 Reca Marian Caballero
Reca Marian Caballero Ivonne González-Gamboa
Ivonne González-Gamboa Stephen L. Craig3
Stephen L. Craig3 Nicole F. Steinmetz
Nicole F. Steinmetz
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Virol., 08 June 2023
Sec. Systems Virology
Volume 3 - 2023 | https://doi.org/10.3389/fviro.2023.1184095
Plant virus-based nanoparticles (VNPs) offer a bioinspired approach to the delivery of drugs and imaging agents. The chemical addressability, biocompatibility, and scalable manufacturability of VNPs make them a promising alternative to synthetic delivery platforms. However, VNPs, just like other proteinaceous or synthetic nanoparticles (NPs), are readily recognized and cleared by the immune system and mechanisms such as opsonization and phagocytosis. Shielding strategies, such as PEGylation, are commonly used to mitigate premature NP clearance. Here, we investigated polyethylene glycol (PEG) coatings on the tobacco mosaic virus (TMV), which was used as a model nanocarrier system. Specifically, we evaluated the effects of linear and multivalent PEG coatings at varying chain lengths on serum protein adsorption, antibody recognition, and macrophage uptake. Linear and multivalent PEGs of molecular weights 2,000 and 5,000 Da were successfully grafted onto the TMV at ≈ 20%–60% conjugation efficiencies, and the degree of cross-linking as a function of PEG valency and length was determined. PEGylation resulted in the modulation of TMV–macrophage interactions and reduced corona formation as well as antibody recognition. Linear and multivalent PEG 5,000 formulations (but not PEG 2,000 formulations) reduced α-TMV antibody recognition, whereas shorter, multivalent PEG coatings significantly reduced α-PEG recognition—this highlights an interesting interplay between the NP and the PEG itself in potential antigenicity and should be an important consideration in PEGylation strategies. This work provides insight into the PEGylation of VNPs, which may improve the possibility of their implementation in clinical applications.
Nanoparticles (NPs) are emerging as promising tools in drug delivery and diagnostic applications, overcoming the limitations of small-molecule therapeutics or contrast agents. Free drug molecules or contrast agents are limited by their passive diffusion through circulation, giving rise to off-target adverse side effects and limiting the achievement of efficacious drug dosages (1). NPs help to overcome these limitations by facilitating delivery to selective cell types, improving cargo stability and solubility, and prolonging circulation times, thus enhancing the efficacy and safety of free drugs (2). NPs have been designed to carry and deliver large payloads of small molecules (3–5), proteins (6, 7), including antigens (8, 9), and imaging agents (10, 11) in a variety of indications including but not limited to diabetes (6, 12), cancer (13–15), and Alzheimer’s disease (16). The co-attachment of targeting agents onto NPs is currently being explored to enhance cell- and tissue-specific delivery (17–19).
Though there are promising NPs in the developmental pipeline, systemically delivered NPs must overcome biological barriers to successfully deliver their payloads to their target site. The mononuclear phagocytic system (MPS) is composed of monocytes, tissue-resident macrophages, granulocytes, and dendritic cells. The system serves as a critical immune defense mechanism. The MPS clears and degrades viruses, pathogens, and NPs (20). In circulation, NPs (and pathogens) are opsonized by antibodies, complement proteins, and other serum proteins, which leads to a protein corona to tag these materials for phagocytosis by cells in the MPS (21). Thus, efficacious shielding methods must be utilized to evade such biological barriers for improved systemic delivery.
One major approach to particle shielding is coating the NPs or other therapeutic agents with polyethylene glycol (PEG), termed PEGylation. PEGylation has been demonstrated to impart “stealth” properties, such as increasing systemic circulation time, decreasing serum protein adsorption, and reducing interactions with cells of the MPS (22–24). The shielding ability of PEG arises from its molecular and physical properties. PEG, an inert and exceedingly hydrophilic polymer, forms a hydration shell with a large, excluded volume that sterically hinders blood components from interacting with the NP core (25). Furthermore, the high levels of flexibility and mobility of PEG chains grafted onto the surface of NPs render interactions between the NP and circulating biomacromolecules entropically unfavorable, as they would reduce the conformational freedom of the PEG chains (25). The success of PEGylation has led to over 10 FDA-approved PEGylated drugs to date, such as Janssen’s liposomal doxorubicin (Doxil®) and Merrimack’s liposomal irinotecan (Onivyde®)—both nanoparticle formulations are used for cancer therapy (26, 27). A wide variety of PEGs with molecular weights (MWs) ranging from < 1 to 40 kDa in linear or branched conformations have been included in the list of FDA-approved PEGylated drugs (26).
In this work, we examined tobacco mosaic virus (TMV), a plant virus nanoparticle (VNP), as a nanocarrier platform. VNPs are highly monodisperse nanoparticles, which are produced by fermentation or molecular farming. Plant VNPs are non-infectious to humans. Their high degrees of stability, biocompatibility, and biodegradability make VNPs an attractive alternative to synthetic nanotechnologies. Specifically, the TMV is a rigid, rod-shaped plant virus measuring 300 nm in length and 18 nm in width and consists of 2,130 identical coat proteins that self-assemble around a single-stranded, positive-sense RNA molecule. Key features of TMV are its high aspect ratio and its anisotropic shape, which confers different advantages over spherical counterparts. Rod-shaped and filamentous structures have been demonstrated to impart tumor-homing properties by enhanced margination to vessel walls and increased penetration into tumor tissues compared with nanospheres (28). High-aspect-ratio nanoparticles also present prolonged circulation times because their shape better evades macrophages (29, 30). Anisotropic nanocarriers provide a higher surface area to increase drug loading and delivery while also improving cell binding and target cell interactions (31). However, manufacturing high-aspect-ratio particles precisely and consistently at the nanoscale is technically demanding when using synthetic materials. Carbon nanotubes (32) and gold nanorods (33) have been developed, but they are restricted by their cytotoxicity; silica nanorods (34) and polymeric filomicelles (31) are more biocompatible, but the preparation of well-ordered structures proves to be difficult. TMV as a nanocarrier successfully addresses these shortcomings because its biological nature renders it as having high monodispersity, biocompatibility, and manufacturing scalability. Furthermore, TMV is unique because its length can be precisely tailored through its RNA cargo, and complex shapes can be directed by RNA-controlled assembly (35, 36).
TMV has been harnessed for chemotherapy, photothermal immunotherapy, and the delivery of thrombolytics, as well as for bioimaging and theranostic applications (37–41). The inner and outer surfaces of TMV are amenable to genetic manipulation or chemical conjugation via available tyrosine and glutamic acid residues (42). The well-established T158K mutant (TMVLys) yields addressable, solvent-exposed lysine residues for further functionalization (43, 44). Although TMV proves an appealing nanocarrier for biomedical applications, the fact that the half-life of the “naked” TMV is 3.5 min in mice and that pre-existing anti-TMV antibodies are found in humans necessitates shielding strategies (45, 46). To address this, we developed and characterized TMV-PEG constructs that considered PEG chains of various lengths and conformations, particularly linear and multivalent PEGs with molecular weights of 2,000 and 5,000 Da. We thoroughly examined the conjugation efficiencies of each PEG onto the surface of TMV and determined to which degree intraparticle cross-linking occurred as a function of PEG valency and length. We investigated how linear vs. multivalent PEGylation affects TMV’s biological properties such as particle stability, antibody evasion, protein corona formation, and interactions with immune cells.
The tobacco mosaic virus was propagated by the mechanical inoculation of Nicotiana benthamiana leaves and purified as previously described to yield ≈ 1 mg of virus per 1 g of infected leaf material (42). For all described studies, the well-characterized T158K mutant of TMV, TMVLys, was used and for simplicity is referred to as TMV (44).
Linear, bivalent, and four-arm PEGs with molecular weights of 2,000 Da and 5,000 Da (Nanocs Inc.) were externally grafted onto TMV particles using N-hydroxysuccinimide (NHS)-activated esters. PEGs with MWs of 2,000 and 5,000 Da were dissolved in dimethyl sulfoxide (DMSO) at 250 and 350 mg mL−1, respectively, and added to TMV at 10, 20, 40, and 60 equivalents per coat protein (CP). The mixtures were allowed to agitate overnight at room temperature (RT). The reactions were carried out at 2 mg mL−1 of TMV in 0.01 M potassium phosphate (KP) buffer at pH 8.0, with a final DMSO concentration not exceeding 10% (v/v; volume to volume). TMV-PEG nanoparticles were purified by ultracentrifugation at 52,000 rpm using a TLA-55 rotor (Beckman Coulter) for 1 h on a 30% (w/v; weight to volume) sucrose cushion. The pellets were resuspended in 0.01 M KP buffer at pH 7.0 and stored at 4°C. The conjugation efficiency of each PEG per TMV particle was quantified by a densitometric analysis of protein bands in denaturing protein gels after staining with GelCode Blue Stain reagent (Thermo Fisher Scientific) using ImageJ software.
To prepare fluorescent TMV formulations for imaging, the remaining free lysine residues on TMV and TMV-PEG nanoparticles were labeled with NHS-activated esters of sulfo-Cy5 (Lumiprobe). Sulfo-Cy5 at 1.8 and 3 equivalents per CP were added to 2 mg mL−1 TMV and TMV-PEG particles, respectively, in 0.01 M KP buffer at pH 8.0 containing 10% (v/v) DMSO. The reactions were carried out in the dark with agitation overnight at RT and purified via ultracentrifugation. Excess dye was further removed using a 100-kDa molecular weight cut-off (MWCO) centrifugal filter unit (Millipore) until a clear flow through was achieved. Dye loading per particle was quantified using UV/visible spectroscopy and the specific extinction coefficient for sulfo-Cy5 (ϵ647 nm = 271,000 L mol−1 cm−1).
Protein concentrations were determined using a NanoDrop 2000 spectrophotometer (Thermo Scientific). The concentration of TMV in each sample was calculated using the Beer–Lambert law and the extinction coefficient of TMV at 260 nm (3 mg−1mL cm−1). A ratio of 260 to 280 nm and an absorbance of ≈ 1.2 was used to confirm structural integrity of TMV and TMV-PEG particles.
TMV and TMV-PEG particles (10 μg) were denatured in a gel loading buffer (1 X lithium dodecyl sulfate sample buffer, Life Technologies, with 1 X NuPAGE sample reducing agent, Invitrogen, in 0.01 M KP buffer) and boiled at 100°C for 5 min. Protein samples and standards (SeeBlue Plus2 pre-stained protein standards, Invitrogen) were loaded on 4%–12% or 12% NuPAGE gels (Thermo Fisher Scientific) and run in a 1 X MOPS buffer at 200 V and 120 mA for 35 min. Protein bands were visualized using GelCode Blue Stain reagent (Thermo Fisher Scientific) and imaged under a FluorChem E system (ProteinSimple). Cy5-labeled TMV and TMV-PEG particles were analyzed and imaged under the 620/40 nm red light on the FluorChem E system.
The hydrodynamic diameter and ζ-potential of TMV and TMV-PEG particles were measured using a Zetasizer Nano ZSP Zen 5600 instrument (Malvern Panalytical) and a scattering angle of 90°. The samples were diluted to 1 mg mL−1 in 0.01 M KP and deionized water for DLS and ζ-potential, respectively, and run at RT.
TMV and TMV/PEG particles (15 μL at 0.5–1 mg mL−1) were negatively stained with 2% (v/v) uranyl acetate (Electron Microscopy Sciences) for 1.5 min on a Formvar/carbon-coated copper grid (Electron Microscopy Sciences). Samples were visualized using a JEOL 1400 Plus transmission electron microscope at 120 kV.
Three μL of a rabbit polyclonal α-TMV antibody (Agdia), a rabbit polyclonal α-CPMV antibody (Pacific Immunology) at 50 μg mL−1, and a rat monoclonal α-PEG antibody (BioVision) at 100 μg mL−1 were spotted on a nitrocellulose membrane. The blots were briefly dried and blocked in 5% (w/v) bovine serum albumin (BSA) (Sigma-Aldrich) in PBS overnight at 4°C. The blots were then washed three times for 5 min each in PBS and incubated with 5 μg mL−1 Cy5-labeled particles in 5% (w/v) BSA in PBS for 2 h at RT. After the blots were subsequently washed in PBS-T three times for 15 min each and a final wash in PBS for 5 min, the blots were dried and imaged for fluorescence using a FluorChem E system (ProteinSimple).
To form hard protein coronas, TMV and TMV-PEG particles were incubated in 2 mL 100% human plasma (Sigma-Aldrich) in dH2O at 0.3 mg mL−1 VNPs for 1 h at RT with agitation. The samples were diluted in 18 mL PBS and purified by ultracentrifugation at 42,000 rpm using a 50.2 Ti rotor (Beckman Coulter) for 2.5 h on a 40% (w/v) sucrose cushion. The pellets were washed twice with 20 mL PBS to remove loosely bound proteins. The samples were denatured in 100–150 μL gel loading buffer, and proteins were separated and visualized by SDS-PAGE, as previously mentioned.
RAW 264.7 murine macrophages (ATCC) in DMEM (Corning) containing 10% (v/v) FBS and 1% (v/v) penicillin/streptomycin at 37°C and 5% CO2 were grown to confluency and seeded into T-25 flasks at 1.5 × 106 cells per flask and allowed to adhere overnight. For the TMV–cell interaction assay, 500,000 VNPs cell−1 were added to each flask and incubated for 24 h at 37°C and 5% CO2. To remove free VNPs, the cells were washed with PBS. The cells were gently scraped and spun down at 500 × g for 5 min, resuspended in PBS, and transferred to a 96-well v-bottom plate. The cells were spun down at 300 × g for 5 min between each of the following steps. Cells were washed again in PBS and then fixed in 100 μL 4% (v/v) paraformaldehyde (PFA) (Electron Microscopy Sciences) in PBS for 10 min at RT. Following fixation, the cells were washed twice in PBS and resuspended in 200 μL FACS buffer (2% (v/v) FBS, 0.09% (w/v) sodium azide (Sigma-Aldrich) in PBS). Cells were analyzed using a BD Accuri C6 Plus Flow Cytometer and 50,000 events were collected. Data were analyzed using FlowJo v10.8.1 software. All experiments were done in triplicate.
To gain insight into whether TMV or TMV-PEG particles are membrane-bound or internalized by macrophages, confocal microscopy was performed. RAW 264.7 murine macrophages were seeded at 1 × 105 cells mL−1 on 18 mm glass coverslips in a 12-well plate and allowed to attach overnight in Dulbecco’s modified Eagle medium (DMEM, Corning) containing 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin/streptomycin at 37°C and 5% CO2. Then, 500,000 VNPs cell−1 were added to each well and incubated for 24 h at 37°C. The cells were then fixed with 5% (v/v) PFA (Electron Microscopy Sciences) for 10 min at RT. Five per cent (v/v) goat serum albumin (GSA) was used to dilute all staining agents. Following fixation, cell membranes were stained with 1:1,000 Alexa Fluor 555-conjugated wheat germ agglutinin (WGA, Invitrogen) for 20 min at RT. The cells were then permeabilized with 0.2% (v/v) Triton X-100 for 2 min followed by blocking in 10% (v/v) goat serum in PBS for 1 h at RT. TMV particles were stained using 1:500 rabbit polyclonal α-TMV antibody (Agdia) for 2 h at RT and 1:2000 Alexa Fluor 647-conjugated goat anti-rabbit secondary antibody for 1 h at RT. The cells were washed three times in PBS between each step. Coverslips were mounted onto slides using Fluoroshield with DAPI (Sigma-Aldrich) and imaged using a Nikon A1R confocal microscope with a 100X oil objective. Images were analyzed using Nikon NIS-Elements software.
TMV is a proteinaceous high-aspect-ratio 300 nm × 18 nm nanorod composed of 2,130 identical coat proteins with a molecular weight of 17.5 kDa (42). For this study, TMVLys was used; TMVLys is a well-characterized T158K TMV mutant, shown in Figure 1A, that has solvent-exposed lysine side chains for chemical addressability. Linear and multivalent PEGs and fluorescent dyes (Sulfo-Cy5) were conjugated to TMVLys using N-hydroxysuccinimide (NHS) activated esters (Figure 1B).
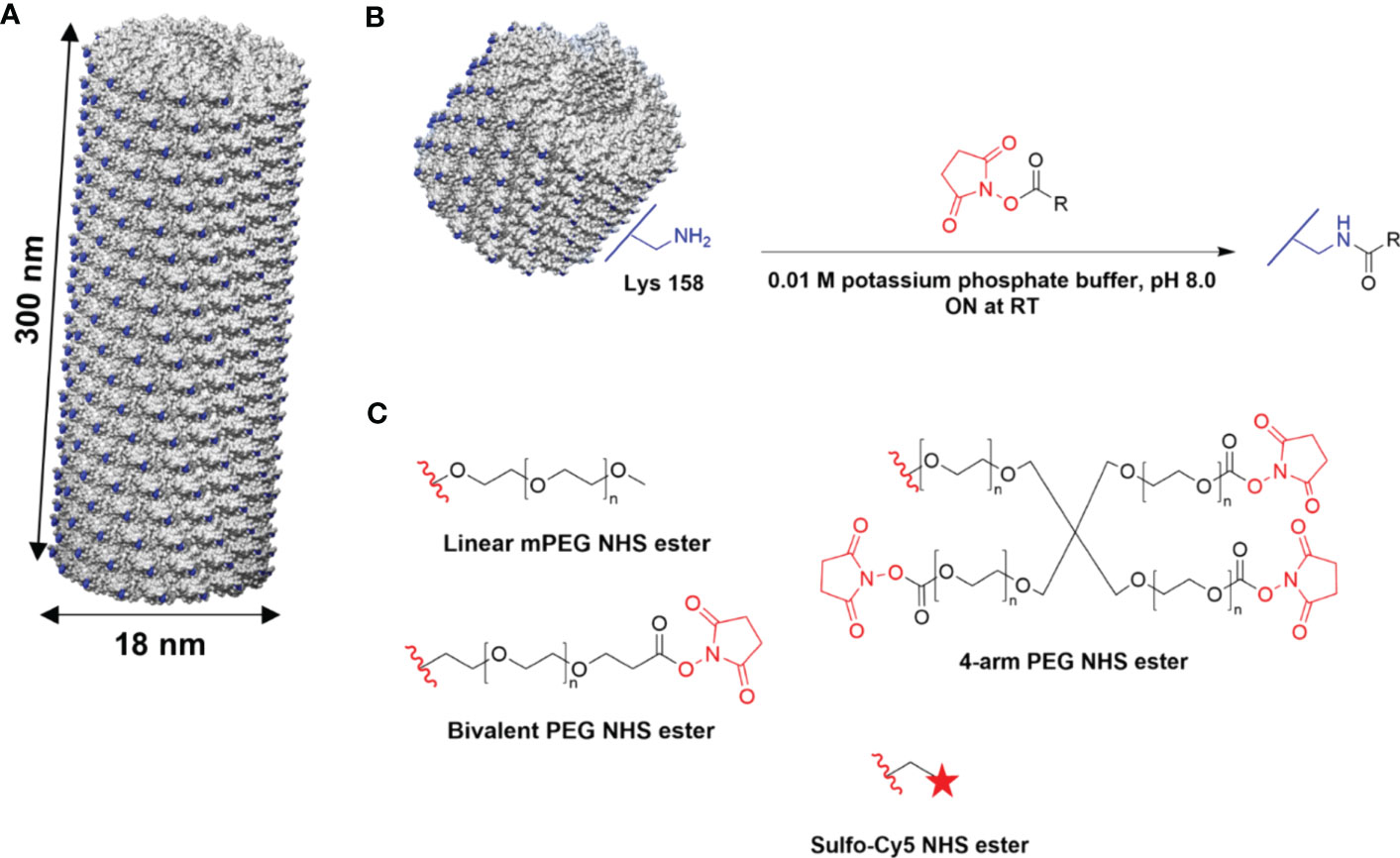
Figure 1 Chemical modification of TMV. (A) Atomic model of TMVLys, a T158K mutant of TMV. One virion comprises 2,130 identical coat proteins; solvent-exposed lysine residues shown in blue. Atomic models were created on UCSF Chimera (47). (B) Schematic of external conjugation of PEGs and fluorescent dyes to lysine residues using NHS-activated esters. R denotes the target molecule. (C) Conjugation reagents (Linear, Bivalent, and four-arm PEG, Sulfo-Cy5); the red wavy line denotes the leaving group. ON, overnight.
To explore PEG length and valency and their role in shielding from immune recognition, linear, bivalent, and four-arm PEGs of MW 2,000 or 5000 Da were conjugated to TMV (Figure 1C). While linear PEG coatings on TMV have been reported, bivalent and four-arm PEG coatings have not yet been explored (48). The multivalent PEGs have multiple NHS handles that can interact with TMV coat proteins—intraparticle cross-linking can lead to a denser packing closer to the surface of TMV. We hypothesized that such denser coatings would provide benefits in shielding, as was shown previously for potato virus X (PVX) (49). Furthermore, multi-arm PEG coatings have been demonstrated to avoid the accelerated blood clearance (ABC) phenomenon and produce fewer anti-PEG IgM levels than their linear counterparts (50, 51). By the same token, we aimed to parse out whether the multivalent PEG coatings on the surface of TMV could achieve more efficacious particle shielding.
To maximize PEG grafting density on TMV, conjugations using molar excesses of 10:1, 20:1, 40:1, and 60:1 PEG to CP ratios were tested. PEGylation to TMV was quantified by SDS-PAGE band densitometry analysis from three independent experiments (Figure 2A), and a representative SDS-PAGE gel of the TMV-PEG formulations is shown in Figure 2B (for additional gels see Figure S1). TMV-PEG formulations were abbreviated as follows: 2L = TMV-PEG2K-Linear; 2B = TMV-PEG2K-Bivalent; 2R = TMV-PEG2K-4arm; 5L = TMV-PEG5K-Linear; 5B = TMV-PEG5K-Bivalent; and 5R = TMV-PEG5K-4arm. SDS-PAGE shows the characteristic TMV CP band at ≈ 19 kDa (Figure 2B); it is notable that SDS-PAGE does not have the resolution to resolve proteins at their exact molecular weight, which explains why the observed and theoretical molecular weights differ. Higher molecular weight bands indicated the successful attachment of PEG to TMV coat proteins. PEG conjugation to CP monomers resulted in bands at ≈ 22 kDa and ≈ 28 kDa, for PEG 2,000 and PEG 5,000 formulations, respectively. Any additional bands of higher molecular weight were considered to be PEGylated dimers or PEGylated multimers, i.e., CPs covalently cross-linked with PEG, PEGylated CPs entangled with non-PEGylated CPs, PEGylated CPs entangled with PEGylated CPs, and mixtures thereof. Particularly for the 5B construct, a band corresponding to non-PEGylated CP dimers was detectable, which was most likely due to the entanglement of two neighboring coat proteins—this is a consistent phenomenon and has been observed before (52–54). Bands ranging from ≈ 40 to 45 kDa were present for the PEG 2,000 and multivalent 5,000 formulations, which we considered to be PEG-interlinked dimers. Bands indicating PEGylated multimers (> ≈ 57 kDa) could also be observed for the four-arm formulations, which was expected as these PEGs had four NHS handles to react with up to four TMV coat proteins leading to more complex structures and CP entanglement. We noted that bands indicating PEGylated tetramers in the 5R formulation was not present in Figure 2B but was shown faintly in gels used to calculate PEG grafting (Figure S1); this discrepancy is likely explained by the incomplete staining of the gel in Figure 2B.
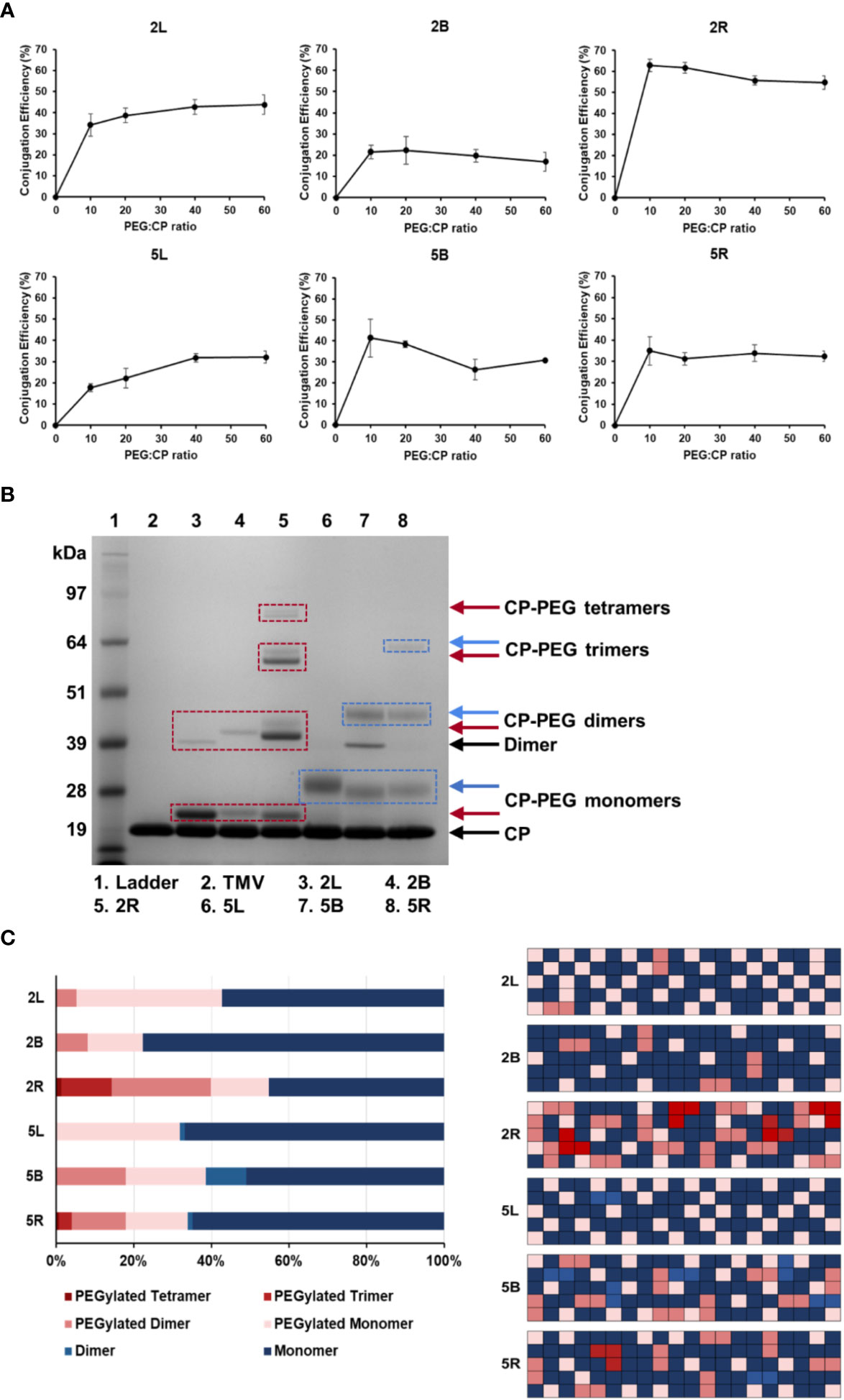
Figure 2 PEGylation of TMV nanoparticles. (A) Quantification of PEG grafting to TMV CPs as a function of molar excess used. Values were averaged from three independent experiments; error bars represent the standard deviation. (B) Representative SDS-PAGE gel of the PEGylated TMV particles. Molecular weight bands above the CP band (the TMV CP was ≈ 17.5 kDa and runs at ≈ 19 kDa in the gel; SDS-PAGE is a size approximation, and the band position varies by gel percentage and buffers used) and dimer bands indicate PEG 2K (red boxes) or PEG 5K (blue boxes) conjugation. (C) Percentage of TMV that was grafted vs. ungrafted for each TMV-PEG formulation (n = 3) quantified by densitometry analysis (left); images were analyzed using ImageJ software using a band analysis tool. Graphical representation of percentage of TMV that was grafted vs. ungrafted assuming 100 TMV coat proteins and random distribution of PEG (right); this was to visualize the PEG density on the TMV rod. TMV-PEG formulations were abbreviated as following: 2L, TMV-PEG2K-Linear; 2B, TMV-PEG2K-Bivalent; 2R, TMV-PEG2K-4arm; 5L, TMV-PEG5K-Linear; 5B, TMV-PEG5K-Bivalent; and 5R, TMV-PEG5K-4arm.
Here, conjugation efficiency was defined as the percentage of TMV CPs labeled by PEG. For linear PEG 2,000 and 5,000, the conjugation efficiency plateaued near 43% and 32%, respectively, at a molar excess of 40:1 CP (Figure 2A). With 2,130 TMV coat proteins, this approximated to 920 and 680 linear PEG 2,000 and PEG 5,000 conjugated per particle, respectively. For the multivalent PEGs, maximum PEG grafting was achieved at a molar excess of 10:1 CP as PEGylation dropped or plateaued at higher molar excesses. We speculate that conjugation efficiency may decrease at higher molar excesses, particularly for the multivalent PEGs, due to increased steric hindrance and lack of available space for free NHS groups to react with neighboring coat proteins. Overall, it should be noted that 10:1 to 20:1 excess of PEG to CP was sufficient to reach maximum labeling; only the 5L formulation required a higher excess of PEG. This may be explained by the longer and linear PEG chain being more entangled, therefore making the NHS reactive group less accessible.
For the remainder of this study, optimized protocols for PEG labeling (see Methods) were used. Each TMV-PEG formulation had a coverage of at least ≈ 20% PEG conjugation, with 2R having the highest percent modification at ≈ 55% (1,170 PEGs/TMV). PEG grafting was probably limited by the effects of steric hindrance, and our results are in good agreement with Lee et al.’s estimation of 25%–50% PEG conjugation efficiency onto PVX, another high-aspect-ratio plant virus, in which there was estimated to be one grafted PEG for every three lysine residues (49).
Interestingly, the 5B formulation had ≈ 16% higher labeling than its PEG 2,000 counterpart (Figure 2C): 5B averaged ≈ 810 PEG per TMV vs. ≈ 470 PEG per TMV for 2B, particularly forming a higher number of PEGylated dimers (≈ 100 more for 5B than 2R). This could possibly be explained by PEG 5,000 having longer ‘arms’ to bypass the steric hindrance of the initial attachment site and link a distal coat protein. Considering the ‘stretched out’ PEG, 5B displayed an arm length of 39 nm vs. 16 nm for 2B. The Flory dimension (RF) of 5B was estimated to be 5.9 nm vs. 3.4 nm for 2B. The 300 × 18 nm TMV rod has a surface area of ATMV = 17,465 nm2 with each of the 2,130 CPs providing a surface area of ACP = 8.2 nm2. Assuming the TMV CP was a square, the distance between two lysine residues was estimated to be 2.9 nm. The bivalent PEG 5,000 formulation provided a longer arm length and larger RF that could better facilitate interactions with neighboring lysine residues from the initial PEG attachment site, contributing to higher numbers of interlinked CP dimers. However, in the case of the four-arm PEGs, 2R had markedly higher labeling than 5R in which more PEGylated dimers and multimers were formed. Although 5R had longer arms, we speculate that the conformation of 5R may be more susceptible to the effects of steric hindrance and lack of available space on TMV’s surface compared with 2R.
Purified TMV-PEG nanoparticles were characterized by ultraviolet-visible spectroscopy (UV-Vis), dynamic light scattering (DLS), zeta-potential measurements, and transmission electron microscopy (TEM) to confirm structural integrity. Using the UV-Vis spectra of the TMV-PEG formulations (Figure 3A), the 260 and 280 nm absorbances, corresponding to RNA and protein signals of the particles, respectively, were measured. A 260:280 nm ratio of 1.2 indicates pure, intact solution of TMV (42). Indeed, all formulations present a 260:280 nm ratio near 1.2. This was also consistent with the SDS-PAGE, where no protein contaminants were detectable.
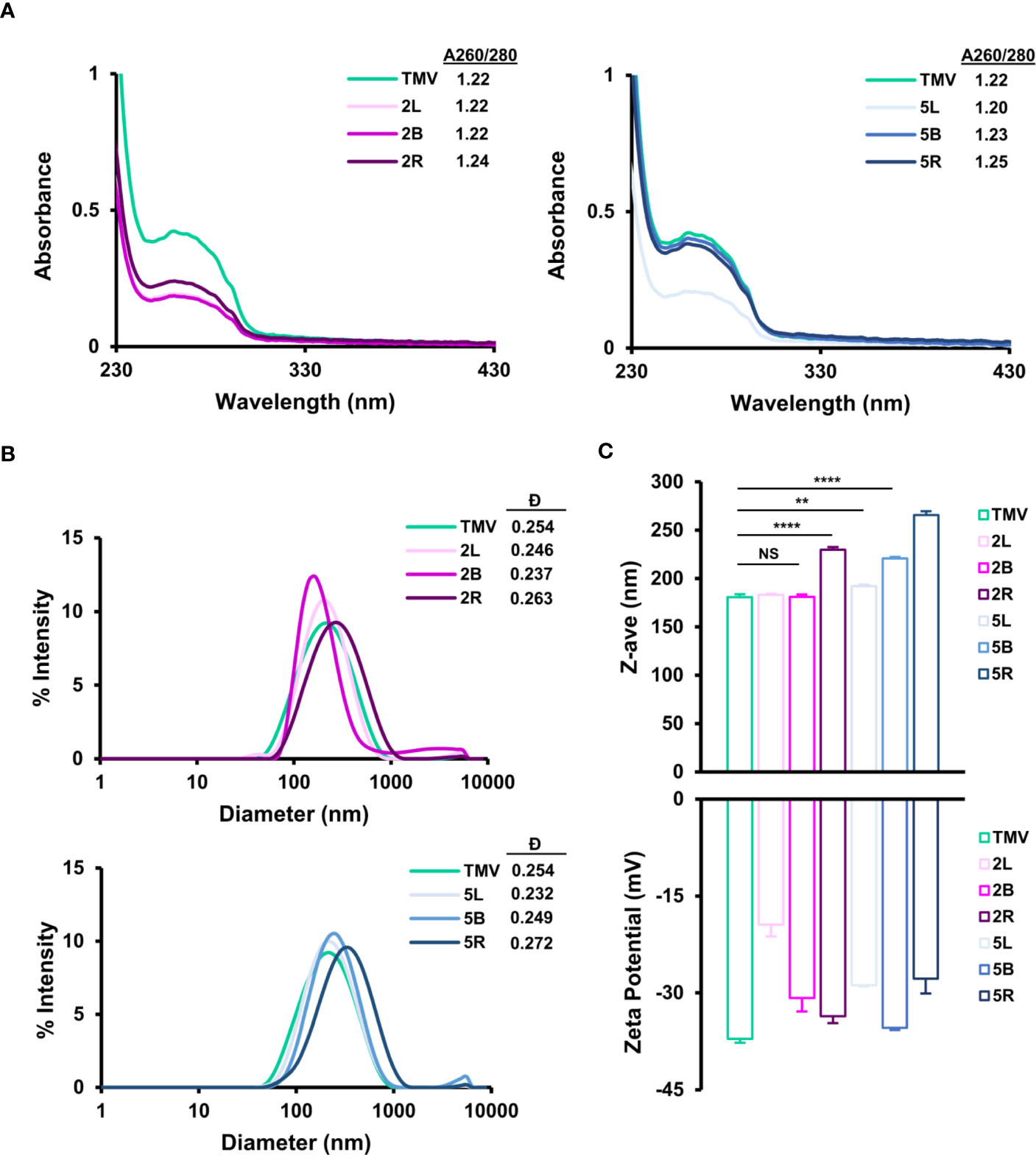
Figure 3 Characterization of TMV-PEG particles. (A) UV-Vis spectra and 260/280 nm absorbance ratios of TMV-PEG2K (left) and TMV-PEG5K (right) particles. (B) DLS spectra of TMV-PEG2K (top) and TMV-PEG5K (bottom) particles. (C) Z-average (top) and zeta-potential (bottom) measurements for each virus formulation. Statistical analyses were performed using one-way ANOVA testing with Tukey’s multiple comparison test. **p < 0.01, ****p < 0.0001, NS, not significant.
The particles were then characterized by DLS (Figures 3B, C); it is important to note that DLS is not accurate for sizing of high-aspect-ratio materials but is useful to analyze relative trends. The z-average of the particles ranged from 181 nm for uncoated TMV to up to 266 nm for the 5R formulation. DLS measurements confirmed a significant increase in size for the 2R, 5L, 5B, and 5R formulations. The most significant size increase was observed for 2R, 5B, and 5R (p < 0.0001). It is plausible that longer and multivalent PEG grafting altered the surface structure of TMV such that its translational diffusion speed was reduced resulting in a larger hydrodynamic diameter (55); increases in hydrodynamic diameter upon PEGylation (MW 5,000) on nanoparticles have also been reported (56). All TMV-PEG formulations were found to be monodispersed: dispersity values ranged from 0.232 to 0.272, with negligible aggregation, as shown in the DLS spectra (Figure 3B). TEM imaging further supports the DLS data as no apparent aggregation was observed for any of the formulations (Figures S2, S3).
Zeta-potential measurements indicated that TMV (as a reminder, for this study TMVLys was used) exhibits a surface charge of –37 mV (Figure 3C); it seems that the true zeta-potential of TMVLys remains ambiguous as other studies have reported a surface charge of ≈ –60 and –14.5 mV, and there may have been differences in experimental procedures such as particle concentration and solutions (57, 58). Thus, zeta-potential measurements here were used to compare relative trends only. For all formulations, PEGylation led to a less negative zeta-potential, demonstrating a reduction in overall negative surface charge consistent with other reports (59, 60); this can be explained by PEG coatings shifting the position of the slipping/shear plane outward from the particle surface resulting in a weaker electrostatic potential (61). Increases in PEG grafting density and length have been shown to reduce zeta-potential (61). It is also of note that grafting PEG chains onto lysine residues on TMV via NHS chemistry would theoretically remove the positive surface charge attributed by such lysine residues and as a result modify zeta-potential as well. The formulation with the greatest amount of PEGylation, 2R, did not lead to the greatest reduction in zeta-potential compared with native TMVLys. Thus, no clear trend was observed between PEG chain length or conformation on zeta-potential in Figure 3C. It is likely that PEG length and conformation and number of PEG labels play a role in zeta-potential modification and further experiments are necessary to probe these factors.
In addition to the structural characterization, we confirmed thermal and chemical stability of the TMV formulations; the PEG coatings did not impact the stability of TMV and all formulations remained structurally sound when stored in buffer mixtures for up to 1 week at 4°C to 37°C and in 50% (v/v) DMSO for up to 24 h (Figure S2).
A primary challenge to viral-based NP delivery methods is neutralization and opsonization by neutralizing antibodies, where virus-specific antibodies bind and biologically inactivate viruses and/or tag them for clearance by phagocytic cells, respectively. PEGylation of (viral) NPs can reduce immune recognition by antibodies (49, 62). To investigate whether and to what degree PEG coatings on TMV exhibit immune shielding, immuno dot-blots were performed. Immuno dot-blots were prepared by spotting α-TMV, α-CPMV, and α-PEG antibodies in triplicate on a nitrocellulose membrane. α-Cowpea mosaic virus (CPMV) antibodies were used as a negative control as there are no structural similarities between CPMV and TMV (52). Upon blocking the membranes with 5% (w/v) BSA, fluorescent Cy5-labeled TMV and TMV-PEG particles were incubated; the characterization of dual-labeled Cy5-TMV-PEG particles are included in Figure S3. The membranes were then washed, dried, and analyzed (see Figure 4A). This method was preferred over blotting the nanoparticles directly onto the membrane followed by the addition of antibodies; the indirect capture via immobilized antibodies helped to avoid protein spreading on the nitrocellulose membrane, which may have induced conformational changes in PEG coatings. Antibody recognition was determined based on a fluorescence readout using a FluorChem E system and quantified by densitometry analysis of the blots using ImageJ (Figures 4B, C).
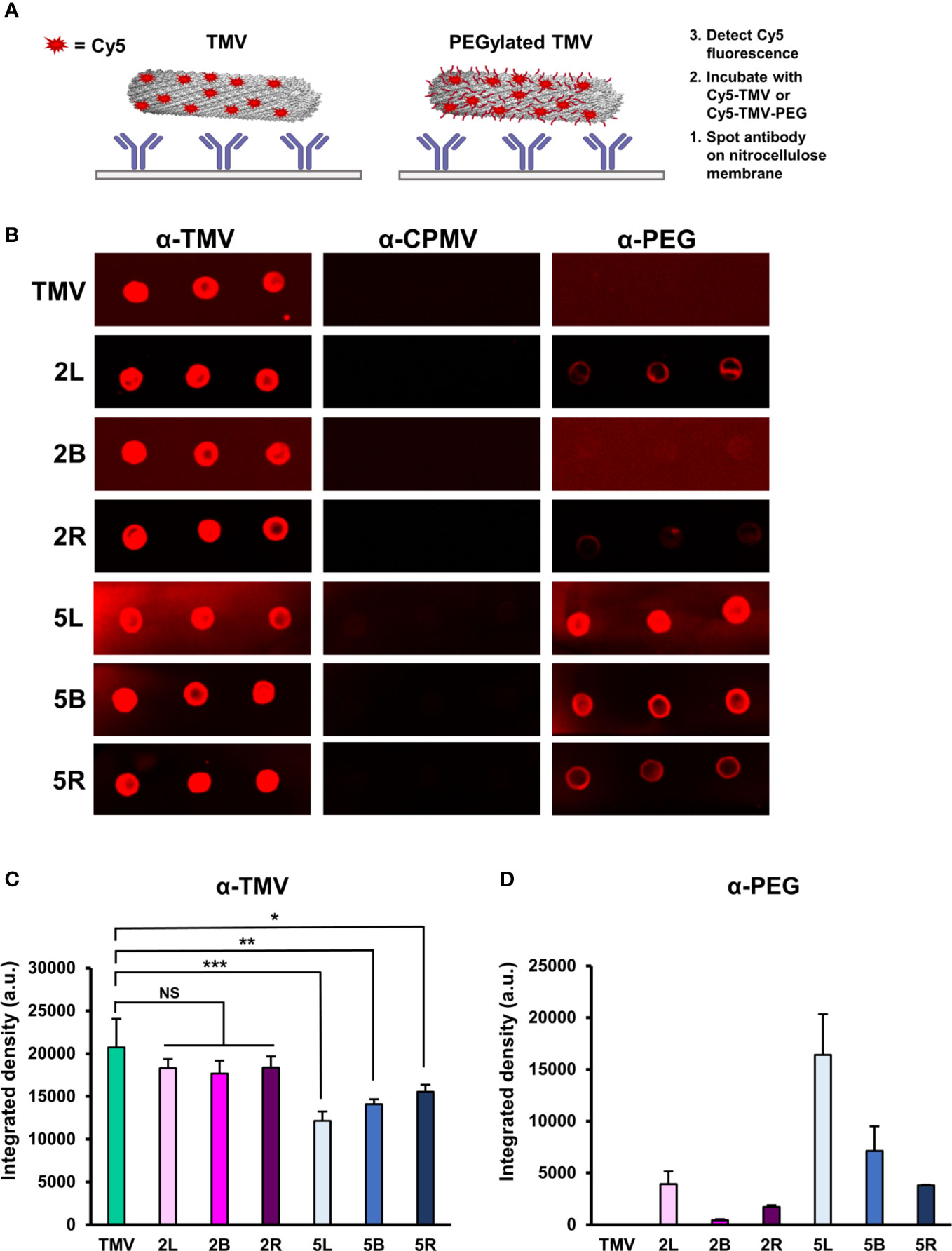
Figure 4 Immune recognition of TMV-PEG constructs. (A) Schematic of the immune dot-blot protocol to detect antibody recognition of TMV and TMV-PEG constructs. (B) Dot blots of α-TMV, α-CPMV, and α-PEG recognition of fluorescent TMV-PEG constructs. (C) Densitometry quantification of fluorescent TMV signal corresponding to antibody recognition by α-TMV antibodies from (B). (D) Densitometry quantification of fluorescent TMV signal corresponding to antibody recognition by α-PEG antibodies from (B). Statistical analyses were performed using one-way ANOVA testing with Tukey’s multiple comparison test. *p < 0.05, **p < 0.01, ***p < 0.001, NS, not significant.
As expected, PEGylation did not result in complete shielding from α-TMV antibody recognition, but successfully reduced antibody binding. Based on the densitometry analysis of the dot blots (Figure 4C), formulations of PEG 5,000 in any conformation, but not PEG 2,000, provided shielding benefits by reducing the binding of α-TMV antibodies to TMV. Data suggest that molecular weight generally plays a larger role in antibody shielding than conformation, but it is important to note that the statistical significance of antibody shielding decreased as multivalency in PEG 5,000 increased. It is also of note that a background Cy5 signal on the nitrocellulose membranes could be observed, especially for the 5L α-TMV blot. Background signal was not taken into account in the densitometry analysis (additional data are shown in Figure S4). The 2L and 2R presented the greatest grafting densities (≈ 40%–55%) yet did not impart α-TMV antibody shielding (Figure 2C, Figure 4C). Thus, the effects of PEG grafting density on α-TMV shielding remain elusive and merit further research. Further studies comparing PEG coatings at similar grafting densities or exploring PEGs with longer backbones are probably necessary to better understand and overcome antibody recognition of the nanoparticle.
Interestingly, α-PEG antibody recognition of the PEG 2,000 formulations was strikingly lower than the PEG 5,000 formulations (Figure 4D). This could probably be attributed to the α-PEG antibody used to detect the linear backbone of PEG, in which case the PEG 5,000 formulations had a ≈ 2.5-times longer backbone than PEG 2,000, allowing for better access for binding. This has implications in rising pre-existing α-PEG antibodies due to the extensive use of PEG in cosmetic products or accelerated blood clearance due to α-PEG antibody formation upon repeat administration of PEGylated therapies (63). The data suggest that α-PEG antibodies recognize shorter and multivalent PEG structures less readily (Figure 4D), building on other studies in which a PEG formulation of lower MW while also providing sufficient immune shielding of the NP would be ideal in clinically relevant applications (52). However, assessing α-PEG antibody binding via dot-blot analysis was a limitation due to its in vitro nature, and, thus, further research involving in vivo models would provide more insights on α-PEG antibody formation and antigenicity of these TMV-PEG constructs. Nonetheless, we recommend taking into account the potential antigenicity caused by the nanoparticle and PEG itself if PEGylation approaches are utilized.4
Next, we analyzed the protein corona. When nanoparticles enter the bloodstream, serum proteins readily recognize the NPs and adsorb to the surface, forming a protein corona. The protein corona inevitably plays a role in NP biological fate and may pose another biological barrier to delivery as the protein corona may specifically or non-specifically increase uptake of NPs in non-target cells, reduce circulation time, and weaken targeting capabilities of functionalized NPs (20). Previous work from our lab has demonstrated that the protein corona of plant virus nanoparticles presents an abundance of immune system proteins, such as complement proteins and immunoglobulins, which would likely contribute to VNP clearance (58). PEG grafting is known to reduce protein adsorption on the surface of NPs, as the hydrophilic nature of PEG facilitates a hydrated cloud that sterically hinders blood components (20). To test whether PEGylation of TMV reduces or alters the protein corona surrounding TMV, we incubated the particles in ≈ 100% human plasma and allowed the protein corona to form. Protein corona can be segregated into hard vs. soft corona, which is determined by the affinity of the adsorbed proteins on the nanoparticle surface; the soft corona is constituted by proteins that are loosely bound to the NP surface while proteins of the hard corona are tightly and often irreversibly bound (64). Upon protein corona formation, the samples were briefly washed to remove the soft corona (see Methods); only the hard protein corona was considered for analysis as the proteins directly adsorbed to the NP surface for prolonged periods and largely dictated the biological properties of the NP (58).
SDS-PAGE analysis confirmed that TMV indeed acquires a protein corona (Figure 5A) with profiles as previously published (58). PEG grafting did not eliminate protein adsorption; rather, each TMV-PEG formulation presented a slightly different protein corona composition. Notably, for the “naked” TMV, large aggregates were detected in the well of the gel. This aggregation was not apparent for the PEGylated TMV samples. Furthermore, the ≈ 70 kDa band has previously been identified as consisting of the complement protein C3/C4, plasminogen, and IgM (58); the protein corona of the 2L, 2B, 2R, and 5R did not contain this band, which suggests that PEGylation prevented adsorption of these serum proteins. While the protein corona of 5L and 5B seemed to be present in this ≈ 70 kDa band, it was at a much lower intensity than that of the corona of TMV.
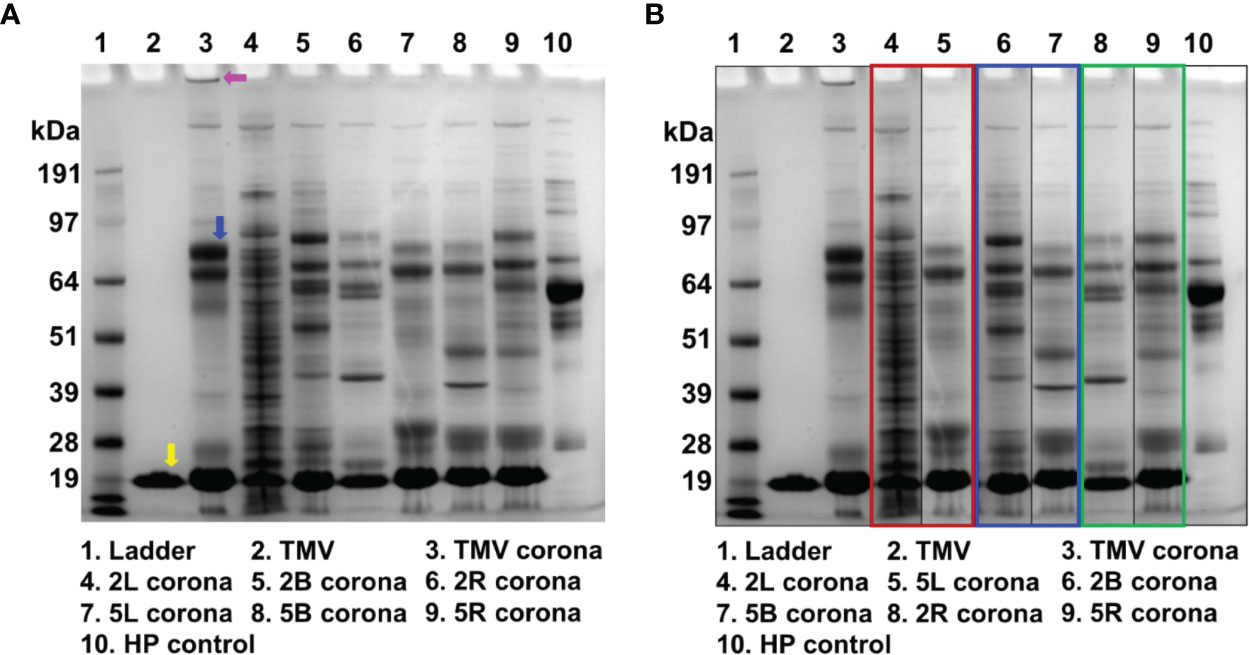
Figure 5 The protein corona of uncoated TMV and TMV-PEG constructs. (A) SDS-PAGE analysis of the protein corona formed around the VNPs after incubating the VNPs in ≈ 100% human plasma for 1 h at RT. The yellow arrow indicates the TMV coat protein. The pink arrow indicates the band of aggregation in the “naked” TMV protein corona. The blue arrow indicates the ≈ 70 kDa protein band (complement C3/C4, plasminogen, and IgM). (B) SDS-PAGE analysis of the gel in (A) organized by PEG multivalency: linear (red box), bivalent (blue box), and four-arm (green box). HP, human plasma.
In Figure 5B, the lanes are organized by PEG valency instead of molecular weight. The 2L and 5L’s corona composition widely differ, so it is likely that the conformation of these PEGs was distinct and affected the interaction with serum proteins. The protein corona of 2B and 5B do not pose striking differences. Interestingly, the protein corona of 2R and 5R are the most similar, implying that the Structure-functional relationships of these PEGs is comparable in serum protein interactions. While insightful, this protein corona study gives only a qualitative understanding of the protein corona of uncoated vs. PEGylated TMV. To gain a more quantitative understanding of protein corona abundance and composition for each virus formulation, mass spectrometry analysis would be required, as has been previously described (65, 66).
Lastly, we assayed the effects of PEGylation on TMV–immune cell interactions. Systemically administered NPs are often prematurely cleared by the mononuclear phagocytic system (MPS) (20); here, we used RAW 264.7 murine macrophages as a model system for phagocytic cells and compared uptake of “naked” and PEGylated TMV using flow cytometry and confocal microscopy. Cy5-labeled TMV was used and flow cytometry analysis confirmed that TMV is taken up by macrophages (Figure 6A). Interestingly, the histogram profile of TMV showed a main peak in the Cy5 signal, but a tail off the main peak was also present, signaling that there is heterogeneity in the amount of TMV that the macrophages interact with. We speculate that the presence of the tail could indicate differences in cellular localization or non-specific uptake of the particles. With the exception of the 2B formulation, PEGylation results in main peak shifted but also lost the tail (Figures 6A, C). This implied that PEGylation modulates TMV–immune cell interactions; while the PEG coatings seemed to promote the uptake of TMV in the main cell population, they also reduced the heterogeneity and overall amount of particle uptake as observed in mean fluorescent intensity values (Figure 6B). Surprisingly, the 2B construct resulted in no significant change in macrophage interactions and even its histogram and contour profiles (Figures 6A, C) resembled “naked” TMVs. This could be explained by insufficient PEG grafting as the 2B construct has the lowest grafting density at ≈ 20% (Figure 2C). Besides the 2B construct, no notable trends were observed between PEG conformation and macrophage interactions. The data highlight that there are nuanced structure–functional relationships in the PEG conformation that need to be elucidated.
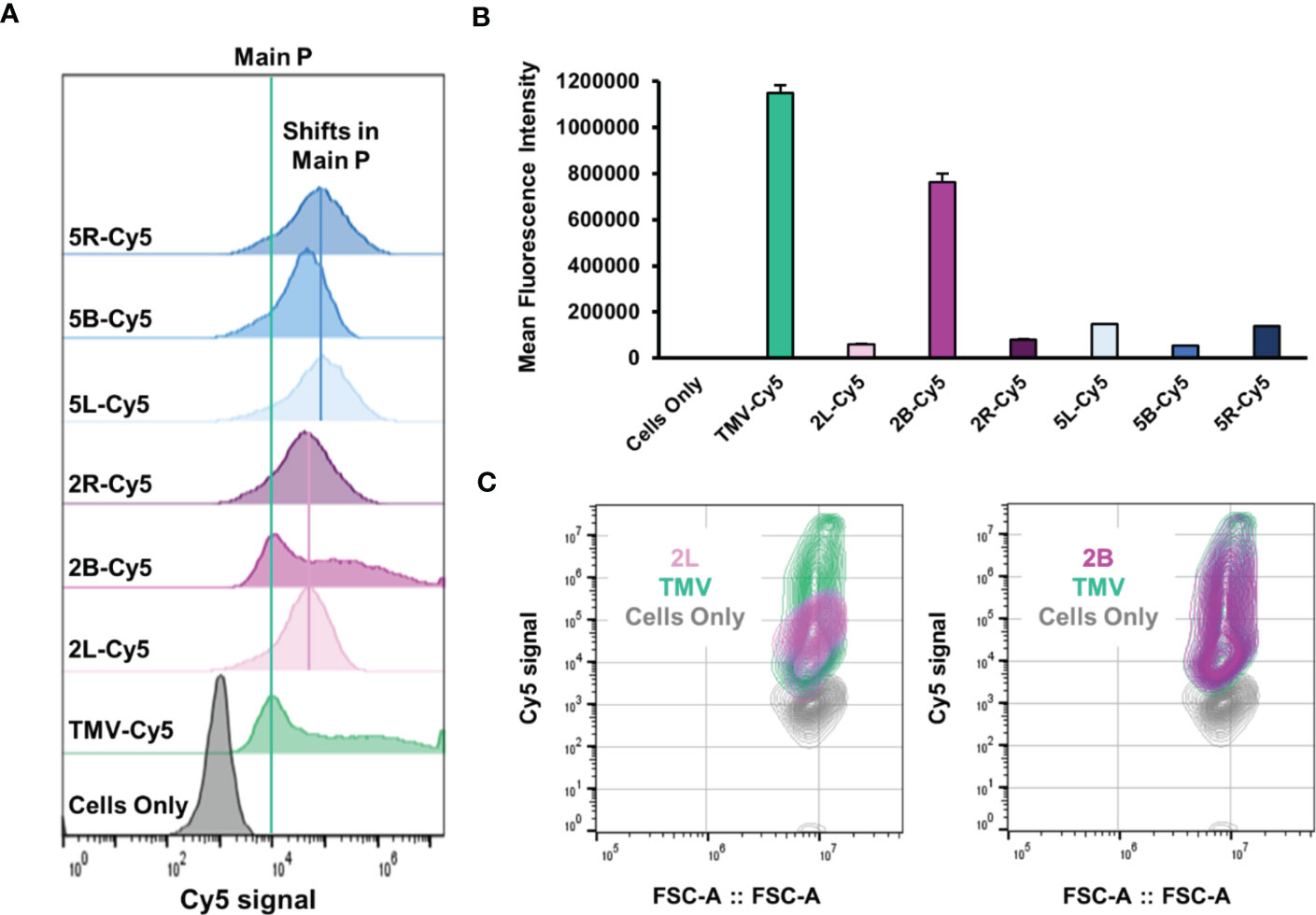
Figure 6 In vitro cellular interactions between TMV-PEG and RAW 264.7 macrophages. (A) Flow cytometry histograms of macrophage uptake of “naked” and PEGylated TMV constructs. The particles were tracked by conjugated Cy5 fluorophores. The green line indicates the main peak of Cy5 signal from TMV, while the pink and blue lines indicate peak shifts in PEG 2,000 and PEG 5,000 formulations, respectively. (B) Quantitative flow cytometry analysis of the same constructs in (A). (C) Contour plots of 2L uptake (left) and 2B uptake (right).
Next, to probe whether signal from the “naked” vs. PEGylated TMV came from internalized vs. cell membrane-bound particles, confocal microscopy was performed. The 2L and 2B constructs were considered for confocal microscopy because 2L, along with the remaining TMV-PEG constructs, modulated TMV–macrophage interactions but 2B did not, so we aimed to characterize any apparent differences between such formulations. The z-stack analysis of confocal microscopy images (Figure 7) showed that both “naked” TMV and PEGylated formulations are internalized by RAW 264.7 murine macrophages after 24 h post-incubation. Furthermore, cell surface binding is not specific to PEGylated particles as both native and PEGylated particles lodged into the cell membrane. Time course or time-lapse studies may elucidate differences in the rate of internalization between native vs. PEGylated particles; for example, PEGylated particles may stay lodged in the cell membrane longer than “naked” TMV before being internalized.
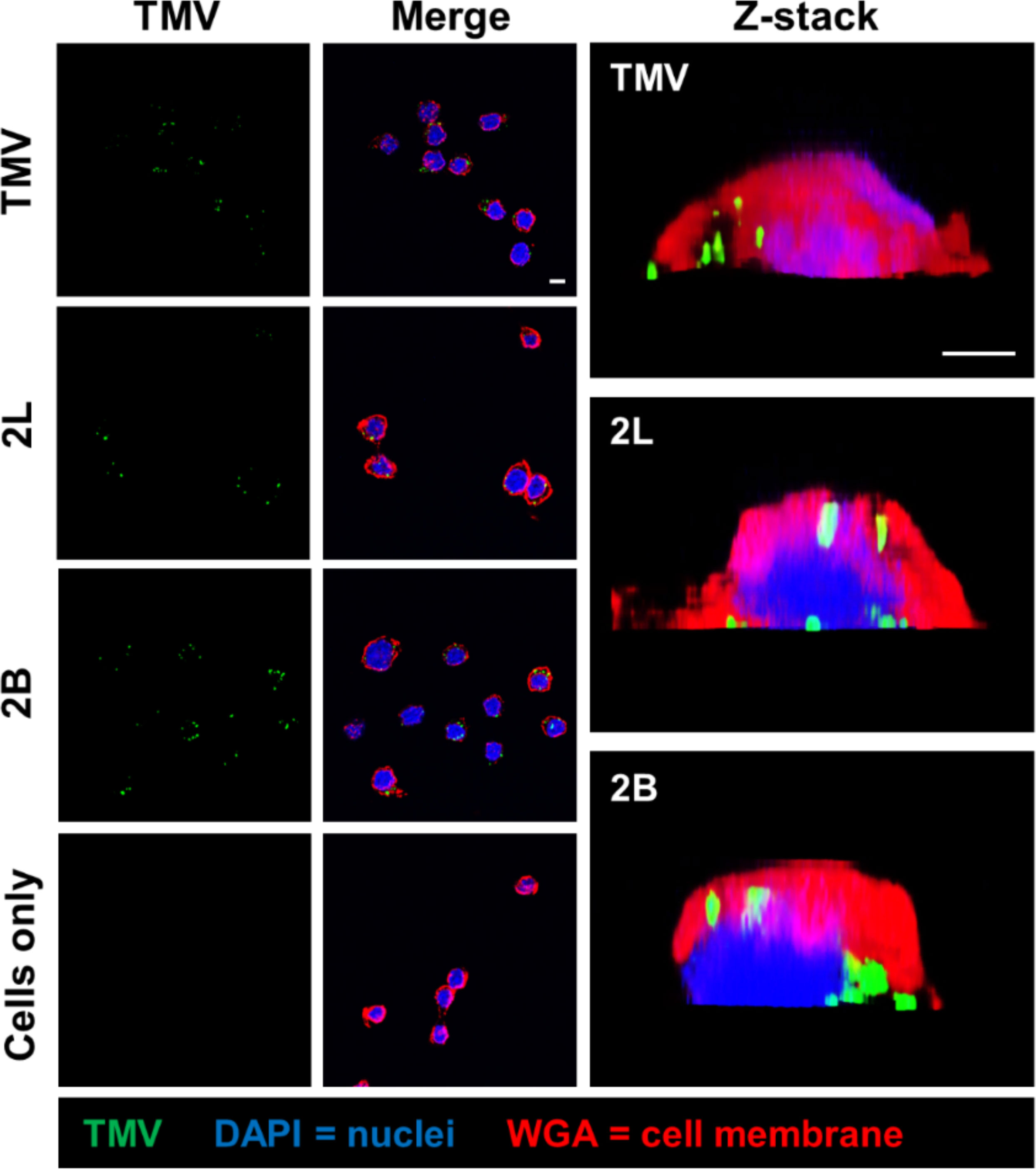
Figure 7 Cellular localization of TMV and TMV-PEG particles in RAW 264.7 macrophages. Confocal microscopy of TMV and TMV-PEG uptake by RAW 264.7 macrophages after 24 h. TMV is labeled in green, cell membrane marker WGA in red, and cell nuclei (DAPI) in blue. Z-stack cross-sections of RAW 264.7 cells are shown in the last column. Scale bars = 10 μm. Additional data are shown in Figure S5 (Supplementary Material).
In this work, we investigated linear vs. multivalent PEG coatings to overcome immune defense mechanisms against nanoparticle delivery platforms. We demonstrated successful PEGylation of TMV, a plant virus nanoparticle widely studied as a candidate material for drug delivery and therapeutic and bioimaging applications. Conjugation efficiencies of ≈ 20%–60% were achieved for linear, bivalent, and four-arm PEGs of MW 2,000 and 5,000 Da onto TMV, and we detailed the formation of higher order PEG-multimer structures upon multivalent PEG grafting. TMV-PEG formulations demonstrated a high degree of thermal and chemical stability with no apparent sign of broken particles or aggregation. PEG 5,000, but not PEG 2,000, coatings provided shielding against α-TMV antibody recognition in vitro, underscoring the importance of PEG chain length in NP stealthing. α-TMV antibody evasion decreased as PEG multivalency increased for the PEG 5,000 formulations. Shorter, multivalent PEG coatings preferentially prevented α-PEG antibody binding. Based on this study, we recommend that potential antigenicity attributed by the NP and PEG itself must be considered if PEGylation strategies are implemented in NP design. All PEG coatings reduced aggregation with serum proteins and yielded differences in the composition of their protein corona, particularly reducing the adsorption of complement proteins and IgM. Apart from the 2B construct, TMV-PEG constructs modulated interactions with macrophages. We report no obvious trends between linear vs. multivalent PEGylation in macrophage uptake and serum protein adsorption, highlighting that each PEG had distinct structure–functional relationships.
In summary, we developed linear and multivalent PEGylated plant virus nanoparticles in which PEG coatings contributed advantageous stealth properties. This work provides insight into NP shielding strategies and can be expanded to proteins and therapeutics. This study also contributes to the research and development of multivalent or branched PEG-grafted nanocarriers. Finally, efficient strategies for polymer conjugation open the door for using TMV and related virus-like particles as high-multivalent junctions in molecularly based polymer networks. The size and aspect ratio of TMV, in combination with the atomic precision of its structure, make it an attractive candidate for nanoscale analogs of fiber-reinforced composites and related microscale materials. The results obtained here provide a foundation for future investigations of the synthesis and properties of such materials.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
NS and SC conceptualized the study. RC, IG-G, and NS designed the study and experiments. RC conducted all experiments, analyzed the data, and wrote the manuscript with guidance from NS. All authors edited the manuscript. NS oversaw the study. All authors contributed to the article and approved the submitted version.
This work was supported by the NSF Center for the Chemistry of Molecularly Optimized Networks (MONET), CHE-2116298. RC was supported in part by the NSF Graduate Research Fellowship under grant no. DGE-2038238.
The authors appreciate Andrea G. Monroy-Borrego and Anthony O. Omole for their guidance on TMVLys propagation/purification and in vitro assays, respectively. The authors acknowledge the University of California, San Diego - Cellular and Molecular Medicine Electron Microscopy Core (UCSD-CMM-EM Core, RRID : SCR_022039) for equipment access and technical assistance. The UCSD-CMM-EM Core is supported in part by the National Institutes of Health Award number S10OD023527. Selected molecular graphics were created on Biorender.com.
NS is a co-founder of, has equity in, and has a financial interest with Mosaic ImmunoEngineering Inc. NS serves as Director, Board Member, and Acting Chief Scientific Officer, and paid consultant to Mosaic.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2023.1184095/full#supplementary-material
1. Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol (2015) 33:941–51. doi: 10.1038/nbt.3330
2. Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discovery (2021) 20:101–24. doi: 10.1038/s41573-020-0090-8
3. Guo F, Fu Q, Zhou K, Jin C, Wu W, Ji X, et al. Matrix metalloprotein-triggered, cell penetrating peptide-modified star-shaped nanoparticles for tumor targeting and cancer therapy. J Nanobiotechnol. (2020) 18:48. doi: 10.1186/s12951-020-00595-5
4. Rao L, Yu G, Meng Q, Bu L, Tian R, Lin L, et al. Cancer cell membrane-coated nanoparticles for personalized therapy in patient-derived xenograft models. Adv Funct Mat. (2019) 29:1905671. doi: 10.1002/adfm.201905671
5. Li K, Lu L, Xue C, Liu J, He Y, Zhou J, et al. Polarization of tumor-associated macrophage phenotype via porous hollow iron nanoparticles for tumor immunotherapy in vivo. Nanoscale (2020) 12:130–44. doi: 10.1039/C9NR06505A
6. Liu X, Li C, Lv J, Huang F, An Y, Shi L, et al. Glucose and h 2 O 2 dual-responsive polymeric micelles for the self-regulated release of insulin. ACS Appl Bio Mat. (2020) 3:1598–606. doi: 10.1021/acsabm.9b01185
7. Clegg JR, Irani AS, Ander EW, Ludolph CM, Venkataraman AK, Zhong JX, et al. Synthetic networks with tunable responsiveness, biodegradation, and molecular recognition for precision medicine applications. Sci Adv (2019) 5. doi: 10.1126/sciadv.aax7946
8. Knight FC, Gilchuk P, Kumar A, Becker KW, Sevimli S, Jacobson ME, et al. Mucosal immunization with a pH-responsive nanoparticle vaccine induces protective CD8 + lung-resident memory T cells. ACS Nano (2019) 13:10939–60. doi: 10.1021/acsnano.9b00326
9. Dölen Y, Valente M, Tagit O, Jäger E, van Dinther EAW, van Riessen NK, et al. Nanovaccine administration route is critical to obtain pertinent iNKt cell help for robust anti-tumor T and b cell responses. Oncoimmunology (2020) 9. doi: 10.1080/2162402X.2020.1738813
10. Xu C, Gao F, Wu J, Niu S, Li F, Jin L, et al. Biodegradable nanotheranostics with hyperthermia-induced bubble ability for ultrasound imaging–guided chemo-photothermal therapy. Int J Nanomed. (2019) 14:7141–53. doi: 10.2147/IJN.S213518
11. Pant K, Neuber C, Zarschler K, Wodtke J, Meister S, Haag R, et al. Active targeting of dendritic polyglycerols for diagnostic cancer imaging. Small (2020) 16:1905013. doi: 10.1002/smll.201905013
12. Volpatti LR, Matranga MA, Cortinas AB, Delcassian D, Daniel KB, Langer R, et al. Glucose-responsive nanoparticles for rapid and extended self-regulated insulin delivery. ACS Nano (2020) 14:488–97. doi: 10.1021/acsnano.9b06395
13. Gao Y, Jia L, Wang Q, Hu H, Zhao X, Chen D, et al. pH/Redox dual-responsive polyplex with effective endosomal escape for codelivery of siRNA and doxorubicin against drug-resistant cancer cells. ACS Appl Mat. Interfaces (2019) 11:16296–310. doi: 10.1021/acsami.9b02016
14. Zhang F, Parayath NN, Ene CI, Stephan SB, Koehne AL, Coon ME, et al. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat Commun (2019) 10:3974. doi: 10.1038/s41467-019-11911-5
15. Wei L, Zhao Y, Hu X, Tang L. Redox-responsive polycondensate neoepitope for enhanced personalized cancer vaccine. ACS Cent Sci (2020) 6:404–12. doi: 10.1021/acscentsci.9b01174
16. Zhang Y, Meng S, Ding J, Peng Q, Yu Y. Transition metal-coordinated graphitic carbon nitride dots as a sensitive and facile fluorescent probe for β-amyloid peptide detection. Analyst (2019) 144:504–11. doi: 10.1039/C8AN01620H
17. Cheng Q, Wei T, Farbiak L, Johnson LT, Dilliard SA, Siegwart DJ. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–cas gene editing. Nat Nanotechnol (2020) 15:313–20. doi: 10.1038/s41565-020-0669-6
18. Valcourt DM, Dang MN, Scully MA, Day ES. Nanoparticle-mediated Co-delivery of notch-1 antibodies and ABT-737 as a potent treatment strategy for triple-negative breast cancer. ACS Nano (2020) 14:3378–88. doi: 10.1021/acsnano.9b09263
19. Brown SB, Wang L, Jungels RR, Sharma B. Effects of cartilage-targeting moieties on nanoparticle biodistribution in healthy and osteoarthritic joints. Acta Biomater (2020) 101:469–83. doi: 10.1016/j.actbio.2019.10.003
20. Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Delivery Rev (2016) 99:28–51. doi: 10.1016/j.addr.2015.09.012
21. Tenzer S, Docter D, Kuharev J, Musyanovych A, Fetz V, Hecht R, et al. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat Nanotechnol (2013) 8:772–81. doi: 10.1038/nnano.2013.181
22. Klibanov AL, Maruyama K, Torchilin VP, Huang L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett (1990) 268:235–7. doi: 10.1016/0014-5793(90)81016-H
23. Miteva M, Kirkbride KC, Kilchrist Kv., Werfel TA, Li H, Nelson CE, et al. Tuning PEGylation of mixed micelles to overcome intracellular and systemic siRNA delivery barriers. Biomaterials (2015) 38:97–107. doi: 10.1016/j.biomaterials.2014.10.036
24. Bazile D, Prud’homme C, Bassoullet M, Marlard M, Spenlehauer G, Veillard M, et al. PEG-PLA nanoparticles avoid uptake by the mononuclear phagocytes system. J Pharm Sci (1995) 84:493–8. doi: 10.1002/jps.2600840420
25. Yang Q, Lai SK. Anti-PEG immunity: emergence, characteristics, and unaddressed questions. Wiley Interdiscip Rev Nanomed Nanobiotechnol (2015) 7:655–77. doi: 10.1002/wnan.1339
26. Swierczewska M, Lee KC, Lee S. What is the future of PEGylated therapies? Expert Opin Emerg Drugs (2015) 20:531–6. doi: 10.1517/14728214.2015.1113254
27. Anselmo AC, Mitragotri S. Nanoparticles in the clinic: an update. Bioeng Transl Med (2019) 4:e10143. doi: 10.1002/btm2.10143
28. Lee KL, Hubbard LC, Hern S, Yildiz I, Gratzl M, Steinmetz NF. Shape matters: the diffusion rates of TMV rods and CPMV icosahedrons in a spheroid model of extracellular matrix are distinct. Biomater Sci (2013) 1:581. doi: 10.1039/c3bm00191a
29. Champion JA, Mitragotri S. Role of target geometry in phagocytosis. Proc Natl Acad Sci (2006) 103:4930–4. doi: 10.1073/pnas.0600997103
30. Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, et al. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat Nanotechnol (2007) 2:249–55. doi: 10.1038/nnano.2007.70
31. Lagarrigue P, Moncalvo F, Cellesi F. Non-spherical polymeric nanocarriers for therapeutics: the effect of shape on biological systems and drug delivery properties. Pharmaceutics (2022) 15:32. doi: 10.3390/pharmaceutics15010032
32. Zare-Zardini H, Hatamizadeh N, Haddadzadegan N, Soltaninejad H, Karimi-Zarchi M. Advantages and disadvantages of using carbon nanostructures in reproductive medicine: two sides of the same coin. JBRA Assist Reprod (2022) 26:142–4. doi: 10.5935/1518-0557.20210070
33. Khan NU, Lin J, Younas MR, Liu X, Shen L. Synthesis of gold nanorods and their performance in the field of cancer cell imaging and photothermal therapy. Cancer Nanotechnol (2021) 12:20. doi: 10.1186/s12645-021-00092-w
34. Jafari S, Derakhshankhah H, Alaei L, Fattahi A, Varnamkhasti BS, Saboury AA. Mesoporous silica nanoparticles for therapeutic/diagnostic applications. Biomed. Pharmacother. (2019) 109:1100–11. doi: 10.1016/j.biopha.2018.10.167
35. Eber FJ, Eiben S, Jeske H, Wege C. RNA-Controlled assembly of tobacco mosaic virus-derived complex structures: from nanoboomerangs to tetrapods. Nanoscale (2015) 7:344–55. doi: 10.1039/C4NR05434B
36. Wege C, Koch C. From stars to stripes: RNA-directed shaping of plant viral protein templates–structural synthetic virology for smart biohybrid nanostructures. WIREs Nanomed. Nanobiotechnol. (2020) 12. doi: 10.1002/wnan.1591
37. Zhao Z, Simms A, Steinmetz NF. Cisplatin-loaded tobacco mosaic virus for ovarian cancer treatment. Biomacromolecules (2022) 23:4379–87. doi: 10.1021/acs.biomac.2c00831
38. Shin MD, Ortega-Rivera OA, Steinmetz NF. Multivalent display of ApoAI peptides on the surface of tobacco mosaic virus nanotubes improves cholesterol efflux. Bioconjug Chem (2022) 33:1922–33. doi: 10.1021/acs.bioconjchem.2c00371
39. Nkanga CI, Ortega-Rivera OA, Steinmetz NF. Photothermal immunotherapy of melanoma using TLR-7 agonist laden tobacco mosaic virus with polydopamine coat. Nanomedicine (2022) 44:102573. doi: 10.1016/j.nano.2022.102573
40. Wu Z, Zhou J, Nkanga CI, Jin Z, He T, Borum RM, et al. One-step supramolecular multifunctional coating on plant virus nanoparticles for bioimaging and therapeutic applications. ACS Appl Mat. Interfaces (2022) 14:13692–702. doi: 10.1021/acsami.1c22690
41. Nkanga CI, Chung YH, Shukla S, Zhou J, Jokerst Jv., Steinmetz NF. The in vivo fate of tobacco mosaic virus nanoparticle theranostic agents modified by the addition of a polydopamine coat. Biomater Sci (2021) 9:7134–50. doi: 10.1039/D1BM01113H
42. Bruckman MA, Steinmetz NF. Chemical modification of the inner and outer surfaces of tobacco mosaic virus (TMV). Methods Mol Biol (2014) 1108:173–85. doi: 10.1007/978-1-62703-751-8_13
43. Shukla S, Eber FJ, Nagarajan AS, DiFranco NA, Schmidt N, Wen AM, et al. The impact of aspect ratio on the biodistribution and tumor homing of rigid soft-matter nanorods. Adv Healthc Mat. (2015) 4:874–82. doi: 10.1002/adhm.201400641
44. Geiger FC, Eber FJ, Eiben S, Mueller A, Jeske H, Spatz JP, et al. TMV nanorods with programmed longitudinal domains of differently addressable coat proteins. Nanoscale (2013) 5:3808. doi: 10.1039/c3nr33724c
45. Bruckman MA, Randolph LN, VanMeter A, Hern S, Shoffstall AJ, Taurog RE, et al. Biodistribution, pharmacokinetics, and blood compatibility of native and PEGylated tobacco mosaic virus nano-rods and -spheres in mice. Virology (2014) 449:163–73. doi: 10.1016/j.virol.2013.10.035
46. Liu R, Vaishnav RA, Roberts AM, Friedland RP. Humans have antibodies against a plant virus: evidence from tobacco mosaic virus. PloS One (2013) 8:e60621. doi: 10.1371/journal.pone.0060621
47. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF chimera–a visualization system for exploratory research and analysis. J Comput Chem (2004) 25:1605–12. doi: 10.1002/jcc.20084
48. Pitek AS., Jameson SA., Veliz FA., Shukla S, Steinmetz NF. Serum albumin ‘camouflage’ of plant virus based nanoparticles prevents their antibody recognition and enhances pharmacokinetics. Biomaterials (2016) 89:89–97. doi: 10.1016/j.biomaterials.2016.02.032
49. Lee KL, Shukla S, Wu M, Ayat NR,el, Sanadi CE, Wen AM, et al. Stealth filaments: polymer chain length and conformation affect the in vivo fate of PEGylated potato virus X. Acta Biomater (2015) 19:166–79. doi: 10.1016/j.actbio.2015.03.001
50. Liu M, Zhao D, Yan N, Li J, Zhang H, Liu M, et al. Evasion of the accelerated blood clearance phenomenon by branched PEG lipid derivative coating of nanoemulsions. Int J Pharm (2022) 612:121365. doi: 10.1016/j.ijpharm.2021.121365
51. Liu M, Li J, Zhao D, Yan N, Zhang H, Liu M, et al. Branched PEG-modification: a new strategy for nanocarriers to evade of the accelerated blood clearance phenomenon and enhance anti-tumor efficacy. Biomaterials (2022) 283:121415. doi: 10.1016/j.biomaterials.2022.121415
52. Gulati NM, Pitek AS, Czapar AE, Stewart PL, Steinmetz NF. The in vivo fates of plant viral nanoparticles camouflaged using self-proteins: overcoming immune recognition. J Mat. Chem B (2018) 6:2204–16. doi: 10.1039/C7TB03106H
53. Pitek AS, Park J, Wang Y, Gao H, Hu H, Simon DI, et al. Delivery of thrombolytic therapy using rod-shaped plant viral nanoparticles decreases the risk of hemorrhage. Nanoscale (2018) 10:16547–55. doi: 10.1039/C8NR02861C
54. McCormick AA, Corbo TA, Wykoff-Clary S, Palmer KE, Pogue GP. Chemical conjugate TMV–peptide bivalent fusion vaccines improve cellular immunity and tumor protection. Bioconjug Chem (2006) 17:1330–8. doi: 10.1021/bc060124m
55. (2012). Malvern Instruments. Dynamic Light Scattering: An Introduction in 30 Minutes. DLS Technical Note, MRK656.
56. Perry JL, Reuter KG, Kai MP, Herlihy KP, Jones SW, Luft JC, et al. PEGylated PRINT nanoparticles: the impact of PEG density on protein binding, macrophage association, biodistribution, and pharmacokinetics. Nano Lett (2012) 12:5304–10. doi: 10.1021/nl302638g
57. Altintoprak K, Seidenstücker A, Welle A, Eiben S, Atanasova P, Stitz N, et al. Peptide-equipped tobacco mosaic virus templates for selective and controllable biomineral deposition. Beilstein J Nanotechnol. (2015) 6:1399–412. doi: 10.3762/bjnano.6.145
58. Pitek AS, Wen AM, Shukla S, Steinmetz NF. The protein corona of plant virus nanoparticles influences their dispersion properties, cellular interactions, and In vivo fates. Small (2016) 12:1758–69. doi: 10.1002/smll.201502458
59. Pamujula S, Hazari S, Bolden G, Graves RA, Chinta DD, Dash S, et al. Cellular delivery of PEGylated PLGA nanoparticles. J Pharm Pharmacol (2011) 64:61–7. doi: 10.1111/j.2042-7158.2011.01376.x
60. Li Y-P, Pei Y-Y, Zhang X-Y, Gu Z-H, Zhou Z-H, Yuan W-F, et al. PEGylated PLGA nanoparticles as protein carriers: synthesis, preparation and biodistribution in rats. J Controlled Release (2001) 71:203–11. doi: 10.1016/S0168-3659(01)00218-8
61. Rabanel J-M, Hildgen P, Banquy X. Assessment of PEG on polymeric particles surface, a key step in drug carrier translation. J Controlled Release (2014) 185:71–87. doi: 10.1016/j.jconrel.2014.04.017
62. O’Riordan CR, Lachapelle A, Delgado C, Parkes V, Wadsworth SC, Smith AE, et al. PEGylation of adenovirus with retention of infectivity and protection from neutralizing antibody in vitro and in vivo. Hum Gene Ther (1999) 10:1349–58. doi: 10.1089/10430349950018021
63. Armstrong JK, Hempel G, Koling S, Chan LS, Fisher T, Meiselman HJ, et al. Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer (2007) 110:103–11. doi: 10.1002/cncr.22739
64. García-Álvarez R, Vallet-Regí M. Hard and soft protein corona of nanomaterials: analysis and relevance. Nanomat. (Basel) (2021) 11. doi: 10.3390/nano11040888
65. Blume JE, Manning WC, Troiano G, Hornburg D, Figa M, Hesterberg L, et al. Rapid, deep and precise profiling of the plasma proteome with multi-nanoparticle protein corona. Nat Commun (2020) 11:3662. doi: 10.1038/s41467-020-17033-7
Keywords: plant virus, tobacco mosaic virus, PEGylation, cell uptake, immune evasion, protein corona
Citation: Caballero RM, González-Gamboa I, Craig SL and Steinmetz NF (2023) Linear and multivalent PEGylation of the tobacco mosaic virus and the effects on its biological properties. Front. Virol. 3:1184095. doi: 10.3389/fviro.2023.1184095
Received: 11 March 2023; Accepted: 12 May 2023;
Published: 08 June 2023.
Edited by:
Mauricio Comas-Garcia, Autonomous University of San Luis Potosí, MexicoReviewed by:
Trevor Douglas, Indiana University Bloomington, United StatesCopyright © 2023 Caballero, González-Gamboa, Craig and Steinmetz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicole F. Steinmetz, bnN0ZWlubWV0ekB1Y3NkLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.