 Lucy R. Stewart
Lucy R. Stewart Kristen Willie2†
Kristen Willie2† Hong Hanh Tran
Hong Hanh Tran- 1Foreign Disease-Weed Science Research Unit, United States Department of Agriculture, Agricultural Research Service (USDA, ARS), Frederick, MD, United States
- 2Corn, Soybean and Wheat Quality Research Unit, United States Department of Agriculture, Agricultural Research Service (USDA, ARS), Wooster, OH, United States
- 3Department of Plant Pathology, The Ohio State University, Wooster, OH, United States
Plant viruses classified in the genus Waikavirus, family Secoviridae, are positive sense single-stranded RNA viruses that include important pathogens of maize (maize chlorotic dwarf virus; MCDV) and rice (rice tungro spherical virus; RTSV). Many aspects of the molecular biology of waikaviruses remain unexplored because of experimental challenges and lack of infectious clones for low titer, phloem-limited, and obligately vector-transmitted waikaviruses. Here we report the first development of waikavirus infectious clones for two MCDV strains, MCDV-S and MCDV-M1, and insect-free launching of infections from these clones in maize by vascular puncture inoculation. We further developed a green fluorescent protein (GFP)-tagged MCDV clone by replacing the viral p27-encoding sequence with GFP-encoding sequence. GFP-tagged virus moved systemically in plants and caused symptomatic infection similar to wild type virus, with vascular expression of GFP. Development of waikavirus infectious clones is a major advance for this group of agriculturally significant viruses.
1 Introduction
Waikaviruses are a unique group of viruses, several with major economic impacts to agriculture, but which have been recalcitrant to molecular biological studies. Virus species classified within the genus Waikavirus, family Secoviridae, are monopartite viruses with ca. 12,000 nt ss(+)RNA genomes. Waikaviruses of agricultural importance include rice tungro spherical virus (RTSV), and maize chlorotic dwarf virus (MCDV). RTSV is a leafhopper-transmitted virus that also enables leafhopper transmission of the unrelated pararetrovirus, rice tungro bacilliform virus (RTBV), with which it causes the synergistic rice tungro disease in plants infected with both [reviewed in (1)]. MCDV and its accompanying disease peaked in known disease impact in the United States in the 1970s (2, 3). Sequenced isolates of MCDV show distinct genotypes which we characterize as strains, including the mild (MCDV-M1) and severe (MCDV-S) strains that share only 59% polyprotein amino acid sequence identity [reviewed in (4)]. Both of these, but predominantly the mild strain are still ubiquitous in Ohio in the overwintering reservoir host, Johnsongrass (Sorghum halepense), but found only rarely in nearby maize (5), even though popular cultivars were readily infected in the laboratory (Stewart and Todd, unpublished data). Partial MCDV sequences most similar to MCDV-M1 were identified in an Oklahoma grass survey (6), and a near complete sequence from Oklahoma maize tissue was submitted more recently, with 99% aa identity to severe isolates (GenBank accession no. MW727468.1). In the first instances of identification outside of the United States, near complete MCDV isolate sequences were recently reported from Brazil and Mexico. These include an isolate from Brazil (MCDV-BR; GenBank accession no. MH713426.1), collected from Brachiaria spp. in 2013 which shares 74% predicted amino acid identity to MCDV-M1; and an isolate from Mexico (MCDV-CD-W; GenBank accession no. MN203201.1), collected from unspecified weedy leaf tissue in 2014 and sharing 91-92% aa identity with severe strains MCDV-S and MCDV-T. With the increased use of high throughput sequencing for virus identification, sequences of new and putative waikaviruses have been reported in a variety of plant hosts, sometimes associated with symptoms, but rarely with demonstrated pathology or transmission. These include bellflower vein chlorosis virus [BVCV (7)]; blackcurrant waikavirus A [BCWVA (8)], Brassica napus RNA virus 1 [BnRV1 (9)], red clover associated virus 1 [RCaV1 (10)], and others [for example, see (11)].
Notwithstanding the agricultural importance of RTSV and MCDV, which have a long history of biological research, many aspects of waikavirus molecular biology remain unknown. Some of the salient open questions include: what is the complete protein expression complement of waikaviruses, and what are the functions of expressed proteins in infection [see (12–16)] what is the role of the waikavirus-specific ORFX (17); what are the viral determinants of vector transmission, including the putative helper component of MCDV (18–20), and is it the same component that enables RTSV and RTBV transmission by leafhoppers (21)); and what determines waikavirus pathogenicity (e.g. what determines the mild versus severe symptoms of MCDV isolates, and the synergistic interaction of RTSV and RTBV in rice tungro disease)?
A major hurdle in waikavirus molecular studies has been lack of infectious clones, important tools for molecular dissection of virus sequence functions. Both the generation of virus infectious clones and launching infection from such clones in natural host plants are significant challenges for many viruses. These challenges are well known for negative sense and ambisense RNA viruses such as rhabdoviruses, tospoviruses, and emaraviruses, for which the genomic RNA is not sufficient to launch infection. The first clones of these viruses that can launch in planta infections have only very recently been reported (22–26). Major technical challenges have also been problematic for many (+) RNA viruses because of bacterial toxicity, as has been observed by virologists for many years [for some examples, see (27–30)]. In addition, delivery of RNA viruses to host plants often relies on agrobacterium (Rhizobium radiobacter) or other DNA-based delivery, introducing nuclear processing steps for which cytoplasmically replicating RNA viruses are not naturally adapted, and requires another host-microbe interaction for bacterial delivery. Delivery of phloem-limited viruses to infection-competent cells is also often challenging. High bacterial selection against MCDV sequences, its large genome size, challenges in full-length sequence amplification, and the necessity of phloem delivery have all impeded development of MCDV infectious clones by multiple scientists for decades. Here we demonstrate that using an improved plasmid backbone, high efficiency in vitro transcription for quality full-length virus, and improved vector-free delivery by vascular puncture inoculation (VPI), a delivery method invented by Dr. Raymond Louie (31), we have developed and delivered infectious clones of both MCDV-S and MCDV-M1, and a green fluorescent protein (GFP) tagged MCDV-S clone. These are the first reported infectious clones for waikaviruses.
2 Materials and methods
2.1 Virus isolates and leafhopper colony
MCDV-S and MCDV-M1 isolates corresponding to GenBank sequences AY362551.1 (32) and AY829112.1 were originally collected near Friendship, Ohio [see (19, 33)] and maintained by leafhopper transmission and vascular puncture inoculation (31). The Graminella nigrifrons colony utilized was developed from insects collected near Wooster, Ohio and maintained as previously described (34).
2.2 5’ and 3’ RACE of MCDV-S and MCDV-M1
Total RNA was extracted from plants infected with MCDV-S and MCDV-M1 using Direct-zol RNA miniprep kits (Zymo Research, Irvine, CA). One µg of RNA from each was used for first strand cDNA synthesis with Superscript III Reverse Transcriptase (ThermoFisher Scientific, Waltham, MA) followed by RNaseH treatment according to the manufacturer’s instructions. For 5’ RACE, cDNA was purified using a Monarch PCR and DNA cleanup column (New England Biolabs, Ipswich, MA), quantified, and tailed in four separate reactions with 0.25 mM of dATP, dCTP, or dGTP for MCDV-S and dATP, dGTP and dTTP for MCDV-M1 using terminal transferase (New England Biolabs, Ipswich, MA). Tailed cDNA was used for nested polymerase chain reaction (PCR) amplification with one of the anchor primers WX27, WX28, WX197, or WX198 and a 5’ RACE reverse primer followed by amplification with primers WX29 and a nested 5’RACE reverse primer (see Table S1). All PCR reactions were carried out using PrimeSTAR DNA polymerase (Takara Bio USA, San Jose, CA) with a 98°C 15s denaturation followed by 30 cycles of 98°C for 10 s, 52°C for 20 s, and 72°C for 30s, ending with a 5 min. 72°C extension. Amplicons purified using a Monarch PCR and DNA cleanup column (New England Biolabs, Ipswich, MA) were either directly Sanger sequenced using primer WX29 or cloned into pMINIT 2.0 (New England Biolabs, Ipswich, MA), then sequenced using primers WX202 and WX203 (Table S1).
For 3’ RACE, cDNA was synthesized from total RNA from plants infected with either MCDV-S or MCDV-M1 with primer WX292 (Table S1). First round PCR was conducted using primers WX212 and WX29 for MCDV-M1 and WX295 and WX293 for MCDV-S (Table S1). A second (nested) PCR was conducted using primers WX294 and WX29 for MCDV-M1 and WX296 and WX29 for MCDV-S (Table S1). PCR conditions and amplicon sequencing were performed as for 5’ RACE.
2.3 Cloning cDNA of MCDV-S and MCDV-M1
Full-length cDNA from MCDV-S was produced using SuperScript III First-Strand Synthesis System (ThermoFisher Scientific, Waltham, MA) according to the manufacturer’s protocol with the gene specific primer LRS881 (Table S2). For cloning, MCDV-S cDNA was amplified in two fragments using PrimeSTAR GXL DNA Polymerase (Takara Bio USA, San Jose, CA) and primers WX344 and LRS922 for Fragment 1, and LRS923 and WX345 for Fragment 2 (Table S2). Fragments 1 and 2 were assembled into a linear pCB301-2µ-HDV plasmid backbone fragment, which had been amplified with primers WX342 and WX343, using NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs, Ipswich, MA) according to manufacturer’s instructions. Plasmid pCB301-2µ-HDV was kindly provided by the late Dr. Michael Goodin, University of Kentucky, with permission from developer Dr. Zhenghe Li (29). Resulting cloned products were transformed into NEB Stable chemically competent Escherichia coli. PCR screening for full-length clones was done using primers WX186 and WX199 (Table S2) with PrimeSTAR GXL DNA Polymerase (Takara Bio USA, San Jose, CA) following the manufacturer’s 2-step PCR procedure.
A similar approach was used to develop a full-length clone of MCDV-M1. One µg total nucleic acid extracted from MCDV-M1-infected plants was first annealed with primer oligo(dT)20, then used for first strand cDNA synthesis using Superscript III reverse transcriptase (ThermoFisher Scientific, Waltham, MA). Overlapping approximate halves of MCDV-M1 sequence were amplified from cDNA with primers incorporating backbone pCB301-2µ-HDV junction sequences, WX346 and WX196 and WX458 and WX347 (Table S2), and both fragments were cloned into pCB301-2µ-HDV as for MCDV-S full-length clones using NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs, Ipswich, MA). Full-length clones pWX73-pWX76 were recovered and verified by PCR screening using MCDV-M1 specific primer pairs including WX216/LRS176, LRS177/WX194, WX216/WX196, WX458/WX189, WX459/WX189, WX216/WX207, WX212/WX189, WX212/WX562, and WX563/WX207 (Table S2).
2.4 Design and construction of MCDV-S-GFPΔp27
The 744 nt sequence encoding green fluorescent protein (GFP) was inserted using NEBuilder HiFi Assembly Master Mix (New England Biolabs, Ipswich, MA) in-frame to replace 669 nucleotides of the 723 nt MCDV-S p27 coding sequence between nt 1816-2512, leaving nine 5’ and nine 3’ codons to retain the protease cleavage sites for p27 fused to translated GFP (Figure S1). Primers WX389 and WX392 (Table S2) were used to amplify the pCB301-2µ-HDV/MCDV-S backbone sequence as a template (pKJW12), and WX390 and WX391 (Table S2) were used to amplify the full-length GFP sequence (35) from plasmid pRTL2sGFP kindly provided by Dr. Tim Sit, North Carolina State University. The GFP-encoding gene fragment was assembled into pCB301-2µ-HDV/MCDV-S backbone template using NEBuilder® HiFi DNA Assembly Master Mix (New England Biolabs, Ipswich, MA). Clones from expected full-length assemblies amplified were further tested for infectivity.
2.5 In vitro RNA transcript production and inoculation
In vitro RNA transcripts were synthesized from full-length virus amplicons from cDNA clones, an approach previously described for low-copy number maize dwarf mosaic virus clones (36, 37). 2.5 ng plasmid template was used to amplify full-length virus sequences in 50 µl reactions containing 1.5 µl 10 µM primers adding 5’ T7 promoter sequences and 3’ poly(A) tails with 2X PrimeStar GXL DNA polymerase premix (Takara Bio USA, San Jose, CA) with PCR conditions: 98°C/30s for denaturing followed by 35 cycles of 98°C/10s and 68°C/10 min. For MCDV-S and MCDV-S-GFPΔp27, templates were amplified using primers WX576 and WX199 (Table S2). MCDV-M1 full-length clone template was amplified using primers WX572 and WX190 (Table S2). Resulting PCR products were purified using Monarch PCR and DNA cleanup kit (New England Biolabs, Ipswich, MA) according to the manufacturer’s instructions.
Transcript RNA was synthesized from PCR-amplified templates using HiScribe™ T7 ARCA mRNA Kit (New England Biolabs, Ipswich, MA). Resulting transcription reactions were cleaned using the Monarch RNA cleanup kit (New England Biolabs, Ipswich, MA). Transcript quantity was estimated by NanoDrop (ThermoFisher Scientific, Waltham, MA) and quality was assessed on non-denaturing 1% agarose 1X TBE (0.089 M Tris, 0.089 M boric acid, 0.002 M EDTA) gels. VPI was used to inoculate maize seed lines OH28, Silver Queen, Va35, and Wf9xOH51A with 2.0 µg (for initial clone screening only) or 4.0 µg RNA transcript per seed as previously described (31, 37, 38). Briefly, seeds were treated with 30% Clorox bleach by immersion for 3 minutes and then soaked in 30°C tap water for 2 hours to initiation germination. Soaked seed were inoculated using tattoo pins mounted on an engraving tool to pass through inoculum on both sides of each seed’s embryo at an approximately 45° angle. Inoculated seeds were incubated for 2 days at 30°C on moist paper towels to germinate before planting in greenhouse or growth chamber. Plants were evaluated for symptoms after 14-28 days.
2.6 ELISA confirmation of infection
Leaf samples collected from plants inoculated by VPI with MCDV-S transcripts were tested for the presence of MCDV-S using protein A sandwich enzyme-linked immunosorbent assay (PAS-ELISA) and in-house virion antisera, anti-MCDV-S 03-01, at a dilution of 1:1,500. Sample tissue was ground at 1:5 (wt:vol) in phosphate-buffered saline (0.02 M NaP04 and 0.15 M NaCl), 2% polyvinylpyrrolidone, and 0.2% bovine serum albumin. Samples were scored as positive if the absorbance slope at 405 nm was greater than twice the slope of the negative controls (based on linear range of absorbance over time), and if sample absorbance at 405 nm was greater than three times the average absorbance of within-plate negative control readings.
2.7 RT-PCR confirmation of infection
Inoculated plants were individually tested between 19 and 22 days post-inoculation (dpi) by grinding tissue 1:20 in GEB (0.015 M sodium carbonate, 0.035 M sodium bicarbonate, 2% polyvinylpyrrolidone, 0.2% bovine serum albumin, 0.05% Tween 20). Extracts were then denatured by diluting the extract 1:12.5 in GES (0.1 M glycine-NaOH, pH 9.0, 50 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 1 mM dithiothreitol (added fresh)) and heating samples at 95°C for 10 minutes. Denatured samples were placed directly on ice for a minimum of 5 minutes. The denatured extracts were then added 1:12.5 to a 25 µl final volume of reverse transcription-polymerase chain reaction (RT-PCR). RT-PCR was performed as previously reported (39) using an annealing temperature of 58°C. Primers LRS167 and LRS168 were used to confirm MCDV-S and MCDV-S-GFPΔp27 infections, and primers WX212 and WX189 (Table S2) were used to confirm MCDV-M1 infection.
2.8 Microscopy
Samples from infected plants were collected for GFP visualization at 21 dpi. Water-mounted 5x10 mm leaf sections were examined using an Infinity 3S-1UR ultra-sensitive digital camera mounted on a motorized Olympus CKX53 microscope (Olympus Corp., Center Valley, PA) with a GFP filter (470/40 nm excitation and 525/50 nm emission bandpass). Camera and microscope settings were controlled using Lumenera Infinity Analyzer software v. 6.5.4. Fluorescence and brightfield images were taken at 10X magnification at exposures of 100 ms and 20 ms, respectively. All fluorescent images were adjusted identically in PowerPoint (Microsoft Office, Redmond, WA) to increase both brightness and contrast by 40%.
Plants inoculated with pKJW12 and MCDV-S-GFPΔp27 were used to assess virion formation. Leaves from maize plants inoculated with pKJW12 and MCDV-S-GFPΔp27 with MCDV-S symptoms, and GFP expression for MCDV-S-GFPΔp27, were cut into small pieces with a new razor blade in 200 ml 0.1 M potassium phosphate buffer, pH 7.0. Extract was incubated for 5 min at room temperature; then, one 25 ml drop was placed on a 300-mesh formvar-coated copper grid and incubated at room temperature for 20 min. After negative staining with 2% uranyl acetate, samples were viewed and images taken with a Hitachi H-7500 electron microscope (Molecular and Cellular Imaging Center, Wooster, OH).
2.9 Dot blot immunoassay
Maize leaves were selected from plants inoculated with MCDV-S-GFPΔp27 that were RT-PCR positive and symptomatic. Selected leaves were ground 1:10 in GEB, and 5 µl of liquid was spotted onto a nitrocellulose membrane for each sample (GE Healthcare, Chicago IL) and GFP protein (Abcam, Cambridge, England) as a positive control. GFP was detected using Anti-GFP antibodies (Abcam, Cambridge, England) at a dilution of 1:3000 and Goat Anti-Rabbit IgG AP Conjugate (Sigma Aldrich, St. Louis, MO) at a dilution of 1:30,000.
2.10 Leafhopper transmission of virus launched from infectious clones
The MCDV leafhopper vector, Graminella nigrifrons, (Hemiptera : Cicadellidae) was reared on oats (Avena sativa, cv. Armor, Ohio Certified Seeds). Leafhoppers were housed in 40 cm3 aluminum- framed cages with a plexiglass door for access and kept in a temperature-controlled room at 25°C, 15 h light:9 h dark. To prepare cohorts of synchronous leafhoppers, several hundred adult females were allowed to oviposit on oats for 7 days, after which the plants were moved to a new cage where the eggs hatched and leafhoppers matured. For virus acquisition, source plants had their asymptomatic leaves removed and were then caged with adult leafhoppers. The multiple inoculation method developed by Louie and Anderson (34) was used to maximize transmission opportunity within the 48-hour retention period of MCDV while minimizing damage to plants by large leafhopper cohorts. Briefly, three sequential waves of leafhoppers were used for each set of acquisition and inoculation plants: leafhoppers were placed on a plant with clone-launched virus infection for a 48-hour acquisition access periods (AAP) and then removed and placed on healthy transmission test seedlings for a 48-hour inoculation access period (IAP) and removed, then this process was repeated with another cohort of leafhoppers for a total of three inoculations from the same source plants to test plants in 2-day intervals. Test plants were initially inoculated when their first leaf was visible but not fully emerged, at a rate of 5 leafhoppers/plant, 25 plants per treatment, and this rate was consistent for each AAP/IAP, so that the final insect pressure was 3 sequential sets of 5 leafhoppers/plant. 48 hours after the final inoculation, transmission test plants were fumigated with dichlorvos (NuVan ProStrips®, AMVAC Chemical Corp., Newport Beach, CA) for 5 hours to kill any remaining leafhoppers, and then taken either to a greenhouse (wild type virus) or an environmental chamber (modified virus) for development.
3 Results
3.1 MCDV-S and MCDV-M1 5’ RACE and development of full-length clones
To confirm authentic ends of viruses, 5’ and 3’ RACE was performed for MCDV-S and MCDV-M1 lab isolates using multiple 5’ tailing nucleotides. From MCDV-S 5’ RACE, 20 sequences were obtained: three from dGTP tailing, nine from dATP tailing, and eight from dCTP tailing. Although four of the dCTP tailed sequences had different 5’ sequences showing variability in 5’ C or T nucleotides, the MCDV-S 5’ consensus sequence from all RACE reactions was the same as the published sequence (AY362551.1; Figure S2A). However, the eight 5’ RACE sequences for MCDV-M1, four from dTTP tailing, one from dGTP tailing, and three from dATP tailing, identified 17 5’ nucleotides missing from the published NCBI sequence and differences in the sequence for an additional 7 bases compared to the original GenBank submission (AY829112.1; Figure 1B), with no variability among the RACE sequences except for an additional 5’ G in one of the dATP-tailed samples (Figure S1B). 3’ RACE for both MCDV-S and MCDV-M1 showed the same sequences reported in published sequences (data not shown). The GenBank sequence for MCDV-M1 was revised to include the 5’ RACE-validated nucleotides (AY829112.2).
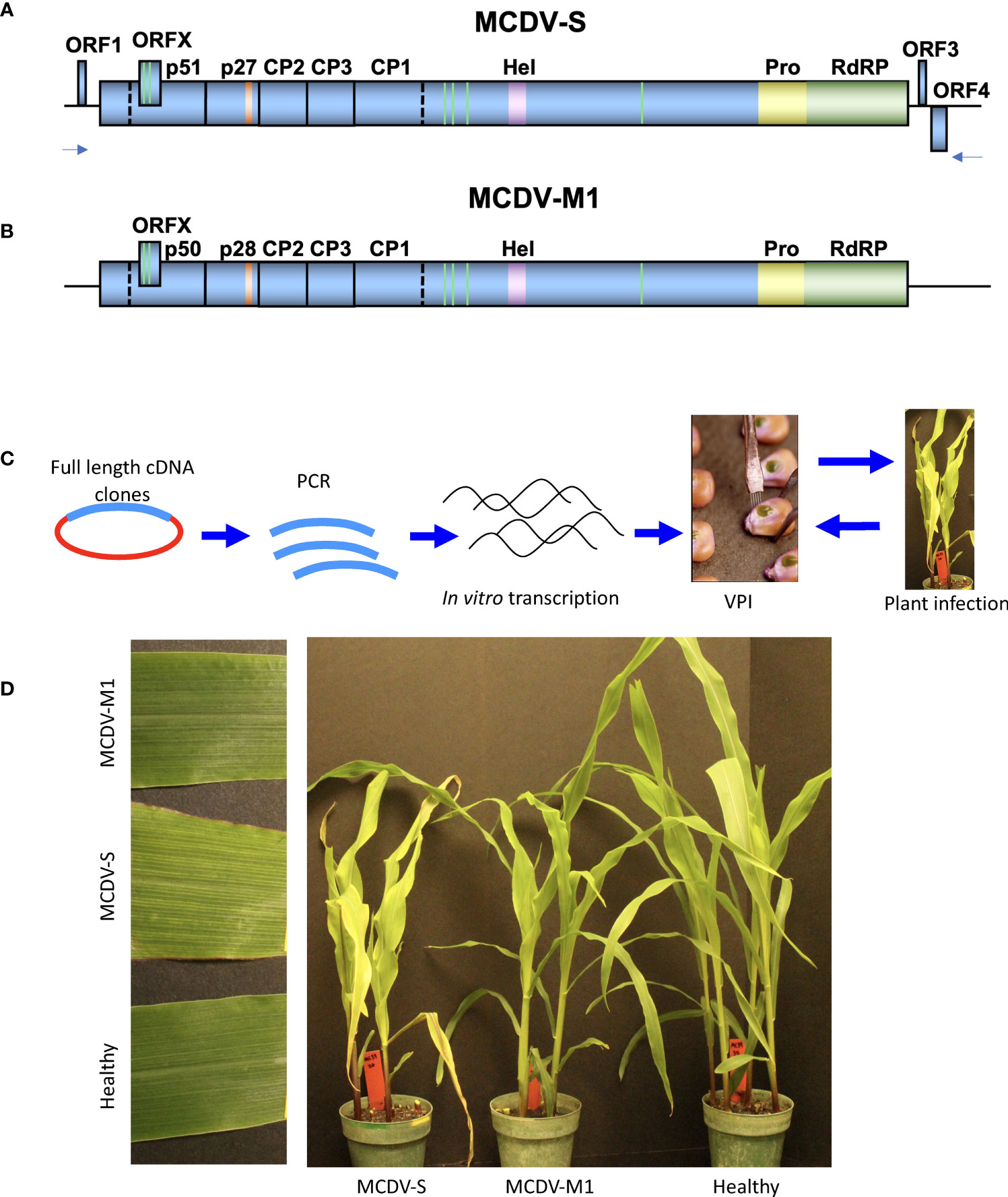
Figure 1 Genome maps of MCDV-S (A) and MCDV-M1 (B). Methodology for inoculation from full-length clones using amplification of cloned virus sequence followed by in vitro transcription and vascular puncture inoculation (C). Infection launched from clones in plants (D).
Since previous attempts at cloning full-length MCDV were strongly selected against in a variety of plasmids, indicative of virus sequence toxicity in E. coli (data not shown), full-length clones of MCDV-S and MCDV-M1 (Figures 1A, B) were constructed in the pCB301-2µ-HDV plasmid backbone (29) designed for development of infectious clones of virus sequences with bacterial toxicity. Nine apparently full-length MCDV-S clones were recovered (pKJW2, 5, 6, 11-16) and four MCDV-M1 full-length clones were recovered (pWX73-76), as determined by PCR amplification of ca. 12 kb virus sequence from recovered clones.
3.2 Infections launched by vascular puncture inoculation of clone in vitro transcripts
Screening was performed by IVT/VPI, in which in vitro transcripts (IVTs) generated from full-length virus PCR amplicon templates from recovered full-length virus clones were tested for infectivity by VPI (Figure 1C). Characteristic symptoms of MCDV-S infection were detected in a subset of inoculated plants (Figure 1D), and infection confirmed in selected plants by ELISA and/or RT-PCR. In initial IVT/VPI screening, only two of the nine MCDV-S clones produced infection confirmed by ELISA: pKJW6 (1/22; 4.6% infection) in one of the three experiments, and pKJW12 which launched infection in each of the three experiments (2/35 [5.7%], 1/17 [5.9%], and 1/34 [2.9%], respectively). Clone pKJW12 was used for all subsequent experiments. Test inoculations of transcripts of MCDV-M1 clones pWX73-pWX76 resulted in 0/23 infected plants confirmed by RT-PCR from pWX73, 2/16 (12.5%) from pWX74, 1/32 (3.1%) from pWX75, and 4/30 (13.3%) from pWX76. pWX76 was selected for all subsequent experiments for MCDV-M1.
Since infection rates were low in initial clone screening experiments, an improved forward primer for MCDV-S was developed (WX576 instead of WX186, with reverse primer WX199; Table S2), higher amounts of inoculum (4.0 µg transcript per seed) and four susceptible maize genotypes were tested with pKJW12 transcripts to optimize infection launching. Among genotypes, the best infection rates were observed for Wf9XOH51A maize (25% overall), while infection rates of 8-13% were observed for Oh28, Silver Queen, and Va35, compared to infection rates by genotype of 60-73% from sap of plants infected with uncloned virus in control inoculations (Table 1). Substantial variability in infection rates between inoculation experiments was observed, indicative of variability between inoculations and/or inocula: infection rates ranged from 3-28% across experiments with independent transcript preparations, and 50-81% infectivity for inoculations with independent MCDV-S extracts for each experiment (Table 1).
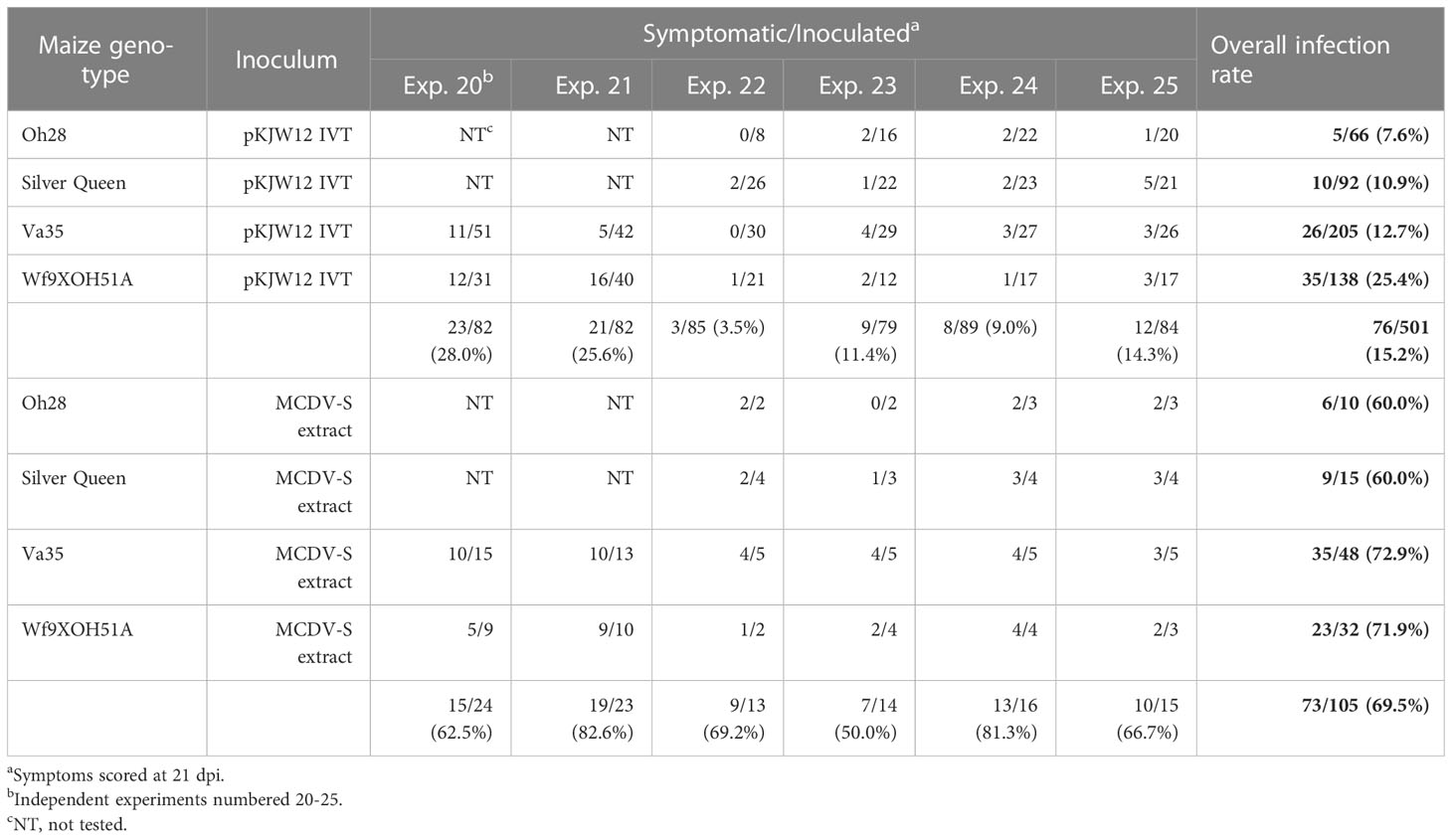
Table 1 Infectivity of pKJW12 MCDV-S transcripts (4 μg transcript RNA/seed generated using primers WX576/WX199) by vascular puncture inoculation.
For MCDV-M1, infection is asymptomatic at most time points, therefore infections were confirmed by a sensitive RT-PCR assay at 27-28 days post-VPI. VPI of transcripts from an MCDV-M1 clone resulted in a 7.1% overall infection rate compared with 53.5% infection rate from MCDV-M1 infected plant sap (Table 2). Typical MCDV symptoms including interveinal chlorosis were observed for plants inoculated with transcript from cloned MCDV-S, while mild or no symptoms were observed for MCDV-M1 (Figure 1D).
Table 2 Infectivity of MCDV-M1 pWX76 transcripts (4 µg/seed, generated using primers WX572/WX190) by vascular puncture inoculation to Silver Queen maize.
3.3 Leafhopper transmission rates of MCDV-S and MCDV-M1 launched from clones similar to those from uncloned virus
Rates of VPI infection with transcript were approximately an order of magnitude lower than from infected plant sap, matching previous research comparing nucleic acid and infected plant sap inoculations for maize viruses (38). However, cloned and uncloned MCDV-S and MCDV-M1 viruses had similar transmission rates by their natural vector G. nigrifrons once infections were launched in plants. Overall experimental leafhopper transmission rates were: 92.6% versus 78.6% for MCDV-S clone-launched versus uncloned virus, respectively (Table 3) and 86.3% versus 89.5% for MCDV-M1 clone-launched versus uncloned virus, respectively (Table 4).
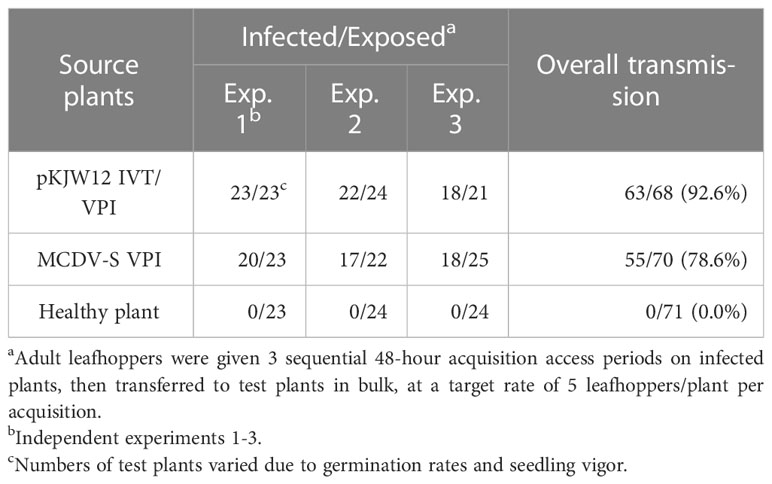
Table 3 Transmission of clone-launched MCDV-S by Graminella nigrifrons.
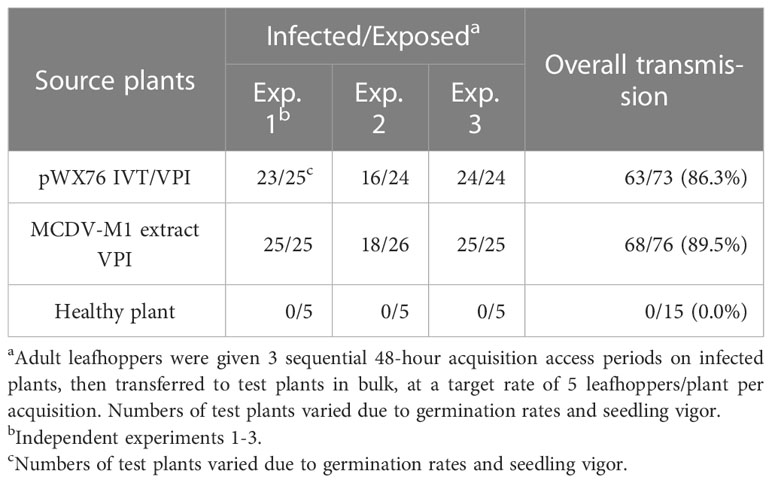
Table 4 Transmission of clone-launched MCDV-M1 by Graminella nigrifrons.
3.4 Systemic movement, symptom induction, and vascular GFP expression by MCDV-S-GFPΔp27
MCDV forms icosahedral virions and is expected to have packaging constraints as a result. To develop a GFP-labeled infectious clone, we replaced sequence encoding a protein of unknown function, p27, with the similar length coding sequence for GFP, with the polyprotein protease excision sites conserved, to generate pMCDV-S-GFPΔp27 (Figures 2A, S1). pMCDV-S-GFPΔp27 clone 3 IVT/VPI resulted in a 4.0% overall infection rate, much lower than that observed for wild type cloned MCDV-S or MCDV-M1 (Table 5). Plants infected with MCDV-S-GFPΔp27 developed typical MCDV-S symptoms in the same time frame as wild type MCDV-S, with veinal chlorosis apparent by 10 days post VPI and leaf twisting and tearing apparent by 35 dpi (Figure 2B). GFP was visible in the vascular tissue of infected plants in systemic leaves (Figure 3).

Figure 2 Genome map of MCDV-S-GFPΔp27 (A). Plants infected with MCDV-S-GFPΔp27 and MCDV-S launched from clones compared to healthy control (B). Top left panel shows plants inoculated in experiment MC47 at 35 dpi, bottom left shows individual leaves from inoculated plants from the same experiment at 22 dpi. Right panels show characteristic vein chlorosis in plants inoculated with MCDV-S-GFPΔp27.
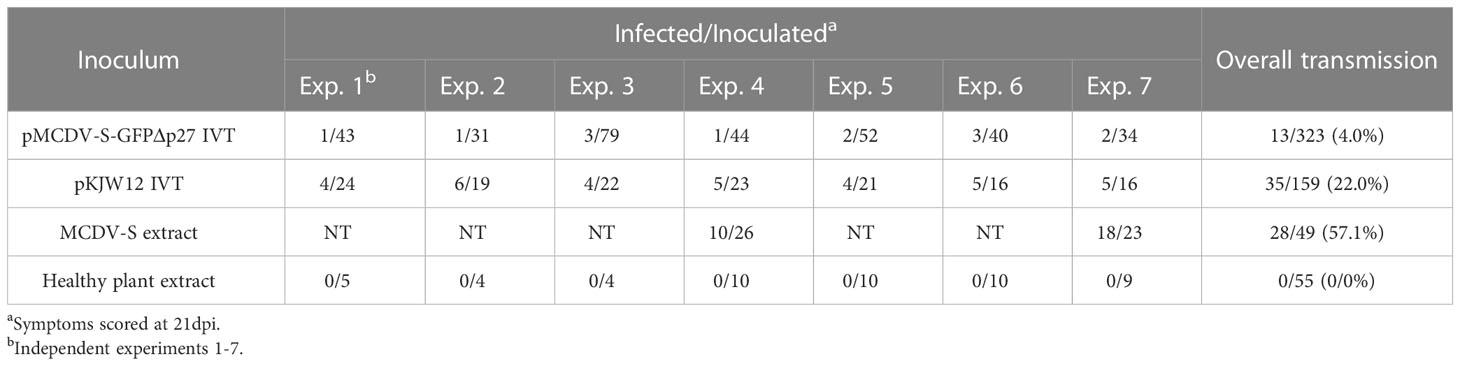
Table 5 Infectivity of pMCDV-S-GFPΔp27 transcripts (4 μg transcript RNA/seed generated using primers WX576/WX199) by vascular puncture inoculation to Silver Queen maize.
Figure 3 Fluorescent and corresponding brightfield images showing green fluorescent protein (GFP) in veins of plants inoculated with MCDV-S-GFPΔp27 transcript. Control comparisons show leaf inoculated with wild type MCDV-S clone pKJW12 transcript, extract from MCDV-S-infected plant (uncloned virus), and healthy control. Samples were collected 21 dpi. Size bar (white) represents 100 µm and scale is identical for all images.
3.5 No leafhopper transmission of MCDV-S-GFPΔp27 and reduced VPI passaging transmission compared to wild type cloned virus
Replicated experiments to test leafhopper transmission of MCDV-S-GFPΔp27 from systemically infected, symptomatic plants resulted in no transmission of this modified virus (Table 6). Since few plants were infected initially with transcript from pMCDV-S-GFPΔp27, we tested whether VPI passaging from infected plants could scale up infections, especially given the much higher infection rates from encapsidated virus compared to naked genomic RNA (37, 38). Using sap from plants infected by cloned virus IVT/VPI to passage by VPI, we compared infection rates of MCDV-S-GFPΔp27, cloned MCDV-S, and wild type uncloned MCDV-S. Sap inoculum from plants with infection launched from cloned MCDV-S had similar infectivity as sap from plants with infection launched from uncloned wild type MCDV-S as expected (Table 7). However, launching infection in plants did not restore the VPI infectivity for MCDV-S-GFPΔp27, for which the overall VPI passaging infection rate (6.3%) remained comparable to the rate of infection by transcript inoculation (Table 7). The low leafhopper and VPI passaging transmission rates for MCDV-S-GFPΔp27 led us to examine whether virion formation was disrupted for this construct. Transmission electron microscopy showed virus-like particles in sap from symptomatic plants inoculated with pMCDV-S-GFPΔp27 transcripts as well as for those inoculated with wild type MCDV-S transcripts (Figure 4) indicating that virions were indeed formed. Whether these virions from pMCDV-S-GFPΔp27 were qualitatively or quantitatively equivalent to those of wild type MCDV-S requires further analysis.
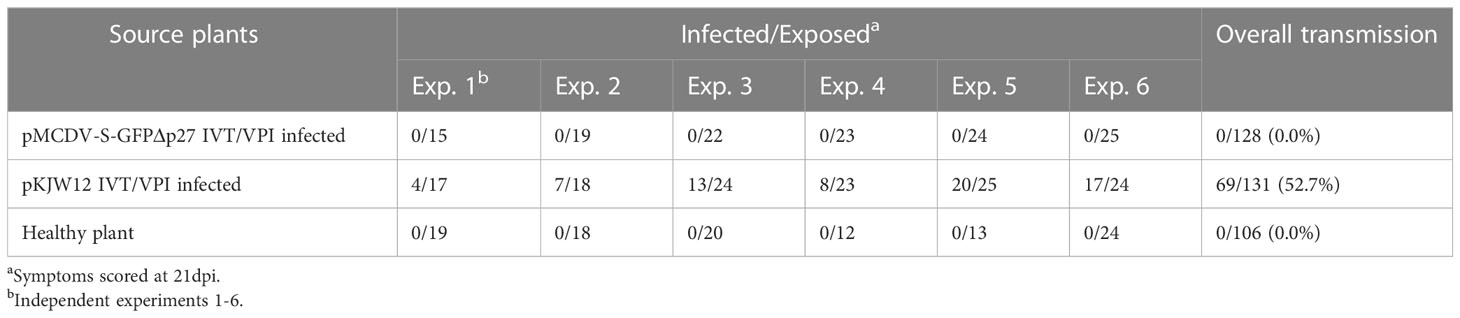
Table 6 Transmission of MCDV-S-GFPΔp27 versus MCDV-S launched from clones by Graminella nigrifrons.
Table 7 Vascular puncture inoculation passaging of clone-launched MCDV-S and MCDV-S-GFPΔp27 scored by symptom development and RT-PCR with primers LRS167/168 at 19-21 dpi.

Figure 4 Virus-like particles formed from infection launched from clones, comparing MCDV-S-GFPΔp27 (A) with MCDV-S wild type clone pKJW12 (B). 60K magnification, 3s exposure images.
4 Discussion
In this manuscript, we describe the first development of waikavirus infectious clones, for MCDV severe and mild (S and M1) strains. Full-length clones were used to amplify full-length virus sequences of MCDV-S and MCDV-M1, from which in vitro transcripts were then delivered by vascular puncture inoculation. Creation of these tools represents a major advance towards understanding waikavirus biology by enabling studies of viral gene functions through mutagenesis. As a first step towards utilizing MCDV infectious clones to understand waikavirus biology, we developed a GFP-tagged MCDV-S by replacement of the gene encoding a protein of unknown function, p27, with the similarly sized GFP gene. In doing so, we successfully developed a GFP-tagged virus construct and identified major clues in the function of MCDV p27. We learned that the gene encoding p27 in the MCDV polyprotein, a protein of unknown function released by virus 3C-like protease cleavage of the leader protease (13), is dispensable for systemic infection and symptom development in plants, since plants infected with virus lacking p27 developed MCDV-S symptoms and systemic infection at the same rate as plants inoculated with cloned wild type MCDV-S. In MCDV-S-GFPΔp27 infections, GFP was observed in plant vasculature as expected for expression from a phloem-limited virus.
The tools of infectious clones provide means to examine further questions of gene function for p27 and other waikavirus genes. Our data indicate that MCDV-S-GFPΔp27 is not transmissible by leafhoppers, suggesting its possible role as the elusive MCDV helper component (18, 19). However, low transmission by VPI passaging suggests alternative explanations for loss of leafhopper transmission that cannot be excluded without further experimentation. These other possibilities include sequence loss or changes during replication for the MCDV-S-GFPΔp27 RNA that impact infectivity, qualitative differences in encapsidation or capsid stability, an infrequent mutation in replication required for MCDV-S-GFPΔp27 establishment, or reduced fitness resulting in lower titer for the modified virus. Preliminary data support the possibility of lower titer of MCDV-S-GFPΔp27 compared to MCDV-S, even though symptom development and timing is comparable in infected plants. Further study is needed to understand the molecular basis for the whole plant infectivity outcomes.
Development of MCDV infectious clones has been attempted by several researchers over many years without previous success, making this an important breakthrough. We attribute our success to incremental and quantum improvements across multiple technical facets: improved and more user-friendly backbone plasmid vectors and hosts for E. coli-toxic viral sequences, footprint-free precise cloning methodology (Gibson assembly), improved high fidelity and progressive polymerases enabling amplification of complete virus genomes even of structured viruses such as MCDV that were recalcitrant to full-length amplification a decade ago, increased yield and quality of in vitro transcripts, and improved rates infection by VPI. Together, these technical improvements enabled successful development of clones that enabled GFP tagging and mutagenesis studies of these low titer, phloem-limited, and obligately leafhopper transmitted waikaviruses for which such research was formerly impossible.
Data availability statement
The RACE sequence data presented in the study are deposited in the GenBank repository, accession number AY829112.2. Original RACE sequence data can be found in Supplementary Figure 2.
Author contributions
LS designed and led experiments and made initial cloning strategy to replace p27 gene with GFP coding sequence. KW constructed MCDV-S clones and first identified infectivity of full-length clones, screened all other full-length clones by IVT/VPI, performed TEM, ELISA, and RT-PCR assays, provided materials and methods sections, and contributed edits to the manuscript. WX designed primers and performed 5’ and 3’ RACE for MCDV-S and MCDV-M1, designed and constructed MCDV-M1 and MCDV-S-GFPΔp27 infectious clones and contributed methods data and critical revisions of the manuscript. JT performed leafhopper transmission assays and provided methods section, transmission results tables and critical revisions of the manuscript. HT performed fluorescent and transmitted light microscopy imaging and manuscript editing. All authors contributed to the article and approved the submitted version.
Funding
LS, KW, and JT were supported by USDA-ARS. This research was supported in part by the U.S. Department of Agriculture, Agricultural Research Service. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer. Part of this research (supporting WX and HT) was performed as part of an Insect Allies team funded under cooperative agreement number HR0011-17-2-0054. The views expressed are those of the author and should not be interpreted as representing the official views or policies of the U.S. Government. The U.S. Government is authorized to reproduce and distribute reprints for Government purposes notwithstanding any copyright notation hereon.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2023.1216285/full#supplementary-material
Supplementary Figure 1 | Sequences showing cloning strategy for replacement of MCDV-S p27 with GFP, retaining nine N- and nine C-terminal amino acids for proteolytic release. MCDV-S p27 encoding cDNA sequence, with nucleotides for nine terminal codons in bolded red text (A). MCDV-S p27 241 amino acid sequence showing residues retained for fusion with GFP sequence in bolded red text (B). Nucleotide sequence of inserted construct with GFP encoding sequence shown in blue text with and codons from p27 in bolded red text on each side (C). Translated fusion sequence of nine p27 N- and C-terminal residues with GFP insertion (D).
Supplementary Figure 2 | 5’ RACE confirmation of MCDV-S and MCDV-M1 sequences using three different tailing nucleotides for each. MCDV-S 5’ end showing consensus sequence the same as published sequence (GenBank accession no. AY362551.1; (A). MCDV-M1 published 5’ sequence compared with MCDV-S and 5’ RACE showing additional 17 nt at the 5’ end and different consensus sequence for the 7 subsequent nucleotides compared to published sequence (GenBank accession no. AY829112.1; (B). Note that T is used rather than U found in RNA to compare cDNA sequences.
Supplementary Table 1 | RACE primers.
Supplementary Table 2 | Primers used for clone construction, template production, and RT-PCR detection.
Abbreviations
IVT, in vitro transcription; VPI, vascular puncture inoculation; IVT/VPI, in vitro transcription/vascular puncture inoculation.
References
1. Hull R. Molecular biology of rice tungro viruses. Annu Rev Phytopathol (1996) 34:275–97. doi: 10.1146/annurev.phyto.34.1.275
2. Gingery RE. Properties of maize chlorotic dwarf virus and its ribonucleic-acid. Virology (1976) 73(2):311–8. doi: 10.1016/0042-6822(76)90392-5
3. Gordon DT, Nault LR. Involvement of maize chlorotic dwarf virus and other agents in stunting diseases of Zea mays the United States. Phytopathology (1977) 67(1):27–36. doi: 10.1094/Phyto-67-27
4. Stewart LR. Waikaviruses: studied but not understood. APS Features (2011). Available at: https://www.apsnet.org/edcenter/apsnetfeatures/Pages/waikavirus.aspx.
5. Stewart LR, Teplier R, Todd JC, Jones MW, Cassone BJ, Wijeratne S, et al. Viruses in maize and johnsongrass in southern Ohio. Phytopathology (2014) 104(12):1360–9. doi: 10.1094/Phyto-08-13-0221-R
6. Thapa V, Melcher U, Wiley GB, Doust A, Palmer MW, Roewe K, et al. Detection of members of the Secoviridae in the tallgrass prairie preserve, Osage county, Oklahoma, USA. Virus Res (2012) 167(1):34–42. doi: 10.1016/j.virusres.2012.03.016
7. Seo JK, Kwak HR, Lee YJ, Kim J, Kim MK, Kim CS, et al. Complete genome sequence of bellflower vein chlorosis virus, a novel putative member of the genus waikavirus. Arch Virol (2015) 160(12):3139–42. doi: 10.1007/s00705-015-2606-9
8. Thekke-Veetil T, Ho T, Postman JD, Tzanetakis IE. Blackcurrant waikavirus a, a new member of the genus waikavirus, and its phylogenetic and molecular relationship with other known members. Eur J Plant Pathol (2020) 157(1):59–64. doi: 10.1007/s10658-020-01978-y
9. Park D, Hahn Y. A novel waikavirus (the family Secoviridae) genome sequence identified in rapeseed (Brassica napus). Acta Virol (2019) 63(2):211. doi: 10.4149/av_2019_205
10. Koloniuk I, Franova J. Complete nucleotide sequence and genome organization of red clover associated virus 1 (Rcav1), a putative member of the genus waikavirus (Family Secoviridae, order picornavirales). Arch Virol (2018) 163(12):3447–9. doi: 10.1007/s00705-018-4005-5
11. Sidharthan VK, Rajeswari V, Baranwal VK. Analysis of public domain plant transcriptomes expands the phylogenetic diversity of the family Secoviridae. Virus Genes (2022) 58(8):598–604. doi: 10.1007/s11262-022-01931-7
12. Chaouch-Hamada R, Redinbaugh MG, Gingery RE, Willie K, Hogenhout SA. Accumulation of maize chlorotic dwarf virus proteins in its plant host and leafhopper vector. Virology (2004) 325(2):379–88. doi: 10.1016/j.virol.2004.04.039
13. Stewart LR, Jarugula S, Zhao Y, Qu F, Marty D. Identification of a maize chlorotic dwarf virus silencing suppressor protein. Virology (2017) 504:88–95. doi: 10.1016/j.virol.2016.11.017
14. Stewart LR, Willman M, Marty D, Cole A, Willie K. Critical residues for proteolysis activity of maize chlorotic dwarf virus (Mcdv) 3c-like protease and comparison of activity of orthologous waikavirus proteases. Virology (2022) 567(2):57–64. doi: 10.1016/j.virol.2021.12.008
15. Thole V, Hull R. Rice tungro spherical virus: nucleotide sequence of the 3' genomic half and studies on the two small 3' open reading frames. Virus Genes (1996) 13(3):239–46. doi: 10.1007/Bf00366984
16. Thole V, Hull R. Rice tungro spherical virus polyprotein processing: identification of a virus-encoded protease and mutational analysis of putative cleavage sites. Virology (1998) 247(1):106–14. doi: 10.1006/viro.1998.9225
17. Firth AE, Atkins JF. Bioinformatic analysis suggests that a conserved orf in the waikaviruses encodes an overlapping gene. Arch Virol (2008) 153(7):1379–83. doi: 10.1007/s00705-008-0119-5
18. Creamer R, Nault LR, Gingery RE. Biological factors affecting leafhopper transmission of purified maize chlorotic dwarf machlovirus. Entomol Exp Appl (1993) 67(1):65–71. doi: 10.1111/j.1570-7458.1993.tb01652.x
19. Hunt RE, Nault LR, Gingery RE. Evidence for infectivity of maize chlorotic dwarf virus and for a helper component in its leafhopper transmission. Phytopathology (1988) 78(4):499–504. doi: 10.1094/Phyto-78-499
20. Ammar ED, Gordon DT, Nault LR. Ultrastructure of maize chlorotic dwarf virus-infected maize and viruliferous leafhopper vectors. Phytopathology (1987) 77(12):1743.
21. Hibino H. Transmission of 2 rice tungro-associated viruses and rice waika virus from doubly or singly infected source plants by leafhopper vectors. Plant Dis (1983) 67(7):774–7. doi: 10.1094/Pd-67-774
22. Feng MF, Cheng RX, Chen ML, Guo R, Li LY, Feng ZK, et al. Rescue of tomato spotted wilt virus entirely from complementary DNA clones. P Natl Acad Sci USA (2020) 117(2):1181–90. doi: 10.1073/pnas.1910787117
23. Gao Q, Xu WY, Yan T, Fang XD, Cao Q, Zhang ZJ, et al. Rescue of a plant cytorhabdovirus as versatile expression platforms for planthopper and cereal genomic studies. New Phytol (2019) 223(4):2120–33. doi: 10.1111/nph.15889
24. Verchot J, Herath V, Urrutia CD, Gayral M, Lyle K, Shires MK, et al. Development of a reverse genetic system for studying rose rosette virus in whole plants. Mol Plant Microbe In (2020) 33(10):1209–21. doi: 10.1094/Mpmi-04-20-0094-R
25. Wang Q, Ma XN, Qian SS, Zhou X, Sun K, Chen XL, et al. Rescue of a plant negative-strand rna virus from cloned cdna: insights into enveloped plant virus movement and morphogenesis. PloS Pathog (2015) 11(10):e1005223. doi: 10.1371/journal.ppat.1005223
26. Kanakala S, Xavier CAD, Martin KM, Tran HH, Redinbaugh MG, Whitfield AE. Rescue of the first alphanucleorhabdovirus entirely from cloned complementary DNA: an efficient vector for systemic expression of foreign genes in maize and insect vectors. Mol Plant Pathol (2022) 24(10):788–800. doi: 10.1111/mpp.13273
27. Lai MMC. The making of infectious viral rna: no size limit in sight. P Natl Acad Sci USA (2000) 97(10):5025–7. doi: 10.1073/pnas.97.10.5025
28. Nagyova A, Subr Z. Infectious full-length clones of plant viruses and their use for construction of viral vectors. Acta Virol (2007) 51(4):223–37.
29. Sun K, Zhao D, Liu Y, Huang C, Zhang W, Li Z. Rapid construction of complex plant rna virus infectious cdna clones for agroinfection using a yeast-e. coli-agrobacterium shuttle vector. Viruses (2017) 9(11):332. doi: 10.3390/v9110332
30. Tuo DC, Fu LL, Shen WT, Li XY, Zhou P, Yan P. Generation of stable infectious clones of plant viruses by using rhizobium radiobacter for both cloning and inoculation. Virology (2017) 510:99–103. doi: 10.1016/j.virol.2017.07.012
31. Louie R. Vascular puncture of maize kernels for the mechanical transmission of maize white line mosaic-virus and other viruses of maize. Phytopathology (1995) 85(2):139–43. doi: 10.1094/Phyto-85-139
32. Chaouch R, Redinbaugh MG, Marrakchi M, Hogenhout SA. Genomics of the severe isolate of maize chlorotic dwarf virus. Plant Prot Sci (2004) 40):113–9. doi: 10.17221/469-PPS
33. Gingery RE, Nault LR. Severe maize chlorotic dwarf disease caused by double infection with mild virus strains. Phytopathology (1990) 80(8):687–91. doi: 10.1094/Phyto-80-687
34. Louie R, Anderson RJ. Evaluation of maize chlorotic dwarf virus resistance in maize with multiple inoculations by Graminella nigrifrons (Homoptera, cicadellidae). J Econ Entomol (1993) 86(5):1579–83. doi: 10.1093/jee/86.5.1579
35. Chiu W, Niwa Y, Zeng W, Hirano T, Kobayashi H, Sheen J. Engineered gfp as a vital reporter in plants. Curr Biol (1996) 6(3):325–30. doi: 10.1016/s0960-9822(02)00483-9
36. Stewart LR, Bouchard R, Redinbaugh MG, Meulia T. Complete sequence and development of a full-length infectious clone of an Ohio isolate of maize dwarf mosaic virus (Mdmv). Virus Res (2012) 165(2):219–24. doi: 10.1016/j.virusres.2012.02.004
37. Xie WS, Marty DM, Xu JH, Khatri N, Willie K, Moraes WB, et al. Simultaneous gene expression and multi-gene silencing in zea mays using maize dwarf mosaic virus. BMC Plant Biol (2021) 21(1):208. doi: 10.1186/s12870-021-02971-1
38. Redinbaugh MG, Louie R, Ngwira P, Edema R, Gordon DT, Bisaro DM. Transmission of viral rna and DNA to maize kernels by vascular puncture inoculation. J Virol Methods (2001) 98(2):135–43. doi: 10.1016/S0166-0934(01)00369-X
39. Stewart LR, Todd J, Willie K, Massawe D, Khatri N. A recently discovered maize polerovirus causes leaf reddening symptoms in several maize genotypes and is transmitted by both the corn leaf aphid (Rhopalosiphum maidis) and the bird cherry-oat aphid (Rhopalosiphum padi). Plant Dis (2020) 104(6):1589–92. doi: 10.1094/PDIS-09-19-2054-SC
Keywords: Waikavirus, Secoviridae, infectious clone, virus vector, maize, leafhopper transmission
Citation: Stewart LR, Willie K, Xie W, Todd J and Tran HH (2023) First waikavirus infectious clones and vascular expression of green fluorescent protein from maize chlorotic dwarf virus. Front. Virol. 3:1216285. doi: 10.3389/fviro.2023.1216285
Received: 03 May 2023; Accepted: 08 June 2023;
Published: 29 June 2023.
Edited by:
Judith Kay Brown, University of Arizona, United StatesReviewed by:
Nicolas Bejerman, National Scientific and Technical Research Council (CONICET), ArgentinaPedro Luis Ramos-González, Biological Institute of São Paulo, Brazil
Copyright © 2023 This work is authored by Lucy R. Stewart, Kristen Willie, and Jane Todd on behalf of the U.S. Government and as regards, Dr. Stewart, Ms. Willie and Ms. Todd, and the U.S Government, is not subject to copyright protection in the United States. Foreign and other copyrights may apply. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lucy R. Stewart, bHVjeS5zdGV3YXJ0QHVzZGEuZ292
†These authors have contributed equally to this work