 Jun Arii
Jun Arii
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Virol. , 05 March 2024
Sec. Fundamental Virology
Volume 4 - 2024 | https://doi.org/10.3389/fviro.2024.1378054
This article is part of the Research Topic The Viral Egress: Strategies for Therapeutic Interference View all articles
Enveloped viruses complete their replication cycle by forming virions that bud from infected cells through membrane scission. The mechanisms by which this is achieved are less well-understood than the well-characterized membrane scission of vesicles budding inwards into the cytosol. The scission of vesicles that bud away from the cytosol is mediated by machinery of the endosomal sorting complexes required for transport (ESCRT)-III, which is highjacked by viruses of several different families. Other groups of viruses can bud independently of ESCRT-III activity. It has not been fully elucidated how the latter achieve this in the absence of host ESCRT-III, but it is known that some viral proteins directly mediate membrane scission. The Herpesviridae constitute a family of highly diverse viruses that bud at the inner nuclear membrane and cytoplasmic membranes in infected cells. Many investigators have attempted to determine the mechanism of membrane scission during herpesvirus budding, and have found this to be complex, not exactly conforming to either of the two methods. The present review attempts to synthesize the disparate findings into a model of herpesvirus egress based on both ESCRT-mediated and viral protein-mediated mechanisms.
Enveloped viruses must produce viral particles by deforming the host cell membrane and then pinching off from the membrane in a scission step. In eukaryotic cells, vesicles can bud inwards into the cytosol or outward away from the cell (Figure 1A) (1). Types of vesicles budding into the cytosol include clathrin-, coat protein I (COPI)- and COPII-coated vesicles (2). In this case, the bud neck is surrounded by cytoplasm and the BAR and dynamin family fission machineries bind outside the bud to constrict and pinch off the membrane necks. This process has been well characterized (Figure 1A) (2).
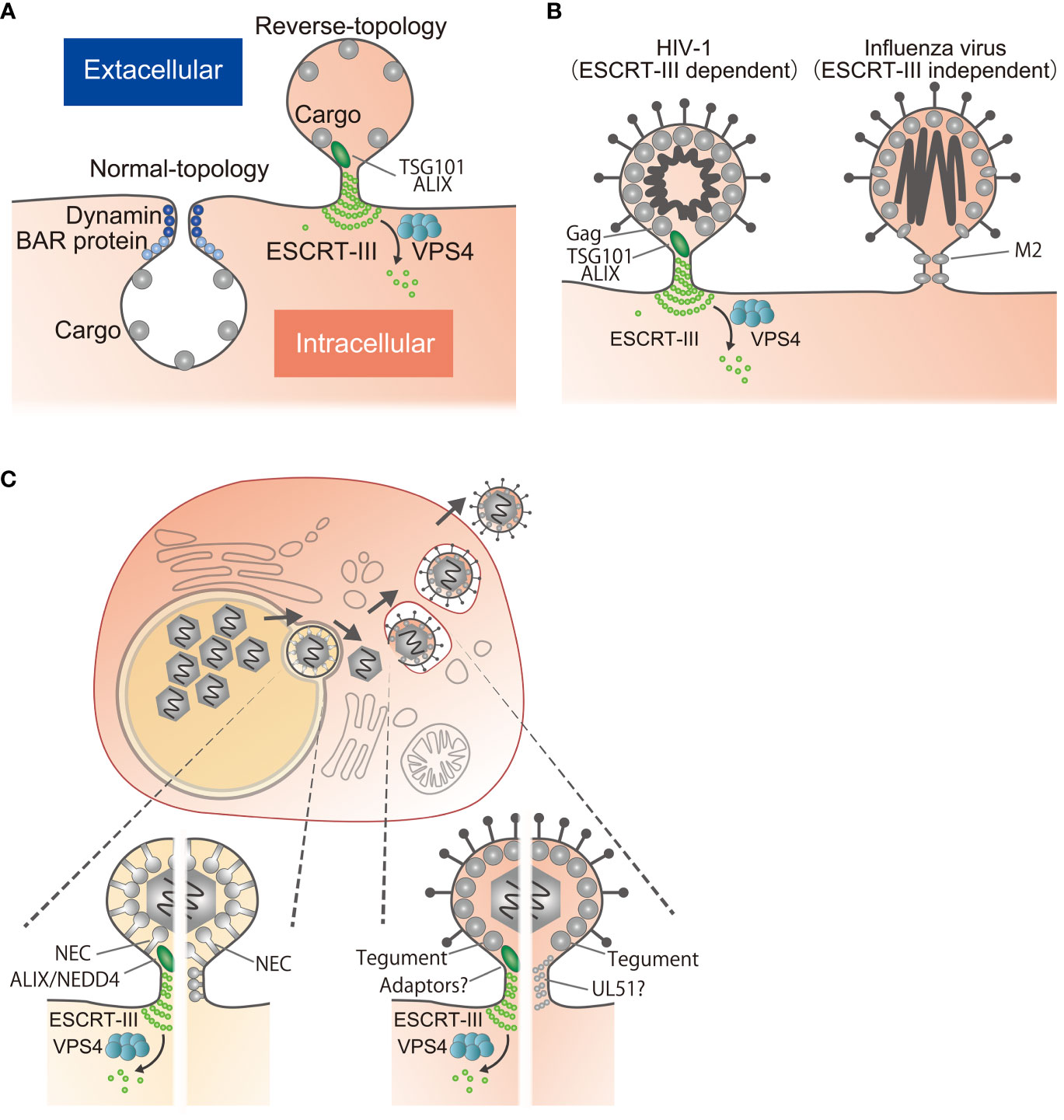
Figure 1 Reverse-topology scission involved in viral egress. (A) Normal-topology scission (left), carried out by dynamin and BAR proteins, for the biogenesis of vesicles directed inwards into the cytosol. Reverse-topology scission (right), carried out by ESCRT-III, functions in vesicle budding away from the cytosol. Note that only the cytosolic side of the membrane neck is accessible for the protein scaffolding and scission machinery. (B) HIV-1 Gag recruits ESCRT-III via TSG101 or ALIX to mediate scission (left). Influenza virus M2 mediates scission by virtue of its own activity through lipid re-ordering (right). (C) Egress of herpesviruses. After genome replication in the nucleus, viral capsids in the nucleus bud through the INM to form vesicles in the perinuclear space that fuse with the ONM to release the capsids into the cytoplasm. Capsids bud again into cytoplasmic vesicles to produce infectious virions, followed by release to the extracellular space. During budding at the INM, the NEC mediates scission by recruitment of ESCRT-III via ALIX/NEDD4 or by itself through its polymerization. During budding at the cytoplasm, tegument proteins may mediate scission by recruitment of ESCRT-III via multiple interactions or by polymerization as is the case for proteins such as UL51.
Vesicles budding outwards away from the cytosol include intraluminal vesicles (ILVs) and extracellular vesicles as well as enveloped viruses (1). It is less clear how this “reverse-topology scission” is directed from the inner surface of the membrane itself. The machinery of the endosomal sorting complexes required for transport (ESCRT)-III molecules is the sole mechanism thus far identified that is responsible for such reverse-topology scission (Figure 1A) (1, 3, 4). Because of the particular topology of viral budding (Figure 1B), studies of viral egress have been focused on whether or not the process is ESCRT-III dependent (5, 6). Nevertheless, for some viruses including herpesviruses, there is no consensus regarding the dependence of viral egress on ESCRT-III.
Herpesviruses are enveloped double-stranded DNA viruses, with a mature virion consisting of three elements: an icosahedral capsid with a linear double-stranded DNA genome, a host-membrane-derived envelope spiked with viral glycoproteins and the tegument, a proteinaceous layer between the capsid and envelope (7). The Herpesviridae family is subdivided into the Alphaherpesvirinae, Betaherpesvirinae, and Gammaherpesvirinae subfamilies, based on their molecular and biological properties (7). Herpes simplex virus 1 (HSV-1) is the prototype of the alphaherpesvirus subfamily and causes a variety of conditions such as mucocutaneous disease, keratitis, skin disease and encephalitis in humans (8). Herpesviruses share a common virion morphology and approximately 40 conserved genes (7). Tegument proteins form a group of structural components playing an important role in virion assembly (9), similar to the matrix proteins of other viruses.
Herpesviruses replicate their genomes and package nascent viral progeny genomes into capsids in the host cell nucleus. These capsids must first acquire envelopes by budding through the inner nuclear membrane (INM) into the perinuclear space between the INM and the outer nuclear membrane (ONM). This is followed by fusion with the ONM to release capsids into the cytoplasm, which then bud again into cytoplasmic vesicles to produce infectious virions (10) (Figure 1C). This variety of viral factors and the complexity of the budding processes make it difficult to understand their relationship to ESCRT-III. Here, I review mechanisms of membrane scission for viral egress, focusing on herpesviruses, which have unique properties.
ESCRT proteins were originally identified by virtue of their essential role in the formation of ILVs in budding yeast (11). Members of the ESCRT family can be classified into five groups: ESCRT-0, -I, -II -III and VPS4 complex (3, 12). Of these, ESCRT-III catalyzes membrane fission and the VPS4 complex mediates ESCRT disassembly (3, 12). ESCRT-III proteins are recruited onto the membrane by ESCRT-II or adaptors such as ALIX. This is followed by the formation of membrane-binding spirals that mediate membrane deformation and scission, in cooperation with the ATPase VPS4 (1, 3). VPS4 ATPases disassemble ESCRT-III filaments into their constituent subunits using the energy of ATP hydrolysis (13, 14). The activity of VPS4 is crucial for the ESCRT-III assembly cycle, as shown by severe defects in ESCRT-III-mediated membrane scission on expression of a dominant negative VPS4 allele. ESCRT-III appears to mediate reverse-topology scission events in a wide range of cellular processes, including vesicle budding from the cytoplasmic membranes, autophagy, membrane repair and cytokinesis (1, 4).
A unique mechanism of scission during viral egress was originally revealed by the finding that disruption of a short tetrapartite motif (the L domain) found in retroviral proteins typically results in defects during the late stages of budding, particularly at the final step of vesicle fission (15). Similar motifs have been identified in a variety of different viruses (5, 15). These motifs mediate the recruitment and interaction of the ESCRT proteins to facilitate virus egress. Briefly, the PTAP motif binds the ESCRT-I protein TSG101 and the YPXL motif binds the ESCRT-III adaptor ALIX, whereas the PPXY motif binds NEDD4 ubiquitin ligase family proteins (16–21). The ESCRT proteins are rich in ubiquitin-binding domains, reflecting their prominent role in sorting ubiquitinated proteins into ILVs (3, 15). In the case of NEDD4-dependent budding, ESCRT-III is thought to be recruited via ubiquitin on the viral protein complex (15).
Different viral species exhibit a wide variety of functional L domains and hence different ESCRT proteins are required for budding. For example, human immunodeficiency virus 1 (HIV-1) Gag protein recruits TSG101 and ALIX through its PTAP and YPXL motifs, respectively (16, 17, 19, 20) (Figure 1B). Equine infectious anemia virus recruits ALIX via the YPXL motif (19–21) and Rous sarcoma virus uses NEDD4 through the PPXY motif (18).
There are also other ways to recruit ESCRTs in addition to the three traditional L domains. The matrix (M) protein of the paramyxovirus parainfluenza virus 5 (PIV5) lacks the well-defined PTAP, YPXL and PPXY motifs but mediates budding through ubiquitination and the ESCRT-III pathway (22). An FPIV motif within the PIV5 M protein is essential for viral budding and can functionally compensate for the absence of the L domain during HIV-1 budding. Similarly, NEDD4L ubiquitin ligase allows the release of HIV-1 mutants lacking the PTAP and YPXL motif (23–25). HIV-1 Gag does not interact with NEDD4L directly but the cellular protein Angiomotin links HIV-1 Gag and NEDD4L to facilitate budding (26, 27). ESCRT-III appears to mediate egress of viral progeny in many different families of viruses, including retroviruses, filoviruses, rhabdoviruses, arenaviruses, paramyxoviruses, flaviviruses, bunyaviruses, poxviruses and hepadnaviruses as well as herpesviruses (5, 28–30).
It was reported early on that the expression of a VPS4 dominant negative allele in HSV-1-infected cells suppressed envelopment in the cytoplasm (31–34). The canonical motifs of the L domains are present in various tegument proteins, but siRNA for TSG101 or ALIX had no effect on HSV-1 reproduction (35). This led to the conclusion that HSV-1 recruits ESCRT-III machinery via complex interactions between multiple viral proteins and ESCRT proteins at the cytoplasm in a redundant fashion (Figure 1C). Another group showed that one of the ESCRT-III components, CHMP4C, has a predominant role in HSV-1 envelopment in the cytoplasm (36). A role for ESCRT-III in the life cycle of the betaherpesvirus human cytomegalovirus (HCMV) has also been reported; expression of dominant negative mutants of VPS4 or ESCRT-III protein severely impaired HCMV replication (37).
In contrast to the redundant role of tegument proteins in the cytoplasm, the nuclear egress complex (NEC) is essential for the envelopment of capsids at the INM (38, 39). Herpesvirus NEC, which consists of nuclear matrix and nuclear membrane proteins, forms a complex on the intranuclear side of the INM (38). Ectopic expression of the NEC results in the generation of characteristic vesicles without capsids located between the INM and ONM, suggesting that the NEC itself can induce membrane deformation and scission in the absence of any other viral factors (40). Our group showed that the NEC of HSV-1 interacts with ALIX to recruit ESCRT-III to the INM (41, 42). Furthermore, depletion of ESCRT-III components severely impaired envelopment at the INM and impaired nucleocytoplasmic transport (41). Similarly, another group showed that the NEC of the gammaherpesvirus Epstein-Barr virus (EBV) interacts with ALIX and the NEDD4 protein to recruit ESCRT-III to the INM (43, 44). These experiments reveal that budding at the INM requires ESCRT-III function in herpesvirus-infected cells (Figure 1C) (39, 45).
During viral infection, different types of extracellular vesicles (EVs) appear to be released by host cells, including so-called L-particles composed of virus envelope and tegument proteins but lacking the viral genome and viral capsid proteins. Although L-particles are themselves non-infectious, they were shown to facilitate HSV-1 infection, at least in cell cultures, most likely by delivering viral and/or cellular proteins to the target cells that are needed for virus replication and suppression of antiviral responses (46). In contrast, HSV-1 infection influences the cargo and functions of EVs released by infected cells; these EVs then negatively impact a subsequent HSV-1 infection (47–49). Because ESCRT-III mediates scission of EVs and is incorporated into them (1, 4), it is conceivable that it affects viral replication via EV biogenesis.
Recently, compounds binding the ESCRT-I protein TSG101 were analyzed in the context of viral infection. These compounds target the N-terminal domain of TSG101, disrupting its ubiquitin binding and inhibiting HIV-1 replication. Electron microscopy revealed a defect in HIV-1 Gag assembly in the cytoplasm. The authors concluded that Tsg101 acts as a chaperone for HIV-1 Gag that is independent of its interaction with the PTAP motif (50). These compounds also impaired the replication of members of the filo-, alpha- and herpesvirus families but not the flaviviruses (51). In agreement with the results of experiments depleting ESCRT proteins, these compounds impair HSV-1, HSV-2 and EBV nuclear egress (52, 53). Thus, the ESCRT machinery supports maturation of herpesvirus virions at the INM in addition to its role in scission. Studies on nucleocytoplasmic transport of capsids contributes anti-viral therapies.
ESCRT-III-independent egress has been reported for several members of the groups of orthomyxoviruses, paramyxoviruses, coronaviruses, togaviruses, and herpesviruses (31, 54–58), although the responsible mechanisms have mostly not been well-studied. The best investigated ESCRT-III-independent egress is that of influenza viruses, which can still replicate in cells with a dominant negative allele of VPS4 (55). The influenza M2 protein is a proton-selective ion channel protein, crucial for the scission step during viral egress (59). In vitro, purified M2 protein alters the membrane curvature and generates vesicles inside giant unilamellar vesicles (GUVs), a process dependent on its cytoplasmic amphipathic helix (60). Thus, upon binding, the M2 amphipathic helix forms clusters and induces membrane curvature and lipid ordering, constricting and destabilizing the membrane neck, causing fission of liposomes (60–62). Based on these observations, it has been concluded that influenza virus M2 protein directly mediates membrane scission (Figure 1B). This model is attractive as the M2 protein at the neck of the bud will be released from the cells together with the virions and recycling of M2 proteins is not required for the next round of budding.
In the past, viral egress was mainly considered in terms of its dependency on ESCRT-III or VPS4 activity. However, the distinction between ESCRT-III-dependent and -independent budding is not clear for some viruses, including herpesviruses. Following the discovery of the intrinsic functions of influenza virus M2 proteins for membrane deformation/scission, similar assays were adapted to investigate the NEC. This documented the ability of NEC to produce vesicles inside the GUV in vitro (63). Purified NECs form hexagonal lattices that might reflect the driving force for particle formation because mutation at the contact site of these lattices severely impaired vesicle formation both at the GUV and in infected cells (63–67). Similar to the situation with influenza M2 proteins, it could be concluded that NEC mediates membrane scission at the bud during egress from the INM (Figure 1C). At the present time, it is unclear whether NECs can trigger INM scission in infected cells in the absence of ESCRT-III, or whether NEC scission activity and ESCRT-III machinery act independently in parallel.
Confusion about the dependence of ESCRT-III has also arisen regarding envelopment in the cytoplasm. HSV-1 UL51 protein and its homologues are conserved tegument proteins which are important for envelopment in the cytoplasm (68–71). Unexpectedly, the crystal structure of the UL51 protein was found to resemble the host ESCRT-III component. As UL51 forms ESCRT-III-like filaments in vitro, it has been proposed that it promotes membrane scission directly at the cytoplasm (72) (Figure 1C). Whether UL51 mediates scission by itself or modifies ESCRT-III assembly in infected cells has not yet been clarified.
Reports on HCMV imply an even greater degree of complexity. In contrast to the previous report (37), others reported that expression of dominant negative mutants of VPS4 or ESCRT-III protein had no effect on the envelopment of HCMV capsids but reduced the efficacy of viral spread, perhaps due to effects on exosome-mediated signaling (73).
Thus far, there is no explanation for these discrepancies regarding the role of ESCRT-III in the life cycle of herpesviruses. Inhibition of ESCRT-III severely inhibits cell division, resistance to cell death, biogenesis of EVs and membrane repair (1, 4). Hence, the true contribution of ESCRT-III for budding per se is difficult to establish in experiments performed under conditions that avoid cytotoxicity. Cytotoxic effects of ESCRT-III deficiency need to be analyzed more carefully, especially in the case of relatively slowly replicating viruses such as herpesviruses.
The model where a single viral factor cleaves the membrane by clustering of viral protein and/or lipid structures might be based on in vitro analyses showing that the ESCRT-III protein CHMP4 forms a helical polymer on the GUVs which act as the executor of membrane scission (74, 75). This approach has documented that the other ESCRT-III components CHMP2 and CHMP3 are located at the termination site of the CHMP4 polymer to recruit VPS4 ATPase (74, 76). Based on these observations, it was proposed that the polymerization of CHMP4 protein itself cleaves the membrane in a reverse-topology manner and that the enzymatic activity of VPS4 recycles the ESCRT-III proteins from the complex (74, 75) (Figure 2A). On the other hand, imaging experiments have shown that VPS4 recruitment precedes membrane scission (77–79). Therefore, there has been a persistent belief that the GUV assay does not adequately reflect reverse-topology membrane scission in the cell. As GUVs are less stiff than cellular membranes, it might be easier to achieve scission in the GUV system than in cells, and thus generate artifacts (80). Likewise, it is uncertain whether viral proteins alone can mediate reverse-topology scission from the cytoplasmic or nuclear space independent of any cellular machinery in infected cells.
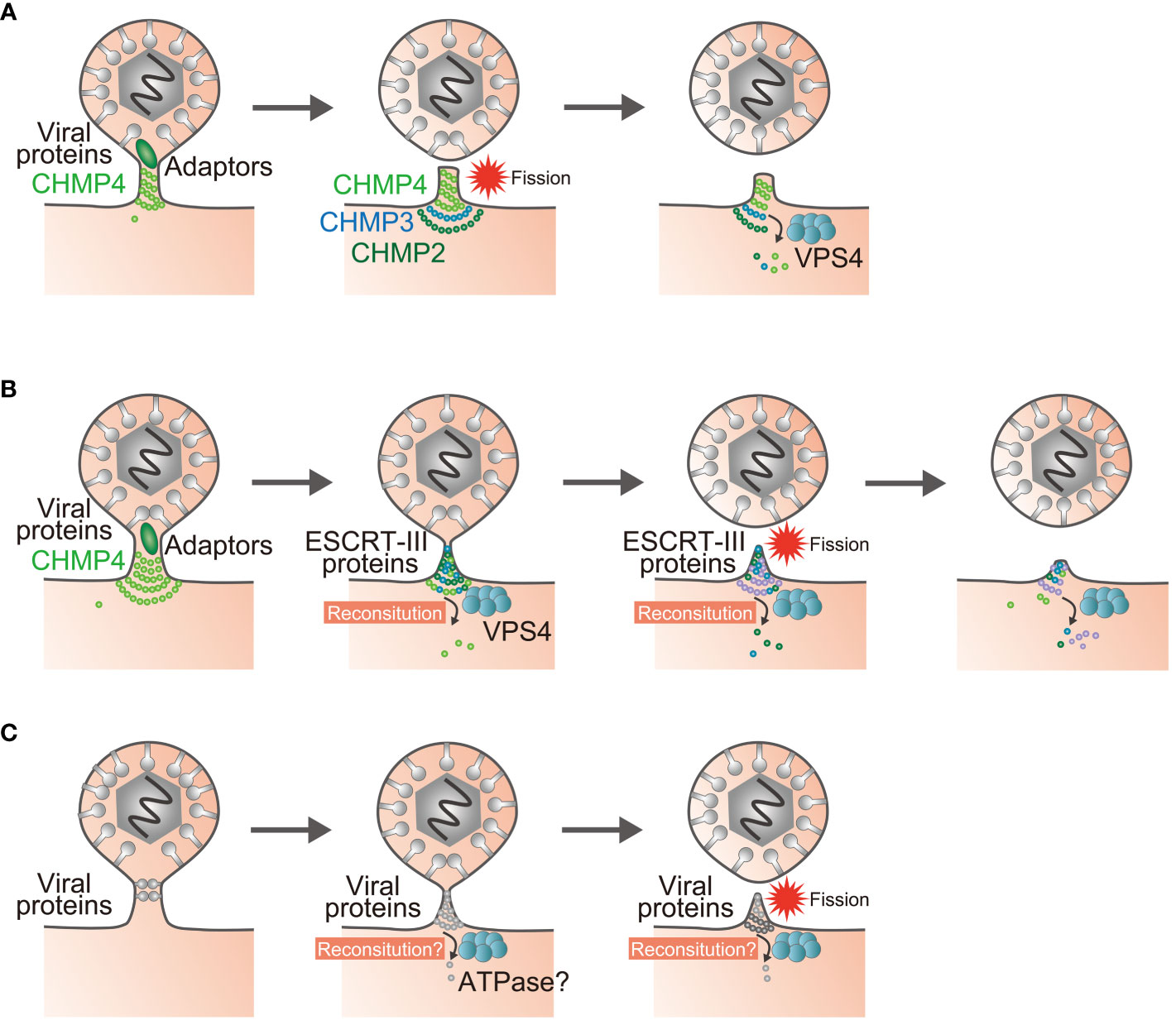
Figure 2 Models of reverse-topology membrane scission in infected cells. (A) Polymerization of the ESCRT-III protein CHMP4 mediates membrane scission, and the enzymatic activity of VPS4 recycles the ESCRT-III proteins. (B) The sequential recruitment of ESCRT-III components, polymerization, and replacement of different filament subunits driven by VPS4 result in constriction and scission of the membrane. (C) Proposed model of ESCRT-III-independent membrane scission. Viral proteins may mediate membrane scission by themselves. Question marks indicate unknown steps.
In an effort to resolve this disparity, GUV assays were modified to create membrane nanotubes under controlled tension and force (80). ESCRT-III/VPS4 assembly was reconstituted using these membrane nanotubules, with the result that it appeared to be the case that VPS4 activity and polymerization of ESCRT-III subunits could generate forces within the nanotubes that led to their constriction and to membrane scission (81, 82). Accordingly, a model was proposed in which sequential polymerization of ESCRT-III subunits, driven by a recruitment cascade and by continuous subunit-turnover by VPS4, induces membrane deformation and fission (81–83) (Figure 2B). These modifications to the reverse-topology membrane scission model with ESCRT-III will need to be addressed for other viral factors as well. It would be important to determine whether reconstitution of viral protein complexes at the bud is necessary for, and whether any ATPases are required for, ESCRT-III-independent viral egress (Figure 2C).
Since the historical report that scission in HIV-1 budding is dependent on ESCRT proteins (16, 17), viral budding has been considered mainly in terms of interactions with ESCRT-III. However, there are many conflicting reports regarding the degree of ESCRT-III dependence for viral budding. Due to the wide range of ESCRT-III functions (1, 4), direct effects of ESCRT-III on viral budding may often be underestimated or overestimated. It is especially difficult to separate the roles of ESCRT-III for budding of virion and EVs, as the latter can promote or inhibit viral replication through cell-cell communication. Furthermore, some viruses mediate membrane scission through multiple mechanisms that might act in a redundant fashion. Of these, herpesviruses employ an extraordinarily complex process, making it impossible to determine the contribution of ESCRT-III at each stage, given the paucity of published reports in the field at this time. As our understanding of membrane scission by ESCRT-III advances, based on biochemical and structural analysis, it is valuable to update consensus knowledge of viral egress. In particular, there are many viruses that apparently egress independently of ESCRT-III, but the details of the responsible membrane scission processes are largely unknown, and it is unclear whether there is a common principle. Improved awareness of the different mechanisms of viral membrane scission will contribute greatly to our understanding of reverse-topology scission, which is difficult to explain in physico-chemical terms.
JA: Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Support for this review was provided by Grants for Scientific Research from the Japan Society for the Promotion of Science (JSPS), the Ministry of Education, Culture, Science, Sports and Technology of Japan (MEXT) Leading Initiative for Excellent Young Researchers Grant, contract research fund of Precursory Research for Innovative Medical Care (PRIME, 22gm6410022h) from the Japan Agency for Medical Research and Development (AMED) and grants from the Takeda Science Foundation, MSD Life Science Foundation and Shionogi Infectious Disease Research Promotion Foundation.
The author is thankful to the lab members for helpful comments.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Vietri M, Radulovic M, Stenmark H. The many functions of escrts. Nat Rev Mol Cell Biol. (2020) 21:25–42. doi: 10.1038/s41580-019-0177-4
2. Mettlen M, Chen PH, Srinivasan S, Danuser G, Schmid SL. Regulation of clathrin-mediated endocytosis. Annu Rev Biochem. (2018) 87:871–96. doi: 10.1146/annurev-biochem-062917-012644
3. McCullough J, Colf LA, Sundquist WI. Membrane fission reactions of the mammalian escrt pathway. Annu Rev Biochem. (2013) 82:663–92. doi: 10.1146/annurev-biochem-072909-101058
5. Chen BJ, Lamb RA. Mechanisms for enveloped virus budding: can some viruses do without an escrt? Virology. (2008) 372:221–32. doi: 10.1016/j.virol.2007.11.008
6. Votteler J, Sundquist WI. Virus budding and the escrt pathway. Cell Host Microbe. (2013) 14:232–41. doi: 10.1016/j.chom.2013.08.012
7. Pellet PE, Roizman B. Herpesviridae. In: Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, et al, editors. Fields virology, 6th ed. Lippincott-Williams &Wilkins, Philadelphia, PA (2013). p. 1802–22.
8. Roizman B, Knipe DM, Whitley RJ. Herpes simplex viruses. In: Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, et al, editors. Fields virology, 6th ed. Lippincott-Williams &Wilkins, Philadelphia, PA (2013). p. 1823–97.
9. Owen DJ, Crump CM, Graham SC. Tegument assembly and secondary envelopment of alphaherpesviruses. Viruses. (2015) 7:5084–114. doi: 10.3390/v7092861
10. Johnson DC, Baines JD. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol. (2011) 9:382–94. doi: 10.1038/nrmicro2559
11. Bryant NJ, Stevens TH. Vacuole biogenesis in saccharomyces cerevisiae: protein transport pathways to the yeast vacuole. Microbiol Mol Biol Rev. (1998) 62:230–47. doi: 10.1128/MMBR.62.1.230-247.1998
12. Olmos Y. The escrt machinery: remodeling, repairing, and sealing membranes. Membranes (Basel). (2022) 12:633. doi: 10.3390/membranes12060633
13. Lata S, Schoehn G, Jain A, Pires R, Piehler J, Gottlinger HG, et al. Helical structures of escrt-iii are disassembled by vps4. Science. (2008) 321:1354–7. doi: 10.1126/science.1161070
14. Babst M, Wendland B, Estepa EJ, Emr SD. The vps4p aaa atpase regulates membrane association of a vps protein complex required for normal endosome function. EMBO J. (1998) 17:2982–93. doi: 10.1093/emboj/17.11.2982
15. Bieniasz PD. Late budding domains and host proteins in enveloped virus release. Virology. (2006) 344:55–63. doi: 10.1016/j.virol.2005.09.044
16. Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, et al. Tsg101 and the vacuolar protein sorting pathway are essential for hiv-1 budding. Cell. (2001) 107:55–65. doi: 10.1016/s0092-8674(01)00506-2
17. Martin-Serrano J, Zang T, Bieniasz PD. Hiv-1 and ebola virus encode small peptide motifs that recruit tsg101 to sites of particle assembly to facilitate egress. Nat Med. (2001) 7:1313–9. doi: 10.1038/nm1201-1313
18. Kikonyogo A, Bouamr F, Vana ML, Xiang Y, Aiyar A, Carter C, et al. Proteins related to the nedd4 family of ubiquitin protein ligases interact with the L domain of rous sarcoma virus and are required for gag budding from cells. Proc Natl Acad Sci U.S.A. (2001) 98:11199–204. doi: 10.1073/pnas.201268998
19. von Schwedler UK, Stuchell M, Muller B, Ward DM, Chung HY, Morita E, et al. The protein network of hiv budding. Cell. (2003) 114:701–13. doi: 10.1016/s0092-8674(03)00714-1
20. Strack B, Calistri A, Craig S, Popova E, Gottlinger HG. Aip1/alix is a binding partner for hiv-1 P6 and eiav P9 functioning in virus budding. Cell. (2003) 114:689–99. doi: 10.1016/s0092-8674(03)00653-6
21. Martin-Serrano J, Yarovoy A, Perez-Caballero D, Bieniasz PD. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc Natl Acad Sci U.S.A. (2003) 100:12414–9. doi: 10.1073/pnas.2133846100
22. Schmitt AP, Leser GP, Morita E, Sundquist WI, Lamb RA. Evidence for a new viral late-domain core sequence, fpiv, necessary for budding of a paramyxovirus. J Virol. (2005) 79:2988–97. doi: 10.1128/JVI.79.5.2988-2997.2005
23. Chung HY, Morita E, von Schwedler U, Muller B, Krausslich HG, Sundquist WI. Nedd4l overexpression rescues the release and infectivity of human immunodeficiency virus type 1 constructs lacking ptap and ypxl late domains. J Virol. (2008) 82:4884–97. doi: 10.1128/JVI.02667-07
24. Usami Y, Popov S, Popova E, Gottlinger HG. Efficient and specific rescue of human immunodeficiency virus type 1 budding defects by a nedd4-like ubiquitin ligase. J Virol. (2008) 82:4898–907. doi: 10.1128/JVI.02675-07
25. Weiss ER, Popova E, Yamanaka H, Kim HC, Huibregtse JM, Gottlinger H. Rescue of hiv-1 release by targeting widely divergent nedd4-type ubiquitin ligases and isolated catalytic hect domains to gag. PloS Pathog. (2010) 6:e1001107. doi: 10.1371/journal.ppat.1001107
26. Mercenne G, Alam SL, Arii J, Lalonde MS, Sundquist WI. Angiomotin functions in hiv-1 assembly and budding. Elife. (2015) 4:e03778. doi: 10.7554/eLife.03778
27. Rheinemann L, Thompson T, Mercenne G, Paine EL, Peterson FC, Volkman BF, et al. Interactions between amot ppxy motifs and nedd4l ww domains function in hiv-1 release. J Biol Chem. (2021) 297:100975. doi: 10.1016/j.jbc.2021.100975
28. Barbosa NS, Mendonca LR, Dias MVS, Pontelli MC, da Silva EZM, Criado MF, et al. Escrt machinery components are required for orthobunyavirus particle production in golgi compartments. PloS Pathog. (2018) 14:e1007047. doi: 10.1371/journal.ppat.1007047
29. Huttunen M, Samolej J, Evans RJ, Yakimovich A, White IJ, Kriston-Vizi J, et al. Vaccinia virus hijacks escrt-mediated multivesicular body formation for virus egress. Life Sci Alliance. (2021) 4. doi: 10.26508/lsa.202000910
30. Corless L, Crump CM, Griffin SD, Harris M. Vps4 and the escrt-iii complex are required for the release of infectious hepatitis C virus particles. J Gen Virol. (2010) 91:362–72. doi: 10.1099/vir.0.017285-0
31. Crump CM, Yates C, Minson T. Herpes simplex virus type 1 cytoplasmic envelopment requires functional vps4. J Virol. (2007) 81:7380–7. doi: 10.1128/JVI.00222-07
32. Calistri A, Sette P, Salata C, Cancellotti E, Forghieri C, Comin A, et al. Intracellular trafficking and maturation of herpes simplex virus type 1 gb and virus egress require functional biogenesis of multivesicular bodies. J Virol. (2007) 81:11468–78. doi: 10.1128/JVI.01364-07
33. Barnes J, Jordan BA, Wilson DW. An escrt/vps4 envelopment trap to examine the mechanism of alphaherpesvirus assembly and transport in neurons. J Virol. (2022) 96:e0217821. doi: 10.1128/jvi.02178-21
34. Kharkwal H, Smith CG, Wilson DW. Blocking escrt-mediated envelopment inhibits microtubule-dependent trafficking of alphaherpesviruses in vitro. J Virol. (2014) 88:14467–78. doi: 10.1128/JVI.02777-14
35. Pawliczek T, Crump CM. Herpes simplex virus type 1 production requires a functional escrt-iii complex but is independent of tsg101 and alix expression. J Virol. (2009) 83:11254–64. doi: 10.1128/JVI.00574-09
36. Russell T, Samolej J, Hollinshead M, Smith GL, Kite J, Elliott G. Novel role for escrt-iii component chmp4c in the integrity of the endocytic network utilized for herpes simplex virus envelopment. mBio. (2021) 12:e02183-20. doi: 10.1128/mBio.02183-20
37. Tandon R, AuCoin DP, Mocarski ES. Human cytomegalovirus exploits escrt machinery in the process of virion maturation. J Virol. (2009) 83:10797–807. doi: 10.1128/JVI.01093-09
38. Mettenleiter TC. Vesicular nucleo-cytoplasmic transport-herpesviruses as pioneers in cell biology. Viruses. (2016) 8:266. doi: 10.3390/v8100266
39. Arii J. Host and viral factors involved in nuclear egress of herpes simplex virus 1. Viruses. (2021) 13:754. doi: 10.3390/v13050754
40. Klupp BG, Granzow H, Fuchs W, Keil GM, Finke S, Mettenleiter TC. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc Natl Acad Sci U.S.A. (2007) 104:7241–6. doi: 10.1073/pnas.0701757104
41. Arii J, Watanabe M, Maeda F, Tokai-Nishizumi N, Chihara T, Miura M, et al. Escrt-iii mediates budding across the inner nuclear membrane and regulates its integrity. Nat Commun. (2018) 9:3379. doi: 10.1038/s41467-018-05889-9
42. Arii J, Takeshima K, Maruzuru Y, Koyanagi N, Nakayama Y, Kato A, et al. Role of the arginine cluster in the disordered domain of herpes simplex virus 1 ul34 for the recruitment of escrt-iii for viral primary envelopment. J Virol. (2022) 96:e0170421. doi: 10.1128/JVI.01704-21
43. Lee CP, Liu PT, Kung HN, Su MT, Chua HH, Chang YH, et al. The escrt machinery is recruited by the viral bfrf1 protein to the nucleus-associated membrane for the maturation of epstein-barr virus. PloS Pathog. (2012) 8:e1002904. doi: 10.1371/journal.ppat.1002904
44. Lee CP, Liu GT, Kung HN, Liu PT, Liao YT, Chow LP, et al. The ubiquitin ligase itch and ubiquitination regulate bfrf1-mediated nuclear envelope modification for epstein-barr virus maturation. J Virol. (2016) 90:8994–9007. doi: 10.1128/JVI.01235-16
45. Rose KM. When in need of an escrt: the nature of virus assembly sites suggests mechanistic parallels between nuclear virus egress and retroviral budding. Viruses. (2021) 13:1138. doi: 10.3390/v13061138
46. Kalamvoki M, Deschamps T. Extracellular vesicles during herpes simplex virus type 1 infection: an inquire. Virol J. (2016) 13:63. doi: 10.1186/s12985-016-0518-2
47. Kalamvoki M, Du T, Roizman B. Cells infected with herpes simplex virus 1 export to uninfected cells exosomes containing sting, viral mrnas, and micrornas. Proc Natl Acad Sci U.S.A. (2014) 111:E4991–6. doi: 10.1073/pnas.1419338111
48. Dogrammatzis C, Saleh S, Deighan C, Kalamvoki M. Diverse populations of extracellular vesicles with opposite functions during herpes simplex virus 1 infection. J Virol. (2021) 95:e02357-20. doi: 10.1128/JVI.02357-20
49. Deschamps T, Kalamvoki M. Extracellular vesicles released by herpes simplex virus 1-infected cells block virus replication in recipient cells in a sting-dependent manner. J Virol. (2018) 92:e01102-18. doi: 10.1128/JVI.01102-18
50. Strickland M, Ehrlich LS, Watanabe S, Khan M, Strub MP, Luan CH, et al. Tsg101 chaperone function revealed by hiv-1 assembly inhibitors. Nat Commun. (2017) 8:1391. doi: 10.1038/s41467-017-01426-2
51. Watanabe SM, Ehrlich LS, Strickland M, Li X, Soloveva V, Goff AJ, et al. Selective targeting of virus replication by proton pump inhibitors. Sci Rep. (2020) 10:4003. doi: 10.1038/s41598-020-60544-y
52. Leis J, Luan CH, Audia JE, Dunne SF, Heath CM. Ilaprazole and other novel prazole-based compounds that bind tsg101 inhibit viral budding of hsv-1/2 and hiv from cells. J Virol. (2021) 95:e00190-21. doi: 10.1128/JVI.00190-21
53. Mannemuddhu SS, Xu H, Bleck CKE, Tjandra N, Carter C, Bhaduri-McIntosh S. Prazoles targeting tsg101 inhibit release of epstein-barr virus following reactivation from latency. J Virol. (2021) 95:e0246620. doi: 10.1128/JVI.02466-20
54. Chen BJ, Leser GP, Morita E, Lamb RA. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J Virol. (2007) 81:7111–23. doi: 10.1128/JVI.00361-07
55. Bruce EA, Medcalf L, Crump CM, Noton SL, Stuart AD, Wise HM, et al. Budding of filamentous and non-filamentous influenza a virus occurs via a vps4 and vps28-independent pathway. Virology. (2009) 390:268–78. doi: 10.1016/j.virol.2009.05.016
56. Utley TJ, Ducharme NA, Varthakavi V, Shepherd BE, Santangelo PJ, Lindquist ME, et al. Respiratory syncytial virus uses a vps4-independent budding mechanism controlled by rab11-fip2. Proc Natl Acad Sci U.S.A. (2008) 105:10209–14. doi: 10.1073/pnas.0712144105
57. Taylor GM, Hanson PI, Kielian M. Ubiquitin depletion and dominant-negative vps4 inhibit rhabdovirus budding without affecting alphavirus budding. J Virol. (2007) 81:13631–9. doi: 10.1128/JVI.01688-07
58. Miura K, Suzuki Y, Ishida K, Arakawa M, Wu H, Fujioka Y, et al. Distinct motifs in the E protein are required for sars-cov-2 virus particle formation and lysosomal deacidification in host cells. J Virol. (2023) 97:e0042623. doi: 10.1128/jvi.00426-23
59. Rossman JS, Lamb RA. Viral membrane scission. Annu Rev Cell Dev Biol. (2013) 29:551–69. doi: 10.1146/annurev-cellbio-101011-155838
60. Rossman JS, Jing X, Leser GP, Lamb RA. Influenza virus M2 protein mediates escrt-independent membrane scission. Cell. (2010) 142:902–13. doi: 10.1016/j.cell.2010.08.029
61. Martyna A, Bahsoun B, Badham MD, Srinivasan S, Howard MJ, Rossman JS. Membrane remodeling by the M2 amphipathic helix drives influenza virus membrane scission. Sci Rep. (2017) 7:44695. doi: 10.1038/srep44695
62. Sutherland M, Tran N, Hong M. Clustering of tetrameric influenza M2 peptides in lipid bilayers investigated by (19)F solid-state nmr. Biochim Biophys Acta Biomembr. (2022) 1864:183909. doi: 10.1016/j.bbamem.2022.183909
63. Bigalke JM, Heuser T, Nicastro D, Heldwein EE. Membrane deformation and scission by the hsv-1 nuclear egress complex. Nat Commun. (2014) 5:4131. doi: 10.1038/ncomms5131
64. Bjerke SL, Cowan JM, Kerr JK, Reynolds AE, Baines JD, Roller RJ. Effects of charged cluster mutations on the function of herpes simplex virus type 1 ul34 protein. J Virol. (2003) 77:7601–10. doi: 10.1128/JVI.77.13.7601-7610.2003
65. Bigalke JM, Heldwein EE. Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J. (2015) 34:2921–36. doi: 10.15252/embj.201592359
66. Hagen C, Dent KC, Zeev-Ben-Mordehai T, Grange M, Bosse JB, Whittle C, et al. Structural basis of vesicle formation at the inner nuclear membrane. Cell. (2015) 163:1692–701. doi: 10.1016/j.cell.2015.11.029
67. Arii J, Takeshima K, Maruzuru Y, Koyanagi N, Kato A, Kawaguchi Y. Roles of the interhexamer contact site for hexagonal lattice formation of the herpes simplex virus 1 nuclear egress complex in viral primary envelopment and replication. J Virol. (2019) 93:e00498-19. doi: 10.1128/JVI.00498-19
68. Klupp BG, Granzow H, Klopfleisch R, Fuchs W, Kopp M, Lenk M, et al. Functional analysis of the pseudorabies virus ul51 protein. J Virol. (2005) 79:3831–40. doi: 10.1128/JVI.79.6.3831-3840.2005
69. Womack A, Shenk T. Human cytomegalovirus tegument protein pul71 is required for efficient virion egress. mBio. (2010) 1. doi: 10.1128/mBio.00282-10
70. Song MJ, Hwang S, Wong WH, Wu TT, Lee S, Liao HI, et al. Identification of viral genes essential for replication of murine gamma-herpesvirus 68 using signature-tagged mutagenesis. Proc Natl Acad Sci U.S.A. (2005) 102:3805–10. doi: 10.1073/pnas.0404521102
71. Oda S, Arii J, Koyanagi N, Kato A, Kawaguchi Y. The interaction between herpes simplex virus 1 tegument proteins ul51 and ul14 and its role in virion morphogenesis. J Virol. (2016) 90:8754–67. doi: 10.1128/JVI.01258-16
72. Butt BG, Owen DJ, Jeffries CM, Ivanova L, Hill CH, Houghton JW, et al. Insights into herpesvirus assembly from the structure of the pul7:Pul51 complex. Elife. (2020) 9:e53789. doi: 10.7554/eLife.53789
73. Streck NT, Carmichael J, Buchkovich NJ. Nonenvelopment role for the escrt-iii complex during human cytomegalovirus infection. J Virol. (2018) 92:e02096-17. doi: 10.1128/JVI.02096-17
74. Wollert T, Wunder C, Lippincott-Schwartz J, Hurley JH. Membrane scission by the escrt-iii complex. Nature. (2009) 458:172–7. doi: 10.1038/nature07836
75. Saksena S, Wahlman J, Teis D, Johnson AE, Emr SD. Functional reconstitution of escrt-iii assembly and disassembly. Cell. (2009) 136:97–109. doi: 10.1016/j.cell.2008.11.013
76. Wollert T, Hurley JH. Molecular mechanism of multivesicular body biogenesis by escrt complexes. Nature. (2010) 464:864–9. doi: 10.1038/nature08849
77. Baumgartel V, Ivanchenko S, Dupont A, Sergeev M, Wiseman PW, Krausslich HG, et al. Live-cell visualization of dynamics of hiv budding site interactions with an escrt component. Nat Cell Biol. (2011) 13:469–74. doi: 10.1038/ncb2215
78. Jouvenet N, Zhadina M, Bieniasz PD, Simon SM. Dynamics of escrt protein recruitment during retroviral assembly. Nat Cell Biol. (2011) 13:394–401. doi: 10.1038/ncb2207
79. Elia N, Sougrat R, Spurlin TA, Hurley JH, Lippincott-Schwartz J. Dynamics of endosomal sorting complex required for transport (Escrt) machinery during cytokinesis and its role in abscission. Proc Natl Acad Sci U.S.A. (2011) 108:4846–51. doi: 10.1073/pnas.1102714108
80. Remec Pavlin M, Hurley JH. The escrts - converging on mechanism. J Cell Sci. (2020) 133:jcs240333. doi: 10.1242/jcs.240333
81. Pfitzner AK, Mercier V, Jiang X, Moser von Filseck J, Baum B, Saric A, et al. An escrt-iii polymerization sequence drives membrane deformation and fission. Cell. (2020) 182:1140–55 e18. doi: 10.1016/j.cell.2020.07.021
82. Schoneberg J, Pavlin MR, Yan S, Righini M, Lee IH, Carlson LA, et al. Atp-dependent force generation and membrane scission by escrt-iii and vps4. Science. (2018) 362:1423–8. doi: 10.1126/science.aat1839
Keywords: viral egress, budding, ESCRT, herpesvirus, membrane scission
Citation: Arii J (2024) ESCRT-III-dependent and -independent egress of herpesviruses. Front. Virol. 4:1378054. doi: 10.3389/fviro.2024.1378054
Received: 29 January 2024; Accepted: 22 February 2024;
Published: 05 March 2024.
Edited by:
Takayuki Murata, Fujita Health University, JapanReviewed by:
Arianna Calistri, University of Padua, ItalyCopyright © 2024 Arii. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Arii, amFyaWlAbWVkLmtvYmUtdS5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.