 Mahamadou Diakite
Mahamadou Diakite Kathryn Shaw-Saliba
Kathryn Shaw-Saliba Chuen-Yen Lau
Chuen-Yen Lau- 1University Clinical Research Center, University of Sciences, Techniques, and Technologies, Bamako, Mali
- 2Collaborative Clinical Research Branch, Division of Clinical Research, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Bethesda, MD, United States
- 3HIV Dynamics and Replication Program, National Cancer Institute (NCI), National Institutes of Health (NIH), Bethesda, MD, United States
The burden of malignancy related to viral infection is increasing in Sub-Saharan Africa (SSA). In 2018, approximately 2 million new cancer cases worldwide were attributable to infection. Prevention or treatment of these infections could reduce cancer cases by 23% in less developed regions and about 7% in developed regions. Contemporaneous increases in longevity and changes in lifestyle have contributed to the cancer burden in SSA. African hospitals are reporting more cases of cancer related to infection (e.g., cervical cancer in women and stomach and liver cancer in men). SSA populations also have elevated underlying prevalence of viral infections compared to other regions. Of 10 infectious agents identified as carcinogenic by the International Agency for Research on Cancer, six are viruses: hepatitis B and C viruses (HBV and HCV, respectively), Epstein-Barr virus (EBV), high-risk types of human papillomavirus (HPV), Human T-cell lymphotropic virus type 1 (HTLV-1), and Kaposi’s sarcoma herpesvirus (KSHV, also known as human herpesvirus type 8, HHV-8). Human immunodeficiency virus type 1 (HIV) also facilitates oncogenesis. EBV is associated with lymphomas and nasopharyngeal carcinoma; HBV and HCV are associated with hepatocellular carcinoma; KSHV causes Kaposi’s sarcoma; HTLV-1 causes T-cell leukemia and lymphoma; HPV causes carcinoma of the oropharynx and anogenital squamous cell cancer. HIV-1, for which SSA has the greatest global burden, has been linked to increasing risk of malignancy through immunologic dysregulation and clonal hematopoiesis. Public health approaches to prevent infection, such as vaccination, safer injection techniques, screening of blood products, antimicrobial treatments and safer sexual practices could reduce the burden of cancer in Africa. In SSA, inequalities in access to cancer screening and treatment are exacerbated by the perception of cancer as taboo. National level cancer registries, new screening strategies for detection of viral infection and public health messaging should be prioritized in SSA’s battle against malignancy. In this review, we discuss the impact of carcinogenic viruses in SSA with a focus on regional epidemiology.
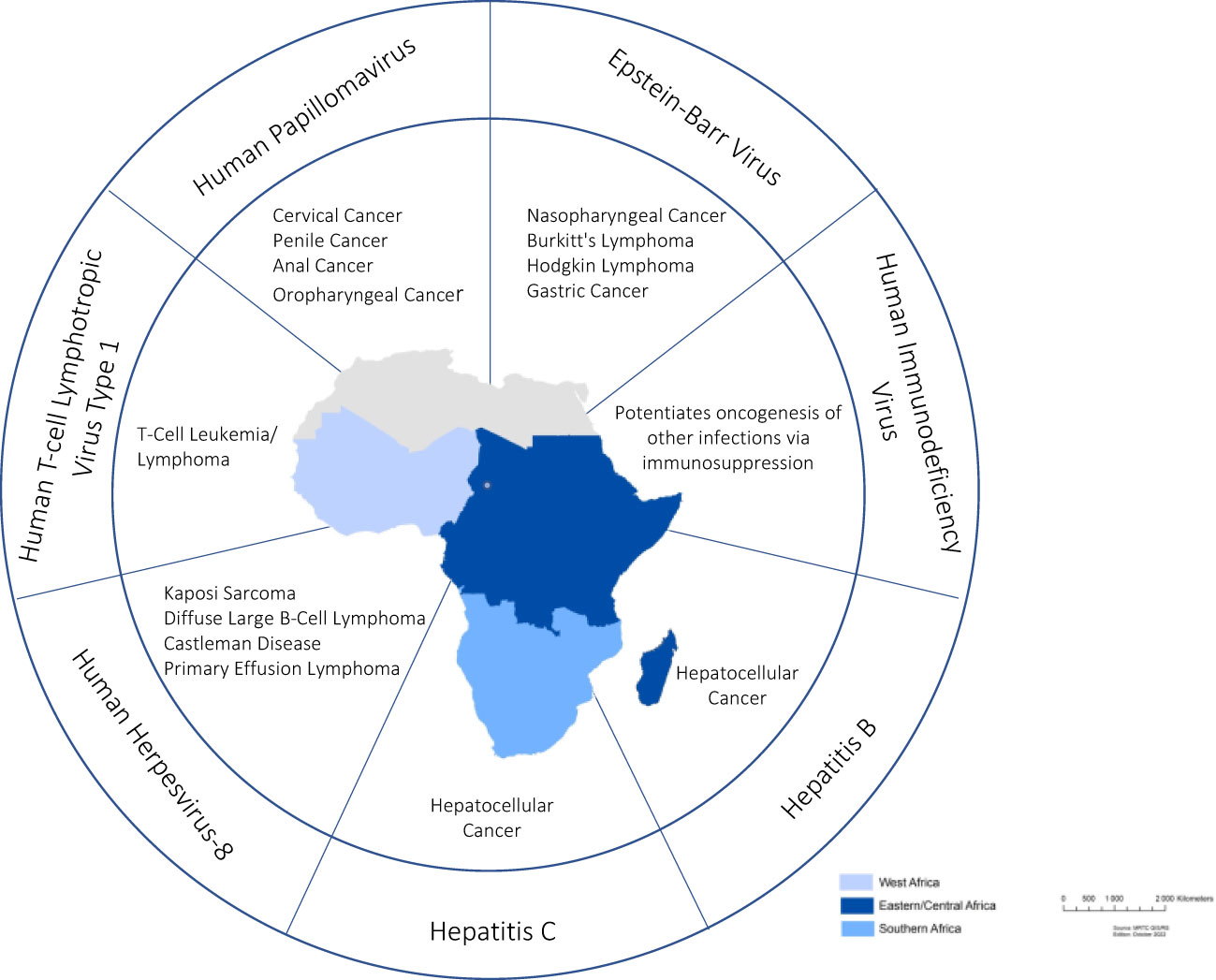
Graphical Abstract Oncogenic Viruses in Sub-Sahara Africa. The outer circle shows the six viruses classsified as oncogenic by the international Agency for Research on Cancer plus Human Immunodeficiency Virus, which potentiates oncogenesis. Examples of malignancies attributable to each virus are shown in the inner circle.
Introduction
According to the World Health Organization (WHO), approximately six of every 100 cancer deaths in developed countries are linked to infection (1). Malignancies related to infection are a much more serious problem in developing regions, particularly Africa. For example, cervical cancer and non-Hodgkin lymphoma associated with human papillomavirus (HPV) and Epstein-Barr virus (EBV) infections, respectively, are disproportionately common in Africa (2). Malignancy due to infection is preventable. Thus, oncogenic viruses are an important cancer control target.
Africa hosts tremendous genetic diversity (3, 4) in conjunction with high infectious disease burden and socio-cultural diversity. This combination presents great opportunity for understanding epidemiology and pathogenesis of oncogenic viruses, but also presents challenges for prevention and control of associated malignancy. Yet the study of viral oncogenesis specific to SSA (West, Central, Eastern and Southern Africa) is relatively young. The earliest arguments linking viruses and cancer have their official origins in the discovery of two bird viruses. The first, avian leukemia virus, was discovered by Vilhelm Ellerman and Olaf Bang, two biologists at the University of Copenhagen in 1908 (5, 6). Leukemias were not considered malignancies at that time, so this discovery was overshadowed by contemporaneous work by Francis Peyton Rous in the United States. In 1911, Rous described a form of avian sarcoma, a well-known tumor, and demonstrated its association with Rous sarcoma virus (7). These earliest oncogenic viruses were retroviruses (8), the study of which laid the foundation for our current understanding of cancer.
Cancer is the second leading cause of death worldwide (after ischemic heart disease). As of 2020, the global burden of cancers caused by infection was estimated at 15.4%. The estimate is over 30% for SSA, making infections amongst the most important causes of cancer in the region (9, 10). Cancer-causing infections are responsible for approximately 30% of cancer cases in low- and lower-middle-income countries. Infection as an etiology of cancer may be increasing, as only 13% of cancers diagnosed in 2018 globally were considered attributable to carcinogenic infections, including Helicobacter pylori, HPV, HBV, HCV, and EBV (2, 11).
Characterizing the link between infection and cancer is an active area of research, particularly in Africa. Elucidating the relationship in SSA is exceptionally challenging due to a high frequency of co-infections. Furthermore, seroprevalence estimates are difficult due to regional heterogeneity of patient populations, size of studies and serodiagnostic methods employed. Temporal uncertainties also complicate the picture, as infections detectable at time of cancer diagnosis may differ from those which played a causal role. Additionally, oncogenic viruses appear to be the etiology of malignancy in a minority of cases, with genetics and lifestyle factors playing a key role.
We will discuss the oncogenic viruses HBV, HCV, EBV, HPV, HTLV-1, HHV-8, HIV, and their impact in SSA in this review. While we will focus on epidemiology, pathogenesis, socio-cultural contributors, and suggestions for additional investigation will also be addressed. Understanding these viruses and their respective malignancies will facilitate effective public policy and advancement of prevention and treatment programs in SSA.
Hepatitis B virus
HBV genomic organization and protein products
HBV is the smallest human DNA virus with 3,200 base pairs. Its double-stranded circular genome contains four overlapping open reading frames (ORFs) and encodes 4 genes: C, S, P and X. The C gene, with a pre-C zone, encodes the 21 kDa core (HBc) antigen (Ag) and HBeAg. The S gene, with a pre-S1 zone and a pre-S2 zone, encodes the surface antigen (HBsAg) (12). HBsAg exists in small 24 kDa, medium 33kDa, and large 39 kDa forms depending on whether it derives from expression of S, pre-S2 + S, or pre-S1 + pre-S2 + S genes. The P gene encodes the 90 kDa DNA polymerase, which serves as a classic DNA-dependent DNA polymerase, a retrotranscriptase, and has RNase H activity. The X gene encodes X protein (HBx), which performs transactivating functions, regulates the viral lifecycle and is required for replication (13). HBx is likely involved in HBV carcinogenesis (14, 15). The HBV genome also includes four promoters, two enhancer elements (EN1 and EN2), a polyadenylation site for viral RNA transcription, and several cis-acting signals for DNA replication.
To optimize its limited capacity, HBV’s genes overlap. The P gene, the longest of the four, comprises three-quarters of the genome and overlaps entirely with the S gene and partially with the C and X genes (16). All HBV RNA transcripts terminate at the same polyadenylation site located in the C ORF (17). A posttranslational cis regulatory element (PRE), which overlaps with part of the X ORF, promotes nuclear export of unspliced HBV RNA to the cytoplasm (18).
This strictly human Hepadnavirus consists of an envelope bearing HBsAg with an interior nucleocapsid. HBV viral glycoproteins (HBsAg) constitute the envelope surrounding the viral capsid in mature virions; HBsAg contains the main HBV antigenic domains and the attachment site to target hepatocytes (19). HBcAg forms the core or capsid and is expressed on the surface of hepatocytes, where it induces cytolysis via CD8+ T lymphocytes. Unlike HBsAg, it does not appear in serum. Conversely, the smaller HBeAg is present in the serum during active viral multiplication (20). Expression of 10 amino acids from the pre-C region on HBeAg enable its passage through the reticuloendothelial system and excretion in serum (21). HBsAg, HBcAg and HBeAg and their respective antibodies are markers of HBV infection status.
HBsAg has at least 5 main antigenic sites. The major determinant (a) is common to all strains and corresponds to production of HBsAb. There are also two pairs of mutually exclusive subtype determinants which define 4 subtypes of HBsAg: adw, ayw, adr and ayr (22). The HBsAg subtypes have geographical specificity. For example, adw predominates in northern Europe, North and South America, and Australia, while ayr is found in northern and eastern Africa, the eastern Mediterranean, eastern Europe, northern and central Asia, and India. HBV genotyping now allows distinguishing genotypes from A to H by PCR. Characteristics of HBV genotypes and subtypes have been used to investigate maternal transmission, familial clustering, and geographic distribution of HBV strains (23).
HBV was identified in 1967. Since then, it has come to be recognized as a cause of hepatitis, cirrhosis, and hepatocellular carcinoma. HBV is a leading cause of malignancy in SSA.
HBV prevalence and distribution in SSA
Approximately 296 million people worldwide are living with HBV and 820,000 die annually from related complications (24). Though effective vaccination has reduced incidence, HBV affects 8%-18% of the SSA population, with 5-10% of the adult population being chronically infected (25). Propensity for chronicity (~2%) makes it the leading cause of cirrhosis and hepatocellular carcinoma in the region. Most chronic HBV infections result from perinatal transmission (26). The epidemiology of HBV in Africa is also intertwined with that of HIV/AIDS, given the overlap in risk factors.
HBV genotypes A, D and E are circulating in Africa. Genotype A is predominant in southern Africa. African subtypes A1 and Aa are associated with early HBe seroconversion and lower viral loads compared to European subtypes A2 and Ae. Genotype D prevails in North Africa. Genotype D is also associated with early HBe seroconversion and high rates of HBeAg negative active hepatitis. Genotype E is observed in the West and Central African regions of SSA (27).
HBV is transmitted via bodily fluids parenterally, sexually, and perinatally. Vertical infection is the most common route of transmission in high endemicity areas, including in SSA. Perinatal transmission is associated with greatest risk of chronic carriage and is more common the later a woman is infected during pregnancy (28, 29). Despite availability of accurate testing and an efficacious vaccine, access to affordable diagnostics and vaccination coverage remain suboptimal in SSA (30, 31). Regional awareness of HBV infection status remains very low, further fueling spread of infection and unchecked disease progression (32).
HBV has a broad spectrum of clinical manifestations including asymptomatic disease, fulminant hepatitis, and hepatocellular carcinoma (HCC). Disease can be acute or chronic, though acute disease is typically self-limited (33). Chronic disease is much more common after perinatal transmission versus acquisition in adulthood. When left untreated, HBV infection can engender liver fibrosis, cirrhosis, and eventually liver cancer. Low rates of detection in SSA result in untreated infections and higher rates of HCC. The confluence of pathogenic mechanisms and epidemiologic risks have made HBV the leading cause of HCC in SSA.
HBV-associated malignancies
HBV leads to HCC typically after many years of infection and in the setting of fibrosis. Longitudinal studies of untreated people with chronic HBV show a 5-year risk for developing cirrhosis of 8-20% (34–38). Amongst those with cirrhosis, the annual risk of hepatic decompensation is approximately 20% and the annual incidence of HBV-related HCC ranges from <1%-5% (39). Untreated decompensated cirrhosis carries a poor prognosis, with survival of 15-40% at five years (38–40). Several host and viral factors, particularly co-infections with HIV, HCV and hepatitis D virus (HDV), as well as alcohol consumption and underlying liver disease, increase the rate of disease progression and the risk of developing HCC (35–38, 41). Other risk factors include age, sex, serum alanine aminotransferase (ALT) levels, serum HBV DNA levels, and HBeAg levels (42).
Integration of HBV DNA into host chromosomes has been found in >80% of HBV associated HCC (43, 44). Specific integration hotspots have been identified (45), particularly within the X gene (46–48). HBV DNA integration occurs early in infection (49), persists, and can activate or disrupt gene expression to promote chromosomal instability and development of cancer, metastases, and angiogenesis (50).
Low engagement in routine preventive care in SSA means that disease progression typically goes unchecked. Approximately 80% of newly diagnosed HBV in Africa is linked to chronic disease (51), and hence risk of HCC. Males with HBV are 5-7 times more likely than females to progress to HCC (52), which is in part attributable to positive feedback through the androgen receptor (53, 54) and differing levels of chronic liver inflammation. Further epidemiologic characterization of the HBV epidemic in SSA is needed to determine if HCC associations will reflect those seen in other populations.
Future directions
The true extent of HBV infection in the African region has not been fully characterized. National and subnational level data is lacking; hepatitis surveillance programs are performing poorly; and thus (55), priorities are difficult to determine for targeted measures and resource allocation (56, 57). Several approaches for prevention and control of viral hepatitis, integration of HBV vaccination into national immunization programs, and routine vaccination of healthcare workers have been proposed (58, 59). With increasing accessibility of HBV vaccines, more affordable diagnostics, and effective antiviral drugs, control of SSA’s HBV epidemic is becoming more feasible.
Addressing the interplay between HBV and HIV co-infection will continue to be important, as both infections impact SSA and risk factors overlap (60). Co-infection can accelerate progression of both infections and inhibit HBV clearance via effects on the host immune response, thus contributing to increased rates of cirrhosis and hepatocellular carcinoma (61–71). All people with HBV should be screened for HIV and vice versa. Accurate assessment of infection will enable appropriate use of antivirals. For example, HIV/HBV co-infected people should be treated with two anti-retrovirals that suppress HBV as part of the HIV treatment regimen to minimize progression and transmission of both infections (72). Detection and management of co-infections should be integrated into current public health systems.
HBV prevention activities in Africa are improving but remain limited. Full implementation of perinatal HBV vaccination has been slow in SSA (73). HBV vaccination is a safe and effective HCC prevention method and must be prioritized (33, 74). Scalable screening programs are also lacking in SSA, where <5% of people with chronic hepatitis are aware of their HBV status (30, 55, 75, 76). Broad implementation of screening should be accompanied by improved access to indicated follow up and management when infections are detected (77, 78). Training of healthcare providers and overcoming structural barriers that perpetuate vulnerability, marginalization of affected populations and inequitable access to services will be critical. Although many SSA countries do not screen or only partially screen donated blood products, transfusion surveillance (79) indicates that broader screening of blood products will be needed to reduce transfusion associated transmission (80, 81–83).
SSA countries should establish a rigorous viral hepatitis surveillance system. Research on prevention and management strategies that consider SSA biology and socio-economic structure should be expanded to complement surveillance. Table 1 shows key information on factors related to acquisition of infection, disease course, and prevention strategies for HBV.
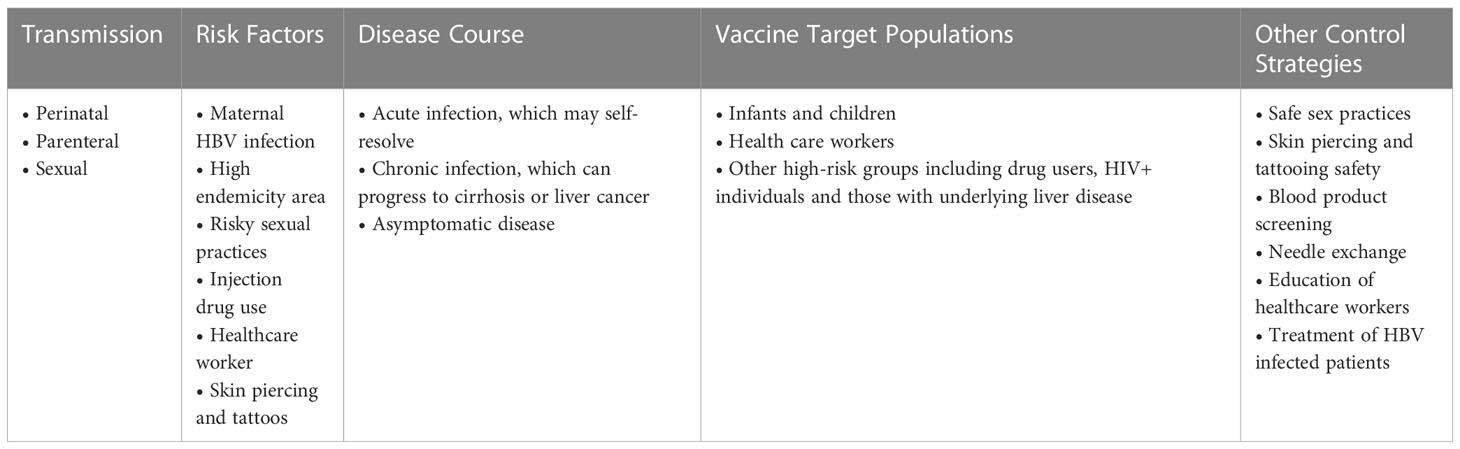
Table 1 HBV.
Hepatitis C virus
HCV genomic organization and protein products
Hepatitis C Virus (HCV) is single positive stranded RNA virus in the family Flaviviridae (84). It contains a 5’ non-translated region (NTR), a long open reading frame encoding a polyprotein, and a 3’ NTR. The polyprotein is cleaved by host enzymes and viral proteases into >10 products. Core (C) is an RNA binding protein which forms the nucleocapsid. E1 and E2 are highly glycosylated type 1 transmembrane proteins which comprise stable heterodimers embedded into the lipid envelope around the viral nucleocapsid. P7 is a small hydrophobic peptide which may be a virioporin involved in release of virions from infected cells. The nonstructural proteins NS2 - NS5B are primarily required for multiplication of viral RNA. NS2 and the amino terminal domain of NS3 constitute the NS2/3 autoproteinase responsible for NS2/3 junction cleavage. NS3 has three different enzymatic functions (85). The amino-terminal domain forms a serine-type proteinase responsible for processing at the carboxy terminal of NS3 and the carboxy terminal domain carries NTPase/helicase activities.
While hepatocytes are the primary host cell, replication also occurs in peripheral blood mononuclear cells (PBMCs) and B and T cell lines. When HCV binds the host cell, viral RNA is released from the nucleocapsid into the cytoplasm, the genome is translated, and the resulting polyprotein is cleaved into individual proteins. These proteins form a higher-order multi-protein complex tightly associated with intracellular membranes. Within this complex, the positive strand RNA genome is copied into a negative strand RNA intermediate that serves as a template for synthesis of positive strand progeny (84). These positive strands may be used for synthesis of new negative strands, translated, or encapsulated into virions.
HCV has six major genotypes, though other lesser contributors have been identified. Each genotype can have multiple subtypes, for example 1a, 1b, 2a and 2b (85, 86). There is high variability of genotypes across SSA, particularly in g4 and g7 strains (87). Furthermore, the ΔG frame shift allele which encodes an open reading frame of IFN-λ4 is a major variant in Africans and influences clinical outcomes through modulation of IFN stimulated gene expression (88). Though initially studied with relation to IFN-based treatment, this allele is also associated with delayed response to currently used DAAs (89). Genotypic predominance not only varies geographically but is also temporally dynamic in association with population interactions.
HCV prevalence and distribution in SSA
Prevalence of chronic HCV varies from 0.2% to 26% across countries globally (56). HCV prevalence on the African continent is estimated to be 6.1%; SSA boasts the largest number of infections at 78 million (55, 90). Within SSA, Central Africa has the highest adult prevalence of chronic HCV at 9.7%, followed by West Africa with a prevalence of 8.3%. Within Central Africa, Burundi and Cameroon have the highest prevalence of 11.3% and 13.8%, respectively. Eastern and Southern SSA have chronic HCV prevalence of 5.5% and 3.8%, respectively (91). Prevalence of chronic HCV is lowest in North Africa at 2.8% (92). An estimated 71.1 million people worldwide are chronic HCV carriers, including approximately 18 million in Africa (76, 91). HCV prevalence increases with age, with the highest rate being reported amongst those >40 years. Chronic carriers are at risk of developing HCC with increasing age.
Like HBV, transmission of HCV is multi-modal. The most common route of transmission in SSA is parenteral. Although injection drug use is not as common in Africa as in many other parts of the world, it does present a major route of transmission. Historically an inadequately screened blood supply and reuse of needles in the medical setting also led to many infections (93, 94). In conjunction with high rates of sickle cell disease, blood transfusion has been a major route of acquisition (95). Vertical transmission is low, but more significant in the setting of coinfection with HIV. Prevalence of pediatric HCV varies from 0.05% to 0.36% in developed countries and from 1.8% to 5% in the developing world (96). Incidence of HCV vertical transmission has been shown to be 3% - 10% (97–99) and higher in infants born to mothers coinfected with HIV.
While multiple genotypes are present in SSA, genotypes 1 and 4 predominate. Prevalence of specific genotypes also varies by route of transmission and demographic characteristics (100). Treatment of HCV is guided by genotype and presence of cirrhosis. Thus, understanding genotype epidemiology is critical for planning of management programs in SSA. Although a vaccine for HCV is not yet approved, highly effective treatment has been available since 2014 when the first direct-acting antiviral (DAA) therapy ledipasvir/sofosbuvir was approved (101). Most of the DAAs available inhibit the HCV NS3/4A protease, NS5A protein, and NS5B polymerase. Prior to availability of DAAs, treatment with ribavirin and interferon-α was standard and had much lower cure rates. Provision of DAAs for treatment of HCV is critical for prevention of hepatocellular cancer and should be supported by national programs. Selection of DAAs for programs in SSA must be guided by local epidemiology.
HCV-associated malignancies
Chronic HCV is a well-established risk factor for HCC, increasing the risk by 10–20 fold. HCV is the second major risk factor for HCC in Africa after HBV (102). HCC is the fourth most common cancer in Africa and has varying regional epidemiology (103). While HCV promotes hepatic (104, 105) and B-cell lymphoproliferative diseases (106, 107), it may not be the primary driver of tumorigenesis because it does not integrate into the host’s genome. HCV likely promotes tumorigenesis through repetitive damage, regeneration, and fibrosis seen during progression of cirrhosis (108). Annual incidence of HCC in persons with HCV-related cirrhosis ranges from 0.5–10% (109). The epidemiology of HCV disease progression to HCC in SSA has not yet been well-characterized but may follow trends seen elsewhere, which suggest a current increase in HCV associated HCC cases (109).
Risk factors for progression from chronic HCV to HCC have not been thoroughly characterized in the African population. Broader studies have shown an association with male sex, Hispanic ethnicity, HCV genotype 3, longer duration of infection, co-infection with HBV or HIV, markers of fibrosis, insulin resistance, obesity, diabetes, tobacco, and alcohol (51, 109) (110). HCV treatment with achievement of sustained virologic response (SVR) has the greatest impact on reducing progression (111). Several large studies of DAAs and meta-analyses (112) have demonstrated that HCC risk, while not eliminated, is reduced by 50–80% among persons who achieve SVR (113, 114). While HCC risk is reduced with SVR, the rates do not revert to baseline, especially among persons with cirrhosis. Long term follow-up of HCV infected patients who achieved SVR with DAAs has shown cumulative 1, 2, and 3-year risks of HCC of 1.1%, 1.9% and 2.8%, respectively. These incidence rates are at or below the threshold for cost-effective HCC surveillance (115).
Future directions
HCV-associated HCC is a major public health problem in Africa. SSA health systems should anticipate an increasing number of HCC patients over the next several years. It remains unclear when the epidemic will peak, though its dynamics will likely trail that of HCV-associated HCC in more developed regions. National screening and prevention programs have been implemented and should be further expanded. International organizations should support local efforts through training and establishment of regionally sustainable facilities. Policy makers must prioritize control of viral hepatitis and its sequelae on the African continent.
The high prevalence of HCV infection in Africa necessitates augmentation of primary prevention efforts including vaccine development, as well as new approaches to reduce the burden of chronic liver disease. Vertical transmission of HCV in SSA must also be addressed. Mother-to-child transmission contributes to the SSA epidemic proportionally more than in other regions, where IDU tends to be the leading cause of infection. Standard screening of the blood supply must also be enforced.
While DAAs are highly effective for treatment of HCV infection, an estimated 95% of HCV infections remain undiagnosed world-wide (116, 117). Furthermore, prior HCV infection does not protect against HCV re-infection (118). Thus, development of a preventive HCV vaccine is essential to managing the epidemic; therapeutic vaccines to treat existing infection may also offer advantages of DAAs. Development of new assay methodologies and animal models facilitated discovery of partially effective antibody based vaccines in the eighties (119). Trials of viral vector vaccines were initiated in the nineties (120, 121). More recently studied vaccine platforms include HCV-like particles, recombinant proteins, DNA constructs, peptides, and novel viral vectors (122–124) (125) An effective HCV vaccine will likely need to induce robust neutralizing antibodies as well as multi-specific cellular immune responses. Advances in understanding of HCV pathogenesis in conjunction with innovative technologies have given rise to candidate vaccines currently in clinical trials (126). Deployment of an effective vaccine will be integral to HCV control in SSA.
Education programs targeting healthcare providers and high-risk populations should increase awareness about HCV infection, sequelae, prevention, and treatment. Societal knowledge expansion will be critical for engaging affected populations and breaking the cycle of disease transmission. Future research on interruption of vertical transmission, HCV vaccines and effective use of DAAs in the SSA context will be helpful. Table 2 shows key information on factors related to acquisition of infection, disease course, and control strategies for HCV.

Table 2 HCV.
Epstein-Barr virus
EBV genomic organization and protein products
Epstein-Barr virus (EBV) is a Herpesviridae whose tropism for lymphocytes has led to it being classified in the subfamily Gammaherpesvirinae (127). It was discovered in malignant B-lymphocyte proliferations of children’s jaws by Epstein and Barr in 1964 (128), consistent with its now established propensity for infection of the oropharynx and B cells (129). EBV is an approximately 172 kb double-stranded DNA virus composed of an icosahedral capsid surrounded by a cell membrane derived envelope into which viral glycoproteins are inserted. The genome encodes approximately 100 proteins that regulate gene expression, DNA replication, and immune responses; roles of several EBV proteins are still being characterized (130). The genome is divided by 0.5 kb terminal direct repeat (TRs) and internal repeat (IR) sequences into short and long sequence domains.
Several strains of EBV have been sequenced (131–134). However, open reading frames (ORFs), genes, and transcription or RNA processing sites are frequently referenced to specific BamHI fragments, from A to Z in descending order of fragment size, because the first sequenced EBV strain B95-8 was sequenced from a cloned library of EBV DNA BamHI fragments (135). Sequence analysis has been essential for characterizing EBV. This has enabled identification of four previously unrecognized ORFs whose functions are still being elucidated: BFRF1A is likely involved in DNA packaging by homology with other herpesviruses; BGLF3.5 may be an integumental protein; functions of BVLF1 and BDLF3.5 are less apparent (133).
EBV’s lifecycle starts with primary infection, then proceeds through latent and lytic phases. The virus enters a new host oropharyngeally and then infects naïve B lymphocyte by binding between the major viral envelope glycoprotein gp350/220 and the CD21 receptor (136) (137) Alternatively, CD35 can serve as receptor for EBV in the absence of CD21 (138). Viral glycoproteins gHgL and gp42 interact with HLA on the target cell surface to facilitate membrane fusion (138, 139). Infection of naïve B cells engenders polyclonal lymphoblastoid proliferation and expression of activation markers such as CD23, CD30, CD39 and CD70. The virus then enters “latency III” (cell proliferation), during which it expresses the antigens of the latency cycle: the 6 EBNAs (Epstein-Barr nuclear antigens): EBNA-1, EBNA-2, EBNA-3A, EBNA-3B, EBNA-3C and EBNA-LP; the 3 LMPs (latent membrane proteins): LMP-1, LMP-2A and LMP-2B; two non-polyadenylated non-coding RNAs, EBER1 and 2 (EBV-encoded RNA transcribed); and transcripts of the BamH1 A region of the genome, BARTS (139, 140). Details of these protein functions have been published (139, 141–144). Of note, the LMP-1 region has been an attractive locus to characterize EBV’s biological, genetic, and epidemiological properties (145, 146). LMP-1 has been linked to biological changes that influence transmission, transformation, and tumor microenvironment (147).
As “latency III” progresses, B lymphocytes differentiate into memory B cells with somatic hypermutation of immunoglobulin (Ig) genes and form a germinal center. This marks the beginning of “latency II” (cell differentiation). When the B lymphocyte divides, episomal EBV will also replicate. It will then be in “latency I” and expressing EBNA1. A healthy immune system will control these lifecycle events such that the virus will enter “latency 0” during which B cells become long-lived resting CD27+ memory cells. Gene expression, with the possible exception of some EBNA1, ceases (139, 140, 148). EBV persists for life in B lymphocytes as a few copies of closed circular genomic DNA, or episomes, which duplicate with each mitosis due to EBNA1 (130, 140). It is estimated that 1 to 50 cells per million B cells are infected with EBV (140).
The lytic phase is divided into three phases by regulated gene expression: immediate-early, early, and late. Emergence from latency involves simultaneous expression of the immediate-early genes BZLF1 and BRLF1 (149–151). These genes encode the transactivating proteins ZEBRA (152) (also known as EB1 (153), Zta (154), and BZLF1) and RTA [also known as BRLF1 (155)], respectively. Both proteins can be expressed from a 2.9 kb R-Z RNA bicistron and a 3.8 kb minor R-Z RNA bicistron transcribed from the promoter R (Rp). ZEBRA is also expressed from a smaller 0.9 kb mRNA from the downstream Z promoter (Zp). In addition, another 0.9 KB message encoding putative fusion protein RAZ, which is composed of parts of RTA and ZEBRA, is transcribed at a low level (156). RAZ may be a ZEBRA inhibitor, though its role is still under investigation (157, 158). Synergy between ZEBRA and RTA suggests that low levels of both proteins can trigger the lytic cascade. The BRRF1 gene product has recently been shown to activate Zp and cooperate with RTA to induce lytic infection (159).Transcription of immediate-early genes does not require de novo protein synthesis, implying that physiological signals from the host may activate the lytic cycle (160). Early gene products include enzymes necessary for viral DNA replication. DNA amplification marks the beginning of the late phase, during which viral structural proteins are expressed and assembled into viral particles. DNA is packaged into these particles before release of infectious virions to infect new naïve B lymphocytes and new epithelial cells. Healthy asymptomatic people perpetuate the infection cycle by reactivating and excreting virus abundantly in their saliva (130, 140, 142).
EBV prevalence and distribution in SSA
EBV is a globally ubiquitous virus. Spread of the virus through saliva means that exposure is nearly inevitable. Most people are infected at some point in their lives, often during childhood. Timing of primary infection is related to hygiene, socioeconomic and demographic factors. Infection is thought to occur later in more developed countries (161). Primary infection during early childhood is common in SSA, which is consistent with the role of EBV in childhood malignancy there.
Although most of the population is infected with EBV, manifestations vary across geographic regions. The high impact of EBV in SSA is reflected by its discovery in the region. In 1964, Denis Burkitt identified a tumor that was the most common childhood malignancy in equatorial Africa (162). The tumor, which subsequently became known as endemic Burkitt lymphoma, was found to harbor herpesvirus particles later recognized as EBV (128, 163). Endemic Burkitt is mainly found in areas of Africa and Papua New Guinea with increased malaria prevalence. Risk of progression to endemic Burkitt lymphoma may be related to severity of past malaria infection (164). The annual incidence of Burkitt lymphoma is 4-5/100,000 amongst children less than 18 years old from equatorial Africa, where 50% of tumors diagnosed during childhood and 90% of lymphoma cases are attributable to Burkitt lymphoma (165) (166).
As immunodeficiency related Burkitt lymphoma predominantly affects HIV-infected populations, it is most common where HIV prevalence is highest. Diffuse large B-cell lymphoma, for which up to 90% of cases in HIV-infected patients are associated with EBV infection, also follows HIV epidemiology (167). Such malignancies tend to occur in more advanced HIV. Given that SSA has the highest global prevalence of HIV and has been unable to control the HIV epidemic thus far, immunodeficiency related Burkitt lymphoma, diffuse large B-cell lymphoma (DLBCL), and other malignancies facilitated by immune-suppression will be more common in the region.
Of the 83,087 cases of Hodgkin lymphoma reported in 2020, 10,815 (13%) were in Africa. The region accounted for 4,315 (18.5%) of the 23,376 attributable deaths. Age-standardized incidence and mortality rates were close to the global average (168). A study using EBV-encoding RNA in situ hybridization found an EBV prevalence of 54% in Hodgkin lymphoma in Rwanda. Prevalence did vary across subtypes (169).
Geographic distribution of EBV subtypes is highly variable, as has been shown for other viral carcinogens, including HPV (170, 171) and HBV (27). These patterns are important for public health, biology, and diagnosis. They also reflect underlying immunological pressures that drive diversification through host-pathogen adaptations in disparate populations. Variation in EBNA-2 and EBNA-3 genes that underly classification of EBV into types 1 and 2 (133, 172) highlight geographic diversity, though Burkitt lymphoma and nasopharyngeal carcinoma (NPC) exhibit geographic distributions and age-specific patterns unexplained by simple EBV epidemiology (173–175). For example, NPC occurs with high incidence in Eastern and South-Eastern-Asia and in some areas of the Middle East and North Africa (176).
Lack of effective EBV infection control strategies and healthcare infrastructure requirements for effective management mean that SSA will continue to shoulder a significant burden of EBV associated malignancy. To further complicate matters, an individual can be infected with multiple EBV variants based on LMP-1 sequencing (177). The most practical approach for SSA may be to control risk factors, including HIV and malaria infection, that contribute to progression of EBV disease.
EBV associated malignancies
Epstein–Barr virus (EBV) is a ubiquitous human lymphotropic herpesvirus with a well-established causal role in several cancers. It is associated with two general types of cancer: lymphoproliferative, which is mainly B-cell but may also affect T and NK cells; and epithelioproliferative, which is mostly nasopharyngeal but also includes certain gastric, mammary and pulmonary carcinomas (130). We will focus on malignant presentations in this paper, but one should also be aware of non-malignant presentations. For example, primary infection in healthy children <5 years old is typically asymptomatic, whereas 50% of adults and adolescents may develop infectious mononucleosis (IMN) (140, 178). Association of EBV with other conditions such as rheumatoid arthritis, multiple sclerosis, myasthenia gravis, and chronic fatigue syndrome remains under investigation (130).
Burkitt lymphoma is one of the most concerning EBV associated malignancies due to its rapid doubling time. It occurs when one of three translocations dysregulates the c-myc oncogene and is associated with proliferation of CD19+, CD20+, CD21+, CD10+, CD77+ and BCL6+ B cells whose origin is the germinal center (127). There are three types of Burkitt lymphoma: endemic, sporadic, and HIV related (140, 178). The endemic or “African” Burkitt preferentially affects bones of the face, most commonly the jaw. Endemic Burkitt lymphoma is most common in children 4 to 7 years old and is found in areas with high malaria endemicity (127). 90–95% of cases are EBV positive. Sporadic Burkitt affects young adults and is localized to the gastrointestinal or respiratory tract. It is associated with EBV in 15% of cases. HIV-related Burkitt localizes to the lymph nodes and bone marrow and it is associated with EBV in 30-40% of cases (179). Approximately 20% of lymphomas in PLWH are attributed to Burkitt and the risk of developing Burkitt has been estimated to be 200 to 1,000 times higher in PLWH compared to the general population (179). Treatment of Burkitt lymphoma is chemotherapy. Since EBV is in latency I in Burkitt lymphoma, expressing only EBNA1 and EBERs, adoptive immunotherapy is ineffective because CTLs do not recognize EBNA1 (180).
The role of EBV in Hodgkin’s disease had long been suspected based on increased incidence of Hodgkin’s in the setting of elevated anti-EBV antibodies, within 5 years of IMN, and in patients with IMN (179). The link was confirmed by identification of EBV DNA in Reed Sternberg cells (HRS). HRS are germinal center B cells that have undergone functional rearrangement of Ig genes but lack Ig transcription. Survival of HRS depends on presence of EBV (179). There are four histological types of Hodgkin’s lymphoma: nodular sclerosing, mixed cellularity, lymphocyte depleted and predominantly lymphocytic. Hodgkin’s lymphoma is associated with EBV in 40 to 65% of cases depending on histological type, age, and geography (181). Cells are in type II latency with expression of EBNA1, LMP1 and LMP2A proteins. The NF-KB pathway is activated due to presence of LMP1 in HRS or inactivation of NF-KB inhibitors when HRS cells are EBV-negative (140, 143, 178). Clinically, Hodgkin’s disease presents with fever, night sweats, asthenia, weight loss, lymphadenopathy, and hepatosplenomegaly.
Undifferentiated nasopharyngeal carcinoma (NPC) is an epithelial tumor responsible for approximately 50,000 deaths per year. The annual mortality rate in Southeast Asia and North Africa is 4 to 8/100,000 compared to 25/100,000 in Southern China where it is the leading cause of cancer regionally and amongst those who emigrate to other areas (127). Genetic and dietary factors (carcinogen ingestion) have been implicated in development of the disease in the setting of EBV infection. Malignant NPC epithelial cells uniformly contain the EBV genome. Peter Clifford demonstrated this association in Kenyan children in 1972 (182), though EBV was not classified as a group 1 carcinogen until 1997 (183). In NPC, the virus is in latency II, with expression of EBNA1 proteins and EBERs, and less consistently LMP1 and 2A (140, 143). MicroRNAs from the BART cluster are also found in plasma of patients with NPC. These microRNAs may play a role in the pathogenesis of NPC and have been proposed as a prognostic marker independent of EBV viral load (184).
Burkitt lymphoma, Hodgkin’s disease and nasopharyngeal carcinoma are examples of EBV associated malignancies seen in SSA. EBV can additionally cause several other malignancies. All have associations with specific phases of EBV latency. Diagnosis typically occurs after symptomatic clinical presentation; screening is not standard. Management may include radiation, chemotherapy, biologics, and immune reconstitution. EBV related malignancies present a significant burden across age groups in SSA.
EBV future directions
High global prevalence of EBV in combination with elevated risk of progression to malignancy in SSA make EBV associated malignancies a public health priority for the region. Both prevention of infection and progression to malignancy need to be addressed. Unfortunately, exposure to EBV is almost unavoidable as it is transmitted by bodily fluids, particularly saliva. With no EBV vaccine currently available, most of the population will be infected. Effective strategies for preventing progression to malignancy are also lacking, though control of malaria could reduce incidence of endemic Burkitt lymphoma and control of the HIV epidemic could reduce incidence of immunosuppression associated Burkitt.
Further elucidation of the relationship between EBV-associated cancer and malaria will likely reveal prevention and mitigation opportunities. Epidemiologic associations have long been recognized, including high geographic correlation between Burkitt lymphoma incidence and intensity of P. falciparum transmission; correlation of Burkitt lymphoma incidence by age with age of acquisition of peak malaria immunoglobulin levels; decreased incidence of Burkitt lymphoma where malaria mortality has decreased; and older age of onset amongst people who migrated from low to high-intensity malaria areas compared to those born in high-intensity areas (185, 186). Such associations gave rise to discovery of EBV as a human oncogenic virus (187, 188) (189).
As EBV is not transmitted by mosquitoes, molecular mechanisms underlying the relationship between P. falciparum and EBV are of great interest. One study demonstrated that the cysteine-rich interdomain alpha region (CIDR1α) of P. falciparum membrane protein 1 acts as a polyclonal activator of B cells. CIDR1α increases B cell survival, preferentially activates the memory compartment where EBV persists, and induces virus production in latently infected primary B cells from healthy carriers and children with endemic Burkitt lymphoma (190). Studies have also shown that EBV can be activated by P. falciparum via cytidine deaminase-induced activation (AID) (191), which increases risk of DNA damage and lymphoma in murine models (192). This activation may facilitate preservation of B-cells with dysregulated c-myc expression that would normally undergo apoptosis (193). Thus, P. falciparum appears to be a risk factor for endemic Burkitt lymphoma through AID activation facilitating DNA damage and persistence of c-myc translocations. Additional research on mechanisms underlying the association between malaria and Burkitt lymphoma should be performed to identify disease intervention targets.
Research on the drivers of latency and its transitions in non-malaria endemic settings will also be useful. Analyses using recombinant forms of EBV may facilitate a better understanding of the role specific viral genes play in the EBV life cycle. Such information might lead to recognition of new vaccine targets (194) or therapeutic interventions targeting the function of essential EBV latent genes. Drugs that prevent binding of EBNA1 to oriP or interaction of FTTs with LMP1 could be developed (172, 195).
Development of an effective EBV vaccine should be prioritized. A vaccine that prevents infection would be ideal, though a vaccine that only reduces progression to malignancy would still have great impact. In conjunction with an improved understanding of mechanisms of EBV oncogenesis and development of an EBV vaccine, malaria vaccines currently being deployed and continuous improvement of the HIV prevention and care continuum could reduce associated Burkitt lymphoma cases in SSA. Fortunately, both malaria and HIV control efforts are supported through local and international programs.
Further understanding of mechanisms regulating EBV cell growth, survival and differentiation could also improve clinical management of those with EBV-associated disease. Adoptive transfer of EBV-specific CSF has proven useful in the treatment of immunoblastic B-cell lymphomas; this and other treatment strategies are currently being evaluated in patients with HD or NPC (196). Gene therapy strategies that exploit transcriptional regulation of the EBV genome or target the functional effects of latent EBV genes also offer possibilities for development of therapeutic and preventive strategies. Furthermore, continued refinement of longitudinal EBV viral load monitoring in transplant recipients presents opportunities for early intervention (194).
EBV genetic variation and host factors likely influence disease course. Further study on the mechanisms underlying epidemiologic associations may reveal risk modification strategies. Pathogenic differences in EBV strains may facilitate prioritization of vaccination targets. Genome deletions and polymorphisms may also be useful as predictive biomarkers.
SSA is highly affected by EBV related malignancies. Manifestations of EBV in the region are influenced by host factors and other regional conditions such as malaria and HIV. Inadequacy of healthcare infrastructure also means that cases may be missed or present with more advanced disease. Mitigating the socio-economic burden of EBV will require a combination of public health approaches, clinical research, and basic science investigation. Table 3 shows key information on factors related to acquisition of infection, EBV associated malignancies, and control strategies for EBV.
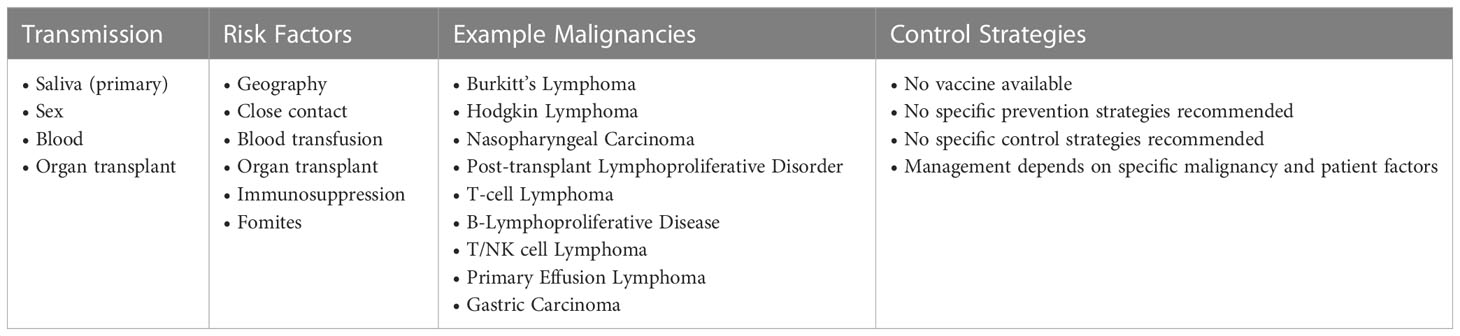
Table 3 EBV.
Human papilloma virus
Genomic organization and protein products
HPV is a small, non-enveloped virus. There are over 120 types that have been characterized by whole genome sequencing. These are divided into two major types: α (mucosal infectivity) and β (cutaneous). Not all types are oncogenic (197). Mucosal HPVs are split into “high risk” and “low risk” depending on if they cause potentially malignant or benign lesions. High risk types includes HPV 16 and 18, which cause ~70% of cervical cancer (198). Cutaneous HPVs, such as HPV 5, are associated with non-melanoma skin cancers.
All types of HPV have a ~8 kb double-stranded DNA genome. The genome is organized into early and late regions (including the long control region (LCR)). Most HPV genomes encode 8 proteins, 6 in the early region and 2 in the late region. The early proteins (E1, E2, E4, E5, E6 and E7) are highly expressed during the infectious or replicative cycle of HPV. E proteins are essential for genome replication, cytoskeleton remodeling, immune modulation, and modulation of the cell cycle. Two E proteins, E6 and E7, are oncogenic (199). However, E5 and E2/4/5 can also play a role in early oncogenic processes and an alternative carcinogenic pathway (200). Some HPV types have different isoforms of the E proteins that may play important roles in modulation of the host cell cycle, the viral lifecycle and cancer progression (201).The late proteins (L1 and L2) are essential for formation of the viral capsid and virus assembly.
Unlike many other viruses, there is no viremia or cell-death with HPV and most infections are asymptomatic; replication is exclusively intraepithelial and depends on the on the state of differentiation of the epithelial cell that the virus infects (202). Thus, most individuals may not know they are infected, which contributes to HPV being one of the most common sexually transmitted infections (203). Almost all adults have had at least one HPV infection; multiple types may integrate into the human genome (204). Given the asymptomatic nature of HPV infections, cancer may be the initial clinical presentation.
HPV prevalence and distribution in SSA
SSA has a very high burden of HPV related cancers; 15.8% of all cancers for men and women can be attributed to HPV in the region (205). SSA is estimated to have the highest prevalence of cervical cancer rates in the world (age standardized incidence rate of 31.0 per 100,000 women) (206, 207). While HPVs are found both in men and women, women are disproportionally affected by HPV cancer (208). Data on HPV cancer trends in SSA is limited, with cervical cancer being best characterized. Incidence rates or HPV-associated oropharyngeal cancer, anal and penile cancer vary across the region; penile cancer rates are rising in some parts of Africa while the incidence of anal and oropharyngeal cancer seems to have remained low (209). Whether these data reflect actual incidence or changes in screening is unclear.
Throughout much of the region, there is a general lack of education about the risks of sexual activity and lack of resources for screening programs (such as cytology) or early treatment. For instance, in Mali, only 4.8% of women of reproductive age have been screened for HPV and cervical cancer. Lack of screening has resulted in a roughly 80% mortality rate for those diagnosed with cervical cancer (206). One study from the Democratic Republic of Congo showed that the highest risk group was women under the age of 30, particularly those who were HIV-positive (210). HIV-positive individuals in general have a higher risk for any type of HPV than HIV-negative individuals (211).
The distribution of HPVs is not uniform in SSA (208, 212). The high-risk HPV 16 is consistently found across SSA; predominance of other types varies regionally (212). Moreover, distribution may also be impacted by HIV status. In one study of women attending a clinic for cervical cancer screening in Sikasso, Mali, 63% of women were infected with high-risk HPVs (213). Interestingly, distribution of types differed by HIV status: HPV35/31/51-52-56 were associated with HIV-positive women while HPV31/56/52 were associated with HIV-negative women.
HPV associated malignancies
It has been estimated that roughly 30% of infection-associated cancers are caused by HPV (214). Most HPV cells are cleared by the immune response. If the response fails, HPV infection becomes chronic, and the viral genome can replicate. As this occurs, early proteins E6 and E7 increase expression (199). E6 and E7 induce immunosuppression so infected cells are not cleared. Moreover, E6 and E7 are oncogenic proteins that result in genomic instability, disruption of the cell cycle, cell proliferation, immortalization, and malignant transformation (215).
Of the mucosal HPVs, 14 cause malignancies, with the majority being caused by HPV 16 and 18 (198). There are several different malignancies, the most common being cervical cancer in females. Females can also experience vulvar and vaginal cancer. Males can experience penile cancer. Both males and females can experience anal, oropharynx, and other head and neck cancers. It should be noted that some types of HPV only cause non-malignant warts, which will not be addressed in this topic on malignancies.
As cervical cancer is the fourth most common cancer amongst females globally and a leading cause of death (216), it is the-HPV associated malignancy that has garnered the most attention. HPV 16 is linked to approximately 50% of cases, while HPV18 is linked to another 20%. HPV 31, 33, 45, 52, and 58 are responsible for an additional 19% (217, 218). Additional HPV types are also classified as high-risk due to their role in cervical cancer, though they cause a much smaller proportion of disease (219). Additionally, men who have sex with men are at high-risk for anal infections and cancers; incidence of these cancer types has been increasing in this population (220) (221). PLWH also represent a unique population when it comes to HPV. PLWH clear the virus less efficiently than the general population and the overall incidence of HPV derived-invasive cancer is higher (222). They can also be infected with multiple HPV types (223). Further, in PLWH, the distribution of affected anatomical sites, HPV-types (224), and HPV derived cancers differs from that of the general population (225).
With increasing HPV vaccination coverage, the epidemiology of cervical cancer and other HPV associated malignancies may shift.
Future directions
Currently, the best protection against high-risk cancer causing HPVs is vaccination (203). Widespread vaccine coverage, especially in areas where screening and early treatment programs are lacking could be an overall public health benefit and could lead to decreased incidence. Currently, it is estimated that only 20% of girls in SSA is vaccinated (226), which is one of the lowest rates in the world (227). Moreover, access to the nonavalent HPV vaccine is extremely limited, with the mono- and bivalent vaccines being most available. Furthermore, HPV related malignancy can be caused by HPV types not covered by the vaccines and HPV vaccination is not often prioritized as part of national vaccination programs in SSA (228). One study estimated that 90% vaccine coverage would reduce peak prevalence by 82% for Mali (229). HPV vaccines have evolved from bivalent products active only against HPV 16 and 18 to the current nonavalent vaccine. Current vaccines are effective against HPV 16 and 18 (206) and low-risk HPV 6 and 11 that cause the majority of anogenital warts. In addition to the aforementioned HPV types covered by the quadrivalent vaccine, the nonavalent vaccine also protects against HPV types 31, 33, 45, 52 and 58. The nonavalent product is indicated for prevention of cervical, vulvar, vaginal, anal, oropharyngeal and other head and neck cancers, genital warts, and some precancerous lesions caused by HPV (230).
While there are many different HPVs prevalent in the region, especially in Sub-Saharan Africa, the majority of cervical cancer is still caused by HPV 16 and 18, which means broader vaccination could have a major impact on HPV associated cancer and warts (211). Nonetheless, distribution of high-risk HPV types and associated malignancies could change with pressure from vaccines and societal factors. It is also likely to be different than the distribution seen in other geographic locations. Thus, ongoing regional characterization of HPV epidemiology will be needed to guide public health efforts.
Given the burden of cervical cancer in SSA, the WHO launched a campaign to increase vaccination coverage and eliminate cervical cancer (231). Following an educational program on HPV, it was found that vaccine acceptance was high in Bamako, Mali for both men and woman (206). However, disruptions to educational efforts and social mobilization have impacted such campaigns, resulting in disruptions to vaccine coverage, vaccine doses, and a downward trend in acceptability (231). It is important to reinvigorate such efforts. Combining these with related endeavors on primary and secondary prevention would further increase the impact (232). Additionally, secondary screening for cervical cancer could help decrease incidence (233). Secondary screening can be performed by visual assessment in resource limited settings such as SSA; when more resources are available, genotyping and cytology can be used to guide follow-up (234). The recommended frequency of screening, which varies with age and comorbidities, would be difficult to fully implement in the current SSA setting. Therefore, a multi-pronged approach is especially important given that HPV distribution is not uniform and current vaccines do not protect against all types of high-risk HPVs present in SSA (208, 212). Communication regarding the safety of vaccines is also essential.
Given the challenges to vaccination and that vaccination does not cover all types of HPV, there are many opportunities for research. For instance, we need to better understand how most HPVs are spontaneously cleared and to develop immunomodulatory vaccines or small molecule inhibitors that would be effective across all types (235). Additionally, vaccination is only effective prior to infection with HPV so development of immunomodulatory vaccines that could clear previous infections would be highly desirable. Table 4 shows key information on factors related to acquisition of infection, disease course, and prevention strategies for HPV.

Table 4 HPV.
Human T-lymphotropic virus type 1
HTLV-1 genomic organization and protein products
Human T-lymphotropic virus type 1 (HTLV-1), also called Human T-cell leukemia virus type 1, is a single stranded RNA retrovirus. Study of HTLV-1, the first oncogenic human retrovirus described, began in 1977 (236) (237) (238). Like HIV, the most common retrovirus, HTLV-1 targets T cells and reverse transcribes its RNA to make double stranded DNA that integrates into the host genome as a provirus. Unlike HIV, its primary mode for maintaining copy number during natural infection is clonal expansion as opposed to infection of new cells (239, 240). Analogous to HIV, the HTLV-1 genome contains gag, pol and env genes and is flanked by long terminal repeats (LTR) at the 5’ and 3’ ends. It has additional genes, particularly tax and HBZ, which regulate persistence and expansion of HTLV-1 infected cells. Activity varies depending on the balance of sense and anti-sense transcription (241). At this time, genomic variation is not fully characterized and association of mutations with specific clinical phenotypes is unclear (242). While pathogenic mechanisms of HTLV-1 remain under investigation, induction of cellular proteins that induce transformation plays a role; pure insertional mutagenesis or capture of a cellular proto-oncogene are unlikely (243).
Tax is a transcriptional activator/repressor capable of modulating expression of multiple cellular genes. It also interacts directly with diverse proteins, is antiapoptotic and promotes cell proliferation (244, 245). The tax protein can immortalize cells in vitro and forced expression in transgenic mice leads to development of leukemia/lymphoma (246, 247). The primary mechanism of tax transformation is related to cell cycle reprogramming and inhibition of DNA repair (248). Tax also induces NFκB activity, which stimulates expression of cytokines and their receptors, including those of IL-13, IL-15, IL-2, IL-2Rα and co-stimulatory surface receptors (OX40/OX40L) (249–251). This activity mimics the chronic inflammatory process, which promotes oncogenic progression of many cancers, and triggers proliferation of T-cells to amplify the pool of HTLV-1 infected cells. Unlike other cancers in which the inflammatory process is mediated by immune cells in response to an oncogenic insult, HTLV-1 progression is directly induced by Tax. In addition to NFkB promoters, Tax also regulates expression of cellular transcriptional promoters through interaction with cyclic response element binding protein-AMP (CREB) and serum response factor (SRF) (252).
HBZ is located on the minus strand of the HTLV-1 provirus. It encodes a Basic Leucine Zipper Domain (bZIP) protein called HTLV-1 bZIP factor (HBZ). HBZ regulates 5’LTR transcription and modulates several cellular signaling pathways involved in cell growth, immunologic responses, and T-cell differentiation. It has been known to promote leukemic cell proliferation and induce T-cell lymphoma and systemic inflammation (253).
Compared to other retroviruses, HTLV-1 is genetically conserved and evolves slowly via accumulation of point mutations and recombination (254). Molecular studies suggest an evolutionary rate between 5.6 x10−7 and 1.5 x 10-6 substitutions/site/year. This slow rate has been attributed to persistence by clonal expansion as opposed to new infectious cycles using reverse transcriptase (255).
Phylogenetic analyses of the LTR region have demonstrated several HTLV-1 genotypes and subgroups which reflect geographic distribution (256–259). The three major molecular genotypes are: Cosmopolitan a-genotype, Central African b-genotype, and Australo-Melanesian c-genotype. The minor genotypes d, e, f, and g have also been characterized in Central Africa (260, 261), particularly in local pygmies (257, 262, 263). The Cosmopolitan genotype HTLV-1a can be subdivided into geographically related subgroups: Transcontinental (a-TC), Japanese (a-Jpn), West-African (a-WA), North-African (a-NA), and Senegalese (a-Sen). Genetic diversity is low within subgroups. Genotype HTLV-1b is found in Central Africa, and is the major genotype in Gabon, Cameroon, and Democratic Republic of Congo. Strains from the HTLV-1d genotype constitute a few percent of strains in Central African countries; genotypes e, f, and g have been sporadically reported in Cameroon, Gabon, and the Central African Republic. HTLV-1c, the most divergent genotype, is only in Australo-Melanesia (255).
HTLV-1 prevalence and distribution in SSA
WHO estimates that 5-10 million people worldwide are infected with HTLV-1 (238), though this is probably an underestimate due to inadequate data (264, 265). While HTLV-1 has spread globally, its geographical distribution is not uniform. The virus is endemic in Africa (266, 267) and is frequently found in Melanesia, Papua New Guinea (268), Solomon Islands and Australian aborigines (264). The greatest genetic diversity is observed in SSA, where six of the seven genotypes have been found; five of these are mainly in Central Africa (261).
Studies representing a total of 42,297 participants have shown seroprevalences of 4.16% (95% CI 2.43-6.31%), 2.66% (95% CI 1.80-3.68%) and 1.56% (95% CI 0.48-3.15%) in Central, West and Southern Africa, respectively. Of note, prevalence varied significantly by year (269, 270). In terms of specific populations, seropositivity rates range from 1 to >5% of West African blood donors and pregnant women in Ghana (271), Benin (272, 273), Mali (274) and Guinea-Bissau (275). One serologic survey in Guinea-Bissau in 2000 showed a prevalence of 9.3% amongst blood donors and 3.6% in the general population (276); a prior survey showed a prevalence of 3.3% of pregnant women (264). Although serologic data has been the foundation for prevalence estimates, it may be confounded by antibody cross-reactivity with malaria antigens (277–279).
HTLV-1 is transmitted primarily through infected body fluids, including blood, breast milk and semen. Risk factors include unprotected sex, injection drug use, and transplantation of tissue, blood, and blood products (280–282). Transfusion of HTLV-infected blood is the most effective mode of transmission due to presence of infected lymphocytes. Estimated seroconversion rates range from 27% to 63% after exposure to seropositive cellular blood components. Fortunately testing of blood donations for HTLV-1 in several high-income countries has greatly reduced transfusion-transmitted HTLV-1 (283). Expansion of blood product screening, particularly in SSA, presents an opportunity for improving control of HTLV-1 and its sequelae.
Interruption of mother-to-child transmission is also an intervention target. The estimated mother-to-child transmission rate is 18-30% (284, 285), which occurs primarily through breast-feeding as opposed to before or during birth. Breastfeeding longer than 6 months has been associated with transmission, leading to the hypothesis that shortening the duration of breastfeeding may reduce HTLV-I transmission. However, vertical transmission is more likely multi-factorial as approximately 3% of children who were not breastfed still became infected in intervention studies (286–289). As breast-feeding is nearly ubiquitous in SSA, control of vertical transmission will be extremely difficult in the region.
Of HTLV-1 infected patients, an estimated 6.6% of males and 2.1% of females develop adult T-cell leukemia/lymphoma (ATL) (264). There is also a potential association of both HTLV-1 infection and its sequelae with familial clusters, with up to seven people in a family affected (290, 291). ATL and HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) were the most frequently reported sequelae within families. Clustering may be related to familial transmission of HTLV-1, though other factors may be involved (292). Characteristics of gender and family dynamics may hold clues to HTLV-1 associated malignancy in Sub-Saharan.
HTLV-1-associated malignancy
Approximately 10% of HTLV-1 infected individuals develop associated disease; most people remain asymptomatic (261, 293). The virus is best known for causing ATL and HAM/TSP, though it is also associated with infectious dermatitis, uveitis, crusted scabies, inflammatory rheumatoid conditions, peripheral neuropathies, myositis, and atherosclerosis (264, 294, 295). This section will focus on ATL because it is the only malignancy associated with HTLV-1.
ATL was the first identified HTLV-1 associated disease, hence the virus’ name. It is a form of leukemia/lymphoma that derives from clonal expansion of infected CD4 T cells. These CD4 cells are presumably part of a long-lived pool of memory T cells with stem cell properties, though tumors express other T-cell associated antigens (296). ATL cases do not have distinct molecular or karyotypic abnormalities. Nonetheless, all cases have clonally integrated HTLV-1, with the most clinically aggressive variants manifesting the most extensive chromosomal alterations (297). Peripheral blood cells may have distinct hyperlobulated nuclei (298). ATL is classified into four clinical subtypes: acute (60%), lymphomatous (20%), chronic (10%) and smoldering (10%); primary cutaneous is an additional provisional subtype. Bone marrow infiltration, lymphadenopathy, skin involvement, hypercalcemia and immunosuppression occur to varying degrees (299, 300). Progression from chronic and smoldering forms to acute disease can occur (301). Diagnosis is based on immunophenotype, cell morphology, clinical features and presence of HTLV-1 (302).
Treatment of ATL depends upon clinical subtype. Intervention may be deferred for non-unfavorable chronic and smoldering subtypes, thought patients should remain under surveillance. Acute, lymphomatous, and unfavorable chronic ATL should undergo chemotherapy. Patients are at risk for hypercalcemia, tumor lysis syndrome and opportunistic infections during treatment. Allogeneic hematopoietic cell transplantation can be considered for those with an appropriate donor; this strategy may engender a beneficial graft-versus-leukemia effect (303). Regardless of treatment, prognosis is guarded.
Regions with highest prevalence of HTLV-1 infection have higher incidence of ATL. This includes Japan, intertropical Africa, the Caribbean, Central and South America, Romania, and northern Iran (304). Japan has the highest regional incidence, with annual rates approaching 60/100,000 HTLV-1 carriers and an estimated lifetime risk of 6-7% amongst men and 2-3% amongst women (305). Most cases stem from HTLV-1 contracted by breast-feeding and during early childhood (306). It has been estimated that the risk of ATL in perinatally infected people approaches 25% and that 2-4% of people will develop ATL within 30 years of HTLV-1 infection (307).
Although not a malignancy, the HTLV-1 associated condition HAM/TSP merits mention here. This is a chronic and progressive condition of adults in equatorial areas. It causes weakness, muscle stiffness and spasms, sensory disturbances, and sphincter dysfunction. The lifetime risk of HAM/TSP in HTLV-1 carriers is approximately 0.25-3%, which is lower than the risk of developing ATL (308, 309). There is no specific treatment, and the condition is associated with high morbidity and mortality (310, 311). As noted above, HTLV-1 is also associated with several other non-malignant manifestations.
Future directions
There is currently no effective and scalable prevention or cure for HTLV-1 infection. Asymptomatic infection is not treated, and management of associated disease is suboptimal. Transmission can occur from asymptomatic carriers through contact and organ or blood donation. The severity of HTLV-1-associated diseases and limited treatment options highlight the need for preventative vaccines and new therapeutic interventions (312). Unfortunately, no HTLV-1 vaccine candidate has yet progressed to efficacy studies in humans. Screening of blood and organ donations is helpful but does not address other major modes of transmission; parenteral, sexual, and vertical transmission must also be interrupted. Public health entities should support community-based strategies to increase awareness and reduce viral transmission.
While research on prevention of HTLV-1 infection and management of associated diseases is ongoing, clinicians can focus on early detection, particularly of ATL and HAM/TSP. This will be a challenge in SSA, where HTLV-1 infection is endemic and healthcare resources are severely limited. As with many medical conditions in SSA, people often rely on scarce public health resources which they access infrequently. Consequently, cases commonly present in advanced stages. Strategies for risk prediction should thus be developed. While proviral load has been suggested as a potential indicator, no single biological correlate of progression has been identified. In addition to proviral load, age, family history of ATL and HTLV-1 testing were identified as risk factors for development of ATL in a prospective study in Japan (313). Additional investigation can facilitate optimization of screening approaches.
In addition to research on prevention and management strategies, further epidemiologic characterization should be conducted. Understanding of subtype distribution, transmission patterns and factors that influence risk of progression will enable targeting of resources where impact will be greatest. A multipronged approach will provide the best prospect for SSA to mitigate HTLV-1 infection and its consequences. Table 5 shows key information on factors related to acquisition of infection, the associated malignancy, and control strategies for HTLV-1.

Table 5 HTLV-1 infection in humans.
Human herpesvirus-8 or Kaposi sarcoma-associated herpesvirus
HHV-8 genomic organization and protein products
HHV-8 is a double-stranded DNA virus of approximately 140 kilobases. Its genome is structured like other herpesviruses, with a single long unique region flanked by GC-rich terminal repeats at both the 3’ and 5’ ends (314). While the central region of the genome is highly conserved, both ends demonstrate significant variability. Sequencing has revealed 86 genes, 22 of which encode immunomodulatory proteins (315). ORF-K1 at the 5’ end encodes a glycosylated transmembrane protein with roles in signal transduction, viral reactivation, endothelial cell immortalization, and host immunity (316, 317). High variability of the ORF-K1 distinguishes the six main HHV-8 clades, which are A, B, C, D, E and F (317–319). The K15 gene at the 3’ end encodes and integral membrane protein and gives rise to the P (predominant), M (minor), and N genotypes. Nine additional loci with less variability than K1 and K15 can be used for subtype characterization (320, 321). Although the HHV-8 gene map has changed little since it was first characterized in 1996 (322), additional evaluation of HHV-8 genotype nomenclature based on evolving methodology is underway (323).
Consistent with other herpesviruses, the HHV-8 life cycle includes lytic and latent phases. Latency is the default pathway; it has been seen in nearly all tissues (324, 325). During this phase, the viral DNA constitutes a non-integrated plasmid and has limited expression. One critically expressed gene used for diagnosis of KS and Primary Effusion Lymphoma (PEL) is latency-associated nuclear antigen (LANA) from ORF73 (326–328). LANA can repress transcription of lytic genes and recruit other transcriptional repressors (329). Lifelong latency is established primarily in B lymphocytes that reside in tissues, with genomes being difficult to detect in plasma or PBMC (330).
Reactivation of HHV-8 from latency is initiated when expression of the replication and transcription activator (RTA) protein is stimulated. RTA is known as the master lytic switch protein (331). It competes with LANA for binding to suppressive co-factors, leading to transactivation of 34 lytic genes (332). Transactivation of the RTA promoter creates a feed-forward loop to overcome repression and further increase lytic gene expression (333). Environmental stimuli such as hypoxia and calcium signaling can promote RTA activation, which may influence anatomic localization of associated lesions (334, 335).
Entry of HHV-8 to the lytic phase engenders oncogenesis. Thus, addressing HHV-8 infection and associated disease is essential for prevention of morbidity and mortality.
HHV-8 prevalence and distribution in SSA
While HHV-8 causes significant morbidity and mortality, most infections remain latent. Thus, prevalence is much higher than reflected by cases of associated malignancy. Estimates based on seroprevalence in adult populations in Africa or of African origin suggest that 36-90% of people have been infected. Some studies showed increasing seroprevalence with age (336–339). In Gabon, where seroprevalence based on plasma anti-LANA antibodies was only 36%, locality, age, sex and ethnic group were not correlated (340). The generally high prevalence of HHV-8 infection in SSA means that a large proportion of the population is at risk of HHV-8 associated disease.
Within SSA, HHV-8 genotypes A and B predominate, along with a lower proportion of F (341). Within genotype A, clade A5 is most prevalent (342). Phylogenetic analyses indicate that circulating strains are dynamic, with those circulating today being distinct from those circulating at the height of the HIV epidemic. However, unavailability of banked samples makes it difficult to elucidate temporal evolution in Africa (314).
As HHV-8 genotype influences risk of disease progression, it is important to understand the regional epidemiology of underlying infection. Analysis of ORF K1-VR1 sequences in blood, serum, and saliva of 38 patients with Kaposi Sarcoma (KS) found that genotype A was associated with rapid progression (12/17 cases) and that C was most common in slow progressors (6/7 cases). Genotype A was also associated with higher viral load in the blood (343). A South African study that performed ORF-K1 subtyping of tissue biopsies from 86 KS patients, 81 of whom had AIDS and 5 of whom had African endemic-KS, showed that subtypes A5 (38 AIDS + 4 African endemic/86) and B2 (16 AIDS/86) predominate. A5 and B were found in African blacks and individuals of mixed ancestry. 62/86 (72%) had >10 lesions at presentation; A5 was associated with >10 KS lesions in patients with AIDS. Thus HHV-8 subtypes A5 may be associated with more extensive disease (318).
Genomic polymorphisms impact disease presentation and may vary by anatomic site. Whole genome sequencing of Zambian KSHV revealed phylogenetic segregation from Western sequences, including polymorphisms in the more conserved genes. Such geographic diversity may engender differences in pathogenesis (319). Conversely, analysis of KSHV genomes from patients and controls in Cameroon revealed that recombination is common, suggesting multiple KSHV infections can occur. Yet sequence variation did not correlate with disease risk (344). A Ugandan study of KSHV genomes from tumors and oral swabs found that genomes were identical at the point mutation level within individuals but intra-host tumor-associated KSHV mutations and genome sequences impacting protein coding were present (345). On the host side, an association likely exists between HLA polymorphisms and susceptibility to KSHV infection and related diseases (346).
Given the high seroprevalence of HHV-8 subtypes associated with more severe disease in SSA, prevention, screening, and management strategies are critical. Saliva serves as the main mode of transmission and infection is common within families. Mother to child transmission and sex also contribute to varying degrees in different populations (347). Screening for HHV-8 is not standard, with infection typically being evaluated only after someone presents with a disease manifestation. Management varies from reconstitution of the immune system to multiple cycles of chemotherapy. Public health strategies to address HHV-8 in SSA must balance the challenge of prevention, cost of disease management and socioeconomic context.
HHV-8 associated malignancies
Major malignancies associated with HHV-8 include KS, PEL and Multicentric Castleman’s Disease (MCD) (348). HHV-8 positive diffuse large B-cell lymphoma and germinotropic lymphoproliferative disorder also occur. These cancers are usually found in immunodeficient patients, but can affect immunocompetent individuals (349) Manifestations typically arise many years after acquisition of infection and only in a subset of those who are infected.
Primary HHV-8 infection is usually asymptomatic (350), with most people seroconverting without progression to malignancy. However, manifestations vary amongst populations by age and risk factors (351, 352). Immunocompromised people can develop more generalized manifestations as well as rapid onset KS (353).
KS is the most common and well-studied malignant manifestation of HHV-8 infection. It remains one of the most frequent cancers amongst HIV-infected patients (354) and children from endemic regions, including Central, Eastern, and Southern Africa (355). There were 34,270 new cases of KS and 15,086 associated deaths reported worldwide in 2020. Actual morbidity and mortality are probably greater. Africa shouldered the greatest burden, with 25,010 (73%) of the cases; 15,457 of those were in the Eastern Africa region. 13,066 (87%) of the deaths occurred in Africa, with 9,121 of them in the Eastern Africa region (356).
KS is an angioproliferative, cytokine-driven neoplasm which frequently presents with peripheral skin lesions (357). Lesions also affect the mucosal membranes and viscera, where manifestations may be due to mass effect or obstruction. Pediatric KS in endemic regions is known to present in lymph nodes and without skin findings (358, 359). Four types of KS are recognized. Classic KS is a slowly progressive cutaneous disease mainly seen in elderly Mediterranean and Jewish males (360). Endemic KS affects all ages and is particularly common in SSA (361). This form is not immunosuppression-associated and is more aggressive, disseminating to lymph nodes, bone and skin (362). Solid organ transplant-associated KS occurs post-transplant and may be due to HHV-8 transmission during transplant. Disease duration is usually related to immunosuppression, with regression occurring when the immune system reconstitutes (363). Epidemic KS, also known as AIDS-related KS, is >20,000 times more common in AIDS patients compared to the general population. It can also occur in other immunosuppressed hosts, but is still much more common in AIDS patients (364).
KS-immune reconstitution inflammatory syndrome (KS-IRIS) can occur when the immune system of an immunosuppressed patient with KS recovers. Symptoms such as increase in lesions, fever, inflammation, and pain occur when the immune system suddenly recognizes antigens to which it was previously not responding. KS-IRIS is typically seen in advanced HIV patients who initiate ART and had KS, though lesions may have been subclinical (365). It is associated with significant morbidity and mortality, particularly in Africa where adjunctive therapies may be less available (366). A study of 58 KS IRIS patients in SSA and the UK found that KS associated mortality was 3.3 fold higher in SSA compared to the UK and was predicted by KS-IRIS, lack of chemotherapy, pre-ART CD4 <200 cells/μL, and detectable baseline KSHV DNA. All 19 study deaths occurred in the SSA cohort (367).
KS-Associated Herpesvirus Inflammatory Cytokine Syndrome (KICS) is a consequence of HHV-8 seen in HIV+ patients with elevated plasma HHV-8 viral loads. It entails systemic inflammatory manifestations and high levels of IL-6 and IL-10 (368, 369). KICS overlaps somewhat with other HHV-8 associated conditions and has high mortality rate.
MCD is an uncommon lymphoproliferative disorder that can present with generalized lymphadenopathy, hepatosplenomegaly, and constitutional symptoms including fever, night sweats, weight loss, and fatigue. Patients are typically cytopenic and can have hypoalbuminemia, polyclonal hypergammaglobulinemia and elevated inflammatory markers. MCD often occurs in the setting of HIV or other etiologies of immunosuppression. It involves multiple areas of lymphadenopathy that is histopathologically on the spectrum of hyaline vascular to plasmacytic (370, 371). HHV-8 causes approximately half of the MCD cases (372). It should be noted that the unicentric form of Castelman’s Disease, which affects a single anatomic region, is not associated with HHV-8 infection.
Although specific diagnostic criteria are not available, MCD is usually identified through histopathologic examination of lymph node biopsy. Fluorodeoxyglucose positron emission tomography (FDG-PET) combined with computed tomography (CT) will reveal activity at multiple sites, though activity is lower than seen with aggressive lymphomas (373). This diagnostic approach is not broadly feasible in resource limited settings. It is likely that many cases of MCD go unrecognized in SSA. Management of MCD depends on the presence of concomitant KS, extent of organ involvement and underlying immunosuppression. Patients with HIV will use ART. Rituximab can be used for simple MCD without organ failure or KS. Chemotherapy is used in more extensive cases. 5-year survival is >90% with proper management, though patients remain at risk for developing non-Hodgkin lymphoma (374).
MCD in the African context has unique features and management challenges. Characterization of MCD in SSA has been limited due to incomplete case recognition. Underdiagnosis of MCD in SSA was suggested by immunohistochemistry positive LANA and viral IL-6 in 3/64 reactive appearing lymph nodes from a Ugandan study (375) and identification of only 35 cases in South Africa from 1990-2014 (376). The first case of MCD in Malawi was reported in 2014 (377). The same group subsequently reported on a Malawian cohort of MCD, lymph node KS and NHL. MCD was typically diagnosed late and associated with high mortality (378). Building on this study, the first well-characterized prospective cohort of MCD in the context of HIV SSA revealed only HIV+ KSHV+ MCD, which differs from the heterogenous epidemiology of MCD in high-income settings where cases can be HIV- and KSHV-. This is not surprising given the high regional prevalence of both HIV and KSHV (376).
PEL is a rare and aggressive B-cell lymphoma that typically occurs in HIV-infected patients, though it can occur in other immunosuppressive conditions (379, 380). Tumor growth depends on HHV-8 infection, and sometimes co-infection with EBV (381, 382). In addition to containing HHV-8 genetic material, the malignant monoclonal B cells express surface CD38 (383). PEL tends to occur on serosal surfaces in body cavities such as the peritoneal, pleural, and pericardial spaces, which is why it is also known as body cavity based lymphoma. Clinical presentation is usually due to fluid accumulation, though presentation with solid tumors is possible (384). Treatment is chemotherapy with ART and prognosis is generally poor, with survival being approximately 3-6 months (385). The burden of PEL in SSA remains unclear, as cases are likely much more frequent than recognized in this endemic area (386, 387).
Future directions
SSA’s confluence of high HHV-8 infection prevalence, high proportion of more aggressive HHV-8 genotypes, burden of HIV and lack of healthcare infrastructure have made HHV-8 associated malignancy a major cause of morbidity and mortality in the region. While KS, the most common malignant manifestation of HHV-8, may be recognized clinically, less common manifestations such as PEL and MCD are inevitably underdiagnosed. Thus, diagnosis and treatment strategies must be developed within the context of SSA. For example, rapid case ascertainment, the expeditious evaluation of a potentially rapidly fatal condition shortly after diagnosis, is feasible for investigation KS in East Africa but is mostly used in resource rich settings (388). One study using rapid case ascertainment of newly diagnosed KS amongst PLWH in Kenya and Uganda showed that most patients had advanced disease despite recommendations for ART for all PLWH. Advanced disease was heterogeneous and raised questions about the possible need for different treatment strategies in this group (389). Also, the most effective chemotherapeutic agents are rarely used in SSA due to high cost. Yet, modeling can reveal the impact of therapies on life expectancy and their cost effectiveness to inform efficient allocation of resources. Given the high prevalence of HIV in SSA, modeling for AIDS-associated KS is particularly salient (390). In the same context, T-cell sparing treatments of KSHV associated diseases should be considered (391). Evaluation of treatment outcomes with respect to epidemiology of KSHV associated conditions in children and adolescents should also be enhanced. A study in Southwestern Tanzania showed favorable outcomes in a varied cohort, but generalizability of these findings is unclear (392).
As appropriate management requires evaluations of the malignancy and host factors that may not be feasible given available resources, prevention of HHV-8 infection would seem to be a desirable goal. Minimization of progression to malignancy would be another key target. Effective HHV-8 infection prevention strategies remain unclear, particularly because routes of transmission have not been fully elucidated. Saliva is the main route, though transmission through sex, blood and organ transplant also occur. Transmission via saliva in combination with high underlying prevalence in SSA make exposure likely in the general population. Furthermore, there is no vaccine for HHV-8.
Once someone is infected with HHV-8, likelihood of progression to malignancy depends on host factors. The main risk is immunosuppression, most often associated with HIV infection. SSA has the greatest worldwide burden of HIV and sub-optimal, though improving, rates of identification and effective management of PLWH. Thus, prevention of progression to malignancy will largely hinge upon controlling the HIV epidemic. Fortunately, progress is being made in this area, including expansion of public health programs with international support.
Considering the limited options for prevention of HHV-8 infection and progression to malignancy in those who are infected, research should be expanded in these areas. Development of an effective HHV-8 vaccine should be prioritized. The vaccine would ideally prevent infection by all genotypes but would still be helpful if it just covered the high-risk genotypes prevalent in SSA. It should induce sterilizing antibody responses to prevent primary infection along with T-cell responses to inhibit cancer development in those who get infected (393). Even a vaccine that does not completely prevent infection, but prevents complications, would be valuable. Platforms used in the recent rapid development of vaccines for COVID-19, and other infections may facilitate development of an HHV-8 vaccine. Other areas of research could include scalable strategies for diagnosing less common HHV-8 associated malignancies, treatment for resource limited settings, and transmission prevention techniques. These would be enhanced by further characterization of HHV-8 epidemiology, continued investigation of pathogenesis, understanding of host factors including genetic and epigenetic contributors, and environmental modifiers.
HHV-8 is a common infection in SSA, as well as a significant cause of morbidity and mortality. Much work remains to be done on prevention of infection, treatment of complications and novel research to improve control. Table 6 shows key information on factors related to acquisition of infection, associated malignancies, and control strategies for HHV-8.

Table 6 HHV-8.
Human immunodeficiency virus
HIV genomic organization and protein products
While HIV-1 and HIV-2 are genetically distinct, their genome, proteins and end stage disease have many similarities. In this section, HIV refers to both HIV-1 and HIV-2; discussion relevant to only one type of HIV will be specified accordingly.
HIV is an RNA virus with an approximately 9kb genome containing nine genes and encoding fifteen proteins. gag, pol and env are the three major genes constituting most of the genome. gag encodes the structural proteins Matrix, Capsid, Nucleocapsid, and p6; pol encodes the enzymes Protease, Reverse transcriptase, and Integrase; env encodes envelope proteins gp120 and gp41. The smaller remaining genes code for regulatory proteins Tat and Rev, as well as accessory proteins Vif, Vpr, Vpu/Vpx, and Nef (394, 395). Infected cells have integrated DNA copies of the virus, or proviruses, which are non-defective or defective. Viruses produced from non-defective proviruses and proteins produced from defective proviruses can facilitate oncogenesis.
Treatment with antiretroviral therapy (ART) targets specific HIV proteins at key steps in the viral lifecycle, e.g., reverse transcriptase inhibitors, protease inhibitors, and integrase inhibitors. Both HIV-1 and HIV-2 are treated similarly with ART. However, the non-nucleoside reverse transcriptase inhibitors (NNRTI) cannot be used in HIV-2 due to structural differences in the NNRTI binding pocket of HIV-2 reverse transcriptase (396). Although three HIV-1 cases have achieved sterilizing cure through stem cell transplant, lifelong ART is generally required because there is no scalable HIV cure. If ART is stopped, the virus rebounds from the integrated proviral DNA reservoir (397). While ART has toxicities that engender concern about promotion of oncogenesis, it inhibits the oncogenic effects of HIV by reducing production of oncogenic proteins and controlling viral load.
HIV prevalence and distribution in SSA
Of the estimated 37.7 million people living with HIV (PLWH) globally in 2020, approximately 25.3 million were in SSA. Most of the infections are HIV-1, with HIV-2 accounting for an estimated 1-2 million cases (398). Eastern and Southern Africa shouldered the greatest burden with 20.6 million PLWH compared to 4.7 million in Western and Central Africa. Access to ART was around 78% for the general adult population, but lower for pregnant women in Western and Central Africa and higher for pregnant women in Eastern and Southern Africa. The only UNAIDS region with greater adult access to ART is Western and Central Europe and North America, where 83% of adult PLWH have ART access. Women and girls accounted for 63% of new HIV infections in SSA during 2020. Amongst adolescents 15-19 years old, approximately 85% of new infections were in females (399). Evidence suggests substantial variation in HIV prevalence across SSA localities. A high-resolution study of adult epidemiology showed generally higher prevalence in southern regions, with pockets of high prevalence in other areas. Risk factors did not consistently align with prevalence; condom use was higher in southern areas and male circumcision was higher in northern and central regions (400).
While HIV prevalence is decreasing in SSA, most infections continue to occur amongst females. Heterosexual sex is the primary mode of transmission. Populations disproportionately affected include sex workers, men who have sex with men (MSM), injection drug users (IDU), transgender persons and prisoners (401). Prevalence is particularly high in areas of high economic activity, likely related to seasonal work and commercial sex. High prevalence areas are clustered around high traffic regions including border crossings, major highways, ports, and areas of migration (402). SSA’s already high HIV prevalence is exacerbated by the local social, political, and economic climate. High levels of poverty, lower levels of education, and poor healthcare infrastructure hinder achievement of the UNAIDS “95-95-95” HIV cascade of care targets, under which 95% of PLWH know their status, 95% of those diagnosed are on ART and 95% of those on ART achieve viral suppression by 2030 (403).
Young adults have especially inadequate access to sexual health resources. Differences between male and female sexual debut, age disparate sex, transactional sex, multiple partnerships, low condom use and high background STI rates contribute to vulnerability, particularly amongst young women (404). While HIV pre-exposure prophylaxis (PrEP) consisting of oral ARV taken regularly or around sexual activity is approved, uptake in SSA has been inadequate. Challenges including target population engagement, use associated stigma, adherence requirements, healthcare infrastructure, provider unfamiliarity, community awareness and accessibility highlight opportunities for national public health program development (405, 406). Recent approval of injectable cabotegravir as PrEP may alleviate some barriers (407).
While HIV-1 and HIV-2 share transmission routes and cause AIDS, it should be noted that HIV-2 has lower rates of transmission and slower clinical progression. HIV-2 is also more geographically localized than HIV-1. Most people with HIV-2 are in West Africa, or countries with strong ties to the region such as France, Spain, and Portugal (398). Furthermore, HIV-2 prevalence in West Africa seems to be declining, likely due to lower virulence and transmissibility (408). The lower prevalence and pathogenicity of HIV-2 contribute to it being less well studied and underly the focus of HIV control efforts on HIV-1.
Understanding the HIV epidemic in SSA, and control strategies, will be key to minimizing HIV associated complications. PLWH are at increased risk for morbidity and mortality, including development of malignancy. Prevention of HIV and associated cancers will reduce socio-economic burden on the region.
HIV-associated malignancies
HIV is well recognized for enabling oncogenic potential of other viruses through decimation of immunologic function that typically suppresses oncogenic activity. While both HIV-1 and HIV-2 are endemic to SSA, they carry different risks for malignancy. This is primarily due to varying rates of disease progression, as the propensity for malignancy is mostly related to degree of immunosuppression (409). Since HIV-2 progresses much more slowly than HIV-1, AIDS defining conditions often take decades to manifest even without ART. Research on HIV-1 and malignancy has been more extensive due to greater prevalence and societal impact. For example, the first case of multicentric Castelman’s disease associated with KSHV in HIV-2 was not reported until 2007, well after it was identified in HIV-1 (410).
HIV associated malignancies considered AIDS defining illnesses include Kaposi Sarcoma, select non-Hodgkin lymphomas, and cervical cancer in certain populations. AIDS defining illnesses typically occur at very low CD4 counts. HPV, which most notoriously causes cervical cancer, can be oncogenic against any immunologic backdrop, but persists longer and causes more disease in HIV infected people. There has been uncertainty about whether HIV-2 may be slightly more associated with high-grade lesions and cervical cancer than HIV-1 or whether HPV type and persistence are the determining factors (411–413). Progression of HHV-8, the etiology of Kaposi Sarcoma, may be augmented in HIV-1 compared to HIV-2 (414).EBV underlies some types of non-Hodgkin lymphoma, which has been better characterized in HIV-1 versus HIV-2 (415, 416). Nonetheless, mechanisms of oncogenesis are similar and the below discussion about HIV-associated malignancies pertains to both HIV-1 and HIV-2, except as otherwise noted. As specific malignancies associated with EBV, HHV-8 and HPV have been previously addressed, this section will focus on how HIV acts at the cellular level to potentiate oncogenicity of these and other viruses, including evidence for direct oncogenesis.
PLWH are at increased risk of developing malignancies despite effective ART, demonstrating that factors other than CD4 depletion and exhaustion of lymphopoiesis contribute. Non-AIDS defining malignancies, including hepatocellular cancer, brain cancer and squamous cell carcinomas occur at increased frequency and/or severity in PLWH compared to HIV-uninfected people (417–419). These malignancies would be expected later in the course of HIV-2 due to its slower progression. HIV facilitates development of malignancy via dysregulation of innate immunity, persistent immune activation, dysfunction of the inflammatory response and immunosenescence (420, 421). Causes of abnormal immune activation and inflammation are multifactorial, related to persistent antigen stimulation by viral proteins and microbial translocation, inadequate generation of memory cells, and exhaustion of effector cells (422). B and T cell defects along with aberrant monocyte and natural killer cell function perpetuate the dysfunction (423–425). Accordingly, CD4:CD8 ratio has been associated with cancer risk amongst PLWH (426).
Chronic immune activation and dysregulation, reflected by elevated inflammatory markers (427), enhance oncogenesis through several mechanisms. Since HIV replicates in activated CD4 cells, replication and spread increase. CD4 activation also upregulates CCR5 co-receptor expression, enhancing susceptibility to HIV infection (428). Impaired immune surveillance simultaneously occurs due to HIV-induced CD4 lymphopenia and ineffective CD8 responses (429, 430). CD4 lymphopenia engenders inadequate production of high-affinity antibodies, and is associated with high-risk HPV progression (431). T-cell senescence reflected by reduced T-cell polyfunctionality and T-cell receptor diversity portend poorer prognosis in certain HIV-associated malignancies (432). Ongoing inflammation is associated with HIV disease progression (433, 434) and non-AIDS comorbidities (435). These mechanisms underly the role of immunotherapy, including ART, check point inhibitors, monoclonal antibodies to specific cell types, IL-7, IL-12, and IL-15 in management of HIV-associated malignancies (436).
In addition to immunomodulatory effects, HIV proteins cause direct oncogenesis through induction of oxidative stress, integration into cancer associated and cell cycle regulatory genes, and interaction with other oncogenic viruses. The HIV proteins tat, gp120, nef, reverse transcriptase, and matrix protein p17 have been implicated in specific interactions that provoke cellular transformation, tumor propagation, and reactive oxygen species (437). For example, HCV and HIV synergistically increase reactive oxygen species in stem cells and hepatocytes. This promotes activation of kinases controlling cell growth, differentiation and apoptosis, which stimulate factors promoting inflammation and cell death, ultimately potentiating hepatocellular cancer (438). Furthermore, proviruses in CD4 cells from individuals on long-term ART can integrate into cell growth regulatory genes to influence persistence and clonal expansion of HIV-infected cells (439). A study of 534 HIV integration sites and 63 adjacent HIV env sequences from three individuals on ART for 11.3-12.7 years demonstrated identical viral sequences integrated at the same position in multiple cells consistent with infected-cell proliferation. Integrations were overrepresented in cancer-associated genes (440).
HIV may also promote oncogenesis via enhancement of age-related clonal hematopoiesis, which is associated with hematologic malignancies (441–443). In a prospective cohort study of 220 HIV-1+ and 226 HIV- participants ≥55 years, clonal hematopoiesis occurred in 28.2% of HIV-1+ participants and 16.8% of HIV- participants (p = 0.004); the adjusted odds ratio for occurrence of clonal hematopoiesis in HIV was 2.16 (95% CI 1.34-3.48, p = 0.002). Clonal hematopoiesis and HIV-1 infection were independently associated with inflammatory biomarkers, suggesting a selective advantage for emergence of clonal hematopoiesis in the context of chronic HIV and associated inflammation (444).
HIV not only enhances oncogenesis but may also be impacted by malignancy. Concurrent cancers can induce cytokines that activate cells (445) containing HIV proviral reservoirs. This may enhance production of replication competent virus and defective viral proteins, which stimulate the inflammatory cascade. Cancer treatments may also interact with ART, leading to reduced efficacy or impaired CD4 recovery and function (446, 447). Thus malignancy and HIV act synergistically to promote both diseases.
Future directions
Preventing new HIV infections, identifying existing infections, effectively treating PLWH and developing strategies to mitigate the oncogenic effects of HIV will reduce the impact of HIV-associated malignancies. These approaches require augmentation of existing infrastructure in SSA and targeted public health programs, including addressing differences in HIV-1 versus HIV-2. As problem-based visits often take priority over routine health maintenance in resource limited settings, opportunities for prevention and screening of the general healthy population are limited. Point of care HIV testing and education should be offered at least once to all presenting for medical care, with regular testing for at-risk populations. Mobile screening, education of local opinion leaders and increasing awareness about prevention strategies could help reduce transmission. For PLWH, cancer screening should be standardized, and infrastructure should be developed to support treatment. National registries to ensure continuity of care should also be established, while protecting confidentiality.
Prevalence of HIV associated malignancies in SSA will reflect control of the HIV epidemic. In high-income countries, the proportion of HIV-related deaths due to cancer is increasing but has transitioned toward non-AIDS defining cancers in conjunction with improving ART coverage and HIV control at cancer diagnosis. While assessment of trends in resource limited settings is more difficult due to lack of infrastructure and data, they will likely follow the same pattern as better control of HIV is achieved and cancer screening expands (448). A 2020 review of clinical trials for treatment and prevention of HIV-associated malignancies in SSA found 11 studies specific to PLWH (449). Several trials were under the auspices of the National Cancer Institute supported AIDS Malignancy Consortium, which continues to expand efforts. AIDS Malignancy Consortium research areas for Africa include hematologic malignancies, HPV, Kaposi’s sarcoma and solid tumors (450). Expansion of such multi-national research networks will be key to addressing the situation in SSA. Future studies should fully characterize the epidemiology of HIV associated malignancies, investigate pathogenic mechanisms, and evaluate treatment, prevention, and screening strategies. Table 7 shows HIV associated malignancies, affected populations, and approaches to prevention and treatment of associated malignancies.

Table 7 HIV-associated malignancies.
Conclusions
Malignancies related to viral infection are increasing in SSA, where prevalence of viral infections is elevated compared to other regions. All six viruses classified as oncogenic (EBV, HBV, HCV, KSHV, HTLV-1 and HPV) per the International Agency for Research on Cancer impact the region. HIV-1, for which SSA has the greatest global burden, has also been linked to increasing risk of malignancy. Public health approaches to prevention of infection, such as vaccination, safer injection practices, screening of blood products, antimicrobial treatments and safer sexual practices could reduce the burden of cancer in Africa.
To combat viral associated malignancies in SSA, policy makers must prioritize public health programs that increase awareness, provide prevention resources, offer screening, and administer treatment. Cultural nuances and socioeconomic constraints must be considered during implementation of control programs. Contemporary lifestyle practices, including diet and activity patterns, will need to be addressed. Social stigma associated with malignancy will need to be overcome to facilitate discourse and encourage engagement with available resources. National level infection and cancer registries will be helpful for elucidation of local epidemiology and targeting of efforts.
We anticipate substantial increases in the burden of virally mediated cancers will occur over the next decade. It is unclear if this will be due to true increases in incidence or better detection strategies. Additional research should be conducted to characterize the dynamic epidemiology of viral oncogenesis more fully, optimize intervention strategies and accommodate behavioral-social nuances related to implementation of public health programs in SSA.
Author contributions
MD, KS-S and C-YL designed and contributed equally to the writing of this article. All authors contributed to the article, reviewed the manuscript, and approved the submitted version.
Funding
This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institute of Health, under Contract No. 75N91019D00024, Task Order No. HHSN2612015000031, Task Order No. 21X143F1. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. World Health Organization. Assessing national capacity for the prevention and control of noncommunicable diseases: report of the 2019 global survey. Geneva: World Health Organization (2020).
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin May (2021) 71(3):209–49. doi: 10.3322/caac.21660
3. Reed FA, Tishkoff SA. African Human diversity, origins and migrations. Curr Opin Genet Dev (2006) 16(6):597–605. doi: 10.1016/j.gde.2006.10.008
4. Paschou P, Lewis J, Javed A, Drineas P. Ancestry informative markers for fine-scale individual assignment to worldwide populations. research support, non-U.S. gov’t research support, U.S. gov’t, non-P.H.S. J Med Genet (2010) 47(12):835–47. doi: 10.1136/jmg.2010.078212
5. Vogt PK. Retroviral oncogenes: a historical primer. Nat Rev Cancer (2012) 12(9):639–48. doi: 10.1038/nrc3320
6. Fan H, Johnson C. Insertional oncogenesis by non-acute retroviruses: implications for gene therapy. Viruses (2011) 3(4):398–422. doi: 10.3390/v3040398
7. Rubin H. The early history of tumor virology: Rous, RIF, and RAV. Proc Natl Acad Sci U.S.A. (2011) 108(35):14389–96. doi: 10.1073/pnas.1108655108
8. Javier RT, Butel JS. The history of tumor virology. Cancer Res (2008) 68(19):7693–706. doi: 10.1158/0008-5472.CAN-08-3301
9. Parkin DM, Hammerl L, Ferlay J, Kantelhardt EJ. Cancer in Africa 2018: The role of infections. Int J Cancer (2020) 146(8):2089–103. doi: 10.1002/ijc.32538
10. Griesel M, Seraphin TP, Mezger NCS, Hämmerl L, Feuchtner J, Joko-Fru WY, et al. Cervical cancer in Sub-Saharan Africa: A multinational population-based cohort study of care and guideline adherence. Oncologist May (2021) 26(5):e807–16. doi: 10.1002/onco.13718
11. Asasira J, Lee S, Tran TXM, Mpamani C, Wabinga H, Jung S, et al. Infection-related and lifestyle-related cancer burden in Kampala, Uganda: projection of the future cancer incidence up to 2030. BMJ Open Mar 16 (2022) 12(3):e056722. doi: 10.1136/bmjopen-2021-056722
12. Robinson WS. The genome of hepatitis b virus. Annu Rev Microbiol (1977) 31:357–77. doi: 10.1146/annurev.mi.31.100177.002041
13. Lucifora J, Arzberger S, Durantel D, Belloni L, Strubin M, Levrero M, et al. Hepatitis b virus X protein is essential to initiate and maintain virus replication after infection. J Hepatol Nov (2011) 55(5):996–1003. doi: 10.1016/j.jhep.2011.02.015
14. Koike K. Hepatitis b virus X gene is implicated in liver carcinogenesis. Cancer Lett (2009) 286(1):60–8. doi: 10.1016/j.canlet.2009.04.010
15. Murata M, Matsuzaki K, Yoshida K, Sekimoto G, Tahashi Y, Mori S, et al. Hepatitis b virus X protein shifts human hepatic transforming growth factor (TGF)-beta signaling from tumor suppression to oncogenesis in early chronic hepatitis b. Hepatology (2009) 49(4):1203–17. doi: 10.1002/hep.22765
16. Ou JH, Bao H, Shih C, Tahara SM. Preferred translation of human hepatitis b virus polymerase from core protein- but not from precore protein-specific transcript. J Virol (1990) 64(9):4578–81. doi: 10.1128/JVI.64.9.4578-4581.1990
17. Ganem D, Varmus HE. The molecular biology of the hepatitis b viruses. Annu Rev Biochem (1987) 56:651–93. doi: 10.1146/annurev.bi.56.070187.003251
18. Huang ZM, Yen TS. Role of the hepatitis b virus posttranscriptional regulatory element in export of intronless transcripts. Mol Cell Biol (1995) 15(7):3864–9. doi: 10.1128/MCB.15.7.3864
19. Le Seyec J, Chouteau P, Cannie I, Guguen-Guillouzo C, Gripon P. Infection process of the hepatitis b virus depends on the presence of a defined sequence in the pre-S1 domain. J Virol (1999) 73(3):2052–7. doi: 10.1128/JVI.73.3.2052-2057.1999
20. Ou JH, Laub O, Rutter WJ. Hepatitis b virus gene function: the precore region targets the core antigen to cellular membranes and causes the secretion of the e antigen. Proc Natl Acad Sci U.S.A. (1986) 83(6):1578–82. doi: 10.1073/pnas.83.6.1578
21. Lin CL, Kao JH. The clinical implications of hepatitis b virus genotype: Recent advances. J Gastroenterol Hepatol (2011) 26 Suppl 1:123–30. doi: 10.1111/j.1440-1746.2010.06541.x
22. Croagh CM, Desmond PV, Bell SJ. Genotypes and viral variants in chronic hepatitis b: A review of epidemiology and clinical relevance. World J Hepatol (2015) 7(3):289–303. doi: 10.4254/wjh.v7.i3.289
23. Lin CL, Kao JH, Chen BF, Chen PJ, Lai MY, Chen DS. Application of hepatitis b virus genotyping and phylogenetic analysis in intrafamilial transmission of hepatitis b virus. Clin Infect Dis (2005) 41(11):1576–81. doi: 10.1086/497837
24. World Health Organization. Global progress report on HIV, viral hepatitis and sexually transmitted infections, 2021 (2022) (Accessed 10 May 2022).
25. Ott JJ, Stevens GA, Groeger J, Wiersma ST. Global epidemiology of hepatitis b virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine (2012) 30(12):2212–9. doi: 10.1016/j.vaccine.2011.12.116
26. Dionne-Odom J, Njei B, Tita ATN. Elimination of vertical transmission of hepatitis b in Africa: A review of available tools and new opportunities. Clin Ther (2018) 40(8):1255–67. doi: 10.1016/j.clinthera.2018.05.016
27. Shi W, Zhang Z, Ling C, Zheng W, Zhu C, Carr MJ, et al. Hepatitis b virus subgenotyping: history, effects of recombination, misclassifications, and corrections. Infect Genet Evol (2013) 16:355–61. doi: 10.1016/j.meegid.2013.03.021
28. McMahon BJ. The natural history of chronic hepatitis b virus infection. Semin Liver Dis (2004) 24 Suppl 1:17–21. doi: 10.1055/s-2004-828674
29. Hoofnagle JH, Doo E, Liang TJ, Fleischer R, Lok AS. Management of hepatitis b: summary of a clinical research workshop. Hepatology (2007) 45(4):1056–75. doi: 10.1002/hep.21627
30. Spearman CW, Afihene M, Ally R, Apica B, Awuku Y, Cunha L, et al. Hepatitis b in sub-Saharan Africa: strategies to achieve the 2030 elimination targets. Lancet Gastroenterol Hepatol (2017) 2(12):900–9. doi: 10.1016/S2468-1253(17)30295-9
31. de Villiers MJ, Nayagam S, Hallett TB. The impact of the timely birth dose vaccine on the global elimination of hepatitis b. Nat Commun (2021) 12(1):6223. doi: 10.1038/s41467-021-26475-6
32. Quadri NS, Shah SM, Rodin H, Debes JD. Promoting hepatitis b awareness: Evaluating an educational approach through health care workers in Tanzania. Ann Glob Health (2021) 87(1):22. doi: 10.5334/aogh.3045
33. Lavanchy D. Hepatitis b virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J Viral Hepat (2004) 11(2):97–107. doi: 10.1046/j.1365-2893.2003.00487.x
34. Petrick JL, Florio AA, Koshiol J, Pfeiffer RM, Yang B, Yu K, et al. Prediagnostic concentrations of circulating bile acids and hepatocellular carcinoma risk: REVEAL-HBV and HCV studies. Int J Cancer (2020) 147(10):2743–53. doi: 10.1002/ijc.33051
35. Valery PC, Laversanne M, Clark PJ, Petrick JL, McGlynn KA, Bray F. Projections of primary liver cancer to 2030 in 30 countries worldwide. Hepatology (2018) 67(2):600–11. doi: 10.1002/hep.29498
36. Petrick JL, Florio AA, Znaor A, Ruggieri D, Laversanne M, Alvarez CS, et al. International trends in hepatocellular carcinoma incidence, 1978-2012. Int J Cancer (2020) 147(2):317–30. doi: 10.1002/ijc.32723
37. Golabi P, Fazel S, Otgonsuren M, Sayiner M, Locklear CT, Younossi ZM. Mortality assessment of patients with hepatocellular carcinoma according to underlying disease and treatment modalities. Med (Baltimore) (2017) 96(9):e5904. doi: 10.1097/MD.0000000000005904
38. Zhang X, El-Serag HB, Thrift AP. Sex and race disparities in the incidence of hepatocellular carcinoma in the united states examined through age-Period-Cohort analysis. Cancer Epidemiol Biomarkers Prev (2020) 29(1):88–94. doi: 10.1158/1055-9965.EPI-19-1052
39. Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology (2004) 127(5 Suppl 1):S35–50. doi: 10.1053/j.gastro.2004.09.014
40. Yang JD, Mohamed EA, Aziz AO, Shousha HI, Hashem MB, Nabeel MM, et al. Characteristics, management, and outcomes of patients with hepatocellular carcinoma in Africa: a multicountry observational study from the Africa liver cancer consortium. Lancet Gastroenterol Hepatol (2017) 2(2):103–11. doi: 10.1016/S2468-1253(16)30161-3
41. Rapti I, Hadziyannis S. Risk for hepatocellular carcinoma in the course of chronic hepatitis b virus infection and the protective effect of therapy with nucleos(t)ide analogues. World J Hepatol May 18 (2015) 7(8):1064–73. doi: 10.4254/wjh.v7.i8.1064
42. Tseng TC, Liu CJ, Yang HC, Su TH, Wang CC, Chen CL, et al. High levels of hepatitis b surface antigen increase risk of hepatocellular carcinoma in patients with low HBV load. Gastroenterology (2012) 142(5):1140–1149.e3. doi: 10.1053/j.gastro.2012.02.007
43. Brechot C, Pourcel C, Louise A, Rain B, Tiollais P. Presence of integrated hepatitis b virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nature (1980) 286(5772):533–5. doi: 10.1038/286533a0
44. Zhao LH, Liu X, Yan HX, Li WY, Zeng X, Yang Y, et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat Commun (2016) 7:12992. doi: 10.1038/ncomms12992
45. Brechot C. Pathogenesis of hepatitis b virus-related hepatocellular carcinoma: old and new paradigms. Gastroenterology (2004) 127(5 Suppl 1):S56–61. doi: 10.1053/j.gastro.2004.09.016
46. Ma NF, Lau SH, Hu L, Xie D, Wu J, Yang J, et al. COOH-terminal truncated HBV X protein plays key role in hepatocarcinogenesis. Clin Cancer Res (2008) 14(16):5061–8. doi: 10.1158/1078-0432.CCR-07-5082
47. Peng Z, Zhang Y, Gu W, Wang Z, Li D, Zhang F, et al. Integration of the hepatitis b virus X fragment in hepatocellular carcinoma and its effects on the expression of multiple molecules: a key to the cell cycle and apoptosis. Int J Oncol (2005) 26(2):467–73. doi: 10.3892/ijo.26.2.467
48. Ou J, Rutter WJ. Hybrid hepatitis b virus-host transcripts in a human hepatoma cell. Proc Natl Acad Sci USA (1985) 82(1):83–7. doi: 10.1073/pnas.82.1.83
49. Pollicino T, Caminiti G. HBV-integration studies in the clinic: Role in the natural history of infection. Viruses (2021) 13(3). doi: 10.3390/v13030368
50. Lee J, Tsai KN, Ou JJ. Mechanisms of hepatitis b virus-induced hepatocarcinogenesis. Recent Results Cancer Res (2021) 217:47–70. doi: 10.1007/978-3-030-57362-1_3
51. Zhu RX, Seto WK, Lai CL, Yuen MF. Epidemiology of hepatocellular carcinoma in the Asia-pacific region. Gut Liver (2016) 10(3):332–9. doi: 10.5009/gnl15257
52. Wang SH, Chen PJ, Yeh SH. Gender disparity in chronic hepatitis b: Mechanisms of sex hormones. J Gastroenterol Hepatol (2015) 30(8):1237–45. doi: 10.1111/jgh.12934
53. Sunbul M. Hepatitis b virus genotypes: global distribution and clinical importance. World J Gastroenterol (2014) 20(18):5427–34. doi: 10.3748/wjg.v20.i18.5427
54. Yang WJ, Chang CJ, Yeh SH, Lin WH, Wang SH, Tsai TF, et al. Hepatitis b virus X protein enhances the transcriptional activity of the androgen receptor through c-src and glycogen synthase kinase-3beta kinase pathways. Hepatology (2009) 49(5):1515–24. doi: 10.1002/hep.22833
55. World Health Organization. Regional office for Africa prevention, care and treatment of viral hepatitis in the African region: Framework for action, 2016 - 2020. Geneva: Licence: CC BY-NC-SA 3.0 IGO (2017).
56. Te HS, Jensen DM. Epidemiology of hepatitis b and c viruses: a global overview. Clin Liver Dis (2010) 14(1):1–21. doi: 10.1016/j.cld.2009.11.009
57. World Health Organization. Global hepatitis report, 2017. (2015) Global Hepatitis Programme Department of HIV/AIDS 20, avenue Appia 1211 Geneva 27 Switzerland. p. 83, ISBN: ISBN: 978-92-4-156545-5.
58. Chauhan RP, Dessie ZG, Noreddin A, El Zowalaty ME. Systematic review of important viral diseases in Africa in light of the ‘One health’ concept. Pathogens (2020) 9(4). doi: 10.3390/pathogens9040301
59. Breakwell L, Marke D, Kaiser R, Tejada-Strop A, Pauly MD, Jabbi S, et al. Assessing the impact of the routine childhood hepatitis b immunization program and the need for hepatitis b vaccine birth dose in Sierra Leone, 2018. Vaccine (2022) 40(19):2741–8. doi: 10.1016/j.vaccine.2022.03.049
60. Koblin BA, Taylor PE, Rubinstein P, Stevens CE. Effect of duration of hepatitis b virus infection on the association between human immunodeficiency virus type-1 and hepatitis b viral replication. Hepatology (1992) 15(4):590–2. doi: 10.1002/hep.1840150406
61. McGovern BH. The epidemiology, natural history and prevention of hepatitis b: implications of HIV coinfection. Antivir Ther (2007) 12 Suppl 3:H3–13. doi: 10.1177/135965350701203S02
62. Di Martino V, Thevenot T, Colin JF, Boyer N, Martinot M, Degos F, et al. Influence of HIV infection on the response to interferon therapy and the long-term outcome of chronic hepatitis b. Gastroenterology Dec (2002) 123(6):1812–22. doi: 10.1053/gast.2002.37061
63. Thio CL, Netski DM, Myung J, Seaberg EC, Thomas DL. Changes in hepatitis b virus DNA levels with acute HIV infection. Clin Infect Dis (2004) 38(7):1024–9. doi: 10.1086/382534
64. Salmon-Ceron D, Rosenthal E, Lewden C, Bouteloup V, May T, Burty C, et al. Emerging role of hepatocellular carcinoma among liver-related causes of deaths in HIV-infected patients: The French national mortalite 2005 study. J Hepatol (2009) 50(4):736–45. doi: 10.1016/j.jhep.2008.11.018
65. Hleyhel M, Hleyhel M, Bouvier AM, Belot A, Tattevin P, Pacanowski J, et al. Risk of non-AIDS-defining cancers among HIV-1-infected individuals in France between 1997 and 2009: results from a French cohort. AIDS (2014) 28(14):2109–18. doi: 10.1097/QAD.0000000000000382
66. Robbins HA, Shiels MS, Pfeiffer RM, Engels EA. Epidemiologic contributions to recent cancer trends among HIV-infected people in the united states. AIDS (2014) 28(6):881–90. doi: 10.1097/QAD.0000000000000163
67. Shiels MS, Engels EA. Evolving epidemiology of HIV-associated malignancies. Curr Opin HIV AIDS (2017) 12(1):6–11. doi: 10.1097/COH.0000000000000327
68. Joshi D, O’Grady J, Dieterich D, Gazzard B, Agarwal K. Increasing burden of liver disease in patients with HIV infection. Lancet (2011) 377(9772):1198–209. doi: 10.1016/S0140-6736(10)62001-6
69. Koziel MJ. The immunopathogenesis of HBV infection. Antivir Ther (1998) 3(Suppl 3):13–24. doi: 10.1177/135965359800301S01
70. Park JS, Saraf N, Dieterich DT. HBV plus HCV, HCV plus HIV, HBV plus HIV. Curr Gastroenterol Rep (2006) 8(1):67–74. doi: 10.1007/s11894-006-0066-9
71. Soriano V, Vispo E, Labarga P, Medrano J, Barreiro P. Viral hepatitis and HIV co-infection. Antiviral Res (2010) 85(1):303–15. doi: 10.1016/j.antiviral.2009.10.021
72. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in adults and adolescents with HIV. department of health and human services (2023) (Accessed 1/25/2023).
73. Kramvis A, Clements CJ. Implementing a birth dose of hepatitis b vaccine for home deliveries in Africa–too soon? Vaccine (2010) 28(39):6408–10. doi: 10.1016/j.vaccine.2010.07.042
74. Lavanchy D. Worldwide epidemiology of HBV infection, disease burden, and vaccine prevention. J Clin Virol Dec (2005) 34 Suppl 1:S1–3. doi: 10.1016/s1386-6532(05)00384-7
75. World Health Organization. Guidelines for the prevention, care and treatment of persons with chronic hepatitis b infection Geneva. Geneva: World Health Organization (2015), ISBN: ISBN-13: 978-92-4-154905-9.
76. Sonderup MW, Dusheiko G, Desalegn H, Lemoine M, Tzeuton C, Taylor-Robinson SD, et al. Hepatitis b in sub-Saharan Africa-how many patients need therapy? J Viral Hepat (2020) 27(6):560–7. doi: 10.1111/jvh.13247
77. Luma HN, Eloumou S, Noah DN, Eyenga BA, Nko'Ayissi G, Taku TS, et al. Hepatitis c continuum of care in a treatment center in Sub-Saharan Africa. J Clin Exp Hepatol (2018) 8(4):335–41. doi: 10.1016/j.jceh.2018.01.001
78. Boyer S, Baudoin M, Nishimwe ML, Santos M, Lemoine M, Maradan G, et al. Cost-utility analysis of four WHO-recommended sofosbuvir-based regimens for the treatment of chronic hepatitis c in sub-Saharan Africa. BMC Health Serv Res (2022) 22(1):303. doi: 10.1186/s12913-021-07289-0
79. Apata IW, Averhoff F, Pitman J, Bjork A, Yu J, Amin NA, et al. Progress toward prevention of transfusion-transmitted hepatitis b and hepatitis c infection–sub-Saharan Africa, 2000-2011. MMWR Morb Mortal Wkly Rep (2014) 63(29):613–9.
80. McGlynn KA, Petrick JL, London WT. Global epidemiology of hepatocellular carcinoma: an emphasis on demographic and regional variability. Clin Liver Dis (2015) 19(2):223–38. doi: 10.1016/j.cld.2015.01.001
81. M’Baya B, Jumbe V, Samuel V, M’Bwana R, Mangani C. Seroprevalence and trends in transfusion transmissible infections among voluntary non-remunerated blood donors at the Malawi blood transfusion service-a time trend study. Malawi Med J (2019) 31(2):118–25. doi: 10.4314/mmj.v31i2.3
82. Loua A, Nikiema JB, Sougou A, Kasilo OJM. Transfusion in the WHO African region. Transfus Clin Biol (2019) 26(3):155–9. doi: 10.1016/j.tracli.2019.06.191
83. Peliganga LB, Mello VM, de Sousa PSF, Horta MAP, Soares AD, da Silva Nunes JP, et al. Transfusion transmissible infections in blood donors in the province of bie, Angola, during a 15-year follow-up, imply the need for pathogen reduction technologies. Pathogens (2021) 10(12). doi: 10.3390/pathogens10121633
84. Pozzetto B, Bourlet T, Grattard F, Bonnevial L. Structure, genomic organization, replication and variability of hepatitis c virus. Nephrol Dial Transplant (1996) 11 Suppl 4:2–5. doi: 10.1093/ndt/11.supp4.2
85. Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, et al. Expanded classification of hepatitis c virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology Jan (2014) 59(1):318–27. doi: 10.1002/hep.26744
86. Simmonds P, Bukh J, Combet C, Deléage G, Enomoto N, Feinstone S, et al. Consensus proposals for a unified system of nomenclature of hepatitis c virus genotypes. Hepatology (2005) 42(4):962–73. doi: 10.1002/hep.20819
87. Davis C, Mgomella GS, da Silva Filipe A, Frost EH, Giroux G, Hughes J, et al. Highly diverse hepatitis c strains detected in Sub-Saharan Africa have unknown susceptibility to direct-acting antiviral treatments. Hepatology (2019) 69(4):1426–41. doi: 10.1002/hep.30342
88. Rosenberg BR, Freije CA, Imanaka N, Chen ST, Eitson JL, Caron R, et al. Genetic variation at IFNL4 influences extrahepatic interferon-stimulated gene expression in chronic HCV patients. J Infect Dis (2017) 217(4):650–5. doi: 10.1093/infdis/jix593
89. Meissner EG, Bon D, Prokunina-Olsson L, Tang W, Masur H, O'Brien TR, et al. IFNL4-ΔG genotype is associated with slower viral clearance in hepatitis c, genotype-1 patients treated with sofosbuvir and ribavirin. J Infect Dis (2014) 209(11):1700–4. doi: 10.1093/infdis/jit827
90. Riou J, Ait Ahmed M, Blake A, Vozlinsky S, Brichler S, Eholié S, et al. Hepatitis c virus seroprevalence in adults in Africa: a systematic review and meta-analysis. J Viral Hepat (2016) 23(4):244–55. doi: 10.1111/jvh.12481
91. Jefferies M, Rauff B, Rashid H, Lam T, Rafiq S. Update on global epidemiology of viral hepatitis and preventive strategies. World J Clin cases (2018) 6(13):589–99. doi: 10.12998/wjcc.v6.i13.589
92. Bigna JJ, Kenne AM, Hamroun A, Ndangang MS, Foka AJ, Tounouga DN, et al. Gender development and hepatitis b and c infections among pregnant women in Africa: a systematic review and meta-analysis. Infect Dis Poverty (2019) 8(1):16. doi: 10.1186/s40249-019-0526-8
93. Garozzo A, Falzone L, Rapisarda V, Marconi A, Cinà D, Fenga C, et al. The risk of HCV infection among health-care workers and its association with extrahepatic manifestations (Review). Mol Med Rep (2017) 15(5):3336–9. doi: 10.3892/mmr.2017.6378
94. Pozzetto B, Memmi M, Garraud O, Roblin X, Berthelot P. Health care-associated hepatitis c virus infection. World J Gastroenterol (2014) 20(46):17265–78. doi: 10.3748/wjg.v20.i46.17265
95. Dakurah OB, Tamandjou CRT, Zunza M, Preiser W, Maponga TG. Viral hepatitis associated hepatocellular carcinoma on the African continent, the past, present, and future: a systematic review. BMC Cancer (2021) 21(1):715. doi: 10.1186/s12885-021-08426-y
96. Yeung CY, Lee HC, Chan WT, Jiang CB, Chang SW, Chuang CK. Vertical transmission of hepatitis c virus: Current knowledge and perspectives. World J Hepatol Sep 27 (2014) 6(9):643–51. doi: 10.4254/wjh.v6.i9.643
97. Le Campion A, Larouche A, Fauteux-Daniel S, Soudeyns H. Pathogenesis of hepatitis c during pregnancy and childhood. Viruses (2012) 4(12):3531–50. doi: 10.3390/v4123531
98. Kanaan T, Liu A, Leroi M, Nanan R. A multicentre survey of hepatitis c awareness in a high-risk population. J Paediatr Child Health (2013) 49(8):649–53. doi: 10.1111/jpc.12259
99. Shiraki K, Ohto H, Inaba N, Fujisawa T, Tajiri H, Kanzaki S, et al. Guidelines for care of pregnant women carrying hepatitis c virus and their infants. Pediatr Int (2008) 50(1):138–40. doi: 10.1111/j.1442-200X.2007.02518.x
100. Sonderup MW, Afihene M, Ally R, Apica B, Awuku Y, Cunha L, et al. Hepatitis c in sub-Saharan Africa: the current status and recommendations for achieving elimination by 2030. Lancet Gastroenterol Hepatol (2017) 2(12):910–9. doi: 10.1016/S2468-1253(17)30249-2
101. Drug approval package: Harvoni (ledipasvir and sofosbuvir) tablets (2014) (Accessed 18 May 2022).
102. Lin L, Yan L, Liu Y, Qu C, Ni J, Li H. The burden and trends of primary liver cancer caused by specific etiologies from 1990 to 2017 at the global, regional, national, age, and sex level results from the global burden of disease study 2017. Liver Cancer (2020) 9(5):563–82. doi: 10.1159/000508568
103. Zakharia K, Luther CA, Alsabbak H, Roberts LR. Hepatocellular carcinoma: Epidemiology, pathogenesis and surveillance - implications for sub-Saharan Africa. S Afr Med J (2018) 108(8b):35–40. doi: 10.7196/SAMJ.2018.v108i8b.13499
104. Denniston MM, Jiles RB, Drobeniuc J, Ward JW, McQuillan GM, Holmberg SD. Chronic hepatitis c virus infection in the united states, national health and nutrition examination survey 2003 to 2010. Ann Intern Med (2014) 160(5):293–300. doi: 10.7326/M13-1133
105. Bartosch B, Thimme R, Blum HE, Zoulim F. Hepatitis c virus-induced hepatocarcinogenesis. J Hepatol (2009) 51(4):810–20. doi: 10.1016/j.jhep.2009.05.008
106. Cacoub P, Comarmond C, Domont F, Savey L, Desbois AC, Saadoun D. Extrahepatic manifestations of chronic hepatitis c virus infection. Ther Adv Infect Dis (2016) 3(1):3–14. doi: 10.1177/2049936115585942
107. Libra M, Polesel J, Russo AE, De Re V, Cinà D, Serraino D, et al. Extrahepatic disorders of HCV infection: a distinct entity of b-cell neoplasia? Int J Oncol (2010) 36(6):1331–40. doi: 10.3892/ijo_00000618
108. Stockdale AJ, Chaponda M, Beloukas A, Beloukas A, Phillips RO, Matthews PC, et al. Prevalence of hepatitis d virus infection in sub-Saharan Africa: a systematic review and meta-analysis. Lancet Glob Health (2017) 5(10):e992–e1003. doi: 10.1016/S2214-109X(17)30298-X
109. Amougou MA, Noah DN, Moundipa PF, Pineau P, Njouom R. A prominent role of hepatitis d virus in liver cancers documented in central Africa. BMC Infect Dis (2016) 16(1):647. doi: 10.1186/s12879-016-1992-2
110. Cooke GS, Andrieux-Meyer I, Applegate TL, Atun R, Burry JR, Cheinquer H. Accelerating the elimination of viral hepatitis: a lancet gastroenterology & hepatology commission. Lancet Gastroenterol Hepatol (2019) 4(2):135–84. doi: 10.1016/S2468-1253(18)30270-X
111. Ott JJ, Stevens GA, Wiersma ST. The risk of perinatal hepatitis b virus transmission: hepatitis b e antigen (HBeAg) prevalence estimates for all world regions. BMC Infect Dis (2012) 12:131. doi: 10.1186/1471-2334-12-131
112. Breakwell L, Tevi-Benissan C, Childs L, Mihigo R, Tohme R. The status of hepatitis b control in the African region. Pan Afr Med J (2017) 27(Suppl 3):17. doi: 10.11604/pamj.supp.2017.27.3.11981
113. Ott JJ, Horn J, Krause G, Mikolajczyk RT. Time trends of chronic HBV infection over prior decades - a global analysis. J Hepatol (2017) 66(1):48–54. doi: 10.1016/j.jhep.2016.08.013
114. Tamandjou CR, Maponga TG, Chotun N, Preiser W, Andersson MI. Is hepatitis b birth dose vaccine needed in Africa? Pan Afr Med J (2017) 27(Suppl 3):18. doi: 10.11604/pamj.supp.2017.27.3.11546
115. Chang MH, You SL, Chen CJ, Liu CJ, Lai MW, Wu TC, et al. Long-term effects of hepatitis b immunization of infants in preventing liver cancer. Gastroenterology (2016) 151(3):472–480 e1. doi: 10.1053/j.gastro.2016.05.048
116. Collins LF, Chan A, Zheng J, Chow SC, Wilder JM, Muir AJ, et al. Direct-acting antivirals improve access to care and cure for patients with HIV and chronic HCV infection. Open Forum Infect Dis (2018) 5(1):ofx264. doi: 10.1093/ofid/ofx264
117. Lyons MS, Kunnathur VA, Rouster SD, Hart KW, Sperling MI, Fichtenbaum CJ, et al. Prevalence of diagnosed and undiagnosed hepatitis c in a Midwestern urban emergency department. Clin Infect Dis (2016) 62(9):1066–71. doi: 10.1093/cid/ciw073
118. Shoukry NH. Hepatitis c vaccines, antibodies, and T cells. Front Immunol (2018) 9:1480. doi: 10.3389/fimmu.2018.01480
119. Kuo G, Choo QL, Alter HJ, Gitnick GL, Redeker AG, Purcell RH, et al. An assay for circulating antibodies to a major etiologic virus of human non-a, non-b hepatitis. Science (1989) 244(4902):362–4. doi: 10.1126/science.2496467
120. Choo QL, Kuo G, Ralston R, Weiner A, Chien D, Van Nest G, et al. Vaccination of chimpanzees against infection by the hepatitis c virus. Proc Natl Acad Sci U.S.A. (1994) 91(4):1294–8. doi: 10.1073/pnas.91.4.1294
121. Ralston R, Thudium K, Berger K, Kuo C, Gervase B, Hall J, et al. Characterization of hepatitis c virus envelope glycoprotein complexes expressed by recombinant vaccinia viruses. J Virol (1993) 67(11):6753–61. doi: 10.1128/JVI.67.11.6753-6761.1993
122. Zhao Q, He K, Zhang X, Xu M, Zhang X, Li H. Production and immunogenicity of different prophylactic vaccines for hepatitis c virus (Review). Exp Ther Med (2022) 24(1):474. doi: 10.3892/etm.2022.11401
123. Hartlage AS, Kapoor A. Hepatitis c virus vaccine research: Time to put up or shut up. Viruses (2021) 13(8). doi: 10.3390/v13081596
124. Frey SE, Houghton M, Coates S, Abrignani S, Chien D, Rosa D, et al. Safety and immunogenicity of HCV E1E2 vaccine adjuvanted with MF59 administered to healthy adults. Vaccine Aug 31 (2010) 28(38):6367–73. doi: 10.1016/j.vaccine.2010.06.084
125. Firbas C, Boehm T, Buerger V, Schuller E, Sabarth N, Jilma B, et al. Immunogenicity and safety of different injection routes and schedules of IC41, a hepatitis c virus (HCV) peptide vaccine. Vaccine Mar 11 (2010) 28(12):2397–407. doi: 10.1016/j.vaccine.2009.12.072
126. Elbahrawy A, Atalla H, Alboraie M, Alwassief A, Madian A, El Fayoumie M, et al. Recent advances in protective vaccines against hepatitis viruses: A narrative review. Viruses (2023) 15(1). doi: 10.3390/v15010214
127. Amiel C. Epstein-Barr Virus (EBV): Pathogenesis and diagnosis. Rev Francophone Des Laboratoires (2013) 456):47–55. doi: 10.1016/S1773-035X(13)72223-8
128. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from burkitt’s lymphoma. Lancet (1964) 1(7335):702–3. doi: 10.1016/s0140-6736(64)91524-7
129. Moghaddam A, Rosenzweig M, Lee-Parritz D, Annis B, Johnson RP, Wang F. An animal model for acute and persistent Epstein-Barr virus infection. Science (1997) 276(5321):2030–3. doi: 10.1126/science.276.5321.2030
130. Odumade OA, Hogquist KA, Balfour HH Jr. Progress and problems in understanding and managing primary Epstein-Barr virus infections. Clin Microbiol Rev (2011) 24(1):193–209. doi: 10.1128/CMR.00044-10
131. Parker BD, Bankier A, Satchwell S, Barrell B, Farrell PJ. Sequence and transcription of raji Epstein-Barr virus DNA spanning the B95-8 deletion region. Virology (1990) 179(1):339–46. doi: 10.1016/0042-6822(90)90302-8
132. Zeng L, Liu YP, Tao YG, Ai MD, Zhao XR, Cao Y. [Cross-talk between c-Jun/Ets1 involved in EB virus-encoded latent membrane protein 1 regulates expression of matrix metalloproteinase-9 in nasopharyngeal carcinoma]. Zhonghua Zhong Liu Za Zhi (2005) 27(4):204–8.
133. Dolan A, Addison C, Gatherer D, Davison AJ, McGeoch DJ. The genome of Epstein-Barr virus type 2 strain AG876. Virology (2006) 350(1):164–70. doi: 10.1016/j.virol.2006.01.015
134. Pizzo PA, Magrath IT, Chattopadhyay SK, Biggar RJ, Gerber P. A new tumour-derived transforming strain of Epstein-Barr virus. Nature (1978) 272(5654):629–31. doi: 10.1038/272629a0
135. Baer R, Bankier AT, Biggin MD, Deininger PL, Farrell PJ, Gibsonet TJ, et al. DNA Sequence and expression of the B95-8 Epstein-Barr virus genome. Nature (1984) 310(5974):207–11. doi: 10.1038/310207a0
136. Tonoyan L, Vincent-Bugnas S, Olivieri CV, Doglio A. New viral facets in oral diseases: The EBV paradox. Int J Mol Sci (2019) 20(23). doi: 10.3390/ijms20235861
137. Crawford DH. Biology and disease associations of Epstein-Barr virus. Philos Trans R Soc Lond B Biol Sci (2001) 356(1408):461–73. doi: 10.1098/rstb.2000.0783
138. Ogembo JG, Kannan L, Ghiran I, Nicholson-Weller A, Finberg RW, Tsokos GC, et al. Human complement receptor type 1/CD35 is an Epstein-Barr virus receptor. Cell Rep (2013) 3(2):371–85. doi: 10.1016/j.celrep.2013.01.023
139. Young LS, Rickinson AB. Epstein-Barr Virus: 40 years on. Nat Rev Cancer (2004) 4(10):757–68. doi: 10.1038/nrc1452
140. Cohen JI. Epstein-Barr Virus infection. N Engl J Med (2000) 343(7):481–92. doi: 10.1056/NEJM200008173430707
141. Saha A, Robertson ES. Epstein-Barr Virus-associated b-cell lymphomas: pathogenesis and clinical outcomes. Clin Cancer Res (2011) 17(10):3056–63. doi: 10.1158/1078-0432.CCR-10-2578
142. Hadinoto V, Shapiro M, Sun CC, Thorley-Lawson DA. The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PloS Pathog (2009) 5(7):e1000496. doi: 10.1371/journal.ppat.1000496
143. Edwards RH, Marquitz AR, Raab-Traub N. Epstein-Barr Virus BART microRNAs are produced from a large intron prior to splicing. J Virol (2008) 82(18):9094–106. doi: 10.1128/JVI.00785-08
144. Edwards RH, Marquitz AR, Raab-Traub N. Changes in expression induced by Epstein-Barr virus LMP1-CTAR1: Potential role of bcl3. mBio (2015) 6(2). doi: 10.1128/mBio.00441-15
145. Pratt ZL, Zhang J, Sugden B. The latent membrane protein 1 (LMP1) oncogene of Epstein-Barr virus can simultaneously induce and inhibit apoptosis in b cells. J Virol (2012) 86(8):4380–93. doi: 10.1128/JVI.06966-11
146. Eliopoulos AG, Young LS. LMP1 structure and signal transduction. Semin Cancer Biol (2001) 11(6):435–44. doi: 10.1006/scbi.2001.0410
147. Wang L, Ning S. New look of EBV LMP1 signaling landscape. Cancers (Basel) (2021) 13(21). doi: 10.3390/cancers13215451
148. Thorley-Lawson DA, Gross A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N Engl J Med Mar 25 (2004) 350(13):1328–37. doi: 10.1056/NEJMra032015
149. Countryman J, Miller G. Activation of expression of latent Epstein-Barr herpesvirus after gene transfer with a small cloned subfragment of heterogeneous viral DNA. Proc Natl Acad Sci U.S.A. (1985) 82(12):4085–9. doi: 10.1073/pnas.82.12.4085
150. Miller G, Heston L, Countryman J. P3HR-1 Epstein-Barr virus with heterogeneous DNA is an independent replicon maintained by cell-to-cell spread. J Virol Apr (1985) 54(1):45–52. doi: 10.1128/JVI.54.1.45-52.1985
151. Sinclair AJ, Brimmell M, Shanahan F, Farrell PJ. Pathways of activation of the Epstein-Barr virus productive cycle. J Virol (1991) 65(5):2237–44. doi: 10.1128/JVI.65.5.2237-2244.1991
152. Countryman J, Jenson H, Seibl R, Wolf H, Miller G. Polymorphic proteins encoded within BZLF1 of defective and standard Epstein-Barr viruses disrupt latency. J Virol (1987) 61(12):3672–9. doi: 10.1128/JVI.61.12.3672-3679.1987
153. Chavrier P, Gruffat H, Chevallier-Greco A, Buisson M, Sergeant A. The Epstein-Barr virus (EBV) early promoter DR contains a cis-acting element responsive to the EBV transactivator EB1 and an enhancer with constitutive and inducible activities. J Virol (1989) 63(2):607–14. doi: 10.1128/JVI.63.2.607-614.1989
154. Lieberman PM, Hardwick JM, Sample J, Hayward GS, Hayward SD. The zta transactivator involved in induction of lytic cycle gene expression in Epstein-Barr virus-infected lymphocytes binds to both AP-1 and ZRE sites in target promoter and enhancer regions. J Virol (1990) 64(3):1143–55. doi: 10.1128/JVI.64.3.1143-1155.1990
155. Germini D, Sall FB, Shmakova A, Wiels J, Dokudovskaya S, Drouet E, et al. Oncogenic properties of the EBV ZEBRA protein. Cancers (Basel) (2020) 12(6). doi: 10.3390/cancers12061479
156. Manet E, Gruffat H, Trescol-Biemont MC, Moreno N, Chambard P, Giot JF, et al. Epstein-Barr Virus bicistronic mRNAs generated by facultative splicing code for two transcriptional trans-activators. EMBO J (1989) 8(6):1819–26. doi: 10.1002/j.1460-2075.1989.tb03576.x
157. Furnari FB, Zacny V, Quinlivan EB, Kenney S, Pagano JS. RAZ, an Epstein-Barr virus transdominant repressor that modulates the viral reactivation mechanism. J Virol (1994) 68(3):1827–36. doi: 10.1128/JVI.68.3.1827-1836.1994
158. Segouffin C, Gruffat H, Sergeant A. Repression by RAZ of Epstein-Barr virus bZIP transcription factor EB1 is dimerization independent. J Gen Virol (1996) 77(Pt 7):1529–36. doi: 10.1099/0022-1317-77-7-1529
159. Hong Y, Jeong H, Park K, Lee S, Shim JY, Kim H, et al. STING facilitates nuclear import of herpesvirus genome during infection. Proc Natl Acad Sci U.S.A. (2021) 118(33). doi: 10.1073/pnas.2108631118
160. Biggin M, Bodescot M, Perricaudet M, Farrell P. Epstein-Barr Virus gene expression in P3HR1-superinfected raji cells. J Virol (1987) 61(10):3120–32. doi: 10.1128/JVI.61.10.3120-3132.1987
161. Dowd JB, Palermo T, Brite J, McDade TW, Aiello A. Seroprevalence of Epstein-Barr virus infection in U.S. children ages 6-19, 2003-2010. PloS One (2013) 8(5):e64921. doi: 10.1371/journal.pone.0064921
162. Burkitt D. A sarcoma involving the jaws in African children. Br J Surg (1958) 46(197):218–23. doi: 10.1002/bjs.18004619704
163. Epstein MA, Barr YM. Cultivation in vitro of human lymphoblasts from burkitt’s malignant lymphoma. Lancet (1964) 1(7327):252–3. doi: 10.1016/s0140-6736(64)92354-2
164. Redmond LS, Ogwang MD, Kerchan P, Kerchan P, Reynolds SJ, Tenge CN, et al. Endemic burkitt lymphoma: a complication of asymptomatic malaria in sub-Saharan Africa based on published literature and primary data from Uganda, Tanzania, and Kenya. Malar J (2020) 19(1):239. doi: 10.1186/s12936-020-03312-7
165. Saleh K, Michot JM, Camara-Clayette V, Vassetsky Y, Ribrag V. Burkitt and burkitt-like lymphomas: a systematic review. Curr Oncol Rep (2020) 22(4):33. doi: 10.1007/s11912-020-0898-8
166. Hammerl L, Colombet M, Rochford R, Ogwang DM, Parkin DM. The burden of burkitt lymphoma in Africa. Infect Agent Cancer (2019) 14:17. doi: 10.1186/s13027-019-0236-7
167. Wong Y, Meehan MT, Burrows SR, Doolan DL, Miles JJ. Estimating the global burden of Epstein-Barr virus-related cancers. J Cancer Res Clin Oncol (2022) 148(1):31–46. doi: 10.1007/s00432-021-03824-y
168. International Agency for Research on Cancer, World Health Organization. Hodgkin Lymphoma (2020). Cancer Fact Sheets (Accessed 30 June 2022).
169. Mpunga T, Clifford GM, Morgan EA, Milner DA Jr, de Martel C, Munyanshongore C, et al. Epstein-Barr Virus prevalence among subtypes of malignant lymphoma in Rwanda, 2012 to 2018. Int J Cancer (2022) 150(5):753–60. doi: 10.1002/ijc.33840
170. Ou Z, Chen Z, Zhao Y, Lu H, Liu W, Liet W, et al. Genetic signatures for lineage/sublineage classification of HPV16, 18, 52 and 58 variants. Virology (2021) 553:62–9. doi: 10.1016/j.virol.2020.11.003
171. Schiffman M, Rodriguez AC, Chen Z, Wacholder S, Herrero R, Hildesheim A, et al. A population-based prospective study of carcinogenic human papillomavirus variant lineages, viral persistence, and cervical neoplasia. Cancer Res (2010) 70(8):3159–69. doi: 10.1158/0008-5472.CAN-09-4179
172. Cohen JI, Fauci AS, Varmus H, Nabel GJ. Epstein-Barr Virus: an important vaccine target for cancer prevention. Sci Transl Med (2011) 3(107):107fs7. doi: 10.1126/scitranslmed.3002878
173. Henle G, Henle W, Diehl V. Relation of burkitt’s tumor-associated herpes-ytpe virus to infectious mononucleosis. Proc Natl Acad Sci U.S.A. (968) 59(1):94–101. doi: 10.1073/pnas.59.1.94
174. de-The G, Day NE, Geser A, Lavoué MF, Ho JH, Simons MJ, et al. Sero-epidemiology of the Epstein-Barr virus: preliminary analysis of an international study - a review. IARC Sci Publ (1971) (1975) 11 Pt 2):3–16.
175. Hossain A. Seroepidemiology of Epstein-Barr virus infections in a developing country. J Trop Pediatr (1987) 33(5):257–60. doi: 10.1093/tropej/33.5.257
176. Chang CM, Yu KJ, Mbulaiteye SM, Hildesheim A, Bhatia K. The extent of genetic diversity of Epstein-Barr virus and its geographic and disease patterns: a need for reappraisal. Virus Res (2009) 143(2):209–21. doi: 10.1016/j.virusres.2009.07.005
177. Walling DM, Brown AL, Etienne W, Keitel WA, Ling PD. Multiple Epstein-Barr virus infections in healthy individuals. J Virol (2003) 77(11):6546–50. doi: 10.1128/jvi.77.11.6546-6550.2003
178. Tsuchiya S. Diagnosis of Epstein-Barr virus-associated diseases. Crit Rev Oncol Hematol (2002) 44(3):227–38. doi: 10.1016/s1040-8428(02)00114-2
179. Macsween KF, Crawford DH. Epstein-Barr Virus-recent advances. Lancet Infect Dis (2003) 3(3):131–40. doi: 10.1016/s1473-3099(03)00543-7
180. Martorelli D, Muraro E, Merlo A, Merlo A, Turrini R, Faè DA, et al. Exploiting the interplay between innate and adaptive immunity to improve immunotherapeutic strategies for Epstein-barr-virus-driven disorders. Clin Dev Immunol (2012) 2012:931952. doi: 10.1155/2012/931952
181. Bakkalci D, Jia Y, Winter JR, Lewis JE, Taylor GS, Stagg HR. Risk factors for Epstein Barr virus-associated cancers: a systematic review, critical appraisal, and mapping of the epidemiological evidence. J Glob Health Jun (2020) 10(1):10405. doi: 10.7189/jogh.10.010405
182. Clifford P. Carcinogens in the nose and throat: nasopharyngeal carcinoma in Kenya. Proc R Soc Med (1972) 65(8):682–6. doi: 10.1177/003591577206500809
183. Proceedings of the IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Epstein-Barr Virus and kaposi’s sarcoma Herpesvirus/Human herpesvirus 8. IARC Monogr Eval Carcinog Risks Hum (1997) 70:1–492.
184. Gourzones C, Ferrand FR, Amiel C, Vérillaud B, Barat A, Guérin M, et al. Consistent high concentration of the viral microRNA BART17 in plasma samples from nasopharyngeal carcinoma patients–evidence of non-exosomal transport. Virol J (2013) 10:119. doi: 10.1186/1743-422X-10-119
185. Morrow RH Jr. Epidemiological evidence for the role of falciparum malaria in the pathogenesis of burkitt’s lymphoma. IARC Sci Publ (1985) 60:177–86. doi: 10.1136/jcp.39.3.349-b
186. Burkitt D. A children’s cancer dependent on climatic factors. Nature (1962) 194:232–4. doi: 10.1038/194232a0
187. Henle W, Diehl V, Kohn G, Zur Hausen H, Henle G. Herpes-type virus and chromosome marker in normal leukocytes after growth with irradiated burkitt cells. Science (1967) 157(3792):1064–5. doi: 10.1126/science.157.3792.1064
188. Epstein MA, Achong BG, Barr YM, Zajac B, Henle G, Henle W. Morphological and virological investigations on cultured burkitt tumor lymphoblasts (strain raji). J Natl Cancer Inst (1966) 37(4):547–59. doi: 10.1093/jnci/37.4.547
189. Magrath I. The pathogenesis of burkitt’s lymphoma. Adv Cancer Res (1990) 55:133–270. doi: 10.1016/s0065-230x(08)60470-4
190. Chene A, Donati D, Orem J, Mbidde ER, Kironde F, Wahlgren M, et al. Endemic burkitt’s lymphoma as a polymicrobial disease: new insights on the interaction between plasmodium falciparum and Epstein-Barr virus. Semin Cancer Biol (2009) 19(6):411–20. doi: 10.1016/j.semcancer.2009.10.002
191. Torgbor C, Awuah P, Deitsch K, Kalantari P, Duca KA, Thorley-Lawson DA. A multifactorial role for p. falciparum malaria in endemic burkitt’s lymphoma pathogenesis. PLoS Pathog (2014) 10(5):e1004170. doi: 10.1371/journal.ppat.1004170
192. Robbiani DF, Deroubaix S, Feldhahn N, Oliveira TY, Callen E, Wang Q, et al. Plasmodium infection promotes genomic instability and AID-dependent b cell lymphoma. Cell (2015) 162(4):727–37. doi: 10.1016/j.cell.2015.07.019
193. Thorley-Lawson DA, Allday MJ. The curious case of the tumour virus: 50 years of burkitt’s lymphoma. Nat Rev Microbiol (2008) 6(12):913–24. doi: 10.1038/nrmicro2015
194. Bollard CM, Cohen JI. How I treat T-cell chronic active Epstein-Barr virus disease. Blood (2018) 131(26):2899–905. doi: 10.1182/blood-2018-03-785931
195. Messick TE, Smith GR, Soldan SS, McDonnell ME, Deakyne JS, Malecka KA, et al. Structure-based design of small-molecule inhibitors of EBNA1 DNA binding blocks Epstein-Barr virus latent infection and tumor growth. Sci Transl Med (2019) 11(482). doi: 10.1126/scitranslmed.aau5612
196. Louis CU, Straathof K, Bollard CM, Ennamuri S, Gerken C, Lopez TT, et al. Adoptive transfer of EBV-specific T cells results in sustained clinical responses in patients with locoregional nasopharyngeal carcinoma. J Immunother (2010) 33(9):983–90. doi: 10.1097/CJI.0b013e3181f3cbf4
197. FDA U. HPV (human papillomavirus). FDA Office of Women’s Health, Food and Drug Administration Office of Women's Health, 10903 New Hampshire Ave WO32-2333, Silver Spring, MD (2017).
198. Graham SV. Human papillomavirus: gene expression, regulation and prospects for novel diagnostic methods and antiviral therapies. Future Microbiol (2010) 5(10):1493–506. doi: 10.2217/fmb.10.107
199. Gravitt PE, Winer RL. Natural history of HPV infection across the lifespan: Role of viral latency. Viruses (2017) 9(10). doi: 10.3390/v9100267
200. Ren S, Gaykalova DA, Guo T, Favorov AV, Fertig EJ, Tamayo P, et al. HPV E2, E4, E5 drive alternative carcinogenic pathways in HPV positive cancers. Oncogene (2020) 39(40):6327–39. doi: 10.1038/s41388-020-01431-8
201. Olmedo-Nieva L, Muñoz-Bello JO, Contreras-Paredes A, Lizano M. The role of E6 spliced isoforms (E6*) in human papillomavirus-induced carcinogenesis. Viruses (2018) 10(1). doi: 10.3390/v10010045
202. Stanley MA. Epithelial cell responses to infection with human papillomavirus. Clin Microbiol Rev (2012) 25(2):215–22. doi: 10.1128/CMR.05028-11
203. CDC. HPV infection. Division of STD Prevention, National Center for HIV, Viral Hepatitis, STD, and TB Prevention, Centers for Disease Control and Prevention (2022).
204. Garza-Rodríguez ML, Oyervides-Muñoz MA, Pérez-Maya AA, Sánchez-Domínguez CN, Berlanga-Garza A, Antonio-Macedo M, et al. Analysis of HPV integrations in Mexican pre-tumoral cervical lesions reveal centromere-enriched breakpoints and abundant unspecific HPV regions. Int J Mol Sci (2021) 22(6):3242. doi: 10.3390/ijms22063242
205. de Martel C, Plummer M, Vignat J, Franceschi S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int J Cancer (2017) 141(4):664–70. doi: 10.1002/ijc.30716
206. Poole DN, Tracy JK, Levitz L, Rochas M, Sangare K, Yekta S, et al. A cross-sectional study to assess HPV knowledge and HPV vaccine acceptability in Mali. PloS One (2013) 8(2):e56402. doi: 10.1371/journal.pone.0056402
207. Louie KS, de Sanjose S, Mayaud P. Epidemiology and prevention of human papillomavirus and cervical cancer in sub-Saharan Africa: a comprehensive review. Trop Med Int Health (2009) 14(10):1287–302. doi: 10.1111/j.1365-3156.2009.02372.x
208. De Vuyst H, Alemany L, Lacey C, Chibwesha CJ, Sahasrabuddhe V, Banura C, et al. The burden of human papillomavirus infections and related diseases in sub-saharan Africa. Vaccine (2013) 31 Suppl 5(0 5):F32–46. doi: 10.1016/j.vaccine.2012.07.092
209. Lekoane KMB, Kuupiel D, Mashamba-Thompson TP, Ginindza TG. Evidence on the prevalence, incidence, mortality and trends of human papilloma virus-associated cancers in sub-Saharan Africa: systematic scoping review. BMC Cancer (2019) 19(1):563. doi: 10.1186/s12885-019-5781-3
210. Mutombo AB, Benoy I, Tozin R, Bogers J, Geertruyden J-PV, Jacquemyn Y. Prevalence and distribution of human papillomavirus genotypes among women in Kinshasa, the democratic republic of the Congo. J Global Oncol (2019) 5):1–9. doi: 10.1200/jgo.19.00110
211. Okoye JO, Ofodile CA, Adeleke OK, Obioma O. Prevalence of high-risk HPV genotypes in sub-Saharan Africa according to HIV status: a 20-year systematic review. Epidemiol Health (2021) 43:e2021039. doi: 10.4178/epih.e2021039
212. Seyoum A, Assefa N, Gure T, Seyoum B, Mulu A, Mihret A. Prevalence and genotype distribution of high-risk human papillomavirus infection among Sub-Saharan African women: A systematic review and meta-analysis. review. Front Public Health (2022) 10:890880. doi: 10.3389/fpubh.2022.890880
213. Jary A, Teguete I, Sidibé Y, Kodio A, Dolo O, Burrel S, et al. Prevalence of cervical HPV infection, sexually transmitted infections and associated antimicrobial resistance in women attending cervical cancer screening in Mali. Int J Infect Dis (2021) 108:610–6. doi: 10.1016/j.ijid.2021.06.024
214. Araldi RP, Sant’Ana TA, Módolo DG, de Melo TC, Spadacci-Morena DD, de Cassia Stocco R, et al. The human papillomavirus (HPV)-related cancer biology: An overview. Biomedicine Pharmacother (2018) 106:1537–56. doi: 10.1016/j.biopha.2018.06.149
215. Vonsky M, Shabaeva M, Runov A, Lebedeva N, Chowdhury S, Palefsky JM, et al. Carcinogenesis associated with human papillomavirus infection. mechanisms and potential for immunotherapy. Biochem (Moscow) (2019) 84(7):782–99. doi: 10.1134/S0006297919070095
217. de Sanjose S, Quint WG, Alemany L, Geraets DT, Klaustermeier JE, Lloveras B, et al. Human papillomavirus genotype attribution in invasive cervical cancer: a retrospective cross-sectional worldwide study. Lancet Oncol (2010) 11(11):1048–56. doi: 10.1016/S1470-2045(10)70230-8
218. Serrano B, Alemany L, Tous S, Bruni L, Clifford GM, Weiss T, et al. Potential impact of a nine-valent vaccine in human papillomavirus related cervical disease. Infect Agent Cancer (2012) 7(1):38. doi: 10.1186/1750-9378-7-38
219. Munoz N, Bosch FX, de Sanjose S, Herrero R, Castellsagué X, Shah KV, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med (2003) 348(6):518–27. doi: 10.1056/NEJMoa021641
220. Koyalta D, Mboumba Bouassa RS, Maiga AI, Balde A, Bagendabanga JB, Alinity AA, et al. Correction: High prevalence of anal oncogenic human papillomavirus infection in young men who have sex with men living in Bamako, Mali. Infect Agent Cancer (2021) 16(1):54. doi: 10.1186/s13027-021-00394-z
221. Nyitray AG, Masunaga KIA, Nyoni J, Ross MW. Prevalence of and factors associated with anal high-risk human papillomavirus in urban Tanzanian men who have sex with men, 2011-2012. Int J STD AIDS (2022) 33(7):672–9. doi: 10.1177/09564624221094456
222. Perez-Gonzalez A, Cachay E, Ocampo A, Poveda E. Update on the epidemiological features and clinical implications of human papillomavirus infection (HPV) and human immunodeficiency virus (HIV) coinfection. Microorganisms (2022) 10(5). doi: 10.3390/microorganisms10051047
223. Camargo M, Del Rio-Ospina L, Soto-De Leon SC, Sánchez R, Pineda-Peña AC, Sussmann O, et al. Association of HIV status with infection by multiple HPV types. Trop Med Int Health (2018) 23(11):1259–68. doi: 10.1111/tmi.13142
224. Chowdhury S, Darragh TM, Berry-Lawhorn JM, Isaguliants MG, Vonsky MS, Hilton JF, et al. HPV type distribution in benign, high-grade squamous intraepithelial lesions and squamous cell cancers of the anus by HIV status. Cancers (2023) 15(3):660. doi: 10.3390/cancers15030660
225. Lekoane KMB, Kuupiel D, Mashamba-Thompson TP, Ginindza TG. The interplay of HIV and human papillomavirus-related cancers in sub-Saharan Africa: scoping review. Systematic Rev 2020/04/22 (2020) 9(1):88. doi: 10.1186/s13643-020-01354-1
226. Lubeya MK, Mwanahamuntu M, Chibwesha C, Mukosha M, Wamunyima MM, Kawonga M. Implementation strategies to increase human papillomavirus vaccination uptake for adolescent girls in sub-Saharan Africa: A scoping review protocol. PLoS One (2022) 17(8):e0267617. doi: 10.1371/journal.pone.0267617
227. Amponsah-Dacosta E, Kagina BM, Olivier J. Health systems constraints and facilitators of human papillomavirus immunization programmes in sub-Saharan Africa: a systematic review. Health Policy Planning (2020) 35(6):701–17. doi: 10.1093/heapol/czaa017
228. Asempah E. Cervical cancer prevalence in sub-Saharan Africa and HPV vaccination policy: A public health grand challenge? J Cancer Immunol (2021) 3(2):87–97. doi: 10.33696/cancerimmunol.3.043
229. Tracy L, Gaff HD, Burgess C, Sow S, Gravitt PE, Tracy JK. Estimating the impact of human papillomavirus (HPV) vaccination on HPV prevalence and cervical cancer incidence in Mali. Clin Infect Diseases (2011) 52(5):641–5. doi: 10.1093/cid/ciq190
231. Amponsah-Dacosta E, Blose N, Nkwinika VV, Chepkurui V. Human papillomavirus vaccination in south Africa: Programmatic challenges and opportunities for integration with other adolescent health services? Front Public Health (2022) 10:799984. doi: 10.3389/fpubh.2022.799984
232. Haque A, Kouriba B, Aïssatou N, Pant A. Eliminating cervical cancer in Mali and Senegal, two Sub-Saharan countries: Insights and optimizing solutions. Vaccines (Basel) (2020) 8(2). doi: 10.3390/vaccines8020181
233. Jeronimo J, Castle PE, Temin S, Denny L, Gupta V, Kim JJ, et al. Secondary prevention of cervical cancer: ASCO resource-stratified clinical practice guideline. J Global Oncol (2017) 3(5):635–57. doi: 10.1200/jgo.2016.006577
234. Shastri SS, Temin S, Almonte M, Basu P, Campos NG, Gravitt PE, et al. Secondary prevention of cervical cancer: ASCO resource–stratified guideline update. JCO Global Oncol (2022) 8):e2200217. doi: 10.1200/go.22.00217
235. Shanmugasundaram S, You J. Targeting persistent human papillomavirus infection. Viruses (2017) 9(8). doi: 10.3390/v9080229
236. Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. Detection and isolation of type c retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci U.S.A. (1980) 77(12):7415–9. doi: 10.1073/pnas.77.12.7415
237. Tagaya Y, Gallo RC. The exceptional oncogenicity of HTLV-1. Front Microbiol (2017) 8:1425. doi: 10.3389/fmicb.2017.01425
238. World Health Organization. Human T-lymphotropic virus type 1 Key facts. WHO: Geneva, Switzerland (2021).
239. Bangham CRM. Human T cell leukemia virus type 1: Persistence and pathogenesis. Annu Rev Immunol (2018) 36:43–71. doi: 10.1146/annurev-immunol-042617-053222
240. Matsuoka M, Jeang KT. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat Rev Cancer (2007) 7(4):270–80. doi: 10.1038/nrc2111
241. Matsuo M, Ueno T, Monde K, Sugata K, Tan BJY, Rahman A, et al. Identification and characterization of a novel enhancer in the HTLV-1 proviral genome. Nat Commun (2022) 13(1):2405. doi: 10.1038/s41467-022-30029-9
242. Araujo THA, Barreto FK, Menezes ADL, de Lima CPS, de Oliveira RS, Lemos PDS, et al. Complete genome sequence of human T-cell lymphotropic type 1 from patients with different clinical profiles, including infective dermatitis. Infect Genet Evol (2020) 79:104166. doi: 10.1016/j.meegid.2019.104166
243. Ernzen KJ, Panfil AR. Regulation of HTLV-1 transformation. Biosci Rep (2022) 42(3):BSR20211921. doi: 10.1042/BSR20211921
244. Giam CZ, Semmes OJ. HTLV-1 infection and adult T-cell Leukemia/Lymphoma-a tale of two proteins: Tax and HBZ. Viruses (2016) 8(6). doi: 10.3390/v8060161
245. Yoshida M. Multiple viral strategies of HTLV-1 for dysregulation of cell growth control. Annu Rev Immunol (2001) 19:475–96. doi: 10.1146/annurev.immunol.19.1.475
246. Grassmann R, Dengler C, Muller-Fleckenstein I, Fleckenstein B, McGuire K, Dokhelar MC, et al. Transformation to continuous growth of primary human T lymphocytes by human T-cell leukemia virus type I X-region genes transduced by a herpesvirus saimiri vector. Proc Natl Acad Sci U.S.A. (1989) 86(9):3351–5. doi: 10.1073/pnas.86.9.3351
247. Grassmann R, Berchtold S, Radant I, Alt M, Fleckenstein B, Sodroski JG, et al. Role of human T-cell leukemia virus type 1 X region proteins in immortalization of primary human lymphocytes in culture. J Virol (1992) 66(7):4570–5. doi: 10.1128/JVI.66.7.4570-4575.1992
248. Marriott SJ, Semmes OJ. Impact of HTLV-I tax on cell cycle progression and the cellular DNA damage repair response. Oncogene (2005) 24(39):5986–95. doi: 10.1038/sj.onc.1208976
249. Li XH, Gaynor RB. Mechanisms of NF-kappaB activation by the HTLV type 1 tax protein. AIDS Res Hum Retroviruses (2000) 16(16):1583–90. doi: 10.1089/08892220050192994
250. Harhaj EW, Harhaj NS, Grant C, Mostoller K, Alefantis T, Sun SC, et al. Human T cell leukemia virus type I tax activates CD40 gene expression via the NF-kappa b pathway. Virology (2005) 333(1):145–58. doi: 10.1016/j.virol.2004.12.008
251. Mori N, Fujii M, Cheng G, Ikeda S, Yamasaki Y, Yamada Y, et al. Human T-cell leukemia virus type I tax protein induces the expression of anti-apoptotic gene bcl-xL in human T-cells through nuclear factor-kappaB and c-AMP responsive element binding protein pathways. Virus Genes (2001) 22(3):279–87. doi: 10.1023/a:1011158021749
252. Azran I, Schavinsky-Khrapunsky Y, Aboud M. Role of tax protein in human T-cell leukemia virus type-I leukemogenicity. Retrovirology (2004) 1:20. doi: 10.1186/1742-4690-1-20
253. Zhao T, Matsuoka M. HBZ and its roles in HTLV-1 oncogenesis. Front Microbiol (2012) 3:247. doi: 10.3389/fmicb.2012.00247
254. Lemey P, Pybus OG, Van Dooren S, Vandamme AM. A Bayesian statistical analysis of human T-cell lymphotropic virus evolutionary rates. Infect Genet Evol (2005) 5(3):291–8. doi: 10.1016/j.meegid.2004.04.005
255. Afonso PV, Cassar O, Gessain A. Molecular epidemiology, genetic variability and evolution of HTLV-1 with special emphasis on African genotypes. Retrovirology (2019) 16(1):39. doi: 10.1186/s12977-019-0504-z
256. Zanella L, Riquelme I, Reyes ME, Tapia MJ, Reyes S, Mora B, et al. Phylogenetic dating analysis of HTLV-1 from Chile suggests transmissions events related to ancient migrations and contemporary expansion. Int J Infect Dis Oct (2020) 99:186–9. doi: 10.1016/j.ijid.2020.07.037
257. Yamashita M, Ido E, Miura T, Hayami M. Molecular epidemiology of HTLV-I in the world. J Acquir Immune Defic Syndr Hum Retrovirol (1996) 13 Suppl 1:S124–31. doi: 10.1097/00042560-199600001-00021
258. Gasmi M, Farouqi B, d’Incan M, Desgranges C. Long terminal repeat sequence analysis of HTLV type I molecular variants identified in four north African patients. AIDS Res Hum Retroviruses (1994) 10(10):1313–5. doi: 10.1089/aid.1994.10.1313
259. Van Dooren S, Gotuzzo E, Salemi M, Watts D, Audenaert E, Duwe S, et al. Evidence for a post-Columbian introduction of human T-cell lymphotropic virus [type I] [corrected] in Latin America. J Gen Virol (1998) 79(Pt 11):2695–708. doi: 10.1099/0022-1317-79-11-2695
260. Gessain A, Cassar O. Epidemiological aspects and world distribution of HTLV-1 infection. Front Microbiol (2012) 3:388. doi: 10.3389/fmicb.2012.00388
261. Verdonck K, Gonzalez E, Van Dooren S, Vandamme AM, Vanham G, Gotuzzo E. Human T-lymphotropic virus 1: recent knowledge about an ancient infection. Lancet Infect Dis (2007) 7(4):266–81. doi: 10.1016/S1473-3099(07)70081-6
262. Salemi M, Van Dooren S, Audenaert E, Delaporte E, Goubau P, Desmyter J, et al. Two new human T-lymphotropic virus type I phylogenetic subtypes in seroindeterminates, a mbuti pygmy and a gabonese, have closest relatives among African STLV-I strains. Virology (1998) 246(2):277–87. doi: 10.1006/viro.1998.9215
263. Mahieux R, Ibrahim F, Mauclere P, Herve V, Michel P, Tekaia F, et al. Molecular epidemiology of 58 new African human T-cell leukemia virus type 1 (HTLV-1) strains: identification of a new and distinct HTLV-1 molecular subtype in central Africa and in pygmies. J Virol Feb (1997) 71(2):1317–33. doi: 10.1128/JVI.71.2.1317-1333.1997
264. Proietti FA, Carneiro-Proietti AB, Catalan-Soares BC, Murphy EL. Global epidemiology of HTLV-I infection and associated diseases. Oncogene (2005) 24(39):6058–68. doi: 10.1038/sj.onc.1208968
265. de The G, Kazanji M. An HTLV-I/II vaccine: from animal models to clinical trials? J Acquir Immune Defic Syndr Hum Retrovirol (1996) 13 Suppl 1:S191–8. doi: 10.1097/00042560-199600001-00029
266. Wiktor SZ, Piot P, Mann JM, Nzilambi N, Francis H, Vercauteren G, et al. Human T cell lymphotropic virus type I (HTLV-I) among female prostitutes in Kinshasa, Zaire. J Infect Dis (1990) 161(6):1073–7. doi: 10.1093/infdis/161.6.1073
267. Delaporte E, Dupont A, Peeters M, Josse R, Merlin M, Schrijvers D, et al. Epidemiology of HTLV-I in Gabon (Western equatorial Africa). Int J Cancer (1988) 42(5):687–9. doi: 10.1002/ijc.2910420509
268. Yanagihara R, Jenkins CL, Alexander SS, Mora CA, Garruto RM. Human T lymphotropic virus type I infection in Papua new Guinea: high prevalence among the hagahai confirmed by western analysis. J Infect Dis (1990) 162(3):649–54. doi: 10.1093/infdis/162.3.649
269. Ngoma AM, Omokoko MD, Mutombo PB, Mvika ES, Muwonga JM, Nollet KE, et al. Population-based prevalence of human T-lymphotropic virus type 1 in sub-Saharan Africa: a systematic review and meta-analysis. Trop Med Int Health (2019) 24(11):1277–90. doi: 10.1111/tmi.13305
270. Ngoma AM, Omokoko MD, Mutombo PB, Nollet KE, Ohto H. Seroprevalence of human T-lymphotropic virus (HTLV) in blood donors in sub-Saharan Africa: a systematic review and meta-analysis. Vox Sang (2019) 114(5):413–25. doi: 10.1111/vox.12779
271. Armah HB, Narter-Olaga EG, Adjei AA, Asomaning K, Gyasi RK, Tettey Y. Seroprevalence of human T-cell lymphotropic virus type I among pregnant women in Accra, Ghana. J Med Microbiol (2006) 55(Pt 6):765–70. doi: 10.1099/jmm.0.46426-0
272. Dumas M, Houinato D, Verdier M, Zohoun T, Josse R, Bonis J, et al. Seroepidemiology of human T-cell lymphotropic virus type I/II in Benin (West Africa). AIDS Res Hum Retroviruses (1991) 7(5):447–51. doi: 10.1089/aid.1991.7.447
273. Collenberg E, Ouedraogo T, Ganame J, Fickenscher H, Kynast-Wolf G, Becher H, et al. Seroprevalence of six different viruses among pregnant women and blood donors in rural and urban Burkina Faso: A comparative analysis. J Med Virol (2006) 78(5):683–92. doi: 10.1002/jmv.20593
274. Fouchard N, Mahe A, Huerre M, Fraitag S, Valensi F, Macintyre E, et al. Cutaneous T cell lymphomas: mycosis fungoides, sezary syndrome and HTLV-i-associated adult T cell leukemia (ATL) in Mali, West Africa: a clinical, pathological and immunovirological study of 14 cases and a review of the African ATL cases. Leukemia (1998) 12(4):578–85. doi: 10.1038/sj.leu.2400956
275. Melbye M, Poulsen AG, Gallo D, Pedersen JB, Biggar RJ, Larsen O, et al. HTLV-1 infection in a population-based cohort of older persons in Guinea-Bissau, West Africa: risk factors and impact on survival. Int J Cancer (1998) 76(3):293–8. doi: 10.1002/(sici)1097-0215(19980504)76:3<293::aid-ijc1>3.0.co;2-q
276. Larsen O, Andersson S, da Silva Z, Hedegaard K, Sandström A, Nauclér A, et al. Prevalences of HTLV-1 infection and associated risk determinants in an urban population in Guinea-Bissau, West Africa. J Acquir Immune Defic Syndr (2000) 25(2):157–63. doi: 10.1097/00042560-200010010-00010
277. Mahieux R, Horal P, Mauclere P, Mercereau-Puijalon O, Guillotte M, Meertens L, et al. Human T-cell lymphotropic virus type 1 gag indeterminate western blot patterns in central Africa: relationship to plasmodium falciparum infection. J Clin Microbiol (2000) 38(11):4049–57. doi: 10.1128/JCM.38.11.4049-4057.2000
278. Elm J, Desowitz R, Diwan A. Serological cross-reactivities between the retroviruses HIV and HTLV-1 and the malaria parasite plasmodium falciparum. P N G Med J Mar (1998) 41(1):15–22. doi: 10.1097/01.qai.0000242465.50947.5f
279. Biggar RJ, Gigase PL, Melbye M, Kestens L, Sarin PS, Bodner AJ, et al. ELISA HTLV retrovirus antibody reactivity associated with malaria and immune complexes in healthy africans. Lancet Sep 7 (1985) 2(8454):520–3. doi: 10.1016/s0140-6736(85)90461-1
280. Khabbaz RF, Onorato IM, Cannon RO, Hartley TM, Roberts B, Hosein B, et al. Seroprevalence of HTLV-1 and HTLV-2 among intravenous drug users and persons in clinics for sexually transmitted diseases. N Engl J Med (1992) 326(6):375–80. doi: 10.1056/NEJM199202063260604
281. Murphy EL, Figueroa JP, Gibbs WN, Brathwaite A, Holding-Cobham M, Waters D, et al. Sexual transmission of human T-lymphotropic virus type I (HTLV-I). Ann Intern Med (1989) 111(7):555–60. doi: 10.7326/0003-4819-111-7-555
282. Manns A, Wilks RJ, Murphy EL, Haynes G, Figueroa JP, Barnett M, et al. A prospective study of transmission by transfusion of HTLV-I and risk factors associated with seroconversion. Int J Cancer (1992) 51(6):886–91. doi: 10.1002/ijc.2910510609
283. Donegan E, Lee H, Operskalski EA, Shaw GM, Kleinman SH, Busch MP, et al. Transfusion transmission of retroviruses: human T-lymphotropic virus types I and II compared with human immunodeficiency virus type 1. Transfusion (1994) 34(6):478–83. doi: 10.1046/j.1537-2995.1994.34694295061.x
284. Manns A, Hisada M, La Grenade L. Human T-lymphotropic virus type I infection. Lancet (1999) 353(9168):1951–8. doi: 10.1016/s0140-6736(98)09460-4
285. Wiktor SZ, Pate EJ, Rosenberg PS, Barnett M, Palmer P, Medeiros D, et al. Mother-to-child transmission of human T-cell lymphotropic virus type I associated with prolonged breast-feeding. J Hum Virol (1997) 1(1):37–44.
286. Shimizu K. High prevalence of HTLV-I infection among the family members of a patient with adult T-cell leukemia/lymphoma from northeastern Japan. Am J Hematol (1999) 61(1):78–81. doi: 10.1002/(sici)1096-8652(199905)61:1<78::aid-ajh13>3.0.co;2-w
287. Kashiwagi K, Furusyo N, Nakashima H, Kubo N, Kinukawa N, Kashiwagi S, et al. A decrease in mother-to-child transmission of human T lymphotropic virus type I (HTLV-I) in Okinawa, Japan. Am J Trop Med Hyg (2004) 70(2):158–63. doi: 10.4269/ajtmh.2004.70.158
288. Ando Y, Matsumoto Y, Nakano S, Saito K, Kakimoto K, Tanigawa T, et al. Long-term follow up study of vertical HTLV-I infection in children breast-fed by seropositive mothers. J Infect (2003) 46(3):177–9. doi: 10.1053/jinf.2002.1107
289. Hino S, Katamine S, Miyata H, Tsuji Y, Yamabe T, Miyamoto T. Primary prevention of HTLV-I in Japan. J Acquir Immune Defic Syndr Hum Retrovirol (1996) 13 Suppl 1:S199–203. doi: 10.1097/00042560-199600001-00030
290. Manns A, Miley WJ, Wilks RJ, Morgan OS, Hanchard B, Wharfe G, et al. Quantitative proviral DNA and antibody levels in the natural history of HTLV-I infection. J Infect Dis (1999) 180(5):1487–93. doi: 10.1086/315088
291. Shoeibi A, Etemadi M, Moghaddam Ahmadi A, Amini M, Boostani R. “HTLV-I infection” twenty-year research in neurology department of mashhad university of medical sciences. Iran J Basic Med Sci (2013) 16(3):202–7. doi: 10.4269/ajtmh.2004.70.158
292. Alvarez C, Gotuzzo E, Vandamme AM, Verdonck K. Family aggregation of human T-lymphotropic virus 1-associated diseases: A systematic review. Front Microbiol (2016) 7:1674. doi: 10.3389/fmicb.2016.01674
293. Kannian P, Green PL. Human T lymphotropic virus type 1 (HTLV-1): Molecular biology and oncogenesis. Viruses (2010) 2(9):2037–77. doi: 10.3390/v2092037
294. Goncalves DU, Proietti FA, Ribas JG, Araújo MG, Pinheiro SR, Guedes AC, et al. Epidemiology, treatment, and prevention of human T-cell leukemia virus type 1-associated diseases. Clin Microbiol Rev (2010) 23(3):577–89. doi: 10.1128/CMR.00063-09
295. Yamanashi H, Koyamatsu J, Nagayoshi M, Shimizu Y, Kawashiri SY, Kondo H, et al. Human T-cell leukemia virus-1 infection is associated with atherosclerosis as measured by carotid intima-media thickness in Japanese community-dwelling older people. Clin Infect Dis (2018) 67(2):291–4. doi: 10.1093/cid/ciy168
296. Karube K, Ohshima K, Tsuchiya T, Yamaguchi T, Kawano R, Suzumiya J, et al. Expression of FoxP3, a key molecule in CD4CD25 regulatory T cells, in adult T-cell leukaemia/lymphoma cells. Br J Haematol (2004) 126(1):81–4. doi: 10.1111/j.1365-2141.2004.04999.x
297. Chadburn A, Athan E, Wieczorek R, Knowles DM. Detection and characterization of human T-cell lymphotropic virus type I (HTLV-I) associated T-cell neoplasms in an HTLV-I nonendemic region by polymerase chain reaction. Blood (1991) 77(11):2419–30. doi: 10.1182/blood.V77.11.2419.2419
298. Jaffe ES, Blattner WA, Blayney DW, Bunn PA, Cossman J, Robert-Guroff M, et al. The pathologic spectrum of adult T-cell leukemia/lymphoma in the united states. Hum T-cell leukemia/lymphoma virus-associated lymphoid malignancies. Am J Surg Pathol (1984) 8(4):263–75. doi: 10.1097/00000478-198404000-00003
299. Katsuya H, Ishitsuka K, Utsunomiya A, Hanada S, Eto T, Moriuchi Y, et al. Treatment and survival among 1594 patients with ATL. Blood (2015) 126(24):2570–7. doi: 10.1182/blood-2015-03-632489
300. Oshiro A, Tagawa H, Ohshima K, Karube K, Uike N, Tashiro Y, et al. Identification of subtype-specific genomic alterations in aggressive adult T-cell leukemia/lymphoma. Blood (2006) 107(11):4500–7. doi: 10.1182/blood-2005-09-3801
301. Tsukasaki K, Tanosaki S, DeVos S, Hofmann WK, Wachsman W, Gombart AF, et al. Identifying progression-associated genes in adult T-cell leukemia/lymphoma by using oligonucleotide microarrays. Int J Cancer (2004) 109(6):875–81. doi: 10.1002/ijc.20028
302. Tsukasaki K, Hermine O, Bazarbachi A, Ratner L, Ramos JC, Harrington W Jr, et al. Definition, prognostic factors, treatment, and response criteria of adult T-cell leukemia-lymphoma: a proposal from an international consensus meeting. J Clin Oncol (2009) 27(3):453–9. doi: 10.1200/JCO.2008.18.2428
303. Cook LB, Fuji S, Hermine O, Bazarbachi A, Ramos JC, Ratner L, et al. Revised adult T-cell leukemia-lymphoma international consensus meeting report. J Clin Oncol (2019) 37(8):677–87. doi: 10.1200/JCO.18.00501
304. Mehta-Shah N, Ratner L, Horwitz SM. Adult T-cell Leukemia/Lymphoma. J Oncol Pract (2017) 13(8):487–92. doi: 10.1200/JOP.2017.021907
305. Iwanaga M, Watanabe T, Yamaguchi K. Adult T-cell leukemia: a review of epidemiological evidence. Front Microbiol (2012) 3:322. doi: 10.3389/fmicb.2012.00322
306. Percher F, Jeannin P, Martin-Latil S, Gessain A, Afonso PV, Vidy-Roche A, et al. Mother-to-Child transmission of HTLV-1 epidemiological aspects, mechanisms and determinants of mother-to-Child transmission. Viruses (2016) 8(2):40. doi: 10.3390/v8020040
307. Malik B, Taylor GP. Can we reduce the incidence of adult T-cell leukaemia/lymphoma? cost-effectiveness of human T-lymphotropic virus type 1 (HTLV-1) antenatal screening in the united kingdom. Br J Haematol (2019) 184(6):1040–3. doi: 10.1111/bjh.15234
308. Gessain A, Gout O. Chronic myelopathy associated with human T-lymphotropic virus type I (HTLV-I). Ann Intern Med (1992) 117(11):933–46. doi: 10.7326/0003-4819-117-11-933
309. Gotuzzo E, Cabrera J, Deza L, Verdonck K, Vandamme A-M, Cairampoma R, et al. Clinical characteristics of patients in Peru with human T cell lymphotropic virus type 1-associated tropical spastic paraparesis. Clin Infect Dis (2004) 39(7):939–44. doi: 10.1086/423957
310. Gessain A, Mahieux R. [A virus called HTLV-1. epidemiological aspects]. Presse Med (2000) 29(40):2233–9.
311. Bazarbachi A, Nasr R, El-Sabban ME, Mahé A, Mahieux R, Gessain A, et al. Evidence against a direct cytotoxic effect of alpha interferon and zidovudine in HTLV-I associated adult T cell leukemia/lymphoma. Leukemia (2000) 14(4):716–21. doi: 10.1038/sj.leu.2401742
312. Tu JJ, Maksimova V, Ratner L, Panfil AR. The past, present, and future of a human T-cell leukemia virus type 1 vaccine. Front Microbiol (2022) 13:897346. doi: 10.3389/fmicb.2022.897346
313. Iwanaga M, Watanabe T, Utsunomiya A, Okayama A, Uchimaru K, Koh KR, et al. Human T-cell leukemia virus type I (HTLV-1) proviral load and disease progression in asymptomatic HTLV-1 carriers: a nationwide prospective study in Japan. Blood (2010) 116(8):1211–9. doi: 10.1182/blood-2009-12-257410
314. Moorad R, Juarez A, Landis JT, Pluta LJ, Perkins M, Cheves A, et al. Whole-genome sequencing of kaposi sarcoma-associated herpesvirus (KSHV/HHV8) reveals evidence for two African lineages. Virology Mar (2022) 568:101–14. doi: 10.1016/j.virol.2022.01.005
315. Arias C, Weisburd B, Stern-Ginossar N, Mercier A, Madrid AS, Bellare P, et al. KSHV 2.0: a comprehensive annotation of the kaposi’s sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PloS Pathog (2014) 10(1):e1003847. doi: 10.1371/journal.ppat.1003847
316. de Oliveira Lopes A, Spitz N, Martinelli KG, de Paula AV, de Castro Conde Toscano AL, Braz-Silva PH, et al. Introduction of human gammaherpesvirus 8 genotypes a, b, and c into Brazil from multiple geographic regions. Virus Res (2020) 276:197828. doi: 10.1016/j.virusres.2019.197828
317. Lee BS, Connole M, Tang Z, Harris NL, Jung JU. Structural analysis of the kaposi’s sarcoma-associated herpesvirus K1 protein. J Virol (2003) 77(14):8072–86. doi: 10.1128/jvi.77.14.8072-8086.2003
318. Isaacs T, Abera AB, Muloiwa R, Katz AA, Todd G. Genetic diversity of HHV8 subtypes in south Africa: A5 subtype is associated with extensive disease in AIDS-KS. J Med Virol (2016) 88(2):292–303. doi: 10.1002/jmv.24328
319. Olp LN, Jeanniard A, Marimo C, West JT, Wood C. Whole-genome sequencing of kaposi’s sarcoma-associated herpesvirus from Zambian kaposi’s sarcoma biopsy specimens reveals unique viral diversity. J Virol (2015) 89(24):12299–308. doi: 10.1128/JVI.01712-15
320. Hayward GS, Zong JC. Modern evolutionary history of the human KSHV genome. Curr Top Microbiol Immunol (2007) 312:1–42. doi: 10.1007/978-3-540-34344-8_1
321. Poole LJ, Zong JC, Ciufo DM, Alcendor DJ, Cannon JS, Ambinder R, et al. Comparison of genetic variability at multiple loci across the genomes of the major subtypes of kaposi’s sarcoma-associated herpesvirus reveals evidence for recombination and for two distinct types of open reading frame K15 alleles at the right-hand end. J Virol (1999) 73(8):6646–60. doi: 10.1128/JVI.73.8.6646-6660.1999
322. Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, et al. Nucleotide sequence of the kaposi sarcoma-associated herpesvirus (HHV8). Proc Natl Acad Sci U.S.A. (1996) 93(25):14862–7. doi: 10.1073/pnas.93.25.14862
323. Sallah N, Palser AL, Watson SJ, Labo N, Asiki G, Marshall V, et al. Genome-wide sequence analysis of kaposi sarcoma-associated herpesvirus shows diversification driven by recombination. J Infect Dis (2018) 218(11):1700–10. doi: 10.1093/infdis/jiy427
324. Hosseinipour MC, Sweet KM, Xiong J, Namarika D, Mwafongo A, Nyirenda M, et al. Viral profiling identifies multiple subtypes of kaposi’s sarcoma. mBio (2014) 5(5):e01633–14. doi: 10.1128/mBio.01633-14
325. Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, et al. Lytic growth of kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med (1996) 2(3):342–6. doi: 10.1038/nm0396-342
326. Dittmer D, Lagunoff M, Renne R, Staskus K, Haase A, Ganem D. A cluster of latently expressed genes in kaposi’s sarcoma-associated herpesvirus. J Virol (1998) 72(10):8309–15. doi: 10.1128/JVI.72.10.8309-8315.1998
327. Kedes DH, Lagunoff M, Renne R, Ganem D. Identification of the gene encoding the major latency-associated nuclear antigen of the kaposi’s sarcoma-associated herpesvirus. J Clin Invest (1997) 100(10):2606–10. doi: 10.1172/JCI119804
328. Rainbow L, Platt GM, Simpson GR, Sarid R, Gao SJ, Stoiber H, et al. The 222- to 234-kilodalton latent nuclear protein (LNA) of kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) is encoded by orf73 and is a component of the latency-associated nuclear antigen. J Virol (1997) 71(8):5915–21. doi: 10.1128/JVI.71.8.5915-5921.1997
329. Toth Z, Papp B, Brulois K, Choi YJ, Gao SJ, Jung JU. LANA-mediated recruitment of host polycomb repressive complexes onto the KSHV genome during De novo infection. PloS Pathog (2016) 12(9):e1005878. doi: 10.1371/journal.ppat.1005878
330. Decker LL, Shankar P, Khan G, Freeman RB, Dezube BJ, Lieberman J, et al. The kaposi sarcoma-associated herpesvirus (KSHV) is present as an intact latent genome in KS tissue but replicates in the peripheral blood mononuclear cells of KS patients. J Exp Med (1996) 184(1):283–8. doi: 10.1084/jem.184.1.283
331. Broussard G, Damania B. Regulation of KSHV latency and lytic reactivation. Viruses (2020) 12(9):1034. doi: 10.3390/v12091034
332. Lu CC, Li Z, Chu CY, Feng J, Feng J, Sun R, et al. MicroRNAs encoded by kaposi’s sarcoma-associated herpesvirus regulate viral life cycle. EMBO Rep (2010) 11(10):784–90. doi: 10.1038/embor.2010.132
333. Wang SE, Wu FY, Fujimuro M, Zong J, Hayward SD, Hayward GS. Role of CCAAT/enhancer-binding protein alpha (C/EBPalpha) in activation of the kaposi’s sarcoma-associated herpesvirus (KSHV) lytic-cycle replication-associated protein (RAP) promoter in cooperation with the KSHV replication and transcription activator (RTA) and RAP. J Virol (2003) 77(1):600–23. doi: 10.1128/jvi.77.1.600-623.2003
334. Dalton-Griffin L, Wilson SJ, Kellam P. X-Box binding protein 1 contributes to induction of the kaposi’s sarcoma-associated herpesvirus lytic cycle under hypoxic conditions. J Virol Jul (2009) 83(14):7202–9. doi: 10.1128/JVI.00076-09
335. Zoeteweij JP, Moses AV, Rinderknecht AS, Davis DA, Overwijk WW, Yarchoan R, et al. Targeted inhibition of calcineurin signaling blocks calcium-dependent reactivation of kaposi sarcoma-associated herpesvirus. Blood Apr 15 (2001) 97(8):2374–80. doi: 10.1182/blood.v97.8.2374
336. Crabtree KL, Wojcicki JM, Minhas V, Kankasa C, Mitchell C, Wood C. Association of household food- and drink-sharing practices with human herpesvirus 8 seroconversion in a cohort of Zambian children. J Infect Dis (2017) 216(7):842–9. doi: 10.1093/infdis/jix399
337. Dedicoat M, Newton R. Review of the distribution of kaposi’s sarcoma-associated herpesvirus (KSHV) in Africa in relation to the incidence of kaposi’s sarcoma. Br J Cancer (2003) 88(1):1–3. doi: 10.1038/sj.bjc.6600745
338. Minhas V, Wood C. Epidemiology and transmission of kaposi’s sarcoma-associated herpesvirus. Viruses (2014) 6(11):4178–94. doi: 10.3390/v6114178
339. Plancoulaine S, Abel L, Tregouet D, Duprez R, van Beveren M, Tortevoye P, et al. Respective roles of serological status and blood specific antihuman herpesvirus 8 antibody levels in human herpesvirus 8 intrafamilial transmission in a highly endemic area. Cancer Res (2004) 64(23):8782–7. doi: 10.1158/0008-5472.CAN-04-2000
340. Mamimandjiami AI, Mouinga-Ondeme A, Ramassamy JL, Djuicy DD, Afonso PV, Mahé A, et al. Epidemiology and genetic variability of HHV-8/KSHV among rural populations and kaposi’s sarcoma patients in Gabon, central africa. review of the geographical distribution of HHV-8 K1 genotypes in Africa. Viruses (2021) 13(2):175. doi: 10.3390/v13020175
341. Lopes AO, Spitz N, Reis CRS, de Paula VS. Update of the global distribution of human gammaherpesvirus 8 genotypes. Sci Rep (2021) 11(1):7640. doi: 10.1038/s41598-021-87038-9
342. Tornesello ML, Biryahwaho B, Downing R, Hatzakis A, Alessi E, Cusini M, et al. Human herpesvirus type 8 variants circulating in Europe, Africa and north America in classic, endemic and epidemic kaposi’s sarcoma lesions during pre-AIDS and AIDS era. Virology (2010) 398(2):280–9. doi: 10.1016/j.virol.2009.12.005
343. Mancuso R, Biffi R, Valli M, Bellinvia M, Tourlaki A, Ferrucci S, et al. HHV8 a subtype is associated with rapidly evolving classic kaposi’s sarcoma. J Med Virol (2008) 80(12):2153–60. doi: 10.1002/jmv.21322
344. Marshall VA, Fisher NC, Goodman CA, Castro EMC, Liu I, Khanal S, et al. Systematic analysis of kaposi’s sarcoma (KS)-associated herpesvirus genomes from a KS case-control study in Cameroon: Evidence of dual infections but no association between viral sequence variation and KS risk. Int J Cancer (2022) 151(7):1127–41. doi: 10.1002/ijc.34136
345. Santiago JC, Goldman JD, Zhao H, Pankow AP, Okuku F, Schmitt MW, et al. Intra-host changes in kaposi sarcoma-associated herpesvirus genomes in Ugandan adults with kaposi sarcoma. PloS Pathog (2021) 17(1):e1008594. doi: 10.1371/journal.ppat.1008594
346. Fang Q, Liu Z, Zhang T. Human leukocyte antigen polymorphisms and kaposi’s sarcoma-associated herpesvirus infection outcomes: A call for deeper exploration. J Med Virol (2019) 91(4):541–8. doi: 10.1002/jmv.25342
347. Casper C, Redman M, Huang ML, Pauk J, Lampinen TM, Hawes SE, et al. HIV Infection and human herpesvirus-8 oral shedding among men who have sex with men. J Acquir Immune Defic Syndr (2004) 35(3):233–8. doi: 10.1097/00126334-200403010-00003
348. Dittmer DP, Damania B. Kaposi sarcoma-associated herpesvirus: immunobiology, oncogenesis, and therapy. J Clin Invest (2016) 126(9):3165–75. doi: 10.1172/JCI84418
349. Vega F, Miranda RN, Medeiros LJ. KSHV/HHV8-positive large b-cell lymphomas and associated diseases: a heterogeneous group of lymphoproliferative processes with significant clinicopathological overlap. Mod Pathol (2020) 33(1):18–28. doi: 10.1038/s41379-019-0365-y
350. Lambert M, Gannage M, Karras A, Abel M, Legendre C, Kerob D, et al. Differences in the frequency and function of HHV8-specific CD8 T cells between asymptomatic HHV8 infection and kaposi sarcoma. Blood Dec 1 (2006) 108(12):3871–80. doi: 10.1182/blood-2006-03-014225
351. Andreoni M, Sarmati L, Nicastri E, El Sawaf G, El Zalabani M, Uccella I, et al. Primary human herpesvirus 8 infection in immunocompetent children. JAMA (2002) 287(10):1295–300. doi: 10.1001/jama.287.10.1295
352. Casper C, Wald A, Pauk J, Tabet SR, Corey L, Celum CL. Correlates of prevalent and incident kaposi’s sarcoma-associated herpesvirus infection in men who have sex with men. J Infect Dis (2002) 185(7):990–3. doi: 10.1086/339605
353. Oksenhendler E, Cazals-Hatem D, Schulz TF, Barateau V, Grollet L, Sheldon J, et al. Transient angiolymphoid hyperplasia and kaposi’s sarcoma after primary infection with human herpesvirus 8 in a patient with human immunodeficiency virus infection. N Engl J Med (1998) 338(22):1585–90. doi: 10.1056/NEJM199805283382204
354. Lopes AO, Lima LRP, Tozetto-Mendoza TR, Martinelli KG, Morgado MG, Pilotto JH, et al. Low prevalence of human gammaherpesvirus 8 (HHV-8) infection among HIV-infected pregnant women in Rio de Janeiro, Brazil. J Matern Fetal Neonatal Med (2021) 34(20):3458–61. doi: 10.1080/14767058.2019.1685967
355. Dalla Pria A, Pinato DJ, Bracchi M, Bower M. Recent advances in HIV-associated kaposi sarcoma. F1000Res (2019) 8:F1000. doi: 10.12688/f1000research.17401.1
356. International Agency for Research on Cancer, World Health Organization. Kaposi sarcoma (2020) (Accessed 11 June 2022). Cancer Fact Sheets.
357. Schneider JW, Dittmer DP. Diagnosis and treatment of kaposi sarcoma. Am J Clin Dermatol (2017) 18(4):529–39. doi: 10.1007/s40257-017-0270-4
358. El-Mallawany NK, McAtee CL, Campbell LR, Kazembe PN. Pediatric kaposi sarcoma in context of the HIV epidemic in sub-Saharan Africa: current perspectives. Pediatr Health Med Ther (2018) 9:35–46. doi: 10.2147/PHMT.S142816
359. El-Mallawany NK, Villiera J, Kamiyango W, Peckham-Gregory EC, Scheurer ME, Allen CE, et al. Endemic kaposi sarcoma in HIV-negative children and adolescents: an evaluation of overlapping and distinct clinical features in comparison with HIV-related disease. Infect Agent Cancer (2018) 13:33. doi: 10.1186/s13027-018-0207-4
360. Iscovich J, Boffetta P, Winkelmann R, Brennan P, Azizi E. Classic kaposi’s sarcoma in jews living in Israel, 1961-1989: a population-based incidence study. AIDS (1998) 12(15):2067–72. doi: 10.1097/00002030-199815000-00019
361. Cook-Mozaffari P, Newton R, Beral V, Burkitt DP. The geographical distribution of kaposi’s sarcoma and of lymphomas in Africa before the AIDS epidemic. Br J Cancer Dec (1998) 78(11):1521–8. doi: 10.1038/bjc.1998.717
362. Taylor JF, Templeton AC, Vogel CL, Ziegler JL, Kyalwazi SK. Kaposi’s sarcoma in Uganda: a clinico-pathological study. Int J Cancer (1971) 8(1):122–35. doi: 10.1002/ijc.2910080116
363. Penn I. Kaposi’s sarcoma in transplant recipients. Transplantation (1997) 64(5):669–73. doi: 10.1097/00007890-199709150-00001
364. Litwin MA, Williams CM. Cutaneous pneumocystis carinii infection mimicking kaposi sarcoma. Ann Intern Med (1992) 117(1):48–9. doi: 10.7326/0003-4819-117-1-48
365. Nyirenda M, Ngongondo M, Kang M, Umbleja T, Krown SE, frey C, et al. Early progression and immune reconstitution inflammatory syndrome during treatment of mild-To-Moderate kaposi sarcoma in Sub-Saharan Africa and south America: Incidence, long-term outcomes, and effects of early chemotherapy. J Acquir Immune Defic Syndr (2020) 84(4):422–9. doi: 10.1097/QAI.0000000000002361
366. Volkow P, Cesarman-Maus G, Garciadiego-Fossas P, Rojas-Marin E, Cornejo-Juarez P. Clinical characteristics, predictors of immune reconstitution inflammatory syndrome and long-term prognosis in patients with kaposi sarcoma. AIDS Res Ther (2017) 14(1):30. doi: 10.1186/s12981-017-0156-9
367. Letang E, Lewis JJ, Bower M, Mosam A, Borok M, Campbell TB, et al. Immune reconstitution inflammatory syndrome associated with kaposi sarcoma: higher incidence and mortality in Africa than in the UK. AIDS (2013) 27(10):1603–13. doi: 10.1097/QAD.0b013e328360a5a1
368. Uldrick TS, Wang V, O’Mahony D, Aleman K, Wyvill KM, Marshall V, et al. An interleukin-6-related systemic inflammatory syndrome in patients co-infected with kaposi sarcoma-associated herpesvirus and HIV but without multicentric castleman disease. Clin Infect Dis (2010) 51(3):350–8. doi: 10.1086/654798
369. El-Mallawany NK, Kamiyango W, Villiera J, Peckham-Gregory EC, Scheurer ME, McAtee CL, et al. Kaposi sarcoma herpesvirus inflammatory cytokine syndrome-like clinical presentation in human immunodeficiency virus-infected children in Malawi. Clin Infect Dis (2019) 69(11):2022–5. doi: 10.1093/cid/ciz250
370. Carbone A, Borok M, Damania B, Gloghini A, Polizzotto MN, Jayanthan RK, et al. Castleman disease. Nat Rev Dis Primers (2021) 7(1):84. doi: 10.1038/s41572-021-00317-7
371. Casper C. The aetiology and management of castleman disease at 50 years: translating pathophysiology to patient care. Br J Haematol (2005) 129(1):3–17. doi: 10.1111/j.1365-2141.2004.05311.x
372. Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric castleman’s disease. Blood (1995) 86(4):1276–80. doi: 10.1182/blood.V86.4.1276.bloodjournal8641276
373. Dispenzieri A, Fajgenbaum DC. Overview of castleman disease. Blood Apr 16 (2020) 135(16):1353–64. doi: 10.1182/blood.2019000931
374. Lurain K, Yarchoan R, Uldrick TS. Treatment of kaposi sarcoma herpesvirus-associated multicentric castleman disease. Hematol Oncol Clin North Am (2018) 32(1):75–88. doi: 10.1016/j.hoc.2017.09.007
375. Engels EA, Mbulaiteye SM, Othieno E, Othieno E, Gomez M, Mathew S, et al. Kaposi sarcoma-associated herpesvirus in non-Hodgkin lymphoma and reactive lymphadenopathy in Uganda. Hum Pathol (2007) 38(2):308–14. doi: 10.1016/j.humpath.2006.08.009
376. Tomoka T, Painschab MS, Montgomery ND, Seguin R, Mulenga M, Kaimila B, et al. A prospective description of HIV-associated multicentric castleman disease in Malawi. Haematologica (2019) 104(5):e215–7. doi: 10.3324/haematol.2018.204479
377. Gopal S, Fedoriw Y, Montgomery ND, Kampani C, Krysiak R, Sanders MK, et al. Multicentric castleman’s disease in Malawi. Lancet (2014) 384(9948):1158. doi: 10.1016/S0140-6736(14)61366-0
378. Gopal S, Liomba NG, Montgomery ND, Moses A, Kaimila B, Nyasosela R, et al. Characteristics and survival for HIV-associated multicentric castleman disease in Malawi. J Int AIDS Soc (2015) 18(1):20122. doi: 10.7448/IAS.18.1.20122
379. Jones D, Ballestas ME, Kaye KM, Gulizia JM, Winters GL, Fletcher J, et al. Primary-effusion lymphoma and kaposi’s sarcoma in a cardiac-transplant recipient. N Engl J Med (1998) 339(7):444–9. doi: 10.1056/NEJM199808133390705
380. Klein U, Gloghini A, Gaidano G, Chadburn A, Cesarman E, Dalla-Favera R, et al. Gene expression profile analysis of AIDS-related primary effusion lymphoma (PEL) suggests a plasmablastic derivation and identifies PEL-specific transcripts. Blood (2003) 101(10):4115–21. doi: 10.1182/blood-2002-10-3090
381. Bigi R, Landis JT, An H, Caro-Vegas C, Raab-Traub N, Dittmer DP. Epstein-Barr Virus enhances genome maintenance of kaposi sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A. (2018) 115(48):E11379–87. doi: 10.1073/pnas.1810128115
382. Godfrey A, Anderson J, Papanastasiou A, Takeuchi Y, Boshoff C. Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin RNA. Blood (2005) 105(6):2510–8. doi: 10.1182/blood-2004-08-3052
383. Panaampon J, Kariya R, Okada S. Efficacy and mechanism of the anti-CD38 monoclonal antibody daratumumab against primary effusion lymphoma. Cancer Immunol Immunother (2022) 71(5):1017–31. doi: 10.1007/s00262-021-03054-8
384. Karcher DS, Alkan S. Human herpesvirus-8-associated body cavity-based lymphoma in human immunodeficiency virus-infected patients: a unique b-cell neoplasm. Hum Pathol (1997) 28(7):801–8. doi: 10.1016/s0046-8177(97)90153-2
385. Boulanger E, Daniel MT, Agbalika F, Oksenhendler E. Combined chemotherapy including high-dose methotrexate in KSHV/HHV8-associated primary effusion lymphoma. Am J Hematol (2003) 73(3):143–8. doi: 10.1002/ajh.10341
386. Dhungel BM, Montgomery ND, Painschab MS, Mulenga M, Tomoka T, Kaimila B, et al. ‘Discovering’ primary effusion lymphoma in Malawi. AIDS (2018) 32(15):2264–6. doi: 10.1097/QAD.0000000000001933
387. Zuze T, Ellis GK, Kasonkanji E, Kaimila B, Nyasosela R, Nyirenda R, et al. Modified EPOCH for high-risk non-Hodgkin lymphoma in sub-Saharan Africa. Cancer Med (2020) 9(1):77–83. doi: 10.1002/cam4.2631
388. Semeere A, Byakwaga H, Laker-Oketta M, Freeman E, Busakhala N, Wenger M, et al. Feasibility of rapid case ascertainment for cancer in East Africa: An investigation of community-representative kaposi sarcoma in the era of antiretroviral therapy. Cancer Epidemiol (2021) 74:101997. doi: 10.1016/j.canep.2021.101997
389. Freeman EE, Semeere A, McMahon DE, Byakwaga H, Laker-Oketta M, Regan S, et al. Beyond T staging in the “Treat-all” era: Severity and heterogeneity of kaposi sarcoma in East Africa. J Acquir Immune Defic Syndr (2021) 87(5):1119–27. doi: 10.1097/QAI.0000000000002699
390. Freeman EE, McCann NC, Semeere A, Reddy KP, Laker-Oketta M, Byakwaga H, et al. Evaluation of four chemotherapy regimens for treatment of advanced AIDS-associated kaposi sarcoma in Kenya: a cost-effectiveness analysis. Lancet Glob Health (2022) 10(8):e1179–88. doi: 10.1016/S2214-109X(22)00242-X
391. Ramaswami R, Lurain K, Yarchoan R. Oncologic treatment of HIV-associated kaposi sarcoma 40 years on. J Clin Oncol (2022) 40(3):294–306. doi: 10.1200/JCO.21.02040
392. Campbell LR, El-Mallawany NK, Slone JS, Malingoti BM, Mehta PS, Scheurer ME, et al. Clinical characteristics and successful treatment outcomes of children and adolescents with kaposi sarcoma in southwestern Tanzania. Pediatr Hematol Oncol (2022) 39(1):28–47. doi: 10.1080/08880018.2021.1936315
393. Casper C, Corey L, Cohen JI, Damania B, Gershon AA, Kaslow DC, et al. KSHV (HHV8) vaccine: promises and potential pitfalls for a new anti-cancer vaccine. NPJ Vaccines (2022) 7(1):108. doi: 10.1038/s41541-022-00535-4
394. Frankel AD, Young JA. HIV-1: fifteen proteins and an RNA. Annu Rev Biochem (1998) 67:1–25. doi: 10.1146/annurev.biochem.67.1.1
395. Li G, Piampongsant S, Faria NR, Voet A, Pineda-Peña AC, Khouri R, et al. An integrated map of HIV genome-wide variation from a population perspective. Retrovirology (2015) 12:18. doi: 10.1186/s12977-015-0148-6
396. Ren J, Bird LE, Chamberlain PP, Stewart-Jones GB, Stuart DI, Stammers DK. Structure of HIV-2 reverse transcriptase at 2.35-a resolution and the mechanism of resistance to non-nucleoside inhibitors. Proc Natl Acad Sci U.S.A. (2002) 99(22):14410–5. doi: 10.1073/pnas.222366699
397. Lau CY, Adan MA, Maldarelli F. Why the HIV reservoir never runs dry: Clonal expansion and the characteristics of HIV-infected cells challenge strategies to cure and control HIV infection. Viruses (2021) 13(12):2512. doi: 10.3390/v13122512
398. Campbell-Yesufu OT, Gandhi RT. Update on human immunodeficiency virus (HIV)-2 infection. Clin Infect Dis (2011) 52(6):780–7. doi: 10.1093/cid/ciq248
400. Dwyer-Lindgren L, Cork MA, Sligar A, Steuben KM, Wilson KF, Provost NR, et al. Mapping HIV prevalence in sub-Saharan Africa between 2000 and 2017. Nature (2019) 570(7760):189–93. doi: 10.1038/s41586-019-1200-9
402. Bulstra CA, Hontelez JAC, Giardina F, Steen R, Nagelkerke NJD, Bärnighausen T, et al. Mapping and characterising areas with high levels of HIV transmission in sub-Saharan Africa: A geospatial analysis of national survey data. PloS Med (2020) 17(3):e1003042. doi: 10.1371/journal.pmed.1003042
403. Ehrenkranz P, Rosen S, Boulle A, Eaton JW, Ford N, Fox MP, et al. The revolving door of HIV care: Revising the service delivery cascade to achieve the UNAIDS 95-95-95 goals. PloS Med (2021) 18(5):e1003651. doi: 10.1371/journal.pmed.1003651
404. Kharsany AB, Karim QA. HIV Infection and AIDS in Sub-Saharan Africa: Current status, challenges and opportunities. Open AIDS J (2016) 10:34–48. doi: 10.2174/1874613601610010034
405. Harris TG, Wu Y, Parmley LE, Musuka G, Mapingure MP, Chingombe I, et al. HIV Care cascade and associated factors among men who have sex with men, transgender women, and genderqueer individuals in Zimbabwe: findings from a biobehavioural survey using respondent-driven sampling. Lancet HIV (2022) 9(3):e182–201. doi: 10.1016/S2352-3018(21)00297-6
406. Irungu EM, Baeten JM. PrEP rollout in Africa: status and opportunity. Nat Med (2020) 26(5):655–64. doi: 10.1038/s41591-020-0872-x
407. Landovitz RJ, Donnell D, Clement ME, Hanscom B, Cottle L, Coelho L, et al. Cabotegravir for HIV prevention in cisgender men and transgender women. N Engl J Med (2021) 385(7):595–608. doi: 10.1056/NEJMoa2101016
408. Gottlieb GS, Raugi DN, Smith RA. 90-90-90 for HIV-2? ending the HIV-2 epidemic by enhancing care and clinical management of patients infected with HIV-2. Lancet HIV Jul (2018) 5(7):e390–9. doi: 10.1016/S2352-3018(18)30094-8
409. Ronchetti AM, Matheron S, Galicier L, Damond F, Mahjoub N, Chaghil N, et al. Lymphoma in HIV-2-infected patients in combination antiretroviral therapy era. AIDS (2021) 35(14):2299–309. doi: 10.1097/QAD.0000000000003015
410. Bouvresse S, Marcelin AG, Franck N, Regnier S, Damond F, Tulliez M, et al. The first reported case and management of multicentric castleman’s disease associated with kaposi’s sarcoma in an HIV-2-infected patient. AIDS Jul 11 (2007) 21(11):1492–4. doi: 10.1097/QAD.0b013e3281df33fc
411. Hawes SE, Critchlow CW, Faye Niang MA, Diouf MB, Diop A, Touré P, et al. Increased risk of high-grade cervical squamous intraepithelial lesions and invasive cervical cancer among African women with human immunodeficiency virus type 1 and 2 infections. J Infect Dis (2003) 188(4):555–63. doi: 10.1086/376996
412. Hawes SE, Critchlow CW, Sow PS, Touré P, N'Doye I, Diop A, et al. Incident high-grade squamous intraepithelial lesions in Senegalese women with and without human immunodeficiency virus type 1 (HIV-1) and HIV-2. J Natl Cancer Inst (2006) 98(2):100–9. doi: 10.1093/jnci/djj010
413. Langley CL, Benga-De E, Critchlow CW, Ndoye I, Mbengue-Ly MD, Kuypers J, et al. HIV-1, HIV-2, human papillomavirus infection and cervical neoplasia in high-risk African women. AIDS Apr (1996) 10(4):413–7. doi: 10.1097/00002030-199604000-00010
414. Ariyoshi K, Schim van der Loeff M, Cook P, Whitby D, Corrah T, Jaffar S, et al. Kaposi’s sarcoma in the Gambia, West Africa is less frequent in human immunodeficiency virus type 2 than in human immunodeficiency virus type 1 infection despite a high prevalence of human herpesvirus 8. J Hum Virol (1998) 1(3):193–9.
415. Colebunders R, De Vuyst H, Verstraeten T, Schroyens W, Van Marck E. A non-hodgkin’s lymphoma in a patient with HIV-2 infection. Genitourin Med (1995) 71(2):129. doi: 10.1136/sti.71.2.129
416. Kempf W, Margolin DH, Dezube BJ, Kadin ME, Letvin NL, Koralnik IJ. Clinicopathological characterization of an HIV-2-infected individual with two clonally unrelated primary lymphomas. Am J Hematol (2000) 65(4):302–6. doi: 10.1002/1096-8652(200012)65:4<302::aid-ajh9>3.0.co;2-4
417. Del Valle L, Pina-Oviedo S. HIV Disorders of the brain: pathology and pathogenesis. Front Biosci (2006) 11:718–32. doi: 10.2741/1830
418. Palefsky JM. Human papillomavirus-associated anal and cervical cancers in HIV-infected individuals: incidence and prevention in the antiretroviral therapy era. Curr Opin HIV AIDS Jan (2017) 12(1):26–30. doi: 10.1097/COH.0000000000000336
419. Pinato DJ, Allara E, Chen TY, Trevisani F, Minguez B, Zoli M, et al. Influence of HIV infection on the natural history of hepatocellular carcinoma: Results from a global multicohort study. J Clin Oncol (2019) 37(4):296–304. doi: 10.1200/JCO.18.00885
420. Yang X, Su B, Zhang X, Liu Y, Wu H, Zhang T. Incomplete immune reconstitution in HIV/AIDS patients on antiretroviral therapy: Challenges of immunological non-responders. J Leukoc Biol (2020) 107(4):597–612. doi: 10.1002/JLB.4MR1019-189R
421. Zicari S, Sessa L, Cotugno N, Ruggiero A, Morrocchi E, Concato C, et al. Immune activation, inflammation, and non-AIDS Co-morbidities in HIV-infected patients under long-term ART. Viruses (2019) 11(3):200. doi: 10.3390/v11030200
422. Vigano S, Bobisse S, Coukos G, Perreau M, Harari A. Cancer and HIV-1 infection: Patterns of chronic antigen exposure. Front Immunol (2020) 11:1350. doi: 10.3389/fimmu.2020.01350
423. Amu S, Lantto Graham R, Bekele Y, Nasi A, Bengtsson C, Rethi B, et al. Dysfunctional phenotypes of CD4+ and CD8+ T cells are comparable in patients initiating ART during early or chronic HIV-1 infection. Med (Baltimore) (2016) 95(23):e3738. doi: 10.1097/MD.0000000000003738
424. Amu S, Ruffin N, Rethi B, Chiodi F. Impairment of b-cell functions during HIV-1 infection. AIDS (2013) 27(15):2323–34. doi: 10.1097/QAD.0b013e328361a427
425. Nabatanzi R, Cose S, Joloba M, Jones SR, Nakanjako D. Effects of HIV infection and ART on phenotype and function of circulating monocytes, natural killer, and innate lymphoid cells. AIDS Res Ther (2018) 15(1):7. doi: 10.1186/s12981-018-0194-y
426. Castilho JL, Bian A, Jenkins CA, Shepherd BE, Sigel K, Gill MJ, et al. CD4/CD8 ratio and cancer risk among adults with HIV. J Natl Cancer Inst (2022) 114(6):854–62. doi: 10.1093/jnci/djac053
427. Lv T, Cao W, Li T. HIV-Related immune activation and inflammation: Current understanding and strategies. J Immunol Res (2021) 2021:7316456. doi: 10.1155/2021/7316456
428. Douek DC, Brenchley JM, Betts MR, Ambrozak DR, Hill BJ, Okamoto Y, et al. HIV Preferentially infects HIV-specific CD4+ T cells. Nature (2002) 417(6884):95–8. doi: 10.1038/417095a
429. Okoye IS, Houghton M, Tyrrell L, Barakat K, Elahi S. Coinhibitory receptor expression and immune checkpoint blockade: Maintaining a balance in CD8(+) T cell responses to chronic viral infections and cancer. Front Immunol (2017) 8:1215. doi: 10.3389/fimmu.2017.01215
430. Porichis F, Hart MG, Massa A, Everett HL, Morou A, Richard J, et al. Immune checkpoint blockade restores HIV-specific CD4 T cell help for NK cells. J Immunol (2018) 201(3):971–81. doi: 10.4049/jimmunol.1701551
431. Konopnicki D, Manigart Y, Gilles C, Barlow P, de Marchin J, Feoli F, et al. High-risk human papillomavirus infection in HIV-positive African women living in Europe. J Int AIDS Soc (2013) 16:18023. doi: 10.7448/IAS.16.1.18023
432. Hernandez DM, Valderrama S, Gualtero S, Hernández C, López M, Herrera MV, et al. Loss of T-cell multifunctionality and TCR-vbeta repertoire against Epstein-Barr virus is associated with worse prognosis and clinical parameters in HIV(+) patients. Front Immunol (2018) 9:2291. doi: 10.3389/fimmu.2018.02291
433. Ganesan A, Chattopadhyay PK, Brodie TM, Qin J, Gu W, Mascola JR, et al. Immunologic and virologic events in early HIV infection predict subsequent rate of progression. J Infect Dis (2010) 201(2):272–84. doi: 10.1086/649430
434. Hunt PW, Brenchley J, Sinclair E, McCune JM, Roland M, Page-Shafer K, et al. Relationship between T cell activation and CD4+ T cell count in HIV-seropositive individuals with undetectable plasma HIV RNA levels in the absence of therapy. J Infect Dis (2008) 197(1):126–33. doi: 10.1086/524143
435. Longenecker CT, Sullivan C, Baker JV. Immune activation and cardiovascular disease in chronic HIV infection. Curr Opin HIV AIDS (2016) 11(2):216–25. doi: 10.1097/COH.0000000000000227
436. Puronen CE, Ford ES, Uldrick TS. Immunotherapy in people with HIV and cancer. Front Immunol (2019) 10:2060. doi: 10.3389/fimmu.2019.02060
437. Isaguliants M, Bayurova E, Avdoshina D, Kondrashova A, Chiodi F, Palefsky JM. Oncogenic effects of HIV-1 proteins, mechanisms behind. Cancers (Basel) (2021) 13(2):305. doi: 10.3390/cancers13020305
438. Lin W, Weinberg EM, Chung RT. Pathogenesis of accelerated fibrosis in HIV/HCV co-infection. J Infect Dis (2013) 207 Suppl 1:S13–8. doi: 10.1093/infdis/jis926
439. Maldarelli F. The role of HIV integration in viral persistence: no more whistling past the proviral graveyard. J Clin Invest (2016) 126(2):438–47. doi: 10.1172/JCI80564
440. Wagner TA, McLaughlin S, Garg K, Cheung CYK, Larsen BB, Styrchak S, et al. HIV Latency. proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science (2014) 345(6196):570–3. doi: 10.1126/science.1256304
441. Desai P, Mencia-Trinchant N, Savenkov O, Simon MS, Cheang G, Lee S, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med (2018) 24(7):1015–23. doi: 10.1038/s41591-018-0081-z
442. Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med (2014) 371(26):2477–87. doi: 10.1056/NEJMoa1409405
443. Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med (2014) 20(12):1472–8. doi: 10.1038/nm.3733
444. Dharan NJ, Yeh P, Bloch M, Yeung MM, Baker D, Guinto J, et al. HIV Is associated with an increased risk of age-related clonal hematopoiesis among older adults. Nat Med (2021) 27(6):1006–11. doi: 10.1038/s41591-021-01357-y
445. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454(7203):436–44. doi: 10.1038/nature07205
446. Jiang G, Maverakis E, Cheng MY, Elsheikh MM, Deleage C, Méndez-Lagares G, et al. Disruption of latent HIV in vivo during the clearance of actinic keratosis by ingenol mebutate. JCI Insight (2019) 4(7):e126027. doi: 10.1172/jci.insight.126027
447. Uldrick TS, Adams SV, Fromentin R, Roche M, Fling SP, Gonçalves PH, et al. Pembrolizumab induces HIV latency reversal in people living with HIV and cancer on antiretroviral therapy. Sci Transl Med (2022) 14(629):eabl3836. doi: 10.1126/scitranslmed.abl3836
448. Chinula L, Moses A, Gopal S. HIV-Associated malignancies in sub-Saharan Africa: progress, challenges, and opportunities. Curr Opin HIV AIDS (2017) 12(1):89–95. doi: 10.1097/COH.0000000000000329
449. Lin LL, Lakomy DS, Chiao EY, Strother RM, Wirth M, Cesarman E, et al. Clinical trials for treatment and prevention of HIV-associated malignancies in Sub-Saharan Africa: Building capacity and overcoming barriers. JCO Glob Oncol (2020) 6:1134–46. doi: 10.1200/GO.20.00153
Keywords: virus, infection, malignancy, oncogenesis, Sub-Saharan Africa (SSA)
Citation: Diakite M, Shaw-Saliba K and Lau C-Y (2023) Malignancy and viral infections in Sub-Saharan Africa: A review. Front. Virol. 3:1103737. doi: 10.3389/fviro.2023.1103737
Received: 20 November 2022; Accepted: 13 February 2023;
Published: 06 March 2023.
Edited by:
Binod Kumar, Institute of Advanced Virology (IAV), IndiaReviewed by:
Corey Casper, Access to Advanced Health Institute, United StatesAlessandra Pierangeli, Sapienza University of Rome, Italy
Copyright © 2023 Diakite, Shaw-Saliba and Lau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mahamadou Diakite, bWRpYWtpdGVAaWNlcm1hbGkub3Jn