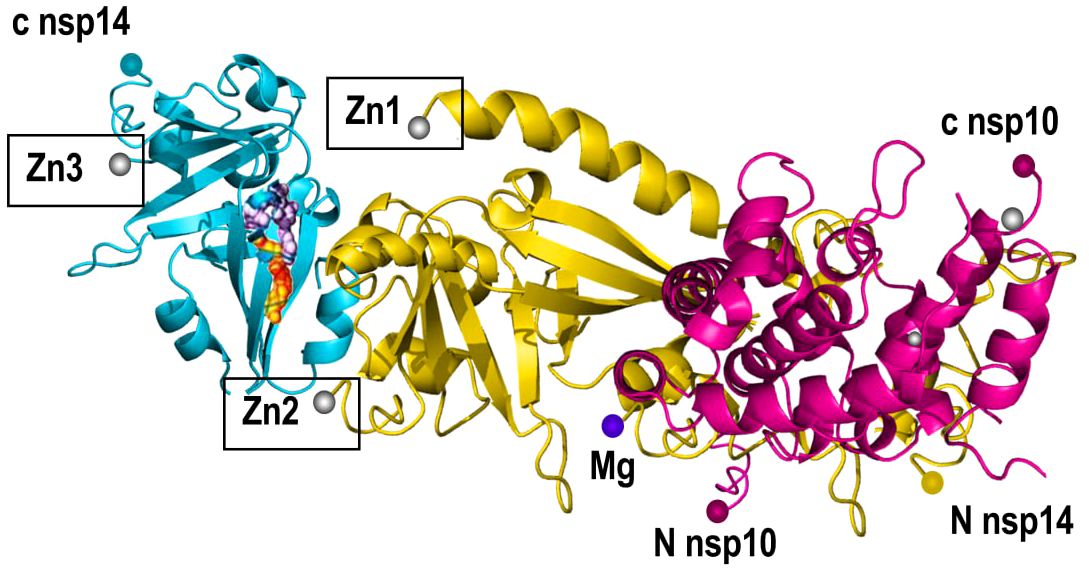
Figure 6 Structure of NSP14. NSP14 (RNA Helicase and Exoribonuclease) includes the N-terminal exonuclease domain (ExoN) and a C-terminal Guanine-N7-methyltransferase (N7-MTase) domain. NSP14 contains zinc fingers (Zn1,2,3) that contribute to the enzyme’s structural integrity and exonuclease activity enhancement.
The C-terminal Guanine-N7-methyltransferase (N7-MTase) domain of the NSP14 protein, when interacting with the NSP10 protein, plays a crucial role in the RNA capping process, which is essential for the stability and function of the viral mRNA. N7-MTase Domain is responsible for the methylation of the guanine base of the nascent RNA cap structure, which is a critical step in the formation of the 5’ cap of the viral mRNA. This cap structure is vital for mRNA stability and efficient translation (107). The interaction with NSP10 enhances the methyltransferase activity of NSP14. NSP10 acts as a cofactor and induces conformational changes in NSP14 that are necessary for the activation of the N7-MTase domain (108). Studies have shown that binding of NSP10 to NSP14 leads to significant structural rearrangements within NSP14, particularly in the ExoN domain. Researchers believe that these changes facilitate the formation of the active site for the N7-MTase domain. The N7-MTase activity of NSP14, enhanced by NSP10, is part of the complex process of viral mRNA capping. This process involves the addition of a methyl group to the N7 position of the guanine cap, which is crucial for the mRNA’s ability to be recognized by the host cell’s translational machinery. In summary, the interaction between NSP14’s N7-MTase domain and NSP10 is a key step in the SARS-CoV-2 life cycle, facilitating the proper capping of viral mRNA, which is essential for the virus’s replication and translation processes (107).
The proofreading mechanism relies on the ExoN activity of NSP14, which performs a critical role in retaining the CoV genome by reducing the amount of mutation of the RDRP of the virus, which is highly prone to error (26). ExoN Domain corrects errors introduced during RNA synthesis by the viral RdRp. It does so by removing mismatched nucleotides from the 3’ end of the nascent RNA strand. Also, NSP14 forms a complex with NSP10, which enhances the nuclease activity but not the methyltransferase activity (107). This interaction induces significant conformational changes within the ExoN domain of NSP14, facilitating the formation of the active site and thus stimulating the nuclease activity. Both the exonuclease and methyltransferase activities of NSP14 are essential to the viral life cycle (107). The exonuclease activity ensures the high fidelity of the viral genome by excising incorrectly incorporated nucleotides, while the methyltransferase activity is crucial for the proper capping of viral mRNA (107). The proofreading function of NSP14 is a key factor in the high replication fidelity of coronaviruses, which is unusual for RNA viruses. Research has found that mutations in NSP14 disrupt proofreading, severely limiting the virus’s function (99, 109–112). Prominently, research has shown that NSP14’s catalytic activity depends on Mg2+ (113).
The NSP14 protein of SARS-CoV-2, which possesses both exoribonuclease and methyltransferase activities, requires Mg2+ for its function. The ExoN domain of NSP14 is a DEDDh-type exonuclease that coordinates divalent Mg2+ at its catalytic center. These Mg2+ ions are crucial for the exonuclease activity, as they are involved in the hydrolysis of RNA, allowing NSP14 to remove mismatched nucleotides during RNA replication (114). Five residues, D90, E92, E191, H268, and D273, make up the catalytic center of the ExoN domain. These residues, D90, E92, E191, H268, and D273, conserve between SARS-CoV and SARS-CoV-2 and play an essential role in coordinating magnesium ions, which are necessary for catalytic activity (114). The magnesium-dependent exonuclease activity of NSP14 is part of the virus’s proofreading mechanism. This activity is critical for maintaining the integrity of the viral genome by correcting errors introduced during RNA synthesis by RdRp (107). In other words, the NSP14 protein consumes Mg2+ because they are integral to the catalytic function of the ExoN domain. This enables the protein to fulfill its role in the viral replication process by ensuring the accuracy of the viral RNA genome (107, 114).
Among the nonstructural proteins, Nsp16 holds great significance within the viral replication cycle owing to its indispensable role in coronavirus’ evasion of the immune system (76, 77, 115). Nsp16 is an enzyme referred to as a 2′-O-methyltransferase (2′-O-MTase) that is a component of the replication-transcription complex (116). The NSP16 protein of SARS-CoV-2, in complex with NSP10, is a 2′-O-methyltransferase that plays a crucial role in the modification of the viral RNA cap structure. NSP16 is comprises twelve β-strands, seven α-helices, and five 3–10 helices (117). These secondary structural elements assemble into a compact fold. NSP16 forms a stable heterodimer with NSP10, which is essential for its methyltransferase activity. NSP10 binds to NSP16 through a significant surface area, enhancing the enzyme’s function (118). The binding forms the active site of NSP16, where the methylation reaction occurs S-adenosyl-L-methionine (SAM) in a highly constricted pocket. This site plays a vital role in transferring a methyl group to the ribose 2′-O position of the nascent RNA cap. The methylation of the viral RNA cap by NSP16 helps the virus evade the host’s immune system by mimicking cellular mRNA, thus preventing recognition by the host innate immunity mechanisms (119). The intricate design of NSP16 enables it to carry out the essential function of RNA cap methylation, which is crucial for ensuring the stability and immune evasion of viral mRNA (117).
The transformation of mRNA molecules from the Cap-0 state to the Cap-1 state requires the creation of an intricate connection between Nsp16 and nsp10. To accomplish this alteration, the ribose 2’-O of the initial nucleotide of the nascent mRNA is methylated using SAM as the provider of methyl groups (72, 120). The recruitment of the translation factor eIF4E is supported by both Cap-0 and Cap-1 forms, but it is the Cap-0 form that specifically promotes IFIT1 binding to viral RNAs (121). Hence, the Cap-1 form not only augments the process of translation but also acts as a preventive measure against the triggering of the natural immune response, facilitated by genes stimulated by interferon, such as IFIT1 (77, 122, 123). When the gene NSP 16 of the SARS-CoV undergoes genetic disruption, there is a substantial reduction in the production of viral RNA by a factor of up to ten (124). Therefore, the cessation of nsp16 function ought to induce an immune response to CoV infection and restrict the progression of the disease (77, 122).
The NSP16 protein of COVID-19 virus, which functions as a 2′-O-methyltransferase, requires Mg2+ for its enzymatic activity. Mg2+ ions are essential cofactors for the catalytic activity of many enzymes, including 2′-O-methyltransferases like NSP16. They facilitate the correct positioning of the substrate and the methyl donor for the transfer reaction (125). During the methylation process, Mg2+ helps stabilize the transition state and the reaction intermediates, making the transfer of the methyl group to the RNA more efficient. Mg2+ is also involved in the binding of SAM, the methyl donor molecule, to NSP16. This binding is crucial for the methyl transfer reaction to occur. Mg2+ can contribute to the structural integrity of the protein, ensuring that the enzyme maintains its proper conformation necessary for its function (125). Although the search results do not provide specific details on NSP16, the general significance of Mg2+ ions in enzymatic reactions consumes how Mg2+, as previously outlined, is relevant for NSP16’s role in the viral life cycle. Mg2+ is vital for the enzyme’s activity, assisting in the process of RNA cap modification, which is crucial for the stability and immune evasion of the viral mRNA (125) (Figure 7).
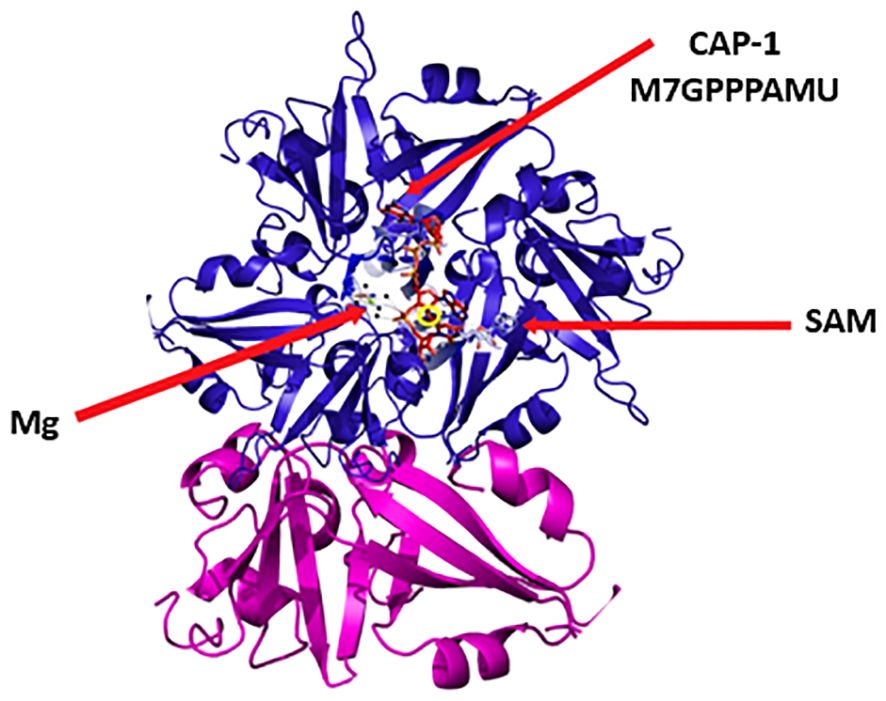
Figure 7 Structure of NSP16. NSP16 is involved in viral RNA capping, a critical process for immune evasion. It catalyzes the transfer of a methyl group from S-adenosyl methionine (SAM) to the 2’-OH group of the first RNA nucleotide. This modification converts the viral RNA cap from a Cap-0 structure to a Cap-1structure, mimicking cellular mRNAs.
In the realm of Coronaviruses, NSP10 serves as an allosteric activator for the process of 2’-O-ribose methylation, which involves the modification of the A1 base by the enzyme nsp16. In addition, NSP10 is responsible for the N7 methylation of the terminal guanine (G0) base in the unmethylated mRNA cap (G0pppA1), a process facilitated by NSP14 (72, 126). The prompt restoration of NSP10 is crucial for the enzymatic activity of NSP16/NSP10 and NSP14/NSP10 enhancing complexes, which becomes evident after methylation of A1 at the 2’-O position and the subsequent guanine-N7-methylation (127).
The genetic data that serves as the major source of information in mRNA is organized into groups of three nucleotides, known as codons (128). The recognition of each codon is done by the anticodon loop of a particular tRNA, which carries unique amino acids at its 3’ ends. Consequently, each codon serves to encode either a particular amino acid or deliver instructions to the translational apparatus, directing the termination of translation. The ribosome is a sophisticated molecular apparatus accountable for the convergence of mRNAs and tRNAs to commence protein synthesis. It employs the information embedded in the mRNAs to construct amino acids, which are furnished by the tRNAs, into an expanding polypeptide chain. This procedure is indispensable for the generation of proteins within cells (129). The ribosome consists of numerous rRNAs and proteins that are meticulously organized into discrete subunits referred to as the small subunit and the large subunit. The phenomenon referred to as translocation encompasses the gradual displacement of ribosomes along the mRNA in the direction from 5’ to 3’. This displacement occurs through the moving of mRNAs through the small subunit of the ribosome systematically, codon by codon. On the outer layer of the small subunit, located within a designated area referred to as the decoding center, the direct engagement between mRNA and tRNAs occurs. In this particular region, the codons of mRNAs establish strong bonds with the anticodon loops of tRNAs. Within the large subunit, the peptidyltransferase center, or PTC, acts as a second functional center. This specific region brings together the 3′ ends of tRNAs, enabling them to closely interact and facilitating the crucial procedure of establishing peptide bonds within an entirely RNA-exclusive environment (130). The binding site for elongation and termination factors forms a crucial functional center, often regarded as the third major one. The complex surface area of this structure consists of subunits that facilitate the attaching of trans-acting GTPases to the ribosome. These GTPases followed two purposes: (1) transporting aminoacyl-tRNAs to the ribosome, and (2) allowing the delivery of translocase/termination factors with a structurally similar (13). The interplay of these variables augments the inherent pace of peptide synthesis and guarantees the progression of ribosomes in the designated direction along the mRNA. Thus, considering the triplet structure of codons, each mRNA can harbor three different translational reading frames (13).
The issue of determining the translational reading frame has historical origins that date back to ancient times. It is noteworthy to mention that the ‘universal’ start signal for protein translation, famously known as AUG, is determined by a solitary codon that encodes methionine. In eukaryotes, the first AUG codon, when read from the 5’ to 3’ direction, functions as the key factor in determining the correct reading frame of a mRNA, marking the start of translation. Conversely, in polycistronic prokaryotic mRNAs harboring multiple ORFs (open reading frames), the process of reinitiation of translation presents a more intricate scenario (131).
Researchers extensively documented the initiation of translation at codons other than AUG and the bypassing of the initial AUG codons, and they typically consider these occurrences as deviations from the general norm. In expanding our comprehension of the overarching principle, scientists have proven that the first AUG codon defines the translational reading frame (132). Recent data from ribosome profiling experiments suggests that a notable proportion of translation initiation events occur at a limited subset of non-AUG codons (133).
To comprehend the establishment of a translational reading frame, it is imperative to grasp the influence of the ribosome’s structure on its function. The small subunit of the ribosome consists of a solitary species of rRNA found in all organisms. This rRNA varies in size, ranging from 16S in bacteria and archaea to 18S in eukaryotes. Additionally, the small subunit contains a minimum of 21 proteins, which increases to a maximum of 33 in more advanced organisms (134). As previously discussed, the decoding center of the small subunit, located near the 3’ end of its rRNA, facilitates base pairing interactions between mRNA codons and tRNA anticodons. On the other hand, the large subunit of bacteria and archaea is composed of two rRNAs, 23S and 5S, along with approximately 31 proteins. In eukaryotes, the proteinaceous component of the LSU has expanded to up to 49 proteins, and a small portion of the 23S rRNA has become detached and transformed into a distinct rRNA known as 5.8S. Interestingly, despite being a separate molecule, structural analyses have revealed that the 5.8S rRNA occupies the same position as its corresponding sequence in bacterial and archaeal ribosomes, situated along the ‘rear’ solvent-accessible side of the large subunit (135–137). Another notable difference in eukaryotes is the expansion of the major large subunit rRNA, which ranges from 25S rRNA in yeast to 28S rRNA in metazoans. The large subunit interacts with the small subunit and possesses three distinct pockets for the binding of tRNAs: The A-site specifically binds aminoacyl-tRNAs, the P-site binds initiator tRNAs, and tRNAs attached to elongating polypeptides (peptidyl-tRNAs), and the E- site binds deacylated tRNAs. Additionally, a tunnel in the PTC facilitates the expulsion of nascent peptides, allowing them to exit from the ‘back’ side of the large subunit (135). Moreover, trans-acting factors play a role in transporting aminoacyl-tRNAs and engaging in termination by being recruited through interactions with both the small and large subunits (138). While these functional centers are the focal points of the ribosome, it is worth mentioning that the ribosome also contains supplementary functional components (13).
After setting up the correct reading frame on mRNA, the ribosome’s primary job is to keep it stable during the rest of the translation process, known as the elongation phase. The elongation process starts with elongator tRNAs at the ribosome, aided by a trans-acting factor called EF-Tu in bacteria/archaea and eEF1A in eukaryotes, along with GTP. This results in the formation of the ternary complex (TC). The decoding center in the A-site of the small subunit receives the anticodon loop of the elongator aa-tRNA. Here, a correct match between the mRNA codon and aminoacyl‐tRNAs anticodon leads to the creation of a mini-helix. Afterward, these additional interactions involving both small subunit rRNA bases and proteins recognize and stabilize this mini-helix (139). This interaction causes a structural rearrangement of the small subunit and aminoacyl‐tRNAs, which in turn transduces information that triggers the hydrolysis of GTP by EF-Tu/eEF1A. Consequently, the elongation factor releases the tRNA (140). The aminoacylated 3’ end of the aminoacyl‐tRNAs then proceeds to move through a structural element referred to as the accommodation corridor, from the exterior of the large subunit, and into the A-site side of the PTC. Concurrently, the ribosome frees EF-Tu/eEF1A + GDP to be re-energized with GTP and aminoacyl-tRNAs (141). In the PTC, catalysis - also known as peptidyltransfer - occurs as the initiator-tRNA transfers methionine to the elongator tRNA. This process involves both steric positioning and active catalysis through a transesterification reaction by the ribosome. Following this process, the 3’ end of the deacylated tRNA moves to the E-site on the large subunit, while the 3’ end of the dipeptidyl-tRNA moves to the P-site of the large subunit. Meanwhile, the anticodon loops of both tRNAs remain bound to the P- and A-sites of the small subunit, respectively. This particular conformation is referred to as the ‘hybrid state’ because the tRNAs occupy one site on the large subunit and another on the small subunit (142). Furthermore, it is at this step that the ribosome undergoes a reorientation from the ‘classical’ or ‘unrotated’ state to the ‘ratcheted’ or ‘rotated’ state. This process involves a complex spatial repositioning of the two subunits relative to one another (143, 144).
The subsequent step in the process involves the introduction of reading frame maintenance, known as translocation. This stage entails the recruitment of a secondary trans-acting factor, EF-G/eEF2, to the ribosome. The hydrolysis of GTP by this protein induces a transitional state for translocation, in which the ribosome disengages from the tRNA-mRNA complexes, permitting the movement of the anticodons of the P and A-site tRNAs. This movement, in conjunction with the release of the elongation factor, leads to the complete occupation of the E and P-sites by the deacylated tRNA and dipeptidyl-tRNA respectively, while the A-site remains empty, thus preparing it for the next aminoacyl‐tRNAs to decode the subsequent codon (141). Scientists theorize that the bipartite structure of the ribosome, composed of a large subunit and a small subunit, separates the movement of the tRNA body on the large subunit from that of the mRNA/tRNA complexes on the small subunit. This segregation allows for the effective preservation of the translational reading frame (144). More recently, advanced techniques with high resolution have uncovered several structural characteristics that are hypothesized to work collectively to ensure the accurate positioning of tRNAs within the ribosome and to precisely limit translocation to three nucleotides (143). Subsequent rounds of elongation repeat this process until the ribosome encounters a termination codon (UUA, UGA, or UAG).
Release factors (RF1 and RF2 in bacteria/archaea, and the eF1-eRF3 complex in eukaryotes) specifically identified termination codons that imitate the structure of the TC and possess a particular affinity for termination codons (145). The absence of an amino donor site in the RFs allows a water molecule to enter the PTC, thus facilitating the hydrolysis of the C-terminus of the nascent polypeptide chain from the peptidyl-tRNA, resulting in the release of the peptide (146).
While it is evident that the translational machinery must faithfully preserve the reading frame, the modification of translational accuracy may prove advantageous in particular situations. Many viruses employ numerous molecular mechanisms, collectively known as translational recoding (147). These mechanisms encompass various strategies, such as redirecting elongating ribosomes to shift into an alternate reading frame, guiding ribosomes to utilize alternative start sites, and bypassing or altering termination codons (132). This becomes especially pertinent when genomic capacity is physically limited, as is the case in viruses where the size of the genome restricts by the volume of the viral particle. In such scenarios, enhancing the information content of a viral mRNA by enabling it to encode multiple proteins could confer a selective advantage. Another hypothesis posits that the ability of a single RNA molecule to encode multiple proteins without modifying its sequence, such as through splicing, might have provided a selective advantage in the prebiotic RNA world (148). Furthermore, the ability to recode mRNAs offers an additional level of control over gene expression. Importantly, these mechanisms are all “programmed” to occur at specific sequences through cis-acting elements present on mRNAs, and they occur at rates that are two or more orders of magnitude higher than non-programmed events (13).
Ribosomal frameshift comprises a biological event that happens along translation that leads to the generation of multiple special proteins by a single mRNA (14). Ribosomal frameshift can be planned by the nucleotide sequence available in mRNA, sometimes influenced by the secondary and tridimensional mRNA complex (149). Typically, -1 ribosomal frameshifting characterizes in viruses (particularly retroviruses, astroviruses, and coronaviruses) bacterial insertion elements, and retrotransposon, and also has been observes in some cellular genes. (150). In other words, -1 ribosomal frameshifting or -1 PRF is a process whereby mRNA cis-acting elements conduct elongating ribosomes to displace the reading frame as much as 1 nucleotide toward 5′. Initially, the Rous sarcoma virus used the -1 ribosomal frame-shifting process for the expression of viral genes (151) followed by some retroviruses (152). By examining a sequence of three nucleotides found on a molecule of messenger RNA, known as codons, the process of protein translation occurs from the 5’ to the 3’ direction of the mRNA strand. This translation process commences with the amino acid methionine, specifically with the initiation codon AUG. Through the process of translation, a per codon becomes an amino acid. The code is designed to be degenerate in nature, allowing for the identification of a particular amino acid through multiple codons. Nevertheless, a relocation of several nucleotides that is not partible upon 3 in the reading format will subsequently next codons to be read variously (153), which impressively alters the ribosomal reading frame.
-1 ribosomal frameshifting can be divided into three distinct sections composed of one slippery site, one connector zone, and an irritating area downstream of the secondary structure of mRNA, usually a mRNA pseudo-knot (13). The initial sequence of the slippery site and its position in association with the input translational reading frame is very important: Must include one of these options N NNW WWZ, Where NNN is tensional of three of the same nucleotides, WWW is either A or U and Z ≠ G. The connector area is less well described. Although it is relatively small, it has a significant impact on determining -1 ribosomal frameshifting. Pausing elongating ribosomes at the stop codon is a crucial step in forming downstream secondary structures, which is essential for inducing efficient -1 ribosomal frameshifting (154). The usually adopted process of -1 ribosomal frameshifting is that the secondary structure of mRNA guides prolonging ribosomes to stop with its A- and P-site attached to aminoacyl- and peptidyl-tRNAs, which on the slippery site are located. The sequence of the slippery site permits for re-coupling of the tRNAs to the -1 framework codons after they “concurrently slippage” by one base toward 5′ in the sequel of the mRNA. The next separation of the downstream secondary structure of mRNA permits the ribosome to carry on the lengthening of the newfangled polypeptide within the new translational reading frame. The downstream irritating components are usually H-type mRNA pseudo-knots. The reason for its name is because they are consists of two superposed with the same axis stem loops whose latter stem comprises base coupling among the sequence in the loop of the first-stem loop, and the supernumerary downstream sequence (155). Because of the presence of a third, inner stem-loop component, the COVID-19 pseudo-knot becomes exceedingly complicated (156–158). Mutations effective in this structure reduced the amount of -1 ribosomal frameshifting and had harmful impacts on virus dissemination, therefore, proposing that may aim for treatment use of small molecules (159, 160). Besides, researchers have recorded the attendance of a hairpin that was promptly deployed 5’ of the slippery site to adjust -1 ribosomal frameshifting by reducing its action (161) (Figure 8).
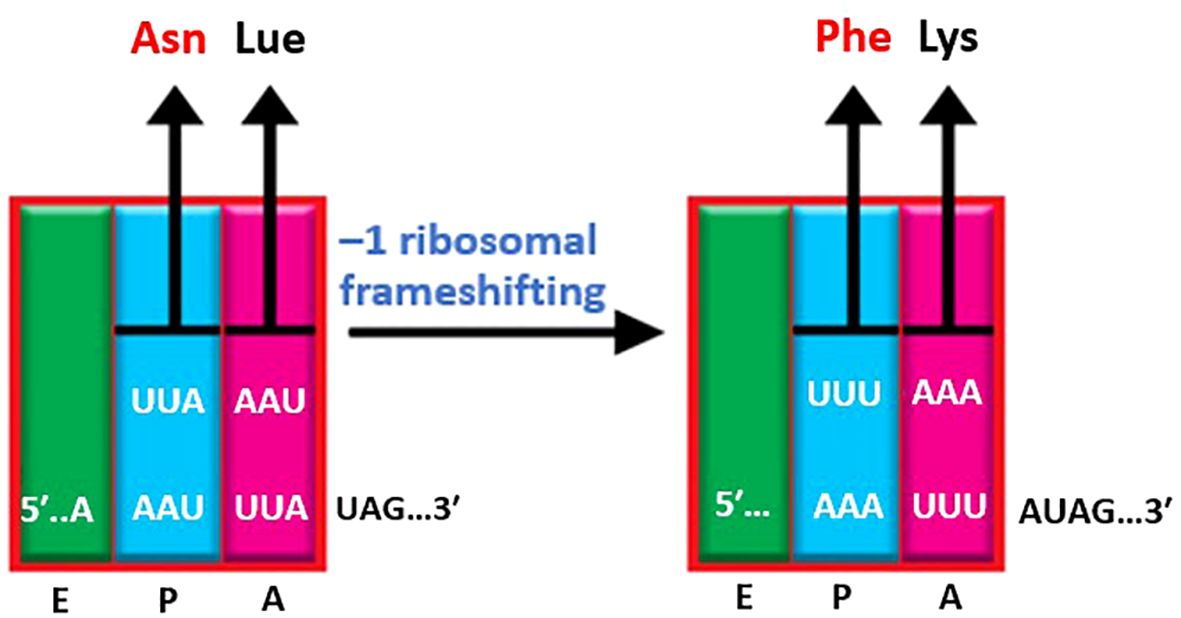
Figure 8 –1 ribosomal frameshifting. Ribosomal frameshifting is an abiological phenomenon in COVID- that occurs during translation. When the ribosome encounters a specific sequence (usually a slippery sequence) in the mRNA, it shifts its reading frame by one nucleotide. As a result, the reading of subsequent codons alters, resulting in the synthesis of a different protein. This process allows a single mRNA to produce multiple unique proteins, contributing to the complexity of cellular processes.
The presence of divalent cation is of significant importance in the regulation of essential cellular processes. Researchers have established that an excessive accumulation of these cations contributes to aging and to neurodegenerative diseases and cancer (162–165). Divalent cation-dependent metalloproteins and metalloenzymes play a central role in regular physiological processes and pathological conditions (166–170). The importance of these proteins is clear in their role in vital cellular functions, for instance, signal transduction (171), cell division (172), excretion (173), transcription of genes (174), immune response regulation (175, 176), and cell adhesion (177, 178). This is why eukaryotic cells possess multiple receptors and ligands that facilitate these divalent cations sorting, transportation, and delivery (179–191). In the realm of host-pathogen interactions, divalent cations assume prominent roles (192–194). It is crucial to regulate the levels of these cations in order to maintain homeostasis, as they have the potential to affect microbial infection (195, 196). The COVID-19 virus relies on Mg2+ for the function of its NSP 13, NSP 14, and NSP 16 proteins. Furthermore, within the array of viruses associated with the -1 ribosomal frameshifting event, HIV stands out as a significant example. Emphasizing the crucial role of Mg2+, specific enzymes in the HIV virus rely heavily on its presence and interaction for efficient functioning.
The role of RT (reverse transcriptase) in HIV (human immunodeficiency virus) is crucial for the process of converting viral RNA into viral DNA. RT consists of two subunits, p66 and p51, which are activated by viral proteases originating from the Gag-Pol polyprotein. Furthermore, the activity of RT necessitates the presence of divalent cations, specifically Mg2+ and Mn2+ (Manganese) (197). The functions of both enzymes of RNAase H and DNA polymerase rely on the presence of both subunits (p66 and p51), resulting in the production of ds-DNA (198, 199). RNAase H (subunit p51) possesses specific sites for the attachment of divalent cations, such as Mn2+ and Mg2+ (200–202).
Metal ions like Mg2+, Na+ (sodium), and K+ (potassium) are of significant importance in various biological processes as they interact with nucleic acids, particularly RNA (203). The specific properties of Mg2+ make it particularly well-suited for neutralizing the negative charge density associated with the RNA phosphate backbone for two reasons (204). Firstly, it is the most abundant multivalent cation found within cells (205). Secondly, it possesses the highest charge density among all biologically available ions due to its relatively small ionic radius of 0.6 Å. The binding of Mg2+ ions to RNA can occur in two different general modes: (1) a “diffuse binding” mode which involves nonspecific long-range electrostatic interactions with Mg2+ hexahydrate, and (2) a “site binding” mode which involves specific coordination of anionic ligands to partially dehydrated Mg2+ (206). Both modes are crucial for the structural stabilization of RNA (204, 207). For years, it has been well recognized that the ribosome’s configuration and function are significantly impacted by the presence of metal ions, especially Mg2+. For instance, the combination of the small and large ribosomal subunits in vitro to create intact ribosomes is heavily reliant on the concentration of Mg2+ ions (208–210). Moreover, Mg2+ deficiency in Escherichia coli cells leads to a depletion of ribosomes (211), thus indicating the vital role of Mg2+ in assembling and structurally stabilizing ribosomes. In line with this, if various polyamines are used to replace Mg2+ in purified preparations of E. coli large ribosomal subunits, it leads to irreversible unfolding and loss of peptidyl transferase activity (212). Mg2+ also influences other ribosomal activities, such as poly-U-directed phenylalanine polymerization (213), the binding of polynucleotides to ribosomes (214), and the attachment of ribosomes to endoplasmic reticulum membranes (215). However, it is challenging to ascertain the extent to which these effects are direct or indirect consequences of structural disturbances to the ribosome (204). Although other divalent cations may exhibit sufficient similarity to Mg2+ to allow for coordination in its place, the divergent characteristics of these cations can have an impact on the biomolecule to which they are bound. For instance, kinases, which typically rely on Mg2+, can form associations with other trace metal ions but experience a decrease in efficacy (216). Divalent metal cations are essential for DNA polymerases, with Mg2+ being the most commonly employed in this capacity. While coordination with alternate metal cofactors such as Mn2+ and Co2+ (Cobalt) can enhance enzymatic activity, it can also have a detrimental effect on fidelity and potentially lead to carcinogenicity (217–219). Ribosomes in eukaryotic cells have the responsibility of conducting protein synthesis and exist as large molecular complexes. Within the process of translation, the smaller 40S subunit and larger 60S subunit come together. The presence of Mg2+ is of utmost importance as they partake in the critical task of stabilizing the structure of the ribosome through their binding to the phosphate backbones of rRNA. Additionally, Mg2+ can enhance the binding of tRNA to the ribosome during the decoding of mRNA, resulting in facilitating precise protein synthesis. They also contribute to the proper folding of rRNA and effectively maintain the active site of the ribosome required for the formation of peptide bonds. Moreover, Mg2+ play a significant role in coordinating ribosomal proteins with rRNA, which in turn assists in the assembly of ribosomal subunits. Additionally, they have a crucial function in the regulation of ribosome biogenesis and the maintenance of pre-ribosomal particle stability throughout maturation (Figure 9).
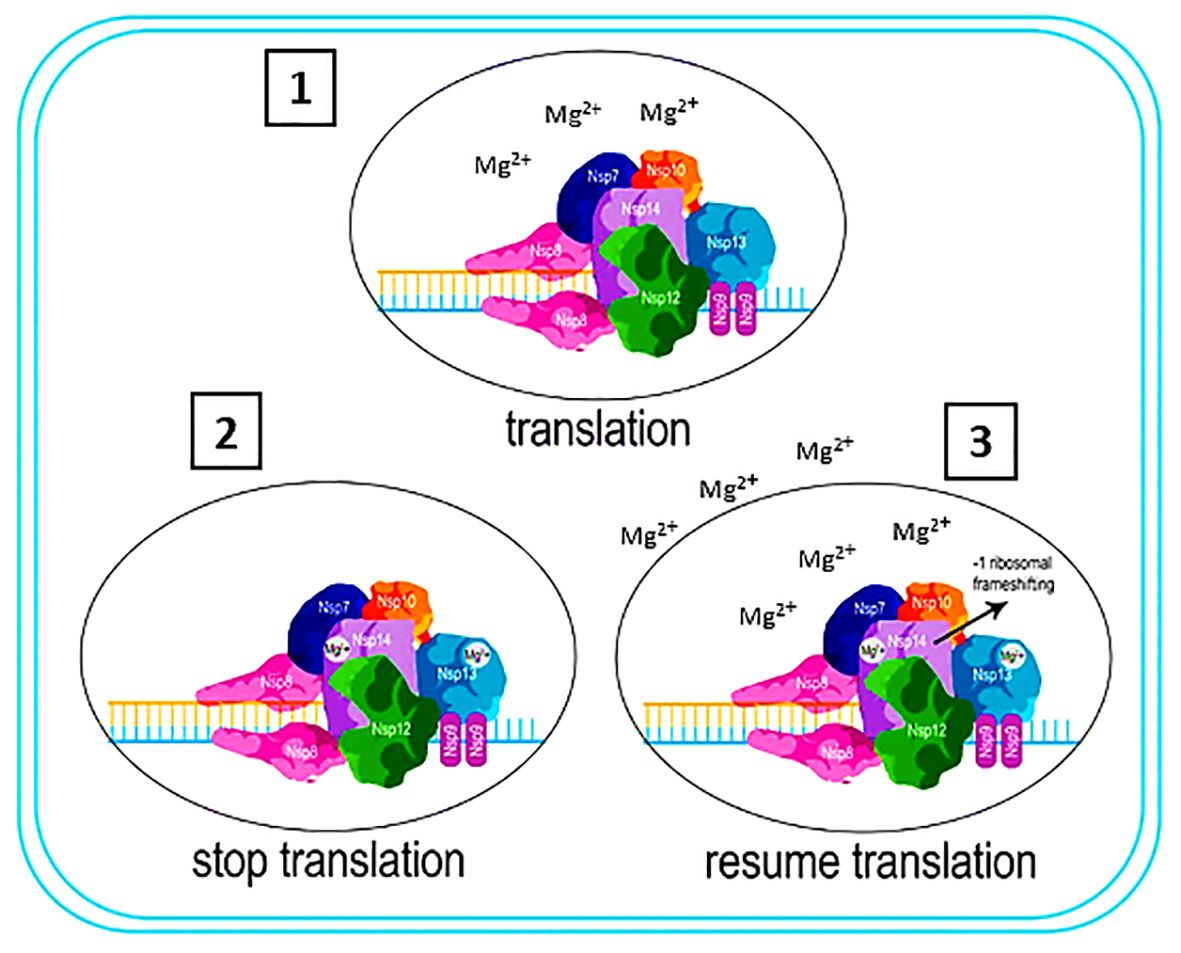
Figure 9 Interaction of NSP13,14, 16 in the pathogenicity of the COVID-19 virus. 1) NSP 13, NSP 14, and NSP 16 are actively involved in the translational process through Mg2+ acquisition from the ribosome. 2) The depletion of Mg2+ by these proteins induces a state of instability within the ribosome, consequently halting the translation process. 3) Ribosome acquires Mg2+ from extracellular sources, allowing the translation process to resume. NSP 14 move back one nucleotide to resume the translation process, resulting in -1 ribosomal frameshifting.
In modern molecular biology, researchers have observed numerous instances where they initially identified fundamental biological regulatory mechanisms in viruses. This is not because of any uniqueness of viruses, but because their compact genomes enhance the signal-to-noise ratios, thus facilitating the process of scientific discovery. Viruses, as intracellular parasites, follow the same rules and regulations that apply to their host cells. Consequently, although researchers initially considered −1 PRF to be specific to viruses, it is now evident that cellular mRNAs also employ this mechanism. The exploration of PRF continues to shed light on our comprehension of the normal maintenance of the reading frame by ribosomes. Generally, −1 PRF involves a heptameric slippery site, a short spacer, and a complex tertiary mRNA structure known as an H‐type pseudoknot. This phenomenon can occur at three distinct stages of translation at the frameshift signal. The pseudoknot can induce a two-nucleotide translocation event either when the ribosome enters or exits the slippery site. Alternatively, the accommodation of the aminoacyl tRNA (aa‐tRNA) into the slippery site results in the downstream mRNA being pulled into the ribosome by a distance of 9Å, which creates a tension between the slippery site and pseudoknot. This tension is subsequently alleviated by decoupling tRNAs from the mRNA, causing the mRNA to slip backward by one base. As mentioned, viruses in the Coronaviridae and Retroviridae families undergo genetic mutations of type -1 ribosomal frameshifting. On the other hand, NSP 13, NSP14 and NSP 16 proteins in COVID-19 virus need Mg2+ for their function. The best source of Mg2+ for these proteins is the ribosome because the stability of the ribosome is dependent on the Mg2+, any interference in the gradient of the Mg2+ concentration will disrupt the function of the ribosome and, as a result, stop the translation process. In other words, NSP13, NSP14, and NSP16 proteins, by consuming Mg2+ present in the ribosome, stop the function of the ribosome and convert it from polysome to monosome, which results in the stop of translation. This stopping of translation plays a role in the initial formation of the -1 ribosomal frameshifting phenomenon. Since the ribosome needs to obtain Mg2+ from various sources to continue the translation process, if it encounters the start codon, NSP14, which has the role of proofreading, pulls back a codon, causing the -1 ribosomal frameshifting phenomenon to occur.
AK: Writing – original draft, Writing – review & editing. AN: Writing – review & editing, Data curation. JH: Writing – review & editing, Data curation. MT: Writing – review & editing. FN: Writing – review & editing. SAH: Writing – review & editing
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by Student Research Committee of Hamadan University of Medical Sciences, Hamadan, Iran.
We would like to extend our thanks to the microbiology department at Hamadan University of Medical Sciences, and we would also like to offer a special recognition to Dr. Babak Asghari for their invaluable support in the preparation of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
CoV, Coronavirus; HCoV, Human coronavirus; ACE2, Angiotensin-converting enzyme 2; RBD, Receptor Binding Domain; RBM, Receptor-Binding Motif; HE, Hemagglutininesterase; NSP, Non-structural protein; TMPRSS2, Transmembrane protease serine 2; NSP, Non-structural protein; ssRNA, single-stranded RNA; ssDNA, single-stranded DNA; TRS, Transcriptional regulating sequence; NiRAN, Nidovirus RdRp-associated nucleotidyltransferase; -1 PRF, −1 programmed ribosomal frameshifting; ZBD, Zinc binding domain; RDRP, RNA polymerase dependent-RNA; EVA, Equine Viral Arteritis; PRRS, Porcine reproductive and respiratory syndrome virus; ORF, Open Reading Frame; PTC, Protein Translocating Channel; TC, Termination Codon; PHB 1, Prohibitin 1; PHB 2, Prohibitin 2; ADRP, Adipose differentiation-related protein; VOI, Variant Of Interest; VOC, Variant Of Concern; eIF4E, Eukaryotic translation initiation factor 4E; SARS‐CoV, severe acute respiratory syndrome coronavirus; SARS‐Cov‐2, severe acute respiratory syndrome coronavirus 2.
1. Lu H, Stratton CW, Tang YW. Outbreak of pneumonia of unknown etiology in Wuhan, China: The mystery and the miracle. J Med Virol. (2020) 92:401. doi: 10.1002/jmv.25678
2. Paraskevis D, Kostaki EG, Magiorkinis G, Panayiotakopoulos G, Sourvinos G, Tsiodras. Full-genome evolutionary analysis of the novel corona virus, (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infection Genet Evol. (2020) 79:104212. doi: 10.1016/j.meegid.2020.104212
3. Sohrabi C, Alsafi Z, O’neill N, Khan M, Kerwan A, Al-Jabir A, et al. World Health Organization declares global emergency: A review of the 2019 novel coronavirus (COVID-19). Int J Surg. (2020) 76:71–6. doi: 10.1016/j.ijsu.2020.02.034
4. Burrell AJ, Pellegrini B, Salimi F, Begum H, Broadley T, Campbell LT, et al. Outcomes for patients with COVID-19 admitted to Australian intensive care units during the first four months of the pandemic. Med J Aust. (2021) 214:23–30. doi: 10.5694/mja2.51314
5. Cherry J, Demmler-Harrison GJ, Kaplan SL, Steinbach WJ, Hotez PJ. Feigin and Cherry’s Textbook of Pediatric Infectious Diseases E-Book: 2-Volume Set. Philadelphia, PA, USA: Elsevier Health Sciences (2013).
7. Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. (2020) 10:766–88. doi: 10.1016/j.apsb.2020.02.008
8. Cui J, Li F, Shi Z-L. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. (2019) 17:181–92. doi: 10.1038/s41579-018-0118-9
9. Payne S. Family coronaviridae. Viruses. (2017) 149:708–717. doi: 10.1016/B978-0-12-803109-4.00017-9
10. Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. nature. (2020) 579:270–3. doi: 10.1038/s41586-020-2172-7
11. Lefkowitz EJ, Dempsey DM, Hendrickson RC, Orton RJ, Siddell SG, Smith DB. Virus taxonomy: the database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. (2018) 46:D708–17. doi: 10.1093/nar/gkx932
12. De Wilde AH, Snijder EJ, Kikkert M, Van Hemert MJ. Host factors in coronavirus replication. Roles Host Gene non-coding RNA Expression Virus infection. (2018) 1–42.
13. Dinman JD. Mechanisms and implications of programmed translational frameshifting. Wiley Interdiscip Reviews: RNA. (2012) 3:661–73. doi: 10.1002/wrna.1126
14. Atkins JF, Loughran G, Bhatt PR, Firth AE, Baranov PV. Ribosomal frameshifting and transcriptional slippage: From genetic steganography and cryptography to adventitious use. Nucleic Acids Res. (2016) 44:7007–78. doi: 10.1093/nar/gkw530
15. Knoops K, Kikkert M, Worm SHVD, Zevenhoven-Dobbe JC, van der Meer Y, Koster AJ, et al. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PloS Biol. (2008) 6:e226. doi: 10.1371/journal.pbio.0060226
16. Lokugamage KG, Narayanan K, Huang C, Makino S. Severe acute respiratory syndrome coronavirus protein nsp1 is a novel eukaryotic translation inhibitor that represses multiple steps of translation initiation. J Virol. (2012) 86:13598–608. doi: 10.1128/JVI.01958-12
17. Cornillez-Ty CT, Liao L, Yates Iii JR, Kuhn P, Buchmeier MJ. Severe acute respiratory syndrome coronavirus nonstructural protein 2 interacts with a host protein complex involved in mitochondrial biogenesis and intracellular signaling. J Virol. (2009) 83:10314–8. doi: 10.1128/JVI.00842-09
18. Lindner HA, Lytvyn V, Qi H, Lachance P, Ziomek E, Ménard R. Selectivity in ISG15 and ubiquitin recognition by the SARS coronavirus papain-like protease. Arch Biochem biophysics. (2007) 466:8–14. doi: 10.1016/j.abb.2007.07.006
19. Saikatendu KS, Joseph JS, Subramanian V, Clayton T, Griffith M, Moy K, et al. Structural basis of severe acute respiratory syndrome coronavirus ADP-ribose-1 ″-phosphate dephosphorylation by a conserved domain of nsP3. Structure. (2005) 13:1665–75. doi: 10.1016/j.str.2005.07.022
20. Frieman M, Ratia K, Johnston RE, Mesecar AD, Baric RS. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-κB signaling. J Virol. (2009) 83:6689–705. doi: 10.1128/JVI.02220-08
21. Hognon C, Miclot T, Garcı́a-Iriepa C, Francés-Monerris A, Grandemange S, Terenzi A, et al. Role of RNA guanine quadruplexes in favoring the dimerization of SARS unique domain in coronaviruses. J Phys Chem Lett. (2020) 11:5661–7. doi: 10.1021/acs.jpclett.0c01097
22. Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus, (2019-nCoV) in vitro. Cell Res. (2020) 30:269–71. doi: 10.1038/s41422-020-0282-0
23. Cottam EM, Whelband MC, Wileman T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy. (2014) 10:1426–41. doi: 10.4161/auto.29309
24. Te Velthuis AJ, Van Den Worm SH, Snijder EJ. The SARS-coronavirus nsp7+ nsp8 complex is a unique multimeric RNA polymerase capable of both de novo initiation and primer extension. Nucleic Acids Res. (2012) 40:1737–47. doi: 10.1093/nar/gkr893
25. Miknis ZJ, Donaldson EF, Umland TC, Rimmer RA, Baric RS, Schultz LW. Severe acute respiratory syndrome coronavirus nsp9 dimerization is essential for efficient viral growth. J Virol. (2009) 83:3007–18. doi: 10.1128/JVI.01505-08
26. Bouvet M, Imbert I, Subissi L, Gluais L, Canard B, Decroly E. RNA 3’-end mismatch excision by the severe acute respiratory syndrome coronavirus nonstructural protein nsp10/nsp14 exoribonuclease complex. Proc Natl Acad Sci. (2012) 109:9372–7. doi: 10.1073/pnas.1201130109
27. Gao Y, Yan L, Huang Y, Liu F, Zhao Y, Cao L, et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. (2020) 368:779–82. doi: 10.1126/science.abb7498
28. Ahn D-G, Choi J-K, Taylor DR, Oh J-W. Biochemical characterization of a recombinant SARS coronavirus nsp12 RNA-dependent RNA polymerase capable of copying viral RNA templates. Arch Virol. (2012) 157:2095–104. doi: 10.1007/s00705-012-1404-x
29. Tanner JA, Watt RM, Chai Y-B, Lu L-Y, Lin MC, Peiris JM, et al. The severe acute respiratory syndrome (SARS) coronavirus NTPase/helicase belongs to a distinct class of 5′ to 3′ viral helicases. J Biol Chem. (2003) 278:39578–82. doi: 10.1074/jbc.C300328200
30. Adedeji AO, Marchand B, Te Velthuis AJ, Snijder EJ, Weiss S, Eoff RL, et al. Mechanism of nucleic acid unwinding by SARS-CoV helicase. PloS One. (2012) 7:e36521. doi: 10.1371/journal.pone.0036521
31. Bianchi M, Benvenuto D, Giovanetti M, Angeletti S, Ciccozzi M, Pascarella S. Sars-CoV-2 envelope and membrane proteins: structural differences linked to virus characteristics? BioMed Res Int. (2020) 2020. doi: 10.1155/2020/4389089
32. Hulswit R, De Haan C, Bosch B-J. Coronavirus spike protein and tropism changes. Adv Virus Res. (2016) 96:29–57. doi: 10.1016/bs.aivir.2016.08.004
33. Thomas S. The structure of the membrane protein of SARS-CoV-2 resembles the sugar transporter SemiSWEET. Pathog Immun. (2020) 5:342. doi: 10.20411/pai.v5i1
34. Walls AC, Park Y-J, Tortorici MA, Wall A, Mcguire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. (2020) 181:281–292.e286. doi: 10.1016/j.cell.2020.02.026
35. Tang T, Bidon M, Jaimes JA, Whittaker GR, Daniel S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Res. (2020) 178:104792. doi: 10.1016/j.antiviral.2020.104792
36. White JM, Delos SE, Brecher M, Schornberg K. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol. (2008) 43:189–219. doi: 10.1080/10409230802058320
37. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. cell. (2020) 181:271–80. doi: 10.1016/j.cell.2020.02.052
38. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. (2020) 367:1260–3. doi: 10.1126/science.abb2507
39. Zou J, Yin J, Fang L, Yang M, Wang T, Wu W, et al. Computational prediction of mutational effects on SARS-CoV-2 binding by relative free energy calculations. J Chem Inf modeling. (2020) 60:5794–802. doi: 10.1021/acs.jcim.0c00679
40. Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med. (2020) 14:185–92. doi: 10.1007/s11684-020-0754-0
41. Law PT, Wong C-H, Au TC, Chuck C-P, Kong S-K, Chan PK, et al. The 3a protein of severe acute respiratory syndrome-associated coronavirus induces apoptosis in Vero E6 cells. J Gen Virol. (2005) 86:1921–30. doi: 10.1099/vir.0.80813-0
42. Tan Y-J, Tham P-Y, Chan DZ, Chou C-F, Shen S, Fielding BC, et al. The severe acute respiratory syndrome coronavirus 3a protein up-regulates expression of fibrinogen in lung epithelial cells. J Virol. (2005) 79:10083–7. doi: 10.1128/JVI.79.15.10083-10087.2005
43. Lu W, Zheng B-J, Xu K, Schwarz W, Du L, Wong CK, et al. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc Natl Acad Sci. (2006) 103:12540–5. doi: 10.1073/pnas.0605402103
44. Minakshi R, Padhan K, Rani M, Khan N, Ahmad F, Jameel S. The SARS Coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PloS One. (2009) 4:e8342. doi: 10.1371/journal.pone.0008342
45. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. (2003) 426:450–4. doi: 10.1038/nature02145
46. Ramsey I, Spibey N, Jarrett O. The receptor binding site of feline leukemia virus surface glycoprotein is distinct from the site involved in virus neutralization. J Virol. (1998) 72:3268–77. doi: 10.1128/JVI.72.4.3268-3277.1998
47. Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. nature. (2020) 581:215–20. doi: 10.1038/s41586-020-2180-5
48. Xu X, Chen P, Wang J, Feng J, Zhou H, Li X, et al. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci China Life Sci. (2020) 63:457–60. doi: 10.1007/s11427-020-1637-5
49. Li Y, Zhou W, Yang L, You R. Physiological and pathological regulation of ACE2, the SARS-CoV-2 receptor. Pharmacol Res. (2020) 157:104833. doi: 10.1016/j.phrs.2020.104833
50. Qiu Y, Zhao Y-B, Wang Q, Li J-Y, Zhou Z-J, Liao C-H, et al. Predicting the angiotensin converting enzyme 2 (ACE2) utilizing capability as the receptor of SARS-CoV-2. Microbes infection. (2020) 22:221–5. doi: 10.1016/j.micinf.2020.03.003
51. Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. (2020) 46:586–90. doi: 10.1007/s00134-020-05985-9
52. Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z, et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. (2020) 181:894–904.e899. doi: 10.1016/j.cell.2020.03.034
53. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. (2020) 367:1444–8. doi: 10.1126/science.abb2762
54. Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. (2020) 5:562–9. doi: 10.1038/s41564-020-0688-y
55. Bosch BJ, Martina BE, van der Zee R, Lepault J, Haijema BJ, Versluis C, et al. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc Natl Acad Sci. (2004) 101:8455–60. doi: 10.1073/pnas.0400576101
56. Xia S, Liu M, Wang C, Xu W, Lan Q, Feng S, et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. (2020) 30:343–55. doi: 10.1038/s41422-020-0305-x
57. Hayashi T, Yaegashi N, Konishi I. Effect of RBD mutation (Y453F) in spike glycoprotein of SARS-CoV-2 on neutralizing antibody affinity. bioRxiv. (2020). doi: 10.1101/2020.11.27.401893
58. Assessment, R.R. Detection of new SARS-CoV-2 variants related to mink. Stockholm, Sweden: Eur. Cent. Dis. Prev. Control (2020).
59. Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KH, Dingens AS, et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. cell. (2020) 182:1295–1310.e1220. doi: 10.1016/j.cell.2020.08.024
60. Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medrxiv. (2020). doi: 10.1101/2020.12.21.20248640
61. Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol. (2021) 19:409–24. doi: 10.1038/s41579-021-00573-0
62. Tian F, Tong B, Sun L, Shi S, Zheng B, Wang Z, et al. N501Y mutation of spike protein in SARS-CoV-2 strengthens its binding to receptor ACE2. elife. (2021) 10:e69091. doi: 10.7554/eLife.69091
63. Akkiz H. Implications of the novel mutations in the SARS-CoV-2 genome for transmission, disease severity, and the vaccine development. Front Med. (2021) 8:636532. doi: 10.3389/fmed.2021.636532
64. Gan HH, Twaddle A, Marchand B, Gunsalus KC. Structural modeling of the SARS-CoV-2 spike/human ACE2 complex interface can identify high-affinity variants associated with increased transmissibility. J Mol Biol. (2021) 433:167051. doi: 10.1016/j.jmb.2021.167051
65. Nelson G, Buzko O, Spilman P, Niazi K, Rabizadeh S, Soon-Shiong P. Molecular dynamic simulation reveals E484K mutation enhances spike RBD-ACE2 affinity and the combination of E484K, K417N and N501Y mutations (501Y. V2 variant) induces conformational change greater than N501Y mutant alone, potentially resulting in an escape mutant. BioRxiv. (2021). doi: 10.1101/2021.01.13.426558
66. Ferrareze P, Franceschi VB, De Menezes Mayer A, Caldana GD, Zimerman RA, Thompson CE. E484K as an innovative phylogenetic event for viral evolution: Genomic analysis of the E484K spike mutation in SARS-CoV-2 lineages from Brazil. Infection Genet Evol. (2021) 93:104941. doi: 10.1016/j.meegid.2021.104941
67. Aleem A, Ab AS, Slenker AK. Emerging variants of SARS-CoV-2 and novel therapeutics against coronavirus (COVID-19). StatPearls (2021).
68. Motozono C, Toyoda M, Zahradnik J, Saito A, Nasser H, Tan TS, et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe. (2021) 29:1124–1136.e1111. doi: 10.1016/j.chom.2021.05.011
69. Kimura I, Kosugi Y, Wu J, Zahradnik J, Yamasoba D, Butlertanaka EP, et al. The SARS-CoV-2 Lambda variant exhibits enhanced infectivity and immune resistance. Cell Rep. (2022) 38. doi: 10.1016/j.celrep.2021.110218
70. Scheepers C, Everatt J, Amoako DG, Mnguni A, Ismail A, Mahlangu B, et al. The continuous evolution of SARS-CoV-2 in South Africa: a new lineage with rapid accumulation of mutations of concern and global detection. Medrxiv. (2021).
71. Brüssow H. COVID-19: emergence and mutational diversification of SARS-CoV-2. Microbial Biotechnol. (2021) 14:756–68. doi: 10.1111/1751-7915.13645
72. Bouvet M, Debarnot C, Imbert I, Selisko B, Snijder EJ, Canard B, et al. In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PloS Pathog. (2010) 6:e1000863. doi: 10.1371/journal.ppat.1000863
73. Walker AP, Fan H, Keown JR, Grimes JM, Fodor E. Identification of guanylyltransferase activity in the SARS-CoV-2 RNA polymerase. bioRxiv. (2021). doi: 10.1101/2021.03.17.435913
74. Yan L, Ge J, Zheng L, Zhang Y, Gao Y, Wang T, et al. Cryo-EM structure of an extended SARS-CoV-2 replication and transcription complex reveals an intermediate state in cap synthesis. Cell. (2021) 184:184–193.e110. doi: 10.1016/j.cell.2021.02.033
75. Chen Y, Guo D. Molecular mechanisms of coronavirus RNA capping and methylation. Virologica Sin. (2016) 31:3–11. doi: 10.1007/s12250-016-3726-4
76. Ramanathan A, Robb GB, Chan S-H. mRNA capping: biological functions and applications. Nucleic Acids Res. (2016) 44:7511–26. doi: 10.1093/nar/gkw551
77. Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. (2010) 468:452–6. doi: 10.1038/nature09489
78. Dong H, Fink K, Züst R, Lim SP, Qin C-F, Shi P-Y. Flavivirus RNA methylation. J Gen Virol. (2014) 95:763–78. doi: 10.1099/vir.0.062208-0
79. Ivanov KA, Thiel V, Dobbe JC, van der Meer Y, Snijder EJ, Ziebuhr J. Multiple enzymatic activities associated with severe acute respiratory syndrome coronavirus helicase. J Virol. (2004) 78:5619–32. doi: 10.1128/JVI.78.11.5619-5632.2004
80. Jia Z, Yan L, Ren Z, Wu L, Wang J, Guo J, et al. Delicate structural coordination of the Severe Acute Respiratory Syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. (2019) 47:6538–50. doi: 10.1093/nar/gkz409
81. Gorbalenya AE, Koonin EV. Viral proteins containing the purine NTP-binding sequence pattern. Nucleic Acids Res. (1989) 17:8413–38. doi: 10.1093/nar/17.21.8413
82. Singleton MR, Wigley DB. Modularity and specialization in superfamily 1 and 2 helicases. J bacteriology. (2002) 184:1819–26. doi: 10.1128/JB.184.7.1819-1826.2002
83. Chen J, Malone B, Llewellyn E, Grasso M, Shelton PM, Olinares PDB, et al. Structural basis for helicase-polymerase coupling in the SARS-CoV-2 replication-transcription complex. Cell. (2020) 182:1560–1573.e1513. doi: 10.1016/j.cell.2020.07.033
84. Yan L, Zhang Y, Ge J, Zheng L, Gao Y, Wang T, et al. Architecture of a SARS-CoV-2 mini replication and transcription complex. Nat Commun. (2020) 11:5874. doi: 10.1038/s41467-020-19770-1
85. Mickolajczyk KJ, Shelton PM, Grasso M, Cao X, Warrington SE, Aher A, et al. Force-dependent stimulation of RNA unwinding by SARS-CoV-2 nsp13 helicase. Biophys J. (2021) 120:1020–30. doi: 10.1016/j.bpj.2020.11.2276
86. Borowski P, Mueller O, Niebuhr A, Kalitzky M, Hwang L-H, Schmitz H, et al. ATP-binding domain of NTPase/helicase as a target for hepatitis C antiviral therapy. Acta Biochim Polonica. (2000) 47:173–80. doi: 10.18388/abp.2000_4075
87. Keller PA, Gordon C. Control of Hepatitis C: A Medicinal Chemistry Perspective. (2005) Hoboken, NJ, USA: Wiley. doi: 10.1002/chin.200516284
88. De Clercq E, Field HJ. Antiviral prodrugs–the development of successful prodrug strategies for antiviral chemotherapy. Br J Pharmacol. (2006) 147:1–11. doi: 10.1038/sj.bjp.0706446
89. Lee N-R, Kwon H-M, Park K, Oh S, Jeong Y-J, Kim D-E. Cooperative translocation enhances the unwinding of duplex DNA by SARS coronavirus helicase nsP13. Nucleic Acids Res. (2010) 38:7626–36. doi: 10.1093/nar/gkq647
90. Florés MJ, Sanchez N, Michel B. A fork-clearing role for UvrD. Mol Microbiol. (2005) 57:1664–75.
91. Patel SS, Picha KM. Structure and function of hexameric helicases. Annu Rev Biochem. (2000) 69:651–97. doi: 10.1146/annurev.biochem.69.1.651
92. Seybert A, Posthuma CC, Van Dinten LC, Snijder EJ, Gorbalenya AE, Ziebuhr J. A complex zinc finger controls the enzymatic activities of nidovirus helicases. J Virol. (2005) 79:696–704. doi: 10.1128/JVI.79.2.696-704.2005
93. Deng Z, Lehmann KC, Li X, Feng C, Wang G, Zhang Q, et al. Structural basis for the regulatory function of a complex zinc-binding domain in a replicative arterivirus helicase resembling a nonsense-mediated mRNA decay helicase. Nucleic Acids Res. (2014) 42:3464–77. doi: 10.1093/nar/gkt1310
94. Tang C, Deng Z, Li X, Yang M, Tian Z, Chen Z, et al. Helicase of type 2 porcine reproductive and respiratory syndrome virus strain HV reveals a unique structure. Viruses. (2020) 12:215. doi: 10.3390/v12020215
95. Jang K-J, Jeong S, Kang DY, Sp N, Yang YM, Kim D-E. A high ATP concentration enhances the cooperative translocation of the SARS coronavirus helicase nsP13 in the unwinding of duplex RNA. Sci Rep. (2020) 10:4481. doi: 10.1038/s41598-020-61432-1
96. Sommers JA, Loftus LN, Jones MP, Lee RA, Haren CE, Dumm AJ, et al. Biochemical analysis of SARS-CoV-2 Nsp13 helicase implicated in COVID-19 and factors that regulate its catalytic functions. J Biol Chem. (2023) 299. doi: 10.1016/j.jbc.2023.102980
97. Newman JA, Douangamath A, Yadzani S, Yosaatmadja Y, Aimon A, Brandão-Neto J, et al. Structure, mechanism and crystallographic fragment screening of the SARS-CoV-2 NSP13 helicase. Nat Commun. (2021) 12:4848. doi: 10.1038/s41467-021-25166-6
98. V’kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. (2021) 19:155–70. doi: 10.1038/s41579-020-00468-6
99. Ogando NS, Zevenhoven-Dobbe JC, van der Meer Y, Bredenbeek PJ, Posthuma CC, Snijder EJ. The enzymatic activity of the nsp14 exoribonuclease is critical for replication of MERS-CoV and SARS-CoV-2. J Virol. (2020) 94. doi: 10.1128/jvi.01246–01220
100. Minskaia E, Hertzig T, Gorbalenya AE, Campanacci V, Cambillau C, Canard B, et al. Discovery of an RNA virus 3′→ 5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc Natl Acad Sci. (2006) 103:5108–13. doi: 10.1073/pnas.0508200103
101. Chen Y, Cai H, Pan JA, Xiang N, Tien P, Ahola T, et al. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc Natl Acad Sci. (2009) 106:3484–9. doi: 10.1073/pnas.0808790106
102. Ma Y, Wu L, Shaw N, Gao Y, Wang J, Sun Y, et al. Structural basis and functional analysis of the SARS coronavirus nsp14–nsp10 complex. Proc Natl Acad Sci. (2015) 112:9436–41. doi: 10.1073/pnas.1508686112
103. Yan L, Yang Y, Li M, Zhang Y, Zheng L, Ge J, et al. Coupling of N7-methyltransferase and 3′-5′ exoribonuclease with SARS-CoV-2 polymerase reveals mechanisms for capping and proofreading. Cell. (2021) 184:3474–3485.e3411. doi: 10.1016/j.cell.2021.09.021
104. Deutscher M, Marlor CW. Purification and characterization of Escherichia coli RNase T. J Biol Chem. (1985) 260:7067–71. doi: 10.1016/S0021-9258(18)88888-3
105. Beese LS, Steitz TA. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J. (1991) 10:25–33. doi: 10.1002/embj.1991.10.issue-1
106. Steitz TA, Steitz JA. A general two-metal-ion mechanism for catalytic RNA. Proc Natl Acad Sci. (1993) 90:6498–502. doi: 10.1073/pnas.90.14.6498
107. Imprachim N, Yosaatmadja Y, Newman JA. Crystal structures and fragment screening of SARS-CoV-2 NSP14 reveal details of exoribonuclease activation and mRNA capping and provide starting points for antiviral drug development. Nucleic Acids Res. (2023) 51:475–87. doi: 10.1093/nar/gkac1207
108. De A, Bhattacharya S, Debroy B, Bhattacharya A, Pal K. Exploring the pharmacological aspects of natural phytochemicals against SARS-CoV-2 Nsp14 through an in silico approach. In Silico Pharmacol. (2023) 11:12. doi: 10.1007/s40203-023-00143-7
109. Deng X, Stjohn SE, Osswald HL, O’brien A, Banach BS, Sleeman K, et al. Coronaviruses resistant to a 3C-like protease inhibitor are attenuated for replication and pathogenesis, revealing a low genetic barrier but high fitness cost of resistance. J Virol. (2014) 88:11886–98. doi: 10.1128/JVI.01528-14
110. Agostini ML, Andres EL, Sims AC, Graham RL, Sheahan TP, Lu X, et al. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. MBio. (2018) 9:10–128. doi: 10.1128/mbio.00221–00218
111. Ogando NS, Ferron F, Decroly E, Canard B, Posthuma CC, Snijder EJ. The curious case of the nidovirus exoribonuclease: its role in RNA synthesis and replication fidelity. Front Microbiol. (2019) 10:475185. doi: 10.3389/fmicb.2019.01813
112. Sheahan TP, Sims AC, Zhou S, Graham RL, Pruijssers AJ, Agostini ML, et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci Trans Med. (2020) 12:eabb5883. doi: 10.1126/scitranslmed.abb5883
113. Ma Z, Pourfarjam Y, Kim I-K. Reconstitution and functional characterization of SARS-CoV-2 proofreading complex. Protein Expression purification. (2021) 185:105894. doi: 10.1016/j.pep.2021.105894
114. Lin S, Chen H, Chen Z, Yang F, Ye F, Zheng Y, et al. Crystal structure of SARS-CoV-2 nsp10 bound to nsp14-ExoN domain reveals an exoribonuclease with both structural and functional integrity. Nucleic Acids Res. (2021) 49:5382–92. doi: 10.1093/nar/gkab320
115. Decroly E, Imbert I, Coutard B, Bouvet M, Selisko B, Alvarez K, et al. Coronavirus nonstructural protein 16 is a cap-0 binding enzyme possessing (nucleoside-2′ O)-methyltransferase activity. J Virol. (2008) 82:8071–84. doi: 10.1128/JVI.00407-08
116. Sawicki SG, Sawicki DL, Younker D, Meyer Y, Thiel V, Stokes H, et al. Functional and genetic analysis of coronavirus replicase-transcriptase proteins. PloS Pathog. (2005) 1:e39. doi: 10.1371/journal.ppat.0010039
117. Lin S, Chen H, Ye F, Chen Z, Yang F, Zheng Y, et al. Crystal structure of SARS-CoV-2 nsp10/nsp16 2′-O-methylase and its implication on antiviral drug design. Signal transduction targeted Ther. (2020) 5:131. doi: 10.1038/s41392-020-00241-4
118. Decroly E, Debarnot C, Ferron F, Bouvet M, Coutard B, Imbert I, et al. Crystal structure and functional analysis of the SARS-coronavirus RNA cap 2′-O-methyltransferase nsp10/nsp16 complex. PloS Pathog. (2011) 7:e1002059. doi: 10.1371/journal.ppat.1002059
119. Vithani N, Ward MD, Zimmerman MI, Novak B, Borowsky JH, Singh S, et al. SARS-CoV-2 Nsp16 activation mechanism and a cryptic pocket with pan-coronavirus antiviral potential. Biophys J. (2021) 120:2880–9. doi: 10.1016/j.bpj.2021.03.024
120. Chen Y, Su C, Ke M, Jin X, Xu L, Zhang Z, et al. Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2′-O-methylation by nsp16/nsp10 protein complex. PloS Pathog. (2011) 7:e1002294. doi: 10.1371/journal.ppat.1002294
121. Habjan M, Hubel P, Lacerda L, Benda C, Holze C, Eberl CH, et al. Sequestration by IFIT1 impairs translation of 2′ O-unmethylated capped RNA. PloS Pathog. (2013) 9:e1003663. doi: 10.1371/journal.ppat.1003663
122. Züst R, Cervantes-Barragan L, Habjan M, Maier R, Neuman BW, Ziebuhr J, et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat Immunol. (2011) 12:137–43. doi: 10.1038/ni.2011.14
123. Hyde JL, Diamond MS. Innate immune restriction and antagonism of viral RNA lacking 2׳-O methylation. Virology. (2015) 479:66–74. doi: 10.1016/j.virol.2015.01.019
124. Almazán F, Dediego ML, Galán C, Escors D, Alvarez E, Ortego J, et al. Construction of a severe acute respiratory syndrome coronavirus infectious cDNA clone and a replicon to study coronavirus RNA synthesis. J Virol. (2006) 80:10900–6. doi: 10.1128/JVI.00385-06
125. Souza ACR, Vasconcelos AR, Dias DD, Komoni G, Name JJ. The integral role of magnesium in muscle integrity and aging: A comprehensive review. Nutrients. (2023) 15:5127. doi: 10.3390/nu15245127
126. Viswanathan T, Arya S, Chan S-H, Qi S, Dai N, Misra A, et al. Structural basis of RNA cap modification by SARS-CoV-2. Nat Commun. (2020) 11:3718. doi: 10.1038/s41467-020-17496-8
127. Viswanathan T, Misra A, Chan S-H, Qi S, Dai N, Arya S, et al. A metal ion orients SARS-CoV-2 mRNA to ensure accurate 2′-O methylation of its first nucleotide. Nat Commun. (2021) 123287. doi: 10.1101/2021.03.12.435174
128. Nirenberg M. Historical review: Deciphering the genetic code–a personal account. Trends Biochem Sci. (2004) 29:46–54. doi: 10.1016/j.tibs.2003.11.009
129. Schmeing TM, Ramakrishnan V. What recent ribosome structures have revealed about the mechanism of translation. Nature. (2009) 461:1234–42. doi: 10.1038/nature08403
130. Leung EKY, Suslov N, Tuttle N, Sengupta R, Piccirilli JA. The mechanism of peptidyl transfer catalysis by the ribosome. Annu Rev Biochem. (2011) 80:527–55. doi: 10.1146/annurev-biochem-082108-165150
131. Kozak M. The scanning model for translation: an update. J Cell Biol. (1989) 108:229–41. doi: 10.1083/jcb.108.2.229
132. Atkins JF, Gesteland RF. Recoding: expansion of decoding rules enriches gene expression. New York, USA: Springer Science & Business Media (2010). doi: 10.1007/978-0-387-89382-2
133. Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. (2011) 147:789–802. doi: 10.1016/j.cell.2011.10.002
134. Taylor DJ, Devkota B, Huang AD, Topf M, Narayanan E, Sali A, et al. Comprehensive molecular structure of the eukaryotic ribosome. Structure. (2009) 17:1591–604. doi: 10.1016/j.str.2009.09.015
135. Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science. (2000) 289:905–20. doi: 10.1126/science.289.5481.905
136. Ben-Shem A, Jenner L, Yusupova G, Yusupov M. Crystal structure of the eukaryotic ribosome. Science. (2010) 330:1203–9. doi: 10.1126/science.1194294
137. Klinge S, Voigts-Hoffmann F, Leibundgut M, Arpagaus S, Ban N. Crystal structure of the eukaryotic 60 S ribosomal subunit in complex with initiation factor 6. Science. (2011) 334:941–8. doi: 10.1126/science.1211204
138. Agirrezabala X, Frank J. Elongation in translation as a dynamic interaction among the ribosome, tRNA, and elongation factors EF-G and EF-Tu. Q Rev biophysics. (2009) 42:159–200. doi: 10.1017/S0033583509990060
139. Ogle JM, Brodersen DE, Clemons WM Jr., Tarry MJ, Carter AP, Ramakrishnan V. Recognition of cognate transfer RNA by the 30 S ribosomal subunit. Science. (2001) 292:897–902. doi: 10.1126/science.1060612
140. Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. (2009) 136:731–45. doi: 10.1016/j.cell.2009.01.042
141. Rodnina MV, Gromadski KB, Kothe U, Wieden H-J. Recognition and selection of tRNA in translation. FEBS Lett. (2005) 579:938–42. doi: 10.1016/j.febslet.2004.11.048
142. Moazed D, Noller HF. Intermediate states in the movement of transfer RNA in the ribosome. Nature. (1989) 342:142–8. doi: 10.1038/342142a0
143. Pisarev AV, Kolupaeva VG, Yusupov MM, Hellen CU, Pestova TV. Ribosomal position and contacts of mRNA in eukaryotic translation initiation complexes. EMBO J. (2008) 27:1609–21. doi: 10.1038/emboj.2008.90
144. Dunkle JA, Cate JH. Ribosome structure and dynamics during translocation and termination. Annu Rev biophysics. (2010) 39:227–44. doi: 10.1146/annurev.biophys.37.032807.125954
145. Nakamura Y, Ito K. tRNA mimicry in translation termination and beyond. Wiley Interdiscip Reviews: RNA. (2011) 2:647–68. doi: 10.1002/wrna.81
146. Brunelle JL, Shaw JJ, Youngman EM, Green R. Peptide release on the ribosome depends critically on the 2′ OH of the peptidyl–tRNA substrate. Rna. (2008) 14:1526–31. doi: 10.1261/rna.1057908
147. Baranov PV, Gesteland RF, Atkins JF. Recoding: translational bifurcations in gene expression. Gene. (2002) 286:187–201. doi: 10.1016/S0378-1119(02)00423-7
148. Dinman JD, Icho T, Wickner RB. A-1 ribosomal frameshift in a double-stranded RNA virus of yeast forms a gag-pol fusion protein. Proc Natl Acad Sci. (1991) 88:174–8. doi: 10.1073/pnas.88.1.174
149. Napthine S, Ling R, Finch LK, Jones JD, Bell S, Brierley I, et al. Protein-directed ribosomal frameshifting temporally regulates gene expression. Nat Commun. (2017) 8:1–11. doi: 10.1038/ncomms15582
150. Ketteler R. On programmed ribosomal frameshifting: the alternative proteomes. Front Genet. (2012) 3:242. doi: 10.3389/fgene.2012.00242
151. Jacks T, Varmus HE. Expression of the Rous sarcoma virus pol gene by ribosomal frameshifting. Science. (1985) 230:1237–42. doi: 10.1126/science.2416054
152. Jacks T, Power MD, Masiarz FR, Luciw PA, Barr PJ, Varmus HE. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature. (1988) 331:280–3. doi: 10.1038/331280a0
153. Ivanov IP, Atkins JF. Ribosomal frameshifting in decoding antizyme mRNAs from yeast and protists to humans: close to 300 cases reveal remarkable diversity despite underlying conservation. Nucleic Acids Res. (2007) 35:1842–58. doi: 10.1093/nar/gkm035
154. Rodnina MV, Korniy N, Klimova M, Karki P, Peng B-Z, Senyushkina T, et al. Translational recoding: canonical translation mechanisms reinterpreted. Nucleic Acids Res. (2020) 48:1056–67. doi: 10.1093/nar/gkz783
155. Puglisi JD, Wyatt JR, Tinoco JI. A pseudoknotted RNA oligonucleotide. Nature. (1988) 331:283–6. doi: 10.1038/331283a0
156. Baranov PV, Henderson CM, Anderson CB, Gesteland RF, Atkins JF, Howard MT. Programmed ribosomal frameshifting in decoding the SARS-CoV genome. Virology. (2005) 332:498–510. doi: 10.1016/j.virol.2004.11.038
157. Plant EP, Pérez-Alvarado GC, Jacobs JL, Mukhopadhyay B, Hennig M, Dinman JD. A three-stemmed mRNA pseudoknot in the SARS coronavirus frameshift signal. PloS Biol. (2005) 3:e172. doi: 10.1371/journal.pbio.0030172
158. Brierley I, Dos Ramos FJ. Programmed ribosomal frameshifting in HIV-1 and the SARS–CoV. Virus Res. (2006) 119:29–42. doi: 10.1016/j.virusres.2005.10.008
159. Plant EP, Rakauskaite R, Taylor DR, Dinman JD. Achieving a golden mean: mechanisms by which coronaviruses ensure synthesis of the correct stoichiometric ratios of viral proteins. J Virol. (2010) 84:4330–40. doi: 10.1128/JVI.02480-09
160. Plant EP, Sims AC, Baric RS, Dinman JD, Taylor DR. Altering SARS coronavirus frameshift efficiency affects genomic and subgenomic RNA production. Viruses. (2013) 5:279–94. doi: 10.3390/v5010279
161. Cho C-P, Lin S-C, Chou M-Y, Hsu H-T, Chang K-Y. Regulation of programmed ribosomal frameshifting by co-translational refolding RNA hairpins. PloS One. (2013) 8:e62283. doi: 10.1371/journal.pone.0062283
162. Anastassopoulou J, Theophanides T. The role of metal ions in biological systems and medicine. In: Bioinorganic Chemistry: An Inorganic Perspective of Life. Dordrecht, Netherlands: Springer (1995). p. 209–18.
163. Delgado R, Vergara C, Wolff D. Divalent cations as modulators of neuronal excitability: emphasis on copper and zinc. Biol Res. (2006) 39:173–82. doi: 10.4067/S0716-97602006000100019
164. Sigel A, Sigel H, Sigel RK. Interrelations between essential metal ions and human diseases. Dordrecht, The Netherlands: Springer (2013). doi: 10.1007/978-94-007-7500-8
165. Bloom AJ. Metal regulation of metabolism. Curr Opin Chem Biol. (2019) 49:33–8. doi: 10.1016/j.cbpa.2018.09.017
166. Reitner J, Thiel V. Encyclopedia of geobiology. Berlin, Germany: Springer Berlin (2011). doi: 10.1007/978-1-4020-9212-1
167. Baier F, Chen J, Solomonson M, Strynadka NC, Tokuriki N. Distinct metal isoforms underlie promiscuous activity profiles of metalloenzymes. ACS Chem Biol. (2015) 10:1684–93. doi: 10.1021/acschembio.5b00068
168. Tan X. Metalloproteins and Metalloenzymes: Roles and Mechanisms of Metals in Functional Proteins. Singapore: World Scientific Publishing Company Pte Limited (2016).
169. Hausinger RP. New metal cofactors and recent metallocofactor insights. Curr Opin Struct Biol. (2019) 59:1–8. doi: 10.1016/j.sbi.2018.12.008
170. Pernil R, Schleiff E. Metalloproteins in the biology of heterocysts. Life. (2019) 9:32. doi: 10.3390/life9020032
171. Knape MJ, Herberg FW. Metal coordination in kinases and pseudokinases. Biochem Soc Trans. (2017) 45:653–63. doi: 10.1042/BST20160327
172. Walker GM, Duffus JH. Magnesium ions and the control of the cell cycle in yeast. J Cell Sci. (1980) 42:329–56. doi: 10.1242/jcs.42.1.329
173. Mackenzie K, Foot NJ, Anand S, Dalton HE, Chaudhary N, Collins BM, et al. Regulation of the divalent metal ion transporter via membrane budding. Cell Discovery. (2016) 2:1–14. doi: 10.1038/celldisc.2016.11
174. Zhang-Keck Z-Y, Eckstein F, Washington L, Stallcup M. A role for divalent cations in specifying the start site for transcription from chromatin templates in vitro. J Biol Chem. (1988) 263:9550–6. doi: 10.1016/S0021-9258(19)76577-6
175. Chaigne-Delalande B, Lenardo MJ. Divalent cation signaling in immune cells. Trends Immunol. (2014) 35:332–44. doi: 10.1016/j.it.2014.05.001
176. Diaz-Ochoa VE, Jellbauer S, Klaus S, Raffatellu M. Transition metal ions at the crossroads of mucosal immunity and microbial pathogenesis. Front Cell infection Microbiol. (2014) 4:2. doi: 10.3389/fcimb.2014.00002
177. Zhang K, Chen J. The regulation of integrin function by divalent cations. Cell adhesion migration. (2012) 6:20–9. doi: 10.4161/cam.18702
178. Stelling MP, Motta JM, Mashid M, Johnson WE, Pavão MS, Farrell NP. Metal ions and the extracellular matrix in tumor migration. FEBS J. (2019) 286:2950–64. doi: 10.1111/febs.14986
179. Aisen P, Listowsky I. Iron transport and storage proteins. Annu Rev Biochem. (1980) 49:357–93. doi: 10.1146/annurev.bi.49.070180.002041
180. Coleman JE. Zinc proteins: enzymes, storage proteins, transcription factors, and replication proteins. Annu Rev Biochem. (1992) 61:897–946. doi: 10.1146/annurev.bi.61.070192.004341
181. Sigel A, Sigel H. Metal ions in biological systems, volume 35: iron transport and storage microorganisms, plants, and animals. Metal-based Drugs. (1998) 5:262–2. doi: 10.1155/MBD.1998.262a
182. Rolfs A, Hediger MA. Metal ion transporters in mammals: structure, function and pathological implications. J Physiol. (1999) 518:1–12. doi: 10.1111/j.1469-7793.1999.0001r.x
183. Hamatake M, Iguchi K, Hirano K, Ishida R. Zinc induces mixed types of cell death, necrosis, and apoptosis, in molt-4 cells. J Biochem. (2000) 128:933–9. doi: 10.1093/oxfordjournals.jbchem.a022844
184. Eide DJ. Zinc transporters and the cellular trafficking of zinc. Biochim Biophys Acta (BBA)-Molecular Cell Res. (2006) 1763:711–22. doi: 10.1016/j.bbamcr.2006.03.005
185. Liang Q, Zhou B. Copper and manganese induce yeast apoptosis via different pathways. Mol Biol Cell. (2007) 18:4741–9. doi: 10.1091/mbc.e07-05-0431
186. Yeo JE, Kang SK. Selenium effectively inhibits ROS-mediated apoptotic neural precursor cell death in vitro and in vivo in traumatic brain injury. Biochim Biophys Acta (BBA)-Molecular Basis Dis. (2007) 1772:1199–210. doi: 10.1016/j.bbadis.2007.09.004
187. Prohaska JR. Role of copper transporters in copper homeostasis. Am J Clin Nutr. (2008) 88:826S–9S. doi: 10.1093/ajcn/88.3.826S
188. Coffin AB, Reinhart KE, Owens KN, Raible DW, Rubel EW. Extracellular divalent cations modulate aminoglycoside-induced hair cell death in the zebrafish lateral line. Hearing Res. (2009) 253:42–51. doi: 10.1016/j.heares.2009.03.004
189. Dribben W, Eisenman L, Mennerick S. Magnesium induces neuronal apoptosis by suppressing excitability. Cell Death Dis. (2010) 1:e63–3. doi: 10.1038/cddis.2010.39
190. Zhao N, Enns CA. Iron transport machinery of human cells: players and their interactions. Curr topics membranes. (2012) 69:67–93. doi: 10.1016/B978-0-12-394390-3.00003-3
191. Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. (2019) 12:1–16. doi: 10.1186/s13045-019-0720-y
192. Botella H, Stadthagen G, Lugo-Villarino G, De Chastellier C, Neyrolles O. Metallobiology of host–pathogen interactions: an intoxicating new insight. Trends Microbiol. (2012) 20:106–12. doi: 10.1016/j.tim.2012.01.005
193. Hood MI, Skaar EP. Nutritional immunity: transition metals at the pathogen–host interface. Nat Rev Microbiol. (2012) 10:525–37. doi: 10.1038/nrmicro2836
194. Veyrier FJ, Cellier MF. Metal economy in host-microbe interactions. Front Media SA. (2015). doi: 10.3389/978-2-88919-497-1
195. Skaar EP, Raffatellu M. Metals in infectious diseases and nutritional immunity. Metallomics. (2015) 7:926–8. doi: 10.1039/C5MT90021B
196. Weiss G, Carver P. Role of divalent metals in infectious disease susceptibility and outcome. Clin Microbiol Infection. (2018) 24:16–23. doi: 10.1016/j.cmi.2017.01.018
197. Sarafianos SG, Marchand B, Das K, Himmel DM, Parniak MA, Hughes SH, et al. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J Mol Biol. (2009) 385:693–713. doi: 10.1016/j.jmb.2008.10.071
198. Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science. (1998) 282:1669–75. doi: 10.1126/science.282.5394.1669
199. Tian L, Kim M-S, Li H, Wang J, Yang W. Structure of HIV-1 reverse transcriptase cleaving RNA in an RNA/DNA hybrid. Proc Natl Acad Sci. (2018) 115:507–12. doi: 10.1073/pnas.1719746115
200. Ben-Artzi H, Zeelon E, Le-Grice SF, Gorecki M, Panet A. Characterization of the double stranded RNA dependent RNase activity associated with recombinant reverse transcriptases. Nucleic Acids Res. (1992) 20:5115–8. doi: 10.1093/nar/20.19.5115
201. Cirino NM, Cameron CE, Smith JS, Roth MJ, Benkovic SJ, Le Grice SF. Divalent cation modulation of the ribonuclease functions of human immunodeficiency virus reverse transcriptase. Biochemistry. (1995) 34:9936–43. doi: 10.1021/bi00031a016
202. Klumpp K, Hang JQ, Rajendran S, Yang Y, Derosier A, Wong Kai In P, et al. Two-metal ion mechanism of RNA cleavage by HIV RNase H and mechanism-based design of selective HIV RNase H inhibitors. Nucleic Acids Res. (2003) 31:6852–9. doi: 10.1093/nar/gkg881
203. Pyle A. Metal ions in the structure and function of RNA. JBIC J Biol Inorganic Chem. (2002) 7:679–90. doi: 10.1007/s00775-002-0387-6
204. Klein DJ, Moore PB, Steitz TA. The contribution of metal ions to the structural stability of the large ribosomal subunit. Rna. (2004) 10:1366–79. doi: 10.1261/rna.7390804
205. Wacker WE. The biochemistry of magnesium. Ann New York Acad Sci. (1969) 162:717–26. doi: 10.1111/j.1749-6632.1969.tb13003.x
206. Misra VK, Draper DE. On the role of magnesium ions in RNA stability. Biopolymers: Original Res Biomolecules. (1998) 48:113–35. doi: 10.1002/(ISSN)1097-0282
207. Shiman R, Draper DE. Stabilization of RNA tertiary structure by monovalent cations. J Mol Biol. (2000) 302:79–91. doi: 10.1006/jmbi.2000.4031
208. Chao F-C, Schachman HK. The isolation and characterization of a macromolecular ribonucleoprotein from yeast. Arch Biochem Biophysics. (1956) 61:220–30. doi: 10.1016/0003-9861(56)90334-4
209. Chao F-C. Dissociation of macromolecular ribonucleoprotein of yeast. Arch Biochem biophysics. (1957) 70:426–31. doi: 10.1016/0003-9861(57)90130-3
210. Tissières A, Watson JD, Schlessinger D, Hollingworth B. Ribonucleoprotein particles from Escherichia coli. J Mol Biol. (1959) 1:221–33. doi: 10.1016/S0022-2836(59)80029-2
211. McCarthy BJ. The effects of magnesium starvation on the ribosome content of Escherichia coli. Biochimica et Biophysica Acta (BBA)-Specialized Section on Nucleic Acids and Related Subjects. (1962) 55(6):880–9. doi: 10.1016/0926-6550(62)90345-6
212. Kimes BW, Morris DR. Cations and ribosome structure. II. Effect of the 50S subunit of substituting polyamines for magnesium ion. Biochemistry. (1973) 12:442–9. doi: 10.1021/bi00727a013
213. Gordon J, Lipmann F. Role of divalent ions in poly U-directed phenylalanine polymerization. J Mol Biol. (1967) 23:23–33. doi: 10.1016/S0022-2836(67)80064-0
214. Moore PB. Polynucleotide attachment to ribosomes. J Mol Biol. (1966) 18:8–20. doi: 10.1016/S0022-2836(66)80072-4
215. Khawaja JA, Raina A. Effect of spermine and magnesium on the attachment of free ribosomes to endoplasmic reticulum membranes in vitro. Biochem Biophys Res Commun. (1970) 41:512–8. doi: 10.1016/0006-291X(70)90536-X
216. Knape MJ, Ballez M, Burghardt NC, Zimmermann B, Bertinetti D, Kornev AP, et al. Divalent metal ions control activity and inhibition of protein kinases. Metallomics. (2017) 9:1576–84. doi: 10.1039/C7MT00204A
217. Ricchetti M, Buc H. E. coli DNA polymerase I as a reverse transcriptase. EMBO J. (1993) 12:387–96. doi: 10.1002/embj.1993.12.issue-2
218. Cowan J. Metal activation of enzymes in nucleic acid biochemistry. Chem Rev. (1998) 98:1067–88. doi: 10.1021/cr960436q
Keywords: COVID-19 virus, proteins s (Spike), RBD, ACE2, -1 ribosomal frameshifting, magnesium
Citation: Khanifar A, Najafi A, Hemmati J, Nouri F, Hosseini SA and Taheri M (2024) Investigation of COVID-19 virus mutagenicity and the effect of the NSP13, NSP14, and NSP16 on the -1 ribosomal frameshifting. Front. Virol. 4:1405680. doi: 10.3389/fviro.2024.1405680
Received: 23 March 2024; Accepted: 10 June 2024;
Published: 16 July 2024.
Edited by:
Angela Wahl, University of Alabama at Birmingham, United StatesCopyright © 2024 Khanifar, Najafi, Hemmati, Nouri, Hosseini and Taheri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohammad Taheri, bW90YWhlcmkzNjBAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.