 Moe Iwata1
Moe Iwata1 Ryoko Kawabata2Nao Morimoto1
Ryoko Kawabata2Nao Morimoto1 Ryosuke F. Takeuchi1
Ryosuke F. Takeuchi1 Takemasa Sakaguchi2
Takemasa Sakaguchi2 Takashi Irie2*
Takashi Irie2* Fumitaka Osakada1,3,4,5,6*
Fumitaka Osakada1,3,4,5,6*- 1Laboratory of Cellular Pharmacology, Graduate School of Pharmaceutical Sciences, Nagoya University, Nagoya, ;Japan
- 2Graduate School of Biomedical and Health Sciences, Hiroshima University, Hiroshima, ;Japan
- 3Laboratory of Neural Information Processing, Institute for Advanced Research, Nagoya University, Nagoya, ;Japan
- 4Institute of Nano-Life-Systems, Institutes of Innovation for Future Society, Nagoya University, Nagoya, ;Japan
- 5Institute for Glyco-core Research, Nagoya University, Nagoya, ;Japan
- 6PRESTO/CREST, Japan Science and Technology Agency, Saitama, ;Japan
Persistent virus infection involves modifying the host immune response and maintaining viral infection. Acute infection with Mononegavirales, such as Sendai viruses (SeVs), can give rise to viruses capable of persistent infection. SeVs establish persistent infection through generating copyback-type defective interfering (cbDI) genomes or acquiring temperature-sensitive mutations. Herein, we identify novel mutations associated with persistent infection and recombinant SeV mutants capable of persistent infection and autonomous production at physiological body temperature, independent of cbDI genomes or temperature-sensitive mutations. Diverse SeV populations were generated by passing the cDNA-recovered SeV Z strain 19 times through embryonated chicken eggs and subsequently infecting LLC-MK2 cells with the SeV populations to finally obtain SeV mutants capable of persistent infection and autonomous production in several types of cultured cells. Sequence analysis identified 4 or 5 mutations in the genome of the persistently infectious SeVs, distinguishing them from other existing strains with persistent infection. Recombinant SeVs carrying 4 or 5 mutations in the Z strain genome (designated SeV-Zpi or SeV-Zpi2, respectively) exhibited persistent infection and autonomous production in LLC-MK2, BHK-21, and Neuro2a cells at 37°C. SeV-Zpi and SeV-Zpi2 consistently produced viral particles even after long-term passages without cbDI particles or temperature-sensitive phenotypes. These results highlight the ability of acute infections of SeVs to spontaneously acquire mutations during replication, thereby endowing persistent infection and autonomous production at body temperature. The vectorization of SeV-Zpi and SeV-Zpi2 will contribute to both basic research and medical applications.
1 Introduction
Viruses exploit cells of infected organisms to enable viral replication while simultaneously interacting with host cells and affecting host functions. Pathogenic viruses can be eliminated by host immunity or may cause host cell death. Intriguingly, certain viruses, including human immunodeficiency virus and hepatitis B virus, persistently infect host cells for extended periods without being eliminated, and persistent infection can be acquired by viruses that typically cause acute infections during repeated replication within host cells (1–4). This phenomenon has been observed in measles viruses (Paramyxoviridae), which upon infecting humans trigger transient fever and rash and can cause viral elimination from the host in many cases. However, in cases where the host immune system is impaired, measles viruses can persist in the central nervous system for long periods. This persistent infection by measles viruses has been suggested to involve the acquisition of envelope and matrix protein mutations that confer persistent infectivity in the nervous system (5–8). These mutations are considered key characteristics directly related to the pathogenicity of subacute sclerosing panencephalitis (SSPE) viruses because they are involved in viral spread between neurons (9–11). Other mononegaviruses, such as Sendai virus (SeV), vesicular stomatitis virus, and rabies virus, have been reported to acquire mutations and produce persistently infectious viruses following continued passaging of acutely infected cells in laboratory settings (1–4). Moreover, acquisition of persistent infectivity in acutely infectious viruses changes the dynamics of virus–host interactions. However, the underlying mechanisms and their significance remain poorly understood. Several examples of mononegaviruses acquiring persistent infectivity through mechanisms different from those observed in SSPE viruses have been reported (2, 12). SeV, which also belongs to the Paramyxoviridae family, has been studied using cultured cells and animal models to elucidate the generation and mechanism of persistently infectious viruses. These studies have revealed at least two mechanisms whereby SeV can establish persistent infection.
The first mechanism of persistent infectivity is through the generation of defective interfering (DI) particles and DI genome contained in them (2, 13). The DI genome is a short RNA that lacks most of the inner genome encoding viral proteins because of errors during viral genome replication in virus-infected cells. Two types of DI genomes are known: the internal deletion-type DI (idDI) genomes, which lack the viral genome sequence between the leader and trailer, and the copyback-type DI (cbDI) genomes, which contain only the 5′ side of the viral genome (14, 15). The idDI genome promoter sequences at both ends are similar to those of the full-length genome, whereas the cbDI genomes have a 5′-end promoter that enables a faster replication rate in the replacement of the 3′-end promoter of the genome (14, 16, 17). Cells infected with high mononegaviral titers are known to occasionally produce virus-like particles encapsulating cbDI genomes and become persistently infected (18, 19). The cbDI genomes are amplified using viral proteins supplied by the intact virus that simultaneously infects the cells (2). However, the generation and accumulation of cbDI genomes inhibit the growth of coexisting intact viruses because of competition between the replication of the two viral genomes (20). Since the replication of the cbDI genome depends on the replication of the intact virus, an extended state of persistent infection is thought to occur with repeated cycles where the intact virus level decreases as the cbDI genome level increases and the cbDI genome level increases as the intact virus level decreases (13, 21). Selective survival of cbDI genome-dominant cells may also contribute to persistent infection by the cbDI genome (22). Tumor necrosis factor (TNF) produced by cbDI genome-dominant cells binds to TNF receptor 1 on neighboring cells to selectively induce apoptosis in cells containing full-length genomes, thereby selectively surviving cells containing cbDI genomes. cbDI genomes arise due to errors during repeated replication of the RNA genome. A single amino acid mutation in the N protein of the Cantell strain of SeV generates the DI genomes, which serve as an RIG-I ligand and induce antiviral responses, including interferon (IFN) production (23).
The second mechanism underlying persistent infection is temperature-sensitive mutations that confer persistent infections independent of the emergence of DI genomes (12, 24, 25). Temperature-sensitive mutants, such as the Cl.151 and ts strains of SeV, arise through specific viral genome mutations during repeated passage under low-temperature conditions (12, 24). Temperature-sensitive mutant viruses can persistently infect host cells at low temperatures because they can replicate at low temperatures but not near physiological temperatures. Due to the loss of viral structural proteins or the instability of viral proteins at physiological temperatures, these temperature-sensitive mutant viruses are unable to produce infectious viral particles and spread infection at physiological temperatures (25–28). In addition, temperature-sensitive mutant SeVs suppress the antiviral response of type I IFNs, in contrast to DI particles (29). Thus, the establishment of persistent infection by temperature-sensitive mutant SeVs may involve a different mechanism compared with the co-existence of DI genomes. In addition, temperature-sensitive mutant SeVs suppress the replication of coinfected wild-type (WT) viruses, potentially by inhibiting protein synthesis of the WT virus by temperature-sensitive mutant SeVs, rather than differences in the replication rate between these.
This study sought to verify whether SeV capable of persistent infection at 37°C can spontaneously emerge, using the Z strain of SeV, which is neither temperature-sensitive nor capable of producing DI genomes. The low fidelity of SeV polymerase allows the SeV Z strain to spontaneously generate a heterogeneous population of viruses carrying various mutations through repeated SeV passages in embryonated chicken eggs. Consequently, we established cell lines in which SeV was persistently infected at 37°C from the highly heterogeneous SeV population and identified mutations in the persistently infected cells. We demonstrated that recombinant SeVs carrying the identified 4-5 mutations established persistent infection and propagated at 37°C. The persistently infectious SeV probably mediates unique mechanisms distinct from DI genomes and temperature-sensitive mutations. Thus, this study reveals the spontaneous generation of persistently infectious SeV from acutely infected SeV at body temperature due to mutations acquired during viral replication.
2 Materials and methods
2.1 Cell culture and virus
LLC-MK2 cells (rhesus macaque monkey kidney-derived cells) (30), BHK-21 expressing T7 polymerase (BHK-T7; 31), HeLa cells (human cervical cancer-derived cells) (32), human embryonic kidney-derived cells expressing simian virus 40 large T antigen (HEK293T; RIKEN BRC Cell Bank), and Vero clone E6 cells stably expressing human transmembrane protease, serine 2 (TMPRSS2) (Vero/TMPRSS2; kindly provided by M. Takeda at the National Institute of Infectious Diseases, Japan) (33) were maintained in Dulbecco’s minimum essential medium (DMEM; Wako) supplemented with 5% fetal bovine serum (FBS; Biosera), penicillin G (100 units/mL, Meiji Seika Pharma, Tokyo, Japan), and streptomycin (100 mg/mL, Meiji Seika Pharma) in 5% CO2 humidified atmosphere at 37°C. Neuro2a cells (RIKEN Cell Bank) were maintained in DMEM containing 10% FBS, penicillin G, and streptomycin. cDNA plasmid of the WT SeV Z strain (pSeVZ(+))was obtained from Kato et al. (31). The Cantell strain of SeV (34) was purchased from ATCC (VR-907), amplified once with embryonic chicken eggs, and titrated for subsequent experiments. The Cl.151 strain was recovered by transfection of genomic plasmid DNA, and titrated for subsequent experiments, as described previously (26).
2.2 Generation of the SeV Z strain from DNA plasmids
For recovery of the Z strain of SeV, 1.0 × 106 cells of BHK-T7 cells were seeded at 1.1 × 103 cells/mm2 in a 6-well plate and infected with vaccinia virus expressing T7 polymerase. The BHK-T7 cells were then transfected with 0.5 µg of pSeVZ(+), 0.46 µg of pGEM-N, 0.46 µg of pGEM-P, and 0.09 µg of pGEM-L plasmids at a ratio of 5.5: 5: 5: 1 using FuGENE HD reagent (Promega) and CombiMag (OZ Biosciences) (31). Transfected BHK-T7 cells were cultured for three days in a humidified atmosphere of 5% CO2 at 37°C and then subjected to a freeze–thaw cycle and filtration. Resulting SeV recombinants were propagated in 10-day-old embryonated chicken eggs at 34°C for 3 days. Allantoic fluids containing the grown SeVs were collected and stored at −80°C as virus stock.
2.3 Virus infectivity titrations
LLC-MK2 cells were infected with SeV at different multiplicities of infection (MOI). Viral titers were measured by immunostaining infected cells using anti-SeV antibody as described previously (35). To prepare infectious virus, the virus in the cell supernatant was treated with trypsin (40 µg/mL). SeVs amplified from embryonated chicken eggs were used without trypsin treatment. Viral titers were calculated as cell infectious units per mL (CIU/mL).
2.4 Diversification of the Z strain
A recombinant SeV Z strain was injected and grown in 10-day-old embryonated chicken eggs. The eggs were incubated at 34°C for 3 days, and then amplified SeV was collected from the allantoic fluids. The amplification process of SeVs in embryonated chicken eggs was repeated 19 times (Z-egg-19).
2.5 Serial passage of SeV-infected cells
LLC-MK2 cells were infected with SeVs and then continuously passaged with TypLE express (Thermo, 1 mL/100-mm dish) every 2-3 days. A small amount of surviving infected cells were maintained in 5% FBS-containing DMEM in 5% CO2 and 37°C.
2.6 Sequencing of the SeV genome
RNA was extracted from cells persistently infected with SeV using the High Pure RNA isolation kit (Roche). A cDNA library of SeV was generated using the QIAGEN OneStep RT-PCR Kit (QIAGEN). Persistently infected SeV genomes were sequenced using the BigDye™ Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). Primers used are listed in Supplementary Table 1.
2.7 Plasmid construction
Four mutations were identified in the genome of viruses isolated from persistently infected LLC-MK2 cells. These mutations were introduced into pSeVZ(+) using the NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs) to generate pSeVZpi(+). One further mutation was identified in the genome of the persistently infected Zpi strain and introduced into pSeVZpi(+) by mutagenesis using the NEBuilder HiFi DNA Assembly to generate pSeVZpi2(+). All plasmids generated in this study were validated by sequencing before cell transfection; primers used are listed in Supplementary Table 1.
2.8 Single-step and multi-step growth curve analyses
The viral growth kinetics of SeVs were evaluated using single-step and multi-step growth curves. The single-step growth curve evaluates viral ability to replicate within infected cells, whereas the multi-step growth curve evaluates viral ability to bud from infected cells and spread infection between cells. For the single-step growth curve, 1 × 106 cells of LLC-MK2 cells were cultured at 1.1 × 103 cells/mm2 in 35-mm dishes for 24 h and infected with either the Z, Zpi, or Zpi2 strains of SeV at an MOI of 5. For the multi-step growth curve, 1 × 106 cells of Vero/TMPRSS2 cells were cultured at 1.1 × 103 cells/mm2 in 35-mm dishes for 24 h and infected with each of the three SeV or Cl.151 strains at an MOI of 0.01. After virus infection, cells were incubated at 37°C for 1 h, washed three times with phosphate-buffered saline (PBS), and then maintained at 37°C with 5% FBS-containing DMEM for single-step growth curves or with FBS-free DMEM containing trypsin (40 µg/mL) for multi-step growth curves. Culture supernatants were collected at 8, 16, 24, 48, and 72 h after infection. The amount of infectious virus in the culture supernatant was determined by infecting LLC-MK2 cells and quantifying the number of infected cells. Log10 values of the final virus titers were obtained for statistical comparison between virus strains.
2.9 Cytotoxicity assay
Cytotoxicity was quantified using the MTT assay. Vero/TMPRSS2 cells (4.0 × 104 cells) were seeded on 96-well plates at a density of 1.2 × 103 cells/mm2. After 18 h, cells were infected with the Z, Zpi, and Zpi2 strains of SeV at 100 µL each at an MOI of 0.01. After 1 h of infection, the plates were rinsed three times with PBS and then incubated in DMEM containing trypsin (40 µg/mL). Cells were cultured for additional 1, 3, and 5 days. For the MTT assay, 10 µL of MTT solution (5 mg/mL, Doujindo) in PBS was applied directly to each well containing 180 µL of the medium. After 2 h of incubation at 37°C, 150 µL of 10% n-sodiumdodecyl sulfate (SDS) was applied to each well of the plates. After 2 h of incubation at 37°C, absorbance was measured at 570 nm (GloMax Discover Microplate Reader, Promega). Survival of virus-infected cells was expressed relative to mock-infected cells at each time point.
2.10 Persistent infectivity in diverse cell types
LLC-MK2, BHK-21/T7, HeLa, Neuro2a, and Vero/TMPRSS2 cells (1 × 106 cells each) were incubated at a density of 1.1 × 103 cells/mm2 in 35-mm dishes for 24 h and then infected with Z, Zpi, and Zpi2 strains of SeVs at an MOI of 0.003. Infected cells were repeatedly passaged every 2–4 days using TrypLE Express (Invitrogen).
2.11 Acquisition of viruses with long-term persistent infection
Cells persistently infected with the Zpi strain were passaged for an extended period, and the culture supernatant of cells from the 101st passage was collected and amplified in 10-day-old embryonated chicken eggs (Zpi-101). Infected cells of the Zpi2 strain were also passaged in the same manner, and the culture supernatant of the 81st passage that had been persistently infected was collected and treated in embryonated chicken eggs (Zpi2-91). Titers were measured in the LLC-MK2 cells.
2.12 Quantitative RT-PCR of cbDI
Total RNA was extracted using High Pure RNA isolation kit (Roche Diagnostics). LLC-MK2 cells were infected with SeV strains Z, Zpi, Zpi2, Zpi-101, Zpi2-91, or Cantell at an MOI of 5 and subjected to RNA isolation at 24 h post-infection (hpi). Levels of viral RNA and DI RNA were quantified by one-step real-time quantitative PCR (RT-qPCR) using QuantiFast SYBR Green RT-PCR (QIAGEN). Primers for the full viral genomes and cbDI genomes are listed in Supplementary Table 1. Viral RNA and actin levels were used as internal controls. The relative level of cbDI genomes to the SeV genome of the Z strain was set as 1. The Cantell strain was used as a positive control for DI genome expression.
2.13 Western blotting for detection of N and M proteins
LLC-MK2 cells infected with SeV strains Z, Zpi, or Zpi2 at an MOI of 5 were lysed in ice-cold sample buffer (125 mM Tris-HCl (pH 6.8), 4.6% SDS, 10% 2-mercaptoethanol, 0.005% bromophenol blue, and 20% glycerol) at 24, 48, and 72 hpi and boiled for 15 min. The samples were electrophoresed on 10% polyacrylamide gels for NP protein detection or on 12% polyacrylamide gels for M protein detection. Separated proteins were transferred to a polyvinylidene difluoride membrane (Millipore) using Trans-Blot Turbo (Bio-Rad). Membranes were blocked with skim milk and probed with rabbit polyclonal anti-NP antibody (1:10000, homemade) or mouse polyclonal anti-M antibody (1:5000, Inoue et al., 2013) overnight at 4°C. Membranes were rinsed and incubated with horseradish peroxidase-conjugated anti-rabbit and anti-mouse IgG goat polyclonal antibodies (1:1000, Santa Cruz Biotechnology) for 2 h. Membranes were treated with the enhanced chemiluminescence kit (Millipore), and immunoreactive bands were visualized.
2.14 Coinfection with wild-type and persistently infectious viruses
The viruses used were Z strain expressing Katushka (Z-K), Z strain expressing Venus (Z-V), Zpi strain expressing Venus (Zpi-V), and Zpi2 strain expressing Venus (Zpi2-V). For single infection, culture supernatants of Vero/TMPRSS2 cells were infected with each of the four viruses and collected at 1, 2, and 3 d post-infection (dpi). Total RNA was isolated from the culture supernatant using a High Pure RNA isolation kit (Roche Diagnostics). Viral RNA and fluorescent protein RNA levels were quantified by QuantiFast SYBR Green RT-PCR (QIAGEN) and one-step RT-qPCR using the primers listed in Supplementary Table 1. A graph was created by plotting the titer measured from the culture supernatant and the Cq value of the quantified fluorescent protein RNA. A standard curve was obtained from the culture supernatants of Z-K- and Z-V-infected cells. A standard curve was drawn from the Cq value and titer calculated by serially diluting the 3-dpi culture supernatant and performing one-step RT-qPCR. For coinfection, LLC-MK2 and Vero/TMPRSS2 cells were infected with Z-K and Zpi-V at a MOI of 0.005 each.
To validate the quantification method of Z-K, Zpi-V, Zpi-V, and Zpi2-V, viral titers and Cq values were measured at different time points (Supplementary Figure S1A). Z strain expressing the red-fluorescent protein Katushka, and Z, Zpi, or Zpi2 strains, each expressing the green-fluorescent protein Venus, were individually infected with Vero/TMPRSS2 cells. Culture supernatants were collected at 1, 2, and 3 dpi for titer measurements. Culture supernatants treated with trypsin (a final concentration of 40 µg/mL) were applied to LLC-MK2 cells, and the number of infected cells was counted by immunostaining with anti-SeV antibody. The culture supernatants at 3 dpi were serially diluted and subjected to titer measurement and RT-qPCR. The genome of each virus in the culture supernatant was also quantified by qPCR for Katushka and Venus sequences. The Cq values and assumed titers were positively correlated for all viruses tested (Supplementary Figure S1B). Coinfected viruses were individually quantified by this established method based on the amount of Katushka and Venus.
2.15 Statistical analysis
Data are expressed as means ± S.E.M. Statistical analyses were performed using GraphPad Prism 9. The statistical significance of differences between groups was determined by one-way analysis of variance (ANOVA) followed by Tukey’s test. Probability values less than 5% were considered significant.
3 Results
3.1 Establishment of cells persistently infected with heterogeneous SeV
Specific strains of SeV can maintain viral infection in infected cultured cells that are serially passaged at a temperature lower than the physiologically relevant temperature of 37°C (e.g. 34°C) (36–39). Therefore, we first investigated whether cultured cells persistently infected with SeV could be established even at 37°C. Several lines of evidence have demonstrated that the Cantell strain of SeV, which has undergone multiple passages and exhibits genetic diversity with various mutations in the viral genome (23), has the potential for persistent infection (40, 41). Accordingly, we infected BHK-21 cells with the Cantell strain of SeV at a high MOI of 5 at 37°C (Figure 1A). At 1 dpi, most infected cells died, whereas a small subset of the infected cells survived and remained attached to the culture dishes (Figure 1A). These surviving cells retained their proliferative potential and were successfully passaged (Figure 1B). Immunofluorescent staining revealed that nearly all of the surviving cells at the 6th passage were positive for SeV, indicating persistent SeV infection in these cells (Figure 1B). To determine whether the Cantell strain allows persistent infection in different cell types, we performed the same sets of experiments for LLC-MK2, HeLa, and Vero cells expressing the signaling lymphocyte activation molecule (Vero/SLAM) cells. All of the cell types yielded persistently infected cells positive for the SeV antigen (Supplementary Figure S2A). These results suggest that a heterogeneous SeV population with diverse mutations, similar to the Cantell strain, can allow persistent infection at 37°C.
Figure 1 Persistent infection was established by heterogeneous populations of the Z strain of SeV at a low MOI. (A) Persistent infection with the Cantell strain. BHK-21 cells infected with Cantell strain at an MOI of 5 were observed over time. (B) Passage of BHK-21 cells persistently infected with the Cantell strain. Infected cells at passages 2 and 6 were immunostained with an anti-SeV antibody for viral infection. (C) Infection by the Z strain at a higher MOI. LLC-MK2 cells infected with the Z strain at an MOI of 5 were observed over time. Infected cells died at 14 dpi. (D) Infection by the Z strain at a lower MOI. LLC-MK2 cells were infected with the Z strain at an MOI of 0.003. A portion of the infected cells were cultured with trypsin at 40 µg/mL; infected cells died at 14 dpi. (E) Generation of heterogeneous SeV populations containing persistently infectious viruses. Amplification of a homogeneous population in embryonated chicken eggs generated viruses with different mutations in each successive passage and increased diversity of the Z strain. Passage of cells infected with heterogeneous viruses at a lower MOI allowed the virus to spread infection with each passage, producing persistently infected cells. (F) Generation of persistently infected cells. The Z strain was grown in embryonated chicken eggs. Images of the 0th and 5th generations of LLC-MK2 cells infected with Z-egg-19 at an MOI of 0.003. (A–D, F) Scale bar = 100 μm.
Next, we investigated whether persistently infectious viruses could arise from a genetically less diverse population of SeV (Figure 1C). Therefore, we examined whether genetically homogeneous SeV recovered from Z strain plasmid DNA by reverse genetics could establish persistent infection in cultured cells at 37°C. The recovered virus carried the green-fluorescent Venus gene in the viral genome for visualization of infected cells (Z-Venus). Initially, we employed the same conditions as those used for the Cantell strain described above, but no viable Venus-positive cells were observed at an MOI of 5 (Figure 1C). To explore alternative conditions (e.g., MOI changes, presence of trypsin in culture) that might allow persistent infection, we infected various cell types with the Z strain at different MOIs and cultured them in DMEM in the presence of 40 µg/mL trypsin, which facilitates F protein activation and viral infection. However, no viable Venus-positive cells were detected under any of these conditions (Figure 1D). Similar results were obtained for other cell types, such as LLC-MK2 cells, HeLa cells, and Vero/SLAM cells when infected with the Z-Venus at an MOI of 5 (Supplementary Figure S2B). These results indicate that plasmid-derived homogeneous SeV does not readily induce persistent infection.
During viral replication, viruses generate error copies in their genomes, leading to altered phenotypes and increased adaptability to their environment (42). The Z strain was passaged 10-19 times in embryonated chicken eggs to obtain diversity in the population and potentially facilitate the emergence of persistently infectious viruses (Figure 1E). The 10th passage of the Z strain was examined similarly, but no Venus-positive viable cells were observed at an MOI of 5. However, by reducing the MOI to 0.003, omitting trypsin during maintenance, and applying trypsin treatment solely during cell passaging, viable Venus-positive cells were successfully passaged and maintained with the 19th passage of Z (Z-egg-19) (Figure 1F). These Venus-positive cells with sustained passaging capability expressed SeV protein as detected via western blotting. Thus, heterogeneous populations of the Z strain grown in embryonated chicken eggs may produce persistently infected cells at a lower MOI at 37°C.
3.2 Characterization of persistently infected cells
Although persistently infected cells were obtained in the above experiment, it remained unclear whether the virus-infected cells replicated via cell division or whether the virus spread from the infected cells to the surrounding uninfected cells. We first examined whether cells persistently infected with Z-egg-19 could produce viral particles in culture supernatants (Figure 2A). We collected culture supernatants from the fifth generation of these cells and assessed them by single-step growth analysis, which determine viral titers of supernatant from LLC-MK2 cells infected with Z-egg-19 over time. Z-egg-19 showed growth kinetics comparable with that of the WT Z strain at all-time points, with a final titer of 6.29 × 107 CIU/mL at 72 hpi. Next, we investigated whether Z-egg-19 can propagate between cells (Figure 2B). We performed a multi-step growth curve analysis to determine whether the viruses budded from infected cells could infect and spread to surrounding uninfected cells. Vero/TMPRSS2, which allow viral infection via F protein cleavage, were infected at an MOI of 0.01, and culture supernatants were collected at the indicated time points for titer measurement. The Z-egg-19, similar to the WT Z strain, exhibited robust growth at all-time points, reaching a final titer of 7.54 × 107 CIU/mL at 72 hpi. These results indicate that Z-egg-19 can bud out of infected cells and spread infectious viral particles to infect surrounding cells. Thus, cells persistently infected with Z-egg-19 can maintain a persistent infection state by producing infectious viruses.
Figure 2 Growth of Z strain-derived persistently infectious SeV. Single-step (A) and multi-step (B) growth curves of Z strain-derived persistently infectious SeV amplified 19 times in embryonated chicken eggs (Z-egg-19). LLC-MK2 cells were infected at an MOI of 5 for the single-step growth assay (A) and 0.01 for the multi-step growth assay (B). Viral titers of culture supernatants at the indicated time points for Z (●) and Z-egg-19 (○) are expressed as mean ± S.E.M.
3.3 Generation and characterization of the persistently infectious Zpi strain
To determine whether the persistently infectious virus had acquired mutations, we analyzed the genome sequences of viruses produced from persistently infected cells. Viral RNA genome was extracted for sequencing analysis from the persistently infected LLC-MK2 cells. From the 10th generation of passages onward, the persistently infected SeV genome was fully sequenced and compared with that of the parental strain (Z strain) to identify 4 mutations (Table 1): A16T in the leader sequence and missense mutations T850C in the M protein, C782A in the F protein, and G1743C in the L protein, which produce amino acid substitutions F284L, S261Y, and R581S, respectively.
Table 1 Comparison of nucleotide and amino acid residues in the viral genome of Z, Zpi, Zpi2, 80th generation of Zpi-infected cells, and 80th generation of Zpi2-infected cells.
We next determined whether the 4 identified mutations could establish persistently infected cells, SeVs with the 4 identified mutations were generated by reverse genetics and evaluated for their ability to establish persistent infection. The 4 mutations were introduced into pSeVZ(+)-Venus, a plasmid containing Venus-expressing parental viral genomic cDNA from the Z strain, to create pSeVZpi(+)-Venus, which served as the viral genome of the Zpi strain (Figure 3A). The Zpi strain virus was generated by transfecting T7 polymerase-expressing BHK-21 cells with pSeVZpi(+)-Venus, pGEM-N, pGEM-P, and pGEM-L. Three days after transfection, cells were harvested, freeze-thawed, filtered through a 0.45-µm filter, and injected into embryonated chicken eggs, to give a titer of 1.9 × 109 CIU/mL. Subsequently, we assessed the persistent infectivity of the Zpi strain by infecting LLC-MK2 cells with the Zpi strain at an MOI of 5 or 0.003 (Figure 3B). Venus expression was observed in infected cells at both MOIs. Cells infected with Zpi at an MOI of 5 died at 14 dpi. However, cells infected at an MOI of 0.003 could be passaged at 37°C, and nearly all cells expressed Venus by the 5th passage. Thus, the Zpi strain with all 4 identified mutations may be capable of establishing persistent infection in LLC-MK2 cells.
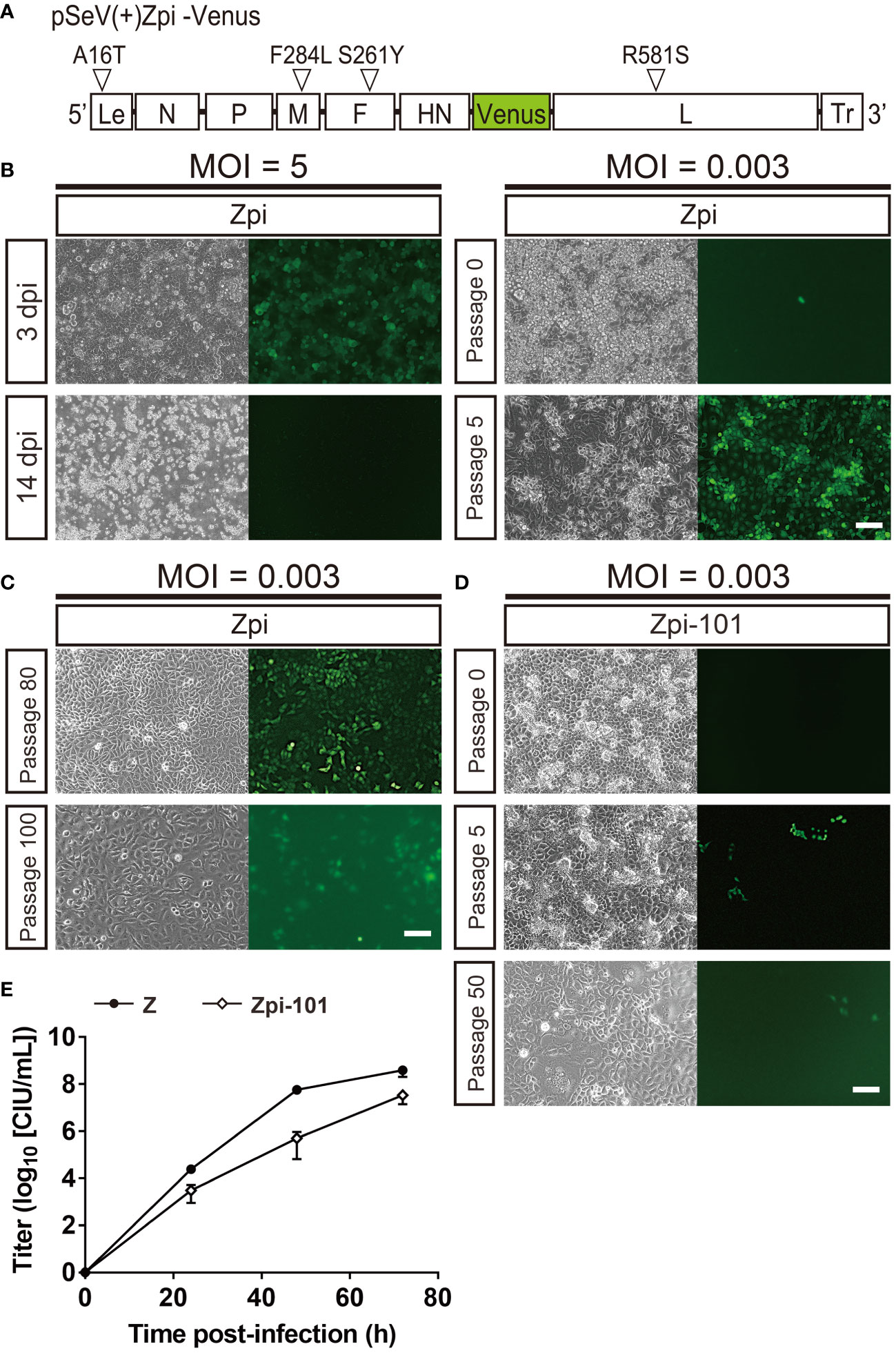
Figure 3 The Zpi strain produced persistently infected cells via viral particle production at 37°C. (A) Schematic representation of the Zpi genome plasmid. This representation shows viral proteins and locations of amino acid mutations and base mutations introduced in this study. Le, Leader region; Tr, Trailer region; white triangles = mutation locations. (B) Generation of persistently infected cells at a lower MOI, but not at a higher MOI. LLC-MK2 cells infected with the Zpi strain at an MOI of 5 expressed virus-derived Venus at 3 dpi and died at 14 dpi. However, passage of LLC-MK2 cells infected with the Zpi strain at an MOI of 0.003 showed a gradual increase in Venus-expressing cells until the fifth generation of passing, resulting in persistent infection. Scale bar = 100 µm. (C) Long-term passages of persistently infected cells with the Zpi strain. LLC-MK2 cells infected with the Zpi strain at an MOI of 0.003 were maintained until 80th and 100th generations. Scale bar = 100 µm. (D) Persistent infection with Zpi-101. LLC-MK2 cells were infected with Zpi-101 at an MOI of 0.003. Venus-expressing cells were maintained up to the 50th generation of passages. Green fluorescence was weak due to mutations in the Venus gene. Left: bright field, right: Venus-expressing cells. Scale bar = 100 µm. (E) Multi-step growth curve of Zpi-101. Vero/TMPRSS2 cells were infected at an MOI of 0.01. Viral titers of culture supernatants were measured at the indicated time points for Z (●) and Zpi-101 (⬦) are expressed as mean ± S.E.M. from three independent sets of experiments.
Furthermore, we examined whether the Zpi strain could establish a persistent infection in different cell types and animal species. Mouse neuroblastoma (Neuro2a), human cervical carcinoma epithelial cells (HeLa), rhesus monkey kidney epithelial cells (Vero), and hamster kidney epithelial cells (BHK-21) were infected with the Zpi strain at an MOI of 5 or 0.003. All cell types infected at an MOI of 5 expressed Venus but showed either cell death after infection or a decrease in the number of infected cells and an increase in the number of uninfected cells with passaging. These results were consistent with the results of Zpi infection of LLC-MK2 cells at an MOI of 5. Conversely, cells infected with the Zpi strain at an MOI of 0.003 expressed Venus and could be passaged in all four cell types (Supplementary Figure S3A). In summary, the Zpi strain with the 4 identified mutations showed persistent infectivity not only in LLC-MK2 cells but also in the other four cell types tested. These results indicate that the Zpi strain can induce persistent infection regardless of cell type and animal species.
Next, we determined how long the Zpi strain could maintain persistent infection (Figure 3C). LLC-MK2 and Neuro2A cells were infected with the Zpi strain at an MOI of 0.003 and passaged. Remarkably, both LLC-MK2 and Neuro2A cells after the 80th passage expressed virus-derived Venus (Figure 3C, Supplementary Figure S3B). However, the fluorescence intensity of Venus of LLC-MK2 cells in the 100th generation was lower than that in the 5th and 80th generation (Figure 3C). Sequence analysis revealed 5 amino acid mutations in the Venus gene of the 100th generation, suggesting that lower intensity of green fluorescence in infected cells was attributable to these mutations in the Venus gene. By titer measurement with anti-SeV antibody, the titer of the supernatant derived from the 100th generation of the Zpi strain (Zpi-100) was 1.80 × 102 CIU/mL. These results indicate that cells persistently infected with the Zpi strain can be passaged up to the 100th generation. The Zpi strain virus that has undergone the 100th passage is referred to as Zpi-100 hereafter.
We then examined whether the proliferative capacity of the Zpi strain was affected by long-term persistent infection. We compared the viral proliferative capacity of Zpi-20 (20th generation of the Zpi strain passaged in LLC-MK2 cells) with that of Zpi-100 (100th generation of the Zpi strain passaged in LLC-MK2 cells). The titers of culture supernatants containing virus produced from persistently infected cells were 6.50 × 103 and 1.80 × 102 CIU/mL for Zpi-20 and Zpi-100, respectively. These results indicate that the Zpi strain retained its proliferative ability even after 100 passages.
Furthermore, we examined whether the Zpi strain with long-term persistent infection could establish persistent infection in fresh LLC-MK2 cells (Figure 3D). Viral supernatant was collected from the 101st passage generation of LLC-MK2 cells persistently infected with the Zpi strain (Zpi-101). LLC-MK2 cells were infected with Zpi-101 at an MOI of 0.003. The infected cells were passable up to another 50th generation, although endogenous green fluorescence signals were dim due to mutations in the Venus gene (Figure 3D). Thus, 101st generation of the Zpi strain maintained persistent infectivity even after long-term passaging.
To determine whether passaged Zpi proliferates at a similar rate as WT, we performed a multi-step growth curve analysis for passaged Zpi (Figure 3E). Vero/TMPRSS2 cells were infected with Zpi-101 or the Z strain at an MOI of 0.01 each, and titers of culture supernatants at 24, 48, and 72 hpi were measured in LLC-MK2 cells by anti-SeV staining. The Zpi-101 virus was detected within 24 hpi with a final titer of 7.54 × 107 CIU/mL at 72 hpi, which was comparable with that of the WT Z strain (Figure 3E). Thus, the 101st generation of the Zpi remained capable of virus budding and infection after long-term passaging.
3.4 An additional mutation acquired by passaged Zpi
To determine whether the Zpi strain acquired additional mutations during persistent infection, we sequenced the viral genome of infected cells from the 10th passage generation (Zpi-10). Five mutations were found compared with the genome of the WT Z strain (Table 1). A16T, T850C, C782A, and G1743C were identical to those found in the Zpi strain, whereas one new mutation, T832C, was found as a nucleotide mutation and produced an amino acid substitution of S278P in the L protein.
To assess whether the 5 mutations acquired by the passaged Zpi were maintained during long-term persistent infection, we sequenced the viral genomes from persistently infected cells at the 80th passage generation (Zpi-80). Sequence analysis demonstrated that the 5 identified mutations remained unchanged compared to the Zpi strain, and no new mutations were detected (Table 1). The 5 mutations acquired by Zpi-10 remained stable even after extended periods of persistent infection (Zpi-80). These results suggest that 4 mutations in Zpi can induce a persistent infection; however, 5 mutations might be required to maintain a persistent infection state.
3.5 Generation and characterization of the persistently infectious Zpi2 strain
To determine the effect of one new mutation identified in the persistently infectious Zpi-10 and Zpi-80 on establishing persistent infection, we generated a mutant SeV with these 5 mutations (referred to as Zpi2 strain hereafter). The pSeVZpi(+)-Venus, which contained 4 mutations, was modified to introduce an additional mutation in the L protein (S278P) to produce pSeVZpi2(+)-Venus with 5 mutations (Figure 4A). Viral particles of the Zpi2 strain were produced using pSeVZpi2(+)-Venus in T7 polymerase-expressing BHK-21 cells. The Zpi2 strain was used to infect LLC-MK2 cells at MOIs of 5 and 0.003. Infection at an MOI of 5 induced cell death. However, infected cells maintained at an MOI of 0.003 did not exhibit morphological changes due to cell fusion, and Venus expression gradually spread to all cells (1.84 × 104 CIU/mL) (Figure 4B). Furthermore, when LLC-MK2 cells infected with the Zpi2 strain were passaged, the number of Venus-expressing cells increased over time without causing cell death. Thus, the Zpi2 strain may establish persistent infection in LLC-MK2 cells.
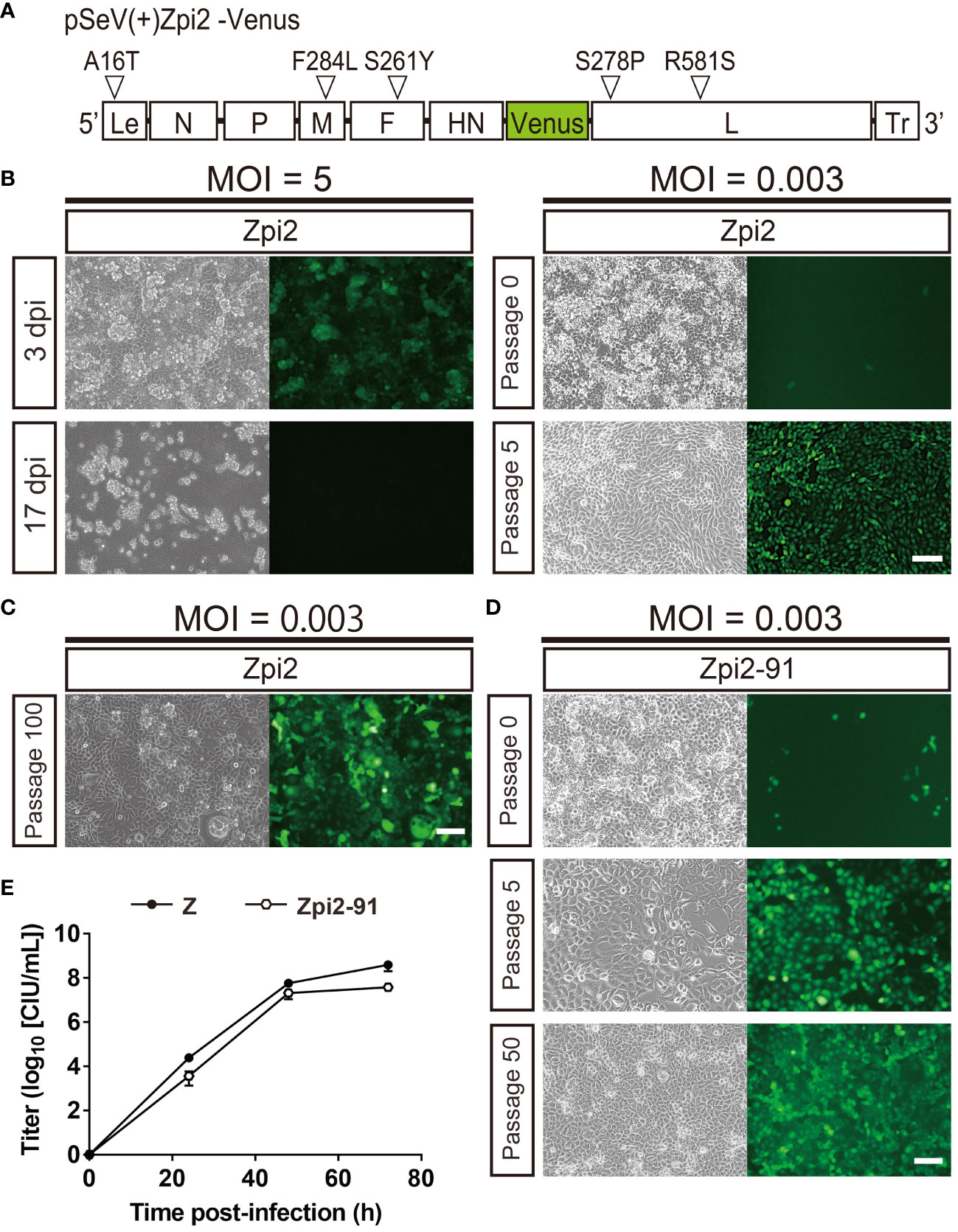
Figure 4 The Zpi2 strain produced persistently infected cells via viral particle production at 37°C. (A) Schematic representation of the Zpi2 genome plasmid. This representation shows viral proteins and locations of amino acid mutations and base mutations introduced in this study. Le, Leader region; Tr, Trailer region; white triangles = mutation locations. (B) Generation of persistently infected cells by the Zpi2 strain at a lower MOI, but not at a higher MOI. LLC-MK2 cells infected with the Zpi2 strain at an MOI of 5 expressed virus-derived Venus at 3 dpi and died at 17 dpi. Passage of LLC-MK2 cells infected with the Zpi2 strain at an MOI of 0.003 increased Venus-expressing cells gradually until the fifth generation of passaging, resulting in persistent infection. Scale bar = 100 µm. (C) Long-term passages of persistent infected cells with Zpi2. LLC-MK2 cells infected with the Zpi2 strain at an MOI of 0.003 were maintained until 100th generation. Scale bar = 100 µm. (D) Persistent infection by Zpi2-91. LLC-MK2 cell infected with the Zpi2-91 at an MOI of 0.003 was passaged until passage 50. Left: bright field, right: Venus-expressing cells. Scale bar = 100 µm. (E) Multi-step growth curve of Zpi2-91. Vero/TMPRSS2 cells were infected at an MOI of 0.01. Viral titers of culture supernatants at the indicated time points are expressed as mean ± S.E.M. from three independent sets of experiments. Z (●) and Zpi2-91 (⬡).
We continuously passaged Zpi2-infected cells to assess whether the Zpi2 strain could maintain persistent infection for an extended period (Figure 4C). Infected cells were maintained up to the 100th generation without cell death, and the virus was detected in the culture supernatant at a titer of 1.34 × 103 CIU/mL. The Zpi2 strain could therefore steadily maintain persistent infection over an extended period.
We then tested whether persistently infectious viruses of the Zpi2 strain in long-term passages can be newly and persistently transmitted to other cells (Figure 4D). The 91st generation of passages (referred to as Zpi2-91) was used to infect LLC-MK2 cells at an MOI of 0.003 to evaluate the persistent infection ability. When the Zpi2-91-infected cells were passaged up to another 50th generation, no cell death occurred, and the SeV virus was detected in the culture supernatant. Thus, the passaged Zpi2 could produce persistent infection after an extended period of passaging.
To assess the proliferative potential of Zpi2-91 after extended passaging, we conducted a multi-step growth curve analysis (Figure 4E). Vero/TMPRSS2 cells were infected with Zpi2-91 and the Z strain at an MOI of 0.01. Culture supernatants at 24, 48, and 72 hpi were collected for titer measurement. Similar to the WT Z strain, Venus-expressing cells increased over time with the Zpi2-91. The virus was detected within 24 hpi. The viral titer in the culture supernatant increased over time and reached 2.20 × 107 CIU/mL at 72 hpi, comparable with the WT Z strain (3.96 × 108 CIU/mL). These results indicate that Zpi2-91 passaged for a prolonged duration retained propagation ability.
3.6 No additional mutations acquired by passaged Zpi2
To determine whether passaged Zpi2 acquired additional mutations during persistent infection, we sequenced the viral genome isolated from persistently infected cells in the 8th generation of passages (Zpi2-8). The 5 mutations identified in the Zpi2 strain were maintained in the Zpi2-8 virus with no further mutations observed (Table 1). Thus, the Zpi2 strain with the 5 mutations could establish persistent infection. To evaluate the long-term stability of the Zpi2-8 mutations, we analyzed the complete genome sequence of the 80th generation virus (Zpi2-80). The Zpi2-80 retained the same 5 mutations as the original Zpi2 strain (Table 1) and also lacked any further mutations. The Zpi2 strain could therefore maintain persistent infection over an extended period without acquiring new mutations.
Collectively, our results demonstrate that the Zpi2 strain with the 5 identified mutations exhibits robust propagation and maintains persistent infection capability. Furthermore, these mutations remained stable throughout prolonged passaging.
3.7 Growth potential of persistently infectious strains
To compare the properties of the Zpi, Zpi2, and WT Z strains, we assessed their growth potential and toxicity. First, we generated single- and multi-step growth curves to determine the growth characteristics of these strains (Figures 5A, B). At 16 hpi of the single-step growth curve, the viral titers of the Zpi and Zpi2 strains were lower than those of the Z strain; however, by 24 and 48 hpi, this difference disappeared (Figure 5A). The final titers at 72 hpi for the Zpi and Zpi2 strains were 9.05 × 106 and 5.84 × 106 CIU/mL, respectively, which were comparable with that of the Z strain (2.07 × 107 CIU/mL).
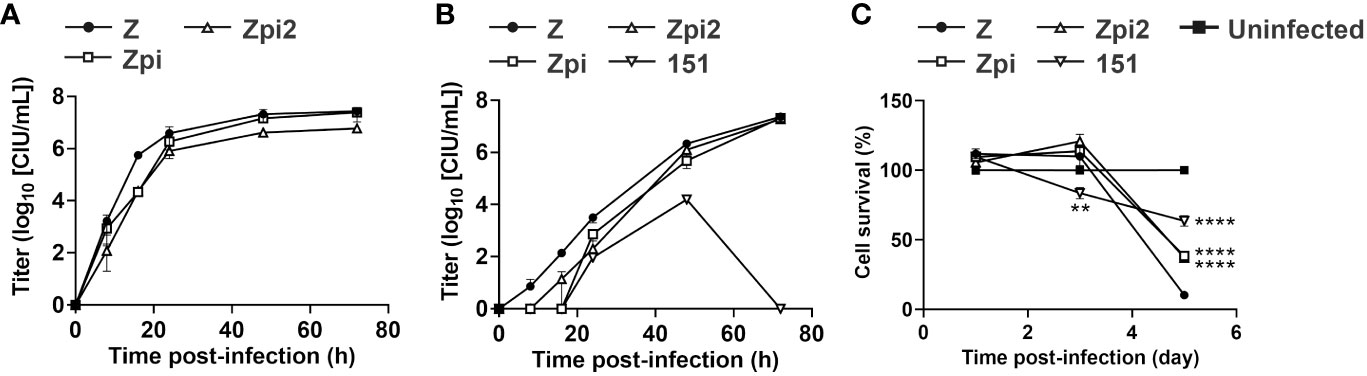
Figure 5 Characterization of persistent infection by the Zpi and Zpi2 strains. (A) Single-step growth of Z, Zpi and Zpi2 strains. LLC-MK2 cells were infected with Z or Zpi or Zpi2 strains at an MOI of 5 for single-step growth assay. Viral titers of culture supernatant at the indicated time points for Z (●), Zpi (□), and Zpi2 (△) are expressed as mean ± S.E.M. from three independent sets of experiments. (B) Multi-step growth of Z, Zpi, Zpi2, and Cl.151 strains. Vero/TMPRSS2 cells were infected with Z, Zpi, Zpi2, or Cl.151 (151) strains at an MOI of 0.01 for multi-step growth assay. Viral titers of culture supernatant at the indicated time points for Z (●), Zpi (□), Zpi2 (△), and 151 (▽) are expressed as mean ± S.E.M. from three independent sets of experiments. (C) Cytotoxicity of the Zpi and Zpi2 strains. Vero/TMPRSS2 cells were infected with Z (●), Zpi (□), Zpi2 (△), or 151 (▽) at an MOI of 0.1. Cell viability was measured by MTT assay at 1, 3, and 5 dpi. Values represent mean ± S.E.M. from 12–15 sets of experiments. **p < 0.01 and ****p < 0.0001 compared with Z (Tukey’s test).
To evaluate multi-step growth curves, we infected Vero/TMPRSS2 cells with the Zpi, Zpi2, or Z strain at an MOI of 0.01 and quantified the amount of virus in the culture supernatant over time (Figure 5B). Existing persistently infectious SeVs have been reported to be temperature sensitive and fail to grow at 37°C. Therefore, we compared the Zpi, Zpi2, and Z strains with a temperature-sensitive mutant strain, Cl.151. At 8 and 16 hpi, the Zpi and Zpi2 strains tended to have lower proliferation rates than the WT strain, although the difference was not statistically significant. At 24 hpi and onward, a similar amount of virus was detected in the supernatant for all three strains. The Cl.151 strain grew transiently until 48 hpi, but no detectable infection was observed at 72 hpi. Notably, both Zpi and Zpi2 strains consistently showed higher titers than the Cl.151 strain at all-time points. Thus, the Zpi and Zpi2 strains showed a proliferative potential comparable with that of the parent Z strain at 37°C and a higher proliferative potential than the temperature-sensitive Cl.151 strain.
Finally, we used multi-step growth curves to assess the proliferative capacity of Zpi-101 and Zpi2-91, which were long-term passaged persistently infectious strains (Figures 3E, 4E). The virus was detected within 24 hpi, and the final titers of Zpi-101 (7.54 × 107 CIU/mL) and Zpi2-91 (2.20 × 107 CIU/mL) were comparable. Therefore, Zpi and Zpi2 strains could maintain the same level of viral proliferative capacity even after long-term persistent infection.
3.8 Cytotoxicity of persistently infectious strains
We determined whether the Zpi and Zpi2 strains reduced the cytotoxicity of the parental Z strain (Figure 5C). Vero/TMPRSS2 cells were infected with the Z, Zpi, Zpi2, and Cl.151 strains at an MOI of 0.1 at 37°C, and cell viability at 1, 3, and 5 dpi was assessed by the MTT assay. The parental Z strain decreased cell viability over time after infection, dropping to 10% at 5 dpi. Conversely, the viability of cells infected with the Zpi and Zpi2 strains was significantly higher than that of the Z strain at 5 dpi, suggesting that the Zpi and Zpi2 strains were less cytotoxic than the Z strain. Compared with the cytotoxicity of the Cl.151 strain, cells infected with the Zpi and Zpi2 strains had higher viability at 3 dpi and lower viability at 5 dpi. The lower cytotoxicity of Cl.151 at a later time point can be attributed to the low replication and budding of the Cl.151 strain at 37°C caused by temperature-sensitive mutations (24, 26). Therefore, Zpi and Zpi2 strains were less cytotoxic than the parental Z strain and more cytotoxic than the Cl.151 strain, a temperature-sensitive persistently infectious virus, at 37°C.
3.9 DI genome levels of persistently infectious strains
We investigated the mechanisms by which the Zpi and Zpi2 strains established persistent infection at 37°C. One cause of persistent infection with SeV is the co-existence of SeV and DI genomes within infected cells. Alternating waves of increasing and decreasing of DI genomes and intact genomes are thought to produce a long-lasting persistent infection state in infected cells (20, 21). In particular, the presence of cbDI genomes is responsible for establishing persistent infection in mononegavirus-infected cells (20). Therefore, we investigated the potential involvement of DI genomes in the persistent infection of the Zpi and Zpi2 strains with the parental WT Z strain and the Cantell strain, which produces cbDI genomes, as positive controls (23) (Figure 6A). LLC-MK2 cells were infected with each virus at an MOI of 5. The cbDI and SeV full genomes in the infected cells at 24 hpi were quantified by RT-qPCR. The Cantell strain produced a high amount of cbDI genomes in the infected cells, whereas the Zpi and Zpi2 strains did not. These results indicate that cbDI genomes were produced by the Cantell strain, consistent with previous reports (43), but not by the Zpi or Zpi2 strains (Figure 6A). Although cbDI genomes can be generated experimentally by infection at a higher MOI (44), RT-qPCR analysis did not detect DI genomes in cells infected with Zpi or Zpi2 even at an MOI as high as 5. In addition, Yoshida et al. (23) demonstrated that the DI genomes were produced only when the DI genome-producing mutant virus emerged by chance and that the probability of new DI emergence was extremely low in the absence of pre-existing DI genomes in infected cells. Notably, the Zpi and Zpi2 strains were capable of establishing persistently infected states without long-term serial passages during which mutations could potentially be generated. Therefore, these results suggest that the production and co-existence of cbDI genomes do not contribute to the establishment of persistent infection with the Zpi and Zpi2 strains.
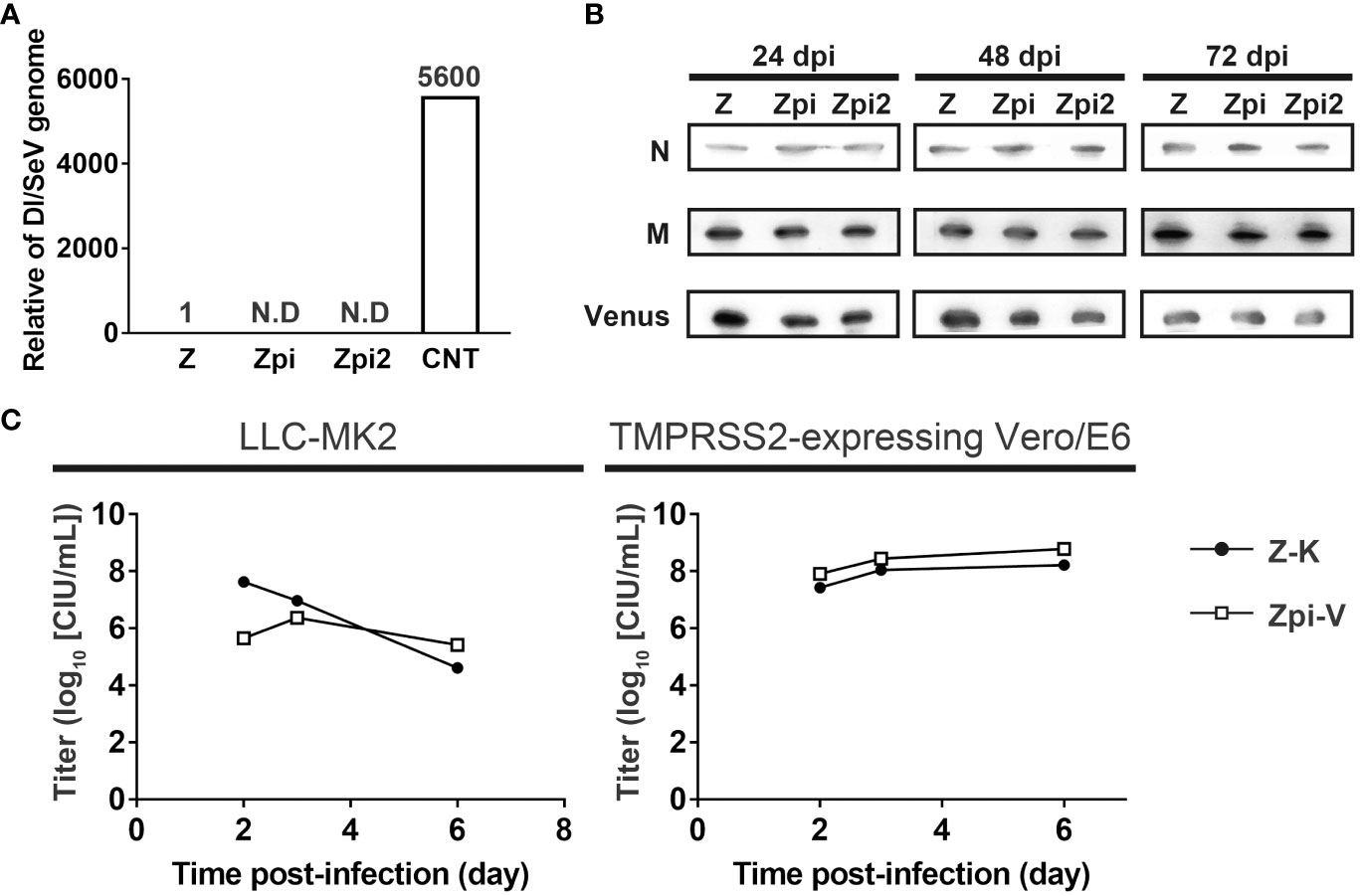
Figure 6 Mechanisms of persistent infection by the Zpi and Zpi2 strains. (A) Zpi and Zpi2 strains did not produce DI genomes. LLC-MK2 cells were infected with Z, Zpi, Zpi2, or Cantell (CNT) strains at an MOI of 5. CNT was used as a positive control. The amount of SeV and DI genome were quantified by RT-qPCR at 24 dpi. The value obtained by dividing the DI genome amount by the SeV genome amount was compared. (B) Zpi and Zpi2 strains did not affect the levels of constituent proteins. LLC-MK2 cells were infected with Z, Zpi, and Zpi2 strains at an MOI of 5, harvested at 24 dpi, and western blotted with anti-SeV-N, anti-SeV-M, and anti-GFP antibodies. (C) Persistently infectious viruses and wild-type viruses did not significantly interact. Cells were coinfected with persistently infectious viruses and wild-type viruses. LLC-MK2 or Vero/TMPRSS2 cells were infected with Z-K and Zpi-V at an MOI of 0.005 each. Titers were measured at the indicated time points. Z-Katusha (Z-K: ●); Zpi-Venus (Zpi-V: □).
3.10 Constitutive protein expression levels in persistently infected strains
Temperature-sensitive mutations in SeV have been reported to contribute to persistent infection. Temperature-sensitive SeV mutants can exhibit growth defects at nonpermissive temperatures, primarily because of reduced expression levels of SeV structural and regulatory proteins, such as N and M proteins, at 37°C (24, 45). Therefore, we investigated the possible involvement of changes in N and M protein expression levels in the persistent infection of Zpi and Zpi2 strains (Figure 6B). We compared the expression levels of N and M proteins among Zpi, Zpi2, and other strains at 37°C. The Zpi, Zpi2, and Z strains were infected at an MOI of 5, and cell lysates were collected at 24, 48, and 72 hpi for western blotting analysis. The expression levels of N and M proteins were comparable across all strains. Furthermore, the expression levels of Venus proteins derived from the viral genome were comparable across all strains. Stability of M proteins is presumed to be independent of MOIs. Notably, these stable SeV protein expressions were consistent with the results indicating that the Zpi and Zpi2 strains replicated at 37°C as well as the parental Z strain (Figures 5A, B). Thus, these results suggest that constitutive protein expression levels do not contribute to persistent infection with the Zpi and Zpi2 strains.
3.11 Interference by coinfection with WT and persistently infectious strains
Coinfection of cells with the WT and temperature-sensitive mutant viruses can suppress protein synthesis and growth of the WT virus (24, 35, 46, 47). This homologous interference phenomenon is considered to be an important factor in establishing a persistent infection. To determine whether persistently infectious SeVs have an interference effect, we quantified WT and persistently infectious viruses separately under overlapping infection conditions (Figure 6C). We used the Z strain expressing the red-fluorescent protein Katushka (Z-K) and the Z, Zpi, and Zpi2 strains, each expressing the green-fluorescent protein Venus (Z-V, Zpi-V, and Zpi2-V, respectively), and quantified the titers of the two viruses via RT-qPCR for Katushka and Venus levels (Supplementary Figures 1A, B).
To compare viral levels in coinfections over time, we collected culture supernatants at 2, 3, and 6 dpi from LLC-MK2 cells and Vero/TMPRSS2 cells coinfected with Z-K and Zpi-V at a MOI of 0.005 each (Figure 6C). The titers of both WT Z-K and Zpi-V in the culture supernatants were comparable at different time points in LLC-MK2 cells. Similarly, in Vero/TMPRSS2 cells, both Zpi and WT strains propagated at the same rate without affecting each other. These results suggest that the WT Z strain and the persistently infectious Zpi strain did not interfere with each other, including growth suppression in LLC-MK2 cells and Vero/TMPRSS2 cells.
4 Discussion
Mononegavirales typically cause acute infection, leading to the elimination of infected cells. However, certain viruses can uniquely establish persistent infections (2, 12). The mechanisms and significance underlying the acquisition of persistent infection by acutely infectious viruses remain poorly understood. Accumulating evidence has demonstrated that either repetition of alternative replication waves of the authentic viral genomes and the cbDI genomes or temperature-sensitive mutant viruses with the inability to propagate at 37°C can enable the establishment of persistently infected cells (12, 13, 21, 24, 25). However, whether other mechanisms are involved in the establishment of persistent infection remains unclear. Therefore, this study sought to elucidate the mechanisms that drive the establishment of persistent infection by acutely infectious viruses at physiological temperature. We identified novel mutations involved in persistent infection and two recombinant SeVs (Zpi and Zpi2) that exhibit both persistent infection and propagation at 37°C. Notably, the Zpi and Zpi2 strains could establish long-term persistent infection, irrespective of host species or cell type, through a mechanism distinct from cbDI genome generation or temperature-sensitive mutations. These findings highlight the inherent potential of SeVs to spontaneously acquire mutations during viral replication, allowing acutely infected SeVs to achieve persistent infection and autonomous production at physiological temperatures.
The emergence of DI and temperature-sensitive mutants of SeV has been observed in vitro but not in vivo (2, 12). Herein, we present a unique scenario where a persistently infectious viral population arose through multiple rounds of viral amplification in embryonated chicken eggs, mimicking viral growth in the natural environment. This viral population demonstrated persistent infectivity by infecting cells at a low MOI with a predominance of persistently infectious viruses. It is reasonable to infer that a persistent infection in our experimental setup was established from the extensive amplification of a pure cDNA-recovered strain in embryonated chicken eggs, generating diverse populations containing persistently infectious viruses harboring multiple mutations.
Multiple lines of evidence have demonstrated that persistent infection by mononegaviruses can be caused by temperature-sensitive mutant viruses, emergence of cbDI genomes, or cells resistant to viral infection (2, 12). Temperature-sensitive mutant strains of SeV can exhibit defects in proliferative capacity and cytotoxicity at nonpermissive temperatures, particularly at 38°C, compared with those of WT SeV (24, 26). Establishment of persistent infection by a temperature-sensitive mutant SeV was associated with a decrease in viral constituent proteins at nonpermissive temperatures (25, 48). Among the temperature-sensitive mutations in SeV, L1618V in L protein (49), but not T116A and A183S in M protein (25), contributed to the establishment of persistent infection. However, we found that the Zpi and Zpi2 strains did not affect the stability of N and M proteins. It is possible that the F284L mutation in M protein of the Zpi and Zpi2 strains does not affect the protein stability. Additionally, the repetitive interplay of authentic and DI genomes and IFNs produced by the DI genomes have also been implicated in establishing persistent infection (20, 21, 50). The Cantell strain has been reported to produce cbDI genomes, which serve as RIG-I ligands and induce IFN production (23). By contrast, the Zpi and Zpi2 strains did not generate DI genomes. It is reasonable to infer that the absence of DI genomes in Zpi- and Zpi2-infected cells may not induce IFN production, considering that DI genomes are recognized by RIG-I as ligands. Furthermore, owing to their evolutionary origin from the Z strain, which is characterized by stable protein levels, absence of DI genomes, and lack of IFN induction in infected cells, it is plausible that the Zpi and Zpi2 strains with only 4 and 5 mutations, respectively, similarly do not affect protein stability, DI genome production, or IFN induction. Furthermore, these Zpi and Zpi2 strains established persistent infection in various cell types, excluding the possibility that the establishment of persistent SeV infection was caused by the emergence of particular cell types resistant to SeV infection. Thus, our study reveals a unique mode of persistent infection by the Zpi and Zpi2 strains, which differs from existing persistently infected strains.
Persistently infectious SeVs, including the Cantell, Cl.151, and pi strains, have a variety of mutations in the genome (26, 28, 49, 51–62). Intriguingly, the Zpi and Zpi2 strains exhibited persistent infectivity with a small set of mutations (4 and 5 mutations, respectively). These mutations were found in 1 mutation in M protein, 1 in F protein, 2 in L protein, and 1 in leader sequence, compared to the Z strain. In contrast, compared to the Z strain, the Cantell strain, which produces cbDI genomes, had 47 amino acid mutations: 7 mutations in N protein, 4 in P protein, 7 in M protein, 7 mutations in F protein, 8 mutations in HN protein, and 14 mutations in L protein. A comparison of these 47 mutations in the Cantell strain with 4 and 5 mutations in the Zpi and Zpi2 strains revealed shared mutations. Specifically, mutation S261Y in F protein and mutation R581S in L protein were identical between the Cantell strain and the Zpi and Zpi2 strains. These shared mutations may be significant in establishing persistent infection. Additionally, the pi strain of SeV, which is characterized by persistent infection through temperature-sensitive mutations, displayed 49 amino acid mutations compared to the Z strain. Of these, mutation R581S in L protein was common to the Zpi and Zpi2 strains. Notably, mutation R581S in L protein was common to all Cantell, Cl.151, pi, Zpi, and Zpi2 strains, suggesting a critical role in the establishment of persistent SeV infection. The L protein serves as an RNA-dependent RNA polymerase, which is pivotal in amplifying and modifying the viral genome (63, 64). Mutation R581S in L protein is located in domain II (aa 503–607), the template recognition site of SeV RNA polymerase. Therefore, this mutation could contribute to a reduced genome replication rate and consequently support persistent infection (65). In addition, the S278P mutation found in the L protein of the Zpi2 strain is located in domain I (aa 225–416), which interacts with the P protein to form a polymerase complex (66). Considering that the domain I of the Mononegavirales L protein functions as the replication domain of the genome as an RNA polymerase, this mutation might affect replication capacity and contribute to persistent infection (67, 68). Furthermore, the leader sequence, functioning as a cis element, plays a promoter role in viral genome replication. Mutations within the leader sequence have been associated with homologous interference, where temperature-sensitive mutant viruses suppress the growth of simultaneously infected WT viruses (24). However, this mutation alone is insufficient to establish persistent infection (47). Moreover, a report demonstrated that disruption of the receptor for the hemagglutinin-neuraminidase protein of the Z strain caused homologous interference (69). Mutations in the F protein of the Zpi and Zpi2 strains may also cause homologous interference by disrupting the infection mechanism. Overall, it is possible that the Zpi and Zpi2 strains characterized by mutations in the leader and F protein sequences could suppress the WT strain via homologous interference, thereby contributing to the development of persistent infection. However, further studies are warranted to clarify the interference effects of Zpi and Zpi2 strains by investigating experimental conditions, such as different MOI and passages after coinfection.
The specific contributions of the mutations introduced into the Zpi and Zpi2 strains to persistent infection remain unclear. The F284L mutation in M protein, the S261Y mutation in F protein, and the S281P mutation L protein that we found in the Zpi and Zpi2 strains have not been identified in other SeV strains. In our study, we generated recombinant viruses bearing 4 or 5 mutations. However, a more detailed approach could help decipher the role of individual mutations in persistent infection. Generating viruses containing individual mutations will allow a comprehensive assessment of the impact of each mutation on viral properties and dynamics. Moreover, the use of minigenomes provides a valuable method for dissecting the function of individual mutant proteins and elucidating the replication and transcription of the mutant genomes (70). Furthermore, given the potential for host adaptation to viral infection, RNAseq analysis of persistently infected cells is expected to reveal changes in host gene expression profiles. The mechanisms underlying persistent infection by the Zpi and Zpi2 strains warrant further investigation.
Notably, temperature-sensitive mutant viruses and DI particles cause cell death, referred to as “crisis”, in the early stages of infection before persistent infection or during subsequent passages (12, 20). Crisis is thought to occur when the virus suddenly disrupts the persistent infection state within persistently infected cells. Interestingly, the Zpi and Zpi2 strains exhibited a marked absence of crisis even after 100 passages. This observation strongly suggests that these strains are less susceptible to a sudden crisis of their persistent infection states, further elucidating the unique characteristics of their persistent infectivity.
The development and maintenance of persistent infection in our study can be postulated as a multi-step process. Initially, a viral population comprising persistently infectious viruses, each carrying unique accidental mutations, emerges and successfully infects cells at a low MOI. Subsequently, these persistently infectious progeny, which arise from some of the infected cells, proceed to infect neighboring noninfected cells and suppress the growth of WT viruses. This dominance enables the persistently infectious progeny to replicate extensively within infected cells, eventually establishing and maintaining the state of persistent infection. This unique mode of viral persistent infection underscores the dynamic interplay between viral populations, mutation dynamics, and the cellular environment in shaping the outcome of viral infections.
SeV causes widespread respiratory diseases in mammals. Recent evidence has even detected SeV in wild mammals, such as civets, highlighting its presence and likely maintenance in natural reservoirs (71). However, the identity of the natural host for SeV remains elusive, and the mechanisms that maintain its persistence in nature over extended periods are poorly understood. Our results provide valuable insights into this enigma. Conceivably, SeVs carrying persistently infectious mutations could spontaneously emerge in nature, thereby increasing genetic diversity within the viral population through continuous replication. These emergent viral strains may possess the capacity for persistent infection, enabling their persistence within individual hosts for extended periods by suppressing WT SeV replication.
Beyond elucidating novel mechanisms of persistent infection, advancing viral vectors based on persistently infectious strains holds promise for future applications. SeVs have been explored as viral vectors for various applications, such as for the establishment of induced pluripotent stem cell lines and gene therapy (72–74). SeV vectors used for gene transfer are based on temperature-sensitive strains and can be removed from infected cells after gene transfer. However, these approaches often have limited versatility. Our study, which identifies novel persistently infectious viruses, has the potential to significantly broaden the spectrum of applications for SeVs, overcoming existing limitations and offering new possibilities in SeV-based research. Viral vectorization of the Zpi and Zpi2 strains as vector platforms can facilitate sustained gene expression with reduced toxicity and enhanced efficiency across organs and cell types. In addition, engineered Zpi or Zpi2 vectors with deletions of F-HN and/or M protein genes can make significant contributions to both basic research and medical applications (75–77). This potential represents a significant step forward in the field of gene delivery and underscores the utility of these persistently infectious strains in biomedicine.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
Ethical approval was not required for the studies on animals in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
MI: Formal Analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. RK: Investigation, Writing - review & editing. NM: Methodology, Writing - review & editing. RT: Methodology, Writing - review & editing. TS: Supervision, Writing - review & editing. TI: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing. FO: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by Grants-in-Aid (TI and FO) from the Japan Society for the Promotion of Science, PRESTO (FO) and CREST (FO) from the Japan Science and Technology Agency, the SRF foundation (FO), and AMED Research Program on Emerging and Re-emerging Infectious Diseases (TI).
Acknowledgments
The authors thank members of the Osakada laboratory (Nagoya University) and the Sakaguchi and Irie laboratory (Hiroshima University) for stimulating discussions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2024.1363092/full#supplementary-material
Supplementary Figure 1 | Validation of qPCR quantification. (A) Multi-step growth curves in mono-infected Vero/TMPRSS2 cells and Cq values of fluorescent protein genes in culture supernatants of mono-infected cells. (B) Infectious titers and Cq values in serially diluted culture supernatants of mono-infected cells at 3 dpi. Z-Katusha (Z-K: X); Z-Venus (Z-V: ●); Zpi-Venus (Zpi-V: □); Zpi2-Venus (Zpi2-V: △).
Supplementary Figure 2 | The Cantell strain, but not the Z strain, induced persistently infected cells at a higher MOI in different cell types. (A) Persistent infection with the Cantell strain in LLC-MK2, HeLa, and Vero/SLAM cells at an MOI of 5. The infected cells expressed Venus by viral replication at 3 dpi and passaged on to the 6th generation. (B) Failed persistent infection with the Z strain in LLC-MK2, HeLa, and Vero/SLAM cells at an MOI of 5. The infected cells expressed virus by viral replication at 3 dpi but died at 9 dpi. Left: bright field; right: Venus-expressing cells. Scale bar = 100 µm.
Supplementary Figure 3 | The Zpi strain established persistent infection in different cell types. (A) Persistent infection of Neuro2a, BHK-21, Vero, and HeLa cells by the Zpi at a lower MOI. BHK-21, HeLa, Vero, and Neuro2a cells were infected with the Zpi strain at an MOI of 0.003. These infected cells expressed Venus at 3 dpi and were passaged to the 5th generation. (B) Long-term passages of persistently infected Neuro2a cells. Neuro2a cells were infected with the Zpi strain at an MOI of 0.003. Persistently infected, Venus-expressing cells of the Zpi strain were passaged up to 80th generations. Left: bright field; right: Venus-expressing cells. Scale bar = 100 µm.
References
1. Herzog S, Rott R. Replication of Borna disease virus in cell cultures. Med Microbiol Immunol. (1980) 168:153–8. doi: 10.1007/BF02122849/METRICS
2. Roux L, Simon AE, Holland JJ. Effects of defective interfering viruses on virus replication and pathogenesis in vitro and in vivo. Adv Virus Res. (1991) 40:181–211. doi: 10.1016/S0065-3527(08)60279-1
3. Calain P, Monroe MC, Nichol ST. Ebola virus defective interfering particles and persistent infection. Virology. (1999) 262:114–28. doi: 10.1006/VIRO.1999.9915
4. Young DF, Wignall-Fleming EB, Busse DC, Pickin MJ, Hankinson J, Randall EM, et al. The switch between acute and persistent paramyxovirus infection caused by single amino acid substitutions in the RNA polymerase P subunit. PloS Pathog. (2019) 15:e1007561. doi: 10.1371/JOURNAL.PPAT.1007561
5. Jabbour JT, Duenas DA, Sever JL, Krebs HM, Horta-Barbosa L. Epidemiology of subacute sclerosing panencephalitis (SSPE): A report of the SSPE registry. JAMA. (1972) 220:959–62. doi: 10.1001/JAMA.1972.03200070051006
6. Coulter JBS, Balch N, Best PV. Subacute sclerosing panencephalitis after drug-induced immunosuppression. Arch Dis Child. (1979) 54:640–2. doi: 10.1136/ADC.54.8.640
7. Cattaneo R, Schmid A, Rebmann G, Baczko K, Ter Meulen V, Bellini WJ, et al. Accumulated measles virus mutations in a case of subacute sclerosing panencephalitis: Interrupted matrix protein reading frame and transcription alteration. Virology. (1986) 154:97–107. doi: 10.1016/0042-6822(86)90433-2
8. Cattaneo R, Rose JK. Cell fusion by the envelope glycoproteins of persistent measles viruses which caused lethal human brain disease. J Virol. (1993) 67:1493–502. doi: 10.1128/JVI.67.3.1493-1502.1993
9. Watanabe S, Shirogane Y, Suzuki SO, Ikegame S, Koga R, Yanagi Y. Mutant fusion proteins with enhanced fusion activity promote measles virus spread in human neuronal cells and brains of suckling hamsters. J Virol. (2013) 87:2648. doi: 10.1128/JVI.02632-12
10. Ayata M, Tanaka M, Kameoka K, Kuwamura M, Takeuchi K, Takeda M, et al. Amino acid substitutions in the heptad repeat A and C regions of the F protein responsible for neurovirulence of measles virus Osaka-1 strain from a patient with subacute sclerosing panencephalitis. Virology. (2016) 487:141–9. doi: 10.1016/J.VIROL.2015.10.004
11. Angius F, Smuts H, Rybkina K, Stelitano D, Eley B, Wilmshurst J, et al. Analysis of a subacute sclerosing panencephalitis genotype B3 virus from the 2009-2010 South African measles epidemic shows that hyperfusogenic F proteins contribute to measles virus infection in the brain. J Virol. (2019) 93:e01700-18. doi: 10.1128/JVI.01700-18
12. Kanda T, Shibuta H. A temperature-sensitive mutant of sendai virus which establishes persistent infection in vero cells without cell crisis. Virology. (1981) 108:318–24. doi: 10.1016/0042-6822(81)90440-2
13. Rezelj VV, Levi LI, Vignuzzi M. The defective component of viral populations. Curr Opin Virol. (2018) 33:74–80. doi: 10.1016/J.COVIRO.2018.07.014
14. Kolakofsky D. Isolation and characterization of Sendai virus DI-RNAs. Cell. (1976) 8:547–55. doi: 10.1016/0092-8674(76)90223-3
15. Leppert M, Kort L, Kolakofsky D. Further characterization of sendai virus DI-RNAs: A model for their generation. Cell. (1977) 12:539–52. doi: 10.1016/0092-8674(77)90130-1
16. Re GG, Kingsbury DW. Nucleotide sequences that affect replicative and transcriptional efficiencies of Sendai virus deletion mutants. J Virol. (1986) 58:578. doi: 10.1128/JVI.58.2.578-582.1986
17. Calain P, Roux L. Functional characterisation of the genomic and antigenomic promoters of sendai virus. Virology. (1995) 212:163–73. doi: 10.1006/VIRO.1995.1464
18. Huang AS, Baltimore D. Defective viral particles and viral disease processes. Nat. (1970) 2265243 226:325–7. doi: 10.1038/226325a0
19. Lazzarini RA, Keene JD, Schubert M. The origins of defective interfering particles of the negative-strand RNA viruses. Cell. (1981) 26:145–54. doi: 10.1016/0092-8674(81)90298-1
20. Roux L, Holland JJ. Role of defective interfering particles of sendai virus in persistent infections. Virology. (1979) 93:91–103. doi: 10.1016/0042-6822(79)90278-2
21. Cave DR, Hendrickson FM, Huang AS. Defective interfering virus particles modulate virulence. J Virol. (1985) 55:366–73. doi: 10.1128/JVI.55.2.366-373.1985
22. Xu J, Sun Y, Li Y, Ruthel G, Weiss SR, Raj A, et al. Replication defective viral genomes exploit a cellular pro-survival mechanism to establish paramyxovirus persistence. Nat Commun. (2017) 81 8:1–13. doi: 10.1038/s41467-017-00909-6
23. Yoshida A, Kawabata R, Honda T, Sakai K, Ami Y, Sakaguchi T, et al. A single amino acid substitution within the paramyxovirus sendai virus nucleoprotein is a critical determinant for production of interferon-beta-inducing copyback-type defective interfering genomes. J Virol. (2018) 92:1–22. doi: 10.1128/JVI.02094-17
24. Yoshida T, Hamaguchi M, Naruse H, Nagai Y. Persistent infection by a temperature-sensitive mutant isolated from a Sendai virus (HVJ) carrier culture: Its initiation and maintenance without aid of defective interfering particles. Virology. (1982) 120:329–39. doi: 10.1016/0042-6822(82)90034-4
25. Kondo T, Yoshida T, Miura N, Nakanishi M. Temperature-sensitive phenotype of a mutant sendai virus strain is caused by its insufficient accumulation of the M protein. J Biol Chem. (1993) 268:21924–30. doi: 10.1016/S0021-9258(20)80629-2
26. Yoshida T, Nagai Y, Maeno K, Iinuma M, Hamaguchi M, Matsumoto T, et al. Studies on the role of M protein in virus assembly using a is mutant of HVJ (Sendai virus). Virology. (1979) 92:139–54. doi: 10.1016/0042-6822(79)90220-4
27. Roux L, Waldvogel FA. Instability of the viral M protein in BHK-21 cells persistently infected with Sendai virus. Cell. (1982) 28:293–302. doi: 10.1016/0092-8674(82)90347-6
28. Nishio M, Tsurudome M, Ito M, Kawano M, Komada H, Ito Y. Characterization of Sendai virus persistently infected L929 cells and Sendai virus pi strain: Recombinant Sendai viruses having Mpi protein shows lower cytotoxicity and are incapable of establishing persistent infection. Virology. (2003) 314:110–24. doi: 10.1016/S0042-6822(03)00404-5
29. Yokoo J, Gotoh B, Komatsu T, Takeuchi K, Miyadai T. Replication-incompetent Sendai virus can suppress the antiviral action of type I interferon. Arch Virol. (1999) 144:1043–55. doi: 10.1007/S007050050568
30. Kato A, Kiyotani K, Hasan MK, Shioda T, Sakai Y, Yoshida T, et al. Sendai virus gene start signals are not equivalent in reinitiation capacity: moderation at the fusion protein gene. J Virol. (1999) 73:9237–46. doi: 10.1128/JVI.73.11.9237-9246.1999
31. Kato A, Sakai Y, Shioda T, Kondo T, Nakanishi M, Nagai Y. Initiation of Sendai virus multiplication from transfected cDNA or RNA with negative or positive sense. Genes to Cells. (1996) 1:569–79. doi: 10.1046/j.1365-2443.1996.d01-261.x
32. Yoshida T, Hamaguchi M, Naruse H, Nishikawa K, Nagai Y. Characterization of the virus isolated from heLa cells persistently infected with sendai virus (HVJ). Microbiol Immunol. (1983) 27:207–11. doi: 10.1111/j.1348-0421.1983.tb03575.x
33. Shirogane Y, Takeda M, Iwasaki M, Ishiguro N, Takeuchi H, Nakatsu Y, et al. Efficient multiplication of human metapneumovirus in vero cells expressing the transmembrane serine protease TMPRSS2. J Virol. (2008) 82:8942–6. doi: 10.1128/JVI.00676-08
34. Cantell K, Hirvonen S. Preparation and assay of Sendai virus. Methods Enzymol. (1981) 78:299–301. doi: 10.1016/0076-6879(81)78131-X
35. Kiyotani K, Takao S, Sakaguchi T, Yoshida T. Immediate protection of mice from lethal wild-type Sendai virus (HVJ) infections by a temperature-sensitive mutant, HVJpi, possessing homologous interfering capacity. Virology. (1990) 177:65–74. doi: 10.1016/0042-6822(90)90460-9
36. Nagata I, Kimura Y, Ito Y, Tanaka T. Temperature-sensitive phenomenon of viral maturation observed in BHK cells persistently infected with HVJ. Virology. (1972) 49:453–61. doi: 10.1016/0042-6822(72)90497-7
37. Kimura Y, Ito Y, Shimokata K, Nishiyama Y, Nagata I. Temperature-sensitive virus derived from BHK cells persistently infected with HVJ (Sendai virus). J Virol. (1975) 15:55–63. doi: 10.1128/JVI.15.1.55-63.1975
38. Ogura H, Sato H, Hatano M. Temperature-sensitive HVJ (Sendai virus) with altered P polypeptide derived from persistently infected cell lines. J Gen Virol. (1981) 55:469–73. doi: 10.1099/0022-1317-55-2-469
39. Adachi A, Kanda T, Shibuta H. Isolation and characterization of temperature-sensitive mutants of Sendai virus. Microbiol Immunol. (1980) 24:1053–68. doi: 10.1111/j.1348-0421.1980.tb02911.x
40. Sato H, Ogura H, Hatano M. An intracellular interaction between a temperature-sensitive mutant and the original wild-type HVJ (Sendai virus) is responsible for the establishment and maintenance of HVJ persistent infection. J Gen Virol. (1981) 55:459–68. doi: 10.1099/0022-1317-55-2-459
41. Strahle L, Garcin D, Kolakofsky D. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology. (2006) 351:101–11. doi: 10.1016/J.VIROL.2006.03.022
42. Domingo E, Perales C. Viral quasispecies. PloS Genet. (2019) 15:e1008271. doi: 10.1371/JOURNAL.PGEN.1008271
43. Yount JS, Kraus TA, Horvath CM, Moran TM, López CB. A novel role for viral-defective interfering particles in enhancing dendritic cell maturation. J Immunol. (2006) 177:4503–13. doi: 10.4049/JIMMUNOL.177.7.4503
44. Kolakofsky D. Studies on the generation and amplification of Sendai virus defective-interfering genomes. Virology. (1979) 93:589–93. doi: 10.1016/0042-6822(79)90263-0
45. Sakaguchi T, Kiyotani K, Kato A, Asakawa M, Fujii Y, Nagai Y, et al. Phosphorylation of the Sendai virus M protein is not essential for virus replication either in vitro or in vivo. Virology. (1997) 235:360–6. doi: 10.1006/viro.1997.8701
46. Kimura Y, Norrby E, Nagata I, Ito Y, Shimokata K. Homologous interference induced by a temperature sensitive mutant derived from an HVJ (Sendai virus) carrier culture. J Gen Virol. (1976) 33:333–43. doi: 10.1099/0022-1317-33-2-333
47. Shimazu Y, Takao S, Irie T, Kiyotani K, Yoshida T, Sakaguchi T. Contribution of the leader sequence to homologous viral interference among Sendai virus strains. Virology. (2008) 372:64–71. doi: 10.1016/J.VIROL.2007.10.026
48. Ito M, Takeuchi T, Nishio M, Kawano M, Komada H, Tsurudome M, et al. Early stage of establishment of persistent Sendai virus infection: unstable dynamic phase and then selection of viruses which are tightly cell associated, temperature sensitive, and capable of establishing persistent infection. J Virol. (2004) 78:11939–51. doi: 10.1128/JVI.78.21.11939-11951.2004
49. Nishio M, Nagata A, Tsurudome M, Ito M, Kawano M, Komada H, et al. Recombinant Sendai viruses with L1618V mutation in their L polymerase protein establish persistent infection, but not temperature sensitivity. Virology. (2004) 329:289–301. doi: 10.1016/J.VIROL.2004.08.023
50. Roux L, Waldvogel FA. Establishment of Sendai virus persistent infection: Biochemical analysis of the early phase of a standard plus defective interfering virus infection of BHK cells. Virology. (1981) 112:400–10. doi: 10.1016/0042-6822(81)90287-7
51. Shioda T, Hidaka Y, Kanda T, Shibuta H, Nomoto A, Iwasaki K. Sequence of 3,687 nudeotides from the 3′ end of sendai virus genome RNA and the predicted amino add sequences of viral NP, P and C proteins. Nucleic Acids Res. (1983) 11:7317–30. doi: 10.1093/nar/11.21.7317
52. Hidaka Y, Kanda T, Iwasaki K, Nomoto A, Shioda T, Shibuta H. Nucleotide sequence of a Sendai virus genome region covering the entire M gene and the 3’ proximal 1013 nucleotides of the F gene. Nucleic Acids Res. (1984) 12:7965–73. doi: 10.1093/NAR/12.21.7965
53. Morgan EM, Re GG, Kingsbury DW. Complete sequence of the sendai virus NP gene from a cloned insert. Virology. (1984) 135:279–87. doi: 10.1016/0042-6822(84)90137-5
54. Blumberg BM, Giorgi C, Rose K, Kolakofsky D. Sequence determination of the Sendai virus fusion protein gene. J Gen Virol. (1985) 66:317–31. doi: 10.1099/0022-1317-66-2-317
55. Miura N, Nakatani Y, Ishiura M, Uchida T, Okada Y. Molecular cloning of a full-length cDNA encoding the hemagglutinin-neuraminidase glycoprotein of Sendai virus. FEBS Lett. (1985) 188:112–6. doi: 10.1016/0014-5793(85)80885-1
56. Shioda T, Iwasaki K, Shibuta H. Determination of the complete nucleotide sequence of the Sendai virus genome RNA and the predicted amino acid sequences of the F, HN and L proteins. Nucleic Acids Res. (1986) 14:1545–63. doi: 10.1093/nar/14.4.1545
57. Tashiro M, Pritzer E, Khoshnan MA, Yamakawa M, Kuroda K, Klenk HD, et al. Characterization of a pantropic variant of Sendai virus derived from a host range mutant. Virology. (1988) 165:577–83. doi: 10.1016/0042-6822(88)90601-0
58. Neubert WJ. Cloning and sequencing of the polymerase gene (P) of Sendai virus (strain Fushimi). Nucleic Acids Res. (1989) 17:10101–1. doi: 10.1093/NAR/17.23.10101
59. Middleton Y, Tashiro M, Thai T, Oh J, Seymour J, Pritzer E, et al. Nucleotide sequence analyses of the genes encoding the HN, M, NP, P, and L proteins of two host range mutants of Sendai virus. Virology. (1990) 176:656–7. doi: 10.1016/0042-6822(90)90040-X
60. Neubert WJ, Eckerskorn C, Homann HE. Sendai virus NP gene codes for a 524 amino acid NP protein. Virus Genes. (1991) 5:25–32. doi: 10.1007/BF00571728
61. Tashiro M, Seto JT, Choosakul S, Yamakawa M, Klenk HD, Rott R. Budding site of sendai virus in polarized epithelial cells is one of the determinants for tropism and pathogenicity in mice. Virology. (1992) 187:413–22. doi: 10.1016/0042-6822(92)90443-S
62. Nishimura K, Segawa H, Goto T, Morishita M, Masago A, Takahashi H, et al. Persistent and stable gene expression by a cytoplasmic RNA replicon based on a noncytopathic variant sendai virus. J Biol Chem. (2007) 282:27383–91. doi: 10.1074/jbc.M702028200
63. Abraham G, Rhodes DP, Banerjee AK. The 5′ terminal structure of the methylated mRNA synthesized in vitro by vesicular stomatitis virus. Cell. (1975) 5:51–8. doi: 10.1016/0092-8674(75)90091-4
64. Testa D, Banerjee AK. Two methyltransferase activities in the purified virions of vesicular stomatitis virus. J Virol. (1977) 24:786–93. doi: 10.1128/JVI.24.3.786-793.1977
65. Smallwood S, Easson CD, Feller JA, Horikami SM, Moyer SA. Mutations in conserved domain II of the large (L) subunit of the sendai virus RNA polymerase abolish RNA synthesis. Virology. (1999) 262:375–83. doi: 10.1006/VIRO.1999.9933
66. Chandrika R, Horikami SM, Smallwood S, Moyer SA. Mutations in conserved domain I of the sendai virus L polymerase protein uncouple transcription and replication. Virology. (1995) 213:352–63. doi: 10.1006/VIRO.1995.0008
67. Horikami SM, Smallwood S, Bankamp B, Moyer SA. An amino-proximal domain of the L protein binds to the P protein in the measles virus RNA polymerase complex. Virology. (1994) 205:540–5. doi: 10.1006/VIRO.1994.1676
68. Holmes DE, Moyer SA. The phosphoprotein (P) binding site resides in the N terminus of the L polymerase subunit of sendai virus. J Virol. (2002) 76:3078–83. doi: 10.1128/JVI.76.6.3078-3083.2002
69. Goto H, Ohta K, Matsumoto Y, Yumine N, Nishio M. Evidence that receptor destruction by the sendai virus hemagglutinin-neuraminidase protein is responsible for homologous interference. J Virol. (2016) 90:7640. doi: 10.1128/JVI.01087-16
70. Su J, Dou Y, You Y, Cai X. Application of minigenome technology in virology research of the Paramyxoviridae family. J Microbiol Immunol Infect. (2015) 48:123–9. doi: 10.1016/J.JMII.2014.02.008
71. Liu P, Chen W, Chen JP. Viral metagenomics revealed sendai virus and coronavirus infection of malayan pangolins (Manis javanica). Viruses. (2019) 11:979. doi: 10.3390/V11110979
72. Bitzer M, Armeanu S, Lauer UM, Neubert WJ. Sendai virus vectors as an emerging negative-strand RNA viral vector system. J Gene Med. (2003) 5:543–53. doi: 10.1002/jgm.426
73. Nakanishi M, Otsu M. Development of sendai virus vectors and their potential applications in gene therapy and regenerative medicine. Curr Gene Ther. (2012) 12:410–6. doi: 10.2174/156652312802762518
74. Yamaki Y, Fukushima T, Yoshida N, Nishimura K, Fukuda A, Hisatake K, et al. Utilization of a novel Sendai virus vector in ex vivo gene therapy for hemophilia A. Int J Hematol. (2021) 113:493–9. doi: 10.1007/S12185-020-03059-6
75. Wickersham IR, Finke S, Conzelmann KK, Callaway EM. Retrograde neuronal tracing with a deletion-mutant rabies virus. Nat Methods. (2007) 4:47–9. doi: 10.1038/nmeth999
76. Nishimura K, Sano M, Ohtaka M, Furuta B, Umemura Y, Nakajima Y, et al. Development of defective and persistent Sendai virus vector: A unique gene delivery/expression system ideal for cell reprogramming. J Biol Chem. (2011) 286:4760–71. doi: 10.1074/jbc.M110.183780
Keywords: Sendai virus, persistent infection, defective interfering genome, temperature sensitive mutation, Mononegavirales, viral vector
Citation: Iwata M, Kawabata R, Morimoto N, Takeuchi RF, Sakaguchi T, Irie T and Osakada F (2024) Evolutionary engineering and characterization of Sendai virus mutants capable of persistent infection and autonomous production. Front. Virol. 4:1363092. doi: 10.3389/fviro.2024.1363092
Received: 29 December 2023; Accepted: 19 February 2024;
Published: 02 April 2024.
Edited by:
Yoon-Seok Chung, Korea Disease Control and Prevention Agencyorea, Republic of KoreaReviewed by:
Shuzo Urata, Nagasaki University, JapanMasatoshi Kakizaki, National Institute of Infectious Diseases (NIID), Japan
Copyright © 2024 Iwata, Kawabata, Morimoto, Takeuchi, Sakaguchi, Irie and Osakada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takashi Irie, dGlyaWVAaGlyb3NoaW1hLXUuYWMuanA=; Fumitaka Osakada, Zm9zYWthZGFAcHMubmFnb3lhLXUuYWMuanA=